

Abstract
RUNX2 is a transcription factor critical for chondrocyte maturation and normal endochondral bone formation. It promotes the expression of factors catabolic to the cartilage extracellular matrix and is upregulated in human osteoarthritic cartilage and in murine articular cartilage following joint injury. To date, in vivo studies of RUNX2 overexpression in cartilage have been limited to forced expression in osteochondroprogenitor cells preventing investigation into the effects of chondrocyte-specific RUNX2 overexpression in postnatal articular cartilage. Here, we used the Rosa26Runx2 allele in combination with the inducible Col2a1CreERT2 transgene or the inducible AcanCreERT2 knock-in allele to achieve chondrocyte-specific RUNX2 overexpression (OE) during embryonic development or in the articular cartilage of adult mice, respectively. RUNX2 OE was induced at E13.5 for all developmental studies. Histology and in situ hybridization analyses suggest an early onset of chondrocyte hypertrophy and accelerated terminal maturation in the limbs of the RUNX2 OE embryos compared to control embryos. For all postnatal studies, RUNX2 OE was induced at 2 months of age. Surprisingly, no histopathological signs of cartilage degeneration were observed even six months following induction of RUNX2 OE. Using the meniscal/ligamentous injury (MLI), a surgical model of knee joint destabilization and meniscal injury, however, we found that RUNX2 OE accelerates the progression of cartilage degeneration following joint trauma. One month following MLI, the numbers of MMP13-positive and TUNEL-positive chondrocytes were significantly greater in the articular cartilage of the RUNX2 OE joints compared to control joints and two months following MLI, histomorphometry and OARSI scoring revealed decreased cartilage area in the RUNX2 OE joints. Collectively, these results suggest that while RUNX2 overexpression alone may not be sufficient to initiate the OA degenerative process, it may predetermine the rate of OA onset and/or progression following traumatic joint injury.
INTRODUCTION
Endochondral ossification is a carefully orchestrated process regulated by a number of growth factors, signaling pathways, and transcription factors. It is responsible for development of the long bones and driven by a process of progressive cell differentiation. Initially, mesenchymal progenitor cells condense and differentiate into committed chondrocytes, whose gene expression is largely controlled by the master chondrogenic transcription factor SOX9(1–3). Committed chondrocytes then begin to rapidly proliferate, building rudimentary skeletal elements rich in ACAN and COL2A1. The cells at the very center of these elements eventually exit the cell cycle and become pre-hypertrophic, expressing Runx2 and Ihh(4–7). These cells further mature into hypertrophic chondrocytes marked by their expression of Col10a1 as well as other factors that promote extracellular matrix (ECM) mineralization and vascular invasion. Finally, late stage hypertrophic chondrocytes express Mmp13. These cells eventually undergo apoptosis, or differentiate into cells of the osteoblast lineage, leaving behind a cartilaginous template upon which bone is formed and the primary ossification center established(8–11).
Postnatally, secondary ossification centers form in the epiphyses to separate the transient growth plate cartilage from the permanent articular cartilage layer that lines the ends of the long bones. In contrast to the highly proliferative growth plate chondrocytes, articular chondrocytes at homeostasis are largely non-proliferative(12,13). Their matrix is composed of distinct zones that vary both biochemically in composition and cellularly with respect to chondrocyte size and gene expression, resembling the layers seen in growth plate cartilage during endochondral bone formation. The superficial zone cells along the surface of the articular cartilage have a flattened morphology and express Prg4, which encodes LUBRICIN to promote joint lubrication(14,15). The middle zone (transitional/radial zone) cells have a rounded morphology and express matrix components COL2A1 and ACAN. The deep zone cells most closely resemble hypertrophic chondrocytes and express COL10A1 and MMP13. The middle and deep zones are separated by a basophilic line called the tidemark; this line segments the unmineralized and mineralized portions of the articular cartilage(16). Despite the fact that the zones of the articular cartilage are reminiscent of those in the growth plate cartilage, articular chondrocytes of the superficial or middle zones do not enter hypertrophy or undergo terminal maturation at homeostasis. However, it has been reported that during the development of osteoarthritis (OA), these articular chondrocytes begin to mimic growth plate chondrocytes and aberrantly express proteins associated with chondrocyte hypertrophy, such as COL10A1 and MMP13(17–20). Whether onset of hypertrophy alone is sufficient to promote OA pathogenesis, though, is unclear.
RUNX2 (also known as CBFA1) is a transcription factor that plays a critical role in induction of chondrocyte hypertrophy and terminal maturation as well as in osteoblast differentiation during endochondral bone formation. RUNX2 is expressed by both prehypertrophic and hypertrophic chondrocytes, as well as by cells within the perichondrium(4,6,21,22). In chondrocytes, direct target genes of RUNX2 include Ihh, Col10a1, and Mmp13, all markers of chondrocyte hypertrophy and maturation(23–26). Global deletion of Runx2 in mice causes a loss or severe delay of chondrocyte hypertrophy and a complete lack of bone formation throughout the skeletons of RUNX2-deficient embryos(21,22,27). Interestingly, Runx2 deletion in osteochondroprogenitor cells leads to a long bone and vertebral phenotype that is similar to that seen in global knockouts, whereas deletion of Runx2 from committed osteoblasts results in no skeletal abnormalities(28,29). Further, studies of RUNX2 overexpression using Col2a1-driven transgenes lead to accelerated and ectopic endochondral ossification due to enhanced chondrocyte hypertrophy and terminal differentiation(6). Overall, these results emphasize the importance of RUNX2 in chondrocytes for normal endochondral bone development.
Based on the well-documented role of RUNX2 in chondrocyte maturation, several studies have examined the postnatal role of RUNX2 in articular cartilage, and in the induction of OA. While baseline RUNX2 expression in the articular cartilage is minimal, it is induced following traumatic joint injury, along with its downstream targets COL10A1 and MMP13(19,30,31). Global RUNX2 haploinsufficiency as well as chondrocyte-specific Runx2 deletion were both shown to be chondroprotective following traumatic knee joint injury(30,32). Expression of matrix degrading enzymes and overall cartilage degeneration were decreased in both models suggesting that RUNX2 expression in chondrocytes contributes to the pace of OA progression. Whether RUNX2 overexpression is sufficient to induce postnatal OA onset, however, has not been able to be addressed by previous genetic models due to perinatal lethality.
In this study, we aimed to explore the ability of chondrocyte-specific RUNX2 overexpression to affect chondrocyte maturation within the postnatal articular cartilage. We used the Rosa26Runx2 allele in combination with the inducible AcanCreERT2 knock-in allele(33) to force expression of RUNX2 specifically in immature chondrocytes of the articular cartilage in adult mice. The Rosa26Runx2 allele was previously shown to rescue bone formation in Runx2−/− embryos carrying the Col2a1Cre transgene(34) and is shown here to promote chondrocyte hypertrophy and terminal maturation during development when combined with the inducible Col2a1CreERT2 transgene(35). RUNX2 overexpression in the articular chondrocytes of adult mice surprisingly did not induce changes to the articular cartilage at homeostasis. Following traumatic joint injury, however, RUNX2 overexpression resulted in more severe articular cartilage degeneration likely due to increased expression of MMP13 and enhanced apoptosis of articular chondrocytes. These data suggest that while increased expression of RUNX2 alone may not be sufficient to promote cartilage degeneration, it can accelerate the development of post-traumatic OA (PTOA) following joint injury.
MATERIALS AND METHODS
Mice
Animal studies used protocols approved by the University of Rochester Committee on Animal Resources. Mice were housed in groups of up to 5 animals in a room using microisolator technology and kept at 21°C-23°C with a 12-hour light/dark schedule. They had ad libitum access to food and water at all times. Col2a1CreERT2, AcanCreERT2, and Rosa26Runx2 (R26Runx2) mice were previously described (33–35). Female R26Runx2/Runx2 mice were bred with male Col2a1CreERT2/+; R26Runx2/+ or AcanCreERT2; R26Runx2/+ mice to generate experimental mice heterozygous or homozygous for the R26Runx2 allele in combination with either the Col2a1CreERT2 transgene AcanCreERT2 knock-in allele. Cre-negative littermate mice were used as controls. To induce RUNX2 overexpression (OE) in embryos, pregnant females were administered tamoxifen once at E13.5 (0.1 mg/g body weight, IP). For postnatal and MLI studies, 2-month-old mice were administered tamoxifen (Sigma) for 5 consecutive days (0.1 mg/g body weight, IP). For the injury studies, meniscal/ligamentous injury (MLI) was performed as previously described on Cre-negative control (Control) and AcanCreERT2/+; R26Runx2/+ (RUNX2 OE) mice at 10 weeks of age (36). The surgeon was blinded to the genotypes of the mice. Briefly, a 5-mm incision was made on the medial aspect of the joint; the medial collateral ligament (MCL) of the right knee was then transected and a segment of the anterior horn of the medial meniscus excised. Contralateral sham joints only received the initial incision (no joint structures were manipulated). Numbers of mice per experimental group are as indicated: E14.5 Col2a1CreERT2 for histology (Cre−, n = 7; Cre+ R26Runx2+/−, n = 6; Cre+ R26Runx2/Runx2, n = 4), E18.5 Col2a1CreERT2 for skeletal preparations (Cre−, n = 4, Cre+ R26Runx2+/−, n = 7; Cre+ R26Runx2/Runx2, n = 6), E18.5 Col2a1CreERT2 for histology (Cre−, n = 6; Cre+ R26Runx2+/−, n = 3; Cre+ R26Runx2/Runx2, n = 9), 3-month-old AcanCreERT2 (Cre−, n = 5, Cre+; R26Runx2+/−, n = 4), 8-month-old AcanCreERT2 (Cre−, n = 9; Cre+ R26Runx2/+, n = 6), 3.5-month-old AcanCreERT2 with MLI at 2.5 months (Cre− males, n = 6; Cre+ males, n = 6; Cre− females, n = 6; Cre+ females, n = 6), 4.5-month-old AcanCreERT2 with MLI at 2.5 months (Cre− male, n = 6; Cre+ male, n = 7; Cre− female, n = 6; Cre+ female, n = 6).
Skeletal preparations, histology, in situ hybridization, and immunohistochemistry
Whole mount Alcian blue/Alizarin red skeletal staining of E18.5 embryos was performed as described(37). For histology, embryonic tissues were fixed in 10% neutral buffered formalin (NBF) for 24 hours followed by decalcification in 14% EDTA (pH 7.3–7.4) for 24 hours. Adult hindlimbs were cleaned of soft tissue and fixed in 10% NBF for 3 days followed by decalcification for 1 week in 14% EDTA (pH 7.4–7.6). All fixation and decalcification steps were carried out at room temperature. Tissues were then processed for paraffin embedding and sectioning at 5 microns with postnatal hindlimbs oriented sagittally. Embryonic tissue sections were stained with Alcian blue Hematoxylin/Orange G and adult tissue sections with Safranin O/Fast green to visualize cartilage, bone, and soft tissues. In situ hybridization of embryonic tissue sections was performed as previously described using 35S-labeled riboprobes to Col10a1 and Mmp13(38,39). After developing, slides were counterstained with Toluidine blue. Final images were prepared by merging brightfield images with red pseudo-colored darkfield images.
For MMP13, COL10A1, and RUNX2 immunohistochemistry, sections were deparaffinized and rehydrated. Antigen retrieval was performed in 4 mg/ml pepsin solution made in 0.01 N HCl for 10 minutes at 37°C (for MMP13 and COL10A1) and in Antigen Unmasking Solution, Citrate Based (Vector Labs) overnight at 65°C (for RUNX2). Following rinses with deionized water, sections were incubated with BLOXALL to quench endogenous peroxidases (Vector Labs) for 10 minutes and rinsed in deionized water. Sections were then blocked in 2.5% normal horse serum for 30 minutes and incubated with primary antibody for 1 hour at room temp (anti-COL10A1 diluted 1:500, Clone X53, Quartett) or overnight at 4°C (anti-MMP13 diluted to 4 μg/ml, ab75606, Abcam; anti-RUNX2 diluted to 0.125 μg/ml, HPA022040, Sigma). Sections were rinsed in phosphate buffered saline + 0.1% tween-20 (PBST) and incubated with ImmPress HRP anti-rabbit IgG peroxidase polymer secondary (anti-MMP13, Vector Labs) or ImmPress HRP anti-mouse IgG peroxidase polymer secondary (anti-COL10A1, Vector Labs) for 30 minutes. After subsequent rinses in PBST and deionized water, COL10A1 or MMP13 were detected by DAB peroxidase HRP substrate (Vector Labs). For RUNX2, the ImmPress Excel HRP anti-rabbit IgG peroxidase polymer secondary kit was used per manufacturer’s instructions (Vector Labs). All slides were counterstained using Mayer’s hematoxylin. For negative control slides for anti-MMP13 and anti-RUNX2 staining, rabbit IgG (Vector Labs) was used at a concentration of 4 μg/ml (MMP13) or 0.125 μg/ml (RUNX2). As the stock concentration of COL10A1 is unknown, negative control slides were incubated with secondary antibody only. TUNEL staining was performed per manufacturer’s instructions using the In Situ Cell Death Detection Kit, Fluorescein (Sigma Roche).
Human cartilage samples
As previously described(40), a tissue microarray was created using 2 to 3 cores from 11 normal cartilage samples and 14 injured cartilage samples. An IRB-approved protocol was used to collect discarded cartilage tissue from orthopaedic surgery patients. Normal cartilage was collected from hip fracture patients. Knee articular cartilage was collected from patients undergoing arthroscopic surgery 4 weeks following meniscal injury. No identifiers are associated with the tissues. Tissue was fixed, decalcified, and processed for embedding into paraffin.
Chondrocyte isolation and culture
Primary chondrocytes from the sterna and ribs of P3 mice were isolated as described(41) and cultured in DMEM (Life Technologies) containing 10% FBS and 1% penicillin/streptomycin (Life Technologies). Ad5-CMV-GFP and Ad5-CMV-Cre adenoviruses were purchased from Baylor Vector Development Laboratory (Houston, TX) and used to infect the chondrocytes at a MOI of 1000. Forty-eight hours following infection, adenovirus was replaced with standard culture media supplemented with 50 μg/ml ascorbic acid. After an additional 48 hours, cells were either fixed with 4% paraformaldehyde and incubated with 1-Step NBT/BCIP Solution (Thermo Fisher Scientific) for detection of Alkaline Phosphatase or harvested for Western blotting or quantitative reverse transcription PCR.
Western blotting
Western blotting was performed as described previously(40). Primary antibodies were used as follows: mouse anti-RUNX2 (1:500, D130–3, MBL) and mouse anti-β-ACTIN (1:5000, AC-74, Sigma-Aldrich).
Isolation of total RNA from mouse articular cartilage
Using scalpels, articular cartilage was shaved from the tibial plateau and frozen over dry ice. Using the Bullet Blender Gold (Next Advance), the cartilage was homogenized in 250 μl of TRIzol (Thermo Fisher) with a mix of 2, 1, and 0.5 mm RNase-free zirconium oxide beads (Next Advance) for 10 minutes. Phases were separated using 100 μl chloroform per TRIzol manufacturer’s instructions (Thermo Fisher), and the aqueous layer and one volume of 70% ethanol were transferred to a RNeasy MinElute Spin Column from the RNeasy Micro Kit (QIAGEN). mRNA was then isolated per Micro Kit instructions (QIAGEN).
Quantitative reverse transcription PCR
Total RNA was isolated from cell cultures using the RNeasy Mini Kit (Qiagen) per manufacturer’s instructions or from mouse tibial cartilage as described above. RNA was reverse-transcribed into cDNA using the iScript cDNA Synthesis Kit (Bio-Rad). Real-time PCR was performed using a Rotor-Gene Q real-time PCR cycler (Qiagen) and the PerfeCTa SYBR Green SuperMix (Quanta Biosciences) according to manufacturer’s instructions. Primer sequences used are as follows (5’−3’): β-actin forward: AGATGTGGATCAGCAAGCAG; β-actin reverse: GCGCAAGTTAGGTTTTGTCA; Runx2 forward: TGATGACACTGCCACCTCTGACTT; Runx2 reverse: ATGAAATGCTTGGGAACTGCCTGG. Col10a1 forward: CTTTGTGTGCCTTTCAATCG; Col10a1 reverse: GTGAGGTACAGCCTACCAGTTTT; Mmp13 forward: GATGACCTGTCTGAGGAAG Mmp13 reverse: ATCAGACCAGACCTTGAAG; Alpl forward: TGACCTTCTCTCCTCCATCC; Alpl reverse: CTTCCTGGGAGTCTCATCCT
Modified OARSI Scoring
Semi-quantitative scoring was performed in a blinded fashion on MLI knee joint sections from mice 1 and 2-months following MLI surgery using the grading system described previously(42). Briefly, 3 different sections from distinct levels per MLI sample were stained with Safranin O/Fast green and randomized for scoring. Four independent scorers performed modified OARSI scoring and scores were averaged for each slide. The average score for the 3 slides from each sample were then averaged to obtain a final score for that sample.
Histomorphometry
Histomorphometry was performed using Visiopharm image analysis software (Version 6.7.0.2590). The measure tool was used to measure limb lengths and an average limb length was calculated from 3–5 embryos per genotype. An image analysis application was designed to measure the area of mineralized (below the tidemark) and unmineralized (above the tidemark) articular cartilage. The measure tool was used as a ruler to measure the same length across the articular surface on each section, and the ROI tool was used to manually circle the relevant cartilage areas within the length measured. The application was then trained to recognize the difference between Safranin O-positive regions (denoted as SafO+) and negative, Fast green, regions (denoted as background) of the cartilage areas using the threshold method. This was done using the Contrast Red-Green feature; areas were marked SafO+ if they met a threshold of −10 pixel intensity or above, and were marked as background if they were below the −10 value. We then applied several post-processing corrections to allow the software to recognize the white interior of chondrocytes not stained by Safranin O as portions of the SafO+ area, rather than background; the change by area feature was used to change any area labeled background 50 μm2 or smaller that was completely surrounded by SafO+ area to SafO+. A second change by area feature was used to change any area labeled SafO+ 50 μm2 or smaller that was completely surrounded by background to background; this allowed Visiopharm to recognize hematoxylin-stained chondrocyte nuclei as a part of the SafO+ area. The software was then able to calculate the area of the manually circled unmineralized and mineralized cartilage, as well as the SafO+ area within those regions. Values were averaged from three sections for both sham and MLI samples, and the MLI sample normalized to the sham for each animal.
For quantification of the COL10A1+ or RUNX2+ area, an image analysis application was designed to determine the amount of tissue stained brown with DAB (denoted as DAB+) within the articular cartilage. As in the SafO application, the measure tool was used, followed by the ROI tool to circle the articular cartilage area within the length measured. The application was then trained to recognize the difference between DAB+ brown tissue and hematoxylin stained tissue (denoted as background) using the threshold method. This was done using the HDAB-DAB feature; areas were marked DAB+ if they were at or below this threshold. The software was then able to calculate the circled cartilage area, as well as the COL10A1+ or RUNX2+ area within this region.
For MMP13+ or RUNX2+ cell counting, the stereology function of Visiopharm was used to manually determine the number of MMP13+ or RUNX2+ cells and total cells. For TUNEL+ cell counting, an image analysis application was designed on Visiopharm to recognize and count TUNEL+ stained cells and FITC+ stained cells within the articular cartilage using the threshold method. As described above, the measure tool was used followed by the ROI tool to manually circle the articular cartilage area within the length measured. The method was programmed to recognize DAPI+ and FITC+ cells using the DAPI W and FITC features. Areas were marked DAPI+ if they had DAPI values at 3000 or above and FITC values lower than 900; areas were marked FITC+ if they had FITC values at or over 900. All areas with DAPI values below 3000 or FITC values below 900 were marked as background. We then applied several post-processing corrections to allow the software to exclude any background positive smaller than the smallest typical area of a cell found within the articular cartilage (less than 10 μm2); using the change by area feature, both DAPI+ and FITC+ areas smaller than 10 μm2 were changed to background. The software was then able to calculate the number of DAPI+ and FITC+ areas within the circled ROI; each positive area was counted as one cell.
Statistics
Data are presented as the mean ± SEM. Statistical significance was determined by Student’s t tests or one-way ANOVA followed by Dunnett’s multiple comparisons test as indicated; p values less than 0.05 were considered significant.
RESULTS
Chondrocyte-specific RUNX2 overexpression during development leads to chondrodysplasia
It is generally accepted that RUNX2 expression in chondrocytes during skeletal development promotes hypertrophy. Therefore, we first evaluated the effects of chondrocyte-specific RUNX2 overexpression from the R26Runx2 allele(34) using a developmental model. Specifically, we generated Col2a1CreERT2/+ embryos(35) with one or two copies of R26Runx2 (Col2a1CreERT2/+; R26Runx2/+ and Col2a1CreERT2/+; R26Runx2/Runx2, respectively), allowing expression of the MASNS isoform of RUNX2 in a Cre-inducible manner. Recombination was induced via administration of tamoxifen at E13.5 and embryos were harvested at E18.5. The RUNX2-overexpressing (OE) embryos had shortened trunks, limbs, and domed skulls with protruding tongues, consistent with chondrodysplasia. Whole mount Alcian blue and Alizarin red staining revealed shorter axial skeletons, smaller rib cages, and shorter forelimbs and hindlimbs (Fig. 1A). Analysis of histological sections revealed decreases in the total length of the femora, tibiae, and humeri of the RUNX2 OE embryos compared to their Cre-negative control littermates (Fig. 1B). Additionally, RUNX2 OE tibiae were bowed with increased endocortical bone (Supplemental Fig. 1A). Histology also showed that the columnar and hypertrophic zones of the RUNX2 OE embryos are hypocellular and disorganized compared to control littermates with some chondrocytes in the hypertrophic zone appearing abnormally large (Fig. 1C). Immunohistochemistry (IHC) for RUNX2 on histological sections confirmed RUNX2 overexpression in the mutant limbs compared to control limbs (Supplemental Fig. 1B).
Fig. 1. Chondrocyte-specific RUNX2 overexpression during development leads to chondrodysplasia.
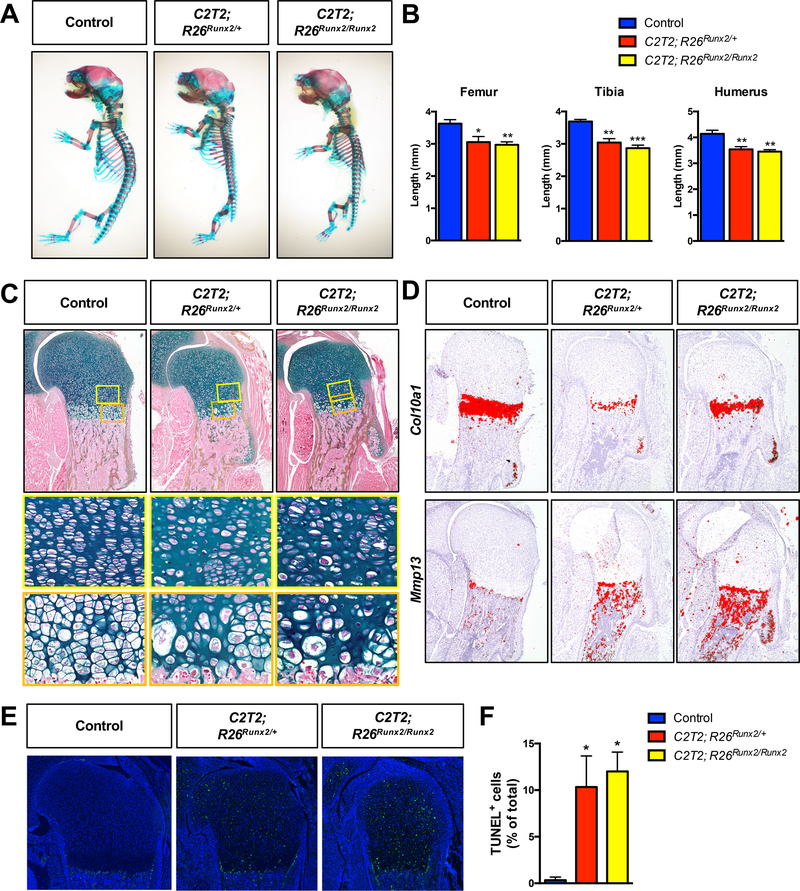
(A) Alcian blue/Alizarin red skeletal staining of E18.5 R26Runx2/+ (Control), Col2a1CreERT2/+; R26Runx2/+ (C2T2; R26Runx2/+), and Col2a1CreERT2/+; R26Runx2/Runx2 (C2T2; R26Runx2/Runx2) embryos. (B) Femur (left), tibia (middle), and humerus (right) lengths from E18.5 Control, C2T2; R26Runx2/+, and C2T2; R26Runx2/Runx2 embryos (n = 3 to 5 for all groups). *p < 0.05, **p < 0.01, ***p < 0.001, one-way ANOVA followed by Dunnett’s multiple comparisons test. (C) Alcian blue Hematoxylin/Orange G staining of humerus growth plate sections from E18.5 Control, C2T2; R26Runx2/+, and C2T2; R26Runx2/Runx2 embryos. Top panels are 5X images of the proximal humerus; middle panels are high magnification images (20X) of the proliferating and pre-hypertrophic zones (yellow boxes); bottom panels are high magnification images (20X) of the hypertrophic zone (orange boxes). (D) In situ hybridization for Col10a1 (top panels) and Mmp13 (bottom panels) on E18.5 Control, C2T2; R26Runx2/+, and C2T2; R26Runx2/Runx2 proximal humerus sections. (E) TUNEL staining of proximal humerus sections from E18.5 Control, C2T2; R26Runx2/+, and C2T2; R26Runx2/Runx2 embryos (5X images). (F) Quantification of TUNEL+ cell number as percentage of total cell number within the growth plate of the proximal humeri of E18.5 Control, C2T2; R26Runx2/+, and C2T2; R26Runx2/Runx2 embryos (n = 3 for all groups). *p < 0.05, one-way ANOVA followed by Dunnett’s multiple comparisons test.
In situ hybridization analyses on E18.5 humerus sections showed that the expression domain of Col10a1 relative to total limb length is unchanged in the RUNX2 OE embryos compared to littermate control embryos, while that of Mmp13 is slightly expanded and maintained in the trabecular bone of the RUNX2 OE embryos (Fig. 1D). Analyses of RUNX2 OE embryos at E14.5, however, revealed an expanded Col10a1 expression domain and early appearance of Mmp13 expression relative to control embryos (Supplemental Fig. 1C), suggesting early onset of hypertrophy and accelerated maturation to late stages of chondrocyte hypertrophy. Primary sternal chondrocytes from R26Runx2/Runx2 mice were infected with adenovirus encoding GFP or Cre recombinase and RUNX2 overexpression was confirmed at both the mRNA and protein levels (Supplemental Fig. 2A, C). Increased Alkaline phosphatase staining and Col10a1, Alpl, and Mmp13 gene expression in these cultures provide in vitro support that the R26Runx2-expressing chondrocytes undergo accelerated hypertrophy and terminal maturation in a cell autonomous manner.
Finally, to investigate whether the hypocellularity in the E18.5 RUNX2 OE limbs could be due to cell death, we performed TUNEL staining on tissue sections. TUNEL staining was visibly increased throughout all regions of the growth plate in the RUNX2 OE limbs and quantification of TUNEL+ cells relative to total cell numbers supported this observation (Fig. 1E, F). Collectively, our data support that chondrocyte-specific overexpression of RUNX2 from the R26Runx2 allele is sufficient to promote chondrocyte hypertrophy and terminal maturation during skeletal development.
Postnatal chondrocyte-specific RUNX2 overexpression alone is not sufficient to induce articular cartilage degeneration
RUNX2 expression is reported to be upregulated in human OA cartilage and in the articular cartilage of mice following traumatic joint injury(30,32,43). Using a human tissue microarray, we also find evidence of increased RUNX2 expression in the articular cartilage of patients just one month following meniscal tear (Fig. 2). Since it is well established that joint injury increases the risk for development of OA(44,45), we decided to examine whether RUNX2 overexpression alone could be sufficient to induce catabolic changes to the joint consistent with the onset of OA. Specifically, we generated AcanCreERT2/+ mice with one copy of the R26Runx2 allele (AcanCreERT2/+; R26Runx2/+). Recombination was induced via administration of tamoxifen at 2 months of age, a time point following the majority of rapid skeletal growth that occurs during early postnatal life. One week following tamoxifen induction, there were no overt changes in cartilage phenotype, but RUNX2 IHC did show increased RUNX2 expression within the articular cartilage of RUNX2 OE mice (Fig. 3A, B). Additionally, quantitative RT-PCR from mRNA isolated from the tibial articular cartilage confirmed this finding, showing a significant increase in Runx2 expression in the RUNX2 OE group (Fig. 3C). Surprisingly, at 8 months of age (6 months following tamoxifen induction), there are still no overt phenotypic changes to the articular cartilage of the RUNX2 OE mice relative to control mice, despite evidence that RUNX2 is still expressed in the articular chondrocytes at this time (Fig. 3D, E). Specifically, significant increases in RUNX2 protein as quantified by RUNX2+ area stained and RUNX2+ cell numbers were determined (Fig. 3F).
Fig. 2. Knee joint injury leads to increased RUNX2 expression in human articular cartilage.

Representative results of RUNX2 immunohistochemistry on a tissue microarray of normal human cartilage or cartilage from patients undergoing arthroscopic surgery 4 weeks following meniscal injury (Post-Injury). Left panels within each group are 10X images; right panels are 20X images of the corresponding boxed regions.
Fig. 3. Chondrocyte-specific RUNX2 overexpression is insufficient to induce phenotypic changes in the articular cartilage.
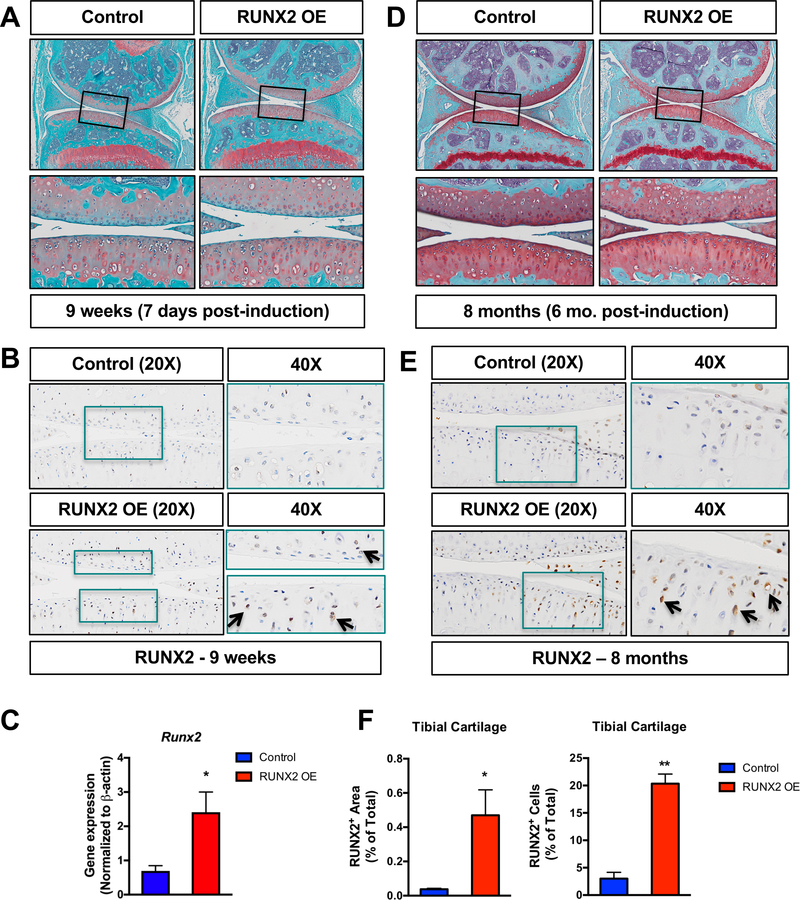
(A, D) Safranin O/Fast green staining of knee joint sections from male R26Runx2/+ (Control) and AcanCreERT2/+; R26Runx2/+ (RUNX2 OE) mice injected with tamoxifen at 2 months of age daily for 5 consecutive days and harvested 48 hours following the last tamoxifen injection at 9 weeks of age (A) or at 8 months of age (D). Top panels are 5X images of the knee joint; bottom panels are high magnification images (20X) of the boxed regions. (B, E) RUNX2 immunohistochemistry on knee joint sections from Control and RUNX2 OE mice at 9 weeks of age (B) and 8 months of age (E). Left panels are 20X images of the articular cartilage; right panels are high magnification images (40X) of the boxed regions. (C) Quantitative RT-PCR from tibial articular cartilage of Control and RUNX2 GOF mice harvested at 3 months of age following tamoxifen injections at 2 months of age (Control, n = 3, RUNX2 OE, n = 3). (F) Quantification of RUNX2+ cartilage area as percentage of total cartilage area and RUNX2+ cell number as percentage of total cell number from Control and RUNX2 OE mice (Control, n = 3, RUNX2 OE, n = 3). *p < 0.05, Student’s t-test.
Postnatal chondrocyte-specific RUNX2 overexpression promotes accelerated articular cartilage degeneration following joint injury
It was shown previously that global haploinsufficiency or chondrocyte-specific loss of Runx2 can inhibit the progression of post-traumatic OA following traumatic joint injury(30,32). Since we did not find that RUNX2 OE alone in articular chondrocytes was sufficient to promote cartilage degeneration, however, we next tested whether chondrocyte-specific RUNX2 OE could affect the progression of OA in the injured joint environment. Specifically, we induced Cre-mediated recombination in both male and female Cre-negative control and RUNX2 OE (AcanCreERT2/+; R26Runx2/+) mice at 2 months of age using tamoxifen and subjected the mice to sham or MLI surgery at 2.5 months of age. Knee joints of control and RUNX2 OE mice were harvested 2 months following injury. Histologically, male RUNX2 OE mice showed enhanced articular cartilage degradation following MLI when compared to control mice, while sham sections did not reveal any detectable differences in the articular cartilage (Fig. 4A). To quantitatively assess the histological differences between male control and RUNX2 OE mice, modified OARSI scoring and manual histomorphometry were performed. MLI sections from RUNX2 OE mice showed significantly higher OARSI scores than those from control mice, and also showed reductions in overall tibial cartilage area (Fig. 4B, C). Tibial histomorphometric changes were seen in both the unmineralized and mineralized regions of the articular cartilage (Fig. 4D). SafO+ cartilage area was specifically reduced in the unmineralized tibial cartilage.
Fig. 4. Postnatal RUNX2 overexpression accelerates articular cartilage degeneration following meniscal-ligamentous injury (MLI) in male mice.
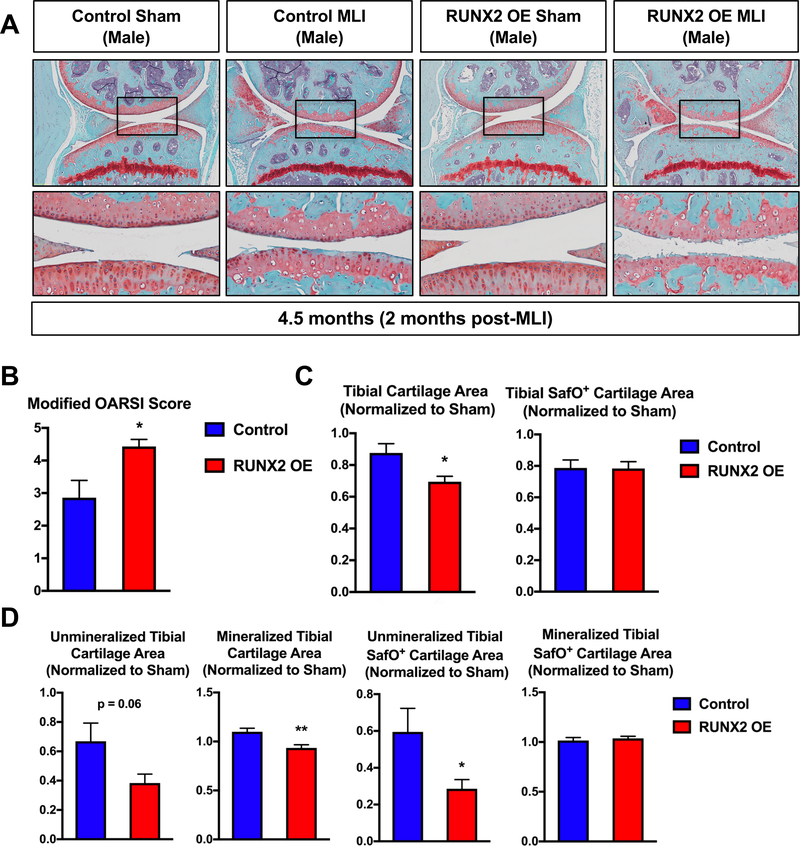
(A) Safranin O/Fast green staining of knee joint sections from male R26Runx2/+ (Control) and AcanCreERT2/+; R26Runx2/+ (RUNX2 OE) mice injected with tamoxifen at 2 months of age and subjected to sham or MLI at 2.5 months of age. Joints were harvested 2 months following injury. Top panels are 5X images of the knee joint; bottom panels are high magnification images (20X) of the boxed regions. (B) Modified OARSI scoring of Control and RUNX2 OE slides from the knee joint receiving MLI. (C, D) Quantitative histomorphometric analyses of total tibial cartilage area (C, left panel), total tibial SafO+ area (C, right panel), unmineralized and mineralized tibial cartilage areas (D, left panels), and unmineralized and mineralized tibial SafO+ areas (D, right panels) where MLI cartilage was normalized to corresponding sham cartilage in knee joint sections from Control (n = 6) and RUNX2 OE (n = 7) mice. *p < 0.05, **p < 0.01, Student’s t-test.
In contrast, female RUNX2 OE mice did not show differences in articular cartilage degradation in response to MLI relative to control mice (Supplemental Fig. 3A). This histological finding was confirmed by OARSI scoring and manual histomorphometry, which revealed no significant changes between the control and RUNX2 OE female mice following injury (Supplemental Fig. 3B-D). To be certain this result was not due to a lower expression of RUNX2 protein levels, we quantified RUNX2+ stained area and RUNX2+ cell numbers in the cartilage of 3.5-month-old sham joints and found similar increases in both males and females (Supplemental Fig. 4).
In order to examine early molecular changes underlying the articular cartilage phenotype seen in the male RUNX2 OE mice 2 months following MLI, we performed a second set of sham or MLI surgeries on control and RUNX2 OE mice and harvested their knee joints one month following injury. Histology revealed decreased Safranin O staining within the tibial articular cartilage of the RUNX2 OE mice relative to controls, but these changes did not result in significant differences in modified OARSI score or manual histomorphometric measurements (Fig. 5A, B). We then performed immunohistochemistry for MMP13 and COL10A1, known downstream targets of RUNX2 in cartilage tissue(24,26,46,47). Quantification of the MMP13 staining revealed a significant increase in MMP13+ chondrocytes present in the tibial articular cartilage of the RUNX2 OE mice (Fig. 5D, E). This increase in MMP13+ cells was primarily due to the increase in positive cells from the unmineralized region of the articular cartilage, as cell numbers were not significantly altered in the mineralized cartilage region (Fig. 3D). There was not a significant change in COL10A1-stained cartilage area in the RUNX2 OE mice.
Fig. 5. Postnatal RUNX2 overexpression induces MMP13 expression in unmineralized articular chondrocytes prior to accelerating articular cartilage degeneration following MLI.
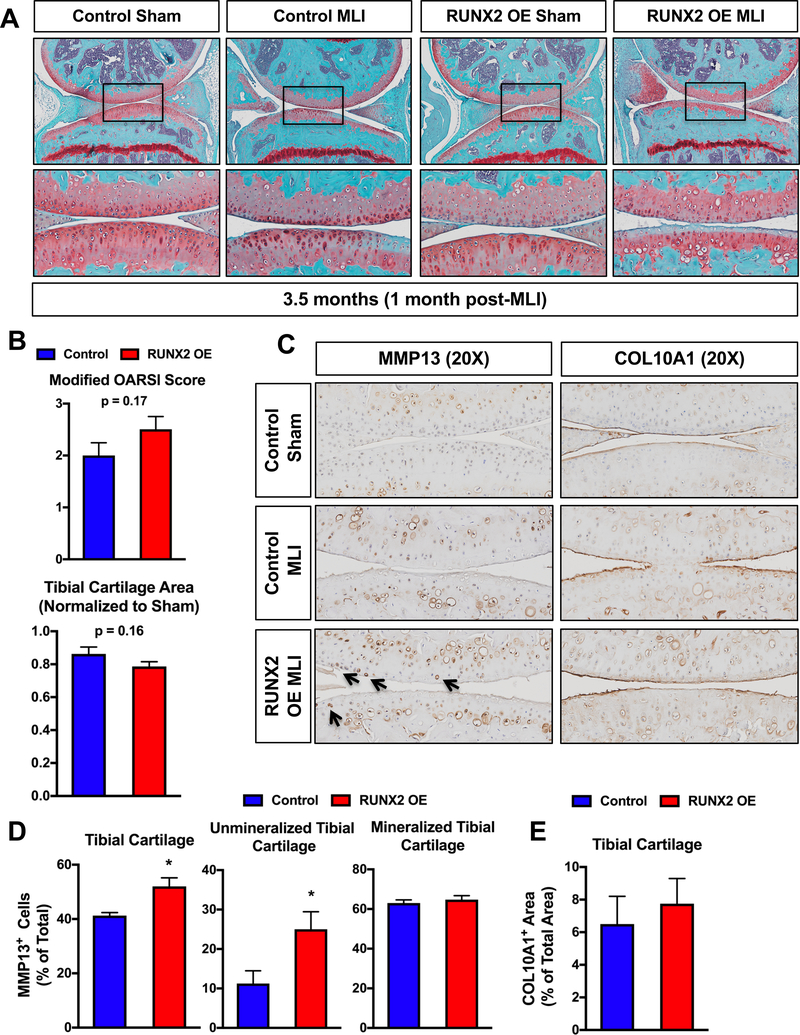
(A) Safranin O/Fast green staining of knee joint sections from R26Runx2/+ (Control) and AcanCreERT2/+; R26Runx2/+ (RUNX2 OE) mice injected with tamoxifen at 2 months of age and subjected to sham or MLI at 2.5 months of age. Joints were harvested one month following injury. Top panels are 5X images of the knee joint; bottom panels are high magnification images (20X) of the boxed regions. (B) Cartilage degeneration in Control (n = 6) and RUNX2 OE (n = 6) mice was evaluated by modified OARSI scoring (top graph) and quantitative histomorphometric analysis (bottom graph, data normalized to contralateral sham control sample). (C) MMP13 (left panels) and COL10A1 (right panels) immunohistochemistry of knee joint sections from Control or RUNX2 OE mice subjected to sham or MLI at 2.5 months of age and harvested 1 month following injury (20X images). (D) Quantification of MMP13+ cell number as percentage of total cell number from Control and RUNX2 OE mice (MLI samples only, n = 4 for both groups). (E) Quantification of COL10A1+ cartilage area as percentage of total cartilage area from Control and RUNX2 OE mice (MLI samples only, n = 4 for both groups). *p < 0.05, Student’s t-test.
Finally, given the increase in chondrocyte apoptosis due to RUNX2 OE during development, we next examined whether RUNX2 OE affected the amount of apoptotic cell death following traumatic joint injury. TUNEL staining was visibly increased within both the tibial and femoral articular cartilage of the RUNX2 OE mice relative to control mice following MLI and quantification of TUNEL+ cells relative to total articular chondrocyte numbers supported this observation (Fig. 6A, B).
Fig. 6. Postnatal RUNX2 overexpression results in enhanced articular chondrocyte apoptosis following meniscal-ligamentous injury (MLI).

(A) TUNEL staining of knee joint sections from R26Runx2/+ (Control) and AcanCreERT2/+; R26Runx2/+ (RUNX2 OE) mice subjected to MLI at 2.5 months of age and harvested 1 month following injury (10X images). (B) Quantification of TUNEL+ cell number as percentage of total cell number from the tibial (left) or femoral (right) articular cartilage of Control or RUNX2 OE mice (MLI samples only, n = 4 for both groups). *p < 0.05, **p < 0.01, Student’s t-test.
DISCUSSION
To our knowledge, this is the first study to assess the effects of exogenous RUNX2 expression in non-hypertrophic chondrocytes of adult mice and to test the hypothesis that expression of RUNX2 alone in this cell population is sufficient to promote the onset of OA. Despite significantly increased detectable expression of RUNX2 protein in the unmineralized region of the articular cartilage of mice immediately following induction and even 6 months later, we observed no difference in joint phenotype when compared to Cre-negative littermate control mice. Following surgical destabilization and meniscal injury of the joint, however, we found a significant difference in the rate of progression of PTOA in male mice. While we interpret these results to mean that RUNX2 expression from the R26Runx2 allele is insufficient to induce articular chondrocyte hypertrophy and onset of OA-like phenotypic changes in adult cartilage, we are hesitant to conclude that no level of RUNX2 expression in this cell population would be able to induce such a phenotype. The R26Runx2 allele was suggested by Tu et. al. to express a relatively low abundance of exogenous Runx2 as they did not observe an increase in Runx2 message by in situ hybridization with one or even two R26Runx2 alleles(34) when driven by the Col2a1-Cre transgene in embryonic limbs. They did observe, however, that the level of expression from the R26Runx2 allele was sufficient to produce a functional protein product capable of rescuing bone formation in Runx2−/− embryos by promoting chondrocyte hypertrophy, perichondrial bone formation, and primary ossification center formation. Here, we detected RUNX2 protein by IHC in the unmineralized region of the articular cartilage in adult AcanCreERT2/+; R26Runx2/+ mice, a region where RUNX2 is not normally expressed(48). Approximately 20% of articular chondrocytes expressed levels detectable by IHC at both 1.5 and 6 months post-induction (Fig. 3F & Supplemental Fig. 4). Whether this number of RUNX2+ cells is simply too low to induce phenotypic changes to the cartilage or whether expression of RUNX2 alone does not lead to RUNX2 activation in this cell population is difficult to determine. What is very clear from our data, however, is that even this modest level of RUNX2 expression can predispose to accelerated onset and progression of PTOA following joint injury.
Eight weeks following surgical induction of injury to the knee joint, OARSI scores and histomorphometric measurements confirmed enhanced cartilage degeneration in male RUNX2 OE mice. While there were no significant differences in OARSI scores or histomorphometric measurements 4 weeks following injury, we did find a significant increase in the number of MMP13+ and TUNEL+ cells suggesting increased cartilage catabolism and chondrocyte death in the injured RUNX2 OE joints. While Mmp13 is a well-established direct downstream target gene of RUNX2(23,24), it is less clear how RUNX2 might be affecting chondrocyte apoptosis. RUNX2 was shown to promote apoptosis in osteosarcoma cells via induction of Bax expression, but it has actually been shown to inhibit apoptosis in some other cell types(44,49,50). Surprisingly, we did not find a significant difference in the amount of COL10A1 present in the cartilage matrix of the control and RUNX2 OE joints. We hypothesize that this might be due to decreased SOX9 expression in the injured joint(51) (data not shown) and the requirement of SOX9 for Col10a1 expression(52).
We employed the MLI surgery to initiate PTOA in these studies. This surgery combines transection of the MCL and excision of a piece of the anteromedial meniscus, which induces a progressive OA-like phenotype resulting in complete loss of cartilage by 20 weeks post-injury(19,36). The MLI is a model of partial meniscectomy, an event known to increase the risk of OA development in humans, and we use it routinely with consistent results in mice(31,53,54). Compared to the more widely used destabilized medial meniscus (DMM) surgery where transection of the medial meniscotibial ligament induces joint instability, OA following MLI progresses at a slightly accelerated rate. While we did find statistically significant differences using the MLI, it is possible that the magnitude of these differences could have been greater had we used a less aggressive model of PTOA such as the DMM.
Among the most important risk factors for the development OA in humans are prior traumatic joint injury and meniscectomy. It is estimated that approximately 12% of all symptomatic OA is related to prior injury and that 50% of all patients with an ACL or meniscus tear will develop knee OA(44,45). A recent study comparing genetic risk variant loci in post-traumatic OA versus non-traumatic OA shows that the genetic contribution to the development of knee OA is just as high following injury as in primary, non-traumatic OA(55). This suggests that underlying genetic variation may contribute to whether a patient will develop OA following injury and may even determine the rapidity of onset. Interestingly, large-scale genome-wide association studies (GWAS) have identified single nucleotide polymorphisms (SNPs) associating with OA near the RUNX2 gene locus(56). Additionally, hypomethylation of the RUNX2 promoter was detected in a genome-wide methylation study comparing normal chondrocytes and OA chondrocytes(57). Furthermore, OA candidate genes identified through GWAS studies include components of the WNT (FRZB, DOT1L), BMP (GDF5), and TGF-β (SMAD3) signaling pathways(58–61). These pathways converge upon RUNX2 to regulate chondrocyte maturation. These findings suggest that genetic or epigenetic changes in Runx2, or in the components of pathways that regulate RUNX2 expression and activity, may predispose to OA. Indeed, the genetic model we present here supports that RUNX2 overexpression in articular chondrocytes can accelerate the progression of OA pathogenesis following traumatic joint injury.
Our studies included both male and female mice, but a significant difference in RUNX2-dependent cartilage degeneration following injury was only seen in male mice. This difference was not simply due to differences in levels of RUNX2 expression as the percentage of RUNX2+ articular chondrocytes was not significantly different between male and female RUNX2 OE mice (Supplemental Figure 4). Female mice, in general, develop less severe OA following traumatic knee joint injury than males and female-specific sex hormones offer protection against the development of post-traumatic OA, as ovariectomized female mice develop more severe OA following injury when compared to intact female mice(62,63). Similarly, in humans, the loss of estrogen following the onset of menopause leads to a dramatic increase in the incidence of OA in women(64,65). Estrogens have been shown to inhibit RUNX2 transcriptional activity in osteoblasts and breast cancer cells via the direct binding of estrogen receptor-α (ER-α) to the DNA-binding domain of RUNX2(66). It may be possible, therefore, that RUNX2 activity is inhibited in the chondrocytes of the RUNX2 OE female mice despite elevated RUNX2 protein expression. ER-α is expressed in the articular chondrocytes of both mice and humans, but whether RUNX2 activity is altered by estrogen signaling in chondrocytes has yet to be determined(67,68).
RUNX2 deletion is protective against the progression of PTOA(30,32). It was, therefore, surprising that RUNX2 overexpression alone did not promote the development of OA without injury. As discussed above, this could be due to technical limitations in the amount of exogenous RUNX2 expression from the R26Runx2 allele, but it may also suggest that there are mechanisms to restrict RUNX2 activity in the unmineralized articular cartilage at homeostasis. The injured joint environment, alternatively, may potentiate RUNX2 activity. RUNX2 is highly regulated through protein-protein interactions and post-translational modifications such as phosphorylation, acetylation, and ubiquitination to control its protein stability and/or transcriptional activity(69–71). Signaling cascades activated by pro-inflammatory cytokines, oxidative stress, and mechanical stress all have the potential to affect RUNX2 activity within chondrocytes following joint injury. For example, p38 and ERK mitogen-activated protein kinases (MAPKs) are both known to positively regulate RUNX2 transcriptional activity in osteoblasts via direct phosphorylation and these kinases are activated in chondrocytes following cartilage injury(72–77). While it is unknown whether they directly phosphorylate RUNX2 in chondrocytes, treatment of human articular chondrocytes with IL-1β leads to an induction of RUNX2-mediated MMP13 expression in a p38-dependent manner, suggesting that it may be possible(78,79). GSK3β can also directly phosphorylate RUNX2 in osteoblasts, but on residues that inhibit transcriptional transactivation(80). Further, treatment of chondrocyte cultures with GSK3β inhibitors was shown to promote nuclear localization of RUNX2 while inhibition of GSK3β in vivo enhanced cartilage degeneration in a murine model post-traumatic OA(81,82). With regard to protein-protein interactions, RUNX2 binds to numerous other transcription factors, co-activators, and co-repressors that modulate its ability to affect gene expression(71). C/EBPβ, for example, is a potent transcriptional partner of RUNX2 in regulation of Mmp13 expression and is only expressed in articular chondrocytes following joint injury in a HIF-2α-dependent manner(23). In contrast, the master chondrogenic transcription factor SOX9 was shown to promote RUNX2 degradation and to strongly suppress its activation through direct interaction(83,84). While SOX9 is expressed in articular chondrocytes at homeostasis, as mentioned above, it is decreased in injured chondrocytes and in response to pro-inflammatory cytokine signaling(51,85,86). Its loss following injury could, therefore, lead to activation of RUNX2 in the RUNX2 OE cartilage. Further studies, of course, will be required to determine if any of these or other potential mechanisms are responsible for the enhanced cartilage degeneration of the RUNX2 OE mice following joint injury.
Finally, the conclusions resulting from our developmental studies support those of the previous studies that investigated the effects of RUNX2 overexpression in non-hypertrophic growth plate chondrocytes(6,27,34). The differences in severity of phenotypes reported here compared to others could be due to timing of induction, cell-type targeting specificity, or simply the level of exogenous RUNX2 produced. We used the Col2a1-CreERT2 transgene to drive expression of the R26Runx2 allele in a chondrocyte-specific manner by allowing induction of Cre-recombination at E13.5 (35,38,87). We found a slight, but significant, decrease in femur, tibia, and humerus length as well as disorganization of the proliferative and hypertrophic growth plate zones at E18.5 using this system. At E14.5, the hypertrophic zone and Col10a1 expression domains were expanded and the Mmp13 expression domain detectable in the R26Runx2-expressing embryos. At E18.5, the Mmp13 expression domain appeared elongated at the chondro-osseous junction with increased expression in the trabecular bone, while the Col10a1 expression domain was no longer expanded relative to total limb length. The increased Mmp13 expression in the trabecular bone is interesting and could be due to continued expression of exogenous RUNX2 in osteoblasts that are originally derived from targeted growth plate chondrocytes, as mounting evidence supports that hypertrophic chondrocytes can become osteoblasts during endochondral bone formation(88–90). Collectively, these data provided functional confirmation that RUNX2 expression from the R26Runx2 allele in non-hypertrophic chondrocytes can accelerate onset of hypertrophy as well as late stages of chondrocyte maturation. Additionally, these results provided a premise for using the R26Runx2 allele in our postnatal studies allowing further investigation into the role of RUNX2 in articular cartilage homeostasis and OA pathogenesis.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by grants from the National Institutes of Health/National Institute of Arthritis and Musculoskeletal and Skin Diseases (T32 AR053459, P30 AR061307, P30 AR069655, R21 AR07928 to JHJ and RE, and R01 AR063071 to MJH). The authors thank Dr. Fanxin Long for his generous gift of the R26Runx2 mouse. We also thank Center for Musculoskeletal Research Histology Core members Sarah Mack and Kathy Maltby for their technical assistance. Finally, we gratefully acknowledge the technical expertise of Dr. Erik Sampson in the creation of the human cartilage tissue microarray as well as Edward Ayoub and Tony Mirando for advice and technical assistance.
Footnotes
DISCLOSURES
Dr. Jonason reports pilot funding from NIH/NIAMS P30 AR061307 and P30 AR069655 grants.
REFERENCES
- 1.Akiyama H, Chaboissier MC, Martin JF, Schedl A, de Crombrugghe B. The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. November 1 2002;16(21):2813–28. Epub 2002/11/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Han Y, Lefebvre V. L-Sox5 and Sox6 drive expression of the aggrecan gene in cartilage by securing binding of Sox9 to a far-upstream enhancer. Mol Cell Biol. August 2008;28(16):4999–5013. Epub 2008/06/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lefebvre V, Huang W, Harley VR, Goodfellow PN, de Crombrugghe B. SOX9 is a potent activator of the chondrocyte-specific enhancer of the pro alpha1(II) collagen gene. Mol Cell Biol. April 1997;17(4):2336–46. Epub 1997/04/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leung VY, Gao B, Leung KK, Melhado IG, Wynn SL, Au TY, et al. SOX9 governs differentiation stage-specific gene expression in growth plate chondrocytes via direct concomitant transactivation and repression. PLoS Genet. November 2011;7(11):e1002356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.St-Jacques B, Hammerschmidt M, McMahon AP. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. August 15 1999;13(16):2072–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takeda S, Bonnamy JP, Owen MJ, Ducy P, Karsenty G. Continuous expression of Cbfa1 in nonhypertrophic chondrocytes uncovers its ability to induce hypertrophic chondrocyte differentiation and partially rescues Cbfa1-deficient mice. Genes Dev. February 15 2001;15(4):467–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vortkamp A, Lee K, Lanske B, Segre GV, Kronenberg HM, Tabin CJ. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science. August 2 1996;273(5275):613–22. [DOI] [PubMed] [Google Scholar]
- 8.Aghajanian P, Mohan S. The art of building bone: emerging role of chondrocyte-to-osteoblast transdifferentiation in endochondral ossification. Bone Res. 2018;6:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karsenty G, Kronenberg HM, Settembre C. Genetic control of bone formation. Annual review of cell and developmental biology. 2009;25:629–48. Epub 2009/07/07. [DOI] [PubMed] [Google Scholar]
- 10.Kronenberg HM. Developmental regulation of the growth plate. Nature. May 15 2003;423(6937):332–6. Epub 2003/05/16. [DOI] [PubMed] [Google Scholar]
- 11.Long F, Ornitz DM. Development of the endochondral skeleton. Cold Spring Harb Perspect Biol. January 1 2013;5(1):a008334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Candela ME, Yasuhara R, Iwamoto M, Enomoto-Iwamoto M. Resident mesenchymal progenitors of articular cartilage. Matrix biology : journal of the International Society for Matrix Biology. October 2014;39:44–9. Epub 2014/09/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu CF, Samsa WE, Zhou G, Lefebvre V. Transcriptional control of chondrocyte specification and differentiation. Seminars in cell & developmental biology. February 2017;62:34–49. Epub 2016/10/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Decker RS, Um HB, Dyment NA, Cottingham N, Usami Y, Enomoto-Iwamoto M, et al. Cell origin, volume and arrangement are drivers of articular cartilage formation, morphogenesis and response to injury in mouse limbs. Dev Biol. June 1 2017;426(1):56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kozhemyakina E, Zhang M, Ionescu A, Ayturk UM, Ono N, Kobayashi A, et al. Identification of a Prg4-expressing articular cartilage progenitor cell population in mice. Arthritis Rheumatol. May 2015;67(5):1261–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zuscik MJ, Hilton MJ, Zhang X, Chen D, O’Keefe RJ. Regulation of chondrogenesis and chondrocyte differentiation by stress. J Clin Invest. February 2008;118(2):429–38. Epub 2008/02/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goldring MB. Chondrogenesis, chondrocyte differentiation, and articular cartilage metabolism in health and osteoarthritis. Ther Adv Musculoskelet Dis. August 2012;4(4):269–85. Epub 2012/08/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kawaguchi H Endochondral ossification signals in cartilage degradation during osteoarthritis progression in experimental mouse models. Mol Cells. February 29 2008;25(1):1–6. Epub 2008/03/06. [PubMed] [Google Scholar]
- 19.Kamekura S, Hoshi K, Shimoaka T, Chung U, Chikuda H, Yamada T, et al. Osteoarthritis development in novel experimental mouse models induced by knee joint instability. Osteoarthritis Cartilage. July 2005;13(7):632–41. Epub 2005/05/18. [DOI] [PubMed] [Google Scholar]
- 20.van der Kraan PM, van den Berg WB. Chondrocyte hypertrophy and osteoarthritis: role in initiation and progression of cartilage degeneration? Osteoarthritis Cartilage. March 2012;20(3):223–32. [DOI] [PubMed] [Google Scholar]
- 21.Inada M, Yasui T, Nomura S, Miyake S, Deguchi K, Himeno M, et al. Maturational disturbance of chondrocytes in Cbfa1-deficient mice. Dev Dyn. April 1999;214(4):279–90. Epub 1999/04/23. [DOI] [PubMed] [Google Scholar]
- 22.Kim IS, Otto F, Zabel B, Mundlos S. Regulation of chondrocyte differentiation by Cbfa1. Mech Dev. February 1999;80(2):159–70. Epub 1999/03/12. [DOI] [PubMed] [Google Scholar]
- 23.Hirata M, Kugimiya F, Fukai A, Saito T, Yano F, Ikeda T, et al. C/EBPbeta and RUNX2 cooperate to degrade cartilage with MMP-13 as the target and HIF-2alpha as the inducer in chondrocytes. Hum Mol Genet. March 1 2012;21(5):1111–23. Epub 2011/11/19. [DOI] [PubMed] [Google Scholar]
- 24.Jimenez MJ, Balbin M, Lopez JM, Alvarez J, Komori T, Lopez-Otin C. Collagenase 3 is a target of Cbfa1, a transcription factor of the runt gene family involved in bone formation. Mol Cell Biol. June 1999;19(6):4431–42. Epub 1999/05/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoshida CA, Yamamoto H, Fujita T, Furuichi T, Ito K, Inoue K, et al. Runx2 and Runx3 are essential for chondrocyte maturation, and Runx2 regulates limb growth through induction of Indian hedgehog. Genes Dev. April 15 2004;18(8):952–63. Epub 2004/04/27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zheng Q, Zhou G, Morello R, Chen Y, Garcia-Rojas X, Lee B. Type X collagen gene regulation by Runx2 contributes directly to its hypertrophic chondrocyte-specific expression in vivo. J Cell Biol. September 1 2003;162(5):833–42. Epub 2003/09/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ueta C, Iwamoto M, Kanatani N, Yoshida C, Liu Y, Enomoto-Iwamoto M, et al. Skeletal malformations caused by overexpression of Cbfa1 or its dominant negative form in chondrocytes. J Cell Biol. April 2 2001;153(1):87–100. Epub 2001/04/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen H, Ghori-Javed FY, Rashid H, Adhami MD, Serra R, Gutierrez SE, et al. Runx2 regulates endochondral ossification through control of chondrocyte proliferation and differentiation. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. December 2014;29(12):2653–65. Epub 2014/05/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takarada T, Hinoi E, Nakazato R, Ochi H, Xu C, Tsuchikane A, et al. An analysis of skeletal development in osteoblast-specific and chondrocyte-specific runt-related transcription factor-2 (Runx2) knockout mice. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. October 2013;28(10):2064–9. [DOI] [PubMed] [Google Scholar]
- 30.Kamekura S, Kawasaki Y, Hoshi K, Shimoaka T, Chikuda H, Maruyama Z, et al. Contribution of runt-related transcription factor 2 to the pathogenesis of osteoarthritis in mice after induction of knee joint instability. Arthritis Rheum. August 2006;54(8):2462–70. Epub 2006/07/27. [DOI] [PubMed] [Google Scholar]
- 31.Sampson ER, Hilton MJ, Tian Y, Chen D, Schwarz EM, Mooney RA, et al. Teriparatide as a chondroregenerative therapy for injury-induced osteoarthritis. Sci Transl Med. September 21 2011;3(101):101ra93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liao L, Zhang S, Gu J, Takarada T, Yoneda Y, Huang J, et al. Deletion of Runx2 in Articular Chondrocytes Decelerates the Progression of DMM-Induced Osteoarthritis in Adult Mice. Scientific reports. May 24 2017;7(1):2371 Epub 2017/05/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Henry SP, Jang CW, Deng JM, Zhang Z, Behringer RR, de Crombrugghe B. Generation of aggrecan-CreERT2 knockin mice for inducible Cre activity in adult cartilage. Genesis. December 2009;47(12):805–14. Epub 2009/10/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tu X, Joeng KS, Long F. Indian hedgehog requires additional effectors besides Runx2 to induce osteoblast differentiation. Dev Biol. February 1 2012;362(1):76–82. Epub 2011/12/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen M, Lichtler AC, Sheu TJ, Xie C, Zhang X, O’Keefe RJ, et al. Generation of a transgenic mouse model with chondrocyte-specific and tamoxifen-inducible expression of Cre recombinase. Genesis. January 2007;45(1):44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sampson ER, Beck CA, Ketz J, Canary KL, Hilton MJ, Awad H, et al. Establishment of an index with increased sensitivity for assessing murine arthritis. J Orthop Res. August 2011;29(8):1145–51. Epub 2011/03/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rigueur D, Lyons KM. Whole-mount skeletal staining. Methods Mol Biol. 2014;1130:113–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dao DY, Jonason JH, Zhang Y, Hsu W, Chen D, Hilton MJ, et al. Cartilage-specific beta-catenin signaling regulates chondrocyte maturation, generation of ossification centers, and perichondrial bone formation during skeletal development. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. August 2012;27(8):1680–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Joeng KS, Regan J, Long F. Radioactive in situ hybridization to detect gene expression in skeletal tissue sections. Methods Mol Biol. 2014;1130:217–32. [DOI] [PubMed] [Google Scholar]
- 40.Zhang Y, Sheu TJ, Hoak D, Shen J, Hilton MJ, Zuscik MJ, et al. CCN1 Regulates Chondrocyte Maturation and Cartilage Development. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. March 2016;31(3):549–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jonason JH, Hoak D, O’Keefe RJ. Primary murine growth plate and articular chondrocyte isolation and cell culture. Methods Mol Biol. 2015;1226:11–8. [DOI] [PubMed] [Google Scholar]
- 42.Gerwin N, Bendele AM, Glasson S, Carlson CS. The OARSI histopathology initiative - recommendations for histological assessments of osteoarthritis in the rat. Osteoarthritis Cartilage. October 2010;18 Suppl 3:S24–34. Epub 2010/10/01. [DOI] [PubMed] [Google Scholar]
- 43.Wang X, Manner PA, Horner A, Shum L, Tuan RS, Nuckolls GH. Regulation of MMP-13 expression by RUNX2 and FGF2 in osteoarthritic cartilage. Osteoarthritis Cartilage. December 2004;12(12):963–73. [DOI] [PubMed] [Google Scholar]
- 44.Brown TD, Johnston RC, Saltzman CL, Marsh JL, Buckwalter JA. Posttraumatic osteoarthritis: a first estimate of incidence, prevalence, and burden of disease. J Orthop Trauma. Nov-Dec 2006;20(10):739–44. [DOI] [PubMed] [Google Scholar]
- 45.Ratzlaff CR, Liang MH. New developments in osteoarthritis. Prevention of injury-related knee osteoarthritis: opportunities for the primary and secondary prevention of knee osteoarthritis. Arthritis Res Ther. 2010;12(4):215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Higashikawa A, Saito T, Ikeda T, Kamekura S, Kawamura N, Kan A, et al. Identification of the core element responsive to runt-related transcription factor 2 in the promoter of human type X collagen gene. Arthritis Rheum. January 2009;60(1):166–78. Epub 2009/01/01. [DOI] [PubMed] [Google Scholar]
- 47.Li F, Lu Y, Ding M, Napierala D, Abbassi S, Chen Y, et al. Runx2 contributes to murine Col10a1 gene regulation through direct interaction with its cis-enhancer. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. December 2011;26(12):2899–910. Epub 2011/09/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kuboki T, Kanyama M, Nakanishi T, Akiyama K, Nawachi K, Yatani H, et al. Cbfa1/Runx2 gene expression in articular chondrocytes of the mice temporomandibular and knee joints in vivo. Arch Oral Biol. July 2003;48(7):519–25. [DOI] [PubMed] [Google Scholar]
- 49.Eliseev RA, Dong YF, Sampson E, Zuscik MJ, Schwarz EM, O’Keefe RJ, et al. Runx2-mediated activation of the Bax gene increases osteosarcoma cell sensitivity to apoptosis. Oncogene. June 5 2008;27(25):3605–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gu L, Zhao J, Zhang S, Xu W, Ni R, Liu X. Runt-related transcription factor 2 (RUNX2) inhibits apoptosis of intestinal epithelial cells in Crohn’s disease. Pathol Res Pract. February 2018;214(2):245–52. [DOI] [PubMed] [Google Scholar]
- 51.Appleton CT, Usmani SE, Bernier SM, Aigner T, Beier F. Transforming growth factor alpha suppression of articular chondrocyte phenotype and Sox9 expression in a rat model of osteoarthritis. Arthritis Rheum. November 2007;56(11):3693–705. [DOI] [PubMed] [Google Scholar]
- 52.Dy P, Wang W, Bhattaram P, Wang Q, Wang L, Ballock RT, et al. Sox9 directs hypertrophic maturation and blocks osteoblast differentiation of growth plate chondrocytes. Dev Cell. March 13 2012;22(3):597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dar QA, Schott EM, Catheline SE, Maynard RD, Liu Z, Kamal F, et al. Daily oral consumption of hydrolyzed type 1 collagen is chondroprotective and anti-inflammatory in murine posttraumatic osteoarthritis. PLoS One. 2017;12(4):e0174705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu Z, Chen J, Mirando AJ, Wang C, Zuscik MJ, O’Keefe RJ, et al. A dual role for NOTCH signaling in joint cartilage maintenance and osteoarthritis. Sci Signal. July 21 2015;8(386):ra71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Valdes AM, Doherty SA, Muir KR, Wheeler M, Maciewicz RA, Zhang W, et al. The genetic contribution to severe post-traumatic osteoarthritis. Ann Rheum Dis. October 2013;72(10):1687–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.arc OC, arc OC, Zeggini E, Panoutsopoulou K, Southam L, Rayner NW, et al. Identification of new susceptibility loci for osteoarthritis (arcOGEN): a genome-wide association study. Lancet. September 1 2012;380(9844):815–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fernandez-Tajes J, Soto-Hermida A, Vazquez-Mosquera ME, Cortes-Pereira E, Mosquera A, Fernandez-Moreno M, et al. Genome-wide DNA methylation analysis of articular chondrocytes reveals a cluster of osteoarthritic patients. Ann Rheum Dis. April 2014;73(4):668–77. [DOI] [PubMed] [Google Scholar]
- 58.Castano Betancourt MC, Cailotto F, Kerkhof HJ, Cornelis FM, Doherty SA, Hart DJ, et al. Genome-wide association and functional studies identify the DOT1L gene to be involved in cartilage thickness and hip osteoarthritis. Proc Natl Acad Sci U S A. May 22 2012;109(21):8218–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Loughlin J, Dowling B, Chapman K, Marcelline L, Mustafa Z, Southam L, et al. Functional variants within the secreted frizzled-related protein 3 gene are associated with hip osteoarthritis in females. Proc Natl Acad Sci U S A. June 29 2004;101(26):9757–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miyamoto Y, Mabuchi A, Shi D, Kubo T, Takatori Y, Saito S, et al. A functional polymorphism in the 5’ UTR of GDF5 is associated with susceptibility to osteoarthritis. Nature genetics. April 2007;39(4):529–33. [DOI] [PubMed] [Google Scholar]
- 61.Valdes AM, Spector TD, Tamm A, Kisand K, Doherty SA, Dennison EM, et al. Genetic variation in the SMAD3 gene is associated with hip and knee osteoarthritis. Arthritis Rheum. August 2010;62(8):2347–52. [DOI] [PubMed] [Google Scholar]
- 62.Ma HL, Blanchet TJ, Peluso D, Hopkins B, Morris EA, Glasson SS. Osteoarthritis severity is sex dependent in a surgical mouse model. Osteoarthritis Cartilage. June 2007;15(6):695–700. [DOI] [PubMed] [Google Scholar]
- 63.Sniekers YH, Weinans H, Bierma-Zeinstra SM, van Leeuwen JP, van Osch GJ. Animal models for osteoarthritis: the effect of ovariectomy and estrogen treatment - a systematic approach. Osteoarthritis Cartilage. May 2008;16(5):533–41. [DOI] [PubMed] [Google Scholar]
- 64.Oliveria SA, Felson DT, Reed JI, Cirillo PA, Walker AM. Incidence of symptomatic hand, hip, and knee osteoarthritis among patients in a health maintenance organization. Arthritis Rheum. August 1995;38(8):1134–41. [DOI] [PubMed] [Google Scholar]
- 65.Srikanth VK, Fryer JL, Zhai G, Winzenberg TM, Hosmer D, Jones G. A meta-analysis of sex differences prevalence, incidence and severity of osteoarthritis. Osteoarthritis Cartilage. September 2005;13(9):769–81. [DOI] [PubMed] [Google Scholar]
- 66.Khalid O, Baniwal SK, Purcell DJ, Leclerc N, Gabet Y, Stallcup MR, et al. Modulation of Runx2 activity by estrogen receptor-alpha: implications for osteoporosis and breast cancer. Endocrinology. December 2008;149(12):5984–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sniekers YH, van Osch GJ, Ederveen AG, Inzunza J, Gustafsson JA, van Leeuwen JP, et al. Development of osteoarthritic features in estrogen receptor knockout mice. Osteoarthritis Cartilage. October 2009;17(10):1356–61. [DOI] [PubMed] [Google Scholar]
- 68.Ushiyama T, Ueyama H, Inoue K, Ohkubo I, Hukuda S. Expression of genes for estrogen receptors alpha and beta in human articular chondrocytes. Osteoarthritis Cartilage. November 1999;7(6):560–6. [DOI] [PubMed] [Google Scholar]
- 69.Bruderer M, Richards RG, Alini M, Stoddart MJ. Role and regulation of RUNX2 in osteogenesis. Eur Cell Mater. October 23 2014;28:269–86. [DOI] [PubMed] [Google Scholar]
- 70.Jonason JH, Xiao G, Zhang M, Xing L, Chen D. Post-translational Regulation of Runx2 in Bone and Cartilage. J Dent Res. August 2009;88(8):693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schroeder TM, Jensen ED, Westendorf JJ. Runx2: a master organizer of gene transcription in developing and maturing osteoblasts. Birth Defects Res C Embryo Today. September 2005;75(3):213–25. [DOI] [PubMed] [Google Scholar]
- 72.Ge C, Xiao G, Jiang D, Yang Q, Hatch NE, Roca H, et al. Identification and functional characterization of ERK/MAPK phosphorylation sites in the Runx2 transcription factor. J Biol Chem. November 20 2009;284(47):32533–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Greenblatt MB, Shim JH, Zou W, Sitara D, Schweitzer M, Hu D, et al. The p38 MAPK pathway is essential for skeletogenesis and bone homeostasis in mice. J Clin Invest. July 2010;120(7):2457–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li Y, Ge C, Long JP, Begun DL, Rodriguez JA, Goldstein SA, et al. Biomechanical stimulation of osteoblast gene expression requires phosphorylation of the RUNX2 transcription factor. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. June 2012;27(6):1263–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chong KW, Chanalaris A, Burleigh A, Jin H, Watt FE, Saklatvala J, et al. Fibroblast growth factor 2 drives changes in gene expression following injury to murine cartilage in vitro and in vivo. Arthritis Rheum. September 2013;65(9):2346–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ding L, Heying E, Nicholson N, Stroud NJ, Homandberg GA, Buckwalter JA, et al. Mechanical impact induces cartilage degradation via mitogen activated protein kinases. Osteoarthritis Cartilage. November 2010;18(11):1509–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pelletier JP, Fernandes JC, Brunet J, Moldovan F, Schrier D, Flory C, et al. In vivo selective inhibition of mitogen-activated protein kinase kinase 1/2 in rabbit experimental osteoarthritis is associated with a reduction in the development of structural changes. Arthritis Rheum. June 2003;48(6):1582–93. [DOI] [PubMed] [Google Scholar]
- 78.Pei Y, Harvey A, Yu XP, Chandrasekhar S, Thirunavukkarasu K. Differential regulation of cytokine-induced MMP-1 and MMP-13 expression by p38 kinase inhibitors in human chondrosarcoma cells: potential role of Runx2 in mediating p38 effects. Osteoarthritis Cartilage. August 2006;14(8):749–58. [DOI] [PubMed] [Google Scholar]
- 79.Rasheed Z, Akhtar N, Haqqi TM. Pomegranate extract inhibits the interleukin-1beta-induced activation of MKK-3, p38alpha-MAPK and transcription factor RUNX-2 in human osteoarthritis chondrocytes. Arthritis Res Ther. 2010;12(5):R195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kugimiya F, Kawaguchi H, Ohba S, Kawamura N, Hirata M, Chikuda H, et al. GSK-3beta controls osteogenesis through regulating Runx2 activity. PLoS One. September 5 2007;2(9):e837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Guidotti S, Minguzzi M, Platano D, Santi S, Trisolino G, Filardo G, et al. Glycogen Synthase Kinase-3beta Inhibition Links Mitochondrial Dysfunction, Extracellular Matrix Remodelling and Terminal Differentiation in Chondrocytes. Scientific reports. September 21 2017;7(1):12059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Litherland GJ, Hui W, Elias MS, Wilkinson DJ, Watson S, Huesa C, et al. Glycogen synthase kinase 3 inhibition stimulates human cartilage destruction and exacerbates murine osteoarthritis. Arthritis Rheumatol. August 2014;66(8):2175–87. [DOI] [PubMed] [Google Scholar]
- 83.Cheng A, Genever PG. SOX9 determines RUNX2 transactivity by directing intracellular degradation. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. December 2010;25(12):2680–9. [DOI] [PubMed] [Google Scholar]
- 84.Zhou G, Zheng Q, Engin F, Munivez E, Chen Y, Sebald E, et al. Dominance of SOX9 function over RUNX2 during skeletogenesis. Proc Natl Acad Sci U S A. December 12 2006;103(50):19004–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Murakami S, Lefebvre V, de Crombrugghe B. Potent inhibition of the master chondrogenic factor Sox9 gene by interleukin-1 and tumor necrosis factor-alpha. J Biol Chem. February 4 2000;275(5):3687–92. [DOI] [PubMed] [Google Scholar]
- 86.Sitcheran R, Cogswell PC, Baldwin AS Jr. NF-kappaB mediates inhibition of mesenchymal cell differentiation through a posttranscriptional gene silencing mechanism. Genes Dev. October 1 2003;17(19):2368–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kohn A, Dong Y, Mirando AJ, Jesse AM, Honjo T, Zuscik MJ, et al. Cartilage-specific RBPjkappa-dependent and -independent Notch signals regulate cartilage and bone development. Development. March 2012;139(6):1198–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yang G, Zhu L, Hou N, Lan Y, Wu XM, Zhou B, et al. Osteogenic fate of hypertrophic chondrocytes. Cell Res. October 2014;24(10):1266–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yang L, Tsang KY, Tang HC, Chan D, Cheah KS. Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proc Natl Acad Sci U S A. August 19 2014;111(33):12097–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhou X, von der Mark K, Henry S, Norton W, Adams H, de Crombrugghe B. Chondrocytes transdifferentiate into osteoblasts in endochondral bone during development, postnatal growth and fracture healing in mice. PLoS Genet. December 2014;10(12):e1004820. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.