

Abstract
G protein-coupled receptors play essential roles in cellular processes such as neuronal signaling, vision, olfaction, tasting, and metabolism. As GPCRs are the most important drug targets, understanding their interactions with ligands is of utmost importance for discovering related new medicines. In many GPCRs, an allosteric sodium ion next to the highly conserved residue D2.50 has been proposed to stabilize the inactive receptor state by mediating interactions between transmembrane helices. Here, we probed the existence of internal and functionally important sodium ions in the dopamine D2 receptor, using molecular dynamics simulations. Besides a new sodium ion at the allosteric ligand binding site, we discovered an additional sodium ion, located close to the orthosteric ligand binding site. Through cell-based activation assays, the signaling of D2 receptor with site-specific mutations was tested against a series of chemically modified agonists. We concluded an important structural role of this newly discovered orthosteric sodium ion in modulating the receptor signaling: It enables the coordination of a polar residue in the ligand binding site with an appropriately designed agonist molecule. An identical interaction was also observed in a recently released high-resolution crystal structure of mu-opioid receptor, which was reresolved in this work. Probably because of similar interactions, various metal ions have been found to increase the signaling of many other GPCRs. This unique principle and strategy could be used to optimize the drug activity of GPCR. Our findings open a new mechanistic opportunity of GPCR signaling and help design the next generation of drugs targeting GPCRs.
Short abstract
A unique strategy was developed to optimize the drug activity of GPCR, which opens a new mechanistic opportunity of GPCR signaling and helps design the next generation of drugs.
Introduction
Na+ ions have been found to influence the functions of many members of class A G protein-coupled receptors (GPCRs), including adrenergic, dopaminergic, adenosine, opioid, and neurotensin receptors.1−9 For example, the binding of certain ligands to these receptors are known to be sensitive to sodium ions.1 For the case of dopamine D2 receptor (D2R), the binding of an agonist leads to the coupling of the receptor to Gi/o protein that in turn inhibits adenylyl cyclase. At physiological concentration, Na+ ions decrease the affinities of agonists including the endogenous agonist dopamine but enhance the affinities for some antagonists. The mutation D80A2.50 or D80E2.50 abolishes Na+ sensitivity. Several high-resolution crystal structures1,3,10,11 indicated that residues in the vicinity of D2.50 form a distinct Na+ mediated coordination network. As an example, in both 2.1 Å high-resolution adenosine A2AR and adrenergic β1AR crystal structures, the side chain oxygen atoms of D2.50 and S3.39 as well as three additional water molecules coordinate with this conserved Na+ ion.12 Such interactions have been proposed to stabilize the inactive state of GPCRs.1,13,14 This phenomenon was also found by computational simulations.15,16
Interestingly, Na+, as well as other metal ions, have been found elsewhere to modulate ligand binding to GPCRs at physiological conditions.13,17−27 However, the molecular basis for this unique observation is still unknown. Here, we investigated the molecular mechanism through which the Na+ ions influence the function of GPCRs. Starting from the recently resolved crystal structure of D2R (pdb: 6CM4),28 we investigated the allosteric interactions between Na+ and the agonist MLS1547 in the ligand binding pocket of D2R13,29 using all-atom molecular dynamics (MD) simulations.30 Surprisingly, our MD simulations revealed an additional Na+ ion in the ligand binding site next to extracellular loop 2 (ECL2). This second Na+ coordinates the interactions between residue H3936.55 and MLS1547 via two water molecules. Our findings were further confirmed by functional analysis of the interactions between the rationally designed synthetic analogues of MLS1547 and the mutants of D2R. A systematic analysis of high-resolution GPCR crystal structures also revealed the newly found coordination motif between Na+ and ligand in other GPCRs. Our findings provide new opportunities for designing novel drugs targeting GPCRs.
Results and Discussion
Allosteric Na+ Ion Next to D802.50 in apo D2R
The allosteric Na+ ion next to D2.50 has been observed in several crystal structures of GPCRs, and is considered to play an essential role in GPCR signaling.1,31 Although no allosteric Na+ ion is observed in the recently published 2.8 Å resolution D2R crystal structure (pdb: 6CM4),28 the signaling of D2R has been shown to depend critically on Na+ ions.20,23,32,33 This raises a question whether an allosteric Na+ ion next to D2.50 may actually be present in the D2R also, but the resolution of the mentioned crystal structure might be too low to detect the electron density of a sodium ion. To evaluate whether an allosteric Na+ ion could exist in D2R, we first performed 3 × 0.5 μs all-atom MD simulations for apo D2R, based on its crystal structure (pdb: 6CM4)28 (Figure 1). The initial D2R structure did not contain any intrinsic sodium ion, and the sodium ions were present in the extramembrane bulk medium for crystal growth. Interestingly, an allosteric Na+ ion was found in all three simulations (Figure 1A and Movie 1). During the initial 0.1 μs simulation period (Figure 1B), Na+ ions fluctuated in the extracellular bulk environment (zone I). During 0.1–0.2 μs time scale, a Na+ ion moved to D1143.32 and formed a transient ionic interaction. After about 0.2 μs, this Na+ ion diffused deeper into the receptor in the vicinity of D802.50 (Figure 1B) and was finally stabilized in this region until the end of the MD simulations. At this position the Na+ ion formed a stable coordination complex with the side chain oxygen atoms of D802.50 and S1213.39 of apo D2R together with three additional water molecules (Figure 1C). This feature is in good agreement with allosteric Na+ ions found in several high-resolution crystal structures of other GPCRs (Figure S1).
Figure 1.

Allosteric Na+ ion next to D802.50 in D2R. (A) Entrance pathway of a Na+ ion from the extracellular receptor interface toward the allosteric site D802.50 of apo D2R. Zone I (gray): extracellular receptor interface at which the Na+ ion is in the bulk environment. Zone II (red): transition zone in the vicinity of D1143.32. Zone III (blue): allosteric site in the vicinity of D802.50. Yellow spheres: Na+ ion positions during MD simulations. (B) Distance between allosteric Na+ ion and D802.50. (C) The final position of the allosteric Na+ ion at the end of MD simulations. The oxygen atoms in D802.50 and S1213.39 as well as those of three water molecules establish a dedicated coordination network with the allosteric Na+ ion.
A New Orthosteric Na+ Ion in the Extracellular Region of D2R
To study the role of Na+ ions within the interaction of a potent agonist, for example, MLS1547, and D2R at the atomic level, we first computationally constructed the D2R-MLS1547 complex by protein–ligand docking. The binding model was identical to that of the dopamine D3 receptor (D3R) crystal structure (pdb: 3PBL, Figure S2). Then, we submitted the D2R-MLS1547 complex to 3 × 4.0 μs all-atom MD simulations (Figure 2). MLS1547 was stable during MD simulations as indicated by its RMSD (root-mean-square deviation) (Figure S3). Because we observed also for this case the existence of the allosteric Na+ ion next to D802.50, we kept it in all subsequent MD simulations from the beginning, especially considering residue D2.50 is deprotonated at the initial activation stage of GPCRs.1,34 Interestingly, we found in these simulations a second Na+ ion captured in the orthosteric site next to the agonist molecule, forming coordination with both MLS1547 and D2R at 1.0–3.2 μs time scale (Figure 2). Quantum mechanics calculation indicated that the −OH group is deprotonated in the quinolin-8-ol moiety of MLS1547, due to the presence of the strong electronegative -Cl atom in the para position. Here, the nitrogen atom and the negatively charged oxygen established a coordination with this orthosteric Na+ ion. Moreover, H3936.55 together with two water molecules from the bulk environment formed coordination bonds with the Na+ ion (Figure 2C and Figure 3A).
Figure 2.

A second Na+ ion has been found in the orthosteric site of D2R using all-atom long-time MD simulations. (A) The entrance pathway of the orthosteric Na+ toward agonist molecule MLS1547. Yellow spheres: Na+ ion trajectories in the MD simulations. Black ball-and-stick: MLS1547. (B) The distance between orthosteric Na+ ion and H3936.55 (C) The final position of the orthosteric Na+ ion at the end of MD simulations. MLS1547 and H3936.55 as well as two water molecules establish a dedicated coordination network with the orthosteric Na+ ion.
Figure 3.

Interaction fingerprint between distinct residues in D2R and MLS1547. (A) The 2D interaction diagram of D2R in complex with MLS1547. Green ball-and-stick: MLS1547. (B) The interaction fingerprint between residues in D2R and MLS1547. It represents the interaction frequency during the MD simulations. (C) Arrestin recruitment for D2R variants showing mutations on key residues predicted by MD simulations. All mutated receptors show noticeably decreased signaling.
We then performed a protein–ligand interaction fingerprint (IFP) analysis on the generated MD simulation trajectories. IFP is a very useful method to quantify the interaction frequency between a protein and a ligand of interest.35−38 The analysis identifies which residues are most important for a specific protein–ligand interaction network. Besides the coordinating interaction with residue H3936.55, IFP analysis on the MD simulation trajectories indicated a few additional interactions of MLS1547 within the receptor including (1) face-to-side π–π stacking interactions with the two aromatic residues F3896.51 and F3906.52, (2) ionic interactions with D1143.32, and finally (3) hydrophobic interactions with the residues I183ECL2 and I184ECL2 in the ECL2 loop (Figure 3B).
To validate this newly discovered binding mode, we modified the D2R site specifically on the key residues indicated by the IFP analysis, and tested the activation of wildtype and mutant proteins by functional analyses. The EC50 value for wildtype receptor activation by MLS1547 was 630 nM for G protein signaling and 250 nM for arrestin signaling. Interestingly, all mutations had a substantial influence on the agonist induced receptor activation (Figure 3C and Table-1). Specifically, mutation variants I83ECL2A, L942.64A, D1143.32 A, F3896.51A, and F3906.52A completely abolished the activations of both G protein and arrestin. Interestingly, the mutation of H3936.55A led to MLS1547 completely inactive toward arrestin signaling, whereas it was still active against G protein signaling (EC50 = 350 nM). In contrast, mutant I84ECL2A was completely inactive toward G protein signaling but still had weak activity toward arrestin signaling (EC50 = 2500 nM). These results echoed the unique binding mode of MLS1547 in the D2R complex.
Table 1. Activities of Agonist MLS1547 against Different D2R variants (EC50, nM).
| WT | L94A | D114A | I83A | I84A | F389A | F390A | H393A | |
|---|---|---|---|---|---|---|---|---|
| G protein | 290 ± 90 | inactive | inactive | inactive | 9000 ± 7600 | inactive | inactive | inactive |
| β-arrestin | 250 ± 60 | inactive | inactive | inactive | 2500 ± 600 | inactive | inactive | inactive |
To further validate our findings, we performed biological testing of dopamine against D2R (Table-S1 and Figure S4). For this case, the interaction between the orthosteric Na+ and dopamine is missing, as indicated by docking (Figure S5). Previous studies found a similar binding mode for dopamine against D2R.39,40 These findings were confirmed by the observation that mutation H3936.55A does not show significant influences on dopamine’s activity: 970 nM (wildtype) vs 1002 nM (mutant).
In contrast, MLS1547 abolished its activity completely. The orthosteric Na+ ion coordinates agonist induced activation of D2R. As indicated in Figure 3A, a Na+ ion is involved in the interaction network of the agonist MLS1547 with residues in the orthosteric binding pocket of D2R and modulates the activation of the receptor. For the stability of the ligand–receptor complex, polar groups in the agonist molecule play a central role.
To further investigate how the chemical structure of the agonist determines the unique binding mode, we synthesized several derivatives of MLS1547 (Figure 4 and Table-S2). We first prepared 5-substituted quinolin-8-ol (see Materials and Methods). In the variant MLS-d1, a fluorine replaced the chlorine in MLS1547. The strong electronegativity of F decreased substantially the pKa value of the hydroxyl group of MLS1547 from 6.3 to 4.2 (Table-S2). Consequently, the group is deprotonated facilitating the interaction with the orthosteric Na+ ion (Figure 3A). This in turn improved the activation of the G protein with a decrease in EC50 values by almost 34 times, to 18.6 nM (Table-S2). Interestingly, the removal of Cl in MLS-d2 increased the pKa only slightly to 6.4 ± 0.7, but resulted also in an improved activation of G protein as shown by the EC50 values of 95 nM (Table-S2). We noticed that the activity of MLS-d2 is even slightly higher than MLS1547. Probably the presence of a -Cl might impose allosteric effects in the binding site as shown in Figure S6. Moreover, the relatively large -Cl is a little bit closer to the negatively charged D1143.32, inducing electrostatic repulsions. We also prepared MLS-d3, in which a bromine replaced the chlorine. In turn, the pKa value increased to 7.4 and the compound was completely inactive (MLS-d3, Table-S2). Moreover, introducing a nitro group in the para position of -OH decreased the pKa substantially to 3.4 (MLS-d4, Table-S2). However, the nitro group is too big to be accommodated in the binding pocket of D2R and therefore this compound did not show activation of G-protein and arrestin (Figure S6). Furthermore, one of the oxygen atoms in the nitro group was too close to the oxygen atom of D1143.32, which might lead to strong unfavorable electrostatic repulsion (Figure S6). In addition, we prepared compound MLS-d5, in which the N atom in quinolin-8-ol ring was replaced by a -CH group. The loss of coordination possibility led MLS-d5 to be completely inactive. Moreover, the estimated relative binding energies of MLS1547 variants correlate well with their EC50 values (Table-S2). This implies that MLS1547 and certain variants enhance the signaling by increasing the ligand binding through unique ion-coordinating interactions (Figures 2C and 3A).
Figure 4.
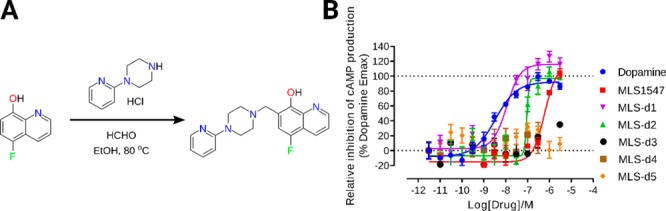
Synthesis and biological activity of MLS1547 derivatives. (A) Scheme for synthesis of compound MLS-d1. (B) Activation of D2R mediated signaling induced by dopamine, MLS1547- and MLS1547-derivatives.
An Allosteric Ion in the Crystal Structure of μ-Opioid Receptor and Potential Application in Other GPCRs
A recent high-resolution crystal structure of μ-opioid receptor (μOR, PDB: 5C1M) showed that there is an “unidentified electron density map” between agonist BU72 and a His residue of μOR.41 We tried filling this part of the electron density map with different ions, including Li, Na, Mg, Ni, and Zn. We found that only Mg fit perfectly to this region (Figures S7 and S8): the FO–FC electron density map was clean and the 2FO–FC map matched model very well, whereas all other ions always result in either negative Fo–Fc maps (ions are too large to fit in) or positive Fo–Fc maps (ions are too small to fit in). Specifically, the Mg coordinates with both the ligand BU72 and μOR, which is identical to the interactions in this work for D2R. More importantly, Mg was confirmed to enhance the signaling of μOR in other previous work.26,27 This further strongly supported that modulating the ligand binding via an orthosteric ion could be a universal strategy for GPCR ligand design.
To further validate whether similar interactions might happen in other GPCRs, a detailed inspection of family A GPCR structures shows that His is a frequent residue in TM6 of GPCRs (Figure S9). Considering that the ligand binding pocket of many family A GPCRs is very large, it is possible that a His in TM6 in other receptors might establish similar interaction networks as reported in the present work. Indeed, the signaling of many other GPCRs have been found to be enhanced by various metal ions elsewhere,15−21 probably because of the similar coordinating interactions as well.
Conclusion and Perspectives
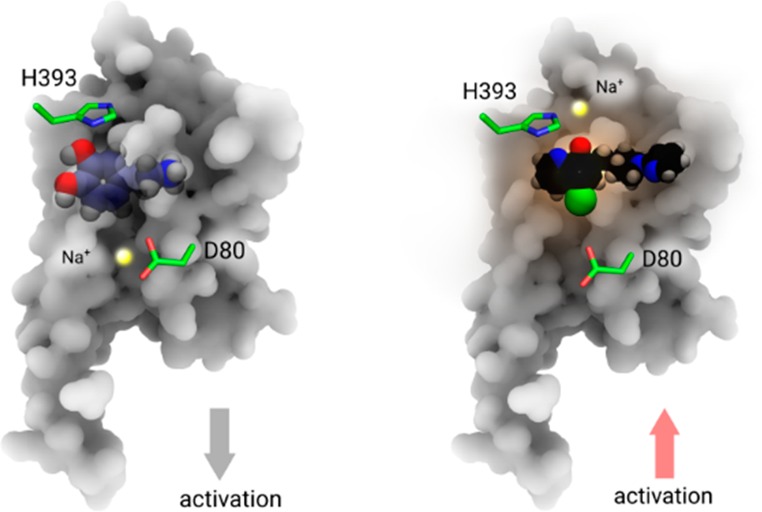
An allosteric sodium ion has been observed in several high-resolution GPCR structures including A2AR, D4R, β1AR, and others.1,3−5 The binding site of the allosteric sodium ion is located next to the highly conserved residue D2.50 in the inner space of the TM region in family A GPCRs. The specific coordination of the allosteric sodium ion strengthen links between different TM helices to enhance the stability of the inactive state of the particular receptors (Figure 5, left panel).1,2,14
Figure 5.
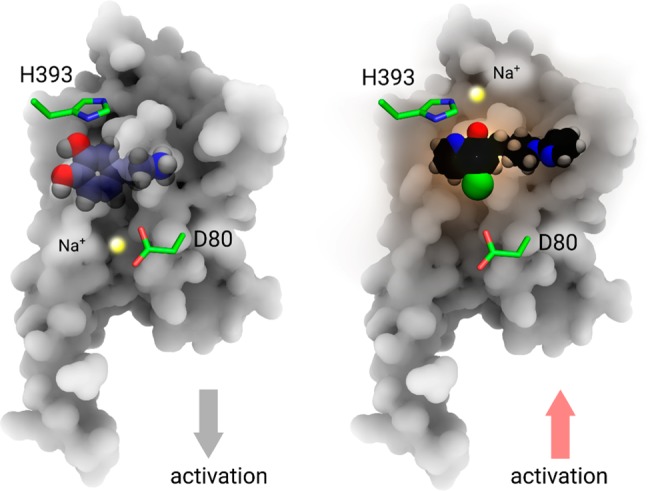
Two different roles of Na+ in D2 receptor activation. Left panel: an allosteric Na+ (yellow sphere) stabilizes the inactive state of receptor. Blue ball-and-sphere: dopamine molecule. Right panel: an orthosteric Na+ enhances the activation of receptor via a coordinating interaction with both H3936.55 and agonist molecule MLS1547 (black ball-and-stick).
Using all-atom MD simulations of apo-D2R, we have identified the potential existence of an allosteric Na+ ion next to D2.50, which resembles allosteric Na+ ions found in crystal structures of other GPCRs.1,2,14,32 For the case of the D2R in complex with an agonist, the MD simulations revealed in an additional Na+ ion located in the orthosteric ligand binding site in the extracellular region (Figure 5, right panel). Different from previous findings, this newly discovered Na+ enhances ligand binding by coordinating with both a His amino acid in TM6 of D2R and a negatively charged group in the ligand (Figure 2C and 3A), which in turn facilitates signaling of agonist molecules. Both site-specific mutagenesis experiments and structure–activity relationship (SAR) studies are in excellent agreement with our proposed model. The complementary site-specific mutations in the ligand binding pocket of the D2R and the properties of the modified agonists are also in agreement with the proposed role of allosteric and orthosteric Na+ ions for the activation of the D2R. An identical interaction was also observed in the high-resolution crystal structure of μOR by reresolving the structure.
Our findings reveal details of the role of sodium ions as well as other metal ions in GPCR signaling. These findings could be used to design unique GPCR drug molecules or to optimize ligand properties.
Materials and Methods
Tango Arrestin Recruitment Assay
The D2R-arrestin recruitment assays were performed as previously described.42 Briefly, HTLA cells expressing the TEV fused-β-arrestin2 were transfected with 1 μg D2R wild-type or mutant DNA per 10 cm dish in 10% dialyzed FBS DMEM. The next day, cells were plated into white 384-well plates at a density of 15 000 cells per well in 20 μL of 1% dialyzed FBS DMEM. After 6 h cells were stimulated with drugs ranging in concentration from 30 μM to 1 pM diluted in drug buffer (20 mM HEPES, 1X HBSS, 0.1% BSA, 0.01% ascorbic acid, pH 7.4) at a 3-fold concentration. After overnight incubation at 37 °C and 5% CO2, media was decanted and 20 μL of BriteGlo (Promega, after 1:20 dilution) was added per well. After 20 min, plates were read on an EnVision (PerkinElmer) at 1 ms per well. Luminescence counts per second (LCPS) were plotted as a function of drug concentration and analyzed using Graphpad Prism 8.0. Data were normalized to percent WT response, which was present in every experiment.
Split Luciferase Biosensor cAMP Assay for Measuring Activation of Gi Protein
To determine Gi-GPCR mediated cAMP production, we used Promega’s split luciferase based GloSensor cAMP biosensor technology. On the first day, 1 μg of target receptor DNA and 1 μg of GloSensor cAMP DNA were cotransfected into ATCC HEK 293T by Lipofectamine 2000. After a minimum incubation of 6 h, cells are seeded into 384-well white clear bottom cell culture plates with F-12 supplemented with 10% FBS at a density of 10–15 000 cells in 40 μL of medium per well. The assay was carried out after overnight incubation. On the day of the assay, supernatant was removed from the plates. Wells are loaded for 60 min at 37°C with 20 μL of 2 mg/mL luciferin prepared in HBSS, pH 7.4. All the following steps were carried out at room temperature. To measure agonist activity at D2R, 10 μL fo 4× test drug solution was added for 15 min before addition of 10 μL of forskolin at a final concentration of 20 μM, followed by counting of the plate for chemiluminescence after 15 min.
Organic Synthesis of Compounds
Experimental Procedures for Organic Synthesis
All commercial
chemicals and solvents were used as obtained from the manufacturer
without further purification. Flash chromatography were run on 200–300
mesh silica gel using a Teledyne CombiFlash instrument. 1H NMR spectra were recorded on a Bruker AVANCE-III spectrometer at
800 MHz. 13C NMR spectra were recorded on a Bruker AVANCE-III
spectrometer at 151 MHz. NMR chemical shifts were reported in δ
(ppm) using residual solvent peaks as standards (CDCl3–7.26
(H), 77.16 (C); CD3OD–3.31 (H), 49.00 (C); DMSO-d6–2.50 (H), 39.52 (C)). Mass spectra
were measured using an LCMS-IT-TOF (Shimadzu) mass spectrometer in
ESI mode. The purity of all final compounds (>95%) were determined
by analytical HPLC (Shim-pack GIST C18 column (250 × 4.6 mm,
particle size 5 μM); 0.05% TFA in H2O/0.05% TFA in
MeOH gradient eluting system; flow rate = 1.0 mL/min). Preparative
HPLC was conducted using Shimadzu HPLC system (Shim-pack GIST C18
column (250 × 20 mm, particle size 5 μM); H2O/MeOH gradient eluting system; flow rate = 10.0 mL/min).
5-Fluoro-7-((4-(pyridin-2-yl)piperazin-1-yl)methyl)quinolin-8-ol (MLS-d1)
To a mixture of compound a1 (300 mg, 1.84 mmol) in anhydrous EtOH (20 mL) was added 1-(pyridin-2-yl)piperazine hydrochloride b (550 mg, 2.76 mmol) and formaldehyde (37% aqueous solution, 0.30 mL, 4.05 mmol). The mixture was stirred at 80 °C for 12 h under N2. Then the pH of the mixture was adjusted to 8.0 by saturated aqueous NaHCO3. The mixture was then extracted with ethyl acetate (30 mL × 2). The combined extracts were washed with brine (10 mL × 2), dried over anhydrous Na2SO4, and concentrated in vacuum to give a residue. The residue was purified by silica gel chromatography (petroleum ether/ethyl acetate = 1/0 to 1/1) to obtain a yellow solid, which was stirred in hexane (2 mL) at room temperature for 2 h. Compound MLS-d1 (110 mg, 17.7% yield) was collected by filtration as a white solid. 1H NMR (800 MHz, CD3OD) δ 8.86 (d, J = 4.1 Hz, 1H), 8.43 (d, J = 8.5 Hz, 1H), 8.08 (d, J = 5.1 Hz, 1H), 7.60–7.53 (m, 2H), 7.22 (d, J = 10.4 Hz, 1H), 6.83 (d, J = 8.6 Hz, 1H), 6.68 (t, J = 6.1 Hz, 1H), 3.92 (s, 2H), 3.58 (t, J = 5.1 Hz, 4H), 2.72 (t, J = 5.1 Hz, 4H). 13C NMR (151 MHz, DMSO-d6) δ 159.0, 149.3, 149.2 (d, J = 243.3 Hz), 147.8 (d, J = 3.1 Hz), 147.6, 137.9 (d, J = 3.1 Hz), 137.5, 129.0, 121.9, 119.5 (d, J = 6.6 Hz), 117.5 (d, J = 18.2 Hz), 113.0, 111.9 (d, J = 20.3 Hz), 107.1, 55.9, 52.4 (2C), 44.7 (2C). HRMS (m/z): [M + H]+ calculated for C19H20FN4O+ 339.1616, found 339.1623.
7-((4-(Pyridin-2-yl)piperazin-1-yl)methyl)quinolin-8-ol (MLS-d2)
To a mixture of compound a2 (250 mg, 1.72 mmol) in anhydrous EtOH (20 mL) was added 1-(pyridin-2-yl)piperazine hydrochloride b (513.4 mg, 2.58 mmol) and formaldehyde (37% aqueous solution, 0.28 mL, 3.79 mmol). The reaction mixture was stirred at 80 °C for 12 h under N2. Then the pH of the mixture was adjusted to 8.0 with saturated aqueous NaHCO3. The mixture was then extracted with ethyl acetate (200 mL × 2). The combined organic extracts were washed with brine (100 mL × 2), dried over anhydrous Na2SO4, and concentrated in vacuum to give a residue, which was purified by preparative HPLC to give compound MLS-d2 (45.7 mg, 8.3% yield) as a gray solid. 1H NMR (800 MHz, CD3OD) δ 8.79 (d, J = 3.1 Hz, 1H), 8.24 (dd, J = 8.3, 1.6 Hz, 1H), 8.08 (dd, J = 5.1, 1.9 Hz, 1H), 7.57–7.54 (m, 1H), 7.48 (dd, J = 8.3, 4.1 Hz, 1H), 7.40 (d, J = 8.3 Hz, 1H), 7.37 (d, J = 8.3 Hz, 1H), 6.82 (d, J = 8.6 Hz, 1H), 6.68 (dd, J = 7.1, 5.0 Hz, 1H), 3.95 (s, 2H), 3.58 (t, J = 5.2 Hz, 4H), 2.73 (t, J = 5.0 Hz, 4H). 13C NMR (151 MHz, DMSO-d6) δ 159.0, 151.3, 148.2, 147.6, 138.2, 137.5, 136.0, 129.1, 127.7, 121.6, 119.7, 117.0, 113.0, 107.1, 56.3, 52.4 (2C), 44.7 (2C). HRMS (m/z): [M + H]+ calculated for C19H21N4O+ 321.1710, found 321.1713.
5-Bromo-7-((4-(pyridin-2-yl)piperazin-1-yl)methyl)quinolin-8-ol (MLS-d3)
To a mixture of a3 (250 mg, 1.12 mmol) in anhydrous EtOH (20 mL) was added 1-(pyridin-2-yl)piperazine hydrochloride b (334 mg, 1.68 mmol) and formaldehyde (37% aqueous solution, 0.18 mL, 2.45 mmol). The mixture was stirred at 80 °C for 12 h under N2. Then the pH was adjusted to 8.0 by saturated aqueous NaHCO3. The mixture was then extracted with ethyl acetate (20 mL × 2). The combined extracts were washed with brine (10 mL × 2), dried over anhydrous Na2SO4, and concentrated in vacuum to give a residue, which was purified by preparative HPLC to give compound MLS-d3 (21.0 mg, 5.8% yield) as a yellow solid. 1H NMR (800 MHz, CD3OD) δ 8.85 (s, 1H), 8.51 (dd, J = 8.5, 1.5 Hz, 1H), 8.08 (dd, J = 5.1, 1.4 Hz, 1H), 7.77 (s, 1H), 7.63 (dd, J = 8.5, 4.2 Hz, 1H), 7.58–7.53 (m, 1H), 6.83 (d, J = 8.6 Hz, 1H), 6.68 (dd, J = 7.1, 5.0 Hz, 1H), 3.93 (s, 2H), 3.58 (t, J = 5.1 Hz, 4H), 2.73 (t, J = 5.1 Hz, 4H). 13C NMR (151 MHz, DMSO-d6) δ 159.0, 151.6, 149.0, 147.6, 139.1, 137.5, 134.9, 132.2, 126.2, 123.1, 121.3, 113.0, 108.1, 107.1, 55.6, 52.4 (2C), 44.7 (2C). HRMS (m/z): [M + H]+ calculated for C19H20BrN4O+ 399.0815, found 399.0827.
5-Nitro-7-((4-(pyridin-2-yl)piperazin-1-yl)methyl)quinolin-8-ol (MLS-d4)
To a mixture of 1-(pyridin-2-yl)piperazine hydrochloride b (0.88 g, 4.41 mmol) in pyridine (3.0 mL) and H2O (3.0 mL) was added formaldehyde (37% aqueous solution, 0.36 mL, 4.41 mmol) at 50 °C. The reaction mixture was stirred at the same temperature for 5 min, and then added to a solution of a4 (400 mg, 2.10 mmol) in pyridine (10.0 mL) at 50 °C under N2. The reaction mixture was stirred at 50 °C for 2 h, and then ethyl acetate (20 mL) was added at room temperature to give a yellow solid, which was collected by filtration and recrystallized from ethanol (20 mL) to give compound MLS-d4 (100 mg, 13.0% yield) as a yellow solid. 1H NMR (800 MHz, DMSO-d6) δ 9.19 (d, J = 8.6 Hz, 1H), 8.75 (d, J = 3.2 Hz, 1H), 8.58 (s, 1H), 8.13 (dd, J = 4.7, 1.4 Hz, 1H), 7.67 (dd, J = 8.7, 4.0 Hz, 1H), 7.58–7.53 (m, 1H), 6.87 (d, J = 8.6 Hz, 1H), 6.68 (dd, J = 6.9, 5.0 Hz, 1H), 4.06 (s, 2H), 3.70 (t, J = 5.0 Hz, 4H), 3.02 (t, J = 5.0 Hz, 4H). 13C NMR (151 MHz, DMSO-d6) δ 169.3, 158.5, 147.6, 147.0, 139.9, 137.8, 132.7, 132.2, 127.1, 124.5, 124.4, 115.6, 113.6, 107.5, 56.1, 51.7 (2C), 43.1 (2C). HRMS (m/z): [M + H]+ calculated for C19H20N5O3+ 366.1561, found 366.1556.
Synthesis of a4

5-Nitrosoquinolin-8-ol (a5)
Concentrated H2SO4 (3 mL) was added slowly into H2O (65 mL), and the resulting solution was stirred vigorously at 15–18 °C. Compound a1 (7.4 g, 50 mmol) was added. A solution of NaNO2 (3.7 g, 54 mmol) in H2O (7 mL) was added dropwise over 30 min. The reaction mixture was stirred at 18–20 °C for 3 h, and then 20% NaOH was added slowly to adjust to pH 10–11, with the temperature kept below 25 °C. The mixture was filtered, and the filtrate was acidified to pH 5–6 with glacial acetic acid. The resulting solid was collected by filtration, washed with H2O (100 mL), and dried to give compound a5 (8.4 g, 95% yield), which was used for the next step without further purification.
5-Nitroquinolin-8-ol (a4)
Fuming HNO3 (0.10 mL) was added dropwise to the suspension of compound a5 (7.5 g, 43 mmol) in acetic acid (23 mL), and the reaction mixture was stirred at 20 °C for 2 h. The mixture was basified at the temperature not exceeding 25 °C using 20% NaOH to pH 10–11, filtered, then the filtrate was acidified to pH 5–6 with glacial acetic acid. The formed precipitate was filtered, washed with H2O (100 mL), acetone (15 mL × 2), and then dried to give the compound a4 (5.7 g, 70% yield).
Loop Modeling and Structural Preparations
Loop Filling and Refinements
The published crystal structures of D2R comprised the engineered receptors and inserted proteins in the intracellular loop ICL2 to facilitate crystallization. Before starting MD simulations, we removed the corresponding inserted proteins from the D2R crystal structures and used the loop refinement protocol in Modeler43 V9.10 to reconstruct and refine the ICL2 region. A total of 10 000 loops were generated for each receptor and the conformation with the lowest DOPE (Discrete Optimized Protein Energy) score was chosen for receptor construction. Repaired models were submitted to Rosetta V3.4 for loop refinement with kinematic loop modeling methods.44 Kinematic closure (KIC) is an analytical calculation procedure inspired by robotics techniques for rapidly determining possible conformations of linked objects subject to constraints. In the Rosetta KIC implementation, 2N – 6 backbone torsions of an N-residue peptide segment (called nonpivot torsions) were set to values drawn randomly from the Ramachandran space of each residue type, and the remaining 6 phi/psi torsions (called pivot torsions) were solved analytically by KIC.
Protein Structure Preparations
All protein models were prepared in Schrodinger suite software under the OPLS_2005 force field.45 Hydrogen atoms were added to the repaired crystal structures at physiological pH (7.4) with the PROPKA46 tool to optimize the hydrogen bond network provided by the Protein Preparation tool in Schrodinger. The highly conserved D802.50 has been found to be deprotonated. Since we only study the initial binding process of D2R, D2.50 in GPCR is always in a deprotnated state as indicated by previous study.34,47 Thus, we kept the deprotonated state for D802.50 during MD simulations. Constrained energy minimizations were carried out on the full-atomic models, until the RMSD of heavy atoms coveraged to 0.4 Å.
Ligand Structure Preparations
All ligand structures were prepared in Schrodinger software. The LigPrep module in Schrodinger 2015 suite software was introduced for geometric optimization by using the OPLS_2005 force field. The ionization state of ligands was calculated with the Epik48 tool employing Hammett and Taft methods in combination with ionization and tautomerization tools.48
Molecular Dynamics Simulations
Membrane systems were built using the membrane building tool g_membed49 in Gromacs with the receptor crystal structure prealigned in the OPM (Orientations of Proteins in Membranes) database.50 Pre-equilibrated 120 POPC lipids coupled with 8000 TIP3P water molecules in a box ∼73 × 73 × 92 Å3 were used to build the protein/membrane/water system. We modeled the protein, lipids, water, and ions using the CHARMM36m force field.51 Ligands were assigned with CHARMM CgenFF force field.52 Ligand geometry was submitted to the GAUSSIAN 09 program53 for optimization at the Hartree–Fock 6-31G* level when generating force field parameters. The system was gradually heated from 0 to 310 K followed by a 1 ns initial equilibration at constant volume with the temperature set at 310 K. The backbone of a particular protein and the heavy atoms of a particular ligand were restrained during the equilibration steps. All bond lengths to hydrogen atoms were constrained with M-SHAKE. Nonbonded interactions were treated using the force switch of 10–12 Å. Long-range electrostatic interactions were computed by the Particle Mesh Ewald (PME) summation scheme. All MD simulations were done in Gromacs.54 The simulation parameter files were obtained from CHARMM-GUI Web site.55 Figures were prepared in PyMOL and Inkscape.56
For the simulation of the apo-GPCR forms, all Na+ ions were presented in the bulk environment. The allosteric Na+ ions diffused into the inner space of D2R at a certain stage of the MD simulations. In the simulation of D2R in complex with MLS1547, we kept a Na+ ion at the allosteric site next to D2.50, which is sampled in the simulation of apo GPCR forms.
Identifying a Mg2+ in the μOR crystal structure
Both coordinates, electron density map and topology files for PDB: 5CM1 were obtained from PDB database. We resolved the structure again with programs Phenix57 and Coot.58 The geometry of molecule BU72 was optimized using quantum mechanical (QM) method at level B3LYP/6-31G* by Jaguar59 in Schrodinger software60 prior to the refinement. The Phenix was used for metal ion identification.57
Acknowledgments
We would like to thank Fei Li from iHuman Institute for repeating some functional analyses. Our calculations were supported by the Interdisciplinary Centre for Mathematical and Computational Modelling in Warsaw (grant nos. GB70-3 & GB71-3). This project was financially supported by the internal funding of Shenzhen Institutes of Advanced Technology, CAS.
Glossary
Abbreviations
- GPCR
G protein-coupled receptor
- MD
molecular dynamics
- ECL2
extracellular loop 2
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.9b01247.
Author Contributions
# (H.C.S.C., Y.X., L.T.) These authors contributed equally to this work. S.Y. initialized and led this work.
The authors declare the following competing financial interest(s): H.C.S.C., H.V., and S.Y. are the cofounders of AlphaMol Science Ltd.
This paper was originally published ASAP on January 23, 2020. The title was corrected, and the paper reposted on January 24, 2020.
Supplementary Material
References
- Katritch V.; Fenalti G.; Abola E. E.; Roth B. L.; Cherezov V.; Stevens R. C. Allosteric sodium in class A GPCR signaling. Trends Biochem. Sci. 2014, 39 (5), 233. 10.1016/j.tibs.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez-de-Teran H.; Massink A.; Rodriguez D.; Liu W.; Han G. W.; Joseph J. S.; Katritch I.; Heitman L. H.; Xia L.; Ijzerman A. P.; et al. The role of a sodium ion binding site in the allosteric modulation of the A(2A) adenosine G protein-coupled receptor. Structure 2013, 21 (12), 2175. 10.1016/j.str.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller-Gallacher J. L.; Nehme R.; Warne T.; Edwards P. C.; Schertler G. F.; Leslie A. G.; Tate C. G. The 2.1 A resolution structure of cyanopindolol-bound beta1-adrenoceptor identifies an intramembrane Na+ ion that stabilises the ligand-free receptor. PLoS One 2014, 9 (3), e92727. 10.1371/journal.pone.0092727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.; Wacker D.; Levit A.; Che T.; Betz R. M.; McCorvy J. D.; Venkatakrishnan A. J.; Huang X. P.; Dror R. O.; Shoichet B. K.; et al. D4 dopamine receptor high-resolution structures enable the discovery of selective agonists. Science 2017, 358 (6361), 381. 10.1126/science.aan5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert T.; Olieric N.; Cheng R.; Brunle S.; James D.; Ozerov D.; Gashi D.; Vera L.; Marsh M.; Jaeger K.; et al. Serial millisecond crystallography for routine room-temperature structure determination at synchrotrons. Nat. Commun. 2017, 8 (1), 542. 10.1038/s41467-017-00630-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y.; Kuybeda O.; de Waal P. W.; Mukherjee S.; Van Eps N.; Dutka P.; Zhou X. E.; Bartesaghi A.; Erramilli S.; Morizumi T.; et al. Cryo-EM structure of human rhodopsin bound to an inhibitory G protein. Nature 2018, 558 (7711), 553. 10.1038/s41586-018-0215-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velgy N.; Hedger G.; Biggin P. C. GPCRs: What Can We Learn from Molecular Dynamics Simulations?. Methods Mol. Biol. 2018, 1705, 133. 10.1007/978-1-4939-7465-8_6. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Sun B.; Feng D.; Hu H.; Chu M.; Qu Q.; Tarrasch J. T.; Li S.; Sun Kobilka T.; Kobilka B. K.; et al. Cryo-EM structure of the activated GLP-1 receptor in complex with a G protein. Nature 2017, 546 (7657), 248. 10.1038/nature22394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst O. P.; Lodowski D. T.; Elstner M.; Hegemann P.; Brown L. S.; Kandori H. Microbial and animal rhodopsins: structures, functions, and molecular mechanisms. Chem. Rev. 2014, 114 (1), 126. 10.1021/cr4003769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W.; Chun E.; Thompson A. A.; Chubukov P.; Xu F.; Katritch V.; Han G. W.; Roth C. B.; Heitman L. H.; IJzerman A. P.; et al. Structural Basis for Allosteric Regulation of GPCRs by Sodium Ions. Science 2012, 337 (6091), 232. 10.1126/science.1219218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenalti G.; Giguere P. M.; Katritch V.; Huang X. P.; Thompson A. A.; Cherezov V.; Roth B. L.; Stevens R. C. Molecular control of delta-opioid receptor signalling. Nature 2014, 506, 191. 10.1038/nature12944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan S.; Hu Z.; Filipek S.; Vogel H. W2466.48 Opens a Gate for a Continuous Intrinsic Water Pathway during Activation of the Adenosine A Receptor. Angew. Chem., Int. Ed. 2014, 54 (2), 556. 10.1002/anie.201409679. [DOI] [PubMed] [Google Scholar]
- Ericksen S. S.; Cummings D. F.; Weinstein H.; Schetz J. A. Ligand selectivity of D2 dopamine receptors is modulated by changes in local dynamics produced by sodium binding. J. Pharmacol. Exp. Ther. 2009, 328 (1), 40. 10.1124/jpet.108.141531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan S.; Vogel H.; Filipek S. The Role of Water and Sodium Ions in the Activation of the mu-Opioid Receptor. Angew. Chem., Int. Ed. 2013, 52 (38), 10112. 10.1002/anie.201302244. [DOI] [PubMed] [Google Scholar]
- Cong X.; Golebiowski J. Allosteric Na(+)-binding site modulates CXCR4 activation. Phys. Chem. Chem. Phys. 2018, 20 (38), 24915. 10.1039/C8CP04134B. [DOI] [PubMed] [Google Scholar]
- Miao Y.; Caliman A. D.; McCammon J. A. Allosteric effects of sodium ion binding on activation of the m3 muscarinic g-protein-coupled receptor. Biophys. J. 2015, 108 (7), 1796. 10.1016/j.bpj.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makman M. H.; Dvorkin B.; Klein P. N. Sodium ion modulates D2 receptor characteristics of dopamine agonist and antagonist binding sites in striatum and retina. Proc. Natl. Acad. Sci. U. S. A. 1982, 79 (13), 4212. 10.1073/pnas.79.13.4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M.; George S. R.; Seeman P. Regulation of anterior pituitary D2 dopamine receptors by magnesium and sodium ions. J. Neurochem. 1985, 45 (6), 1842. 10.1111/j.1471-4159.1985.tb10542.x. [DOI] [PubMed] [Google Scholar]
- Ye L.; Neale C.; Sljoka A.; Lyda B.; Pichugin D.; Tsuchimura N.; Larda S. T.; Pomes R.; Garcia A. E.; Ernst O. P.; et al. Mechanistic insights into allosteric regulation of the A2A adenosine G protein-coupled receptor by physiological cations. Nat. Commun. 2018, 9 (1), 1372. 10.1038/s41467-018-03314-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draper-Joyce C. J.; Verma R. K.; Michino M.; Shonberg J.; Kopinathan A.; Klein Herenbrink C.; Scammells P. J.; Capuano B.; Abramyan A. M.; Thal D. M.; et al. The action of a negative allosteric modulator at the dopamine D2 receptor is dependent upon sodium ions. Sci. Rep. 2018, 8 (1), 1208. 10.1038/s41598-018-19642-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien E. Y. T.; Liu W.; Zhao Q.; Katritch V.; Won Han G.; Hanson M. A.; Shi L.; Newman A. H.; Javitch J. A.; Cherezov V.; et al. Structure of the Human Dopamine D3 Receptor in Complex with a D2/D3 Selective Antagonist. Science 2010, 330 (6007), 1091. 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selley D. E.; Cao C.-C.; Liu Q.; Childers S. R. Effects of sodium on agonist efficacy for G-protein activation in μ-opioid receptor-transfected CHO cells and rat thalamus. Br. J. Pharmacol. 2000, 130 (5), 987. 10.1038/sj.bjp.0703382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neve K. A. Regulation of dopamine D2 receptors by sodium and pH. Mol. Pharmacol. 1991, 39 (4), 570. [PubMed] [Google Scholar]
- Ananthanarayanan V. S.; Kerman A. Role of metal ions in ligand-receptor interaction: insights from structural studies. Mol. Cell. Endocrinol. 2006, 246 (1–2), 53. 10.1016/j.mce.2005.11.023. [DOI] [PubMed] [Google Scholar]
- Strasser A.; Wittmann H. J.; Schneider E. H.; Seifert R. Modulation of GPCRs by monovalent cations and anions. Naunyn-Schmiedeberg's Arch. Pharmacol. 2015, 388 (3), 363. 10.1007/s00210-014-1073-2. [DOI] [PubMed] [Google Scholar]
- Gulya K.; Kova′cs G. L.; Ka′sa P. Regulation of endogenous calcium and magnesium levels by δ opioid receptors in the rat brain. Brain Res. 1991, 547 (1), 32. 10.1016/0006-8993(91)90570-L. [DOI] [PubMed] [Google Scholar]
- Tejwani G. A.; Hanissian S. H. Modulation of mu, delta and kappa opioid receptors in rat brain by metal ions and histidine. Neuropharmacology 1990, 29 (5), 445. 10.1016/0028-3908(90)90166-O. [DOI] [PubMed] [Google Scholar]
- Wang S.; Che T.; Levit A.; Shoichet B. K.; Wacker D.; Roth B. L. Structure of the D2 dopamine receptor bound to the atypical antipsychotic drug risperidone. Nature 2018, 555 (7695), 269. 10.1038/nature25758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Free R. B.; Chun L. S.; Moritz A. E.; Miller B. N.; Doyle T. B.; Conroy J. L.; Padron A.; Meade J. A.; Xiao J.; Hu X.; et al. Discovery and characterization of a G protein-biased agonist that inhibits beta-arrestin recruitment to the D2 dopamine receptor. Mol. Pharmacol. 2014, 86 (1), 96. 10.1124/mol.113.090563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Jonsson A. L.; Beuming T.; Shelley J. C.; Voth G. A. Ligand-dependent activation and deactivation of the human adenosine A(2A) receptor. J. Am. Chem. Soc. 2013, 135 (23), 8749. 10.1021/ja404391q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White K. L.; Eddy M. T.; Gao Z.-G.; Han G. W.; Lian T.; Deary A.; Patel N.; Jacobson K. A.; Katritch V.; Stevens R. C. Structural Connection between Activation Microswitch and Allosteric Sodium Site in GPCR Signaling. Structure 2018, 26 (2), 259. 10.1016/j.str.2017.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton C. L.; Wood M. D.; Strange P. G. Examining the Effects of Sodium Ions on the Binding of Antagonists to Dopamine D2 and D3 Receptors. PLoS One 2016, 11 (7), e0158808. 10.1371/journal.pone.0158808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michino M.; Free R. B.; Doyle T. B.; Sibley D. R.; Shi L. Structural basis for Na(+)-sensitivity in dopamine D2 and D3 receptors. Chem. Commun. 2015, 51 (41), 8618. 10.1039/C5CC02204E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickery O. N.; Carvalheda C. A.; Zaidi S. A.; Pisliakov A. V.; Katritch V.; Zachariae U. Intracellular Transfer of Na(+) in an Active-State G-Protein-Coupled Receptor. Structure 2018, 26 (1), 171. 10.1016/j.str.2017.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vass M.; Kooistra A. J.; Ritschel T.; Leurs R.; de Esch I. J.; de Graaf C. Molecular interaction fingerprint approaches for GPCR drug discovery. Curr. Opin. Pharmacol. 2016, 30, 59. 10.1016/j.coph.2016.07.007. [DOI] [PubMed] [Google Scholar]
- Singh J.; Deng Z.; Narale G.; Chuaqui C. Structural interaction fingerprints: a new approach to organizing, mining, analyzing, and designing protein-small molecule complexes. Chem. Biol. Drug Des. 2006, 67 (1), 5. 10.1111/j.1747-0285.2005.00323.x. [DOI] [PubMed] [Google Scholar]
- Deng Z.; Chuaqui C.; Singh J. Structural interaction fingerprint (SIFt): a novel method for analyzing three-dimensional protein-ligand binding interactions. J. Med. Chem. 2004, 47 (2), 337. 10.1021/jm030331x. [DOI] [PubMed] [Google Scholar]
- Chan H. C. S.; Wang J.; Palczewski K.; Filipek S.; Vogel H.; Liu Z.-J.; Yuan S. Exploring a new ligand binding site of G protein-coupled receptors. Chemical Science 2018, 9, 6480. 10.1039/C8SC01680A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kling R. C.; Clark T.; Gmeiner P. Comparative MD Simulations Indicate a Dual Role for Arg1323.50 in Dopamine-Dependent D2R Activation. PLoS One 2016, 11 (1), e0146612. 10.1371/journal.pone.0146612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kling R. C.; Tschammer N.; Lanig H.; Clark T.; Gmeiner P. Active-state model of a dopamine D2 receptor-Galphai complex stabilized by aripiprazole-type partial agonists. PLoS One 2014, 9 (6), e100069. 10.1371/journal.pone.0100069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W.; Manglik A.; Venkatakrishnan A. J.; Laeremans T.; Feinberg E. N.; Sanborn A. L.; Kato H. E.; Livingston K. E.; Thorsen T. S.; Kling R. C.; et al. Structural insights into micro-opioid receptor activation. Nature 2015, 524 (7565), 315. 10.1038/nature14886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroeze W. K.; Sassano M. F.; Huang X.-P.; Lansu K.; McCorvy J. D.; Giguere P. M.; Sciaky N.; Roth B. L. PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat. Struct. Mol. Biol. 2015, 22 (5), 362. 10.1038/nsmb.3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswar N.; Webb B.; Marti-Renom M. A.; Madhusudhan M. S.; Eramian D.; Shen M. Y.; Pieper U.; Sali A. Comparative protein structure modeling using MODELLER. Current protocols in protein science 2007, 50, 2.9.1. 10.1002/0471140864.ps0209s50. [DOI] [PubMed] [Google Scholar]
- Mandell D. J.; Coutsias E. A.; Kortemme T. Sub-angstrom accuracy in protein loop reconstruction by robotics-inspired conformational sampling. Nat. Methods 2009, 6 (8), 551. 10.1038/nmeth0809-551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivakumar D.; Williams J.; Wu Y. J.; Damm W.; Shelley J.; Sherman W. Prediction of Absolute Solvation Free Energies using Molecular Dynamics Free Energy Perturbation and the OPLS Force Field. J. Chem. Theory Comput. 2010, 6 (5), 1509. 10.1021/ct900587b. [DOI] [PubMed] [Google Scholar]
- Sondergaard C. R.; Olsson M. H. M.; Rostkowski M.; Jensen J. H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pK(a) Values. J. Chem. Theory Comput. 2011, 7 (7), 2284. 10.1021/ct200133y. [DOI] [PubMed] [Google Scholar]
- Zarzycka B.; Zaidi S. A.; Roth B. L.; Katritch V. Harnessing Ion-Binding Sites for GPCR Pharmacology. Pharmacol. Rev. 2019, 71 (4), 571. 10.1124/pr.119.017863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood J. R.; Calkins D.; Sullivan A. P.; Shelley J. C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput.-Aided Mol. Des. 2010, 24 (6–7), 591. 10.1007/s10822-010-9349-1. [DOI] [PubMed] [Google Scholar]
- Wolf M. G.; Hoefling M.; Aponte-Santamaria C.; Grubmuller H.; Groenhof G. g_membed: Efficient insertion of a membrane protein into an equilibrated lipid bilayer with minimal perturbation. J. Comput. Chem. 2010, 31 (11), 2169. 10.1002/jcc.21507. [DOI] [PubMed] [Google Scholar]
- Lomize A. L.; Pogozheva I. D.; Mosberg H. I. Anisotropic solvent model of the lipid bilayer. 1. Parameterization of long-range electrostatics and first solvation shell effects. J. Chem. Inf. Model. 2011, 51 (4), 918. 10.1021/ci2000192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klauda J. B.; Venable R. M.; Freites J. A.; O’Connor J. W.; Tobias D. J.; Mondragon-Ramirez C.; Vorobyov I.; MacKerell A. D.; Pastor R. W. Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 2010, 114 (23), 7830. 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; Raman E. P.; MacKerell A. D. Jr. Automation of the CHARMM General Force Field (CGenFF) II: assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model. 2012, 52 (12), 3155. 10.1021/ci3003649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, Revision A.1; Gaussian, Inc.: Wallingford CT, 2009.
- Pronk S.; Pall S.; Schulz R.; Larsson P.; Bjelkmar P.; Apostolov R.; Shirts M. R.; Smith J. C.; Kasson P. M.; van der Spoel D.; et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29 (7), 845. 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S.; Kim T.; Iyer V. G.; Im W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29 (11), 1859. 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- Yuan S.; Chan H. C.; Filipek S.; Vogel H. PyMOL and Inkscape Bridge the Data and the Data Visualization. Structure 2016, 24 (12), 2041. 10.1016/j.str.2016.11.012. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L. W.; Kapral G. J.; Grosse-Kunstleve R. W.; et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66 (Pt 2), 213. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66 (4), 486. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochevarov A. D.; Harder E.; Hughes T. F.; Greenwood J. R.; Braden D. A.; Philipp D. M.; Rinaldo D.; Halls M. D.; Zhang J.; Friesner R. A. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 2013, 113 (18), 2110. 10.1002/qua.24481. [DOI] [Google Scholar]
- Schrödinger; Schrödinger, LLC, 2018.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.