

Abstract
Rationale
Recent studies have revealed that, in critically ill patients, lung microbiota are altered and correlate with alveolar inflammation. The clinical significance of altered lung bacteria in critical illness is unknown.
Objectives
To determine if clinical outcomes of critically ill patients are predicted by features of the lung microbiome at the time of admission.
Methods
We performed a prospective, observational cohort study in an ICU at a university hospital. Lung microbiota were quantified and characterized using droplet digital PCR and bacterial 16S ribosomal RNA gene quantification and sequencing. Primary predictors were the bacterial burden, community diversity, and community composition of lung microbiota. The primary outcome was ventilator-free days, determined at 28 days after admission.
Measurements and Main Results
Lungs of 91 critically ill patients were sampled using miniature BAL within 24 hours of ICU admission. Patients with increased lung bacterial burden had fewer ventilator-free days (hazard ratio, 0.43; 95% confidence interval, 0.21–0.88), which remained significant when the analysis was controlled for pneumonia and severity of illness. The community composition of lung bacteria predicted ventilator-free days (P = 0.003), driven by the presence of gut-associated bacteria (e.g., species of the Lachnospiraceae and Enterobacteriaceae families). Detection of gut-associated bacteria was also associated with the presence of acute respiratory distress syndrome.
Conclusions
Key features of the lung microbiome (bacterial burden and enrichment with gut-associated bacteria) predict outcomes in critically ill patients. The lung microbiome is an understudied source of clinical variation in critical illness and represents a novel therapeutic target for the prevention and treatment of acute respiratory failure.
Keywords: lung microbiome, acute respiratory distress syndrome, lung injury, prognosis
At a Glance Commentary
Scientific Knowledge on the Subject
Recent studies have revealed that lung microbiota of critically ill patients are altered and correlated with alveolar and systemic inflammation. Altered lung microbiota may propel and perpetuate alveolar inflammation and injury among critically ill patients. Yet no study has determined whether altered lung microbiota predict disease outcomes in this population.
What This Study Adds to the Field
We show that, among mechanically ventilated critically ill patients, variation in lung microbiota at admission predicts ICU outcomes. Two key features of the lung microbiome—bacterial burden and community composition—predict ventilator-free days. Specifically, increased lung bacterial DNA burden and enrichment of the lung microbiome with gut-associated bacterial taxa (e.g., Lachnospiraceae and Enterobacteriaceae families) were predictive of both poor ICU outcomes and the clinical diagnosis of acute respiratory distress syndrome. This correlation validates prior findings and supports the hypothesis that translocation of gut bacteria to the lungs contributes to the pathogenesis of lung injury. Our results confirm the clinical significance of lung microbiota in critically ill patients. The lung microbiome is an important and underappreciated source of clinical heterogeneity among the critically ill, and it represents a novel therapeutic target for the prevention and treatment of lung injury.
In the past decade, advances in culture-independent microbiology have revealed that the lungs, previously considered sterile, harbor complex and dynamic communities of bacteria (1). Lung microbiota are detectable in health (2–4), are altered in disease (5, 6), and correlate with variation in airway and alveolar immunity (2, 4, 7). In numerous chronic respiratory diseases, key features of the lung microbiome are predictive of disease outcomes. The burden of lung bacteria (measured by quantification of bacterial DNA) predicts mortality and disease progression in stable patients with idiopathic pulmonary fibrosis (8, 9) and responsiveness to inhaled antibiotics in patients with bronchiectasis (10). The diversity of sputum microbiota predicts mortality in patients with chronic obstructive pulmonary disease (11), and the community composition of respiratory microbiota predicts exacerbations in bronchiectasis (12) and respiratory infections in infants (13).
The lung microbiota of critically ill patients are profoundly altered compared with those of healthy subjects (7, 14–16), and they correlate with alveolar and systemic inflammation (7, 15). Specifically, among patients with acute respiratory distress syndrome (ARDS), the lung microbiome is enriched with gut-associated bacteria (7), and early enrichment of the lung microbiome with gut-associated bacteria (e.g., species of the Enterobacteriaceae family) is associated with subsequent development of ARDS (15). Altered lung microbiota may propel and perpetuate alveolar inflammation and injury among critically ill patients, but to date, no study has determined whether altered lung microbiota predict disease outcomes in this population.
To determine if lung microbiota at ICU admission predict clinical outcomes in critically ill patients, we performed a prospective, observational cohort study in critically ill patients receiving mechanical ventilation. The primary outcome was ventilator-free days, adjudicated at 28 days after enrollment. We hypothesized that key features of the lung microbiome (bacterial burden, diversity, and community composition) would predict ICU outcomes, even when the analysis was controlled for the presence of clinically appreciated pneumonia.
Methods
Study Design
This study was a secondary analysis of specimens collected from patients in the BASIC (Biomarker Analysis in Septic ICU Patients) study. This study was incorporated into the Molecular Diagnosis and Risk Stratification of Sepsis (MARS) project (NCT01905033) (17–20). The present study was conducted in the ICU of the Academic Medical Center in Amsterdam and was approved by the institutional medical ethics committee (institutional review board no. NL34294.018.10), and written informed consent was obtained from the patient representatives before collection of airway samples via miniature BAL (mini-BAL). Analysis was restricted to baseline specimens and data collected within 24 hours of admission.
Study Population
All patients older than 18 years of age admitted to the ICU with an expected length of stay longer than 24 hours were included in the MARS project. The BASIC study comprised a subset of patients included in the MARS study at the Amsterdam ICU who met at least two systemic inflammatory response syndrome criteria and received no antibiotics in the days preceding ICU admission. The present analysis is limited to consecutive patients who were included between September 12, 2011, and November 7, 2013, received invasive mechanical ventilation, and provided informed consent for distal airway sampling. Adjudication of infection was assessed retrospectively using a 4-point scale (in ascending order of none, possible, probable, and definite) according to the CDC and International Sepsis Forum consensus definitions as previously described (18). ARDS was scored on a daily basis by a team of well-trained clinical researchers according to the American-European consensus criteria. After the publication of the Berlin definition, all cases were reevaluated and scored according to the new definition, as described previously (21). For the purposes of ARDS versus non-ARDS comparisons, we used adjudication at 24 hours after ICU admission. Severity of illness was quantified using the validated Acute Physiology and Chronic Health Evaluation IV (APACHE IV) (22) and Sequential Organ Failure Assessment (23) models.
Specimen Collection and Processing
Miniature BAL specimens were collected using a standard clinical protocol. In short, a 50-cm, 14-French tracheal suction catheter was introduced through the orotracheal tube and inserted until significant resistance was encountered. The catheter was then pulled back 1 cm, and 20 ml of 0.9% saline was injected in 10 seconds and immediately aspirated, after which the catheter was removed. Specimens were stored on ice from the time of specimen collection until processing. DNA was extracted, amplified, and sequenced according to previously published protocols (24–26). Sequencing was performed using the Illumina MiSeq platform. Bacterial DNA was quantified using a QX200 Droplet Digital PCR System (Bio-Rad Laboratories). Additional details are provided in the online supplement.
Statistical Analysis
As detailed in the online supplement, we analyzed microbial ecology using the vegan package 2.4-1 and mvabund in R (27–29) after sequence processing with mothur (30, 31). We prespecified that key features of the microbiome (predictors) would be 1) bacterial DNA burden (as quantified using droplet digital PCR [ddPCR]), 2) bacterial community diversity (as calculated using the Shannon diversity index), and 3) community composition. We determined significance in community composition (e.g., mini-BAL specimens vs. negative sequencing controls and ARDS vs. non-ARDS mini-BAL specimens) using mvabund (model-based approach to analysis of multivariate abundance data). To identify community members driving differences in community composition, we used 1) biplot analysis, 2) rank abundance analysis, and 3) a random forest ensemble learning approach (randomForest package in R version 4.6-14 [32]). For random forest, we determined variable importance using 100 forests. The importance parameter was set to TRUE to enable retrieval of importance metrics: mean decrease in accuracy for classification (ARDS vs. non-ARDS) or percentage increase in mean square error for regression (ventilator-free days). Default settings were used for all other parameters. After model creation, the unscaled feature importance metric was extracted from each forest, assembled into a data frame, ordered by highest feature importance, and displayed in box plots of the most important features across the 100 forests. Our primary index of feature importance was mean decrease in accuracy, which ranks predictors by the relative loss in accuracy that occurs when they are removed from the predictive model. We compared means via Student’s t test (when normally distributed), the Mann-Whitney U test (when nongaussian), and analysis of variance with the Holm-Šidák multiple comparisons test as appropriate. Time-to-event analysis was performed using univariate and multivariate Cox proportional hazards models using ventilator-free days (adjudicated 28 d after enrollment) as a primary outcome; multivariate analysis was adjusted for age, sex, severity of illness (APACHE IV), diagnosis of ARDS, and the presence of clinically suspected pneumonia as determined both by the primary clinical service and via post hoc CDC adjudication criteria. The primary outcome was the proportional hazards ratio (HR) for being alive and liberated from mechanical ventilation, as adjudicated 28 days after admission.
Results
Study Cohort
We obtained mini-BAL specimens from 91 critically ill patients within 24 hours of their ICU admission. The Consolidated Standards of Reporting Trials diagram of the study is shown in Figure E1 in the online supplement. Patient demographics and clinical characteristics are reported in Table 1.
Table 1.
Demographics and Clinical Characteristics of Study Cohort
| Characteristic | Study Cohort (N = 91) |
|---|---|
| Mean age (SD), yr | 60.7 (15.4) |
| Sex, M | 55 (60) |
| Admission type | |
| Medical | 67 (74) |
| Surgical, emergency | 20 (22) |
| Surgical, elective | 4 (4) |
| Severity of illness | |
| Mean SOFA score (SD) | 7.2 (4.1) |
| Mean APACHE IV score (SD) | 82.6 (28.5) |
| Lung injury | |
| ARDS at admission | 17 (19) |
| Mean PaO2/FiO2 (SD) | 262.0 (104.7) |
| Comorbidities | |
| Diabetes mellitus | 13 (14) |
| Malignancy | 11 (12) |
| COPD | 5 (5) |
| Immune deficiency | 4 (4) |
| ICU outcomes | |
| Mean ventilator-free d (SD) | 18.5 (10.5) |
| Mean ICU length of stay (SD), d | 5.6 (4.6) |
| 30-d Mortality | 27 (30) |
Definition of abbreviations: APACHE IV = Acute Physiology and Chronic Health Score IV; ARDS = acute respiratory distress syndrome; COPD = chronic obstructive pulmonary disease; SOFA = Sequential Organ Failure Assessment.
Values are n (%) unless otherwise indicated.
The distribution of admission diagnoses is reported in Table E1. Bacterial quantification and 16S rRNA gene sequencing were performed on all specimens. Details regarding adequacy of sequencing and exclusion of specimens are provided in the online supplement.
Microbiota of Lung Specimens from Critically Ill Patients Are Distinct from Those of Background Sequencing Controls
Low-biomass microbiome studies are vulnerable to contamination due to bacterial DNA present in reagents used in DNA extraction and library preparation (33). Our study used low-volume specimens (“mini-BAL”) from a low–microbial biomass body site (the lower respiratory tract) and had their bacterial burden further decreased via a centrifugation step to remove eukaryotic cells (34). Given these concerns, we first asked whether a bacterial signal could be detected in these mini-BAL specimens that was distinct from that of negative control specimens. We accomplished this by comparing the bacterial DNA in mini-BAL specimens with no-template control specimens (n = 25), AE buffer specimens (n = 8), sterile water used in DNA extraction (n = 4), extraction control specimens (n = 9), and blank sequencing wells (n = 6).
As shown in Figure E2, we found clear evidence of a distinct bacterial signal in mini-BAL specimens despite their low microbial biomass. Using ultrasensitive quantification of the bacterial 16S gene (via ddPCR), we found significantly more bacterial DNA in mini-BAL specimens than in no-template control specimens (P < 0.001) (Figure E2A). The median bacterial DNA burden was 21,246 bacterial gene copies per milliliter (mean, 118,411 ± 707,438 copies). We found a wide span of bacterial burden (6,329–6,713,947 copies/ml), ranging from comparable to no-template control specimens to 1,000-fold greater than background. Using bacterial community analysis (16S rRNA gene amplicon sequencing), we confirmed that the identity of bacteria detected in mini-BAL specimens was distinct from that of negative control specimens (P < 0.0001; mvabund). Principal component analysis revealed distinct clustering of mini-BAL specimens apart from negative control specimens (Figure E2B), though overlap did occur between some mini-BAL specimens and negative control specimens. Rank abundance analysis showed clear differences in relative abundance of taxa in negative control specimens and mini-BAL specimens (Figure E2C). The dominant taxonomic group in negative control specimens (operational taxonomic unit [OTU] 008:Pelomonas) comprised 25.5% of bacterial sequences in negative control specimens but only 2.6% of sequences in mini-BAL specimens. We thus concluded that although highly variable in their bacterial DNA burden and susceptibility to contamination, mini-BAL specimens contained a bacterial signal distinct from that of negative control specimens.
Lung Microbiota of Critically Ill Patients Are Altered in Patients with ARDS and Enriched with Gut-associated Bacteria (Species of Enterobacteriaceae Family)
We next compared the lung microbiota of critically ill patients with and without ARDS. Prior studies have demonstrated that the lung microbiota of patients with ARDS are altered and enriched with gut-associated bacteria. We compared lung bacterial communities in patients with and without physician-adjudicated ARDS. As shown in Figure 1, lung bacterial communities of patients with ARDS differed in the bacterial DNA burden and community composition compared with those of patients without ARDS.
Figure 1.
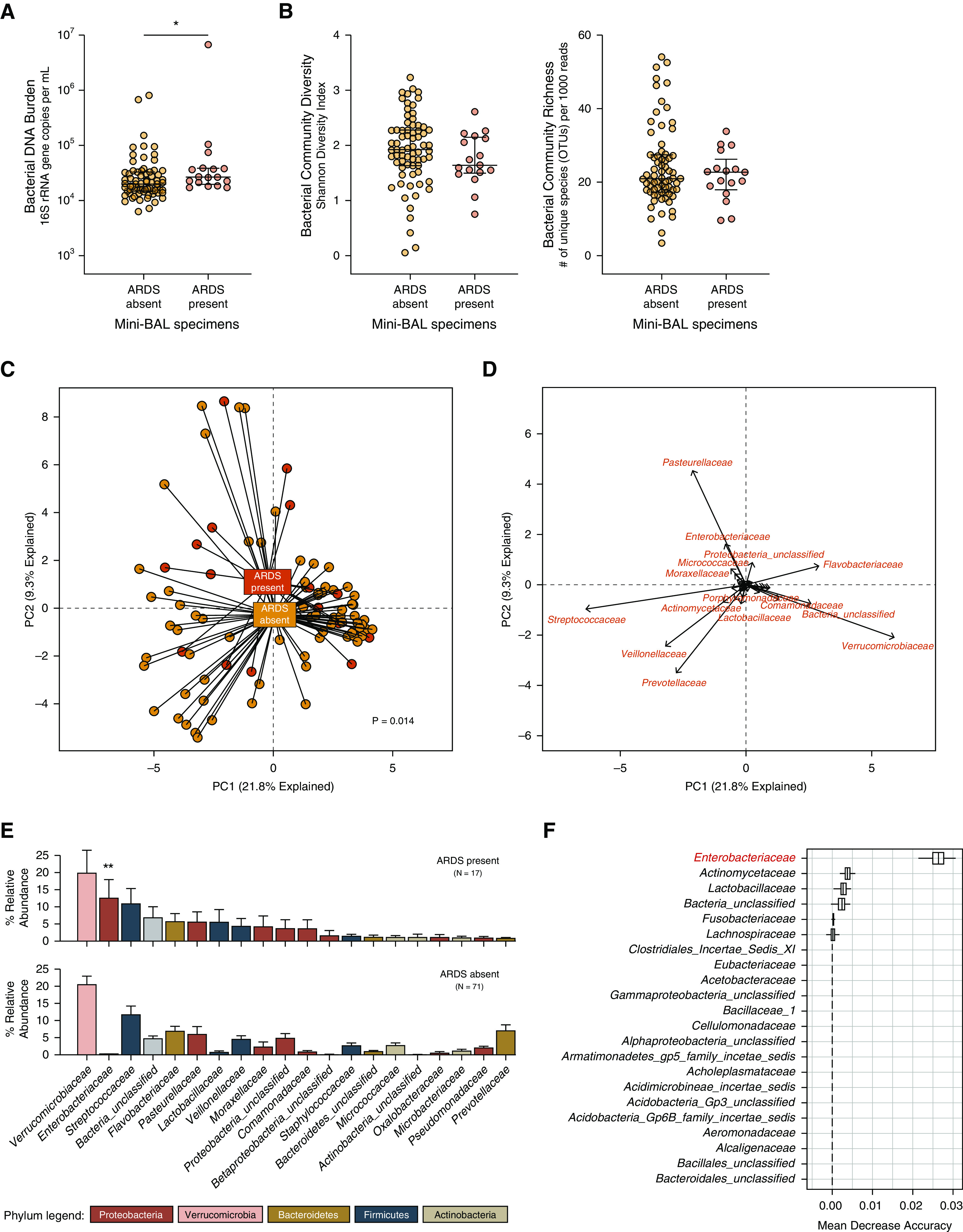
Lung microbiota are altered in patients with acute respiratory distress syndrome (ARDS). (A and B) Compared with patients without ARDS, patients with ARDS had increased lung bacterial burden (A) but no difference in community diversity (B). (C and D) Principal component analysis of bacterial communities (C) revealed that the community composition of lung bacteria was distinct in specimens from patients with ARDS (C), driven by members of the Pasteurellaceae and Enterobacteriaceae families (D). (E and F) Rank abundance analysis (E) identified taxa enriched in ARDS specimens (e.g., Enterobacteriaceae), and random forest analysis (F) confirmed that the Enterobacteriaceae family was the most discriminating taxonomic group between ARDS and non-ARDS specimens. Hypothesis testing was performed using the (A) Mann-Whitney U test, (B) Student’s t test, and (C and E) mvabund. Data in A and B are medians and interquartile ranges. *P ≤ 0.05 and **P = 0.002. OTUs = operational taxonomic units; PC = principal component; rRNA = ribosomal RNA.
We first compared ARDS and non-ARDS specimens in their bacterial DNA burden and community diversity. Though highly variable, the bacterial DNA burden in mini-BAL specimens was greater in patients with ARDS than in those of patients without ARDS (P = 0.014) (Figure 1A). ARDS specimens did not differ in bacterial community diversity, measured either via the Shannon diversity index (P = 0.13) or community richness (P = 0.83) (Figure 1B). With both comparisons (bacterial DNA burden and diversity), within-group variation far exceeded across-group differences.
We next compared the community composition of bacterial communities in ARDS and non-ARDS specimens using complementary approaches. We first visualized communities using principal component analysis (Figure 1C). Although considerable taxonomic overlap was found across ARDS and non-ARDS specimens, there was a detectable separation of specimens according to ARDS status. This collective difference in community composition was confirmed statistically via mvabund and was robust to taxonomic level of comparison (P = 0.014 at the OTU level of taxonomy; P = 0.013 at the family level; and P = 0.003 at the phylum level). We next used biplot analysis to identify specific taxa responsible for this collective difference in community composition (Figure 1D). Clusters of non-ARDS specimens were defined by bacterial taxa commonly detected in healthy lungs (species of the Streptococcaceae, Veillonellaceae, and Prevotellaceae families) and taxa detected in negative sequencing control specimens (species of the Verrucomicrobiaceae and Flavobacteriaceae families), whereas ARDS specimens were more commonly characterized by species of the Pasteurellaceae and Enterobacteriaceae families.
We then used complementary techniques to identify ARDS-associated bacterial taxa. Using rank abundance visualization (Figure 1E), we compared the relative abundance of prominent taxa across ARDS and non-ARDS specimens. Although many taxa were common to both groups, the Enterobacteriaceae family was far more abundant in ARDS specimens than in non-ARDS specimens (12.5% of all bacterial sequences in ARDS specimens compared with 0.18% of all bacterial specimens in non-ARDS specimens). We used unbiased regression-based (mvabund) and ensemble learning (random forest) approaches to identify ARDS-enriched taxa. mvabund, which rigorously controls for multiple comparisons, identified the Enterobacteriaceae family as enriched in ARDS specimens (P = 0.002). Random forest clearly identified the Enterobacteriaceae family as the most important taxonomic feature discriminating ARDS from non-ARDS specimens (Figure 1F).
We next compared our ARDS-associated Enterobacteriaceae taxonomic group with that of an ARDS-associated Enterobacteriaceae taxon in a recently published study of mechanically ventilated patients with trauma (15). We compared the most prominent Enterobacteriaceae-classified OTU in our data set (OTU0005, comprising 61.5% of all Enterobacteriaceae-classified sequences) with the ARDS-associated Enterobacteriaceae identified by Panzer and colleagues (OTU2119418) (15). As shown in Figure E3A, the representative sequence of our study’s ARDS-associated Enterobacteriaceae OTU was 96% aligned with that of the ARDS-associated Enterobacteriaceae OTU identified by Panzer and colleagues, differing in only three base pairs. We compared these ARDS-associated OTUs with the taxonomic classifications of closely aligned sequences from the SILVA rRNA database. As shown in Figure E3B, both OTUs were exclusively identical to Enterobacteriaceae-classified taxa, including Escherichia coli, Enterobacter spp., and Klebsiella pneumoniae. We thus concluded that the lung microbiota of patients with ARDS differ from those of critically ill patients without ARDS, driven by relative enrichment with gut-associated species of the Enterobacteriaceae family.
Lung Microbiota Are Predictive of Clinical Outcomes in Critically Ill Patients
We next asked if key features of the lung microbiome (bacterial burden, diversity, and community composition) predict clinical outcomes in critically ill patients. Our primary outcome was ventilator-free days measured at 28 days after admission.
We first asked if bacterial burden of mini-BAL specimens (quantified using ddPCR of the 16S rRNA gene) predicted ICU outcomes (Table 2). Using univariate analysis, we found that increased baseline lung bacterial DNA burden predicted fewer ventilator-free days, either when analyzed continuously (HR, 0.43; 95% confidence interval, 0.21–0.88; P = 0.022) or when comparing tertiles defined by total lung bacterial DNA burden. In other words, for each additional 10-fold increase in lung bacterial DNA, the HR for favorable outcome (liberation from mechanical ventilation) was 0.43. As shown in Figure 2, the tertile of patients with the highest baseline lung bacterial DNA burden was less likely to be extubated and alive at 7, 14, 21, and 28 days than patients with low bacterial DNA burden (HR, 0.45; 95% confidence interval, 0.25–0.81; P = 0.008).
Table 2.
Predictors of Ventilator-Free Days in Mechanically Ventilated Critically Ill Patients
| Predictor | Univariate |
Multivariate |
|||
|---|---|---|---|---|---|
| Hazard Ratio (95% CI) | P Value | Hazard Ratio (95% CI) | P Value | ||
| Lung bacterial DNA burden, continuous | 0.43 (0.21–0.88) | 0.022 | 0.40 (0.18–0.86) | 0.019 | |
| Lung bacterial DNA burden, middle tertile* | 0.87 (0.50–1.51) | 0.62 | — | — | |
| Lung bacterial DNA burden, highest tertile* | 0.45 (0.25–0.81) | 0.008 | — | — | |
| Shannon diversity index† | 1.27 (0.87–1.86) | 0.21 | — | — | |
| Age, yr | 0.99 (0.98–1.01) | 0.35 | 1.01 (0.99–1.03) | 0.32 | |
| Sex, M | 1.26 (0.78–2.03) | 0.35 | 0.90 (0.54–1.49) | 0.68 | |
| SOFA score | 0.95 (0.90–1.01) | 0.10 | — | — | |
| APACHE IV score | 0.98 (0.98–0.99) | <0.001 | 0.98 (0.97–0.99) | <0.001 | |
| Suspected pneumonia | 1.01 (0.60–1.70) | 0.96 | 0.90 (0.53–1.55) | 0.71 | |
| Pneumonia, post hoc, CDC criteria | 0.48 (0.18–1.33) | 0.16 | — | — | |
| ARDS | 0.51 (0.27–0.98) | 0.044 | 0.61 (0.31–1.21) | 0.16 | |
Definition of abbreviations: APACHE IV = Acute Physiology and Chronic Health Evaluation IV; ARDS = acute respiratory distress syndrome; CI = confidence interval; SOFA = Sequential Organ Failure Assessment.
Versus lowest tertile.
Shannon diversity index, per 1-unit increase.
Figure 2.
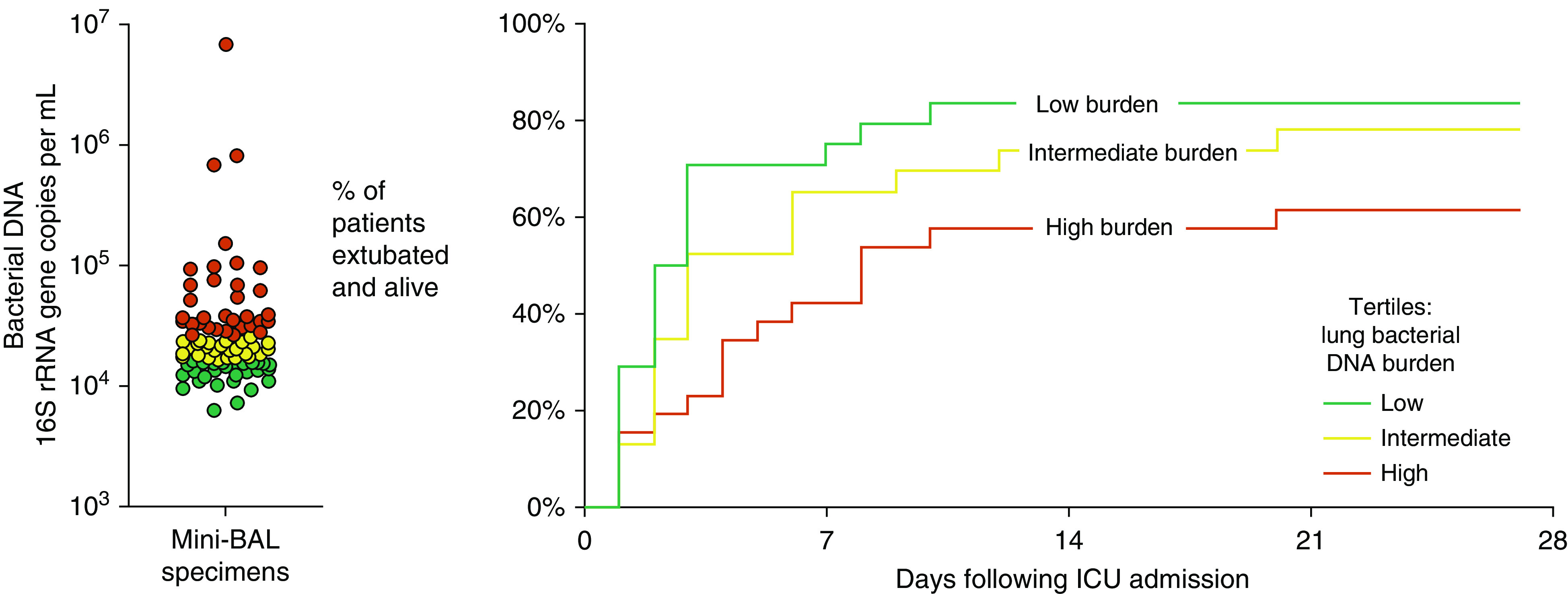
Lung microbiota predict 28-day outcomes in mechanically ventilated critically ill patients. In critically ill patients receiving mechanical ventilation, the burden of bacterial DNA detected in miniature BAL specimens was predictive of total ventilator-free days. Patients with high lung burdens of bacterial DNA were less likely to be extubated and alive than patients with low bacterial DNA burden (P = 0.008). Hypothesis testing was performed using univariate Cox proportional hazards modeling. rRNA = ribosomal RNA.
Pneumonia is common among mechanically ventilated patients and a potential source of confounding in lung microbiome studies. In our study cohort, 27 patients (30%) had suspected pneumonia either at admission or within 48 hours of admission; of these, 7 patients met CDC criteria for probable or confirmed pneumonia via post hoc chart review. When we controlled our outcome analysis of lung bacterial DNA burden for the presence of clinically suspected pneumonia, neither the HR nor the significance of the model was meaningfully changed (HR, 0.43; P = 0.021). Similarly, controlling for post hoc adjudicated pneumonia (using CDC criteria) also did not influence the predictive power of lung bacterial DNA burden (HR, 0.43; P = 0.019). We thus concluded that lung bacterial burden predicts poor outcomes in mechanically ventilated critically ill patients, even when the analysis is controlled for the presence of suspected or confirmed pneumonia.
We then performed multivariate analysis to determine whether lung bacterial DNA burden is independently predictive of poor outcomes. The relationship between increased lung bacterial DNA burden and fewer ventilator-free days remained significant when we controlled the analysis for age, sex, severity of illness (APACHE IV score), the presence of suspected pneumonia, and the presence of ARDS (HR, 0.40; 95% confidence interval, 0.18–0.86; P = 0.019). We thus concluded that lung bacterial DNA burden is an independent predictor of poor outcomes in critically ill patients.
We next asked if bacterial diversity of lung bacteria predicts ICU outcomes (Figure 3A). Bacterial diversity of lung bacteria (as measured by the Shannon diversity index) did not significantly predict ICU outcomes (P = 0.22). Although the most favorable ICU outcomes were observed among patients with high baseline lung bacterial diversity, followed in stepwise manner by patients with intermediate and low diversity, this difference in tertiles was not statistically significant. Other indices of lung bacterial diversity (community richness and community dominance) also were not significantly predictive of ICU outcomes (ventilator-free days; P > 0.05 for all comparisons) (Table E2).
Figure 3.
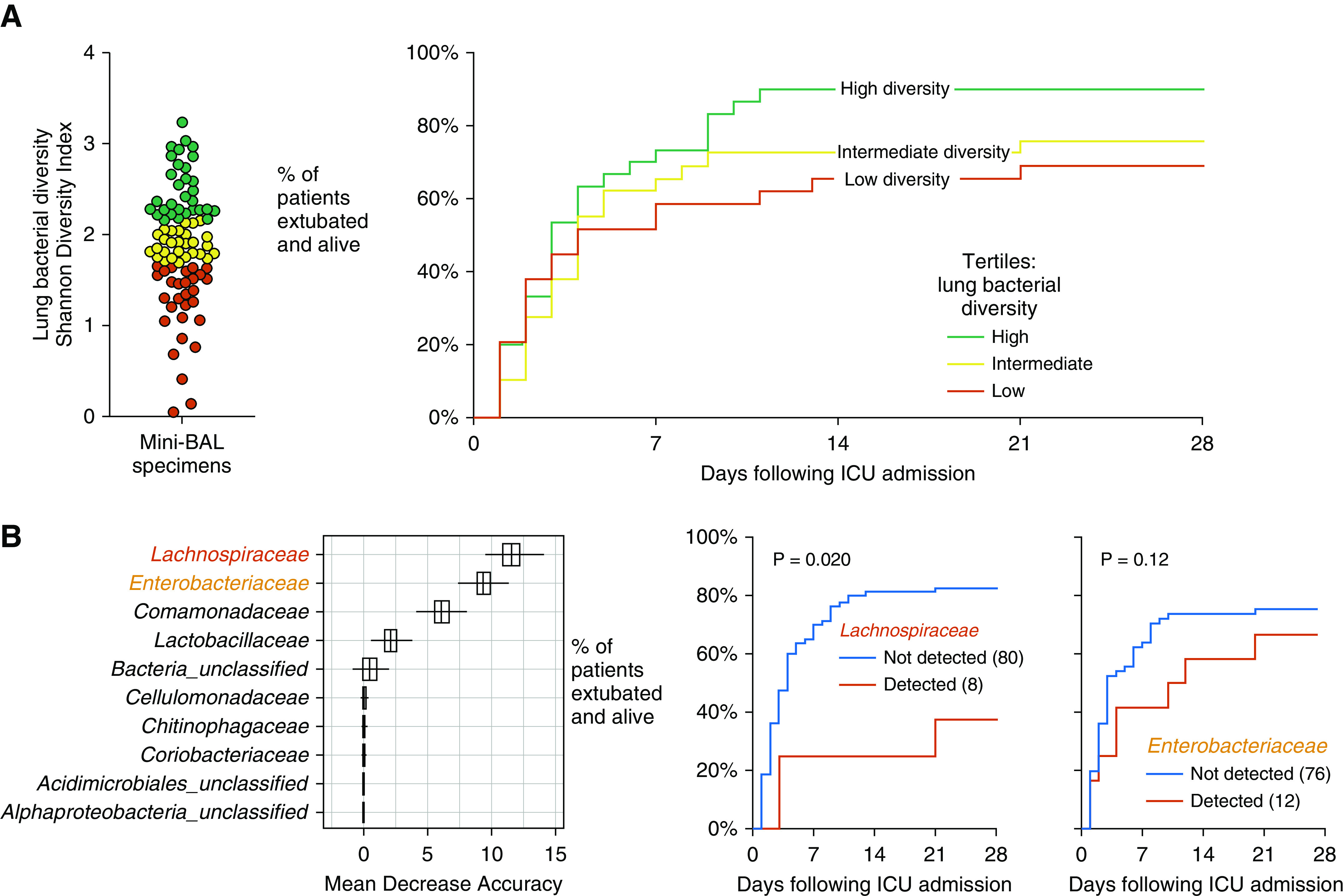
Lung microbiota and 28-day outcomes in mechanically ventilated critically ill patients. (A) Community diversity of lung bacteria was highly variable among patients and did not significantly predict ventilator-free days. (B) Community composition of lung bacteria was significantly predictive of ventilator-free days (P = 0.003; mvabund). Random forest identified the gut-associated Lachnospiraceae and Enterobacteriaceae families as the strongest predictors of ventilator-free days. Hypothesis testing was performed using a Cox proportional hazards model.
Finally, we asked if the community composition of lung bacteria is predictive of ICU outcomes. We compared patient ventilator-free days with lung bacterial community structure using mvabund (model-based approach to analysis of multivariate abundance data). The overall community composition of baseline lung microbiota was significantly predictive of patient ventilator-free days (P = 0.003 at the OTU level of taxonomy and P = 0.004 at the family level). Using random forest to identify taxa associated with poor outcomes, we identified the gut-associated Lachnospiraceae and Enterobacteriaceae families as the taxa most strongly predictive of fewer ventilator-free days (Figure 3B). We then tested the hypotheses that detection of these taxa predicts poor outcomes. As shown in Figure 3B, detection of the Lachnospiraceae family was significantly predictive of worse ICU outcomes (P = 0.020). The relationship between Enterobacteriaceae detection and ventilator-free days was not significant (P = 0.12). We thus concluded that in the lung microbiota of critically ill patients, poor ICU outcomes are predicted both by increased bacterial burden and by community composition (specifically, enrichment with gut-associated taxa).
Discussion
The core finding of this study is that among mechanically ventilated critically ill patients, variation in lung microbiota at admission predicts ICU outcomes. Two key features of the lung microbiome—bacterial burden and community composition—predicted ventilator-free days. Specifically, increased lung bacterial DNA burden and enrichment of the lung microbiome with gut-associated bacterial taxa (e.g., Lachnospiraceae and Enterobacteriaceae families) were predictive of poor ICU outcomes and the clinical diagnosis of ARDS.
To our knowledge, our study is the first to demonstrate that variation in lung microbiota is predictive of clinical outcomes in critically ill patients. This key finding is compatible with broader (non-ICU) lung microbiome studies which have found that lung microbiota are predictive of disease outcomes in idiopathic pulmonary fibrosis (8, 9), chronic obstructive pulmonary disease (11), bronchiectasis (10, 12), and infants susceptible to respiratory infections (13). Although these findings, robust across disease states, confirm that the lung microbiome is a risk factor for disease progression, a crucial and unanswered question is whether lung microbiota are a modifiable risk factor. Animal studies using germ-free and antibiotic-exposed mice suggest that manipulation of the microbiome does influence host susceptibility to lung inflammation, injury, and mortality (9, 35). Yet, the precise role of gut and lung microbiota in the pathogenesis of acute and chronic lung injury has yet to be elucidated. Future studies should interrogate whether the microbiome’s role in lung disease is more attributable to remote (gut–lung) or local (lung–lung) host–microbiome interactions (2).
Our findings both validate several recent studies and provide new insight into the importance of the lung microbiome in critical illness. We have previously reported that the lung microbiome is enriched with gut-associated bacteria in ARDS and correlated with the severity of systemic and alveolar inflammation (7). In a subsequent study of mechanically ventilated patients with trauma, the presence of gut-associated bacteria in endotracheal aspirates (species of the Enterobacteriaceae family) was associated with ARDS onset (15). In our present study, we found that the lung microbiota of patients with ARDS were distinct from those of patients without ARDS, again driven by the presence of gut-associated bacteria (species of the Enterobacteriaceae family). Indeed, the bacterial taxon most strongly correlated with ARDS status in our study (OTU0005:Enterobacteriaceae) was nearly identical to that of the ARDS-associated bacterial taxon found by Panzer and colleagues (15). These multiple findings, now robust across cohorts, sequencing platforms, laboratories, and continents, all provide indirect support for the hypothesis that gut–lung translocation of bacteria contributes to the pathogenesis of lung injury in critically ill patients.
Importantly, our core findings remained significant when controlled for the clinical suspicion or post hoc adjudication of pneumonia. Although this may seem paradoxical (increased lung bacterial burden predicts poor outcomes, and pneumonia is a condition of high lung bacterial burden), recent culture-independent studies have revealed both the complexity of lung bacterial communities in mechanically ventilated patients (14, 16, 36) and the inadequacy of the conventional understanding of pneumonia (37). The lack of concordance between our molecular characterization of lung bacteria and clinical assessment of pneumonia likely reflects several key issues in the microbiology of injured lungs: 1) clinical adjudication of pneumonia, especially in mechanically ventilated patients, is imprecise and unreliable (38); 2) a dichotomous adjudication of pneumonia is too simplistic and reductionistic to meaningfully describe the complex ecological spectrum of respiratory microbiota; and 3) the lung microbiome may play a role in the pathogenesis of disease processes not classically considered infectious (e.g., perpetuating inflammation and injury in ARDS). Our results highlight the need for improved molecular diagnostics to provide clinicians with a more accurate and comprehensive assessment of lung microbiota, as well as a more refined ecological understanding of respiratory infections in critically ill patients.
Our study has several limitations that should prompt further validation and study. Although we detected a distinct bacterial signal in our specimens, the bacterial biomass in these cell-free mini-BAL specimens was low, and in many specimens, it overlapped with background “sequencing noise.” Future studies using larger volumes of whole BAL may find stronger bacterial signals. Our mini-BAL sampling approach was nondirectional; thus, the anatomical site of sampling was not standardized across patients. Although our findings remained significant when controlled for important clinical confounders, we could not control for all potential exposures (e.g., ICU antibiotic exposure or pre-ICU medications), and residual confounding is likely. Finally, although our findings provide indirect support for the hypothesis of gut–lung translocation contributing to lung injury in critically ill patients, our lack of paired gut specimens precludes our determining whether gut-associated taxa (e.g., species of the Enterobacteriaceae and Lachnospiraceae families) were derived from the lower gastrointestinal tract or via another route (e.g., aspiration of altered pharyngeal microbiota). Future prospective studies of critically ill patients, in addition to sampling the lower respiratory tract, should collect time-matched specimens from the lower and upper gastrointestinal tracts.
In conclusion, in this prospective observational cohort study of mechanically ventilated critically ill patients, variation in baseline lung microbiota predicted ICU outcomes. Increased lung bacterial burden and lung enrichment with gut-associated bacteria were predictive of worse outcomes. The lung microbiome is an important and understudied source of variation among critically ill patients and may represent a novel therapeutic target for the prevention and treatment of lung injury.
Acknowledgments
Acknowledgment
The authors thank Carolyn Calfee, Ariane Panzer, and Susan Lynch for sharing the representative sequence of OTU2119418:Enterobacteriaceae.
BASIC Consortium Group members: Amsterdam University Medical Centers, location AMC, Amsterdam, the Netherlands: F. M. de Beer, L. D. Bos, T. A. Claushuis, G. J. Glas, J. Horn, A. J. Hoogendijk, R. T. van Hooijdonk, M. A. Huson, M. D. de Jong, N. P. Juffermans, W. A. Lagrand, T. van der Poll, B. Scicluna, L. R. Schouten, M. J. Schultz, K. F. van der Sluijs, M. Straat, L. A. van Vught, L. Wieske, M. A. Wiewel, and E. Witteveen.
Footnotes
Sample and clinical data collection was supported by the Center of Translational Medicine via the Molecular Diagnosis and Risk Stratification of Sepsis (MARS) Consortium. Additional support was provided by the Academic Medical Center. The authors were supported by the NIH (grants K23HL130641 [R.P.D.], R21AI137669 [R.P.D.], and R01HL144599 [R.P.D.]). Additional support was provided by the University of Michigan Center for Integrative Research in Critical Care (R.P.D.). The funding agencies had no role in the design, conduct, and analysis of the study or in the decision to submit the manuscript for publication. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
A complete list of BASIC Consortium group members may be found before the beginning of the References.
Data availability: Sequences are available via the National Center for Biotechnology Information Sequence Read Archive (accession number PRJNA553560). operational taxonomic unit tables, taxonomy classification tables, and metadata tables are available from github.com/dicksonlunglab/MARS_lung_microbiome.
Author Contributions: Conception and design: R.P.D. and L.D.J.B. Acquisition of data: M.J.S., T.v.d.P., L.R.S., and L.D.J.B. Processing of specimens and generation of data: N.R.F. and J.E.L. Analysis and interpretation of data: R.P.D., M.W.S., C.A.B., R.C., and L.D.J.B. Drafting or revision of the manuscript: R.P.D., M.J.S., T.v.d.P., L.R.S., N.R.F., J.E.L., M.W.S., C.A.B., R.C., G.B.H., and L.D.J.B. Final approval of the manuscript: R.P.D., M.J.S., T.v.d.P., L.R.S., N.R.F., J.E.L., M.W.S., C.A.B., R.C., G.B.H., and L.D.J.B.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.201907-1487OC on January 24, 2020
Author disclosures are available with the text of this article at www.atsjournals.org.
Contributor Information
on behalf of the Biomarker Analysis in Septic ICU Patients (BASIC) Consortium:
F. M. de Beer, L. D. Bos, T. A. Claushuis, G. J. Glas, J. Horn, A. J. Hoogendijk, R. T. van Hooijdonk, M. A. Huson, M. D. de Jong, N. P. Juffermans, W. A. Lagrand, T. van der Poll, B. Scicluna, L. R. Schouten, M. J. Schultz, K. F. van der Sluijs, M. Straat, L. A. van Vught, L. Wieske, M. A. Wiewel, and E. Witteveen
References
- 1. Dickson RP, Erb-Downward JR, Martinez FJ, Huffnagle GB. The microbiome and the respiratory tract. Annu Rev Physiol . 2016;78:481–504. doi: 10.1146/annurev-physiol-021115-105238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dickson RP, Erb-Downward JR, Falkowski NR, Hunter EM, Ashley SL, Huffnagle GB. The lung microbiota of healthy mice are highly variable, cluster by environment, and reflect variation in baseline lung innate immunity. Am J Respir Crit Care Med . 2018;198:497–508. doi: 10.1164/rccm.201711-2180OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dickson RP, Erb-Downward JR, Freeman CM, McCloskey L, Falkowski NR, Huffnagle GB, et al. Bacterial topography of the healthy human lower respiratory tract. mBio . 2017;8:e02287-16. doi: 10.1128/mBio.02287-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Segal LN, Clemente JC, Tsay JC, Koralov SB, Keller BC, Wu BG, et al. Enrichment of the lung microbiome with oral taxa is associated with lung inflammation of a Th17 phenotype. Nat Microbiol . 2016;1:16031. doi: 10.1038/nmicrobiol.2016.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dickson RP, Martinez FJ, Huffnagle GB. The role of the microbiome in exacerbations of chronic lung diseases. Lancet . 2014;384:691–702. doi: 10.1016/S0140-6736(14)61136-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hilty M, Burke C, Pedro H, Cardenas P, Bush A, Bossley C, et al. Disordered microbial communities in asthmatic airways. PLoS One . 2010;5:e8578. doi: 10.1371/journal.pone.0008578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dickson RP, Singer BH, Newstead MW, Falkowski NR, Erb-Downward JR, Standiford TJ, et al. Enrichment of the lung microbiome with gut bacteria in sepsis and the acute respiratory distress syndrome. Nat Microbiol . 2016;1:16113. doi: 10.1038/nmicrobiol.2016.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Molyneaux PL, Cox MJ, Willis-Owen SA, Mallia P, Russell KE, Russell AM, et al. The role of bacteria in the pathogenesis and progression of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med . 2014;190:906–913. doi: 10.1164/rccm.201403-0541OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. O’Dwyer DN, Ashley SL, Gurczynski SJ, Xia M, Wilke C, Falkowski NR, et al. Lung microbiota contribute to pulmonary inflammation and disease progression in pulmonary fibrosis. Am J Respir Crit Care Med . 2019;199:1127–1138. doi: 10.1164/rccm.201809-1650OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sibila O, Laserna E, Shoemark A, Keir HR, Finch S, Rodrigo-Troyano A, et al. Airway bacterial load and inhaled antibiotic response in bronchiectasis. Am J Respir Crit Care Med . 2019;200:33–41. doi: 10.1164/rccm.201809-1651OC. [DOI] [PubMed] [Google Scholar]
- 11. Leitao Filho FS, Alotaibi NM, Ngan D, Tam S, Yang J, Hollander Z, et al. Sputum microbiome is associated with 1-year mortality after chronic obstructive pulmonary disease hospitalizations. Am J Respir Crit Care Med . 2019;199:1205–1213. doi: 10.1164/rccm.201806-1135OC. [DOI] [PubMed] [Google Scholar]
- 12. Rogers GB, Zain NM, Bruce KD, Burr LD, Chen AC, Rivett DW, et al. A novel microbiota stratification system predicts future exacerbations in bronchiectasis. Ann Am Thorac Soc . 2014;11:496–503. doi: 10.1513/AnnalsATS.201310-335OC. [DOI] [PubMed] [Google Scholar]
- 13. Bosch AATM, de Steenhuijsen Piters WAA, van Houten MA, Chu MLJN, Biesbroek G, Kool J, et al. Maturation of the infant respiratory microbiota, environmental drivers, and health consequences: a prospective cohort study. Am J Respir Crit Care Med . 2017;196:1582–1590. doi: 10.1164/rccm.201703-0554OC. [DOI] [PubMed] [Google Scholar]
- 14. Kelly BJ, Imai I, Bittinger K, Laughlin A, Fuchs BD, Bushman FD, et al. Composition and dynamics of the respiratory tract microbiome in intubated patients. Microbiome . 2016;4:7. doi: 10.1186/s40168-016-0151-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Panzer AR, Lynch SV, Langelier C, Christie JD, McCauley K, Nelson M, et al. Lung microbiota is related to smoking status and to development of acute respiratory distress syndrome in critically ill trauma patients. Am J Respir Crit Care Med . 2017;197:621–631. doi: 10.1164/rccm.201702-0441OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zakharkina T, Martin-Loeches I, Matamoros S, Povoa P, Torres A, Kastelijn JB, et al. The dynamics of the pulmonary microbiome during mechanical ventilation in the intensive care unit and the association with occurrence of pneumonia. Thorax . 2017;72:803–810. doi: 10.1136/thoraxjnl-2016-209158. [DOI] [PubMed] [Google Scholar]
- 17. Scicluna BP, Klein Klouwenberg PM, van Vught LA, Wiewel MA, Ong DS, Zwinderman AH, et al. A molecular biomarker to diagnose community-acquired pneumonia on intensive care unit admission. Am J Respir Crit Care Med . 2015;192:826–835. doi: 10.1164/rccm.201502-0355OC. [DOI] [PubMed] [Google Scholar]
- 18. Klein Klouwenberg PM, Ong DS, Bos LD, de Beer FM, van Hooijdonk RT, Huson MA, et al. Interobserver agreement of Centers for Disease Control and Prevention criteria for classifying infections in critically ill patients. Crit Care Med . 2013;41:2373–2378. doi: 10.1097/CCM.0b013e3182923712. [DOI] [PubMed] [Google Scholar]
- 19. van Vught LA, Klein Klouwenberg PM, Spitoni C, Scicluna BP, Wiewel MA, Horn J, et al. MARS Consortium. Incidence, risk factors, and attributable mortality of secondary infections in the intensive care unit after admission for sepsis. JAMA . 2016;315:1469–1479. doi: 10.1001/jama.2016.2691. [DOI] [PubMed] [Google Scholar]
- 20. Scicluna BP, van Vught LA, Zwinderman AH, Wiewel MA, Davenport EE, Burnham KL, et al. MARS consortium. Classification of patients with sepsis according to blood genomic endotype: a prospective cohort study. Lancet Respir Med . 2017;5:816–826. doi: 10.1016/S2213-2600(17)30294-1. [DOI] [PubMed] [Google Scholar]
- 21. Bos LD, Schouten LR, van Vught LA, Wiewel MA, Ong DSY, Cremer O, et al. MARS consortium. Identification and validation of distinct biological phenotypes in patients with acute respiratory distress syndrome by cluster analysis. Thorax . 2017;72:876–883. doi: 10.1136/thoraxjnl-2016-209719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zimmerman JE, Kramer AA, McNair DS, Malila FM. Acute Physiology and Chronic Health Evaluation (APACHE) IV: hospital mortality assessment for today’s critically ill patients. Crit Care Med . 2006;34:1297–1310. doi: 10.1097/01.CCM.0000215112.84523.F0. [DOI] [PubMed] [Google Scholar]
- 23. Jones AE, Trzeciak S, Kline JA. The Sequential Organ Failure Assessment score for predicting outcome in patients with severe sepsis and evidence of hypoperfusion at the time of emergency department presentation. Crit Care Med . 2009;37:1649–1654. doi: 10.1097/CCM.0b013e31819def97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mason KL, Erb Downward JR, Mason KD, Falkowski NR, Eaton KA, Kao JY, et al. Candida albicans and bacterial microbiota interactions in the cecum during recolonization following broad-spectrum antibiotic therapy. Infect Immun . 2012;80:3371–3380. doi: 10.1128/IAI.00449-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci USA . 2011;108:4516–4522. doi: 10.1073/pnas.1000080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol . 2013;79:5112–5120. doi: 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oksanen JF, Blanchet G, Kindt R, Legendre P, Minchin PR, O’Hara RB, et al. Vegan: community ecology package. R package version 2.0-4; 2012.
- 28.R Core Team. Vienna, Austria: R Foundation for Statistical Computing; 2013. R: a language and environment for statistical computing. [Google Scholar]
- 29. Wang Y, Naumann U, Wright ST, Warton DI. mvabund – an R package for model-based analysis of multivariate abundance data. Methods Ecol Evol . 2012;3:471–474. [Google Scholar]
- 30.Schloss PD. MiSeq SOP: mothur; 2015 [accessed 2018 Aug 1]. Available from: http://www.mothur.org/wiki/MiSeq_SOP.
- 31. Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol . 2009;75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liaw A, Wiener M. Classification and regression by randomForest. R News . 2002;2:18–22. [Google Scholar]
- 33. Salter SJ, Cox MJ, Turek EM, Calus ST, Cookson WO, Moffatt MF, et al. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol . 2014;12:87. doi: 10.1186/s12915-014-0087-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dickson RP, Erb-Downward JR, Prescott HC, Martinez FJ, Curtis JL, Lama VN, et al. Cell-associated bacteria in the human lung microbiome. Microbiome . 2014;2:28. doi: 10.1186/2049-2618-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schuijt TJ, Lankelma JM, Scicluna BP, de Sousa e Melo F, Roelofs JJ, de Boer JD, et al. The gut microbiota plays a protective role in the host defence against pneumococcal pneumonia. Gut . 2016;65:575–583. doi: 10.1136/gutjnl-2015-309728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Emonet S, Lazarevic V, Leemann Refondini C, Gaïa N, Leo S, Girard M, et al. Identification of respiratory microbiota markers in ventilator-associated pneumonia. Intensive Care Med . 2019;45:1082–1092. doi: 10.1007/s00134-019-05660-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dickson RP, Erb-Downward JR, Huffnagle GB. Towards an ecology of the lung: new conceptual models of pulmonary microbiology and pneumonia pathogenesis. Lancet Respir Med . 2014;2:238–246. doi: 10.1016/S2213-2600(14)70028-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schurink CAM, Nieuwenhoven CAV, Jacobs JA, Rozenberg-Arska M, Joore HCA, Buskens E, et al. Clinical pulmonary infection score for ventilator-associated pneumonia: accuracy and inter-observer variability. Intensive Care Med . 2004;30:217–224. doi: 10.1007/s00134-003-2018-2. [DOI] [PubMed] [Google Scholar]