

Abstract
Objective -
Retinoic acid (RA) is a ligand for nuclear receptors that modulate gene transcription and cell differentiation. Whether RA controls ectopic calcification in humans is unknown. We tested the hypothesis that RA regulates osteogenic differentiation of human arterial smooth muscle cells (SMC) and aortic valvular interstitial cells (VIC) that participate respectively in atherosclerosis and heart valve disease.
Approach and Results -
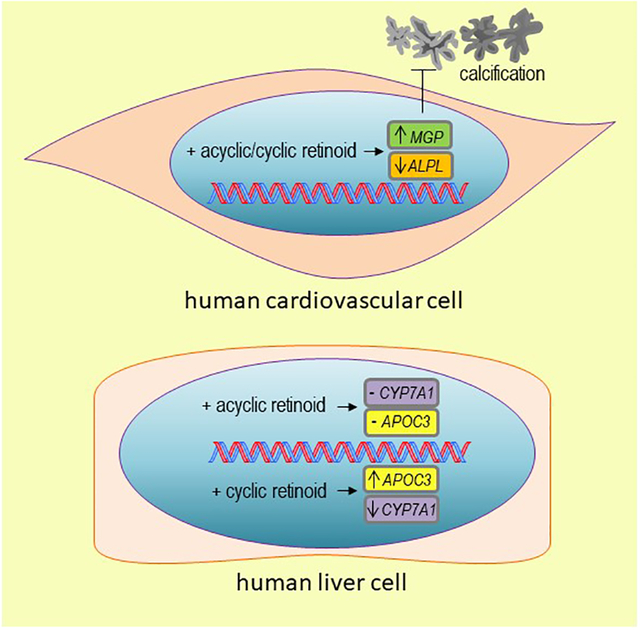
Human cardiovascular tissue contains immunoreactive retinoic acid receptor (RAR), a retinoid-activated nuclear receptor directing multiple transcriptional programs. RA stimulation suppressed primary human cardiovascular cell calcification while treatment with the RAR inhibitor AGN 193109 or RARα siRNA increased calcification. RA attenuated calcification in a coordinated manner – increasing levels of the calcification inhibitor matrix Gla protein while decreasing calcification-promoting tissue non-specific alkaline phosphatase activity. Given that nuclear receptor action varies as a function of distinct ligand structures, we compared calcification responses to cyclic retinoids and the acyclic retinoid, peretinoin. Peretinoin suppressed human cardiovascular cell calcification without inducing either secretion of apolipoprotein-CIII, which promotes atherogenesis, or reducing cytochrome P450 family 7 subfamily A member 1 expression, which occurred with cyclic retinoids all-trans retinoic acid, 9-cis retinoic acid, and 13-cis retinoic acid. Additionally, peretinoin did not suppress human femur osteoblast mineralization, whereas all-trans retinoic acid inhibited osteoblast mineralization.
Conclusions -
These results establish retinoid regulation of human cardiovascular calcification, provide new insight into mechanisms involved in these responses, and suggest selective retinoid modulators, like acyclic retinoids may allow for treating cardiovascular calcification without the adverse effects associated with cyclic retinoids.
Keywords: retinoic acid, matrix Gla protein, cardiovascular calcification, apolipoprotein-CIII
Subject codes: Cell Signaling/Signal Transduction, Vascular Disease, Valvular Heart Disease
Graphical Abstract
Introduction
Cardiovascular calcification predicts risk for coronary heart disease events.1 Arterial calcium deposition impairs vascular elasticity2 while microcalcifications within the fibrous cap of atheromata may promote plaque rupture.3,4 Clinical observations link cardiovascular calcification and bone metabolism although often in a divergent manner:5 bone density and fractures associate inversely with the extent of vascular calcification and cardiovascular risk.6,7 Vascular smooth muscle cells (SMC) and valvular interstitial cells (VIC) differentiate to osteoblast-like cells that express several osteogenic-related genes.8,9,10 Mechanistic studies support this cardiovascular calcification-bone osteogenesis connection, with overlapping pathways observed in cardiovascular and skeletal bone mineralization.11 Retinoic acid (RA), a metabolite of vitamin A, is a mediator linking bone and cardiovascular mineralization through its potent, proximal regulation of transcriptional programs. Despite this, the role of retinoid signaling in cardiovascular calcification, particularly in humans remains poorly defined.
We previously reported deficiency of retinaldehyde dehydrogenase 1 increases bone formation in mice in a specific, restricted pattern.12,13 Vitamin A metabolism produces retinaldehyde, which is further metabolized by retinaldehyde dehydrogenase 1 to generate the key retinoid, all-trans-retinoic acid (ATRA), which exerts its effect in large part by activating the nuclear receptor, retinoid activated receptor (RAR). While RA convincingly increases ectopic calcification in rodents and birds,14,15,16 the role of RA in human cardiovascular cell calcification is unknown. Vitamin A supplementation can suppress inflammatory processes in human atherosclerotic patients;17 however, beta carotene, which is converted into vitamin A does not impact cardiovascular events in women at high risk of cardiovascular disease.18 Previous studies have demonstrated major cell type and species differences in RA-mediated gene expression.16,19,20,21 Given these divergent reports, an understanding of the effects of RA on cardiovascular calcification in human cells and tissues is important. Here, we demonstrate a novel species difference in retinoid regulation of cardiovascular calcification, with retinoids inhibiting human cardiovascular cell calcification. Additionally, this study shows the cyclohexene hydrocarbon moiety present in cyclic retinoids mediates adverse retinoid responses, while acyclic retinoid inhibited cardiovascular cell calcification without adverse effects.
Materials and Methods
The authors declare that all supporting data are available within the article and its online supplementary files.
Cell culture
Human coronary artery SMC (PromoCell, Heidelberg, Germany) were expanded in Smooth Muscle Cell Growth Medium 2 (PromoCell) supplemented with epidermal growth factor (0.5ng/mL), insulin (5μg/mL) basic fibroblast growth factor- B (2ng/mL), and 5% fetal bovine serum, and cells were used between passages 3 and 8. SMC identity was confirmed by α-smooth muscle actin flow cytometry using a BDFACSAria™ II flow cytometer (BD Biosciences, Franklin Lakes, NJ). SMC were cultured up to 21 days in either control medium, CM: DMEM, 4.5g/L glucose, L-glutamine, 10% FBS, 1% penicillin/streptomycin; or osteogenic medium, OM: CM supplemented with 10nmol/L dexamethasone, 10mmol/L β–glycerol phosphate, and 100μmol/L L-ascorbic acid 2-phosphate. Cells were treated with combinations of the following reagents: 0.1% DMSO vehicle; ATRA (Sigma, St. Louis, MO); AGN 193109 (Sigma), 9-cis retinoic acid (Sigma), or peretinoin (MedChem Express LLC, Monmouth Junction, NJ). Human femur bone alkaline phosphatase positive osteoblasts (PromoCell) were expanded and cultured in an identical manner as with human SMC. Vimentin positive primary human VIC were obtained using isolated human aortic valve tissue from patients undergoing valve replacement (Institutional Review Board protocol #2011P001703; written informed consent was obtained for human tissue donors), and cultured similarly to SMC, but with some changes to the calcification inducing media, pro-calcifying media, PM: CM supplemented with 2mmol/L NaH2PO4 (Sigma) and 50μg/mL L-ascorbic (Sigma). Medium was changed every three to four days. For knockdown experiments, siRNA (used at a final concentration 20 nmol/L) was added with each media change according to the Lipofectamine RNAiMAX (Thermo Fisher Scientific, Waltham, MA) protocol. The following validated siRNA (Thermo Fisher Scientific) were used and independently confirmed: RARα siRNA (#s11801), RARβ (#s11804), RARγ siRNA (#s11807), and negative control siRNA (#4390843). HepG2 cells (ATCC, Manassas, VA) were grown in EMEM with 1% penicillin/streptomycin and 10% FBS. For RA stimulation experiments, HepG2 were switched to EMEM with 1% penicillin/streptomycin, and without FBS but containing 1% bovine serum albumin at the time of RA stimulation and cultured for 24 hours.
Calcification analysis
Alizarin red staining was performed after two to three weeks in CM, OM, or PM. Cells were fixed in 4% paraformaldehyde and incubated in 2% Alizarin red solution at room temperature for one hour, and then rinsed three times with water. The stain was extracted by incubating cells in 100mmol/L cetylpyridinium chloride (Sigma) for 30 minutes with gentle shaking, and supernatant absorbance was read at 540nm. Von Kossa staining was performed by incubating fixed cells in 5% silver nitrate (American MasterTech Scientific, Lodi, CA) under ultraviolet light for 30 minutes, followed by rinsing in distilled water and incubating with 5% sodium thiosulfate (American MasterTech Scientific) for 2 minutes, and then rinsed in distilled water. OsteoSense 680 (PerkinElmer, Waltham, MA) staining was performed with a 1:100 dilution and overnight incubation at 37 degrees Celsius, followed by imaging on a Nikon A1 confocal microscope (Nikon Instruments, Melville, NY).
TNAP activity and extracellular vesicles
Cellular TNAP activity was determined by the Alkaline Phosphatase Activity Colorimetric Assay Kit (BioVision, Milpitas, CA) according to the manufacturer’s protocol, and normalized by protein content, determined with the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). Extracellular vesicle TNAP activity was assayed in vesicles in a similar manner after isolating vesicles by centrifuging cell culture media at 2,000 X g to remove any cell debris, and then centrifuging at 100,000G and four degrees Celsius for one hour to pellet vesicles. Vesicle diameter and particle number was determined by nanoparticle tracking analysis using a NanoSight LM10 (Malvern Instruments, Malvern, United Kingdom). Samples were diluted 1:10 in phosphate buffered saline prior to injection, and continuously injected via a syringe pump, with five nanoparticle tracking analysis videos collected for each minute. Camera gain was set at a constant value of 9, and the threshold value for vesicle detection was set to 2. Size and concentration from the data in each video was averaged to generate the distribution per sample.
RNA and protein analysis
Total RNA was extracted with TRIzol (Thermo Fisher Scientific), and cDNA was generated using the qScript cDNA Synthesis Kit (Quanta Biosciences, Gaithersburg, MD). Calcified and non-calcified human tissues were assessed by gross morphology and OsteoSense 680 staining. Quantitative PCR was performed using the following Taqman probes (Thermo Fisher Scientific): Hs01029144_m1 (ALPL); Hs00985000_g1 (APOA1); Hs00163644_m1 (APOC3); Hs00167982_m1 (CYP7A1); Hs01054040_m1 (ENPP1); Hs02758991_g1 (GAPDH); Hs00230853_m1 (HNF4α); Hs00969490_m1 (MGP); Hs05013609_mH (LPA); Hs01016719_m1 (PPARGC1α); Hs00751239_s1 (MSX2); Hs00940445_g1 (RARα);Hs00977140_m1 (RARβ); Hs01559234_m1 (RARγ); Hs00231692_m1 (RUNX2); Hs01001343_g1 (SOX9). Gene expressions were normalized to GAPDH using the ΔΔCt method. Secreted matrix Gla protein (MGP) was measured by ELISA (Lifespan Biosciences Inc, Seattle, WA, #LS-F4628) according to the manufacture’s protocol. APO(a) protein was assessed in HepG2 cells by taking HepG2 treated for 24 hours and lysing in RIPA buffer (Thermo Fisher Scientific) containing 1:100 Halt protease and phosphatase inhibitor (Thermo Fisher Scientific). Cell lysates were run on PROTEAN TGX precast gels (Bio-Rad Laboratories, Hercules, CA) and blotted using APO(a)/Lp(a) antibody (Lee Biosolutions Inc., Maryland Heights, MO, #400–42, 1:1000) and β-actin antibody (Novus Biologicals, Centennial, CO, #NB600–501, 1:5,000). Human liver microsome fraction (Thermo Fisher Scientific, #HMMCPL) was used as an APO(a) positive blotting control.
Secreted apolipoprotein analysis
Conditioned media was collected from HepG2 cells 24 hours post-treatment. ELISA was used to measure lipoproteins according to manufacturer’s protocols: APOA1 (Abcam #ab189576), APO(a) (Cell Biolabs Inc. #STA-359), APOB (Abcam #ab190806), APOC3 (Abcam #ab55984).
Immunohistochemistry
Carotid artery samples were obtained from patients undergoing endarterectomy (Brigham and Women’s Hospital Institutional Review Board protocol #1999P001348) and aortic valve samples were obtained from patients undergoing valve replacement surgery (Institutional Review Board protocol #2011P001703; written informed consent was obtained for human tissue donors). Tissues were embedded in optimum cutting temperature compound, cut into slices with 7μm thickness, and cryosections were fixed in acetone. Following blocking in 4% serum, sections were incubated in RARα antibody (#MA1–810A, Thermo Fisher Scientific, 1:100) with two sections stained per donor. Negative control staining was performed by substituting the primary antibody with an IgG control antibody (#sc-32342, Santa Cruz Biotechnology, Dallas, TX, 1:100). For immunofluorescence, cryosections were incubated with primary antibody overnight (RARα, #MA1–810A, Thermo Fisher Scientific, 1:100; vimentin, #ab92547, Abcam, 1:200; α-smooth muscle actin, #ab5694, 1:200), with two sections stained per donor. Cryosections were then incubated with Alexa Fluor 488 and 594-labelled secondary (Fisher Scientific). 4’,6-diamidino-2-phenylindole (DAPI; Thermo Fisher Scientific) was used for nuclear staining. Imaging was performed with a confocal microscope A1 (Nikon Instruments). Cell culture immunofluorescence was performed by growing cells on 0.1% gelatin coated Lab-Tek II chambered cover glass #1.5 borosilicate slides (Lab-Tek, Rochester, NY), fixing in 4% paraformaldehyde for 15 minutes, followed by permeabilization and blocking using 0.3% Triton X-100 and 5% serum for 30 minutes. Cells were incubated with primary antibody (MGP, #sc-81546, Santa Cruz Biotechnology, 1:200; SOX9, #82630S, Cell Signaling Technology, Danvers, MA, 1:400) overnight at 4 degrees Celsius, and incubated with Alexa Fluor 488-labelled secondary (Fisher Scientific). 4’,6-diamidino-2-phenylindole (DAPI; Thermo Fisher Scientific) was used for nuclear staining. Imaging was performed with a confocal microscope A1 (Nikon Instruments).
Statistical analysis
Analysis of variance (ANOVA) with Tukey’s multiple comparison testing and Student’s t-tests were performed using Prism 6 (GraphPad, La Jolla, CA). Data was analyzed for normality and equal variance to determine whether the applied parametric test was appropriate using Prism 6. Five donors were used for human tissue experiments. Three to nine donors were used at similar passage number for primary human cell culture experiments, and six independent experimental replicates were used for HepG2 cells.
Results
Retinoic acid attenuated human SMC calcification
As retinoic acid regulates SMC differentiation,22 we hypothesized that ATRA the major form of RA in humans, might be a transcriptional mediator of human cardiovascular cell osteogenic differentiation and calcification. ATRA exerts its effects primarily through ligand activation of the retinoic acid receptor-α (RARα) in vascular SMC.23 We examined RARα by immunohistochemistry in human carotid arteries and aortic valves, which stained positive for RARα (Figure 1A and 1B; online-only Data Supplement Figures I and II). Additionally, RARα localized in SMC (α-smooth muscle actin-positive) in human carotid artery and in VIC (vimentin-positive) in human aortic valve, assessed by immunofluorescence (Figure 1A and 1B; online-only Data Supplement Figures I and II). To assess effects of retinoic acid on cellular calcification, we used an established in vitro method with primary human coronary artery SMC cultured for up to three weeks in control media (CM) or osteogenic differentiation media (OM: CM containing β-glycerophosphate, ascorbic acid, and dexamethasone), and primary human VIC cultured for up to three weeks in pro-calcifying media (PM) containing inorganic phosphate and L-ascorbic acid.24,25 RARα and RARβ mRNA levels where not altered in calcified human cardiovascular tissues and primary cells compared to controls (Figure 1C and 1D; online-only Data Supplement Figures I and II). RARγ mRNA levels were unchanged in calcified human cardiovascular tissues and SMC in OM, and slightly increased in VIC in PM (online-only Data Supplement Figures I and II). siRNA against RARα but not RARβ or RARγ induced earlier calcification in human SMC exposed to OM for two weeks (Figure 2A). OM increased SMC mineralized matrix as assessed by three different methods: Alizarin Red staining (Figure 2B and 2C), von Kossa staining (Figure 2D), and OsteoSense 680 near-infrared fluorescence26 (Figure 2E). In contrast, the mineralization induction effect of OM was almost completely suppressed in the presence of ATRA in a concentration-dependent manner: 1 μmol/L (Figure 2B, 2D, and 2E), and to a slightly lesser extent with 0.1 μmol/L ATRA (Figure 2B). We also tested the RA isomer 9-cis RA, which has reported distinct effects in vitro but debated as to its existence in vivo, to assess isomer specificity. In similar experiments, 9-cis retinoic acid also attenuated human SMC calcification (Figure 2C). While ATRA and 9-cis retinoic acid can both bind to RAR, only 9-cis retinoic acid also binds to the retinoid X receptor (RXR). To test whether RA effects on calcification were through RAR, we treated human SMC with a well-established, specific RAR inhibitor AGN 193109, which dose-dependently increased SMC mineralization (Figure 2B). Taken together these data support a selective RARα-mediated mechanism for ATRA-induced inhibition of human SMC calcification.
Figure 1.
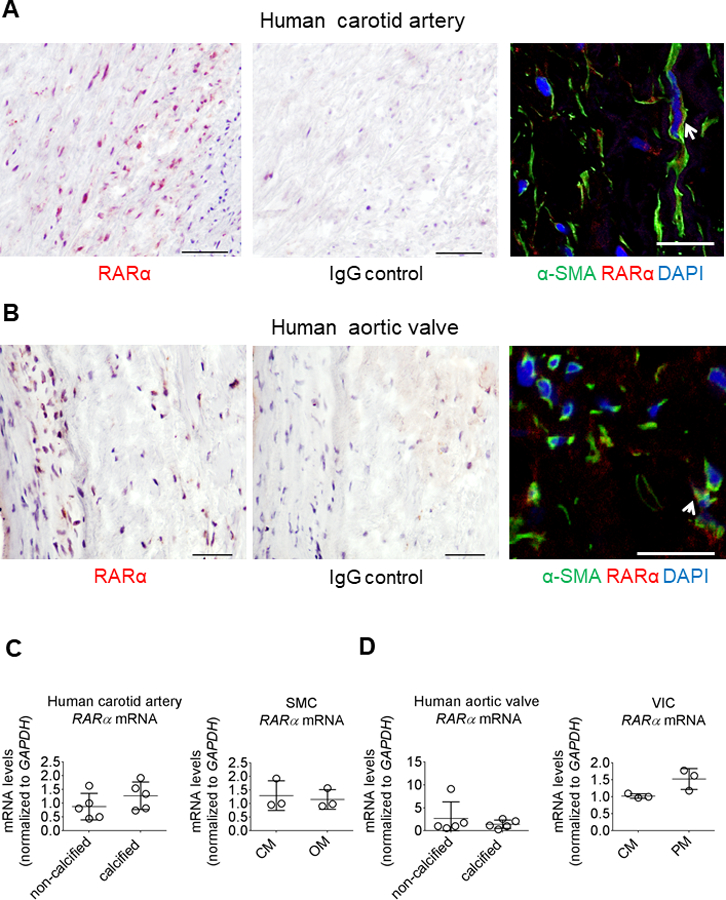
Human cardiovascular tissue contained immunoreactive retinoic acid receptor. (A) Human carotid artery and (B) aortic valve RARα immunohistochemistry (red color); scale bars, 50 μm. IgG control antibody immunohistochemistry staining for matching donor adjacent sections used as negative controls. RARα (red color) immunofluorescence showing localization with α-smooth muscle actin (α-SMA) positive cells (green color) in human carotid artery (white arrow) and with vimentin-positive cells (green color) in human aortic valve (white arrow); scale bars, 20 μm. One of five carotid artery and aortic valve donors shown, with representative RARα immunohistochemistry shown for two additional donors in Figures I and II in the online-only Data Supplement. (C) Human carotid artery, SMC, (D) aortic valve, and VIC RARα mRNA levels in calcified and non-calcified tissues (n = 5 donors) and CM, OM or PM treated cells after two weeks in culture (n = 3 donors). Error bars indicate STDEV of mean. Data analyzed by Student’s t-test.
Figure 2.
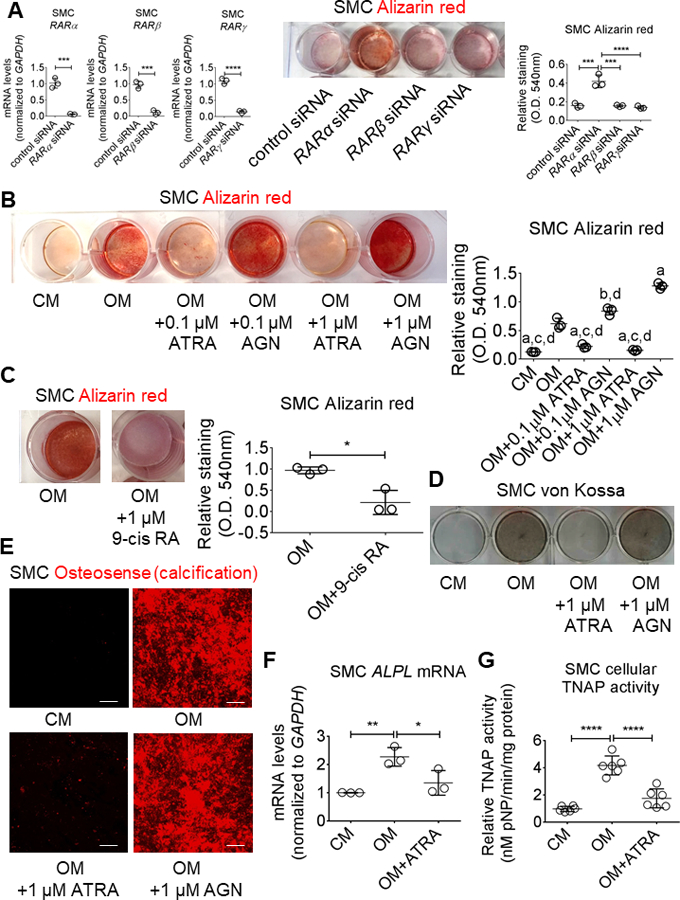
Retinoic acid attenuated human SMC calcification. (A) RAR siRNA knockdown validation by mRNA level quantification, and Alizarin red stain and relative quantification for primary human SMC treated for two weeks in OM with negative control siRNA, RARα siRNA, RARβ siRNA, or RARγ siRNA. (B) Alizarin red stain and relative quantification for primary human artery SMC treated for three weeks in CM or OM with DMSO vehicle (0.1%), 0.1 μmol/L or 1 μmol/L ATRA, 0.1 μmol/L or 1 μmol/L AGN 193109 (AGN), or (C) 1 μmol/L 9-cis retinoic acid (9-cis RA). (D) Von Kossa and (E) OsteoSense 680 stain for SMC treated for three weeks; scale bars, 100 μm. (F) Cellular ALPL mRNA levels and corresponding TNAP enzyme activity (G) for SMC treated for two weeks. Error bars indicate STDEV of mean. aP < 0.0001 vs OM, bP < 0.01 vs OM, cP < 0.0001 vs OM + 0.1 μmol/L AGN, dP < 0.0001 vs OM + 1 μmol/L AGN, *P < 0.05, **P < 0.01, ***P <0.001, ****P < 0.0001 analyzed by ANOVA or Student’s t-test; n = 3 donors, except for TNAP activity in which n = 6 donors.
Retinoic acid suppressed OM-induced TNAP activity in human SMC
We previously reported a novel mechanism identifying tissue non-specific alkaline phosphatase (TNAP protein; ALPL gene) as a key enzyme mediating OM mineralization of SMC through extracellular vesicles (EV).27 To assess whether ATRA inhibited SMC calcification via TNAP regulation, we measured ALPL mRNA levels and TNAP activity in SMC stimulated with ATRA. As expected, OM increased SMC ALPL mRNA levels (Figure 2F), and SMC cellular (Figure 2G) and EV TNAP (Figure 3A) activity; in contrast, ATRA suppressed OM-induced SMC ALPL mRNA levels (Figure 2F) and SMC cellular (Figure 2G) and EV (Figure 3A) TNAP activity, doing so without altering either EV abundance (Figure 3B) or size (Figure 3C). These data demonstrate ATRA suppressed TNAP-mediated calcification in human SMC and EV.
Figure 3.
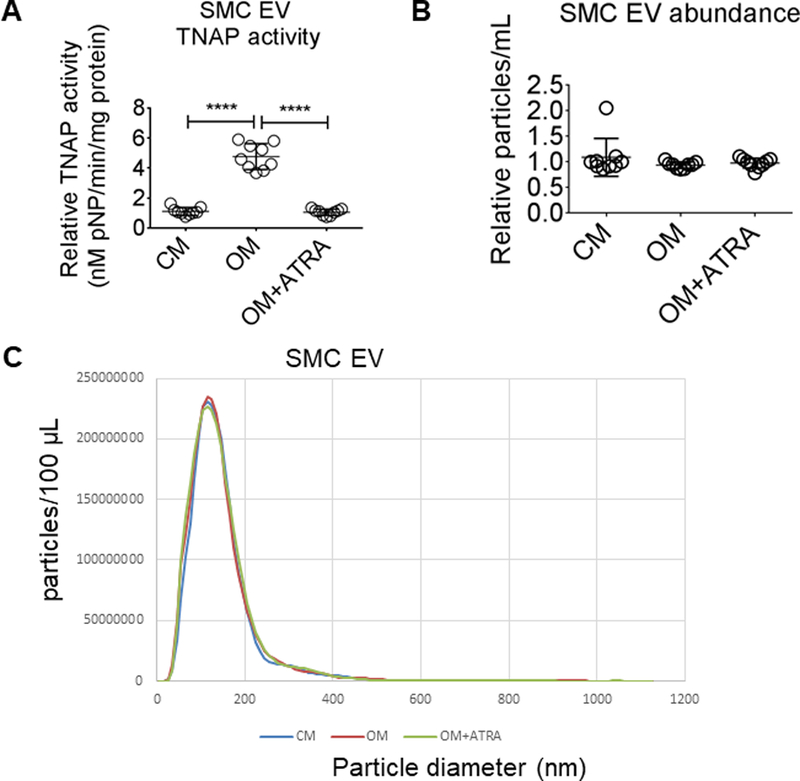
Retinoic acid suppressed SMC EV TNAP activity. (A) EV TNAP activity from primary human coronary artery SMC treated for two weeks in CM or OM with DMSO vehicle (0.1%) or 1 μmol/L ATRA. (B) SMC EV abundance and (C) diameter from SMC treated for two weeks. Error bars indicate STDEV of mean. ****P < 0.0001 analyzed by ANOVA; n = 9 donors.
Retinoic acid attenuated an osteogenic transcriptional program in human SMC
To examine whether ATRA signaling modulates gene expression involved in differentiating SMC to an osteoblast-like cell phenotype with elevated TNAP, we investigated ATRA effects on osteogenic-related transcription factors. As ATRA mediates transcriptional effects through RAR, we first examined ATRA effects on RARα expression. Indeed, ATRA increased RARα mRNA levels in human SMC (Figure 4A). To assess whether ATRA changed the osteogenic transcriptional program of human coronary artery SMC, we measured mRNA levels of three transcription factors strongly implicated in cardiovascular calcification: runt related transcription factor 2 (RUNX2), Msh homeobox 2 (MSX2), and SRY-box 9 (SOX9).28 OM increased human SMC RUNX2 mRNA levels, which was attenuated in the presence of ATRA (Figure 4B). While none of the stimulation conditions altered MSX2 expression in human SMC (Figure 4C), ATRA stimulation did increase SOX9 mRNA levels and SOX9 protein nuclear localization, as visualized by confocal immunofluorescence (Figure 4D). Taken together, these data demonstrate that ATRA signaling inhibits an OM-induced osteogenic transcriptional program in human SMC.
Figure 4.
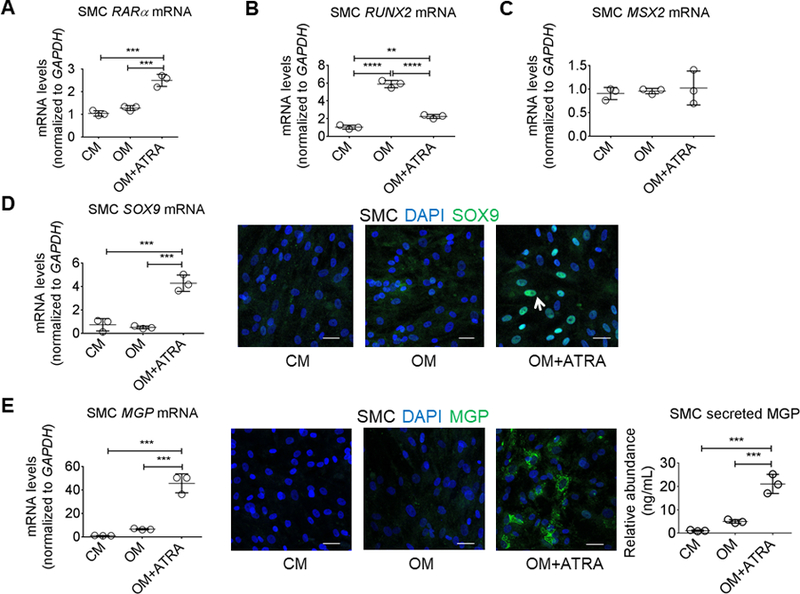
Retinoic acid increased SOX9 and MGP in human SMC. (A) RARα, (B) RUNX2, and (C) MSX2 mRNA levels from primary human coronary artery SMC treated for two weeks in CM or OM with DMSO vehicle (0.1%) or 1 μmol/L ATRA. (D) SOX9 mRNA levels and representative SOX9 and 4’,6-diamidino-2-phenylindole (DAPI) immunofluorescence from SMC treated for two weeks; white arrow depicts an example of SOX9 nuclear localization. (E) MGP mRNA levels, representative MGP and DAPI immunofluorescence, and secreted MGP protein abundance from SMC treated for two weeks. Error bars indicate STDEV of mean. **P < 0.01, ***P < 0.001, ****P < 0.0001 analyzed by ANOVA; n = 3 donors. Scale bars, 50 μm.
Retinoic acid increased matrix Gla protein in human SMC
Given the overlap between bone, SMC, and ATRA responses, we next assessed whether ATRA altered the levels of matrix Gla protein (MGP), a calcification inhibitor29 induced by RA in human osteoblastic cells.19 Exposure to ATRA over two weeks strongly increased human SMC MGP mRNA levels, intracellular MGP protein visualized by confocal microscopy, and MGP protein secretion in human SMCs cultured in the presence of OM (Figure 4E). These data demonstrate that ATRA increased the abundance of the calcification inhibitor MGP in human SMC.
Retinoic acid increased MGP and suppressed human valvular interstitial cell calcification
As calcification in the aortic valve is of clinical relevance in addition to this process in arteries, we investigated ATRA responses in a second human cardiovascular cell type: primary human valvular interstitial cells (VIC). We used an established osteogenic differentiation protocol, with human VIC exposed to PM containing inorganic phosphate and L-ascorbic acid for three weeks.25,30,31 PM was used for VIC studies given our prior report that PM generates more consistent calcification in VIC as compared to OM.31 Unlike OM, PM contains inorganic phosphate and induces VIC calcification independent of TNAP activity.31 This distinction allows assessment of the ATRA impact on mineralization through TNAP-independent calcification mechanisms like MGP. ATRA increased VIC MGP mRNA levels and MGP protein secretion in PM (Figure 5A). Human VIC formed a mineralized matrix after three-week PM treatment, visualized by Alizarin red staining, which was suppressed in the presence of ATRA (Figure 5B). In agreement with our SMC data, siRNA against RARα but not RARβ or RARγ promoted earlier induction of calcification in human VIC cultured for two weeks in PM (Figure 5C). These data demonstrate that in human VIC, ATRA regulates similar transcriptional programs as seen in human SMC, including induced expression and release of the calcification inhibitor MGP.
Figure 5.
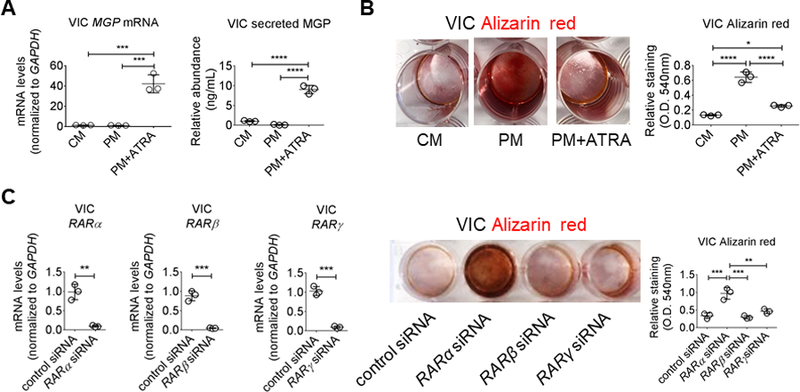
Retinoic acid increased MGP in human VIC. (A) MGP mRNA levels and secreted protein from VIC treated for two weeks in CM or PM with DMSO vehicle (0.1%) or 1 μmol/L ATRA. (B) Alizarin red stain and relative quantification for VIC treated for three weeks. (C) RAR siRNA knockdown validation by mRNA level quantification, and Alizarin red stain and relative quantification for primary human VIC treated for two weeks in PM with negative control, RARα, RARβ, or RARγ siRNA. Error bars indicate STDEV of mean. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 analyzed by ANOVA or Student’s t-test; n = 3 donors.
The acyclic retinoid, peretinoin does not unfavorably alter lipids as do classical retinoids
Therapeutic use of cyclic retinoids like ATRA in humans has been limited by their adverse effects, including increased triglyceride levels via retinoid induction of the lipoprotein lipase inhibitor apolipoprotein CIII (APOC3).32 Increased APOC3 levels have been associated with coronary calcification33 and adverse cardiovascular outcomes, as supported by recent strong human genetic evidence,34 consistent with causality in these responses. Cyclic retinoids also decrease levels of cytochrome P450 family 7 subfamily A member 1 (CYP7A1), a major hepatic enzyme involved in cholesterol metabolism.35 While ATRA is selective for RAR and 9-cis retinoic acid activates both RAR and RXR, naturally occurring retinoids can undergo distinct modifications and interconversion in vivo,32 with distinct ligand structures known to exert divergent biologic effects.36 Therefore, we tested cellular responses consistent with these potential adverse cellular effects in response to three natural retinoid isomers containing a cyclic cyclohexene hydrocarbon, namely ATRA, 9-cis retinoic acid, and 13-cis retinoic acid that is in clinical use, in comparison to the synthetic, clinically-tested acyclic retinoid, peretinoin, which lacks a cyclohexene hydrocarbon moiety (online-only Data Supplement Figure III). As expected, all three cyclic retinoids significantly elevated APOC3 mRNA levels and secreted APOC3 protein in human liver HepG2 cells (Figure 6A). However, in direct contrast, the acyclic retinoid peretinoin did not alter APOC3 mRNA or secreted APOC3 protein levels in HepG2 cells (Figure 6A). A similar pattern was seen in apolipoprotein A1 (APOA1) levels, with cyclic retinoids inducing APOA1 mRNA and secreted APOA1 protein, while peretinoin had no such effect (Figure 6B). Neither cyclic nor acyclic retinoid treatment significantly affected secreted levels of apolipoprotein B (APOB) (online-only Data Supplement Figure IVA) and had no impact on the very low basal expression of apolipoprotein (a) (APO(a)) mRNA as well as APO(a) protein, assessed by Western blotting, or media secretion, measured by ELISA (online-only Data Supplement Figure IVB). Cyclic retinoids can repress CYP7A1 mRNA expression,35 which can alter metabolic responses, prompting us to assess cyclic versus acyclic retinoid regulation of CYP7A1 transcription. Cyclic retinoid stimulation but not the acyclic retinoid peretinoin significantly reduced CYP7A1 mRNA levels in HepG2 cells (Figure 6C). Expression of APOC3 and APOA1 are regulated by several nuclear receptors, including hepatocyte nuclear factor 4 alpha (HNF4α)37,38 and retinoid X receptor (RXR).32,38 CYP7A1 is transcriptionally induced by HNF4α and the co-activator peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PPARGC1α).39 We therefore investigated cyclic versus acyclic retinoid effects on these transcription factors. None of the retinoids tested altered HNF4α and PPARGC1α mRNA levels (Figure 6D). These data demonstrate that cyclic retinoid transcriptional regulation of APOC3, APOA1, and CYP7A1 did not involve increasing HNF4α or decreasing PPARGC1α mRNA levels.
Figure 6.
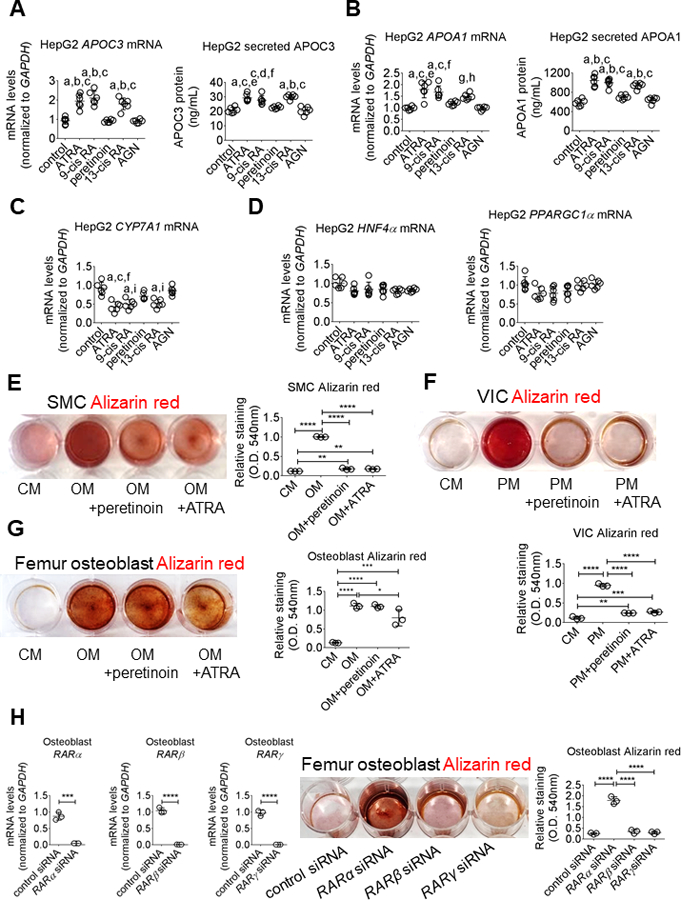
Peretinoin inhibits human cardiovascular cell calcification without inducing liver cell APOC3 secretion. (A) APOC3 and (B) APOA1 mRNA levels and secreted protein in HepG2 treated for 24 hours with 0.1% DMSO vehicle (control), 1 μmol/L ATRA, 1 μmol/L 9-cis retinoic acid (9-cis RA), 1 μmol/L peretinoin, 13-cis retinoic acid (13-cis RA), or 1 μmol/L AGN 193109 (AGN). (C) CYP7A1 and (D) HNF4α, PPARGC1α mRNA levels in HepG2 cells treated for 24 hours. Alizarin red stain and relative quantification for (E) primary human coronary artery SMC and (F) VIC treated for three weeks, and (G) primary human femur osteoblasts treated for one three weeks in CM, OM, or PM with DMSO vehicle (0.01%), 1 μmol/L peretinoin, or 1 μmol/L ATRA. (H) RAR siRNA knockdown validation by mRNA level quantification, and Alizarin red stain and relative quantification for primary human femur osteoblasts treated for two weeks in OM with negative control, RARα, RARβ, or RARγ siRNA. Error bars indicate STDEV of mean. aP < 0.0001 vs control, bP < 0.0001 vs peretinoin, cP < 0.0001 vs AGN, dP < 0.001 vs control, eP < 0.001 vs peretinoin, fP < 0.01 vs peretinoin, gP < 0.01 vs control, hP < 0.01 vs AGN, iP < 0.001 vs AGN, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 analyzed by ANOVA or Student’s t-test; n = 6 independent experiments for HepG2 and n = 3 donors for primary human VIC, SMC, and osteoblasts.
Acyclic retinoid, peretinoin inhibited human cardiovascular cell calcification without inhibiting human femur bone osteoblast mineralization
Since the acyclic retinoid peretinoin differed from cyclic retinoids by not exhibiting adverse effects on genes expected to alter triglyceride levels and other hepatic responses, we next tested if peretinoin retained the inhibition of human cardiovascular cell calcification seen with ATRA. Indeed, peretinoin significantly attenuated OM-induced human SMC calcification to a similar level as seen with ATRA stimulation (Figure 6E). Peretinoin also significantly reduced SMC RUNX2 and ALPL mRNA levels, and increased MPG mRNA levels in SMC, but to a lesser extent than ATRA (online-only Data Supplement Figure VA). In agreement with its effect in SMC, peretinoin attenuated PM-induced human VIC calcification to a similar extent as ATRA (Figure 6F). Peretinoin also significantly increased VIC MGP mRNA levels, although to a lesser extent than ATRA (online-only Data Supplement Figure VIA). Peretinoin did not alter SMC or VIC SOX9 mRNA levels (online-only Data Supplement Figure VA and VIA). We also assessed whether RA worked via ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENNP1) that can repress calcification,2 but found no RA regulation of ENPP1 mRNA expression in SMC or VIC (online-only Data Supplement Figures VB and VIB). Lastly, given evidence for differences in calcification depending on cell and bone contexts,25 we tested the effects of ATRA versus peretinoin on human femur bone osteoblasts. ATRA reduced bone osteoblast mineralization in OM, which was not seen in response to peretinoin (Figure 6G). Furthermore, in agreement with our human SMC and VIC data, siRNA against RARα but not RARβ or RARγ induced earlier calcification in human femur osteoblasts in OM (Figure 6H).
Discussion
We report here the following novel findings: 1) ATRA inhibits human artery SMC and aortic VIC calcification via RARα and subsequent coordinated transcriptional regulation. 2) Likely involvement of the retinoid cyclohexene hydrocarbon in the adverse effects of ATRA and other cyclic retinoids on plasma lipids. Most studies investigating RA action in calcification have focused on its role in bone mineralization, with evidence for RA-mediated repression of bone mineralization through retinoid signaling.12,13,40 Limited and often conflicting data exists regarding retinoic acid and cardiovascular calcification.41 As bone formation and cardiovascular calcification share common biologically-mediated pathways,42 we investigated the effect of retinoids on factors implicated in both processes. Among these was the osteogenic transcription factor RUNX2 that induces vascular calcification.43 Another transcription factor, SOX9, inhibits bone formation44 and valvular calcification,14 while taking part in providing feedback inhibition of RUNX2.45,46 Our finding that ATRA decreased RUNX2 and increased SOX9 mRNA levels in human SMC indicates that ATRA drives a coordinated transcriptional program. This RA program suppressed osteogenic differentiation-mediated calcification via RAR activation, as further supported by findings with both chemical modulators of RAR activity and RARα siRNA, which accelerated calcification in primary human cells.
ATRA suppressed OM-induced cellular and EV TNAP activity in human SMC without altering EV abundance. We and others reported a role for EV47 and EV TNAP27 in mediating increased SMC calcification; these results place retinoid signaling upstream of this novel calcific mechanism, opening new ways in which to consider inputs into TNAP action in EV. To model calcification in human SMC and VIC, the use of two different conditions allowed for isolating potential contributory mechanisms. In OM, human SMC calcify readily through TNAP-mediated metabolism of β-glycerophosphate to generate inorganic phosphate. In human VIC, we used PM that contains inorganic phosphate to promote matrix calcification, thus bypassing any dependency on TNAP for inorganic phosphate formation.31 In both experimental conditions, ATRA suppressed calcification. This result suggested that ATRA induced additional calcification inhibitory mechanisms beyond TNAP suppression. In human SMC and VIC, ATRA induced the transcription of MGP, a well-established calcification inhibitor. In human aortic VIC, knockdown of MGP increases inflammation-induced calcification whereas overexpression of MGP suppresses inflammatory driven calcific responses.48 Post-translational modifications can alter MGP activity by shifting between carboxylated (active) and uncarboxylated MGP (inactive) forms. In a human proof-of-concept study, the slowing of aortic valve calcification seen with vitamin K supplementation correlated with decreased uncarboxylated MGP.49 Thus, increasing MGP abundance and/or MGP activity has been proposed as a potential therapeutic strategy for reducing cardiovascular calcification.50 Of note, MGP inhibition of SMC calcification may involve MGP binding to EV,51 suggesting ways in which these mechanisms may be integrated, as also occurs through the action of ATRA. The data presented here raises the intriguing prospect that combining vitamin K and retinoid supplementation may be particularly efficacious in reducing cardiovascular calcification and warrants further exploration. Complicating assessment of MGP’s role in human cardiovascular calcification is an unclear association between Keutel syndrome and coronary artery disease. Keutel syndrome patients lack functional MGP and have abnormal cartilage calcification.52 While cardiovascular calcification is not a key disease feature, one case report observed coronary, hepatic, renal, meningeal and cerebral artery concentric calcification in a 31-year old male Keutel syndrome patient.53 Additionally, a significant association between the MGP gene rs1800801 polymorphism and increased risk of vascular calcification and atherosclerotic disease in Caucasians has been reported in a meta-analysis.54 Regardless, our data supports that RA inhibition of human cardiovascular cell calcification occurs via both MGP and MGP-independent mechanisms. As the data here establishes, further studies dissecting the contributions of these or other potential mechanisms to retinoid-inhibited cardiovascular calcification are warranted and should consider species-specific differences in response.
Despite evidence for benefit, the clinical use of RA has been complicated. Based on demonstrated improvements, RA is in use for certain dermatologic55 and oncologic conditions56. For cardiovascular disease, the RA data has been limited and often conflicting.41 While some positive results have been seen in observational studies, supplementation with beta-carotene, which can be converted into the ATRA precursor vitamin A, did not reduce cardiovascular events.18 However, beta-carotene, vitamin A and retinoid biology involves multiple distinct components and variables that could impact trial results. Along these lines, our present study demonstrates that species, cell type, and experimental differences may well underlie divergent reports regarding ATRA effects on cardiovascular cells. As shown, we determine definitive ATRA-mediated decreases in calcification in multiple human cell models, with coordinated modulation of distinct cellular targets and functional responses using various methodologies. We found ATRA either increased or had no effect on human cardiovascular cell SOX9. In contrast to these findings, ATRA has been reported to increase mouse heart valve calcification, which was attenuated by Sox9 overexpression.15 In mice, high 10-fold excess vitamin A intake reduced Sox9 expression and promoted aortic valve calcification.14 In avian mitral valve explants, ATRA also increased calcification, a response reversed with a RAR antagonist.14 In contrast to these studies, ATRA decreased 1α,25-dyhydroxycholecalciferol-induced renal calcification in male mice while increasing carboxylated MGP but not Mgp expression,57 results that contrast with the increased MGP expression and activity in human cells shown here. We previously reported species differences in OM-induced calcification between primary human and mouse SMC.25 Differential regulation of several genes also occurs between mice and humans with the peroxisome proliferator-activated receptor-α, a nuclear receptor like RAR.58 In human fibroblasts, chondrocytes, osteoblasts, osteosarcoma cell lines, and rat type II pneumocytes, ATRA increases MGP mRNA levels.19,20 In contrast, in rat kidney cells, rat vascular SMC, and human breast cancer cells ATRA was found to decrease MGP mRNA levels.16,21 RA signaling may act to suppress human cardiovascular cell calcification directly by binding to regulatory elements in the promoter regions of calcification-related genes or through more indirect mechanisms. The RAR/RXR heterodimeric complex binds to the human MGP promoter,21 and the SOX9 promoter contains a RAR response element.59 In mouse preadipocytes, RARγ but not RARα stimulates Sox9 expression;59 whereas selective RARα activation stimulates SOX9 expression in human breast cancer cells.60 RA-mediated regulation of RUNX2 is less clear, as it may likely occur indirectly. In mouse mesenchymal stem cell-like cells, RA stimulates the disassociation of CCAAT/enhancer binding protein β from the Runx2 promoter, which induces Runx2 expression.61 In mouse MC-3T3 pre-osteoblastic cells, RA was reported to inhibit calcification in two separate studies40, 62, but the reported effects on Runx2 mRNA diverged in these studies for unknown reasons. In one MC-3T3 cell line study, 0.4 μmol/L RA did not alter Runx2 mRNA but reduced RUNX2 protein.40 In another report, 0.5 μmol/L RA increased Runx2 mRNA although RUNX2 protein levels were not reported.62 Further investigation into what underlies differences in RA responses in rodent versus human cells may uncover additional mechanisms controlling calcification in humans, studies hopefully prompted by this report.
In humans, in general, cyclic retinoids like ATRA can increase triglyceride levels; postulated mechanisms include ATRA increases in expression of the lipoprotein lipase inhibitor APOC3.32 Of particular relevance here, in patients with diabetes, elevated APOC3 was associated with higher coronary artery calcification.33 Retinoids are also reported to decrease bone mineralization, with ATRA inhibiting bone osteoblast differentiation and mineral deposition.12,13,40,63 These reports regarding adverse effects of ATRA highlight the potential importance of our finding that the acyclic retinoid peretinoin retains decreased calcification, but does not induce cellular responses associated with adverse ATRA effects. Although the structure of peretinoin is similar to ATRA, it lacks the cyclohexene hydrocarbon, which likely precludes this molecule from undergoing isomer conversion. Distinct molecular structures, including retinoid species, are known to differ in their ligand interaction with nuclear receptors, like RAR and RXR, thus altering nuclear receptor conformational states, accessory molecule recruitment or release and biologic responses, as is likely the case for peretinoin. There is a paucity of studies specifically examining RA signaling differences between cyclic and acyclic RA. Our study supports differences in overall magnitude of transcriptional response and induction of SOX9 in OM between cyclic and acyclic RA, but further work is needed to identify additional differences, including in the specific activation of RARα/β/γ and/or RXR in human cells and tissues. The prospect of harnessing distinct molecular structure interaction with nuclear receptors to maintain clinical benefits while removing adverse effects has been pursued in other settings, including the estrogen receptor (selective estrogen receptor modulators)64 and peroxisome proliferator activated receptors (selective PPAR modulators).65 Whether acyclic retinoids might offer such options for retinoid receptors remains unknown but worthy of consideration given the data shown here.
Despite the impact calcification has on common, life-threatening clinical conditions like aortic valve disease and aortic stenosis, currently no agents have been approved for slowing or reversing cardiovascular calcification. Our findings provide new insight into retinoid regulation of human cardiovascular cell calcification and offer new prospects for developing therapies to address the unmet need of treating calcification and avoiding retinoid side effects.
Supplementary Material
Highlights.
Retinoic acid increased mRNA levels and protein secretion of matrix Gla protein, a calcification inhibitory molecule, in human coronary artery and aortic valve cells.
In contrast to non-human models, retinoic acid suppressed calcification in human arterial smooth muscle cells and aortic valvular interstitial cells.
Cyclohexene hydrocarbon moiety in cyclic retinoids likely mediates APOC3 induction, which can be avoided by using acyclic retinoids.
Acknowledgements
We thank Dr. Galina Sukhova for providing carotid endarterectomy specimens, James Gosnell and Svetlana Gorbatov for assisting with the collection of surgical heart valve samples, and Dr. Hong Wang for cell culture and assay assistance.
Sources of Funding
This study was supported by a research grant from Kowa Company, Ltd. (Tokyo, Japan, to M.A.) and the National Institutes of Health grants (R01 HL114805, R01 HL136431, R01 HL147095 to E.A., NHLBI P01 HL048743, R01 NIDDK107239, and Pollin Cardiovascular Research Fund to J.P. and R01 HL134892 to P.L.)
Non-standard Abbreviations and Acronyms:
- 9-cis RA
9-cis retinoic acid
- 13-cis RA
13-cis retinoic acid
- ATRA
all-trans-retinoic acid
- APO(a)
apolipoprotein-(a)
- APOA1
apolipoprotein-A1
- APOB
apolipoprotein-B
- APOC3
apolipoprotein-CIII
- CYP7A1
cytochrome P450 family 7 subfamily A member 1
- CM
control media
- ENNP1
ectonucleotide pyrophosphatase/phosphodiesterase 1
- EV
extracellular vesicles
- HNF4α
hepatocyte nuclear factor 4 alpha
- MGP
matrix Gla protein
- MSX2
Msh homeobox 2
- PGC-1α
peroxisome proliferator-activated receptor gamma coactivator 1-alpha
- OM
osteogenic differentiation media
- RA
retinoic acid
- RAR
retinoic acid receptor
- RUNX2
runt-related transcription factor 2
- RXR
retinoid X receptor
- SMC
vascular smooth muscle cells
- SOX9
SRY-box 9
- TNAP protein; ALPL gene
tissue non-specific alkaline phosphatase
- VIC
aortic valvular interstitial cells
Footnotes
Disclosures
S.G. is an employee of Kowa Company, Ltd and was a visiting scientist at Brigham and Women’s Hospital when the experiments reported in this study were performed. Kowa Company, Ltd had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript. The other authors declare no competing interests.
References
- 1.Greenland P, Bonow RO, Brundage BH, et al. ACCF/AHA 2007 clinical expert consensus document on coronary artery calcium scoring by computed tomography in global cardiovascular risk assessment and in evaluation of patients with chest pain: a report of the American College of Cardiology Foundation Clinical Expert Consensus Task Force (ACCF/AHA Writing Committee to Update the 2000 Expert Consensus Document on Electron Beam Computed Tomography). Circulation. 2007;115:402–426. [DOI] [PubMed] [Google Scholar]
- 2.Demer LL, Tintut Y. Inflammatory, metabolic, and genetic mechanisms of vascular calcification. Arterioscler Thromb Vasc Biol. 2014;34:715–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kelly-Arnold A, Maldonado N, Laudier D, Aikawa E, Cardoso L, Weinbaum S. Revised microcalcification hypothesis for fibrous cap rupture in human coronary arteries. Proc Natl Acad Sci U S A. 2013;110:10741–10746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maldonado N, Kelly-Arnold A, Vengrenyuk Y, Laudier D, Fallon JT, Virmani R, Cardoso L, Weinbaum S. A mechanistic analysis of the role of microcalcifications in atherosclerotic plaque stability: potential implications for plaque rupture. Am J Physiol Heart Circ Physiol. 2012;303:H619–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lampropoulos CE, Papaioannou I, D’Cruz DP. Osteoporosis—a risk factor for cardiovascular disease? Nat Rev Rheumatol 2012;8:587–598. [DOI] [PubMed] [Google Scholar]
- 6.Hyder JA, Allison MA, Wong N, Papa A, Lang TF, Sirlin C, Gapstur SM, Ouyang P, Carr JJ, Criqui MH. Association of coronary artery and aortic calcium with lumbar bone density: the MESA Abdominal Aortic Calcium Study. Am J Epidemiol. 2009;169:186–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi SH, An JH, Lim S, Koo BK, Park SE, Chang HJ, Choi SI, Park YJ, Park KS, Jang HC, Shin CS. Lower bone mineral density is associated with higher coronary calcification and coronary plaque burdens by multidetector row coronary computed tomography in pre- and postmenopausal women. Clin Endocrinol. 2009;71:644–651. [DOI] [PubMed] [Google Scholar]
- 8.Naik V, Leaf EM, Hu JH, Yang HY, Nguyen NB, Giachelli CM, Speer MY. Sources of cells that contribute to atherosclerotic intimal calcification: an in vivo genetic fate mapping study. Cardiovasc Res. 2012;94:545–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Speer MY, Yang HY, Brabb T, Leaf E, Look A, Lin WL, Frutkin A, Dichek D, Giachelli CM. Smooth muscle cells give rise to osteochondrogenic precursors and chondrocytes in calcifying arteries. Circ Res. 2009;104:733–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rajamannan NM, Subramaniam M, Rickard D, Stock SR, Donovan J, Springett M, Orszulak T, Fullerton DA, Tajik AJ, Bonow RO, Spelsberg T. Human aortic valve calcification is associated with an osteoblast phenotype. Circulation. 2003;107:2181–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Giachelli CM. Vascular calcification mechanisms. J Am Soc Nephrol. 2004;15:2959–2964. [DOI] [PubMed] [Google Scholar]
- 12.Nallamshetty S, Wang H, Rhee EJ, Kiefer FW, Brown JD, Lotinun S, Le P, Baron R, Rosen CJ, Plutzky J. Deficiency of retinaldehyde dehydrogenase 1 induces BMP2 and increases bone mass in vivo. PLoS One. 2013;8:e71307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nallamshetty S, Le PT, Wang H, Issacsohn MJ, Reeder DJ, Rhee EJ, Kiefer FW, Brown JD, Rosen CJ, Plutzky J. Retinaldehyde dehydrogenase 1 deficiency inhibits PPARγ-mediated bone loss and marrow adiposity. Bone. 2014;67:281–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huk DJ, Hammond HL, Kegechika H, Lincoln J. Increased dietary intake of vitamin A promotes aortic valve calcification in vivo. Arterioscler Thromb Vasc Biol. 2013;33:285–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peacock JD, Levay AK, Gillaspie DB, Tao G, Lincoln J. Reduced sox9 function promotes heart valve calcification phenotypes in vivo. Circ Res. 2010;106:712–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Farzaneh‐Far A, Weissberg PL, Proudfoot D, Shanahan CM. Transcriptional regulation of matrix Gla protein. Z Kardiol. 2001;90:38–42. [DOI] [PubMed] [Google Scholar]
- 17.Mottaghi A, Ebrahimof S, Angoorani P, Saboor-Yaraghi AA. Vitamin A supplementation reduces IL-17 and RORc gene expression in atherosclerotic patients. Scand J Immunol. 2014;80:151–157. [DOI] [PubMed] [Google Scholar]
- 18.Cook NR, Albert CM, Gaziano JM, Zaharris E, MacFadyen J, Danielson E, Buring JE, Manson JE. A randomized factorial trial of vitamins C, E, and beta-carotene in the secondary prevention of cardiovascular events in women. Arch Intern Med. 2008;167:1610–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cancela ML, Price PA. Retinoic acid induces matrix Gla protein gene expression in human cells. Endocrinology. 1992;130:102–108. [DOI] [PubMed] [Google Scholar]
- 20.Rannels SR, Cancela ML, Wolpert EB, Price PA. Matrix Gla protein mRNA expression in cultured type II pneumocytes. Am J Physiol. 1993;265:L270–L278. [DOI] [PubMed] [Google Scholar]
- 21.Kirfel J, Kelter M, Cancela LM, Price PA, Schule R. Identification of a novel negative retinoic acid responsive element in the promoter of the human matrix Gla protein gene. Proc Natl Acad Sci U S A. 1997;94:2227–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang H, Xie C, Sun X, Ritchie RP, Zhang J, Chen YE. miR-10a contributes to retinoid acid-induced smooth muscle cell differentiation. J Biol Chem. 2010;285:9383–9389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi JH, Zheng B, Chen S, Ma GY, Wen JK. Retinoic acid receptor α mediates all-trans-retinoic acid-induced Klf4 gene expression by regulating Klf4 promoter activity in vascular smooth muscle cells. J Biol Chem. 2012;287:10799–10811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mori K, Shioi A, Jono S, Nishizawa Y, Morii H. Dexamethasone enhances In vitro vascular calcification by promoting osteoblastic differentiation of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 1999;19:2112–2118. [DOI] [PubMed] [Google Scholar]
- 25.Rogers MA, Maldonado-Martinez N, Hutcheson JD, Goettsch C, Goto S, Yamada I, Faits T, Sesaki H, Aikawa M, Aikawa E. Dynamin-Related Protein 1 Inhibition Attenuates Cardiovascular Calcification in the Presence of Oxidative Stress. Circ Res. 2017;121:220–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aikawa E, Nahrendorf M, Figueiredo JL, Swirski FK, Shtatland T, Kohler RH, Jaffer FA, Aikawa M, Weissleder R. Osteogenesis associates with inflammation in early-stage atherosclerosis evaluated by molecular imaging in vivo. Circulation. 2007;116:2841–2850. [DOI] [PubMed] [Google Scholar]
- 27.Goettsch C, Hutcheson JD, Aikawa M, Iwata H, Pham T, Nykjaer A, Kjolby M, Rogers M, Michel T, Shibasaki M, Hagita S, Kramann R, Rader DJ, Libby P, Singh SA, Aikawa E. Sortilin mediates vascular calcification via its recruitment into extracellular vesicles. J Clin Invest. 2016;126:1323–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tyson KL, Reynolds JL, McNair R, Zhang Q, Weissber PL, Shanahan CM. Osteo/chondrocytic transcription factors and their target genes exhibit distinct patterns of expression in human arterial calcification. Aterioscler Thromb Vasc Biol. 2003;23:489–494. [DOI] [PubMed] [Google Scholar]
- 29.Luo G, Ducy P, McKee MD, Pinero GJ, Loyer E, Behringer RR, Karsenty G. Spontaneous calcification of arteries and cartilage in mice lacking matrix Gla protein, Nature. 1997;386:78–81. [DOI] [PubMed] [Google Scholar]
- 30.Bouchareb R, Mahmut A, Nsaibia MJ, et al. Autotaxin derived from lipoprotein(a) and valve interstitial cells promotes inflammation and mineralization of the aortic valve. Circulation. 2015;132:677–690. [DOI] [PubMed] [Google Scholar]
- 31.Goto S, Rogers MA, Blaser MC, Higashi H, Lee LH, Schlotter F, Body SC, Aikawa M, Singh SA, Aikawa E. Standardization of human calcific aortic valve disease in vitro modeling reveals passage-dependent calcification. Front Cardiovasc Med. 2019;6:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vu-Dac N, Gervois P, Torra IP, Fruchart JC, Kosykh V, Kooistra T, Princen HM, Dallongeville J, Staels B. Retinoids increase human apo C-III expression at the transcriptional level via the retinoid X receptor. Contribution to the hypertriglyceridemic action of retinoids. J Clin Invest. 1998;102:625–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qamar A, Khetarpal SA, Khera AV, Qasim A, Rader DJ, Reilly MP. Plasma apolipoprotein C-III levels, triglycerides, and coronary artery calcification in type 2 diabetics. Aterioscler Thromb Vasc Biol. 2015;35:1880–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.TG and HDL Working Group of the Exome Sequencing Project, National Heart, Lung, and Blood Institute, Crosby J, Peloso GM, et al. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med. 2014;371:22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cai SY, He H, Nguyen T, Mennone A, Boyer JL. Retinoic acid represses CYP7A1 expression in human hepatocytes and HepG2 cells by FXR/RXR-dependent and independent mechanisms. J Lipid Res. 2010;51:2265–2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Golczak M, Kuksa V, Maeda T, Moise AR, Palczewski K. Positively charged retinoids are potent and selective inhibitors of the trans-cis isomerization in the retinoid (visual) cycle. Proc Natl Acad Sci U S A. 2005; 102:8162–8167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chan J, Nakabayashi H, Wong NC. HNF-4 increases activity of the rat Apo A1 gene. Nucleic Acids Res. 1993;21:1205–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Transcriptional factors mediating retinoic acid signals in the control of energy metabolism. Int J Mol Sci. 2015;16:14210–14244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bhalla S, Ozalp C, Fang S, Xiang L, Kemper JK. Ligand-activated pregnane X receptor interferes with HNF-4 signaling by targeting a common coactivator PGC-1alpha. Functional implications in hepatic cholesterol and glucose metabolism. J Biol Chem. 2004; 279:45139–45147. [DOI] [PubMed] [Google Scholar]
- 40.Lind T, Sundqvist A, Hu L, Pejler G, Andersson G, Jacobson A, Melhus H. Vitamin a is a negative regulator of osteoblast mineralization. PLoS One. 2013;8:e82388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rhee EJ, Nallamshetty S, Plutzky J. Retinoid metabolism and its effects on the vasculature. Biochim Biophys Acta. 2012;1821:230–240. [DOI] [PubMed] [Google Scholar]
- 42.Duer MJ, Friscić T, Proudfoot D, Reid DG, Schoppet M, Shanahan CM, Skepper JN, Wise ER. Mineral surface in calcified plaque is like that of bone: further evidence for regulated mineralization. Arterioscler Thromb Vasc Biol. 2008;28:2030–2034. [DOI] [PubMed] [Google Scholar]
- 43.Sun Y, Byon CH, Yuan K, Chen J, Mao X, Heath JM, Javed A, Zhang K, Anderson PG, Chen Y. Smooth muscle cell-specific runx2 deficiency inhibits vascular calcification. Circ Res. 2012;111:543–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hattori T, Müller C, Gebhard S, Bauer E, Pausch F, Schlund B, Bösl MR, Hess A, Surmann-Schmitt C, von der Mark H, de Crombrugghe B, von der Mark K. SOX9 is a major negative regulator of cartilage vascularization, bone marrow formation and endochondral ossification. Development. 2010;137:901–911. [DOI] [PubMed] [Google Scholar]
- 45.Zhou G, Zheng Q, Engin F, Munivez E, Chen Y, Sebald E, Krakow D, Lee B. Dominance of SOX9 function over RUNX2 during skeletogenesis. Proc Natl Acad Sci U S A. 2006;103:19004–19009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cheng A, Genever PG. SOX9 determines RUNX2 transactivity by directing intracellular degradation. J Bone Miner Res. 2010;25:2680–2689. [DOI] [PubMed] [Google Scholar]
- 47.Reynolds JL, Joannides AJ, Skepper JN, McNair R, Schurgers LJ, Proudfoot D, Jahnen-Dechent W, Weissberg PL, Shanahan CM. Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: a potential mechanism for accelerated vascular calcification in ESRD. J Am Soc Nephrol. 2004;15:2857–2867. [DOI] [PubMed] [Google Scholar]
- 48.Chiyoya M, Seya K, Yu Z, Daitoku K, Motomura S, Imaizumi T, Fukuda I, Furukawa KI. Matrix Gla protein negatively regulates calcification of human aortic valve interstitial cells isolated from calcified aortic valves. J Pharmacol Sci. 2018;136:257–265. [DOI] [PubMed] [Google Scholar]
- 49.Brandenburg VM, Reinartz S, Kaesler N, Krüger T, Dirrichs T, Kramann R, Peeters F, Floege J, Keszei A, Marx N, Schurgers LJ, Koos R., Slower progress of aortic valve calcification with vitamin K supplementation: results from a prospective interventional proof-of-concept study. Circulation. 2017;135:2081–2083. [DOI] [PubMed] [Google Scholar]
- 50.Rogers MA, Aikawa E. Cardiovascular calcification: artificial intelligence and big data accelerate mechanistic discovery. Nat Rev Cardiol. 2019;16:261–274. [DOI] [PubMed] [Google Scholar]
- 51.Schurgers LJ, Spronk HM, Skepper JN, Hackeng TM, Shanahan CM, Vermeer C, Weissberg PL, Proudfoot D. Post-translational modifications regulate matrix Gla protein function: importance for inhibition of vascular smooth muscle cell calcification. J Thromb Haemost. 2007;5:2503–2511. [DOI] [PubMed] [Google Scholar]
- 52.Munroe PB, Olgunturk RO, Fryns JP, Van Maldergem L, Ziereisen F, Yuksel B, Gardiner RM, Chung E. Mutations in the gene encoding the human matrix Gla protein cause Keutel syndrome. Nat Genet. 1999;21:142–144. [DOI] [PubMed] [Google Scholar]
- 53.Meier M, Weng LP, Alexandrakis E, Ruschoff J, Goeckenjan G. Tracheobronchial stenosis in Keutel syndrome. Eur Respir J. 2001;17:566–569. [DOI] [PubMed] [Google Scholar]
- 54.Sheng K, Zhang P, Lin W, Cheng J, Li J, Chen J. Association of matrix Gla protein gene (rs1800801, rs1800802, rs4236) polymorphism with vascular calcification and atherosclerotic disease: a meta-analysis. Sci Rep. 2017;7:8713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leyden J, Stein-Gold L, Weiss J. Why Topical Retinoids Are Mainstay of Therapy for Acne. Dermatol Ther. 2017;7:293–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tan CK. Peretinoin as an adjuvant therapy for hepatocellular carcinoma. Expert Rev Gastroenterol Hepatol. 2016;10:1201–1201. [DOI] [PubMed] [Google Scholar]
- 57.Fu X, Wang XD, Mernitz H, Wallin R, Shea MK, Booth SL. 9-Cis retinoic acid reduces 1alpha,25-dihydroxycholecalciferol-induced renal calcification by altering vitamin K-dependent gamma-carboxylation of matrix gamma-carboxyglutamic acid protein in A/J male mice. J Nutr. 2008;138:2337–2341. [DOI] [PubMed] [Google Scholar]
- 58.Yang Q, Nagano T, Shah Y, Cheung C, Ito S, Gonzalez FJ. The PPAR alpha-humanized mouse: a model to investigate species differences in liver toxicity mediated by PPAR alpha. Toxicol Sci. 2008;101:132–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Berry DC, DeSantis D, Soltanian H, Croniger CM, Noy N. Retinoic acid upregulates preadipocyte genes to block adipogenesis and suppress diet-induced obesity. Diabetes. 2012;61:1112–11121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Afonja O, Raaka BM, Huang A, Das S, Zhao X, Helmer E, Juste D, Samuels HH. RAR agonists stimulate SOX9 gene expression in breast cancer cell lines: evidence for a role in retinoid-mediated growth inhibition. Oncogene. 2002;21:7850–7860. [DOI] [PubMed] [Google Scholar]
- 61.Wiper-Bergeron N, St-Louis C, Lee JM. CCAAT/Enhancer binding protein beta abrogrates retinoic acid-induced osteoblast differentiation via repression of Runx2 transcription. Mol Endocrinol. 2007;21:2124–2135. [DOI] [PubMed] [Google Scholar]
- 62.Roa LA, Bloemen M, Carels CEL, Wagener FADTG, Von den Hoff JW. Retinoic acid disrupts osteogenesis in pre-osteoblasts by down-regulating WNT signaling. Int J Biochem Cell Biol. 2019;116:105597. [DOI] [PubMed] [Google Scholar]
- 63.Wang A, Ding X, Sheng S, Yao Z. Retinoic acid inhibits osteogenic differentiation of rat bone marrow stromal cells. Biochem Biophys Res Commun. 2008;375:435–439. [DOI] [PubMed] [Google Scholar]
- 64.Riggs BL, Hartmann LC. Selective estrogen-receptor modulators – mechanisms of action and application to clinical practice. N Engl J Med. 2003;348:618–629. [DOI] [PubMed] [Google Scholar]
- 65.Ishibashi S, Yamashita S, Arai H, Araki E, Yokote K, Suganami H, Fruchart JC, Kodama T, K-877–04 Study Group. Effects of K-877, a novel selective PPARα modulator (SPPARMα), in dyslipidaemic patients: A randomized, double blind, active- and placebo-controlled, phase 2 trial. Atherosclerosis. 2016;249:36–43. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.