

Abstract
Gliomas are the most common malignant brain tumors in the pediatric population. Even though great efforts have been made to understand their distinctive molecular characteristics, there has not been any improvements in the median survival in decades. In children, high-grade glial tumors have a median survival of 9–15 months. It has recently been demonstrated that pediatric high-grade gliomas (pHGG) are biologically and molecularly different from the adult counterparts, which could explain why conventional treatments universally fail. The development of an in vivo pHGG model harboring the specific genetic alterations encountered in pediatric gliomas is imperative in order to study the molecular basis that drives the progression and aggressiveness of these tumors. It would also enable harnessing these results for the development of novel therapeutic approaches. Our lab has implemented a method to induce brain tumors using transposon-mediated integration of plasmid DNA into cells of the subventricular zone of neonatal mouse brain. One of the main advantages of this method is that tumors are induced by altering the genome of the host cells, allowing us to recapitulate the salient features of the human disease. In this chapter we describe a method to isolate two cell populations from tumors generated in situ in mice, i.e. one population enriched in tumor cells and another population enriched in CD45+ cells. We also present methodologies as to how tumor infiltrating immune cells can be phenotypically characterized using flow cytometry.
Keywords: high grade glioma, immune cell isolation, magnetic sorting, immune cell characterization, flow cytometry
1. Introduction
Pediatric high grade gliomas
High-grade gliomas in children represent 15 to 20% of all brain tumors and the leading cause of cancer-related mortality in this age group (Braunstein, Raleigh, Bindra, Mueller, & Haas-Kogan, 2017; Faury et al., 2007). These tumors include anaplastic astrocytoma (World Health Organization (WHO) grade III), glioblastomas (WHO grade IV) and diffuse intrinsic pontine glioma (MacDonald, Aguilera, & Kramm, 2011). Seventy to 90% of patients die within 2 years of diagnosis (Paugh et al., 2010) and prognosis has remained largely unchanged despite novel treatment approaches (Braunstein et al., 2017; MacDonald et al., 2011). The spectrum of glioma in pediatric patients is distinctly different than that of adults, suggesting unique pathogenesis and disease progression (Paugh et al., 2010). In children, low grade juvenile pilocytic astrocytomas are the most common brain tumor type (Paugh et al., 2010). Low grade gliomas in the pediatric population rarely progress to a higher grade tumor (Braunstein et al., 2017; Broniscer et al., 2007), whereas the opposite is true in adults where glioblastoma is more common and low grade diffuse gliomas frequently undergo anaplastic progression to a high-grade tumor over time (Furnari et al., 2007; Paugh et al., 2010).
The standard of care for pediatric high grade glioma (pHGG) includes maximal safe surgical resection followed by focal radiation therapy in children usually older than 3 years of age due to cognitive and developmental sequelae from radiation in childhood (MacDonald et al., 2011). The most important prognostic factors are extent of resection and histologic grade (Gajjar et al., 2015). Adjuvant chemotherapeutics used for adult glioma, such as temozolomide, have performed with disappointing results in the pediatric population (Cohen, Heideman, et al., 2011; Cohen, Pollack, et al., 2011; Lashford et al., 2002). Moreover, results from adult clinical trials cannot be extrapolated to children due to the unique differences in pathogenesis and molecular biology between adult and pHGG (Jones et al., 2017). Important molecular signatures in pHGG include the novel oncogenic driver mutations in histones H3.1 (K27) and H3.3 (K27 and G34) as well as in the activin A receptor, type 1 (ACVR1) (Gajjar et al., 2015; Mackay et al., 2017).
Pediatric high grade gliomas harboring H3.3-G34R/V mutation
Recently, high-throughput genomic and epigenomic studies have permitted the classification of pediatric glioma subtypes based on molecular features which, in combination with other characteristics such as clinical data, revealed distinct biological subgroups (Sturm et al., 2014). The analysis of high-throughput data emerging from pHGG samples reveal a number of mutations in genes associated with the regulation of epigenetic processes (Schwartzentruber et al., 2012; Sturm et al., 2012; Wu et al., 2012). In eukaryotes, DNA is packaged around a core octamer of histone proteins which, besides conferring structural integrity to the chromatin, are subjected to posttranslational modifications (PTM) on their N-terminal tail as a mean to regulate the chromatin compaction and gene expression (Jenuwein & Allis, 2001; Strahl & Allis, 2000). Accordingly, mutations in the histone genes themselves or the genes encoding the proteins performing the PTMs have been identified as oncogenic drivers in different types of cancer (Funato & Tabar, 2018).
Two independent studies in 2012 identified mutations in the histone genes H3.1 (HIST1H3B) and H3.3 (H3F3A) in a large proportion of pHGG (Schwartzentruber et al., 2012; Wu et al., 2012). Mutations in the coding regions of the histone tails at lysine 27 to methionine (K27M) in H3.1 and H3.3 were found in diffuse midline glioma, in children with a median age of diagnosis of 9 years, while mutations in H3.3 at glycine 34 to arginine or valine (G34R/V) were found in about 15% of cerebral hemispheric HGG of children and young adults (Mackay et al., 2017).
It has been demonstrated that K27M mutations exhibit a dominant inhibitory effect on the polycomb repressive complex 2 (PRC2), by blocking the active site of enhancer of zeste homolog 2 (EZH2) (Nagaraja et al., 2017; Piunti et al., 2017). These findings were important for the conduct of drug testing experiments and specific clinical trials (Grasso et al., 2015; Jansen, Van Vuurden, Vandertop, & Kaspers, 2012). However, the exact mechanisms by which H3.3-G34R/V drive malignancy of pHGGs remain largely unknown. A more recent meta-analysis showed that most of the pHGG in the H3.3-G34R/V subgroup harbor inactivating mutations in TP53 and ATRX genes (Schwartzentruber et al., 2012; Sturm et al., 2012). Notably, ATRX performs a role as H3.3 depositor (Voon & Wong, 2016), suggesting a positive interaction between these two mutations in cancer (Haase et al., 2018).
H3.3-G34R/V has been shown to lead to a distinctive binding of the H3K36 trimethylation mark throughout the genome (Bjerke et al., 2013). For example, a differential binding of the activating H3K36me3 mark to upregulate genes associated with cortical precursors and the oncogene MYCN has been observed in H3.3-G34V glioma cells (Bjerke et al., 2013). Moreover, this mutation has been associated to a local downregulation of H3K36me3 at specific gene loci (Lewis et al., 2013). It has been suggested that a mechanism by which SETD2, the methyltransferase responsible for generating the H3K36me3 mark, is locally inhibited by G34R/V mutations, leads to reduced levels of the K36me3 mark in cis (Lewis et al., 2013). In conclusion, despite recent advances in the understanding of the H3.3-G34R/V pHGG biology, it is still imperative to develop accurate pre-clinical models in which to implement mechanistic studies that will guide the translation of research findings to the clinical arena.
2. Sleeping Beauty transposase-mediated mouse glioma model
Pediatric high grade glioma (pHGG) is a highly heterogeneous disease entity. Basic and translational research models that accurately recapitulate the genetic aberrations and tumor microenvironment in pHGG are necessary for effective pre-clinical studies. The Sleeping Beauty (SB) transposase method was developed to induce intrinsic brain tumors in mice with non-viral, transposon-mediated integration of DNA into cells of the subventricular zone of neonatal 1-day old mouse pups (Calinescu et al., 2015; Koschmann et al., 2016; Nunez et al., 2019; Wiesner et al., 2009) (see Note 1). Since the SB transposase is encoded in the same plasmid as the Luciferase gene (pT2/SB100x-Luc), and the latter gets inserted into the host genome, the plasmid uptake and disease progression can be monitored using bioluminescence (Calinescu et al., 2015; Koschmann et al., 2016; Nunez et al., 2019). Moreover, the SB plasmids encode not only for the specific genetic alterations, but also for different fluorescent proteins, so that the tumors can be recognized macroscopically using a stereo-zoom microscope equipped with a fluorescent light (Calinescu et al., 2015; Koschmann et al., 2016; Nunez et al., 2019). This model represents a valuable tool to induce tumors from cells original to the animal and to assess the role of candidate genes in the induction and progression of tumors. Also, this model can be used to test novel therapies, without the need of invasive surgical procedures or the risk of cell rejection by the host immune system.
Sleeping Beauty transposase pHGG model harboring H3.3-G34R mutation
-
Prepare the injection solution by mixing the plasmid DNA with the in vivo transfection reagent (Polyethylenimine, PEI) (in vivo-Jet PEI, Polyplus, New York, NY, USA) to create the PEI/DNA complexes for transfection. In our case, we used a 1:2 ratio for the injection of the pT2/SB100x-Luc plasmid and the following plasmids encoding:
H3.3-WT group (control group) H3.3-G34R (case group) NRAS(V12) oncogene NRAS(V12) oncogene shRNA against Tp53 shRNA against Tp53 shRNA against Atrx shRNA against Atrx H3.3-G34R To generate the PEI/DNA complexes for in vivo transfection, prepare:- DNA solution: add 2.85 μg of pT2/SB100x-Luc, 5.75 μg of each plasmid, 10 μl of 10% glucose solution and sterile water to a final volume of 20 μl.
- In vivo-Jet PEI solution: for the H3.3-WT group, add 2.8 μl of in vivo-Jet PEI, 7.2 μl of sterile water, and 10 μl of 10% glucose. For the H3.3-G34R group, add 3.6 μl of in vivo-Jet PEI, 6.4 μl of sterile water, and 10 μl of 10% glucose (see Note 2).
- Add the PEI solution to the DNA solution, mix by pipetting, vortex and incubate at room temperature for 20 min. The solution is now ready for injection.
Place a 10 μl syringe with a 30G hypodermic needle (12.5° bevel) into the micropump of the stereotaxic frame.
With a slurry of dry ice and alcohol, cool the neonatal stereotaxic stage to 2–8°C.
To induce anesthesia, place the 1-day old mouse pup wrapped in gauze on wet ice for 2 minutes (see Note 3).
Fill the syringe with the solution with the PEI/DNA complexes for the in vivo transfection.
Immobilize the pup in the stereotaxic frame, ensuring that the dorsal side of the skull is horizontal, parallel to the surface of the frame and wipe the head with 70% ethanol.
Lower the needle and adjust stereotaxic coordinates until the needle touches the midline intersection of the parietal and occipital bones (lambda).
Lift the needle and adjust stereotaxic coordinates to 0.8 mm lateral and 1.5 mm rostral to the lambda.
Lower the needle in dorsal-ventral direction, until it touches and slightly dimples the skin and lower the needle another 1.5 mm (see Note 4).
Using the automatic injector, inject 0.75 μl of the transfection solution at a rate of 0.5 μl/min.
Keep the pup on the frame for another minute and, in the meantime, anesthetize another pup, for the next injection.
Lift the needle gently and slowly, and place the pup under a heating lamp. Monitor breathing and activity ensuring first breathing.
After 3–5 min, when the pup is warm, looks rosy in color, is breathing regularly and active, return it to its mother (see Notes 5 and 6). Continue with the procedure until all pups have been injected.
3. Mouse perfusion and tumor dissection
Two to 6 weeks after SB injections animals will start developing macroscopic tumors. Monitor tumor progression in mice every week by bioluminescence imaging and look for signs of intracranial tumor burden (see Note 7).
When a mouse displays signs of tumor burden, anesthetize it with an intraperitoneal injection of 120 mg/kg of Ketamine and 0.5 mg/kg of Dexmedetomidine. Examine the animal by toe pinch and tail pinch. When no responses are observed in both tests, consider that the animal is in a deep anesthetic state.
Immobilize the mouse on a dissection board, open the abdominal cavity by an incision, dissect the diaphragm and open the thoracic cavity completely by cutting the ribs to visualize the heart.
Insert a blunt 20-gauge cannula into the left ventricle of the heart. Connect the cannula with plastic tubing passing through a peristaltic pump to a container with oxygenated and heparinized Tyrode’s solution (0.8% NaCl, 0.0264% CaCl2 2H2O, 0.005% NaH2PO4, 0.1% glucose, 0.1%NaHCO3, 0.02% KCl). Tyorde’s solution may be kept at 4 °C for up to one week.
Make a small incision into the right atrium of the heart to permit the drain of blood and Tyrode’s. Perfuse the mouse for ten minutes with cold Tyrode’s solution.
Decapitate the mouse and carefully dissect the brain from the skull. Remove musculature and skeletal matter from around the cranium. Remove the cranium by first flicking off the occipital, then temporal, then frontal cranium.
Remove the brain from skull and place it in a petri dish on ice.
Place the brain under a stereo-zoom microscope equipped with a fluorescent light illumination system. Dissect the tumor from the brain using scalpels.
Place the tumor pieces immediately into a 50 ml conical tube with 10 ml of cold Neurosphere (NS) media (DMEM/F12 supplemented with anti-anti, B27 supplement (1 X), N2 supplement (1 X) (all from Gibco, Thermo Scientific, Waltham, MA, USA), Normocin (100 μg/ml) (InvivoGen, San Diego, CA, USA), EGF and FGF (20 ng/ml) (PeproTech, Rocky Hill, NJ, USA)).
4. Tumor homogenization and magnetic labeling of CD45+ cells
For tumor homogenization, transfer the dissected tumor sections onto a 70 μm nylon strainer. Use a pestle to disaggregate the tumor and obtain a single-cell suspension. Add 10–15 ml of cold NS media through strainer to wash it and to resuspend the cells. Repeat the washing 3 times.
Centrifuge the cells at 300 x g for 4 minutes at room temperature.
For red blood cell lysis, remove the supernatant and add 1 ml of cold Red Blood Cell Lysis Buffer 1 X (BioLegend, San Diego, CA, USA) to the cells. Incubate 1 minute on ice. Immediately add 9 ml of Dulbecco’s Phosphate Buffered Saline 1 X (DPBS) (Gibco, Thermo Scientific, Waltham, MA, USA) to stop the red blood cell lysis reaction (see Note 8).
-
Centrifuge the cells at 300 × g for 5 minutes at 4° C.
For these next steps work quickly, keep the cells cold at all times, and use pre-cooled solutions. This will prevent capping of antibodies on the cell surface and non-specific cell labeling.
Completely remove media and resuspend the cells in 90 μl of cold BSA-EDTA buffer (0.5% Bovine serum albumin (BSA), 2mM Ethylenediaminetetraacetic acid (EDTA) in phosphate buffered saline pH 7.2) (Miltenyi Biotec, Bergisch Gladbach, Germany). Keep buffer cold (4–8 °C).
Add 10 μl of CD45 MicroBeads (Miltenyi Biotec, Bergisch Gladbach, Germany) per 107 total cells (see Notes 9 and 10).
Mix the cell suspension well and incubate for 15 minutes at 4° C (not on ice).
Wash cells by adding 1 ml of cold BSA-EDTA buffer and centrifuge at 300 × g for 5 minutes at 4°C.
Pipet off supernatant and resuspend cell pellet in 500 μl of cold BSA-EDTA buffer. Proceed to magnetic column separation.
5. Magnetic separation of tumor and CD45+ enriched cell fractions
Place MACS column in the magnetic field of a suitable MACS Separator (Miltenyi Biotec, Bergisch Gladbach, Germany) (see Note 11).
Precondition the column by rinsing with appropriate amount of BSA-EDTA buffer. Let all the solution go through the column (see Note 12).
Put a 30 μM nylon mesh on the top of the column and apply cell suspension onto it (see Note 13).
Collect unlabeled cells which flow through the column (500 μl for large columns). Wash the column once with BSA-EDTA buffer (3 ml for large columns) and collect the cells that eluate in the same tube. This is the first tumor cell-enriched eluate (CD45-cells).
Apply the first tumor cell-enriched eluate into a second preconditioned column with the filter on the top and collect the second tumor cell-enriched eluate. Wash once with BSA-EDTA buffer and collect the eluate in the same tube. If using large columns, the total volume should be approximately 6,5 ml. This is the tumor cell-enriched fraction.
Save 500 μl of the tumor cell-enriched fraction for flow cytometry analysis (keep them on ice or at 4 °C), centrifuge the rest for 5 minutes at 300 × g at 4 °C and discard the supernatant. The pellet can now be used for downstream applications; such as flow cytometry or RNA purification.
To collect the CD45+ cells that are magnetically adhered to the first column, first wash this column twice with BSA-EDTA buffer (see Note 14).
Remove the column from the magnetic field and place it on a suitable collection tube, such as a 15 ml tube.
Add BSA-EDTA buffer on top of the column and immediately flush out this fraction with the magnetically labeled CD45+ cells by firmly applying the plunger supplied with the column. In the case of large columns, 5 ml are used for the elution step.
Save 500 μl of the CD45+ cell fraction for flow cytometry analysis (keep them on ice or at 4 °C), centrifuge the rest for 5 minutes at 300 × g at 4 °C and discard the supernatant. The pellet can now be used for downstream applications; such as flow cytometry or RNA purification.
6. Flow cytometry analysis of cell fractions: CD45 and Ter119 assessment
Pellet all the samples that are going to be tested and resuspend them in 200 μl of 2% FBS in Hanks’ Balanced Salt Solution (HBSS, no calcium, no magnesium, no phenol red) (Gibco, Thermo Scientific, Waltham, MA, USA) (flow buffer) (see Note 15).
Add 1 μl of Fc blocking (see Note 16).
Incubate on ice for 15 minutes.
Spin cells down at 300 × g, for 5 minutes at 4 °C.
- Resuspend the pellets in 600 μl of flow buffer and divide them in 3 wells of a 96 V-bottom multiwell plate (200 μl per well) (see Note 17):
- Unstained control
- CD45 only
- Ter119 only
Incubate on ice for 30 minutes, protected from light.
Centrifuge cells down at 300 × g, for 5 minutes at 4 °C.
Wash twice with 100 μl of flow buffer, centrifuging in between at 300 × g, for 5 minutes at 4 °C.
Resuspend all wells in 500 μl of flow buffer and pass the samples to a flow cytometry tube.
Before running the samples, add 2.5 μl of Propidium Iodide (PI) Staining Solution (Invitrogen, Carlsbad, CA, USA) for cell viability assessment (see Note 19). Record at least 20,000 events of the singlet population.
7. Flow cytometry analysis of the tumor microenvironment
Wash the cells that were purified from the brain tissue by magnetic sorting with 600 μl of flow buffer. Check cell viability by performing a cell count using trypan blue and a hemocytometer.
After the cell count, resuspend the cells at a concentration of 2.0 × 106 cells in 200 μl of flow buffer.
Plate the cells at a density of 1.5 × 106 cells in 150 μl volume in a 96 well V-bottom plate for immunostaining.
Combine the remaining 50 μl of cells from all samples and plate 150 μl of the resuspension in a 96 wells V-bottom multiwell plate for staining with the isotype control.
Pellet the cells by centrifuging the plate at 300 × g for 5 minutes at 4 °C.
Discard the supernatant and add 100 μl of Fc block diluted in flow buffer at a 1:200 ratio to each sample. Then, incubate the plate on ice for 15 minutes (see Note 16).
Centrifuge the plate at 300 × g for 5 minutes at 4 °C. Discard the supernatant and wash the cells two times with flow buffer before staining with the cell surface markers.
To identify various immune cells within the tumor microenvironment, prepare fluorophore conjugated primary antibody mixes with the combination of key markers listed in Table 1. Antibodies and isotype controls are diluted in flow buffer at a 1:200 ratio. Catalog numbers and company names of the antibodies used can be found in Table 2.
Add 100 μl of fluorophore conjugated primary antibody mix to the appropriate well and pipette up and down to mix.
Incubate the plate for 30 minutes on ice protected from light.
Centrifuge the plate at 300 × g for 5 minutes at 4 °C. Discard the supernatant and wash the cells three times with flow buffer.
Fix the cells by adding 100 μl of Cytofix/Cytoperm (BD Biosciences, San Jose, CA, USA) solution to each well and incubate the plate for 20 minutes on ice in the dark (see Note 20).
Wash the cells two times with 1 X Perm/Wash (BD Biosciences, San Jose, CA, USA) solution before staining with the intracellular markers (see Note 21).
To evaluate the expression of intracellular markers in the immune cells, prepare fluorophore conjugated primary antibody mix with the combination of key markers listed in Table 1. Antibodies and isotype controls are diluted in 1 X Perm/Wash solution at a 1:200 ratio.
Add 100 μl of fluorophore conjugated primary antibody intracellular mix to the appropriate well.
Incubate the plate for 30 minutes on ice protected from the light.
Centrifuge the plate at 300 × g for 5 minutes at 4 °C. Discard the supernatant and wash the cells three times with 1 X Perm/Wash solution.
Resuspend the cell pellets in 200 μl of 1 X Perm/Wash solution and transfer the cell suspension to flow tubes.
For flow cytometry acquisition, obtain at least 30,000 events of the singlet population.
Table 1.
Mouse surface and intracellular markers for the most common immune cell types.
| Immune Cell | Subtype | Mouse Surface Markers | Mouse Intracellular Markers |
|---|---|---|---|
| Macrophages | CD45+, F4/80+ | - | |
| M1 | CD45+, F4/80+, CD206− | IFNγ+ | |
| M2 | CD45+, F4/80+, CD206+ | Arg1+ | |
| Dendritic Cells | CD45+, CD11c+, MHC II+ | - | |
| Conventional | CD45+, CD11c+, MHC II+, B220− | - | |
| Plasmacytoid | CD45+, CD11c+, MHC II+, B220+ | - | |
| T cells | CD45+, CD3+ | - | |
| Cytotoxic T cells | CD45+, CD3+, CD8+ | IFNγ+, Granzyme B+ | |
| Helper T cells | CD45+, CD3+, CD4+ | - | |
| NK cells | N/A | CD45+, CD3−, NK1.1+ | - |
| MDSC | CD45+, Gr1+, CD11b+ | - | |
| Granulocytic | CD45+, Gr1+, CD11b+, Ly6G+, LygClo | - | |
| Monocytic | CD45+, Gr1+, CD11b+, Ly6G−, LygChi | - |
Table 2.
Catalog numbers and company names of the antibodies used.
| Protein | Catalog number | Company |
|---|---|---|
| CD45 (Pacific blue conjugated) | 103126 | Biolegend |
| F4/80 | 123128 | Biolegend |
| CD206 | 141734 | Biolegend |
| CD11c | 117319 | Biolegend |
| MHCII | 107614 | Biolegend |
| B202 | 103223 | Biolegend |
| CD3 | 100328 | Biolegend |
| CD8 | 100722 | Biolegend |
| CD4 | 100421 | Biolegend |
| NK1.1 | 108708 | Biolegend |
| Gr1 | 108412 | Biolegend |
| CD11b | 101225 | Biolegend |
| Ly6G | 127606 | Biolegend |
| Ly6C | 128018 | Biolegend |
| IFNγ | 113603 | Biolegend |
| Arg1 | IC5868P | R&D Systems |
| Granzime B | 372205 | Biolegend |
| Ter119 | 116232 | Biolegend |
| CD45 (APC conjugated) | 103112 | Biolegend |
8. Concluding remarks
This method represents a valuable tool to obtain two distinct cell populations from genetically engineered high-grade glioma models: one population enriched in tumor cells and the other enriched in immune cells. High grade gliomas are generated intrinsically in 1-day old mouse pups through the injection of a combination of plasmids encoding for the desired genetic lesions in combination with the SB transposase (Figure 1, A and B). Two advantages of this method are that the plasmid uptake and tumor growth can be monitored using bioluminescence and the tumors can easily be identified and excised using a stereo-zoom microscope equipped with a fluorescent light (Figure 1, C). When a tumor bearing mouse displays signs of tumor burden (ataxia, impaired mobility, hunched posture, seizures, and scruffy fur), the mouse is perfused with Tyrode’s solution. After extracting the brain from of the skull, the tumor is dissected to be processed following the described protocol, which is depicted in Figure 2.
Figure 1. Generation of a mouse glioma model using the Sleeping Beauty (SB) Transposase System.
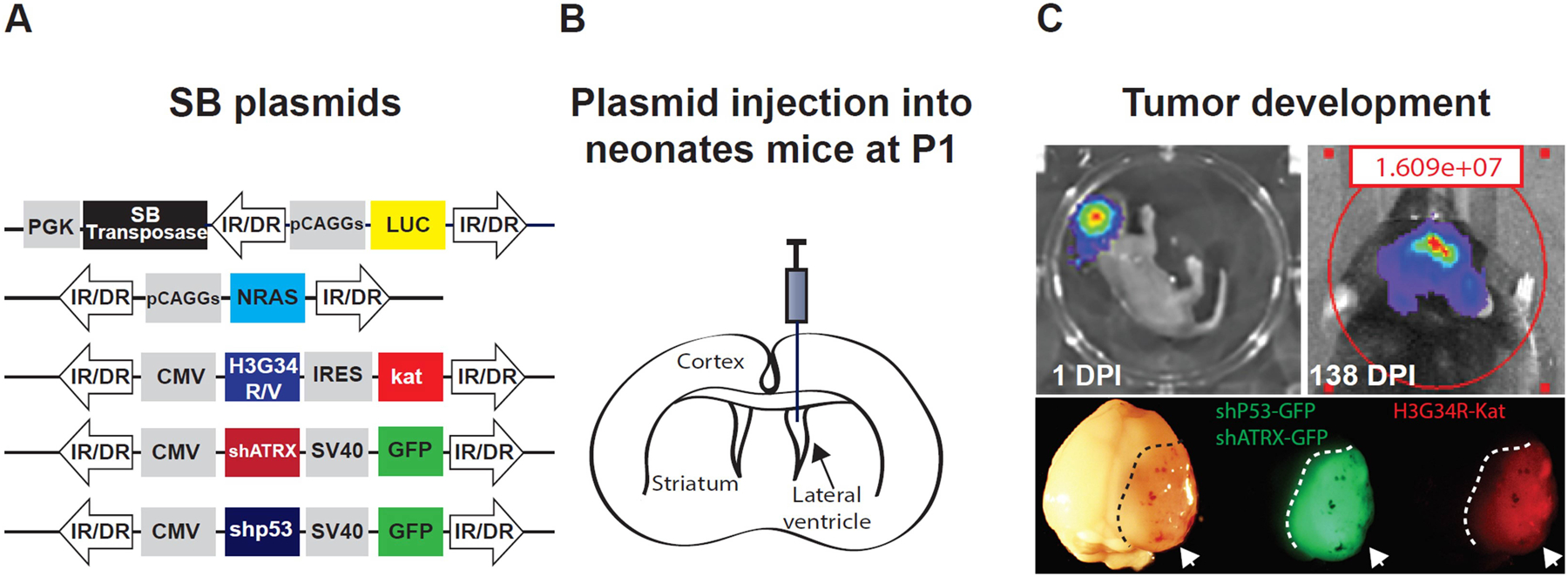
A) Diagram representing the plasmid constructs encoding for the SB transposase with the luciferase and the genetic lesions to be inserted into the host cell genome via the SB transposase (SB plasmids). B) Diagram indicating the brain ventricles and the site for plasmid injection (lateral ventricle), which contains brain stem cells from which the tumor is originated. C) (Upper panel) Plasmid uptake efficiency one day after injection and tumor development monitoring by bioluminescence. (Lower panel). Tumor tissue (white arrows and dashed lines) is identified macroscopically by the expression of fluorophores associated with the respective genetic lesions. H3.3-G34R tumors express both green fluorescent protein (GFP) and Katushka protein (Kat) (a red fluorescent protein), since the shP53 and shATRX plasmids are coupled with GFP coding sequence, whereas the H3.3-G34R coding sequence is coupled with Kat sequence.
Figure 2. Schematic summarizing the technique to obtain two cell fractions from a genetically engineered mouse glioma model.
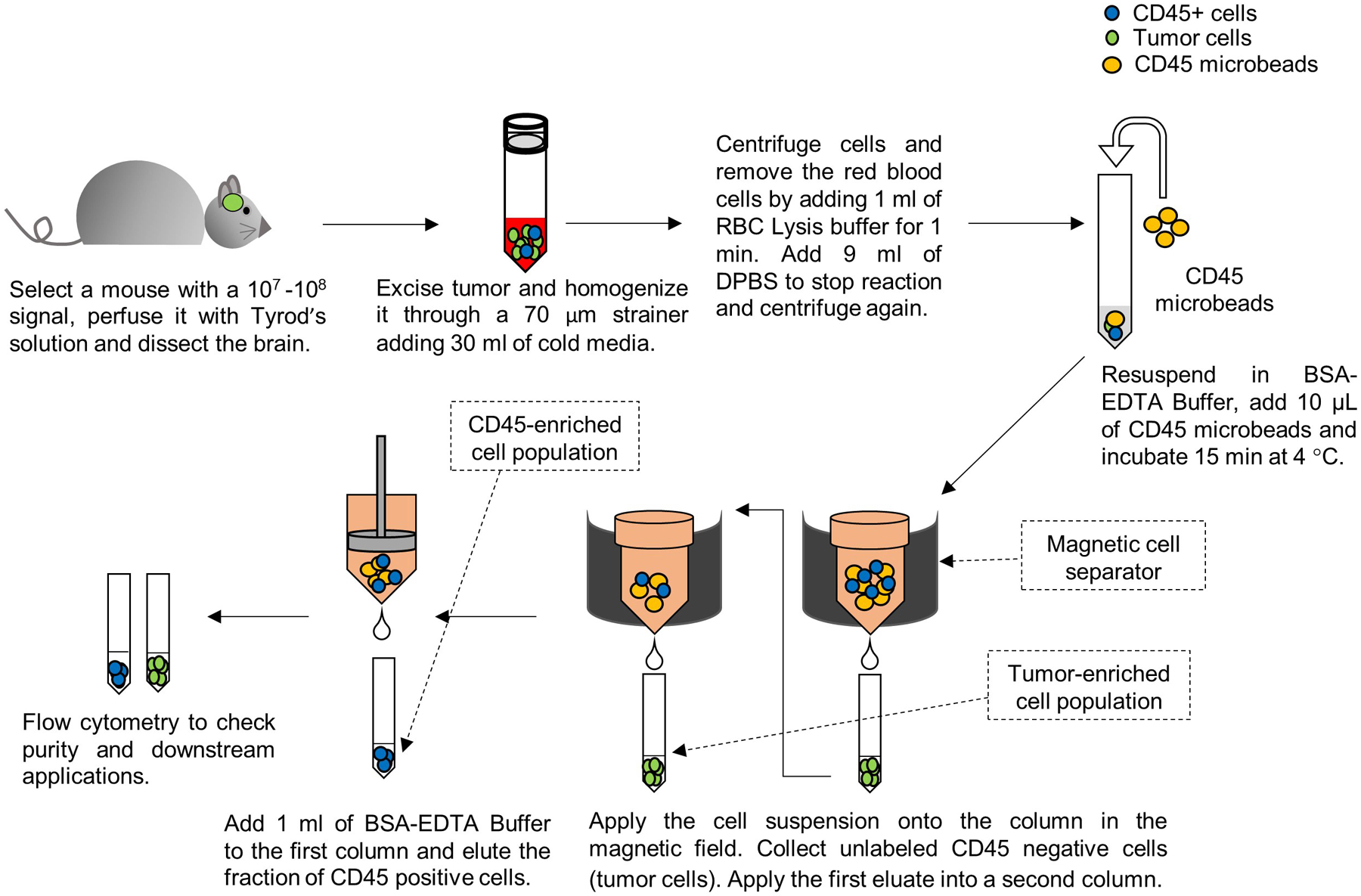
One cell fraction will be enriched in tumor cells and the other in CD45+ cells (immune cells).
This protocol allowed us to isolate two cell populations, enriched in tumor or immune cells. By Flow Cytometry, we check the purity of these isolated populations, by assessing the quantity of CD45+ and Ter119+ cells within the live cell population (Figure 3, A and B). In both fractions, the proportion of viable cells is between 80–90 %. In the tumor cell enriched fraction, the amount of CD45+ cells is 3 % on average, whereas in the tumors without any separation the amount of CD45+ cell is between 10–15 % in the case of the H3.3-WT or H3.3-G34R glioma mouse models (Figure 4). On the contrary, we observed that the amount of CD45+ cells in the immune cell fraction is 8-fold higher in comparison to the amount of CD45+ cells in the tumor homogenate without any enrichment (Figure 4). The amount of Ter119+/CD45− cells (erythroid cells) is 0.4 % on average in both cell fractions after the procedure (Figure 3, A and B).
Figure 3. Flow cytometry plots and histograms showing live, CD45+ and Ter119+ cells.
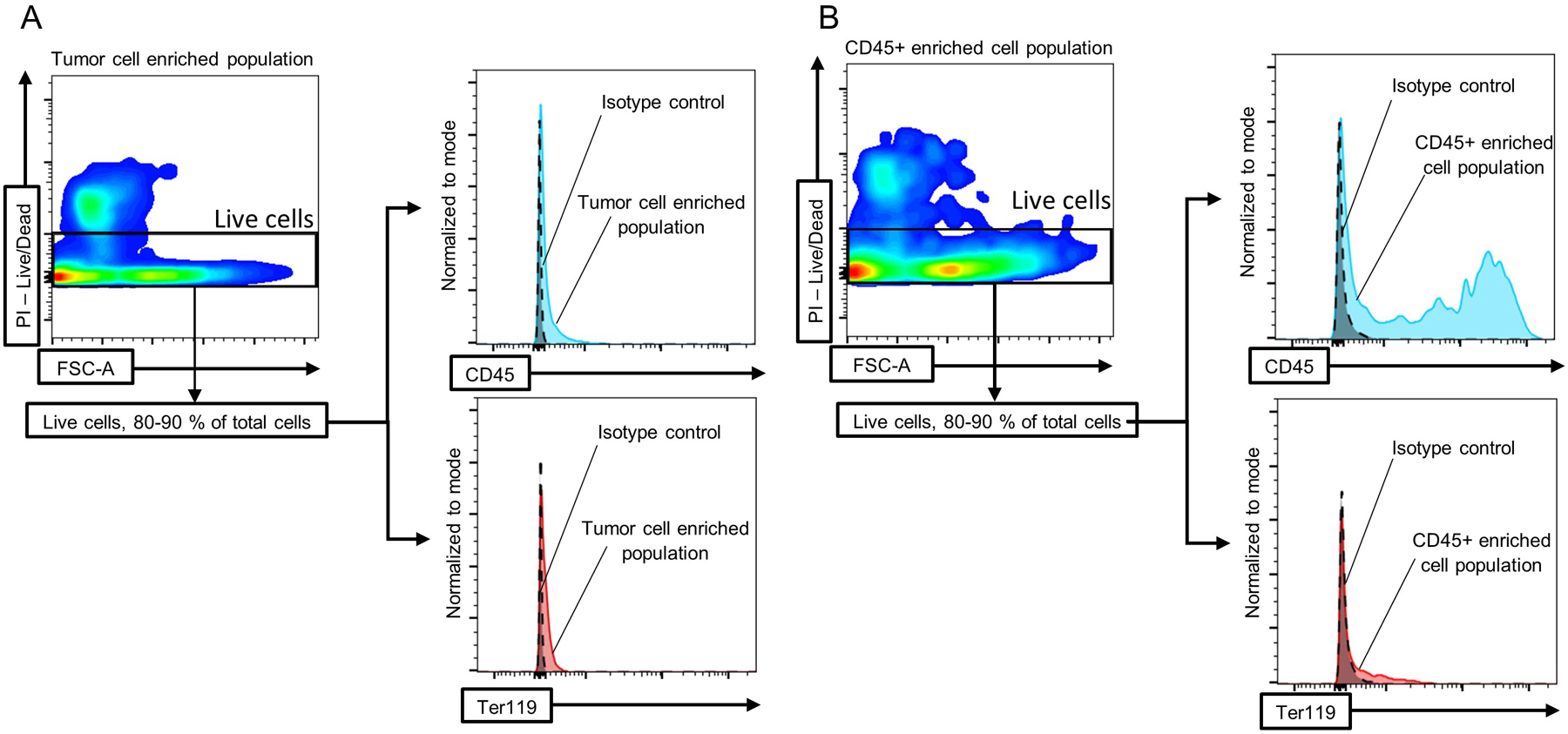
Cell populations enriched in A) tumor cells or B) immune cells were isolated as described in the protocol and flow cytometry was performed to analyze the quantity of CD45+ (immune cells) and Ter119+ (erythroid cells) cells in both fractions.
Figure 4. Surface expression of CD45 marker in the entire tumor homogenate, tumor cell enriched population and CD45+ cell enriched population.
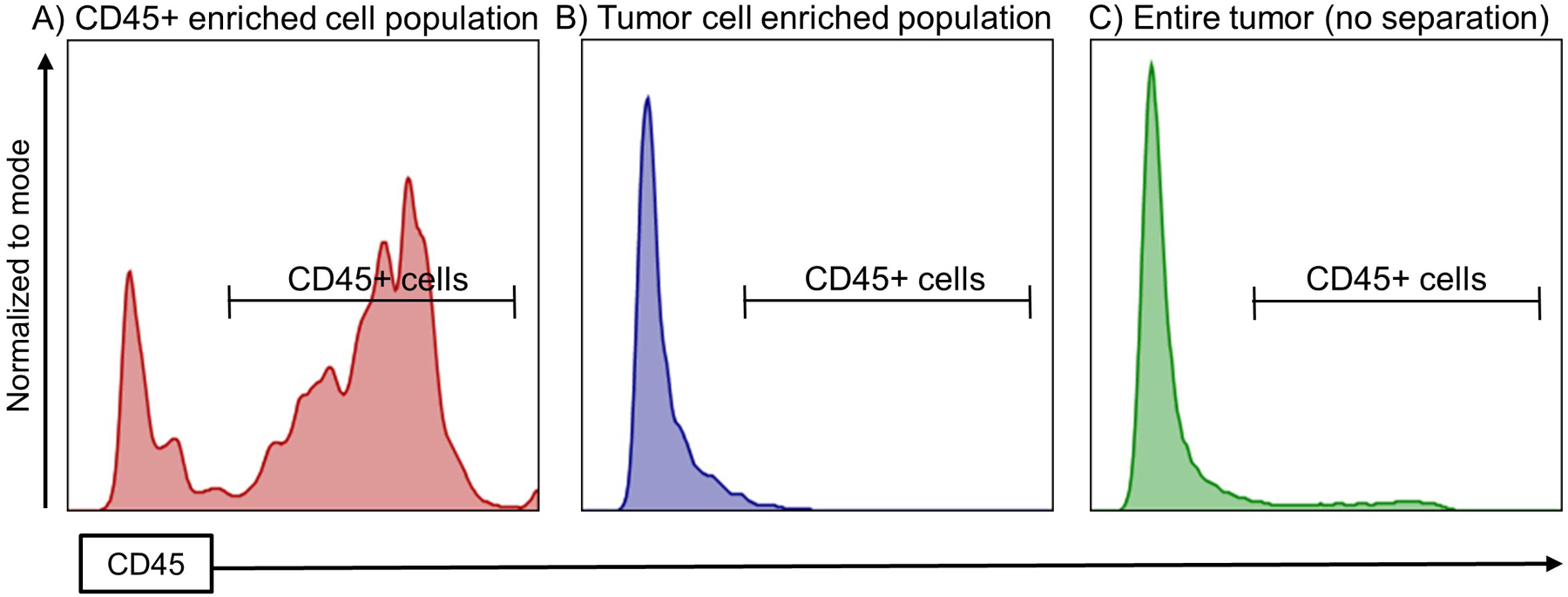
The expression of CD45 was studied to analyze the extent of CD45 depletion or enrichment in the tumor cell enriched population or in the CD45+ cell enriched population, respectively.
In conclusion, this method allowed us to obtain a high yield separation of tumor cells from immune cells from a high-grade glioma mouse model, with a substantial amount of viable cells. These enriched cell fractions can be employed in downstream applications such as RNA purification, DNA purification, flow cytometry analysis, cell culture, and functional studies among others. This procedure can be also applied for isolating immune cells from tumor cells in any other tumor model.
Notes
For a more detailed protocol, including the maps of the plasmids used, the cloning strategy and the intra-ventricular injections, see Calinescu, A. A., Nunez, F. J., Koschmann, C., Kolb, B. L., Lowenstein, P. R., & Castro, M. G. (2015). Transposon mediated integration of plasmid DNA into the subventricular zone of neonatal mice to generate novel models of glioblastoma. J Vis Exp(96). doi:10.3791/52443.
The optimal ratio of nitrogen residues in the polyethyleneimine (PEI) to the phosphate residues in the DNA (N/P ratio) is 7, which ensures the formation of positively charged particle complexes that will bind anionic moieties on cell surfaces and will be endocytosed. The in vivo jet-PEI solution has a concentration of 150 mM expressed as nitrogen residues. Given that one μg DNA has 3 nmol of anionic phosphate, the amount of transfection reagent necessary can be calculated with the formula: μl of in vivo jet-PEI = [(μg DNA × 3) × N/P ratio]/150.
Pups receive continuous anesthesia when placed on the chilled stereotaxic frame during the plasmid injection. Maintaining the temperature of the frame above freezing, between 2–8 °C, will prevent thermal burn injuries for the pups.
The needle will pierce the skin and skull, and pass through the cortex to enter the lateral ventricle.
Keep all the pups together under the heating lamp until all the injections are done. If injections take longer than 30 min, return the pups to the cage after the first 30 min.
Monitor that the mother is responsive and nurturing to her pups. If necessary, a surrogate mother can be placed in the cage. Also, ensure that the interval between placing the pup on ice and placing it under the warming lamp is less than 10 min. Pups survival is better when the procedure is carried out quickly. Usually, the time it takes to anesthetize and inject a pup is 4 min.
Signs of end point intracranial tumor burden consist of weight loss, ruffled fur, hunched posture, impaired mobility or other abnormalities in movement. At this time point the bioluminescence would be usually 107-108 photons/s/cm2/sr.
Longer incubations in the Red Blood Cell Lysis Buffer could cause excessive lysis of the cells.
Microbeads are conjugated to monoclonal anti-non-human primate CD45 antibodies (Isotype: mouse IgG1, Clone: MB4–6D6), (Miltenyi Biotec Inc, Auburn, CA, USA).
The volumes of reagents listed in the protocol are for up to 1 × 107 cells. When working with higher cell numbers, scale up all reagent volumes accordingly.
The size of the column used depends on the size of the tumor and the number of cells collected. The SB method usually generates large tumors and more than 1 × 107 cells can be collected, so large columns were preferentially used.
The volume of BSA-EDTA buffer used for preconditioning and washing the column depends on the size of the column used. We used large columns and followed the manufacturer’s instructions.
It is important to have a suspension of single cells for the optimal performance of the magnetic separation and for preventing the clogging of the column.
CD45+ cells should be collected from the first columns only, after washing.
Using HBSS buffer yielded better cell viability compared to the use of DPBS buffer.
Cells contain numerous Fc receptors that could contribute to non-specific binding and background fluorescence, therefore it is important to block the Fc receptor on the cells.
Due to the low amount of CD31+ cells (endothelial cells, less than 3%) in the tumor microenvironment, we did not include this marker in the regular assessment of the cell fractions’ purity (data not shown).
We used APC conjugated anti-mouse CD45 and Pacific Blue conjugated anti-mouse Ter119.
PI is a dye that binds to double stranded DNA by intercalating between base pairs. Viable cell membranes are impermeant, so the dye is excluded from live cells.
Fixing the cells minimizes deterioration of the sample.
Staining of intra-cellular markers is carried out in 1 X Perm/Wash solution to maintain the cells in a permeabilized state.
Acknowledgements
This work was supported by NIH/NINDS Grants, R37-NS094804, R01-NS105556 and 1R21NS107894 to M.G.C.; NIH/NINDS Grants R01-NS076991, and R01-NS096756 to P.R.L.; NIH/NIBIB: R01-EB022563; NIH/NCI U01CA224160; the Department of Neurosurgery, the Rogel Cancer Center, Program in Cancer Hematopoiesis and Immunology (CHI), the ChadTough Foundation and Leah’s Happy Hearts to M.G.C. and P.R.L. and RNA Biomedicine Grant F046166 to M.G.C.
M.B.G.F, A.C., P.R.L. and M.G.C. conducted and designed the experiments, analyzed the data, and wrote the manuscript. P.K. performed experiments and contributed to the final manuscript. S.H., F.J.N and D.A. contributed to the final manuscript and figure design. P.R.L. and M.G.C. directed the research and generated the funding. All the authors read and edited the manuscript.
References
- Bjerke L, Mackay A, Nandhabalan M, Burford A, Jury A, Popov S, … Jones C (2013). Histone H3.3. mutations drive pediatric glioblastoma through upregulation of MYCN. Cancer Discov, 3(5), 512–519. doi: 10.1158/2159-8290.cd-12-0426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunstein S, Raleigh D, Bindra R, Mueller S, & Haas-Kogan D (2017). Pediatric high-grade glioma: current molecular landscape and therapeutic approaches. J Neurooncol, 134(3), 541–549. doi: 10.1007/s11060-017-2393-0 [DOI] [PubMed] [Google Scholar]
- Broniscer A, Baker SJ, West AN, Fraser MM, Proko E, Kocak M, … Fuller CE (2007). Clinical and molecular characteristics of malignant transformation of low-grade glioma in children. J Clin Oncol, 25(6), 682–689. doi: 10.1200/jco.2006.06.8213 [DOI] [PubMed] [Google Scholar]
- Calinescu A-A, Núñez FJ, Koschmann C, Kolb BL, Lowenstein PR, & Castro MG (2015). Transposon mediated integration of plasmid DNA into the subventricular zone of neonatal mice to generate novel models of glioblastoma. Journal of visualized experiments: JoVE(96). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen KJ, Heideman RL, Zhou T, Holmes EJ, Lavey RS, Bouffet E, & Pollack IF (2011). Temozolomide in the treatment of children with newly diagnosed diffuse intrinsic pontine gliomas: a report from the Children’s Oncology Group. Neuro Oncol, 13(4), 410–416. doi: 10.1093/neuonc/noq205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen KJ, Pollack IF, Zhou T, Buxton A, Holmes EJ, Burger PC, … Heideman RL (2011). Temozolomide in the treatment of high-grade gliomas in children: a report from the Children’s Oncology Group. Neuro Oncol, 13(3), 317–323. doi: 10.1093/neuonc/noq191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faury D, Nantel A, Dunn SE, Guiot MC, Haque T, Hauser P, … Jabado N (2007). Molecular profiling identifies prognostic subgroups of pediatric glioblastoma and shows increased YB-1 expression in tumors. J Clin Oncol, 25(10), 1196–1208. doi: 10.1200/jco.2006.07.8626 [DOI] [PubMed] [Google Scholar]
- Funato K, & Tabar V (2018). Histone Mutations in Cancer. Annual Review of Cancer Biology, 2(1), 337–351. doi: 10.1146/annurev-cancerbio-030617-050143 [DOI] [Google Scholar]
- Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, … Cavenee WK (2007). Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev, 21(21), 2683–2710. doi: 10.1101/gad.1596707 [DOI] [PubMed] [Google Scholar]
- Gajjar A, Bowers DC, Karajannis MA, Leary S, Witt H, & Gottardo NG (2015). Pediatric Brain Tumors: Innovative Genomic Information Is Transforming the Diagnostic and Clinical Landscape. J Clin Oncol, 33(27), 2986–2998. doi: 10.1200/jco.2014.59.9217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasso CS, Tang Y, Truffaux N, Berlow NE, Liu L, Debily MA, … Sun W (2015). Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. 21(6), 555–559. doi: 10.1038/nm.3855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase S, Garcia-Fabiani MB, Carney S, Altshuler D, Nunez FJ, Mendez FM, … Castro MG (2018). Mutant ATRX: uncovering a new therapeutic target for glioma. Expert Opin Ther Targets, 22(7), 599–613. doi: 10.1080/14728222.2018.1487953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen M, Van Vuurden D, Vandertop W, & Kaspers G (2012). Diffuse intrinsic pontine gliomas: a systematic update on clinical trials and biology. Cancer treatment reviews, 38(1), 27–35. [DOI] [PubMed] [Google Scholar]
- Jenuwein T, & Allis CD (2001). Translating the histone code. Science, 293(5532), 1074–1080. [DOI] [PubMed] [Google Scholar]
- Jones C, Karajannis MA, Jones DTW, Kieran MW, Monje M, Baker SJ, … Weiss WA (2017). Pediatric high-grade glioma: biologically and clinically in need of new thinking. Neuro Oncol, 19(2), 153–161. doi: 10.1093/neuonc/now101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koschmann C, Calinescu A-A, Nunez FJ, Mackay A, Fazal-Salom J, Thomas D, … Mulpuri L (2016). ATRX loss promotes tumor growth and impairs nonhomologous end joining DNA repair in glioma. Sci Transl Med, 8(328), 328ra328–328ra328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lashford LS, Thiesse P, Jouvet A, Jaspan T, Couanet D, Griffiths PD, … Frappaz D (2002). Temozolomide in malignant gliomas of childhood: a United Kingdom Children’s Cancer Study Group and French Society for Pediatric Oncology Intergroup Study. J Clin Oncol, 20(24), 4684–4691. doi: 10.1200/jco.2002.08.141 [DOI] [PubMed] [Google Scholar]
- Lewis PW, Muller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, … Allis CD (2013). Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science, 340(6134), 857–861. doi: 10.1126/science.1232245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald TJ, Aguilera D, & Kramm CM (2011). Treatment of high-grade glioma in children and adolescents. Neuro Oncol, 13(10), 1049–1058. doi: 10.1093/neuonc/nor092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay A, Burford A, Carvalho D, Izquierdo E, Fazal-Salom J, Taylor KR, … Jones C (2017). Integrated Molecular Meta-Analysis of 1,000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell, 32(4), 520–537.e525. doi: 10.1016/j.ccell.2017.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraja S, Vitanza NA, Woo PJ, Taylor KR, Liu F, Zhang L, … Monje M (2017). Transcriptional Dependencies in Diffuse Intrinsic Pontine Glioma. Cancer Cell, 31(5), 635–652.e636. doi: 10.1016/j.ccell.2017.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez FJ, Mendez FM, Kadiyala P, Alghamri MS, Savelieff MG, Garcia-Fabiani MB, … Castro MG (2019). IDH1-R132H acts as a tumor suppressor in glioma via epigenetic upregulation of the DNA damage response. Sci Transl Med, 11(479). doi: 10.1126/scitranslmed.aaq1427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paugh BS, Qu C, Jones C, Liu Z, Adamowicz-Brice M, Zhang J, … Baker SJ (2010). Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J Clin Oncol, 28(18), 3061–3068. doi: 10.1200/jco.2009.26.7252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piunti A, Hashizume R, Morgan MA, Bartom ET, Horbinski CM, Marshall SA, … Misharin AV (2017). Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. 23(4), 493–500. doi: 10.1038/nm.4296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, … Jabado N (2012). Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature, 482(7384), 226–231. doi: 10.1038/nature10833 [DOI] [PubMed] [Google Scholar]
- Strahl BD, & Allis CD (2000). The language of covalent histone modifications. Nature, 403(6765), 41. [DOI] [PubMed] [Google Scholar]
- Sturm D, Bender S, Jones DT, Lichter P, Grill J, Becher O, … Pfister SM (2014). Paediatric and adult glioblastoma: multiform (epi)genomic culprits emerge. Nat Rev Cancer, 14(2), 92–107. doi: 10.1038/nrc3655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, … Pfister SM (2012). Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell, 22(4), 425–437. doi: 10.1016/j.ccr.2012.08.024 [DOI] [PubMed] [Google Scholar]
- Voon HP, & Wong LH (2016). New players in heterochromatin silencing: histone variant H3. 3 and the ATRX/DAXX chaperone. Nucleic acids research, 44(4), 1496–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesner SM, Decker SA, Larson JD, Ericson K, Forster C, Gallardo JL, … Low WC (2009). De novo induction of genetically engineered brain tumors in mice using plasmid DNA. Cancer research, 69(2), 431–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, … Baker SJ (2012). Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet, 44(3), 251–253. doi: 10.1038/ng.1102 [DOI] [PMC free article] [PubMed] [Google Scholar]