

Abstract
Objective:
By binding to its high affinity receptor FcεR1, IgE activates mast cells, macrophages, and other inflammatory and vascular cells. Recent studies support an essential role of IgE in cardiometabolic diseases. Plasma IgE level is an independent predictor of human coronary heart disease. Yet, a direct role of IgE and its mechanisms in cardiometabolic diseases remain incompletely understood.
Approach and Results:
Using atherosclerosis prone Apoe−/− mice and IgE-deficient Ige−/− mice, we demonstrated that IgE deficiency reduced atherosclerosis lesion burden, lesion lipid deposition, smooth muscle cell and endothelial cell contents, chemokine MCP-1 expression and macrophage accumulation. IgE deficiency also reduced body weight gain and increased glucose and insulin sensitivities with significantly reduced plasma cholesterol, triglyceride, insulin, and inflammatory cytokines and chemokines, including IL-6, IFN-γ, and MCP-1. From atherosclerotic lesions and peritoneal macrophages from Apoe−/−Ige−/− mice that consumed an atherogenic diet, we detected reduced expression of M1 macrophage markers (CD68, MCP-1, TNF-α, IL-6, and iNOS) but increased expression of M2 macrophage markers (Arg-1 and IL-10) and “macrophage-sterol-responsive-network” (MSRN) molecules (complement C3, lipoprotein lipase, low-density lipoprotein receptor-related protein-1, and transferrin) that suppress macrophage foam cell formation. These IgE activities can be reproduced in bone marrow-derived macrophages from wild-type mice, but muted in cells from FcεR1-deficient mice, or blocked by anti-IgE antibody or complement C3 deficiency.
Conclusions:
IgE deficiency protects mice from diet-induced atherosclerosis, obesity, glucose tolerance, and insulin resistance by regulating macrophage polarization, MSRN gene expression, and foam cell formation.
Keywords: IgE, atherosclerosis, obesity, macrophage polarization, macrophage-sterol-responsive-network
Graphical Abstract
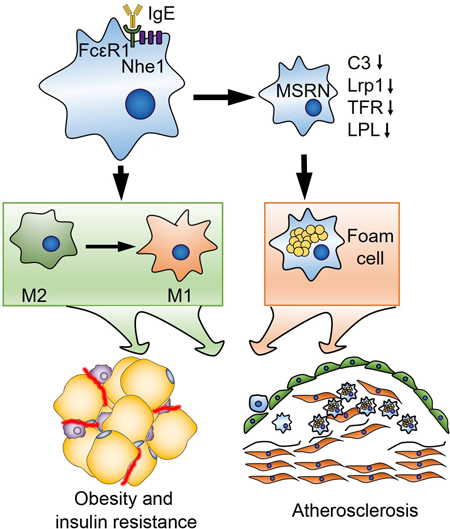
INTRODUCTION
IgE is an important regulator of allergic disease. It binds with high affinity to FcεR1 and with low affinity to FcεR2 (CD23)1,2. In mice and humans, FcεR1 is primarily expressed on mast cells and basophils. In addition to these granulocytes, other leukocytes such as macrophages, eosinophils, monocytes and dendritic cells, and even vascular cells also express this receptor3–5. IgE-mediated activation of macrophages releases inflammatory molecules such as IFN-γ and TNF-α6. From our previous report6 and those of others7,8, plasma IgE level associates with cardiovascular events in mice and humans and serves as an independent predictor for coronary heart disease (CHD). We reported that both IgE and FcεR1 are increased in human and mouse atherosclerotic lesions and colocalize to plaque macrophages6. Deficiency in secreted IgM (sIgM−/−) in low-density lipoprotein receptor-deficient (Ldlr−/−) mice increased plasma IgE and accelerated atherosclerosis9, whereas deficiency of FcεR1 protected apolipoprotein E-deficient (Apoe−/−) mice from diet-induced atherosclerosis6. Anti-IgE antibody reduced atherosclerosis in sIgM−/−Ldlr−/− mice9. In B-cell-deficient Apoe−/−μMT mice, IgE challenge increased atherosclerosis10. It was thought that IgE contributes to atherogenesis by activating mast cells, leading to enhanced atherosclerotic lesion neutrophil accumulation9,10. Yet, IgE may not only activate mast cells, but also other cells that express its receptor during atherogenesis.
Macrophages participate importantly in atherogenesis, but exert different roles depending on the types of macrophages. During atherogenesis, monocytes enter the atheroma and differentiate into M1 macrophages. These cells demonstrate a decreased ability to clear cholesterol and as a result, become foam cells after internalizing lipoprotein particles. Foam cells secrete local inflammatory cytokines, reactive oxygen species, and matrix metalloproteinases that destabilize the atherosclerotic plaques. Foamy macrophages also associate with consequent immune cell infiltration and smooth muscle cell (SMC) activation11, all which are essential to atherosclerotic lesion progression and rupture. In advanced atherosclerotic lesions, dying macrophages promote necrotic core formation and plaque complexity. In contrast, macrophages that initially infiltrate into the plaques may also exhibit an alternative macrophage (M2) phenotype and have a low susceptibility to become foam cells. These cells display high phagocytic activity and produce anti-inflammatory IL-10. As the plaque develops however, M2 macrophages subsequently shift to a M1 phenotype and secrete IL-1β, IL-6 and TNF-α at the advanced stage of the disease12,13.
Patients with type-2 diabetes demonstrate increased risk of having CHD with increased overall plaque burden, high rates of multi-vessel disease and death from vascular complications14,15. Macrophages serve as an important link between type-2 diabetes and atherosclerosis16,17. A major pathway of this link is the IFN-γ-regulated pro-atherogenic “macrophage-sterol-responsive-network” (MSRN). This sterol-responsive network was originally identified from the studies of peritoneal macrophages isolated from the Ldlr−/− mice that consumed a high cholesterol atherogenic diet. In these mice, macrophages convert free cholesterol into cholesteryl ester that accumulates as cytosolic lipid droplets and generate foam cells by perturbing the MSRN interacting proteins during atherogenesis. This network is also dysregulated by diet-induced obesity, predisposes macrophages to cholesterol accumulation, and promotes foam cell formation16,17.
In this study, we demonstrated that IgE deficiency in Apoe−/− mice reduced atherosclerosis by decreasing lesion macrophage content and inflammation. These mice showed altered atherosclerotic lesion and peritoneal macrophage phenotypes and improved obesity and insulin resistance. Mechanistic studies suggested that IgE controlled macrophage polarization, targeted the MSRN proteins, and promoted macrophage cholesterol accumulation, all which depended on the expression of FcεR1 and its associated protein Na+/H+ exchanger-1 (Nhe1).
METHODS
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Mice
Apoe−/−Ige−/− and Apoe−/−Ige+/+ were produced by breeding the Apoe−/− mice (#002052, C57BL/6J, The Jackson Laboratory, Bar Harbor, ME, USA) and Ige−/− mice18. Nhe1+/– mice (#003012, C57BL/6J, The Jackson Laboratory) were bred with Fcer1a−/− mice5 to produce the Fcer1a−/−Nhe1+/– mice. Six-week-old Apoe−/−Ige−/− (C57BL/6J, N=10) and Apoe−/−Ige+/+ (C57BL/6J, N=10) mice consumed a high cholesterol (1.25%) atherogenic diet (#D12108c, Research Diets Inc. New Brunswick, NJ) to induce atherosclerosis. Results from this study are limited to males. It is possible that IgE activity in atherosclerosis may differ in females according to the ATVB council statement19. Their body weights and food intake were measured weekly. After 12 weeks, mice were euthanized by CO2, followed by cardiac puncture blood and peritoneal macrophage collection, heart and aortic arch and root tissue harvest. Liver and adipose tissues, including epididymal adipose tissue, subcutaneous adipose tissue, and brown adipose tissue were isolated and weighed. All animal procedures conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health and was approved by the Brigham and Women’s Hospital Standing Committee on Animals (protocol #2016N000442).
Atherosclerotic lesion characterization and immunohistological analysis
Aortic root and arch were embedded in optimum cutting temperature and 6 μm aortic root serial sections and longitudinal aortic arch serial sections that contained all three branches (brachiocephalic artery, left common carotid artery, left subclavian artery) were prepared to analyze atherosclerotic lesions as previously described6. We designed, executed, and reported mouse atherosclerotic lesion analysis according to the guidelines for experimental atherosclerosis studies described in the AHA Statement20. To determine atherosclerotic lesion sizes, we stained the aortic root and arch sections with oil-red O (ORO). Lesion area (intima) and ORO-positive area were calculated. Toluidine blue (#89640, Sigma-Aldrich, St. Louis, MO) staining detected aortic arch section mast cells as we reported previously21. For immunohistological analysis, serial cryostat sections were prepared and stained for Mac-3 (macrophages, 1:900, #553322, BD Biosciences, San Jose, CA), CD31 (endothelial cells, 1:1500, #553370; BD Biosciences), α-smooth muscle actin (SMC, 1:500, BD Biosciences), myosin heavy chain-11 (1:2000, #702544, Thermo Fisher Scientific, Waltham, MA), major histocompatibility complex class-II (1:250, #556999, BD Biosciences), CD4 (1:90, #553043, BD Biosciences), CD8 (1:100, #14–0081-85; eBiosciences, San Diego, CA) and monocyte chemoattractant protein-1 (MCP-1, 1:100, #AF-479-NA, R&D Systems, Minneapolis, MN) antibodies, followed by appropriate biotin-conjugated secondary antibodies (1:500, Vector Laboratories, Burlingame, CA) and HRP-streptavidin (DAKO, Carpinteria, CA). All images were captured using a Microscope VS120 Whole Slide Scanner (Olympus) and analyzed using the computer-assisted Image-Pro Plus software (Media Cybernetics, Bethesda, MD). For immunofluorescent staining, aortic arch sections were prepared and stained to detect M1 macrophages by rabbit anti-iNOS (1:100, #PA1–036, Thermo Fisher Scientific) and rat anti-Mac-2 (1:100, CL8942LE, Cadarlane Laboratories, Burlington, NC), M2 macrophages by mouse anti-arginase-1 (Arg-1, 1:100, #678802, BioLegend, San Diego, CA) and rat anti-Mac-2 (1:100), and basophils by rat anti-CD49b (1:100, #14–5971-85, eBiosciences) and rabbit anti-FcεR1 (1:50, #06–727, Millipore, Burlington, MA) antibodies, followed by Alexa Fluor 555 or 488-labelled secondary antibody detection. DAPI (#D9542, Sigma-Aldrich) was used to stain the nuclei. All images were collected using the Olymbus Fluoview FV1000 confocal laser scanning microscopy.
Glucose tolerance test (GTT) and insulin tolerance test (ITT)
After 12 weeks of an atherogenic diet, both Apoe−/−Ige−/− and Apoe−/−Ige+/+ mice were also used for GTT and ITT assays after fasting22. For GTT, mice were i.p. injected with D-glucose (1 g/kg bodyweight, Sigma-Aldrich) after 16 hrs of fasting. For ITT, mice were i.p. injected with insulin (1.5 IU/kg bodyweight, Novolin) after 6 hrs of fasting. Blood glucose level was measured from tail veins using a blood glucose meter (Bayer Healthcare LLC, Mishawaka, IN) at 0, 15, 30, 45, 60, 90 and 120 min after injection.
Plasma insulin, inflammatory cytokines and lipoprotein measurement
The plasma levels of insulin (#90080, Crystal Chem, Elk Grove Village, IL), IL-1β (#88–7013-22, Invitrogen, Carlsbad, CA), TNF-α (#88–7324-22, Invitrogen), IL-6 (# 88–7064-88, Invitrogen), MCP-1 (#88–7391-88, Invitrogen), IFN-γ (#88–7314-88, Invitrogen), and histamine (#RE59221, IBL, Hamburg, Germany) were assessed using the mouse ELISA kits according to manufacturers. Plasma total cholesterol, triglyceride, and high-density lipoprotein (HDL) levels were determined by enzymatic methods using reagents from Pointe Scientific (Canton, MI) according to the manufacturer. The low-density lipoproteins (LDL) cholesterol level was determined using the Friedewald’s formula: Plasma LDL cholesterol concentration (mg/dL) = total cholesterol – HDL cholesterol – (triglycerides/5)23. To measure the enzymatic activities of β-hexosaminidase, we treated plasma with 1.3 mg/ml p-nitrophenyl-N-acetyl-β-D-glucosaminide (#N9376, Sigma-Aldrich) in 0.1M sodium citrate (pH 4.5, #S4641, Sigma-Aldrich) for 60 min at 37 °C. The reaction was stopped with 0.2 M glycine (pH 10.7, #G7126, Sigma-Aldrich). Absorbance at 405 nm measured the release of 4-p-nitrophenol.
Mouse plasma lipoprotein profiling was determined using high-resolution size-exclusion chromatography as previously reported24. Briefly, mouse plasma samples were fractionated by fast-performance liquid chromatography (FPLC; Merck-HPLC System) using the Superose 6 HR 10/30 size-exclusion chromatography column (GE Healthcare, Buckinghamshire, UK). The column was equilibrated with 10 mM sodium phosphate buffer, pH 7.4 containing 140 mM NaCl. After equilibration, 400 μl of mouse plasma pool was applied to the column with a flow rate of 0.5 ml/min at room temperature, and 0.5 ml fractions were collected and analyzed for cholesterol and triglyceride. For each group of animals, plasma pools were prepared from a combination of 3 mice. The elution fractions were analyzed for total cholesterol (enzymatic method, CHOD-PAP 1489232) and for triglycerides (enzymatic method, CHOD-PAP 1488872), both from Roche Diagnostics, Mannheim, Germany.
Cell culture
Bone marrow-derived macrophages (BMDMs) were prepared from wild type (WT), Fcer1α−/−, Fcer1α−/−Nhe1+/–, and C3−/− mice. Fresh bone marrow was isolated from the femurs and tibias bone and cultured DMEM supplemented with 10% FBS and 20 ng/ml of macrophage-colony stimulating factor (PeproTech, Rocky Hill, NJ) for 7 days. Mouse peritoneal macrophages were isolated from Apoe−/−Ige−/− and Apoe−/−Ige+/+ mice after consuming an atherogenic diet for 12 weeks by lavaging the peritoneal cavity with phosphate-buffered saline (PBS) containing 2% bovine serum albumin. After red blood cell lysis and washed with PBS, cells were cultured in DMEM with 10% FBS for 24 hrs to adhere prior to use. To assess the purity of peritoneal macrophages, we stained peritoneal cells with anti-CD11b-APC (1:100, #17–0112-82, eBiosciences) and anti- F4/80-PerCP-Cyanine5.5 (1:100, #123128, BioLegend), followed by flow cytometry (FACS) analysis. For macrophage foam cell formation assay, BMDM were incubated for 12 hrs in serum-free DMEM and then cultured with 50 μg/ml oxidized-LDL (ox-LDL, Alfa Aesar, Tewksbury, MA) for 48 hrs with or without 50 μg/ml of IgE (SPE-7, Sigma-Aldrich). After cells were stained with ORO, the lipid content was evaluated by alcohol extraction and absorbance measured at 540 nm. For macrophage polarization assay, BMDM were stimulated with or without lipopolysaccharide (LPS) (Sigma-Aldrich) (100 ng/ml, 24 hrs) or IL-4 (10 ng/ml, 24 hrs) with or without 50 μg/mL IgE. For IgE neutralization, 5 or 50 μg/ml anti-IgE (#93236, BioLegend) antibody was added to BMDM with 100 ng/ml LPS and 50 μg/ml IgE or 10 ng/ml IL-4 and 50 μg/ml IgE.
FACS
Cells from peritoneal cavity and blood were resuspended in PBS with 2% FBS after red blood cell lysis and washed with PBS, and incubated with FACS antibodies for 30 min on ice. Cells were initially selected by size on the basis of forward scatter (FSC) and side scatter (SSC), following separated on the basis of cell-surface markers using a FACS Analyzer LSR (BD Biosciences). To detect macrophage percentage in peritoneal cavity and blood, we stained macrophages with anti-CD45-FITC (1:100, #11–0451-82, eBiosciences), anti-CD11b-APC and anti- F4/80-PerCP-Cyanine5.5. To detect monocyte, we stained cells with anti-CD45-PerCP-Cyanine5.5 (1:100, #45–0451-82, Invitrogen), anti-CD11b-APC, and anti- Ly6C-FITC (1:250, #53–5932-82, Invitrogen). To detect mast cells, we stained cells with anti-CD45-PerCP-Cyanine5.5, anti-c-kit-APC (1:100, #17–1171-82, eBiosciences) and anti-FcεR1-FITC (1:250, #11–5898-82, eBiosciences). To detect basophils, we stained cells with anti-CD45-PerCP-Cyanine5.5, anti-CD63-PE/Cy7 (1:100, #143910, BioLegend), and CD200R3-PE (1:100, #142206, BioLegend).
Real-time PCR
Total RNA was isolated from aorta or cultured cells using Trizol reagent (Invitrogen). cDNA was synthesized from total RNA reverse transcriptase (Invitrogen) according to the manufacturer. The cDNA was used as a template for real-time RT-PCR (CFX Connect™ Real-Time PCR Detection System, Bio-Rad, Hercules, California) in the presence of iTaq™ Universal SYBR® Green Supermix (Bio-rad). All primer sequences are listed in Table S1. Data were processed using the ΔΔCT method. Glyceraldehyde 3-phosphate dehydrogenase was used as the reference gene.
Western blot
For immunoblot analysis, an equal amount of proteins extracted from BMDM of WT, Fcer1α−/−, Fcer1α−/−Nhe1+/–, and C3−/−mice were separated on SDS-PAGE, blotted, and detected with the following antibodies: rabbit anti-iNOS (1:1000, #PA1–036, Thermo Fisher Scientific), rat anti-Mac-2 (1:1000, #CL8942LE, Cadarlane Laboratories), mouse anti-Arg-1 (1:1000, #678802, BioLegend), rabbit anti-complement C3 (1:1000, #PA5–21349, Thermo Fisher Scientific), rabbit anti-pERK (1:1000, #4370), mouse anti-ERK antibody (1:1000, #9107), rabbit anti-pp38 (1:1000, #4631), mouse anti-p38 (1:1000, #9228) and rabbit anti-glyceraldehyde 3-phosphate dehydrogenase (1:2000, #2118S) antibodies from Cell Signaling Technology (Danvers, MA).
Statistics
All data were expressed as mean±SEM. We used non-parametric Mann-Whitney U test followed by Bonferroni correction to compare two-group data that did not pass the normality test. The statistical significance of weekly body weight was determined with a two-way analysis of variance repeated-measures; a Bonferroni post-test was used to obtain the P value for the bracketed pairwise comparison. Non-parametric Kruskal-Wallis test (one-way analysis of variance on ranks) was used for all cell culture data analysis that contained multiple group comparisons and that did not pass the normality test. SPSS16 version was used for analysis and P<0.05 were considered significant.
RESULTS
IgE deficiency reduces atherosclerosis and lesion SMC and microvessel contents in Apoe−/− mice
To test a direct role of IgE in atherogenesis, we fed IgE-deficient Apoe−/−Ige−/− mice and their Apoe−/−Ige+/+ littermates an atherogenic diet for 12 weeks. IgE deficiency reduced atherosclerosis development in the whole aorta from thoracic-abdomen to aortic arch and branches (Figure 1A), aortic arch (Figure I–A), branches (brachiocephalic artery, left common carotid artery, and left subclavian artery) (Figure I–B), and aortic root (Figure I–C), as determined by ORO staining. During the development of these atherosclerotic lesions, SMC and endothelial cell undergo phenotypic switching, pro-inflammatory activation, and abnormal proliferation and migration25,26. IgE deficiency reduced atherosclerotic lesion intima α-smooth muscle actin- or myosin heavy chain-11-positive SMC contents (Figure 1B/1C) and CD31-positive microvessel contents (Figure 1D) in the aortic arch, although we did not analyze those in the thoracic-abdominal aorta or in the aortic root. Reduced atherosclerosis in Apoe−/−Ige−/− mice was also associated with decreased plasma total cholesterol and total triglyceride levels, although plasma LDL and HDL levels did not differ from those in Apoe−/−Ige+/+ mice (Figure 1E). FPLC-based analysis for mouse lipoprotein profiling revealed significantly reduced plasma VLDL-C and VLDL-TG in Apoe−/−Ige−/− mice compared with those in Apoe−/−Ige+/+ mice (Figure 1F/1G). Reduced plasma VLDL can be secondary to the reduced atherosclerosis in Apoe−/−Ige−/− mice. It is still possible that IgE may directly contribute to VLDL synthesis or metabolism, a hypothesis that has not been tested in this study.
Figure 1.
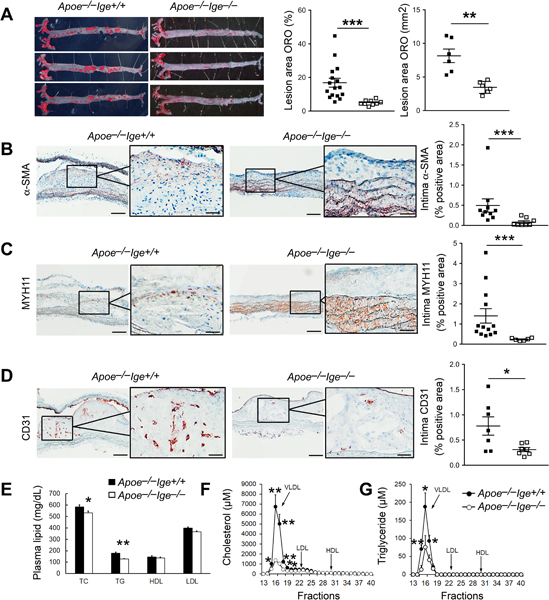
IgE deficiency reduces atherosclerosis in Apoe−/− mice after 12 weeks of an atherogenic diet. A. En face oil-red O staining and quantifications of the whole aortic tree. B. Intima α-SMC-positive area. C. Intima myosin heavy chain-11-positive area. D. Intima CD31-positive area. Representative images are shown to the left. Scale: 200 μm, inset: 70 μm. E. Plasma total cholesterol, total triglyceride, HDL, and LDL levels. F-G. Distribution of cholesterol (E) and triglyceride (F) in lipoprotein particles based on FPLC lipoprotein profiling. n=6~16 mice per group. All data are mean±SEM. *P<0.05, **P<0.01, ***P<0.001.
IgE deficiency reduces systemic and atherosclerotic lesion inflammation and promotes M2 macrophage polarization
Blood-borne leukocyte infiltration into the atherosclerotic lesions, such as T cells, monocytes, and macrophages, increases lesion inflammatory cytokine expression, matrix-degrading protease expression and activation, and promotes atherogenesis. Immunostaining did not detect significant difference of aortic arch lesion CD4+ and CD8+ T-cell numbers between Apoe−/−Ige+/+ and Apoe−/−Ige−/− mice (Figure II–A/II–B). Yet, IgE deficiency decreased lesion major histocompatibility complex class-II (Figure 2A) and Mac-3-positive macrophage contents (Figure 2B), suggesting reduced lesion inflammation in Apoe−/−Ige−/− mice. Reduction of lesion macrophage accumulation in Apoe−/−Ige−/− mice can also be secondary to reduced atherosclerosis of these mice (Figure 1A, I–A–I–C). Alternatively, these mice may have lower numbers of blood monocytes or macrophages or lower levels of lesion or blood chemokine and cytokine levels than those from the Apoe−/−Ige+/+ mice. FACS showed that F4/80+CD11b+ macrophage contents were reduced significantly in the peritoneal cavity in Apoe−/−Ige−/− mice (Figure 2C). Although IgE deficiency did not affect blood macrophage contents (Figure 2D), blood Ly6Clo and Ly6Chi monocytes were all reduced in Apoe−/−Ige−/− mice (Figure 2E), which may contribute to reduced atherosclerotic lesion macrophage contents in these mice27. Chemokine and inflammatory cytokines such as MCP-1, IL-1β, TNF-α and IFN-γ regulate lesion macrophage accumulation and inflammation13. Although we did not detect significant differences in plasma IL-1β or TNF-α levels between Apoe−/−Ige+/+ and Apoe−/−Ige−/− mice, IgE deficiency reduced aortic arch lesion MCP-1 expression (Figure 2F) and plasma cytokines IL-6 and IFN-γ and chemokine MCP-1 in Apoe−/−Ige−/− mice compared with those from Apoe−/−Ige+/+ mice (Figure 2G).
Figure 2.
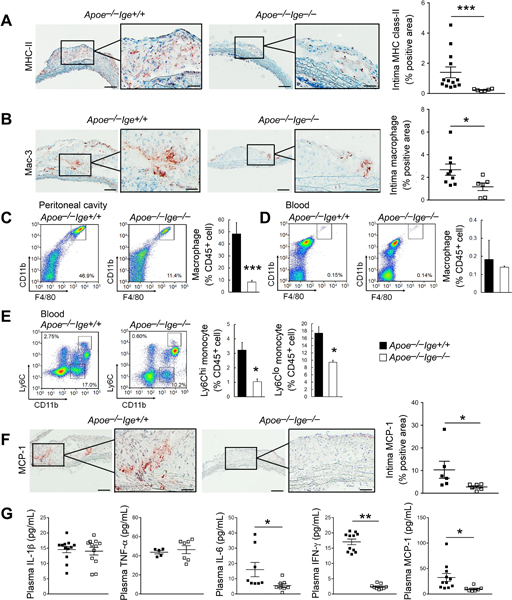
IgE deficiency reduces aortic arch lesion inflammation in Apoe−/− mice after 12 weeks of an atherogenic diet. Intima major histocompatibility complex class-II (A) and Mac-3-positive macrophage contents (B) were presented as positive area versus total intima area in percentage. C-D. FACS analysis of F4/80+CD11b+ macrophages in peritoneal cavity and blood. E. FACS analysis of Ly6ChiCD11b+ and Ly6ClowCD11b+ monocytes in blood. (F) Intima MCP-1 contents were presented as positive area versus total intima area in percentage. G. Plasma IL-1β, TNF-α, IL-6, IFN-γ and MCP-1 levels. n=6~13 mice per group. Representative images for panels A-F are shown to the left. Scales: 200 μm, insets: 70 μm. All data are mean±SEM. *P<0.05, **P<0.01, ***P<0.001.
Mast cells and basophils are the primary FcεR1 expressing cells. We tested whether IgE deficiency affected lesion or peripheral mast cell and basophil contents and their activities3. Toluidine blue staining did not detect significant difference of aortic arch intima and adventitia total (Figure III–A) and degranulated/activated mast cell numbers (Figure III–B) between Apoe−/−Ige+/+ and Apoe−/−Ige−/− mice. FACS analysis detected low blood c-kit+FcεR1+ mast cell progenitors28 from both groups of mice, and significantly fewer mast cells in the peritoneal cavity in Apoe−/−Ige−/− mice than in Apoe−/−Ige+/+ mice after consuming an atherogenic diet for 12 weeks (Figure III–C/III–D). Consistent with these observations, plasma histamine level and β-hexosaminidase activity (OD450nm) from Apoe−/−Ige−/− mice did not differ from those from Apoe−/−Ige+/+ mice (Figure III–E/III–F), suggesting that IgE deficiency did not affect systemic or local mast cell activation and other mechanisms replaced the role of IgE in mast cell activation29. Similarly, immunofluorescent double staining with antibodies against basophil CD49b and FcεR1 revealed few basophils in the aortic arch intima and their numbers did not differ between Apoe−/−Ige+/+ and Apoe−/−Ige−/− mice (Figure IV–A). FACS analysis using basophil activation marker CD6330 and CD220R3 demonstrated that IgE deficiency significantly reduced the numbers of activated basophils in blood but not those in the peritoneal cavity (Figure IV–B/IV–C). Therefore, mast cells and possibly basophils may contribute negligibly to reduced atherosclerosis in Apoe−/−Ige−/− mice.
Importantly, immunofluorescent double staining found that IgE deficiency significantly decreased atherosclerotic lesion intima iNOS+Mac-2+ M1 macrophage contents (Figure 3A) but increase intima Arg-1+Mac-2+ M2 macrophage contents (Figure 3B) in aortic arches. Consistent with these observations, the mRNA levels of macrophage marker CD68, M1 macrophage markers MCP-1, TNF-α, IL-6 and inducible nitric oxide synthase (iNOS) were decreased and the mRNA levels of M2 macrophage markers Arg-1 and IL-10 were increased (Figure 3C) in thoracic-abdominal aortas from IgE-deficient mice. We obtained the same observations from macrophages harvested from the peritoneum of Apoe−/−Ige−/− and Apoe−/−Ige+/+ mice that had consumed an atherogenic diet for 12 weeks. Peritoneal macrophages from Apoe−/−Ige−/− mice showed significantly reduced CD68 and M1 markers MCP-1, TNF-α, and iNOS but increased M2 markers Arg-1 and IL-10, although the IL-6 mRNA level did not differ from that of Apoe−/−Ige+/+ mice (Figure 3D). FACS analysis using CD11b and F4/80 antibodies verified the peritoneal macrophage purity (~75%) (Figure V).
Figure 3.
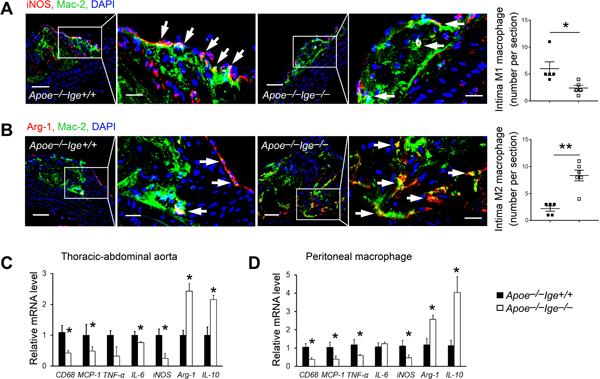
IgE deficiency tilts M2 macrophage polarization in Apoe−/− mice after 12 weeks of an atherogenic diet. Intima iNOS+Mac-2+ M1 macrophages (A) and Arg-1+Mac-2+ M2 macrophages (B) were presented as number per section. Scale: 100 μm, inset: 30 μm. RT-PCR determined the mRNA levels of inflammation and macrophage polarization genes in the thoracic-abdominal aortas (C) and peritoneal macrophages (D). n=5~13 mice per group. All data are mean±SEM. *P<0.05, **P<0.01.
IgE regulates macrophage polarization towards a M1 phenotype
Reduced M1 macrophage content and increased M2 macrophage content in thoracic-abdominal aortas and in peritoneal macrophages from Apoe−/−Ige−/− mice after mice consumed an atherogenic diet for 12 weeks (Figure 3A–3D) could be secondary to reduced atherosclerosis in these mice (Figure 1A, I–A–I–C). Alternatively, IgE activity may directly control macrophage polarization. We previously showed that FcεR1 and Toll-like receptor-4 (TLR-4) formed complexes. IgE failed to induce cytokine and chemokine expression and apoptosis in macrophages from FcεR1- or TLR-4-deficient mice6. We recently showed that FcεR1 also formed complexes with Nhe1. IgE failed to induce macrophage foam cell formation and activate the foam cell formation-associated PI3K-AKT-mTOR signaling pathway in cells from Apoe−/−Nhe1+/– mice31, suggesting that FcεR1 did not work alone. To test a direct role of IgE on macrophage polarization towards the M1 phenotypes, we added IgE to BMDM from both WT and IgE receptor-deficient Fcer1α−/− and Fcer1α−/−Nhe1+/– mice under M1- and M2-polarizing conditions in the presence of LPS or IL-4, respectively. Addition of IgE to WT BMDM under the M1-polarizing media with LPS significantly increased the iNOS, TNF-α, and Mcp-1 mRNA levels (Figure 4A). Such increases remained in cells from Fcer1α−/− mice, but muted in cells from Fcer1α−/−Nhe1+/– mice, although IgE still showed weak activity in enhancing TNF-α expression (Figure 4B/4C). We obtained the same observations in macrophages cultured in the M2-polarizing media with IL-4. IgE blocked M2 macrophage polarization in BMDM from WT mice by reducing the expression of Arg-1, IL-10, and Mrc-1 (Figure 5D). IgE lost its activity in reducing Arg-1, and Mrc-1 expression from BMDM from Fcer1α−/− and Fcer1α−/−Nhe1+/– mice, although IgE still showed its activity in reducing IL-10 expression from these macrophages (Figure 4E/4F).
Figure 4.
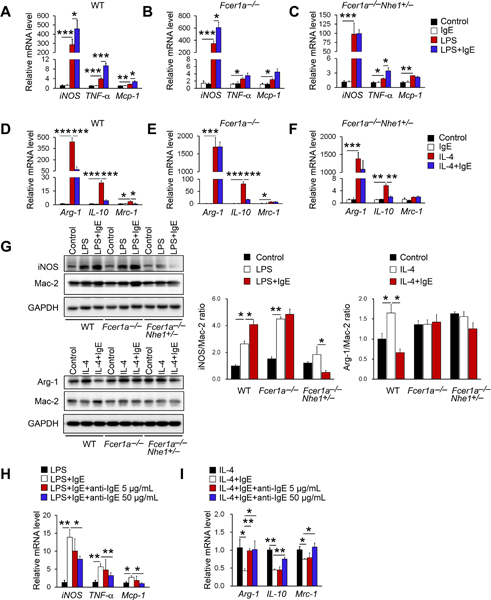
IgE activity in macrophages polarization. RT-PCR analysis of M1 macrophage marker genes after 24 hrs LPS (100 ng/mL) stimulation with or without IgE (50 μg/mL) in BMDM from WT, Fcer1α−/−, and Fcer1α−/−Nhe1+/− mice (A-C). RT-PCR analysis of M2 macrophage marker genes after 24 hrs IL-4 (10 ng/mL) stimulation with or without IgE in BMDM from WT, Fcer1α−/−, and Fcer1α−/−Nhe1+/− mice (D-F). (G) Immunoblots detected the expression of iNOS and Mac-2 and quantification of iNOS to Mac-2 ratio after LPS stimulation or the expression of Arg-1 and Mac-2 and quantification of Arg-1 to Mac-2 ratio after IL-4 stimulation with or without IgE in BMDM from WT, Fcer1α−/−, and Fcer1α−/−Nhe1+/− mice. Representative immunoblot images are shown to the left. RT-PCR analysis of M1 macrophage marker genes after LPS stimulation (H) and M2 macrophage marker genes after IL-4 stimulation (I) with or without IgE and anti-IgE antibody in BMDM from WT mice. Data are mean±SEM from 4 independent experiments. *P<0.05, **P<0.01, ***P<0.001.
Figure 5.
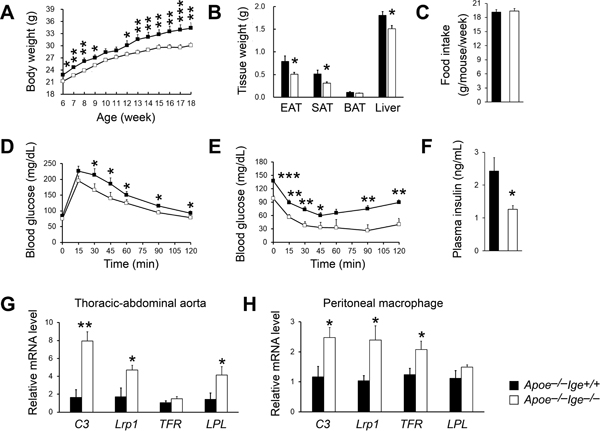
IgE deficiency reduces obesity and insulin resistance and promotes MSRN gene expression in aorta and peritoneal macrophages in Apoe−/− mice after 12 weeks of an atherogenic diet. Body weight gain (A), epididymal adipose tissue, subcutaneous adipose tissue, brown adipose tissue, and liver weight (B), food intake (C), GTT (D), ITT (E), and plasma insulin levels (F) were detected after 12 weeks of an atherogenic diet feeding. RT-PCR analysis of MSRN genes in the thoracic-abdominal aortas (G) and peritoneal macrophages (H). n=10 mice per group. All data are mean±SEM. *P<0.05, **P<0.01, ***P<0.001.
Consistent with the RT-PCR results from WT, Fcer1α−/− and Fcer1α−/−Nhe1+/– mice, immunoblot analyses showed that IgE increased LPS-induced iNOS expression in BMDM from WT mice. Such activity of IgE lost in cells from Fcer1α−/− mice. In contrast, LPS did not induce iNOS expression in BMDM from Fcer1α−/−Nhe1+/– mice (Figure 4G). Similarly, IgE blocked IL-4-induced Arg-1 expression in BMDM from WT mice. IL-4 and IgE both lost their activities in affecting Arg-1 expression in BMDM from Fcer1α−/− and Fcer1α−/−Nhe1+/– mice (Figure 4G). To test the role of IgE on macrophage polarization further, we used two different concentrations of IgE neutralizing antibody to block the IgE activity on BMDM. Results showed that 50 μg/mL IgE antibody effectively blocked the IgE activity in inducing M1 macrophage polarization (Figure 4H) and reversed IgE-reduced M2 macrophage polarization (Figure 4I). Therefore, IgE may use both FcεR1 and Nhe1 to regulate macrophage polarization.
IgE deficiency improves obesity and insulin resistance and increases MSRN gene expression.
Diabetes and obesity are significant risk factors of CHD, including increased overall plaque burden, high rates of multi-vessel disease and death from vascular complications14,15,32. Macrophages serve as an important link between these metabolic diseases and atherosclerosis16,17,33. After 12 weeks on an atherogenic diet, Apoe−/−Ige−/− mice showed reduced bodyweight gain (Figure 5A) together with reduced white adipose tissue and wet liver weight (Figure 5B), compared with those of Apoe−/−Ige+/+ control mice, although these two types of mice did not show significant differences in food intake (Figure 5C). IgE deficiency also improved glucose (Figure 5D) and insulin (Figure 5E) sensitivities with significantly reduced blood insulin levels (Figure 5F). At 6 weeks of age, Apoe−/−Ige+/+ mice were already heavier than the Apoe−/−Ige−/− mice (Figure 5A). To minimize the initial weight difference, we selected mice with comparable weight and monitored their bodyweight gain under a normal laboratory diet. IgE deficiency again reduced bodyweight gain at later time points (Figure VI). Therefore, IgE deficiency improved obesity and insulin sensitivity with concurrent decreases in adipose content and blood insulin levels.
Macrophage IFN-γ-regulated pro-atherogenic MSRN acts as an important link between metabolic diseases and atherosclerosis16,17. MSRN genes are synergistically dysregulated in obesity and macrophage cholesterol accumulation reduces MSRN gene expression16. Using RT-PCR, we compared the expression of genes from this network in atherosclerotic lesion and peritoneal macrophages from Apoe−/−Ige−/− and Apoe−/−Ige+/+ control mice that had consumed an atherogenic diet for 12 weeks. We found that IgE deficiency significantly increased the expression of the MSRN genes complement C3 (C3), low-density lipoprotein-related protein 1 (Lrp1), and lipoprotein lipase (LPL) from thoracic-abdominal aortic lesions from Apoe−/−Ige−/− mice, compared to those from the Apoe−/−Ige+/+ control mice, although the expression of transferrin (TFR) did not differ between the groups (Figure 5G). Peritoneal macrophages yielded the same conclusion. MSRN genes C3, Lrp1, TFR were also significantly upregulated in cells from Apoe−/−Ige−/− mice (Figure 5H).
IgE reduces macrophage IFN-γ-regulated MSRN gene expression and promotes macrophage foam cell formation
Prior study revealed a role of obesity and diabetes in promoting atherosclerosis by dysregulating IFN-γ-regulated MSRN protein expression17. Reduced plasma IFN-γ and increased MSRN protein expression in atherosclerotic lesions and macrophages from Apoe−/−Ige−/− mice support a role of IgE in reducing IFN-γ-mediated macrophage MSRN protein expression and promoting foam cell formation17. When BMDM from WT mice were stimulated with IFN-γ, IgE promoted ox-LDL uptake and macrophage foam cell formation (Figure 6A). In contrast, when BMDM from IgE receptor-deficient Fcer1α−/−Nhe1+/– mice were used, IgE did not affect IFN-γ-induced macrophage foam cell formation (Figure 6B).
Figure 6.
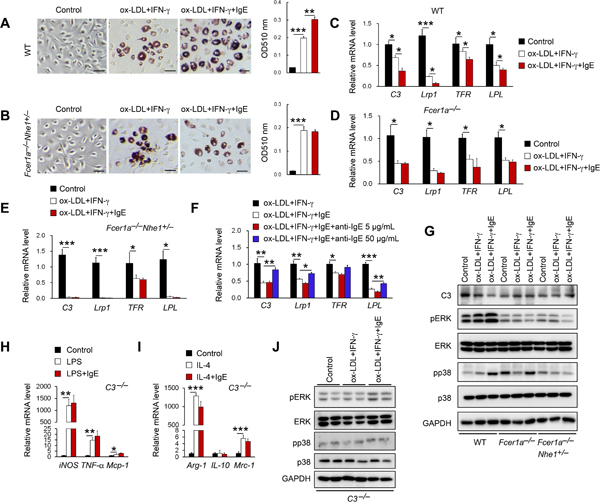
IgE reduces MSRN gene expression and promotes macrophages cholesterol loading. ORO staining and quantification of WT (A) and Fcer1α−/−Nhe1+/− (B) BMDM after 48 hrs of ox-LDL (2 mg/mL) and IFN-γ (12 ng/mL) stimulation with or without IgE (50 μg/mL). Scale: 50 μm. Representative images are shown to the left. RT-PCR analysis of MSRN genes in BMDM from WT (C), Fcer1α−/− (D), and Fcer1α−/−Nhe1+/− BMDM (E) after ox-LDL and IFN-γ stimulation with or without IgE. (F) RT-PCR analysis of MSRN genes after ox-LDL and IFN-γ stimulation with or without IgE and anti-IgE antibody in BMDM from WT mice. (G) Immunoblots detected the expression of C3, pERK, ERK, pp38 and p38 after ox-LDL and IFN-γ stimulation with or without IgE in BMDM from WT, Fcer1α−/− and Fcer1α−/−Nhe1+/− mice. RT-PCR analyses of M1 macrophage marker genes after LPS stimulation (H) and M2 macrophage marker genes after IL-4 stimulation (I) with or without IgE in BMDM from C3−/− mice. (J) Immunoblots detected the expression of pERK, ERK, pp38 and p38 after ox-LDL and IFN-γ stimulation with or without IgE in BMDM from C3−/− mice. Data are mean±SEM of four independent experiments. *P<0.05, **P<0.01, ***P<0.001.
Consistent with the increased expression of MSRN genes in atherosclerotic lesion and macrophages from Apoe−/−Ige−/− mice with atherosclerosis (Figure 5G/5H), IgE enhanced the ox-LDL- and IFN-γ-induced suppression of all tested MSRN genes C3, Lrp1, TFR and LPL in BMDM cells from WT mice (Figure 6C). IgE lost this activity when BMDM cells from Fcer1α−/− mice were used (Figure 6D). When cells from Fcer1α−/−Nhe1+/– mice were used, IFN-γ and ox-LDL blunted the expression of MSRN genes C3, Lrp1, TFR and LPL. Although we did not explore further why BMDM cells from Fcer1α−/−Nhe1+/– mice acted differently from those from Fcer1α−/− mice, IgE showed no effect on IFN-γ and ox-LDL-suppressed TFR expression (Figure 6E). IgE antibody further confirmed the IgE activity in reducing MSRN gene expression. At 50 μg/ml, anti-IgE antibody effectively blocked IgE-induced suppression of MSRN gene expression in BMDM from WT mice (Figure 6F). Immunoblots confirmed this IgE activity that was muted in cells from Fcer1α−/− or Fcer1α−/−Nhe1−/− mice (Figure 6G). Mechanistic studies showed that IgE enhanced ox-LDL- and IFN-γ-induced pERK and pp38 activation6. This activity of IgE also muted in BMDM from Fcer1α−/− or Fcer1α−/−Nhe1−/− mice (Figure 6G). These observations suggest that IgE reduced MSRN gene expression by activating the MAPK signaling pathway.
Using BMDM from C3−/− mice, we demonstrated a role of MSRN C3 molecule in mediating IgE activity in macrophage polarization. IgE lost its activity in increasing M1 macrophage gene expression (TNF-α, iNOS, Mcp-1) and in reducing M2 macrophage gene expression (Arg-1, IL-10, Mrc-1) (Figure 6H/6I) and IgE, ox-LDL, and IFN-γ lost their activity in pERK and pp38 activation in BMDM from C3−/− mice (Figure 6J).
DISCUSSION
Clinical and epidemiological studies link obesity and diabetes to cardiovascular diseases. Deterioration of one condition is often followed by the others14,15,34. Patients with chronic heart failure commonly have diabetes or prediabetes35 and similarly, patients with diabetes are prone to develop heart failure. Hyperglycemia is an important risk factor of heart failture36. Our prior studies used the FcεR1-deficient mice and indirectly suggested a role of IgE in atherosclerosis6. Here, we used IgE-deficient mice and established a direct role of IgE in atherosclerosis, obesity, and diabetes. Mechanistic studies revealed a role of IgE in macrophage polarization and foam cell formation by regulating the expression of the IFN-γ-controlled MSRN genes.
Macrophages are among the major inflammatory cells in atherosclerotic lesions. Macrophage foam cell formation leads to atherogenesis11. Consistent with our previous finding9, lack of IgE decreased macrophage inflammation, including reduced atherosclerotic lesion and peritoneal cavity macrophage accumulation, as well as decreased blood Ly6Chi and Ly6Clo monocytes, likely through a decrease in expression of chemokine MCP-1. A decrease of plasma IFN-γ and IL-6 levels in Apoe−/−Ige−/− mice after mice consuming an atherogenic diet for 12 weeks suggests a role of IgE in promoting systemic inflammation. As a result, macrophages in atherosclerotic lesion from Apoe−/−Ige−/− mice exhibited phenotypic changes from M1 to M2 phenotypes. This activity of IgE depended on the expression of macrophage FcεR1 and Nhe1, likely via the activation of TLR-4 and down stream NF-κB and JNK pathways as we previously described6. Although IgE plays a central role on the activation of mast cells and basophils that are rich in FcεR1 expression, the number and activation status of these innate immune cells in aortic lesions did not differ between Apoe−/−Ige+/+ and Apoe−/−Ige−/− mice. IgE deficiency decreased peritoneal mast cell and blood activated basophils but didn’t affect plasma histamine level and β-hexosaminidase activity. Therefore, we concluded that IgE-mediated activation of mast cells and basophils played a moderate role, but IgE actions on macrophages contributed importantly to diet-induced atherosclerosis.
In this study, we used BMDM from WT, Fcer1α−/−, and Fcer1α−/−Nhe1+/– mice and demonstrated a role of IgE in promoting LPS-induced iNOS, TNF-α, and Mcp-1 expression and reducing IL-4-induced Arg-1, IL-10, and Mrc-1 expression in macrophages from WT. IgE activity in LPS- and IL-4-regulated iNOS, Mcp-1, Arg-1, and Mrc-1 expression was completely muted in cells from Fcer1α−/−Nhe1+/– mice, but not in those from Fcer1α−/− mice. Yet, IgE remained effective in promoting TNF-α expression and reducing IL-10 expression in BMDM from Fcer1α−/−Nhe1+/– mice (Figure 4C/4F). Although we currently do not have an explanation to these observations, it is known that the expression of iNOS and Arg-1 and the expression of IL-10 and TNF-α use different signaling pathways. Macrophage expression of iNOS and Arg-1 uses the JNK/AP1 pathway37,38, whereas the expression of IL-10 and TNF-α is regulated by the MyD88-mediated activation of the MAPK pathway39 and the protein kinase C40 pathway. Here, we demonstrated that the MAPK pathway activation was decreased in BMDM from both Fcer1α−/− mice Fcer1α−/−Nhe1+/– mice (Figure 6G). Our earlier studies reported an interaction between FcεR1 and TLR-4 or Nhe16,31. It is possible that the protein kinase C pathway remained active in macrophages from Fcer1α−/−Nhe1+/– mice. Protein kinase C activation may stimulate the activity of remaining Nhe1 in these cells41 that mediated the IgE activity31, a hypotheses that merits further investigation. Nevertheless, in macrophages from WT mice, anti-IgE antibody effectively blocked IgE-induced TNF-α expression and IgE-suppressed IL-10 expression (Figure 4H/4I). FcεR1 deficiency blocked IgE activity in reducing the expression of MSRN genes C3, Lrp1, TFR and LPL and downstream signaling (pERK and pp38) (Figure 6D–6G). These results suggest that FcεR1 plays a predominant role in response to IgE. IgE regulates MSRN gene expression using FcεR1. IgE may use both FcεR1 and Nhe1 or additional receptors such as TLR-4 to regulate macrophages polarization.
Macrophage foam cell formation contributes critically to the development of advanced atherosclerotic plaques11. Cholesteryl ester loading promotes foam cell formation and atherogenesis through the MSRN16. Diet-induced obesity and insulin resistance produce IFN-γ from inflammatory cells to modulate the MSRN and link this network to atherogenesis17. In this study, we revealed a direct role of IgE in controlling the macrophage expression of the MSRN genes C3, LRP1, LPL and TRF, as a mechanism of IgE activity in macrophage foam cell formation. Consistent with the increased expression of these MSRN genes in atherosclerotic lesions from Apoe−/−Ige−/− mice, macrophages from these mice also demonstrated increased expression of these MSRN genes. This study first linked the IgE activity to IFN-γ-regulated MSRN gene expression reduction and foam cell formation. Yet, this study did not tell whether diet-induced obesity and insulin resistance dysregulated the expression of IFN-γ-regulated MSRN genes that affected atherogenesis, or the other way around. It is possible that the development of atherosclerosis increased systemic IFN-γ release and suppressed the same sets of MSRN genes, thereby promoting the development of obesity and insulin resistance, a hypothesis that has not been tested.
Together, this study revealed a detrimental role of IgE in atherosclerosis, obesity, and diabetes by polarizing macrophages towards a pro-inflammatory M1 phenotype and dysregulating the macrophage sterol response. These findings not only reinforce the role of IgE in atherosclerosis development but also propose a possibility that IgE links CHD to metabolic diseases via the MSRN and explain why patients with obesity and diabetes are prone to develop CHD, or vice versa. Inhibition of IgE activity may become an attractive therapeutic strategy to treat atherosclerosis, obesity, and diabetes.
Supplementary Material
Highlights.
IgE deficiency reduces diet-induced atherosclerosis and lesion contents of macrophages, smooth muscle cells, and microvessels in Apoe−/− mice.
IgE deficiency reduces diet-induced bodyweight gain, glucose tolerance, and insulin resistance.
IgE promotes macrophage polarization towards M1 phenotype.
IgE reduces macrophage-sterol-responsive-network gene expression and promotes macrophage cholesteryl ester intracellular accumulation and foam cell formation.
ACKNOWLEDGEMENTS
The authors thank Ms. Eugenia Shvartz for her technical assistance. The authors also thank Dr. Hans Oettgen from the Division of Immunology, Boston Children’s Hospital, Harvard Medical School for providing the IgE-deficient mice.
SOURCES OF FUNDING
This work was supported by grants from the Finance Science and Technology Projects of Hainan Province (ZDYF2018102 to JG); the National Natural Science Foundation of China (91939107 and 81770487 to JG); the National Institute of Health, USA [HL123568 and HL60942 to GPS]. Dr. Xian Zhang is supported by the American Heart Association Postdoctoral Fellowship #18POST34050043.
NONSTANDARD ABBREVIATIONS AND ACRONYMS
- CHD
coronary heart disease
- SMC
smooth muscle cell
- MSRN
macrophage-sterol-responsive-network
- Nhe1
Na+/H+ exchanger 1
- Arg-1
arginase-1
- iNOS
inducible nitric oxide synthase
- ORO
oil-red O
- GTT
glucose tolerance test
- ITT
insulin tolerance test
- MCP-1
monocyte chemoattractant protein-1
- Mrc-1
macrophage mannose receptor 1
- HDL
high-density lipoprotein
- LDL
low-density lipoprotein
- BMDM
bone marrow-derived macrophage
- PBS
phosphate-buffered saline
- ox-LDL
oxidized-LDL
- LPS
lipopolysaccharide
- WT
wild-type
- C3
complement C3
- Lrp1
low-density lipoprotein receptor-related protein-1
- LPL
lipoprotein lipase
- TFR
transferrin
- TLR-4
toll-like receptor-4
Footnotes
DISCLOSURES
None.
REFERENCES
- 1.Kinet JP. The high-affinity ige receptor (fc epsilon ri): From physiology to pathology. Annu Rev Immunol. 1999;17:931–972. [DOI] [PubMed] [Google Scholar]
- 2.Kraft S, Kinet JP. New developments in fcepsilonri regulation, function and inhibition. Nat Rev Immunol. 2007;7:365–378. [DOI] [PubMed] [Google Scholar]
- 3.Gould HJ, Sutton BJ. Ige in allergy and asthma today. Nat Rev Immunol. 2008;8:205–217. [DOI] [PubMed] [Google Scholar]
- 4.Liu FT, Goodarzi H, Chen HY. Ige, mast cells, and eosinophils in atopic dermatitis. Clin Rev Allergy Immunol. 2011;41:298–310. [DOI] [PubMed] [Google Scholar]
- 5.Wang J, Lindholt JS, Sukhova GK, Shi MA, Xia M, Chen H, Xiang M, He A, Wang Y, Xiong N, Libby P, Wang JA, Shi GP. Ige actions on cd4+ t cells, mast cells, and macrophages participate in the pathogenesis of experimental abdominal aortic aneurysms. EMBO Mol Med. 2014;6:952–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang J, Cheng X, Xiang MX, Alanne-Kinnunen M, Wang JA, Chen H, He A, Sun X, Lin Y, Tang TT, Tu X, Sjoberg S, Sukhova GK, Liao YH, Conrad DH, Yu L, Kawakami T, Kovanen PT, Libby P, Shi GP. Ige stimulates human and mouse arterial cell apoptosis and cytokine expression and promotes atherogenesis in apoe−/− mice. J Clin Invest. 2011;121:3564–3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo X, Yuan S, Liu Y, Zeng Y, Xie H, Liu Z, Zhang S, Fang Q, Wang J, Shen Z. Serum ige levels are associated with coronary artery disease severity. Atherosclerosis. 2016;251:355–360. [DOI] [PubMed] [Google Scholar]
- 8.Sinkiewicz W, Blazejewski J, Bujak R, Kubica J, Dudziak J. Immunoglobulin e in patients with ischemic heart disease. Cardiol J. 2008;15:122–128. [PubMed] [Google Scholar]
- 9.Tsiantoulas D, Bot I, Ozsvar-Kozma M, Goderle L, Perkmann T, Hartvigsen K, Conrad DH, Kuiper J, Mallat Z, Binder CJ. Increased plasma ige accelerate atherosclerosis in secreted igm deficiency. Circ Res. 2017;120:78–84. [DOI] [PubMed] [Google Scholar]
- 10.Wezel A, Lagraauw HM, van der Velden D, de Jager SC, Quax PH, Kuiper J, Bot I. Mast cells mediate neutrophil recruitment during atherosclerotic plaque progression. Atherosclerosis. 2015;241:289–296. [DOI] [PubMed] [Google Scholar]
- 11.Libby P, Ridker PM, Hansson GK, Leducq Transatlantic Network on A. Inflammation in atherosclerosis: From pathophysiology to practice. J Am Coll Cardiol. 2009;54:2129–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khallou-Laschet J, Varthaman A, Fornasa G, Compain C, Gaston AT, Clement M, Dussiot M, Levillain O, Graff-Dubois S, Nicoletti A, Caligiuri G. Macrophage plasticity in experimental atherosclerosis. PLoS One. 2010;5:e8852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Komohara Y, Fujiwara Y, Ohnishi K, Shiraishi D, Takeya M. Contribution of macrophage polarization to metabolic diseases. J Atheroscler Thromb. 2016;23:10–17. [DOI] [PubMed] [Google Scholar]
- 14.Beckman JA, Creager MA, Libby P. Diabetes and atherosclerosis: Epidemiology, pathophysiology, and management. JAMA. 2002;287:2570–2581. [DOI] [PubMed] [Google Scholar]
- 15.Hayward RA, Reaven PD, Emanuele NV, Investigators V. Follow-up of glycemic control and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2015;373:978. [DOI] [PubMed] [Google Scholar]
- 16.Becker L, Gharib SA, Irwin AD, Wijsman E, Vaisar T, Oram JF, Heinecke JW. A macrophage sterol-responsive network linked to atherogenesis. Cell Metab. 2010;11:125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reardon CA, Lingaraju A, Schoenfelt KQ, Zhou G, Cui C, Jacobs-El H, Babenko I, Hoofnagle A, Czyz D, Shuman H, Vaisar T, Becker L. Obesity and insulin resistance promote atherosclerosis through an ifngamma-regulated macrophage protein network. Cell Rep. 2018;23:3021–3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oettgen HC, Martin TR, Wynshaw-Boris A, Deng C, Drazen JM, Leder P. Active anaphylaxis in ige-deficient mice. Nature. 1994;370:367–370. [DOI] [PubMed] [Google Scholar]
- 19.Robinet P, Milewicz DM, Cassis LA, Leeper NJ, Lu HS, Smith JD. Consideration of sex differences in design and reporting of experimental arterial pathology studies-statement from atvb council. Arterioscler Thromb Vasc Biol. 2018;38:292–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daugherty A, Tall AR, Daemen M, Falk E, Fisher EA, Garcia-Cardena G, Lusis AJ, Owens AP, 3rd, Rosenfeld ME, Virmani R, American Heart Association Council on Arteriosclerosis T, Vascular B, Council on Basic Cardiovascular S. Recommendation on design, execution, and reporting of animal atherosclerosis studies: A scientific statement from the american heart association. Arterioscler Thromb Vasc Biol. 2017;37:e131–e157. [DOI] [PubMed] [Google Scholar]
- 21.Liu J, Divoux A, Sun J, Zhang J, Clement K, Glickman JN, Sukhova GK, Wolters PJ, Du J, Gorgun CZ, Doria A, Libby P, Blumberg RS, Kahn BB, Hotamisligil GS, Shi GP. Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nat Med. 2009;15:940–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang X, Zhang QX, Wang X, Zhang L, Qu W, Bao B, Liu CA, Liu J. Dietary luteolin activates browning and thermogenesis in mice through an ampk/pgc1alpha pathway-mediated mechanism. Int J Obes (Lond). 2016;40:1841–1849. [DOI] [PubMed] [Google Scholar]
- 23.Tremblay AJ, Morrissette H, Gagne JM, Bergeron J, Gagne C, Couture P. Validation of the friedewald formula for the determination of low-density lipoprotein cholesterol compared with beta-quantification in a large population. Clin Biochem. 2004;37:785–790. [DOI] [PubMed] [Google Scholar]
- 24.Zhang X, Huang Q, Wang X, Deng Z, Li J, Yan X, Jauhiainen M, Metso J, Libby P, Liu J, Shi GP. Dietary cholesterol is essential to mast cell activation and associated obesity and diabetes in mice. Biochim Biophys Acta Mol Basis Dis. 2019;1865:1690–1700. [DOI] [PubMed] [Google Scholar]
- 25.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. [DOI] [PubMed] [Google Scholar]
- 26.Gimbrone MA, Jr., Garcia-Cardena G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. 2016;118:620–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rahman K, Vengrenyuk Y, Ramsey SA, Vila NR, Girgis NM, Liu J, Gusarova V, Gromada J, Weinstock A, Moore KJ, Loke P, Fisher EA. Inflammatory ly6chi monocytes and their conversion to m2 macrophages drive atherosclerosis regression. J Clin Invest. 2017;127:2904–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dahlin JS, Ekoff M, Grootens J, Lof L, Amini RM, Hagberg H, Ungerstedt JS, Olsson-Stromberg U, Nilsson G. Kit signaling is dispensable for human mast cell progenitor development. Blood. 2017;130:1785–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shi GP, Bot I, Kovanen PT. Mast cells in human and experimental cardiometabolic diseases. Nat Rev Cardiol. 2015;12:643–658. [DOI] [PubMed] [Google Scholar]
- 30.Sanz ML, Sanchez G, Gamboa PM, Vila L, Uasuf C, Chazot M, Dieguez I, De Weck AL. Allergen-induced basophil activation: Cd63 cell expression detected by flow cytometry in patients allergic to dermatophagoides pteronyssinus and lolium perenne. Clin Exp Allergy. 2001;31:1007–1013. [DOI] [PubMed] [Google Scholar]
- 31.Liu CL, Zhang X, Liu J, Wang Y, Sukhova GK, Wojtkiewicz GR, Liu T, Tang R, Achilefu S, Nahrendorf M, Libby P, Guo J, Zhang JY, Shi GP. Na(+)-h(+) exchanger 1 determines atherosclerotic lesion acidification and promotes atherogenesis. Nat Commun. 2019;10:3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ortega FB, Lavie CJ, Blair SN. Obesity and cardiovascular disease. Circ Res. 2016;118:1752–1770. [DOI] [PubMed] [Google Scholar]
- 33.Kralova Lesna I, Petras M, Cejkova S, Kralova A, Fronek J, Janousek L, Thieme F, Tyll T, Poledne R. Cardiovascular disease predictors and adipose tissue macrophage polarization: Is there a link? Eur J Prev Cardiol. 2018;25:328–334. [DOI] [PubMed] [Google Scholar]
- 34.Lovren F, Teoh H, Verma S. Obesity and atherosclerosis: Mechanistic insights. Can J Cardiol. 2015;31:177–183. [DOI] [PubMed] [Google Scholar]
- 35.Kristensen SL, Preiss D, Jhund PS, Squire I, Cardoso JS, Merkely B, Martinez F, Starling RC, Desai AS, Lefkowitz MP, Rizkala AR, Rouleau JL, Shi VC, Solomon SD, Swedberg K, Zile MR, McMurray JJ, Packer M, Investigators P-H, Committees. Risk related to pre-diabetes mellitus and diabetes mellitus in heart failure with reduced ejection fraction: Insights from prospective comparison of arni with acei to determine impact on global mortality and morbidity in heart failure trial. Circ Heart Fail. 2016;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.MacDonald MR, Petrie MC, Varyani F, Ostergren J, Michelson EL, Young JB, Solomon SD, Granger CB, Swedberg K, Yusuf S, Pfeffer MA, McMurray JJ, Investigators C. Impact of diabetes on outcomes in patients with low and preserved ejection fraction heart failure: An analysis of the candesartan in heart failure: Assessment of reduction in mortality and morbidity (charm) programme. Eur Heart J. 2008;29:1377–1385. [DOI] [PubMed] [Google Scholar]
- 37.Bhatt KH, Sodhi A, Chakraborty R. Role of mitogen-activated protein kinases in peptidoglycan-induced expression of inducible nitric oxide synthase and nitric oxide in mouse peritoneal macrophages: Extracellular signal-related kinase, a negative regulator. Clin Vaccine Immunol. 2011;18:994–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tugal D, Liao X, Jain MK. Transcriptional control of macrophage polarization. Arterioscler Thromb Vasc Biol. 2013;33:1135–1144. [DOI] [PubMed] [Google Scholar]
- 39.Guha M, O’Connell MA, Pawlinski R, Hollis A, McGovern P, Yan SF, Stern D, Mackman N. Lipopolysaccharide activation of the mek-erk1/2 pathway in human monocytic cells mediates tissue factor and tumor necrosis factor alpha expression by inducing elk-1 phosphorylation and egr-1 expression. Blood. 2001;98:1429–1439. [DOI] [PubMed] [Google Scholar]
- 40.Niedbala MJ, Stein-Picarella M. Role of protein kinase c in tumor necrosis factor induction of endothelial cell urokinase-type plasminogen activator. Blood. 1993;81:2608–2617. [PubMed] [Google Scholar]
- 41.Soleimani M, Singh G, Dominguez JH, Howard RL. Long-term high osmolality activates na(+)-h+ exchange and protein kinase c in aortic smooth muscle cells. Circ Res. 1995;76:530–535. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.