

Abstract
Combinatorial chemistry drives the biological generation of protein structural diversity in antibodies and T-cell receptors. When applied to nucleic acids, vast engineered random libraries of DNA and RNA strands allow selection of affinity reagents (“aptamers”) against molecular targets. Selection involves cycles rewarding target binding affinity with amplification. Despite the success of this approach, delivery of selected aptamers across cell membranes and to specific subcellular compartments is an unmet need in chemical biology. Here, we address this challenge, demonstrating in vitro selection of DNA aptamers capable of homing to nuclei of cultured cells without transfection agents or viral trans-duction. Selection of such folded karyophilic DNA aptamers (~100 nucleotides) is achieved by a biosensor strategy that rewards exposure to nuclear DNA ligase. Identified DNA molecules are preferentially delivered to cell nuclei within minutes. Related strategies can be envisioned to select aptamers that home to other subcellular compartments.
While in vitro nucleic acid selection has successfully generated aptamer ligands with high affinity for molecular targets,1–6 the delivery of aptamers across cell membranes to intracellular targets is inefficient, typically requiring viruses or toxic transfection agents.7 Several aptamer technologies have exploited affinity to cell surface components to facilitate their own intracellular delivery via receptor-mediated endocytosis,8–12 although it is unclear whether such aptamers can escape endosomes.13,14 In contrast, certain plant viroids are infectious as naked nucleic acids that achieve intracellular delivery by exploiting available cell machinery to access both cytoplasm and nuclei.15,16 We reasoned that similarly robust molecules that gain entry to human cells could be identified by directly leveraging vast random (diversity ~1014) combinatorial DNA libraries and selective pressure to reward exposure to enzyme activities unique to specific subcellular compartments (e.g., nuclear DNA ligase), yielding molecules capable of homing to such compartments (e.g., “karyophilic” DNA aptamers).
To reward unique DNA sequences from a combinatorial library for their ability to access nuclei without the aid of exogenous reagents, a ligase-sensitive DNA construct was engineered (Figure 1A and Figure S1). This ligase reward sensor is formed by the hybridization of three synthetic oligonucleotides: a combinatorial DNA library, a “bait” oligonucleotide, and a “splint” oligonucleotide that positions a nick between library and bait. In the presence of DNA ligase, the free 5′ phosphoryl group of the bait is covalently bonded to the 3′ terminus of the combinatorial library, effectively rewarding the library strand with biotinylation. Following streptavidin bead capture, a stringent alkaline wash step preserves only covalently biotinylated sequences from the DNA library. This ligation-based selection strategy enables specific capture of “winning” sequences so that oligonucleotides that are internalized but not localized to the nucleus are not recovered and is likely more stringent than selection based upon physical partitioning of subcellular components.
Figure 1.

Development and validation of DNA ligase biosensor reward strategy for in vitro selection. (A) Chemistry catalyzed by DNA ligase in the context of an engineered duplex DNA-based biosensor. (B) Experimental approach to evaluate biosensor performance. Biotinylated biosensor is shown in gray (splint) and purple (bait) with 5′-phosphoryl group, hybridized to random library (black) flanked by constant regions (green, gold). (C) qPCR validation experiment demonstrating sensitivity of the engineered biosensor to DNA ligase activities in vitro.
Quantitative PCR (qPCR) was used to validate the ligase reward sensor design (Figure 1B). Both exogenous DNA ligase and ligase activity in cell lysate were shown to be sufficient to covalently biotinylate DNA library in hybrid constructs (Figure 1C). Conversely, constructs exposed to cell culture conditioned media or oligonucleotide configurations inconsistent with ligation were poorly recovered. This validation demonstrates the ability to reward aptamer library sequences on the basis of exposure to DNA ligase in biological contexts.
We reasoned that this approach could be applied to identify naked DNA aptamers capable of reaching nuclei of cultured HEK293T cells. After assembly of the DNA ligase-sensitive library (>1014 sequences), HEK239T cells were briefly exposed to library constructs in cell culture media, followed by lysis, DNA isolation, streptavidin bead capture, and stringent washing. Captured constructs were amplified by PCR and single-stranded libraries were isolated from double-stranded products by denaturing polyacrylamide gel purification. This completes a repeatable in vitro selection cycle. Repetition of this cycle allows identification of DNAs with the ability to gain access to DNA ligase activity in human cells (Figure 2).
Figure 2.
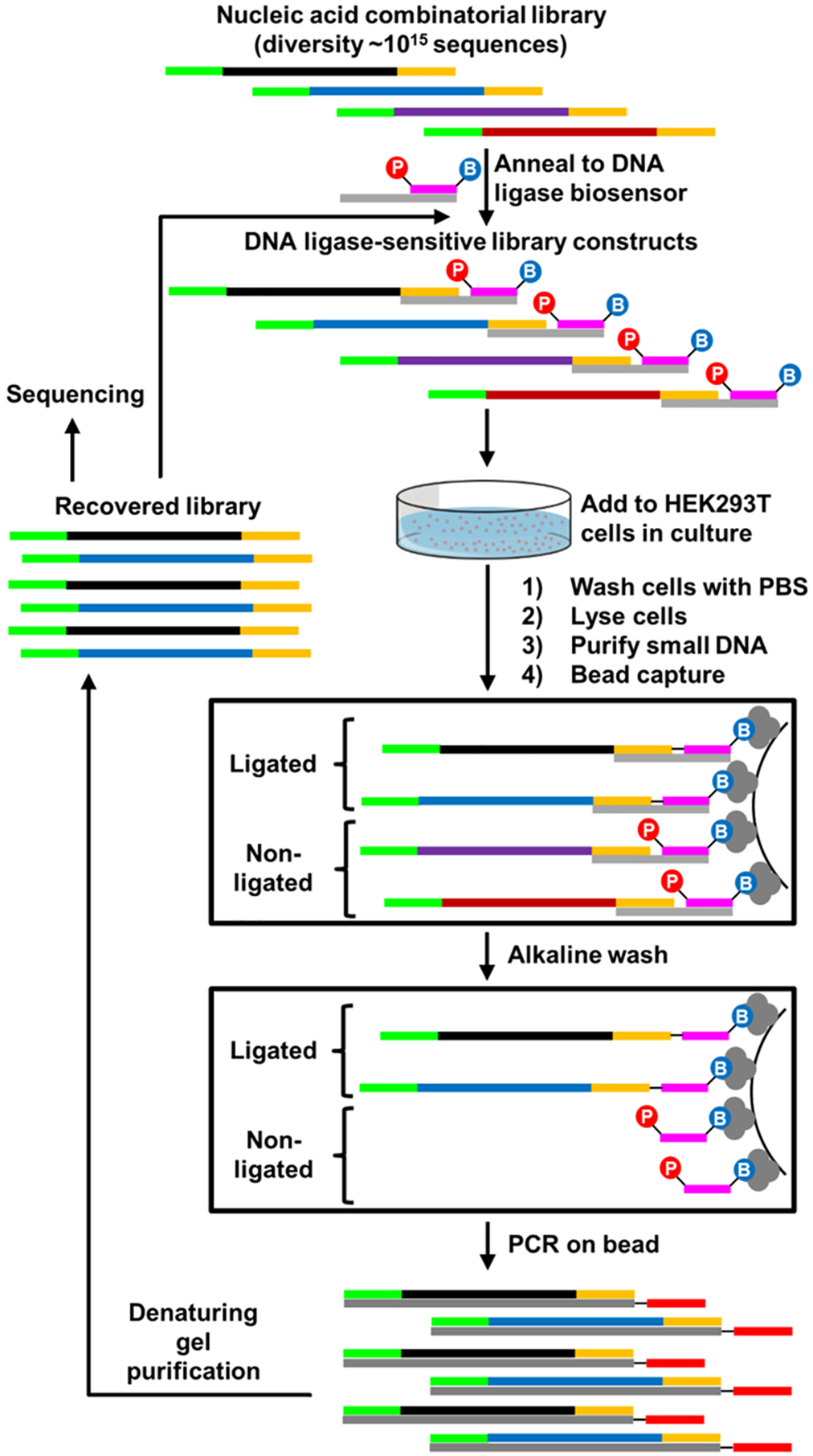
In vitro selection strategy for rewarding aptamers capable of gaining access to DNA ligase activity in cultured cells. A random (N55) DNA library flanked by constant primer regions is annealed to a ligation sensor and exposed to HEK293T cells. Ligated constructs are recovered by capture on magnetic streptavidin beads via 3′-terminal biotin conjugation of the ligation sensor. Recovered molecules are amplified by PCR, and the process is repeated.
High-throughput sequencing and subsequent analysis of recovered aptamer pools yielded two uniquely enriched sequences from the library (Figure S2A–C). Molecules termed clones 1 and 2 (18% and 1%, respectively, of the final recovered pool) each increased in prevalence over eight in vitro selection cycles (Figure S2D), suggesting their propensity for accessing a source of DNA ligase in cultured HEK293T cells. A random sequence from the naïve DNA library was chosen as a negative control in subsequent experiments. To rule out the possibility that clones 1 and 2 are capable of self-ligation, streptavidin capture assays were repeated for these sequences after incubation with adenosine triphosphate and with or without supplemental DNA ligase. This assessment confirmed that these sequences are not self-ligating (Figure S2E).
To facilitate oligonucleotide synthesis, an identical block of 21 nucleotides was removed from the 3′ termini of the unselected control aptamer and each selected clone to create oligonucleotides LJM-5587 (control), LJM-5588 (clone 1), and LJM-5589 (clone 2). Each oligonucleotide is predicted to form stable secondary structures under physiologic ion concentrations with ΔG37°C values of −4.79 kcal/mol (LJM-5587), −9.95 kcal/mol (LJM-5588), and −5.60 kcal/mol (LJM-5589) (Figure S3).17 The stability of folded oligonucleotide structures was additionally empirically characterized by absorption spectroscopy in thermal melting experiments, similarly demonstrating favorable free energies of folding under physiological conditions, with melting temperatures of 53 °C (LJM-5587), 61 °C (LJM-5588), and 48 °C (LJM-5589) (Figure S4). The small difference between predicted and observed folding ΔG37°C is likely attributable to slight differences in ionic conditions and inability of the in silico prediction to account for aptamer tertiary structure. The susceptibility of oligonucleotide folded structures to degradation by cellular nucleases was additionally assessed, with each of the three oligonucleotides displaying approximately the same degree of time-dependent degradation in vitro when exposed to whole cell extract (Figure S5).
Predicted secondary structures of LJM-5588 and LJM-5589 were validated by KMnO4 and Mung Bean nuclease reactivity assay (Figure 3, Figure S6, and Table S1). Thymine bases are sensitive to oxidation by KMnO4, detectable by thermal cleavage. This reactivity is suppressed in double-helical structures. Quantitation of cleavage at each nucleotide under native and denaturing conditions yields a protection ratio, indicative of the structural context of a base. The results support the predicted secondary structures of each molecule (Figure 3 and Figure S6).
Figure 3.

Structural characterization of karyophilic aptamers by KMnO4 reactivity and Mung Bean nuclease enzymatic cleavage. Thymine nucleotides (blue) and associated protection ratios (native/ denatured KMnO4 reactivity) overlaid on predicted mFold aptamer secondary structures for (A) LJM-5588 and (B) LJM-5589. Positions with high protection of thymine under native conditions are indicated in red. Positions susceptible to cleavage by Mung Bean nuclease are indicated with green triangles.
Synthetic aptamers selected for karyophilic behavior were assessed for their ability to specifically localize to HEK293T cell nuclei. This was achieved by exposing cells to aptamers (1 μM) in fresh media, washing, and subjecting cells to isolation of nuclear and cytoplasmic fractions after osmotic lysis and differential centrifugation (Figure S7). Following proteinase K digestion and ethanol precipitation of DNAs, qPCR was employed to quantify aptamer present in nuclear and cytoplasmic fractions. Upon normalization to total protein content in each fraction, it is evident that aptamers LJM-5588 and LJM-5589 both selectively localize to nuclei, while negative control oligonucleotide LJM-5587 does not (Figure 4A). Interestingly, a rough calculation indicates that the fraction of input aptamer recovered from cell nuclei approximates the fraction of total culture volume occupied by cell nuclei.
Figure 4.
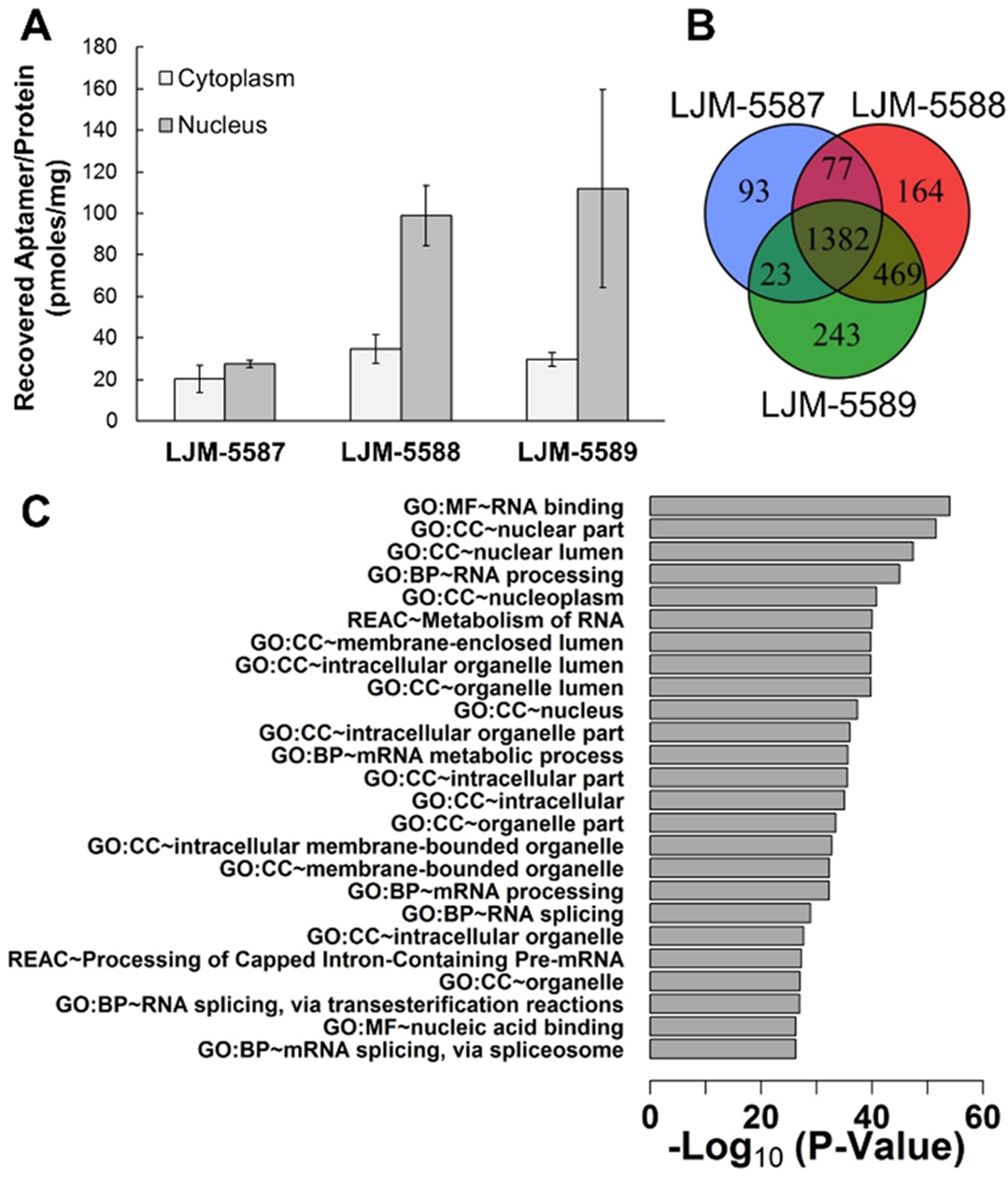
Validation of aptamer karyophilic properties in HEK293T cells. (A) qPCR-based assessment of aptamer localization to cytoplasm and nucleus following addition to cell culture medium. Total aptamer recovered in each cellular fraction is normalized to total protein content from three replicates. (B) Proteomic assessment of aptamer subcellular localization. Venn diagram comparison of interacting proteins for each aptamer. C) Functional term enrichment analysis for the 469 protein interaction partners shared by LJM-5588 and LJM-5589, but not control LJM-5587 (GO: gene ontology, MF: molecular function, CC: cellular component, BP: biological process, REAC: reactome). Degree of statistical enrichment of proteomic interactions for individual functional classes is indicated as −log10(adjusted p value).
To confirm nuclear localization by an independent approach, protein interaction partners for each aptamer were identified by exposing biotinylated aptamers to cells in culture, formaldehyde cross-linking of biotinylated aptamers to any colocalized proteins, purifying aptamer–protein complexes, and identifying copurified proteins via proteomic methods. Shared and unique protein binding partners were identified for each aptamer (Figure 4B). Functional term enrichment analysis of binding partners shared by LJM-5588 and LJM-5589, but not by control LJM-5587, reveals proteins with striking enrichment for nuclear terms (Figure 4C). Examples include “GO:CC ~ nuclear lumen” and “GO:CC ~ nucleoplasm”. This supports the assertion that LJM-5588 and LJM-5589 rapidly and specifically access HEK293T nuclei in culture.
The propensity for aptamer uptake into various other cell lines was then studied via qPCR assay. Notably, HeLa, HepG2, and RPE cell lines do not show similar patterns of enriched nuclear uptake of LJM-5588 and LJM-5589 relative to LJM-5587, suggesting that the aptamers identified here are specific for HEK293T cells (Figure S8). The basis for the specificity was investigated by examining differences in proteomic expression patterns between HEK293, HeLa, and HepG2 cell lines in publically available nontargeted proteomic data.18 Intriguingly, the collection of empirically nominated aptamer-binding partners is collectively expressed at lower level in both HeLa and HepG2 relative to the HEK293 cell line (Figure S9). This suggests that the proteins comprising the biological pathway responsible for karyophilic aptamer uptake may be coordinately regulated in a way that may account for the observed differences in aptamer uptake among the tested cell lines. A hypothesis of coordinated regulation is further supported by analysis of human protein coexpression networks in the STRING database, revealing a higher degree of coexpression among aptamer-binding partners than would be predicted by chance alone (enrichment p value: < 1 × 10−16) (Figure S10).19
LJM-5588, the most abundant aptamer identified in the final round of selection, was selected for analysis of uptake over time, at various doses, and after inhibition of different endocytosis pathways. HEK239T cells were exposed to LJM-5588 (1 μM) for up to 3 h (Figure 5A). Nuclear uptake was detected as early as 0.5 h after treatment and did not increase after 1.5 h. For a 2 h exposure, LJM-5588 nuclear uptake was dose-dependent over a concentration range of 0.1–2.0 μM aptamer (Figure 5B). These observations suggested that nuclear uptake may involve an active endocytosis mechanism.
Figure 5.

Time and dose dependence of LJM-5588 uptake in HEK293T cells. (A) qPCR assessment of aptamer localization to cytoplasm and nucleus following treatment with 1 μM aptamer over 0–3 h. Logistic curve fits are shown (solid line: nucleus, dashed line: cytoplasm). (B) qPCR assessment of aptamer localization to cytoplasm and nucleus following treatment with 0–2 μM aptamer for 2 h. Findings in both panels are normalized to total protein recovered in each fraction.
To explore this mechanism, siRNA knockdown of CLTC (clathrin heavy chain) and DYN2 (dynamin 2) (independently required for clathrin-dependent endocytosis) or PAK1 (required for macropinocytosis), was used to determine uptake dependence on each factor.20 Knockdown of CLTC or DYN2 significantly reduced aptamer internalization to cytoplasmic and nuclear fractions. PAK1 knockdown did not have a statistically significant impact (Figures S11–S13). This suggests that nuclear delivery of LJM-5588 in HEK293T cells is clathrin-dependent. This conclusion is also supported by the observation of LJM-5588 binding to CLTC in proteomic analysis: CLTC is the 11th most prominent cross-linked binding partner by abundance among enriched binding partners versus negative control LJM-5587 (Figure S14).
In summary, we have identified two DNA aptamers, LJM-5588 and LJM-5589, capable of rapidly and specifically accessing nuclei of HEK293T cells. Notably, this activity was specific for HEK293T cells and not observed for several other common cell lines. Each aptamer folds into a structure stable at physiological temperature and ionic conditions. Nuclear localization was confirmed by both qPCR and proteomic assessments, the former also suggesting a clathrin-dependent uptake mechanism for LJM-5588. Thus, the combinatorial chemistry approach described here drives in vitro selection of DNAs capable of self-delivery to HEK293T cell nuclei.
Future investigation will assess the potential for aptamer-mediated nuclear delivery of oligonucleotide or other macromolecular cargos and the modification of this SELEX method to reward homing to other subcellular compartments. Additionally, beyond dependence upon clathrin-dependent endocytosis, the remaining mechanistic details regarding aptamer endosomal escape and localization to cell nuclei are unclear. Intriguingly, a previous report has suggested that clathrin-dependent membrane-associated vesicles are capable of directly delivering macromolecular cargo to cell nuclei.21 It is tantalizing to speculate that karyophilic aptamers may exploit this known biological process to orchestrate their own nuclear delivery. Future mechanistic work will be required to test this hypothesis.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge technical assistance from the laboratory of Mark McNiven and the Mayo Clinic Microscopy and Cell Analysis Core. This work was supported by NIH Grant No. GM128579 (L.J.M.), T32GM065841 (Mayo Clinic Medical Scientist Training Program), F30CA220660 (J.S.), The Mayo Clinic Graduate School of Biomedical Sciences, and an NSF graduate fellowship (B.W.).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b06736.
Materials and methods as well as supplemental figures (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Cho EJ; Lee JW; Ellington AD Applications of aptamers as sensors. Annu. Rev. Anal. Chem 2009, 2, 241–64. [DOI] [PubMed] [Google Scholar]
- (2).Nimjee SM; White RR; Becker RC; Sullenger BA Aptamers as Therapeutics. Annu. Rev. Pharmacol. Toxicol 2017, 57, 61–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Gilboa E; McNamara J; Pastor F Use of Oligonucleotide Aptamer Ligands to Modulate the Function of Immune Receptors. Clin. Cancer Res 2013, 19 (5), 1054–1062. [DOI] [PubMed] [Google Scholar]
- (4).Zhou JH; Rossi J Aptamers as targeted therapeutics: current potential and challenges. Nat. Rev. Drug Discovery 2017, 16 (3), 181–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Thiel KW; Giangrande PH Therapeutic Applications of DNA and RNA Aptamers. Oligonucleotides 2009, 19 (3), 209–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Mondragon E; Maher LJ Anti-Transcription Factor RNA Aptamers as Potential Therapeutics. Nucleic Acid Ther 2016, 26 (1), 29–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Lakhin AV; Tarantul VZ; Gening LV Aptamers: Problems, Solutions and Prospects. Acta Naturae 2013, 5 (4), 34–43. [PMC free article] [PubMed] [Google Scholar]
- (8).Thiel KW; Giangrande PH Intracellular delivery of RNA-based therapeutics using aptamers. Ther. Delivery 2010, 1 (6), 849–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Yoo H; Jung H; Kim SA; Mok H Multivalent comb-type aptamer-siRNA conjugates for efficient and selective intracellular delivery. Chem. Commun. (Cambridge, U. K.) 2014, 50 (51), 6765–7. [DOI] [PubMed] [Google Scholar]
- (10).McNamara JO 2nd; Andrechek ER; Wang Y; Viles KD; Rempel RE; Gilboa E; Sullenger BA; Giangrande PH Cell type-specific delivery of siRNAs with aptamer-siRNA chimeras. Nat. Biotechnol 2006, 24 (8), 1005–15. [DOI] [PubMed] [Google Scholar]
- (11).Chu TC; Twu KY; Ellington AD; Levy M Aptamer mediated siRNA delivery. Nucleic Acids Res. 2006, 34 (10), No. e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Zhou J; Rossi JJ Cell-specific aptamer-mediated targeted drug delivery. Oligonucleotides 2011, 21 (1), 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Zhang K; Sefah K; Tang L; Zhao Z; Zhu G; Ye M; Sun W; Goodison S; Tan W A novel aptamer developed for breast cancer cell internalization. ChemMedChem 2012, 7 (1), 79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Tawiah KD; Porciani D; Burke DH Toward the Selection of Cell Targeting Aptamers with Extended Biological Functionalities to Facilitate Endosomal Escape of Cargoes. Biomedicines 2017, 5 (3), 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Tsagris EM; de Alba AEM; Gozmanova M; Kalantidis K Viroids. Cell. Microbiol 2008, 10 (11), 2168–2179. [DOI] [PubMed] [Google Scholar]
- (16).Ding B The Biology of Viroid-Host Interactions. Annu. Rev. Phytopathol 2009, 47, 105–131. [DOI] [PubMed] [Google Scholar]
- (17).Zuker M Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31 (13), 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Geiger T; Wehner A; Schaab C; Cox J; Mann M Comparative Proteomic Analysis of Eleven Common Cell Lines Reveals Ubiquitous but Varying Expression of Most Proteins. Mol. Cell. Proteomics 2012, 11 (3), M111.014050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Szklarczyk D; Gable AL; Lyon D; Junge A; Wyder S; Huerta-Cepas J; Simonovic M; Doncheva NT; Morris JH; Bork P; Jensen LJ; Mering C STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47 (D1), D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Al Soraj M; He L; Peynshaert K; Cousaert J; Vercauteren D; Braeckmans K; De Smedt SC; Jones AT siRNA and pharmacological inhibition of endocytic pathways to characterize the differential role of macropinocytosis and the actin cytoskeleton on cellular uptake of dextran and cationic cell penetrating peptides octaarginine (R8) and HIV-Tat. J. Controlled Release 2012, 161 (1), 132–141. [DOI] [PubMed] [Google Scholar]
- (21).Chaumet A; Wright GD; Seet SH; Tham KM; Gounko NV; Bard F Nuclear envelope-associated endosomes deliver surface proteins to the nucleus. Nat. Commun 2015, DOI: 10.1038/ncomms9218. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.