

Abstract
We determined whether the structural and functional integrity of amacrine cells (ACs), the largest cohort of neurons in the mammalian retina, are affected in glaucoma. Intraocular injection of microbeads was made in mouse eyes to elevate intraocular pressure as a model of experimental glaucoma. Specific immunocytochemical markers were used to identify AC and displaced (d)ACs subpopulations in both the inner nuclear and ganglion cell layers, respectively, and to distinguish them from retinal ganglion cells (RGCs). Calretinin- and GABA- immunoreactive cells were highly vulnerable to glaucomatous damage, whereas ChAT-positive and glycinergic AC subtypes were unaffected. The AC loss began four weeks after initial microbead injection, corresponding to the time course of RGC loss. Recordings of ERG oscillatory potentials and scotopic threshold responses, which reflect AC and RGC activity, were significantly attenuated in glaucomatous eyes following a time course that matched that of the AC and RGC loss. Moreover, we found that it was the ACs coupled to RGCs via gap junctions that were lost in glaucoma, whereas uncoupled ACs were largely unaffected. Our results suggest that AC loss in glaucoma occurs secondary to RGC death through the gap junction-mediated bystander effect.
Keywords: retina; glaucoma; cell death; neuroprotection; electroretinogram; amacrine cells; ganglion cells; RRIDs: AB_2109797, AB_2167511, AB_10061777, AB_11003211, AB_90893
Graphical Abstract
INTRODUCTION
Glaucoma, the second leading cause of blindness worldwide, is a neurodegenerative disorder commonly associated with elevated intraocular pressure (IOP) and a progressive loss of retinal ganglion cells (RGCs) resulting in visual deficits (Quigley, 1999; Chang and Goldberg, 2012; Tian et al., 2015). While insult appears initially within the optic nerve, the exact mechanism(s) that underlie RGC somatic loss in glaucoma and the extent to which other retinal neurons are affected remains unclear (Schwartz, 2003; Calkins, 2012; Huberman and El-Danaf, 2015). Both human and animal studies of glaucoma have reported significant alterations in components of the scotopic electroretinogram (ERG), including a-wave, b-wave, the scotopic threshold response (STR) and the oscillatory potentials (OPs), suggesting functional deficits in photoreceptors, bipolar cells and amacrine cells (ACs) (Stockton and Slaughter, 1989; Holopigian et al., 1990; Vaegan et al., 1991; Wachtmeister, 1998; Bayer et al, 2001; Velten et al., 2001; Danias et al, 2006; Harazny et al, 2009; Alarcón-Martínez et al., 2010; Heiduschka et al., 2010; Cuenca et al., 2010; Fernández-Sánchez et al., 2014; Georgiou et al., 2014). Taken together, these studies suggest that neurons at every retinal level are affected in glaucoma, but whether all are primary targets or may reflect secondary degenerations following RGC death remains unclear (Schwartz et al., 1996; Levkovitch-Verbin et al., 2001; Lei et al., 2008; Calkins, 2012).
Amacrine cells are the most diverse neuronal type in the retina, differentiable into some 40 different subtypes based on morphology, physiology, and neurochemistry (Kolb, 1997; Wässle and Boycott, 1991; Masland, 2012). In mouse, the major AC subpopulations are formed by glycinergic and GABAergic cells with further divisions identified by specific markers such as calretinin (CR), calbindin (CB), tyrosine hydroxylase (TH), glutamic acid decarboxylase 65 (GAD65), choline acetyltransferase (ChAT), and nitric oxide synthase (NOS) (Pourcho and Goebel, 1985; Voigt, 1986; Brecha et al., 1988; Vaney and Young, 1988; Menger et al., 1998; Haverkamp and Wässle, 2000). The ACs subserve complex circuitry in the inner retina and thereby provide an integration of signals in both the temporal and spatial domain that is essential to construction of the RGC visual messages sent to the brain. It is therefore critical in assessing the mechanisms responsible for loss of visual function in glaucoma to determine the extent of any injury to AC subpopulations.
As aforementioned, changes in the amplitude of the STR and OP components of the ERG in glaucomatous retinas suggest AC vulnerability (Saszik et al., 2002; Heiduschka et al., 2010; Frankfort et al., 2013; Porciatti, 2015). However, there has been only limited study of the effects of glaucoma on AC structural integrity and the existing results are ambiguous (May and Mittag, 2004; Moon et al., 2005; Hernandez et al., 2009; Kunzevitzky et al., 2010). For example, one study of the DBA/2J mouse retina, a model of hereditary glaucoma, reported a marked reduction in the number of GABA- and ChAT-immunoreactive (IR) ACs, but an increase in the NOS-positive ACs and no change in glycinergic ACs (Moon et al., 2005). In contrast, Jacobs et al. (2005) found no detectable reduction in GABA- and ChAT-IR AC subpopulations in 1-year-old DBA/2J mice. While other studies have reported substantial reductions in CR-, GABA-, and ChAT-IR ACs, it was uncertain whether these changes reflected cell death and/or a reduction in antigen expression (Kielczewski et al., 2005; Hernandez et al., 2009; Gunn et al., 2011). The conflicting data may reflect differences in the methodology used to induce experimental glaucoma as well as the difficulty in identifying the large spectrum of AC subtypes as no single labeling method can provide complete coverage due to their neurochemical diversity. Further, displaced ACs (dACs) comprise approximately one-half of the neurons found in the GCL (Jeon et al., 1998; Schlamp et al., 2013) making differentiation from RGCs difficult, but necessary, to assess the effect of glaucoma on a large portion of the total AC population.
In the present study, we used a multidisciplinary approach to determine the structural and functional vulnerability of ACs in a mouse model of glaucoma. Our results indicate that different subpopulations of ACs and dACs show significant loss in glaucoma, corresponding to an attenuation of the OP and STR components of the ERG. Importantly, we found that the most vulnerable ACs/dACs in glaucoma are those that are coupled to RGCs via gap junctions (Völgyi et al., 2009), whereas uncoupled ACs were largely unaffected. We thus posit that most AC loss in glaucoma reflects degeneration secondary to that of RGCs through a gap junction-mediated intercellular mechanism consistent with the bystander effect (Akopian et al., 2014).
MATERIALS and METHODS
Animals
All animal procedures were in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee (IACUC) at the State University of New York College of Optometry. All experiments were performed on adult C57BL/6 mice (total of 20 female and 15 male animals). The mice were originally obtained from Jackson Laboratories (Bar Harbor, ME) and subsequently bred in the animal care facility at the College of Optometry. Animals were kept under a 12-hour light/12-hour dark ambient light cycle and fed ad libitum.
IOP elevation by microbead injection
The intraocular pressure (IOP) was elevated by injection of 10 μm diameter polystyrene microbeads (Invitrogen, Carlsbad, CA) into the anterior chamber as previously described (Sappington et al., 2010). The intracameral injections were performed unilaterally with 2 μl of microbead suspension (containing 7.2 × 106 beads) using a glass micropipette connected to a Hamilton microsyringe. The cornea was gently punctured using a 30-gauge needle prior to intracameral injections. An equivalent volume of phosphate-buffered saline (PBS) was injected into the contralateral eyes to provide control measurements. A second microbead injection was performed during the fourth week, which maintained elevated IOP for at least 8 weeks. All injections were performed on animals anesthetized with an intraperitoneal (IP) injection of ketamine/xylazine mixture and topical application of 0.5% proparacaine (Akorn, Lake Forest, IL). Enucleations were performed at 2 days or 1, 3, 4, or 8 weeks after the initial microbead injection and retinas were assessed for gross structural and cellular damage.
IOP measurements
Measurements of IOP were made weekly using a tonometer (TonoLab; Colonial Medical Supply, Espoo, Finland) as previously described (Sappington et al., 2010). IOP measurements were performed within 1–3 minutes after anesthetizing the mice with an IP injection of ketamine/xylazine mixture and topical application of 0.5% proparacaine. Six measurements were obtained per eye and averaged. IOP measurements were performed between 10 AM and 12 PM, to minimize the effect of diurnal IOP variation.
Retrograde labeling of retinal neurons
The gap junction-permeant tracer Neurobiotin (NB, Vector Labs, Burlingame, CA) was used to retrogradely label RGCs and the ACs that were coupled to them. Isolated globes with attached optic nerves were submersed in oxygenated Ames medium and a drop of NB (4% in 0.1 M Tris buffer) was applied to the cut optic nerve for 40 minutes. The cornea and lens were removed and the remaining retina-eyecups were washed in oxygenated Ames medium for one hour. The eyecups were then fixed with 4% paraformaldehyde in a 0.1 M PBS, pH 7.4, for 30 minutes at room temperature, cryoprotected in 30% sucrose, embedded in Tissue-Tek OCT Compound (Andwin Scientific, Schaumburg, IL), and froze. To identify RGCs, vertical retinal cryosections were immunostained for Brn3a, which is expressed in approximately 86–92% of RGCs in rodent retinas at (Nadal-Nicolás et al., 2009; Galindo-Romero et al 2011). Retinal sections then were counterstained with the nuclear dye DAPI to obtain the total cell count in the GCL and INL. The conventional ACs and displaced ACs (dACs) coupled to RGCs were identified as those cells in the proximal INL and in the GCL, respectively, that were NB-positive, but Brn3a-negative. In contrast, the ACs/dACs not coupled to RGCs were identified as those cells stained with DAPI, but negative for both NB and Brn3a labeling.
Immunohistochemistry
The immunohistochemical methods have been described previously (Akopian et al., 2014). Briefly, vertical retinal sections (10–15 μm thick) were blocked in 0.1 M PBS containing 10% normal donkey serum (NDS), 1% BSA, and 0.5% Triton x-100 for one hour. The sections were then incubated with primary antibodies (Table 1) diluted in 0.1 M PBS containing 3% NDS, 1% BSA, and 0.1% Triton x-100 at room temperature for three hours or overnight at 4°C. After an extensive washing in 0.1 M PBS, sections were incubated for 2 hours in secondary anti-goat/rabbit antibodies conjugated with Alexa-488/633 (1:200, Life Technologies Corporation, NY). Neurobiotin was visualized with Alexa-488/Cy3-conjugated streptavidin (1:200, Invitrogen). Retinal sections were then mounted in Vectashield (Vector Labs, Burlingame, CA) media with DAPI. Images of immunolabeled tissues were taken under an Olympus FV1200 MPE confocal microscope (Tokyo, Japan) with 20x or 40x oil immersion objectives with a focusing step size of 2–3 μm. High resolution (1024 x 1024 pixels) Z-stacked images were compiled to a single plane and analyzed quantitatively. The brightness and contrast of presentation micrographs were adjusted using Adobe Photoshop CS6 (San Jose, CA).
Table 1.
Primary antibodies
| Name | Immunogen | Source, Cat #, species | Concentration |
|---|---|---|---|
| GFAP | Human GFAP fusion protein | Thermo Fisher Scientific, PA1-9565, rabbit, polyclonal, RRID: AB_2109797 | 1:1000 |
| Brn-3a | N-terminus of Brn-3a of human origin | Santa Cruz Biotechnology, sc-31984, goat, polyclonal, RRID: AB_2167511 | 1:500 |
| ChAT | Human placental enzyme | EMD Millipore, AB144P- 1ML, goat, polyclonal, RRID: AB_10061777 | 1:100 |
| GABA | GABA | EMD Millipore, PC213L, rabbit, polyclonal, RRID: AB_2232326 | 1:500 |
| Calretinin | Recombinant full-length mouse calretinin protein | Thermo Fisher Scientific, PA5-16681, rabbit, polyclonal, RRID: AB_11003211 | 1:1000 |
| GlyT1 | Synthetic peptide from the carboxy-terminus as predicted from the cloned rat GLYT1 | EMD Millipore, AB1770, goat, polyclonal, RRID: AB_90893 | 1:10,000 |
Antibody characterization
The primary antibodies (Table 1) used in the present study are well-characterized markers of ACs and RGCs in mouse retina. Astrocytes and Muller cells were labeled with an antibody against glial fibrillary acidic protein (GFAP) as described in previous studies (Yassa 2014; Akopian et al., 2014). The Brn3a antibody has been shown to selectively label RGCs in the rodent retina (Nadal-Nicolas et al., 2009; Galiando-Romero et al 2011). The antibodies for ChAT, GABA, and CR have been widely used to identify select AC subpopulations in the mouse retina (Moon et al., 2005; Jakobs et al., 2005; Pang et al., 2013; Akopian et al., 2014). The glycine transporter-1 (GlyT1) antibody selectively stains membranes of glycinergic ACs in the mouse retina (Menger et al 1998; Pow et al 1999).
Assessment of retinal injury and neuronal death
The detailed methods to assess retinal injury and neuronal loss have been described previously (Akopian et al., 2014). Briefly, cell counts were made manually per unit length of 630 μm (unless stated otherwise) in the INL and GCL of retinal vertical sections counterstained with propidium iodide (PI) or DAPI. Measurements were made from all retinal quadrants and eccentricities and then averaged. Analysis of GFAP immunoreactivity was made by analyzing confocal images with Image J software (NIH, Bethesda, MA, USA). Following binarization and thresholding of the images, 3–4 uniform fields extending from the GCL to the inner limit of the ONL in each image were selected for analysis. We calculated the percent area in each field occupied by immunolabeling by analyzing individual pixels; this parameter was independent of differences in the intensity of label within or across retinas. Values were calculated for 5–10 sections per retina and then averaged across at least 3 control and 3 experimental retinas for each protocol. The GFAP fluorescence intensity profile as a function of distance was generated along a single line extending from the GCL to the ONL of vertical retinal sections. Data were then imported into Sigmaplot software (Systat Software, Inc. CA, USA) and histograms of the mean ± SEM values were constructed.
Electroretinography
Electroretionograms (ERGs) were recorded from mice injected intraocularly with microbeads and from control animals (some sham injected with 0.1 M PBS) at various time points up to 8 weeks after the injections. Animals were anesthetized with an IP injection of ketamine/xylazine (70/7 mg/kg) under dim red light following overnight dark adaptation. Pupils were fully dilated with 2.5% phenylephrine hydrochloride/1% tropicamide and the cornea was kept moist with 1% methylcellulose (Akorn). Body temperature was maintained around 37°C with an electric heating pad. ERGs were recorded from the two eyes simultaneously with platinum electrodes placed in contact with the cornea with needle electrodes inserted into the cheek and skin serving as reference and ground, respectively. Visual stimuli consisted of brief (< 5ms) white full field Ganzfeld flashes generated by an array of light emitting diodes ranging from −6.7 to 2.0 log scot. cd-s/m2 (Espion ColorDome stimulator; Diagnosys LLC, Lowell MA). Responses were averaged over 40–50 trials for weak stimuli and fewer trials for stronger stimuli. Signals were amplified, filtered (1–300 Hz) and digitized at 1 kHz with a resolution of 0.1 μV. The STR and OPs reflect the activity of RGCs and ACs (Sazsik et al., 2002; Wachtmeister, 1998). The STR was elicited in the intensity range of −4.9 to −4.0 log scot. cd-s/m2 and the amplitude of the positive STR (pSTR) was computed from the peak of the pSTR to the trough of the negative STR (nSTR). The OPs, extracted from the b-wave using a band pass filter (50–300 Hz), were elicited in the intensity range of −0.4 to 2.0 log scot. cd-s/m2. OPs were analyzed in response to flashes at the fixed low intensity of −0.4 log scot. cd-s/m2 to prevent contributions from the a-wave that emerges at higher intensities. The overall OP amplitude was computed from the sum of that of the first 5 OP waves (OP1–OP5) measured from trough to peak.
Statistical analysis
Data are presented as mean ± SEM. For cells counts of immunolabeled retinas, data are presented with the following notations: n is the total number of measures; x/y, where x is the number of sections in which the measurements were made and y is the number of retinas. Statistical comparisons were assessed using Student’s 2-sided t-test. Value of p<0.05 were considered statistically significant.
RESULTS
Elevated IOP and experimental glaucoma induce retinal injury
To induce elevated IOP and experimental glaucoma, we adopted and modified a method of injecting polystyrene microbeads into the anterior chamber of mouse eyes (Sappington et al., 2010). One week after injection, the 10 μm-diameter microbeads were found accumulated at the angle of the anterior chamber or within Schlemm’s canal leading to a blockade of aqueous humor outflow. This resulted in a significant increase in the average IOP from a control value of 12.0 ± 0.9 mm Hg to 21.9 ± 1.8 mm Hg (n=10, p<0.001) at 2 weeks following injection of microbeads (Fig. 1A). There was a slight decrease in the average IOP beginning at 3 weeks and so a second microbead injection was regularly performed at 4 weeks to stabilize the elevated IOP to at least 8 weeks after the initial microbead injection (18.9 ± 1.3 mm Hg, n=10). In control experiments, sham injections of PBS were made into the anterior chamber, which did not significantly alter the average IOP from that seen in uninjected mouse eyes (11.4 ±1.5 mm Hg, n=10, p>0.1) (Fig. 1A).
Figure 1.

Intracameral injection of microbeads elevates IOP of mouse eyes. A: An initial injection of microbeads followed by a second injection at 4 weeks (arrow) resulted in a significant elevation of IOP that was maintained for at least 8 weeks (red circles). Mice that received PBS sham injections showed no change in IOP from steady values at week 0 (black circles). B: Elevated IOP reduced the thickness of entire retina as the individual inner nuclear layer (INL), inner plexiform layer (IPL) and ganglion cell layer (GCL). C: In control retinas, GFAP immunofluorescence was confined almost exclusively to astrocytes in the GCL. D: At eight weeks after initial microbead injection, the GFAP labeling was dramatically increased due to expanded expression in Müller cell processes extending across retinal layers. E: Histograms quantify and compare the mean percentage area of GFAP labeling in control and glaucomatous retinas. Measurements were made of fluorescent pixels within a 100 x 100 μm square (in panels in C and D). Three to four area measurements were made and averaged in five control and five microbead-injected retinas. F: The fluorescent intensity profile of GFAP labeling for pixels measured along a single line profile (white lines in panels C and D), shows its upregulation in the IPL and INL of glaucomatous retinas compared to controls, with the largest increase in expression in the IPL. Bars indicate mean value ± SD (in A) and mean ± SEM (applies to panels B and E). *p<0.05; ***p<0.001, Student’s t-test. Scale bars = 50 μm (applies to panels C and D).
We initially assessed gross structural changes of retinas at 8 weeks after the initial microbead injections. Analysis of DAPI-labeled vertical sections revealed a significant decrease in the overall thickness of retinas by 13% (189 ± 8 μm to 165 ± 4 μm, n=12/3, p<0.05) (Fig. 1B). The reduction in overall retinal thickness was reflected in that of individual cellular and synaptic layers, with the largest degree of thinning seen in the inner retina. The IPL, INL, OPL, and ONL in microbead-injected eyes were 25%, 24%, 15%, and 16% thinner, respectively, than those in control retinas (n=12/3, p<0.05 for all comparisons) (Fig. 1B)
To test overall injury of retina with induced experimental glaucoma we analyzed reactive gliosis in the form of upregulated GFAP expression in astrocytes and Müller cells (Wang et al., 2000; Formichella et al., 2014; Chong and Martin, 2015). We performed immunohistochemical analysis of GFAP in the vertical sections of control and glaucomatous retinas and quantified GFAP expression within a square area (100 x 100 μm) running from the proximal edge of the GCL to the proximal edge of the ONL (Fig. 1C,D). In addition, we measured GFAP pixel intensities along a diagonal line running from the proximal edges of the GCL to that of the INL. In control retinas, GFAP immunofluorescence was confined almost exclusively to astrocytes in the GCL (Fig. 1C,F). However, at eight weeks after microbead injection the overall GFAP immunolabeling was dramatically increased as it expanded to Müller cell processes extending vertically through the GCL to INL (n=40/5, p<0.001) (Fig. 1D–F). The increased GFAP expression was seen within the INL, IPL, and GCL, with greatest upregulation seen in the IPL (Fig. 1F). These data revealed a significant injury to the retina at all levels after induction of experimental glaucoma.
Neuronal loss in glaucomatous retinas
We next quantified the loss of neurons in the GCL in the retinas of microbead-injected eyes. Since RGCs comprise only one-half of the cells in the GCL of the mouse retina, with dACs forming the remainder (Jeon et al., 1998; Schlamp et al., 2013), we used the selective RGC marker Brn3a (Nadal-Nicolás et al., 2009), to differentiate RGCs and dACs in vertical sections of control and glaucomatous retinas (Fig. 2A,B). In the retinas of control eyes, the number of dACs, which was computed as the difference in the number of DAPI-positive nuclei and Brn3a-positive cells in the GCL, comprised approximately 45% (n=36/5) of the total cells in the GCL with Brn3a-labeled RGCs forming the remainder. At eight weeks following initial microbead injection we found that the total number of cells in the GCL, total number of RGCs, and total number of dACs were all significantly reduced by 34%, 36% and 28% (n=27/4, p<0.001 for all measures), respectively, from individual control levels (Fig. 2B,C).
Figure 2.

Elevated IOP results in a progressive reduction of dACs and RGCs in the GCL. A,B: Vertical sections of control and bead-injected retinas were labeled for Brn3a to identify RGCs and counterstained with a nuclear dye DAPI to differentiate dACs. C: By eight weeks after initial microbead injection the total number of cells in the GCL (DAPI-positive), total number of RGCs (Brn3a-positive), and total number of dACs (difference of DAPI- and Brn3a-positive cells), were all significantly reduced from control levels. D,E: Histograms quantifying cell counts performed at weekly intervals show a statistically significant decrease in the numbers of both dACs and RGCs beginning at 4 weeks after microbead injection and a further reduction at 8 weeks. Bars indicate mean value ± SEM. *** p<0.001; Student’s t-test. Scale bars =50 μm (applies to panels A and B).
To determine whether the loss of dACs followed the temporal pattern of RGC loss over the 8-week period following initial microbead injection, we performed cell counts at weekly intervals. There was a small, but insignificant reduction in the number of RGCs (7 ± 1% reduction from control levels) and dACs (5 ± 1% reduction) in the GCL up to 3 weeks after the first microbead injection (n=27/4, p=0.06 for both measures) (Fig. 2D,E). In contrast, a significant decrease in the number of RGCs (26 ± 1% reduction) and dACs (21 ± 1% reduction; n=22/4, p<0.001 for both measures) was seen beginning at 4 weeks and was extended at 8 weeks (RGC: 36 ± 1% reduction, dACs: 31±1% reduction; n=45/7, p<0.001, for both measures) after the initial microbead injection (Fig. 2D,E). Thus the loss of RGCs and dACs induced by elevation of mean IOP followed comparable time courses.
Different subpopulations of ACs are lost in experimental glaucoma
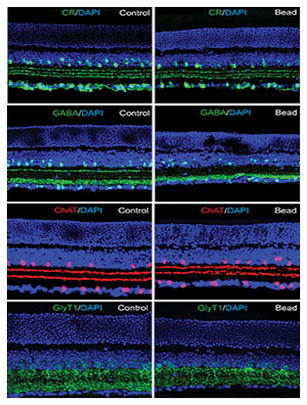
There are over 40 subtypes of ACs in the mammalian retina, which are highly diversified based on morphological, neurochemical, and physiological features (Vaney, 1990; MacNeil and Masland, 1998). To study whether distinct AC subpopulations were differentially affected in experimental glaucoma, we performed histochemical analysis using the specific AC markers CR, ChAT, GABA, and GlyT1 in vertical sections of control and microbead-injected retinas.
Consistent with earlier reports (Haverkamp and Wässle, 2000; Mojumder et al., 2008), we found a large number of cells in the INL and GCL of control retinas immunoreactive to CR with dendritic processes forming a characteristic three bands in the IPL (Fig. 3A). Although there was no change in the number of CR-positive cells at 3 weeks post injection, by 4 weeks we observed a significant reduction in cell number in both the INL (20% loss) and GCL (30% loss) (n=18/3, p<0.001 for both measures) (Fig. 3C,D). At 8 weeks, the number of CR-positive cells in the INL and GCL was reduced by 28% and 27% (n=45/5, p<0.001), respectively, compared with levels in control retinas (Fig. 3B–D).
Figure 3.

Selective subpopulations of ACs are lost in experimental glaucoma. A: In control retinas there was a large number of cells in the INL and GCL that were CR-IR with dendritic processes forming characteristic three bands in the IPL. B,C,D: Eight weeks after initial bead injection there was a significant reduction in the number of CR-positive cells in both the INL and GCL as compared to values in control (Ctr) retinas. Histograms show a significant reduction in CR-positive cells beginning by 4 weeks after microbead injection. E: GABA immunoreactivity was observed in conventional ACs at the inner margin of the INL and in some dACs in the GCL of control retinas. F: The number of GABA-positive ACs and dACs was significantly reduced eight weeks after bead injection. G,H: Histograms show the time course of changes in the number of GABA-positive ACs and dACs in the INL and GCL, respectively. I,J: ChAT-immunoreactive starburst ACs were found in the GCL and proximal INL with two well defined dendritic bands in the IPL. K: There was no significant change in the number of ChAT-positive cells in the INL or GCL at eight weeks after microbead injection (p>0.5). L,M: GlyT1 immunoreactivity was present in a large number of somata in the INL and their dendritic processes within the IPL. N: There was no significant difference in the number of GlyT1-positive ACs in control retinas and in microbead-injected retinas after eight weeks (p>0.1). Bars indicate mean value ± SEM. *** p<0.001; Student’s t-test. Scale bars =50 μm for all micrographs.
In vertical sections of control retinas, GABA-positive ACs formed two rows of somata at the inner margin of the INL, with a few presumed dACs labeled in the GCL (Fig. 3E), consistent with a previous report (Haverkamp and Wässle, 2000). Similar to the finding for CR-positive cells, we found no statistically significant decrease in GABA-positive ACs up to 3 weeks following initial microbead injection (6% loss, n=12/3, p>0.5). After 4 weeks, however, the number of GABA-positive cells was reduced significantly in both the INL (25% loss) and GCL (26% loss, n=20/3, p<0.001 for both measures), respectively (Fig. 3G,H). By 8 weeks, the number of GABA-positive somata was further reduced in both the INL (40% loss) and GCL (47% loss, n=55/5, p<0.001 for both measures) compared to values in control retinas (Fig 3F–H).
Over 15 different GABAergic ACs have been identified in the mouse retina, including the numerous and well-characterized starburst ACs, which also express acetylcholine (Masland, 2005; Pérez De Sevilla Müller et al., 2007). To evaluate the effects of experimental glaucoma on starburst ACs we immunolabeled vertical sections of control and microbead-injected retinas for ChAT. In control retinas, ChAT-IR cells were found in both the GCL and proximal INL with two well defined dendritic bands in the IPL reflecting ON and OFF starburst cell processes (Fig. 3I). Interestingly, even at eight weeks after the initial microbead injection we detected no significant change in the number of ChAT-positive in the INL and GCL (n=36/5, p>0.5) nor in the staining pattern of ChAT in the IPL (Fig. 3J,K).
We next studied the fate of glycinergic ACs in experimental glaucoma. In the mouse retina, approximately one-half of the ACs are glycinergic and can be selectively immunolabeled with antibodies against the glycine transporter GlyT1 (Menger et al., 1998; Pow et al., 1999). In both control and microbead-injected retinas, immunolabeling with anti-GlyT1 revealed strong labeling of AC somatic membranes forming multiple rows in the proximal INL as well as their dendritic processes in the IPL (Fig. 3L,M). Cell counts performed up to eight weeks after induction of experimental glaucoma with microbead injection showed no significant change in the number of GlyT1-positive ACs from that seen in control retinas (48 ± 2 cells/area vs. 51 ± 2; n=35/5, p>0.1) (Fig. 3L–N). The high resistance of glycinergic ACs to glaucoma damage resembled that of the cholinergic starburst ACs. Taken together, these data indicate that the loss of ACs in experimental glaucoma was subtype specific.
ACs coupled to RGCs via gap junctions are selectively vulnerable in glaucoma
In mouse retina, 15 of the 22 subtypes of RGCs are coupled to ACs via gap junctions (Völgyi et al., 2009). We showed recently that the gap junction-mediated bystander effect plays a critical role in promoting cell death across syncytia of coupled retinal neurons under a number of insult conditions (Akopian et al., 2014). We therefore investigated whether ACs that are coupled RGCs may be more vulnerable than uncoupled ACs in experimental glaucoma. To identify ACs coupled to RGCs we injected the gap junction-permeant tracer NB into the optic nerve, which retrogradely labeled RGCs and the ACs that they were coupled to (Fig. 4L). We found that retrograde Retinas were then immunolabeled with the AC markers GABA, CR, ChAT or GlyT1. We found that most somata that were double labeled for NB and AC-specific markers lay in the GCL and so we limited our study to these dACs (Fig. 4). We found that the majority of GABA- and CR-positive dACs in control retinas were also labeled with NB indicating coupling to RGCs (Fig. 4A,C,D,F). However, eight weeks after an initial microbead injection to elevate IOP we observed a significant reduction in the number of GABA- (70% reduction, n=20/3, p<0.001) and CR-positive (33% reduction, n=15/3, p<0.05) dACs that were labeled with NB, whereas the number of NB-negative cells (uncoupled) remained unchanged from control levels (p>0.05 for both measures) (Fig. 4,B,C,E,F). These data indicate that the AC coupled to RGCs were not merely uncoupled in glaucoma, but were lost.
Figure 4.

Amacrine cells coupled to RGCs are selectively vulnerable in glaucoma. A,D: A large number of GABA- and CR-positive dACs in control retinas were also labeled with NB indicating coupling to RGCs. B,C,E,F: At eight weeks after microbead injection the number of GABA- and CR-positive cells that were also labeled with NB (coupled) was significantly reduced, whereas the number of NB-negative cells (uncoupled) remained unchanged from control levels. G,H: ChAT-positive dACs of both control and microbead-injected retinas were rarely labeled with NB indicating that they were not coupled to RGCs. I: At eight weeks after microbead injection the number of ChAT-positive dACs was comparable to that in control retinas. J: GlyT1-labeled ACs were found predominantly in the INL of control retinas. Only a very small number of NB-labeled cells in the INL (asterisks) were also GlyT1-positive (arrowhead), with majority of GlyT1-IR cells being NB-negative (arrows). K: There was no significant change in the number of GlyT1-positive ACs observed eight weeks after microbead injection as compared to control levels. L: Confocal image of a control retinal whole mount in which RGCs and their axons are retrograde labeled with NB delivered through an optic nerve cut. M: Histogram summarizes changes in the number of total dACs and those coupled or not coupled to RGCs in control and microbead-injected retinas. See text for details. Bars indicate mean value ± SEM. p*<0.05, p**<0.01, p***<0.001, Student’s t-test. Scale bars = 200 μm in L and 50 μm for all other micrographs.
In contrast, most ChAT-positive starburst ACs in the GCL of both control and microbead-injected retinas were NB-negative (Fig. 4G–I). This finding is consistent with previous studies reporting that only a small number, at best, of starburst ACs are coupled to RGCs via gap junctions in the mouse retina (Pang et al., 2013; Akopian et al., 2014). We found no change in the number of coupled and uncoupled ChAT-positive cells in the GCL at up to eight weeks after injection of microbeads (Fig. 4I)
As aforementioned, all the GlyT1-positive AC somata formed multiple rows at the proximal edge of INL (Fig. 4J, arrows). After injection of NB into the optic nerve we found a number of somata in the INL that were NB-positive, indicating ACs that were coupled to RGCs via gap junctions (Fig. 4J, asterisks). However, we found only a very small cohort of GlyT1-positive somata that were also labeled with NB (~7%, n=13/3), indicating that few glycinergic ACs were coupled to RGCs (Fig. 4J, arrowhead, K). Like for ChAT-positive ACs, we found no significant change in the number of coupled or uncoupled GlyT1-positive cells at up to eight weeks after induction of experimental glaucoma with microbead injection of the eye.
In the next phase of experiments we combined retrograde NB labeling with immunolabeling of the RGC marker Brn3a, and the counterstaining of cell nuclei with DAPI to clearly differentiate RGCs and dACs in the GCL. This approach allowed us to identify the total number of cells in the GCL (DAPI-positive nuclei), the total number of RGCs (Brn3a-positive cells), the total number of coupled (NB-positive/Brn3a-negative), and uncoupled (NB-negative/Brn3a-negative) dACs in control and glaucomatous retinas. Consistent with the results described above, we found that the total number of dACs was reduced by 27% at 8 weeks after microbead injection as compared to control values (n=20/3, p<0.01). Moreover, the loss of dACs coupled to RGCs reached 62% (n=20/3, p<0.01) at 8 weeks after microbead injection, whereas there was no significant loss of uncoupled dACs (n=20/3, p=0.07) (Fig. 4M). These data provided further support for the selective vulnerability of ACs coupled to RGCs in glaucomatous retinas.
It is important to note that the labeling of cells with NB was dependent on retrograde transport through the optic nerve, which may have been compromised in glaucomatous retinas, thereby affecting relative cell counts. To address this issue, we compared the percent of Brn3a-positive RGCs that were also NB-positive in control and microbead-injected retinas. We found that whereas 89.8% of Brn3a-positive RGCs were retrogradely labeled with NB in control retinas, this value was reduced to 77.5% in retinas at 8 weeks after initial microbead injection. These data suggest that retrograde transport was compromised in glaucomatous retinas, resulting in an approximate 12% overestimate of surviving NB-negative RGCs compared to controls and a corresponding overestimate of uncoupled (NB-negative) ACs. However, considering the tremendous difference (62%) we found in the survivability of uncoupled vs. coupled ACs in glaucoma, any misidentification of cell coupling due to the compromised retrograde transport would not impact our conclusion.
Effect of elevated IOP on the OP and STR components of the ERG
We next compared ERG components recorded from control, PBS sham-injected and microbead-injected retinas to determine whether there were physiological changes that correlated with the retinal injury and cellular losses found in glaucoma. We focused on the OPs and pSTRs believed to represent AC and AC/RGC activity, respectively (Wachtmeister, 1998; Saszik et al., 2002). Digitally filtered (50–300 Hz) raw data obtained from light stimuli of −0.4 log scot. cd-s/m2 generally produced 5–6 major OPs derived from the b-wave of scotopic ERG recordings (Fig. 5A,B). In comparison to control eyes (190.8 ± 17.4 μV, n=12), there was a significant 33% reduction in the OP amplitude at 4 weeks after microbead injection (128.1 ± 15.7 μV, n=12, p<0.05), with a further 6% reduction by 8 weeks (116.8 ± 16.3 μV, n=15, p<0.01) (Fig. 5C). We found a similar reduction in the amplitude of the pSTR in glaucomatous retinas when compared to control levels. For a selected flash intensity of −4.3 log scot. cd-s/m2, the pSTR amplitude was reduced by 34% at 4 weeks (14.8 ± 2.0 μV, n=12, p<0.05) and 53% at 8 weeks (10.5 ± 1.8 μV, n=13, p<0.001) after initial microbead injection as compared to control values (22.5 ± 0.9 μV, n=11) (Fig. 5D,E). Overall, the temporal course of the OP and pSTR reductions paralleled that of the loss of ACs and RGCs in retinas subjected to experimental glaucoma.
Figure 5.

Reduction in the amplitude of ERG oscillatory potentials (OPs) and the positive scotopic threshold response (pSTR) in experimental glaucoma. A,B: Representative scotopic ERG b-wave from dark-adapted control and microbead-injected eyes after eight weeks. The OPs peaks were extracted from first 150 ms of the b-wave (red). Digitally filtered (50Hz – 300Hz) OPs from control and microbead-injected retinas mice show a reduction in the 5 peak amplitudes, labeled as 1,2,3,4, and 5, respectively (red). C: Average of summed OP peak amplitudes ± SEM from control (Ctr) and 4, 5, and 8 weeks after microbead injection showing a clear reduction in amplitude. D: Representative STR responses of control and microbead-injected eyes at eight weeks. E: Bars indicate mean peak amplitude ± SEM. p*<0.05, p**<0.01, p***<0.001; Student’s t-test.
DISCUSSION
While the loss of RGC somata and axonal processes have been well characterized in glaucoma (Quigley, 1999; Chang and Goldberg, 2012; Tian et al., 2015), numerous human and animal studies have suggested that the overall insult extends to other neuronal types. The evidence includes thinning of layers throughout the retina and the apparent loss of bipolar, horizontal, amacrine, and photoreceptor cell somata and/or their synaptic processes (Grozdanic et al., 2003; Harazny et al., 2009; Heiduschka et al., 2010; Cuenca et al., 2010; Guo et al., 2010; Fernández-Sánchez et al., 2014; Georgiou et al., 2014). These extensive structural changes in glaucoma are supported by attenuation of several ERG components, which reflect both inner and outer retinal activity (Vaegan et al., 1995; Saszik et al., 2002; Harazny et al., 2009; Heiduschka et al., 2010; Frankfort et al., 2013; Porciatti, 2015). Consistent with these studies, we observed a reduction in the thickness of all retinal layers by eight weeks after intracameral microbead injection to elevate IOP. These changes were accompanied with a dramatic increase in the GFAP expression in astrocytes and Müller cell processes, indicative of gliosis, as previously reported for primate and rodent models of experimental glaucoma (Tanihara et al., 1997; Wang et al 2002; Woldemussie et al., 2004; Inman and Horner, 2007; Vidal et al., 2010).
A main focus of this study was to determine the extent of AC loss in glaucoma. While a number of studies have reported the loss of ACs in animal models of glaucoma, the reductions reported for different immunolabeled subpopulations have been inconsistent (May and Mittag, 2004; Moon et al., 2005; Hernandez et al., 2009; Kunzevitzky et al., 2010). Further, it was unclear whether the reductions in ACs reflected cell death and/or a decrease in the expression of the different markers (e.g., ChAT, CR, GABA) used to identify ACs (Kielczewski et al., 2005; Hernandez et al., 2009; Gunn et al., 2011). To differentiate dACs from RGCs, we combined labeling with the RGC marker Brn3a with the nuclear marker DAPI to compute the total cell count in the GCL. The finding that the reduction of DAPI-labeled nuclei eight weeks after induction of elevated IOP was greater than the loss of Brn3a-positive cells provided clear evidence for the loss of dACs in glaucomatous retinas. It is important to note that this result could not be explained by a reduction in Brn3a expression rather than RGC loss, as this would have resulted in a lower reduction in the number of DAPI-labeled cells.
Using a number of markers for ACs, we found that the subpopulations were differentially affected in our experimental model of glaucoma. Both CR- and GABA-positive ACs and dACs showed an approximate 30–50% loss in the INL and GCL that began at 4 weeks after the initial microbead injection. Interestingly, we found that the loss of CR- and GABA-positive cells in glaucoma was slightly, but consistently, greater in the GCL than in the INL. This likely reflects the fact that CR also labels a small cohort of RGCs (Haverkamp and Wässle, 2000; Mojumder et al., 2008) and that GABA label can be passed from ACs to RGCs through interconnecting gap junctions (Marc and Jones, 2002; Völgyi et al., 2009, Pang and Wu, 2013). Thus, our calculation of the CR- and GABA-positive cells in GCL lost in glaucoma likely included some RGCs in addition to dACs.
To determine if there was a functional correlation to the structural loss of ACs we recorded the OP and pSTR components of the ERG believed to correspond to AC and AC/RGC activity, respectively (Sazsik et al., 2002; Wachtmeister, 1998). Indeed, we found a significant reduction in the amplitude of these components that began at four weeks after the initial microbead injection, which was extended at eight weeks. Thus, functional impairment indicated by the attenuated ERG components followed a time course that paralleled the loss of CR- and GABA-positive cells. Taken together, our results strongly support the idea that the reduced CR and GABA immunolabeling in glaucomatous retinas reflected true cell loss, as suggested by earlier studies (May and Mittag, 2004; Moon et al., 2005; Hernandez et al., 2009), rather than a downregulation of antigen production as proposed by others (Kielczewski et al., 2005; Hernandez et al., 2009; Gunn et al., 2011).
In contrast, we found that ChAT-positive starburst ACs and glycinergic ACs positive for GlyT1 were not lost over the eight week period we examined tissue after induction of elevated IOP. These data support previous studies suggesting that subpopulations of ACs are differentially affected in glaucoma. A key question then is why is there such heterogeneity in AC loss? One idea stems from the recent work of Akopian et al. (2014), who showed that progressive cell death of both RGCs and ACs results to a large degree from the so-called bystander effect in which dying cells release toxic molecules via gap junctions, leading to the death of neighbors to which they are coupled. With the finding that 15 of the 22 subtypes of RGCs in the mouse retina are coupled to ACs via gap junctions (Völgyi et al., 2009), Akopian et al. (2014) posited that the bystander effect could be a major mechanism for progressive loss of RGCs and ACs under a number of degenerative insults. Further, they reported that whereas RGCs and CR-positive ACs were significantly lost under excitotoxic or ischemic conditions, ChAT-positive ACs were unaffected, paralleling the present results in glaucomatous retinas.
Using retrograde labeling with NB, we found that CR- and GABA-positive cells were coupled to RGCs, whereas both ChAT- and GlyT1-positive cells were rarely coupled, confirming earlier studies (Pang et al 2013; Akopian et al., 2014) and lending support to a bystander effect mechanism. In addition, our finding that the time course of dAC loss paralleled that of RGCs, while not definitive, is consistent with this scenario as the bystander effect would occur as RGCs are dying, but not structurally degenerated as this would compromise gap junction integrity.
Taken together with the current literature on cell death in glaucoma, the present results support the following hypothesis. Under glaucoma, the elevated IOP results initially in damage to vulnerable axons in the optic nerve that subsequently leads to a retrograde degeneration of RGC somata in the GCL (Calkins, 2012; Chang and Goldberg, 2012). This initiates a loss of ACs coupled to the dying RGCs via the gap junction-mediated bystander effect. Our results thus suggest that the majority of AC loss in glaucoma is a secondary consequence of RGC death, which, in turn, could lead to further RGC loss as dying ACs reverse the sequence of somatic damage. This idea is consistent with the proposal that most RGC death following optic nerve injury is associated with secondary death of neighboring cells not directly affected by the initial insult (Schwartz et al., 1996; Levkovitch-Verbin et al., 2001).
Secondary cell death mediated by neuronal gap junctions has been implicated previously in retinal cell loss associated with a number of degenerative conditions, including retinitis pigmentosa, ischemia, excitotoxicity, and glaucoma (Ripps, 2002; Malone et al., 2007; Kerr et al., 2011; Danesh-Meyer et al., 2012; Akopian et al., 2014). Interestingly, a low vulnerability of ChAT-immunoreactive and glycinergic ACs has been reported under a number of pathological situations (Williams et al 2001; Moon et al., 2005; Bernstein and Guo, 2011; Akopian et al 2014). Further, in a rat model of glaucoma, acetylcholine, presumably released from cholinergic starburst ACs, reduced RGC loss (Iwamoto et al., 2014; Mata et al., 2015), suggesting that these less vulnerable cells may provide some protection of neighboring RGCs against glaucomatous damage. Overall, the idea that the gap junction-mediated bystander effect forms a critical mechanism of progressive cell death is consistent with numerous studies of retinal neuron loss under a number of different pathological conditions. Gap junctions may thus represent important targets for novel neuroprotective strategies to prevent the progressive cell loss associated with glaucoma and other degenerative diseases of the retina.
In this study we show that the structural and functional integrity of retinal amacrine cells are compromised in a mouse model of glaucoma. Specific immunocytochemical markers were used to identify amacrine cells subpopulations in both the inner nuclear and ganglion cell layers. Calretinin- and GABA- immunoreactive cells were highly vulnerable to glaucomatous damage, whereas ChAT-positive and glycinergic AC subtypes were largely unaffected. This difference between amacrine cell subtypes appears related to their gap junctional coupling patterns. Amacrine cells that are coupled to ganglion cells were lost in glaucoma, whereas uncoupled amacrine cells were not. Our results suggest that amacrine cell loss in glaucoma occurs secondary to ganglion cell death through the gap junction-mediated bystander effect.
Acknowledgments
Funding: Supported by NIH Grants EY007360 and EY026024 to SAB.
Footnotes
CONFLICT OF INTEREST
The authors have no conflict of interest to declare.
AUTHOR CONTRIBUTIONS
Conceived and designed study – AA, SK, SAB; Carried out experiments – AA, SK, HR, SV; Analysis and interpretation of data – AA, SK, HR, SV, SAB; Wrote the manuscript – AA, SK, HR, SV, SAB
References
- Akopian A, Atlasz T, Pan F, Wong S, Zhang Y, Völgyi B, Paul DL, Bloomfield SA. Gap junction-mediated death of retinal neurons is connexin and insult specific: a potential target for neuroprotection. J Neurosci. 2014;34:10582–10591. doi: 10.1523/JNEUROSCI.1912-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alarcón-Martínez L, Avilés-Trigueros M, Galindo-Romero C, Valiente-Soriano J, Agudo-Barriuso M, de Villa PL, Villegas-Pérez MP, Vidal-Sanz M. ERG changes in albino and pigmented mice after optic nerve transection. Vision Res. 2010;50:2176–2187. doi: 10.1016/j.visres.2010.08.014. [DOI] [PubMed] [Google Scholar]
- Bayer AU, Danias J, Brodie S, Maag KP, Chen B, Shen F, Podos SM, Mittag TW. Electroretinographic abnormalities in a rat glaucoma model with chronic elevated intraocular pressure. Exp Eye Res. 2001;72:667–677. doi: 10.1006/exer.2001.1004. [DOI] [PubMed] [Google Scholar]
- Bernstein SL, Guo Y. Changes in cholinergic amacrine cells after rodent anterior ischemic optic neuropathy (rAION) Invest Ophthalmol Vis Sci. 2011;52:904–910. doi: 10.1167/iovs.10-5247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brecha N, Johnson D, Peichl L, Wässle H. Cholinergic amacrine cells of the rabbit retina contain glutamate decarboxylase and gamma-aminobutyrate immunoreactivity. Proc Natl Acad Sci U S A. 1988;85:6187–6191. doi: 10.1073/pnas.85.16.6187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calkins DJ. Critical pathogenic events underlying progression of neurodegeneration in glaucoma. Prog Retin Eye Res. 2012;31:702–719. doi: 10.1016/j.preteyeres.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang EE, Goldberg JL. Glaucoma 2.0: neuroprotection, neuroregeneration, neuroenhancement. Ophthalmology. 2012;119:979–986. doi: 10.1016/j.ophtha.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong RS, Martin KR. Glial cell interactions and glaucoma. Curr Opin Ophthalmol. 2015;26:73–77. doi: 10.1097/ICU.0000000000000125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuenca N, Pinilla I, Fernández-Sánchez L, Salinas-Navarro M, Alarcón-Martínez L, Avilés-Trigueros M, de la Villa P, Miralles de Imperial J, Villegas-Pérez MP, Vidal-Sanz M. Changes in the inner and outer retinal layers after acute increase of the intraocular pressure in adult albino Swiss mice. Exp Eye Res. 2010;91:273–285. doi: 10.1016/j.exer.2010.05.020. [DOI] [PubMed] [Google Scholar]
- Danesh-Meyer HV, Kerr NM, Zhang J, Eady EK, O’Carroll SJ, Nicholson LF, Johnson CS, Green CR. Connexin43 mimetic peptide reduces vascular leak and retinal ganglion cell death following retinal ischaemia. Brain. 2012;135:506–520. doi: 10.1093/brain/awr338. [DOI] [PubMed] [Google Scholar]
- Danias J, Shen F, Kavalarakis M, Chen B, Goldblum D, Lee K, Zamora MF, Su Y, Brodie SE, Podos SM, Mittag T. Characterization of retinal damage in the episcleral vein cauterization rat glaucoma model. Exp Eye Res. 2006;82:219–228. doi: 10.1016/j.exer.2005.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Sánchez L, Pérez de Sevilla Müller L, Brecha NC. Loss of outer retinal neurons and circuitry alterations in the DBA/2J mouse. Ret Cell Biol. 2014;55:6059–6072. doi: 10.1167/iovs.14-14421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Formichella CR, Abella SK, Sims SM, Cathcart HM, Sappington RM. Astrocyte Reactivity: A Biomarker for Retinal Ganglion Cell Health in Retinal Neurodegeneration. J Clin Cell Immunol. 2014;5 doi: 10.4172/2155-9899.1000188. pii: 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankfort BJ, Khan AK, Tse DY, Chung I, Pang JJ, Yang Z, Gross RL, Wu SM. Elevated intraocular pressure causes inner retinal dysfunction before cell loss in a mouse model of experimental glaucoma. Invest Ophthalmol Vis Sci. 2013;54:762–770. doi: 10.1167/iovs.12-10581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galindo-Romero C, Avilés-Trigueros M, Jiménez-López M, Valiente-Soriano FJ, Salinas Navarro M, Nadal-Nicolás F, Villegas-Pérez MP, Vidal-Sanz M, Agudo-Barriuso M. Axotomy-induced retinal ganglion cell death in adult mice: quantitative and topographic time course analyses. Exp Eye Res. 2011;92:377–387. doi: 10.1016/j.exer.2011.02.008. [DOI] [PubMed] [Google Scholar]
- Georgiou AL, Guo L, Francesca Cordeiro M, Salt TE. Electroretinogram and visual-evoked potential assessment of retinal and central visual function in a rat ocular hypertension model of glaucoma. Curr Eye Res. 2014;39:472–486. doi: 10.3109/02713683.2013.848902. [DOI] [PubMed] [Google Scholar]
- Grozdanic SD1, Betts DM, Sakaguchi DS, Allbaugh RA, Kwon YH, Kardon RH. Laser-induced mouse model of chronic ocular hypertension. Invest Ophthalmol Vis Sci. 2003;44:4337–4346. doi: 10.1167/iovs.03-0015. [DOI] [PubMed] [Google Scholar]
- Gunn DJ, Gole GA, Barnett NL. Specific amacrine cell changes in an induced mouse model of glaucoma. Clin Experiment Ophthalmol. 2011;39:555–563. doi: 10.1111/j.1442-9071.2010.02488.x. [DOI] [PubMed] [Google Scholar]
- Guo L, Normando EM, Nizari S, Lara D, Cordeiro MF. Tracking longitudinal retinal changes in experimental ocular hypertension using the cSLO and spectral domain-OCT. Invest Ophthalmol Vis Sci. 2010;51:6504–6513. doi: 10.1167/iovs.10-5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harazny J, Scholz M, Buder T, Lausen B, Kremers J. Electrophysiological deficits in the retina of the DBA/2J mouse. Doc Ophthalmol. 2009;119:181–197. doi: 10.1007/s10633-009-9194-5. [DOI] [PubMed] [Google Scholar]
- Haverkamp S, Wässle H. Immunocytochemical analysis of the mouse retina. J Comp Neurol. 2000;424:1–23. [PubMed] [Google Scholar]
- Heiduschka P, Julien S, Schuettauf F, Schnichels S. Loss of retinal function in aged DBA/2J mice - New insights into retinal neurodegeneration. Exp Eye Res. 2010;91:779–783. doi: 10.1016/j.exer.2010.09.001. [DOI] [PubMed] [Google Scholar]
- Hernandez M, Rodriguez FD, Sharma SC, Vecino E. Immunohistochemical changes in rat retinas at various time periods of elevated intraocular pressure. Mol Vis. 2009;15:2696–2709. [PMC free article] [PubMed] [Google Scholar]
- Holopigian K, Seiple W, Mayron C, Koty R, Lorenzo M. Electrophysiological and psychophysical flicker sensitivity in patients with primary open-angle glaucoma and ocular hypertension. Invest Ophthalmol Vis Sci. 1990;31:1863–1868. [PubMed] [Google Scholar]
- Huberman AD, El-Danaf RN. Blindness: Assassins of eyesight. Nature. 2015;527:456–457. doi: 10.1038/527456a. [DOI] [PubMed] [Google Scholar]
- Inman DM, Horner PJ. Reactive nonproliferative gliosis predominates in a chronic mouse model of glaucoma. Glia. 2007;55:942–953. doi: 10.1002/glia.20516. [DOI] [PubMed] [Google Scholar]
- Iwamoto K, Birkholz P, Schipper A, Mata D, Linn DM, Linn CL. A nicotinic acetylcholine receptor agonist prevents loss of retinal ganglion cells in a glaucoma model. Invest Ophthalmol Vis Sci. 2014;552:1078–1087. doi: 10.1167/iovs.13-12688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobs TC, Libby RT, Ben Y, John SW, Masland RH. Retinal ganglion cell degeneration is topological but not cell type specific in DBA/2J mice. J Cell Biol. 2005;171:313–325. doi: 10.1083/jcb.200506099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon CJ, Strettoi E, Masland RH. The major cell populations of the mouse retina. J Neurosci. 1998;18:8936–8946. doi: 10.1523/JNEUROSCI.18-21-08936.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr NM, Johnson CS, Green CR, Danesh-Meyer HV. Gap junction protein connexin43 (GJA1) in the human glaucomatous optic nerve head and retina. J Clin Neurosci. 2011;18:102–108. doi: 10.1016/j.jocn.2010.06.002. [DOI] [PubMed] [Google Scholar]
- Kielczewski JL, Pease ME, Quigley HA. The effect of experimental glaucoma and optic nerve transection on amacrine cells in the rat retina. Invest Ophthalmol Vis Sci. 2005;46:3188–3196. doi: 10.1167/iovs.05-0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb H. Amacrine cells of the mammalian retina: neurocircuitry and functional roles. Eye (Lond) 1997;11:904–923. doi: 10.1038/eye.1997.230. [DOI] [PubMed] [Google Scholar]
- Kunzevitzky NJ, Almeida MV, Goldberg JL. Amacrine cell gene expression and survival signaling: differences from neighboring retinal ganglion cells. Invest Ophthalmol Vis Sci. 2010;51:3800–3812. doi: 10.1167/iovs.09-4540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei Y, Garrahan N, Hermann B, Becker DL, Hernandez MR, Boulton ME, Morgan JE. Quantification of retinal transneuronal degeneration in human glaucoma: a novel multiphoton-DAPI approach. Invest Ophthalmol Vis Sci. 2008;49:1940–1945. doi: 10.1167/iovs.07-0735. [DOI] [PubMed] [Google Scholar]
- Levkovitch-Verbin H, Quigley HA, Kerrigan-Baumrind LA, D’Anna SA, Kerrigan D, Pease ME. Optic nerve transection in monkeys may result in secondary degeneration of retinal ganglion cells. Invest Ophthalmol Vis Sci. 2001;42:975–982. [PubMed] [Google Scholar]
- Lin B, Masland RH. Populations of wide-field amacrine cells in the mouse retina. J Comp Neurol. 2006;499:797–809. doi: 10.1002/cne.21126. [DOI] [PubMed] [Google Scholar]
- MacNeil MA, Masland RH. Extreme diversity among amacrine cells: implications for function. Neuron. 1998;20:971–982. doi: 10.1016/s0896-6273(00)80478-x. [DOI] [PubMed] [Google Scholar]
- Malone P, Miao H, Parker A, Juarez S, Hernandez MR. Pressure induces loss of gap junction communication and redistribution of connexin 43 in astrocytes. Glia. 2007;55:1085–1098. doi: 10.1002/glia.20527. [DOI] [PubMed] [Google Scholar]
- Marc RE, Jones BW. Molecular phenotyping of retinal ganglion cells. J Neurosci. 2002;22:413–427. doi: 10.1523/JNEUROSCI.22-02-00413.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masland RH. The many roles of starburst amacrine cells. Trends Neurosci. 2005;28:395–396. doi: 10.1016/j.tins.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Masland RH. The neuronal organization of the retina. Neuron. 2012;76:266–280. doi: 10.1016/j.neuron.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata D, Linn DM, Linn CL. Retinal ganglion cell neuroprotection induced by activation of alpha7 nicotinic acetylcholine receptors. Neuropharmacology. 2015;99:337–346. doi: 10.1016/j.neuropharm.2015.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May CA, Mittag T. Neuronal nitric oxide synthase (nNOS) positive retinal amacrine cells are altered in the DBA/2NNia mouse, a murine model for angle-closure glaucoma. J Glaucoma. 2004;13:496–499. doi: 10.1097/01.ijg.0000137435.83307.fd. [DOI] [PubMed] [Google Scholar]
- Menger N, Pow DV, Wässle H. Glycinergic amacrine cells of the rat retina. J Comp Neurol. 1998;401:34–46. doi: 10.1002/(sici)1096-9861(19981109)401:1<34::aid-cne3>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Mojumder DK, Wensel TG, Frishman LJ. Subcellular compartmentalization of two calcium binding proteins, calretinin and calbindin-28 kDa, in ganglion and amacrine cells of the rat retina. Mol Vis. 2008;14:1600–1613. [PMC free article] [PubMed] [Google Scholar]
- Moon JI, Kim IB, Gwon JS, Park MH, Kang TH, Lim EJ, Choi KR, Chun MH. Changes in retinal neuronal populations in the DBA/2J mouse. Cell Tissue Res. 2005;320:51–59. doi: 10.1007/s00441-004-1062-8. [DOI] [PubMed] [Google Scholar]
- Nadal-Nicolás FM, Jiménez-López M, Sobrado-Calvo P, Nieto-López L, Cánovas-Martínez I, Salinas-Navarro M, Vidal-Sanz M, Agudo M. Brn3a as a marker of retinal ganglion cells: qualitative and quantitative time course studies in naive and optic nerve-injured retinas. Invest Ophthalmol Vis Sci. 2009;50:3860–3868. doi: 10.1167/iovs.08-3267. [DOI] [PubMed] [Google Scholar]
- Pang JJ, Paul DL, Wu SM. Survey on amacrine cells coupling to retrograde-identified ganglion cells in the mouse retina. Invest Ophthalmol Vis Sci. 2013;54:5151–5162. doi: 10.1167/iovs.13-11774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez De Sevilla Müller L, Shelley J, Weiler R. Displaced amacrine cells of the mouse retina. J Comp Neurol. 2007;505:177–189. doi: 10.1002/cne.21487. [DOI] [PubMed] [Google Scholar]
- Porciatti V. Electrophysiological assessment of retinal ganglion cell function. Exp Eye Res. 2015;141:164–170. doi: 10.1016/j.exer.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pourcho RG, Goebel DJ. Immunocytochemical demonstration of glycine in retina. Brain Res. 1985;348:339–342. doi: 10.1016/0006-8993(85)90453-6. [DOI] [PubMed] [Google Scholar]
- Pow DV, Hendrickson AE. Distribution of the glycine transporter glyt-1 in mammalian and nonmammalian retinae. Vis Neurosci. 1999;16:231–239. doi: 10.1017/s0952523899162047. [DOI] [PubMed] [Google Scholar]
- Quigley HA. Neuronal death in glaucoma. Prog Retin Eye Res. 1999;18:39–57. doi: 10.1016/s1350-9462(98)00014-7. [DOI] [PubMed] [Google Scholar]
- Ripps H. Cell death in retinitis pigmentosa: gap junctions and the ‘bystander’ effect. Exp Eye Res. 2002;74:327–336. doi: 10.1006/exer.2002.1155. [DOI] [PubMed] [Google Scholar]
- Sappington RM, Carlson BJ, Crish SD, Calkins DJ. The microbead occlusion model: a paradigm for induced ocular hypertension in rats and mice. Invest Ophthalmol Vis Sci. 2010;51:207–216. doi: 10.1167/iovs.09-3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saszik SM, Robson JG, Frishman LJ. The scotopic threshold response of the dark-adapted electroretinogram of the mouse. J Physiol. 2002;543:899–916. doi: 10.1113/jphysiol.2002.019703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlamp CL, Montgomery AD, Mac Nair CE, Schuart C, Willmer DJ, Nickells RW. Evaluation of the percentage of ganglion cells in the ganglion cell layer of the rodent retina. Mol Vis. 2013;19:1387–1396. [PMC free article] [PubMed] [Google Scholar]
- Schwartz M, Belkin M, Yoles E, Solomon A. Potential treatment modalities for glaucomatous neuropathy: neuroprotection and neuroregeneration. J Glaucoma. 1996;5:427–432. [PubMed] [Google Scholar]
- Schwartz M. Neuroprotection as a treatment for glaucoma: pharmacological and immunological approaches. Eur J Ophthalmol. 2003;13:S27–31. doi: 10.1177/112067210301303s05. [DOI] [PubMed] [Google Scholar]
- Stockton RA, Slaughter MM. B-wave of the electroretinogram. A reflection of ON bipolar cell activity. J Gen Physiol. 1989;93:101–122. doi: 10.1085/jgp.93.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanihara H, Hangai M, Sawaguchi S, Abe H, Kageyama M, Nakazawa F, Shirasawa E, Honda Y. Up-regulation of glial fibrillary acidic protein in the retina of primate eyes with experimental glaucoma. Arch Ophthalmol. 1997;115:752–756. doi: 10.1001/archopht.1997.01100150754011. [DOI] [PubMed] [Google Scholar]
- Tian K, Shibata-Germanos S, Pahlitzsch M, Cordeiro MF. Current perspective of neuroprotection and glaucoma. Clin Ophthalmol. 2015;9:2109–2118. doi: 10.2147/OPTH.S80445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaegan, Graham SL, Goldberg I, Buckland L, Hollows FC. Flash and pattern electroretinogram changes with optic atrophy and glaucoma. Exp Eye Res. 1995;60:697–706. doi: 10.1016/s0014-4835(05)80011-9. [DOI] [PubMed] [Google Scholar]
- Vaney DI, Young HM. GABA-like immunoreactivity in cholinergic amacrine cells of the rabbit retina. Brain Res. 1988;438:369–373. doi: 10.1016/0006-8993(88)91366-2. [DOI] [PubMed] [Google Scholar]
- Vaney DI. The mosaic of amacrine cells in the mammalian retina. In: Osborne N, Chader J, editors. Progress in Retinal Research. Pergamon Press; Oxford: 1990. pp. 49–100. [Google Scholar]
- Velten IM, Horn FK, Korth M, Velten K. The b-wave of the dark adapted flash electroretinogram in patients with advanced asymmetrical glaucoma and normal subjects. Br J Ophthalmol. 2001;85:403–409. doi: 10.1136/bjo.85.4.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal L, Díaz F, Villena A, Moreno M, Campos JG, Pérez de Vargas I. Reaction of Müller cells in an experimental rat model of increased intraocular pressure following timolol, latanoprost and brimonidine. Brain Res Bull. 2010;82:18–24. doi: 10.1016/j.brainresbull.2010.02.011. [DOI] [PubMed] [Google Scholar]
- Voigt T. Cholinergic amacrine cells in the rat retina. J Comp Neurol. 1986;248:19–35. doi: 10.1002/cne.902480103. [DOI] [PubMed] [Google Scholar]
- Völgyi B, Chheda S, Bloomfield SA. Tracer coupling patterns of the ganglion cell subtypes in the mouse retina. J Comp Neurol. 2009;512:664–687. doi: 10.1002/cne.21912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachtmeister L. Oscillatory potentials in the retina: what do they reveal? Prog Retin Eye Res. 1998;17:485–521. doi: 10.1016/s1350-9462(98)00006-8. [DOI] [PubMed] [Google Scholar]
- Wang L, Cioffi GA, Cull G, Dong J, Fortune B. Immunohistologic evidence for retinal glial cell changes in human glaucoma. Invest Ophthalmol Vis Sci. 2002;43:1088–1094. [PubMed] [Google Scholar]
- Wang X, Tay SS, Ng YK. An immunohistochemical study of neuronal and glial cell reactions in retinae of rats with experimental glaucoma. Exp Brain Res. 2000;132:476–484. doi: 10.1007/s002210000360. [DOI] [PubMed] [Google Scholar]
- Wässle H, Boycott BB. Functional architecture of the mammalian retina. Physiol Rev. 1991;71:447–480. doi: 10.1152/physrev.1991.71.2.447. [DOI] [PubMed] [Google Scholar]
- Williams RR, Cusato K, Raven MA, Reese BE. Organization of the inner retina following early elimination of the retinal ganglion cell population: effects on cell numbers and stratification patterns. Vis Neurosci. 2001;18:233–244. doi: 10.1017/s0952523801182088. [DOI] [PubMed] [Google Scholar]
- Woldemussie E, Wijono M, Ruiz G. Müller cell response to laser-induced increase in intraocular pressure in rats. Glia. 2004;47:109–119. doi: 10.1002/glia.20000. [DOI] [PubMed] [Google Scholar]
- Yassa HD. Age-related changes in the optic nerve of Sprague-Dawley rats: an ultrastructural and immunohistochemical study. Acta Histochem. 2014;116:1085–1095. doi: 10.1016/j.acthis.2014.05.001. [DOI] [PubMed] [Google Scholar]