

Abstract
Alzheimer’s disease (AD) is the most common form of neurodegenerative disorder with dementia, accounting for approximately 70% of the all cases. Currently, 5.8 million people in the U.S. are living with AD and by 2050 this number is expected to double resulting in a significant socio-economic burden. Despite intensive research, the exact mechanisms that trigger AD are still not known and at the present there is no cure for it. In recent years, many signaling pathways associated with AD neuropathology have been explored as possible candidate targets for the treatment of this condition including glycogen synthase kinase-3β (GSK3-β). GSK3-β is considered a key player in AD pathophysiology since dysregulation of this kinase influences all the major hallmarks of the disease including: tau phosphorylation, amyloid-β production, memory, neurogenesis and synaptic function. The present review summarizes the current understanding of the GSK3-β neurobiology with particular emphasis on its effects on specific signaling pathways associated with AD pathophysiology. Moreover, it discusses the feasibility of targeting GSK3-β for AD treatment and provides a summary of the current research effort to develop GSK3-β inhibitors in preclinical and clinical studies.
Introduction
Alzheimer’s disease (AD) is the most common form of age-related neurodegenerative disorder with dementia. Currently, the estimated number of people living with this disease in the United Sates is 5.8 million, a number projected to increase rapidly in the coming years due to the aging of the baby-boomer generation and the lack of a cure [1]. In fact, the number of Americans age 65 and older is predicted to double from 52 million in 2018 to 95 million by 2060 representing a potentially substantial social and economic burden for the future society [1,2].
Clinically, AD is characterized by progressive loss of memory and cognitive functions. Early symptoms include difficulty recalling recent conversations, names or events. With the progression of the pathology, patients develop impaired communication skills, confusion, behavioral changes, poor judgment and often experience passiveness and depression [1,3]. The pathological hallmarks of this disorder are extracellular accumulation of amyloid-β (Aβ) plaques composed of Aβ peptides, neurofibrillary tangles (NFTs) composed by hyperphosphorylated tau protein, brain inflammation and atrophy. These changes are thought to begin years before the clinical symptoms are noticeable [1, 3]. With the exception of the familial form which is the result of mutations on specific genes such as the Aβ precursor protein (APP), the presenilin 1 (PS1) and PS2 and which only account for approximately 2–4% of the total cases, AD is mainly a sporadic disorder whose etiology is currently unknown [4]. In fact, despite intensive research, the mechanisms responsible for Aβ accumulation and NFTs formation and deposition are not understood and there is not effective treatment for this condition. For this reason, in the past years several signaling pathways relevant to some of the pathophysiological processes have been investigated and evaluated as possible targets of novel therapeutic approaches for AD treatment.
Glycogen synthase kinase-3 (GSK3) is a ubiquitously expressed and constitutively active serine-threonine kinase involved in the regulation of many key cell biology pathways, some of which have been also implicated in neurodegeneration. There are two isoforms of GSK3, GSK3-α and GSK3-β encoded by two different genes [5]. In the central nervous system (CNS), GSK3-β is the most abundant and its expression levels are known to increase with age [6]. GSK3-β is found hyperactive in the brain of AD patients and compelling evidence supports its contribution to AD pathology by distinct mechanisms [7]. Dysregulation of this kinase have been observed to affect both Aβ and tau metabolism and toxicity in vitro and in vivo AD models. Additionally, GSK3-β activity has been linked to memory consolidation, neurogenesis, synaptic plasticity, long term potentiation and inflammation (Figure 1) [8].
Figure 1. Role of GSK3-β signaling in AD pathology.
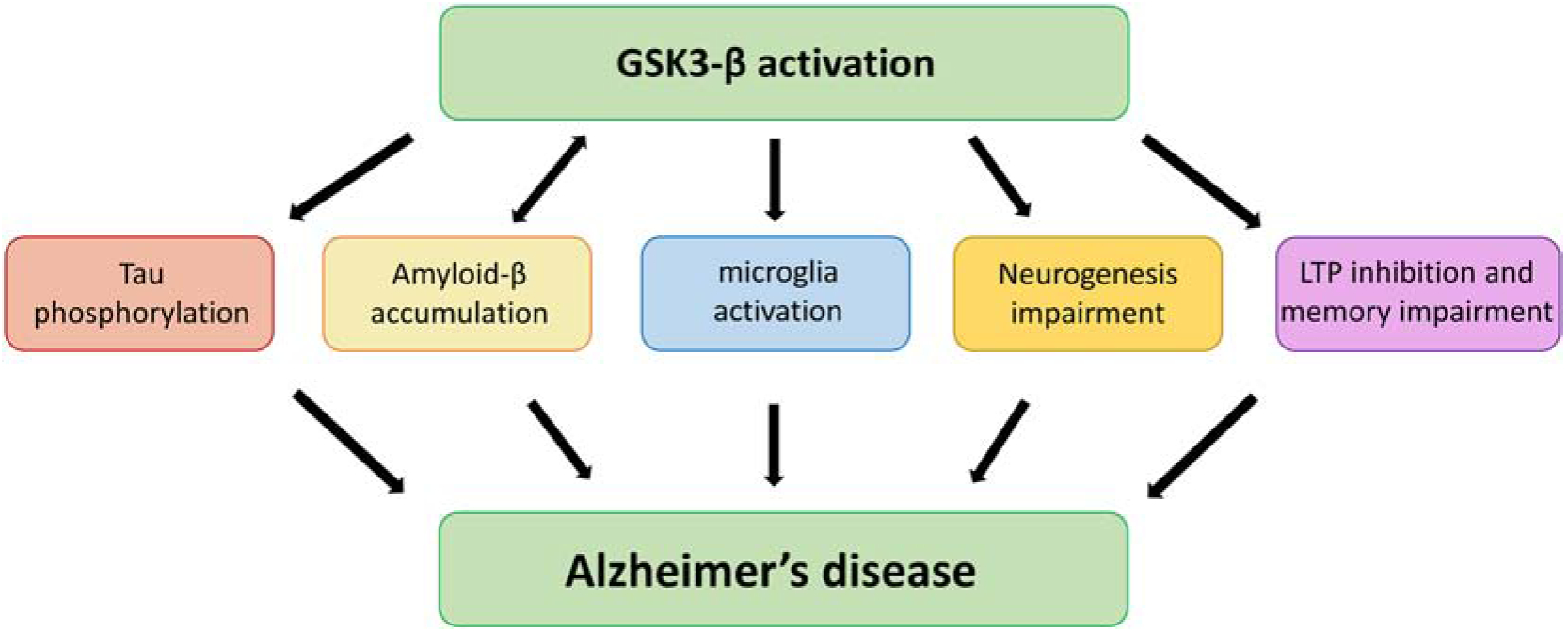
Schematic representation of the potential involvement of GSK3-β in different aspects and pathways relevant to the onset and development of Alzheimer’s disease neuropathology. 1) GSK3-β activation contributes to neurodegeneration by directly promoting tau hyper-phosphorylation. Hyperphosphorylated tau dissociates from the microtubules leading to impaired axonal transport, NFTs formation, neuronal and synaptic dysfunction. 2) GSK3-β promotes amyloid production and accumulation which induces apoptosis and neuronal damage in AD. 3) GSK3-β also displays pro-inflammatory functions; it regulates the biological response of microglia, primary immune cells of the CNS and promotes production of inflammatory molecules. 4) Additionally, GSK3-β play a critical role in the regulation of hippocampal neurogenesis, an important process that supports specific form of learning and memory and known to be affected in AD pathology. 5) Finally, GSK3-β is also involved in synaptic plasticity and memory. Overactivation of this kinase has been linked to inhibition of hippocampal long-term potentiation (LTP), a mechanism required for memory formation.
Given the relevance of GSK3-β signaling to the pathophysiology of AD, the therapeutic potential of its inhibition has been widely investigated in preclinical and clinical studies. This review will focus on GSK3-β physiological function, current data regarding its aberrant activity in AD, and the evidence supporting the beneficial effect of GSK3-β modulation as therapeutic strategy for the treatment of AD.
GSK3β biological function
GSK3 is a kinase that phosphorylates serine and threonine sites of a variety of substrates [9]. It was originally discovered in the 1980s and identified as one of the kinases that modulates glycogen synthase activity through phosphorylation leading to its inhibition [10]. Extracted from the rabbit skeletal muscle [10], GSK3 has been implicated in the regulation of several vital cellular processes including cell apoptosis, differentiation, proliferation and the microtubule morphology [11]. GSK3 is constitutively active and abundantly expressed in the CNS [9]. As the research on GSK3 has expanded greatly following its discovery, two isozymes have been identified, GSK3-α and GSK3-β, which although are coded by different genes on chromosomes 19 and 3 share a striking 98% similarity in structure [12]. Contrary to their similar structure, these isozymes are known to act on different substrates [13]. In fact, while in-vivo studies demonstrate that the complete elimination of GSK3-β can be sufficient to cause death in embryo [14], GSK3-α genetic deletion is found to have no significant impact on survival [15].
Although, constitutively active, GSK3-β function can be regulated by phosphorylation and de-phosphorylation on different sites. For instance, auto-phosphorylation on tyrosine-279/216 mediates GSK3-β activation, while phosphorylation on serine sites 21/9 by AKT, protein kinase A and B (PKA-PKB) in the N-terminal which leads to its inhibition [16]. Moreover, the phosphate groups added to GSK3-β during phosphorylation are removed by protein phosphatase PP2A resulting in GSK3-β reactivation (Figure 2) [17]. From postmortem studies to in-vivo and in-vitro studies, in recent years this kinase has emerged as a potentially active player in several mechanisms relevant to neurodegeneration [18–20].
Figure 2. Regulation of GSK3-β activity.
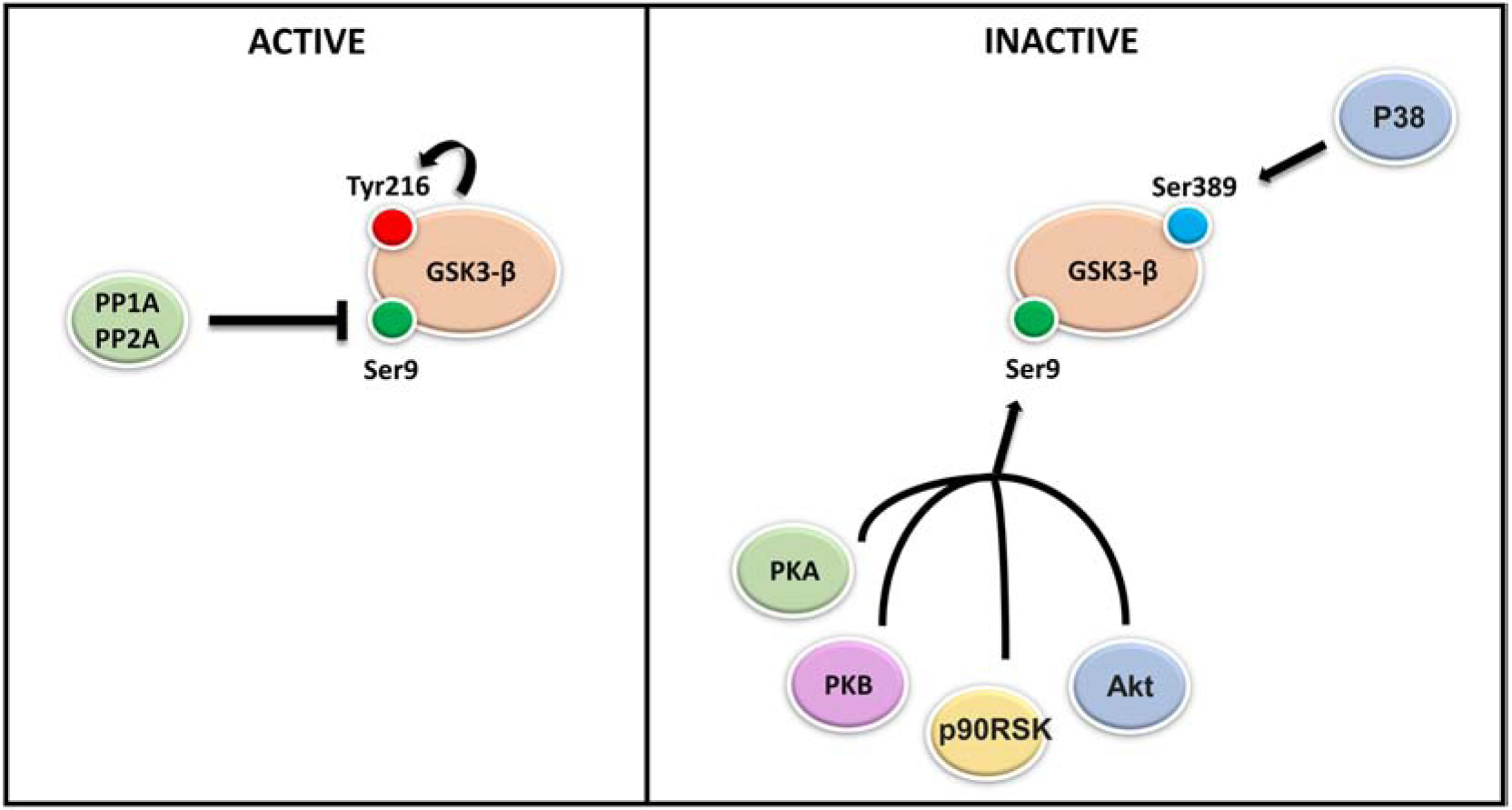
GSK3-β exists in an active and in inactive form depending on its phosphorylation status. Two families of enzymes modulate GSK3-β activity: Serine/Threonine and Tyrosine kinases which attach phosphate groups to GSK3-β and phosphatases which remove phosphate groups from GSK3-β protein in response to external stimuli. In vitro and in vivo evidence have shown that, GSK3-β activation is mediated by auto-phosphorylation on tyrosine-216, while its inhibition is mediated by phosphorylation on serine 9 by several kinases including: AKT, protein kinase A and B (PKA-PKB) and P90RSK. GSK3-β phosphorylation on serine 389 by P38 MAPK kinase instead, has been shown to inhibit GSK3-β. Additionally, the phosphate groups added to GSK3-β during phosphorylation can be removed by protein phosphatase PP2A resulting in GSK3-β re-activation.
GSK3β and Aβ
In vitro and in vivo evidence has shown that GSK3-β activation is involved in Aβ formation and accumulation in the AD brain by modulating the cleavage of APP. In the brain two different pathways, non-amyloidogenic and amyloidogenic, are involved in the cleavage of APP through the action of different proteases [21]. The non-amyloidogenic cleavage of APP is done by α-secretase complex consisting of ADAM-10 and −17 and the γ-secretase. The cleavage done by α-secretase produces peptides that are easily degraded [22]. By contrast, in the amyloidogenic pathway APP is initially cleaved by the β-secretase (BACE-1) enzyme followed by the γ-secretase complex processing which causes the generation of Aβ peptides, which tend to fibrilize and oligomerize and eventually accumulate as Aβ deposits in the AD brain. Previous studies have identified APP and PS1, one of the catalytic components of γ-secretase complex [21], as GSK3-β substrates, suggesting that APP cleavage and PS1 function are affected by elevated GSK3-β activity causing higher Aβ production and subsequent deposition in the AD brain (Figure 3) [23,24].
Figure 3. GSK3-β interferes with Aβ and tau metabolism.
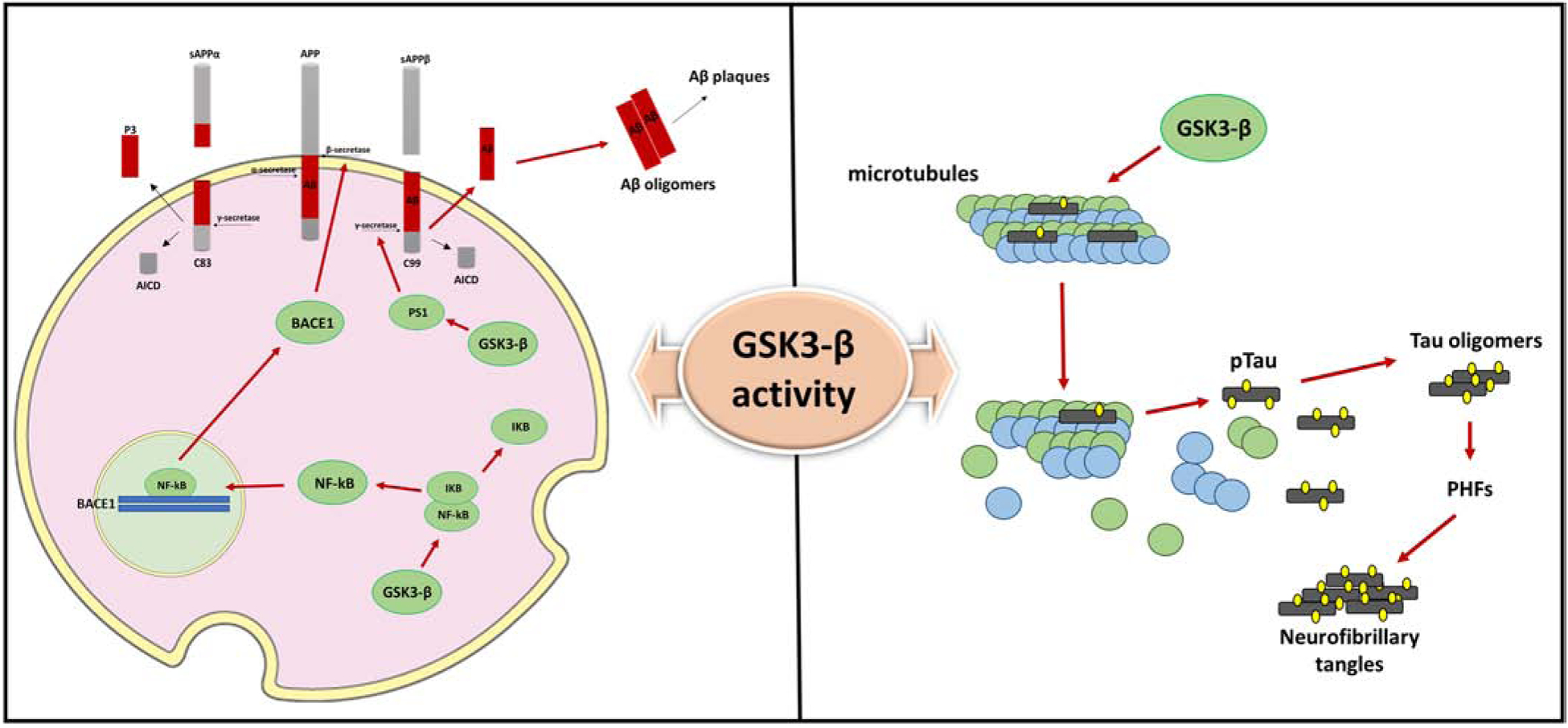
Increased activity of GSK3-β promotes Aβ formation via two distinct mechanisms: GSK3-β induces BACE1 gene expression via upregulation of NFK-β signaling and modulates γ-secretase activity. 1) The human BACE1 promoter region contains two functional NF-kB-binding sites. In vitro and in vivo evidence have shown that GSK3-β activation is involved in NFK-β/p65 nuclear translocation and binding to the BACE1 promoter sites, ultimately resulting in increased BACE1 protein levels and BACE1 mediated-APP processing and Aβ production. 2) GSK3-β modulates γ-secretase activity by direct interaction and regulation of PS1 activity and cellular localization. Upon GSK3-β phosphorylation, PS1 shows lower binding affinity for N-cadherin and reduced cell-surface expression which alter PS1/γ-secretase substrate specificity.
Additionally, GSK3-β is one of the major kinases involved in tau phosphorylation. The addition of a phosphate group on the specific Thr231 residue of tau protein leads to microtubules disassembly and promotes formation of tau oligomers and NFTs contributing to neuronal dysfunction and degeneration.
Some studies exploring the PS1 and GSK3-β relationship suggest that GSK3-β over-activation alters the function of PS1 as well its localization in the CNS [24]. PS1 is localized in Golgi membranes, endoplasmic reticulum and plasma membrane. This gene product is associated with N-cadherin/beta-catenin which together forms a synaptic trimeric complex consisting of serine residues that are subject to GSK3-β phosphorylation. Over-activated GSK3-β can downregulate the efficiency of the PS1/N-cadherin/beta-catenin complex causing deficiencies in synaptic and neuronal viability that leads to AD pathology [24]. Involvement of GSK3-β in plaque formation has been also linked to BACE1 over-activation. Studies exploring the connection between GSK3-β and BACE1 revealed that GSK3-β over-activation or overexpression promotes BACE1 enzymatic cleavage of APP. Moreover, BACE1 expression has been shown to be mediated by GSK3-β, however this event requires NFK-beta signaling in order for BACE1 transcription to take place [16]. Studies showed that both BACE1 and NFK-β expression are upregulated in AD patients and that NFK-β regulates the GSK3-β-induced transcription of BACE1 through manipulation of the cis-acting p65-binding promoter causing abnormal Aβ production (Figure3) [16, 25]. In support of these observations, the inhibition of GSK3-β in APP mutant cell lines has proven to be an effective way to reduce the BACE1 cleavage of APP, which then result in a significant decrease of both Aβ 40–42 peptide levels [26,27].
Beside the direct effect on APP, GSK3-β activity can also induce Aβ pathology via disruption of the insulin signaling pathway. In vivo studies done using insulin-deficient mice models have revealed a positive correlation between Aβ and GSK3-β levels [28]. Additionally, insulin degrading enzyme protein (IDE), a major player for Aβ degradation, has also been identified to be negatively correlated with GSK3-β levels in diabetic mice [28]. Moreover, other findings suggest that brain tissues of insulin deficient rodent models show increased GSK3-β and Aβ levels while IDE is significantly down-regulated [29–31].
Interestingly, some studies have proposed a feedback loop interaction between Aβ and GSK3-β activation in certain pathways that may ultimately lead to tau hyper-phosphorylation. Moreover, Aβ can disrupt the normal Wnt pathway activity that prevents Wnt from inhibiting the GSK3-β [32], and directly activate GSK3- β which will then promote tau phosphorylation [33]. Taken together these findings would support the hypothesis that GSK3-β can be considered as the biological link for both AD neuropathologies (i.e., Aβ and tau) [21].
GSK3β and tau
Tau is a microtubule-associated protein (MAP) that is known to stabilize the microtubules by securing tubulin assembly [34]. Tau is encoded by the MAPT gene located on chromosome 17 and consists of six isoforms generated by alternative splicing [18,19]. Tau function and affinity for the microtubules mainly depends on its phosphorylation status. In AD, tau is hyper-phosphorylated and accumulates in the cytoplasm leading to microtubules disassembly, loss of neuronal integrity and eventually NFTs formation [35]. The majority of early studies on tau hyper-phosphorylation explored the kinase-phosphatase imbalance in the brain identifying GSK3-β as a major tau kinase that may be involved in the development of the AD tau pathology [16, 20]. There are a considerable number of amino-acid residues of the tau protein that are subject to GSK3-β phosphorylation [25]. The localization of the tau phosphorylation sites has also been of interest in AD research. GSK3-β mediated phosphorylation of tau protein is seen in areas that are in close proximity to microtubule binding domains and their amino acid residues [36]. Protein-protein interactions are known to take place in these binding domains, therefore GSK3-β induced tau phosphorylation is prone to self-aggregate in a toxic manner (Figure 3) [33]. One study using a transgenic Drosophila model successfully demonstrated that the aberrant upregulation of GSK3-β positively correlates with accumulation of toxic tau aggregates [36]. Consistent with the in-vivo findings, in-vitro studies also show that human neuroblastoma cells display tau-filament aggregates resulting from GSK3-β mediated phosphorylation strikingly similar to AD tau pathology [37]. For this reason, and because in vitro and in vivo studies have established that GSK3-β inhibition restores hyper-phosphorylation of tau, the majority of drug discovery and development programs against tau pathology tends to target GSK3-β activity [38–40].
Sex differences in GSK3β activity and signaling pathway
Although current knowledge on sex differences for this kinase is insufficient, few studies have explored the effects of male and female hormones on GSK3β activity and AD-related signaling pathways. In the brain of C57Bl6/J mice, activation of the 17β-estradiol pathway has been associated with GSK3β inactivation through serine-9 phosphorylation, and therefore with a significant reduction in Aβ accumulation along with decreases in hyper-phosphorylated tau protein [41], suggesting that estrogen can lower GSK3β initiated p-tau (phosphorylated tau) levels and APP processing. Moreover, 17β-estradiol also prevented GSK3β induced neuronal apoptosis by inducing phosphoinositol-3 kinase (PI-3K) and MAPK3 transduction in hippocampal slice culture [41]. Notably, another study exploring the changes in BACE and GSK3β mRNA levels in AD patients reported a 7% downregulation in BACE mRNA in female post-mortem brain tissue compared to the male subjects but unfortunately, when GSK3β was measured, its levels were below the limits of reliable detection and conclusion could not be drawn [42]. Thus, given all the current available data more work is needed to address this aspect of the GSK3β neurobiology.
GSK3β, synaptic plasticity and memory
Synaptic dysfunction is an early event in AD pathology [43]. The molecular mechanism underlying memory storage involves changes in synaptic strength including Long Term Potentiation (LTP), which defines a long-lasting increase of synaptic strength, and Long Term Depression (LTD), which is the opposite process [44]. Growing evidence indicates that GSK3-β plays a critical role also in synaptic plasticity and memory formation (Figure 1) [43]. GSK3-β is highly expressed in the hippocampus and although is mainly found in the cytosol, a fraction of this kinase is detected in synaptosomes, dendrites and spines [44,45]. Interestingly, GSK3-β can also associate with the AMPA receptor and act like a key regulator of the balance between LTP and LTD [43]. In fact, GSK3-β phosphorylation at the inhibitory Ser9 site is known to increase upon induction of LTP in the dentate gyrus (DG) and in the CA1 region of the hippocampus [45]. Overactivation of GSK3-β instead, impairs LTP and promotes the NMDA receptor-dependent LTD [43]. Consistent with this observation, transgenic mice conditionally overexpressing GSK-3β display impaired LTP and significant deficits in hippocampus-dependent spatial and fear memory formation [43].
Furthermore, GSK3-β inhibition enables stabilization of β-catenin which would be otherwise degraded by the proteasome following GSK3-β phosphorylation [46]. β-catenin present at both pre- and postsynaptic terminals associates with the cytoplasmic domain of cadherin and regulate cell adhesion and influence synaptic size and strength [47–48]. Downregulation of the β-catenin signaling pathway due to increase GSK3-β activity has been observed in AD brains and implicated in the pathophysiology of learning and memory deficits [50,51]. Given the pivotal role of GSK3-β in the regulation of these processes, reduction of its activity obtained via both genetic and pharmacologic approach has been extensively explored and has been reported to rescue cognitive and memory deficits in several animal models of AD [8]. For example, the GSK3-β inhibitor indirubin-3’-monoxime rescued impairments in the Morris water maze task and, attenuated Aβ production and tau hyperphosphorylation in the APP/Presenilin-1 double transgenic mice [52]. The GSK3-β inhibitor 2-methyl-5-(3-{4-[(S)-methylsulfinyl]phenyl}−1-benzofuran-5-yl)-1,3,4-oxadiazole (MMBO) ameliorated impairments in the Y-maze and the novel object recognition test in 3xTg mice [53]. Moreover, 5XFAD mice treated with the selective GSK3-β inhibitor L803-mts had improved contextual fear memory in association with lower Aβ peptide levels [54].
GSK3β and neurogenesis
GSK3-β has also been reported to suppress adult hippocampal neurogenesis and to induce neuronal death in the neurogenic niche of dentate gyrus (DG) (Figure 1) [55]. Neurogenesis is a dynamic process required for memory consolidation and pattern separation and is altered in the CNS of AD patients and animal models of the disease [56,57]. In favor of the hypothesis of GSK3-β as an important regulator of adult neurogenesis, transgenic mice overexpressing this kinase display reduction in proliferation and maturation of new functional DG neurons and significant memory impairments. This depletion of neurogenesis is also associated with activation of microglia which further promotes degeneration [56,58]. Thus, it seems pretty intuitive that, inactivation of this enzyme could be extremely beneficial in order to promote the birth of new functional neurons and booster memory in neuropathology with impaired neurogenesis like AD. In vitro and in vivo data seem to support this hypothesis. GSK3-β inhibition was demonstrated to enhance proliferation, migration, and differentiation of neural stem cells in primary neurosphere cultures in vitro. Moreover, its inhibition in vivo with the NP03112 compound (tideglusib) was shown to promote neural stem cells proliferation and differentiation in the DG of the hippocampus in adult rats [59].
GSK3-β and inflammation
Beside Aβ and tau aggregation, neuroinflammation is another major feature of the AD brain pathology (Figure 1) [60,61]. Extensive activation of microglia and astrocytes are observed around Aβ plaques and dystrophic neurites [62–64]. Once activated these cells release a variety of pro-inflammatory cytokines like interleukin 1 (IL-1), interleukin-6 (IL-6), interleukin-8 (IL-8) and tumor necrosis factor alpha (TNF-α) which can promote neurotoxicity and contribute to neuronal death [65].
Remarkably, GSK3-β has been recently identified as an important regulator of the inflammatory response in microglia via multiple pathways. GSK-3β activation has been demonstrated to promote production of IL-1, IL-6, TNF-α but also to activate JNK-, STAT3/5- and NF-κB signaling [65]. Indeed, GSK-3β can activate NF-κB by phosphorylation of the p65 subunit which results in further expression of proinflammatory cytokines and chemokines [65]. In addition, the binding of GSK3-β to STAT3 promotes STAT3 association with the IFNγ receptor-associated intracellular signaling complex which is required for IFNγ activation [66]. Few studies have also implicated GSK3-β in the regulation of microglia migration. For example, treatment of BV-2 microglia cells with GSK3 inhibitors has been shown to significantly reduce microglia migration in vitro, in a transwell migration assay [67]. Given the substantial evidence for a proinflammatory action of GSK-3β, its inhibition might not only reduce Aβ production and tau phosphorylation, but also exert anti-inflammatory effects in the AD brain.
GSK3β as therapeutic approach
Since GSK-3β signaling has been strongly associated with several AD neuropathological features including tau phosphorylation, Aβ production, neurogenesis, memory and synaptic dysfunction, its inhibition has emerged as a potentially important therapeutic approach for the treatment of AD. Loss of function studies have been conducted on several AD mouse models and have been proven to be efficient in counteracting many aspects of AD-like pathologic phenotype [68,69] The mood stabilizing drug lithium has been shown to reduce phospho-tau and Aβ levels in several mouse models of AD and tauopathies [68–72]. Tideglusib, a non-ATP competitive GSK-3β inhibitor lowered levels of tau phosphorylation, decreased Aβ deposition, astrocytes proliferation and ameliorated memory deficits in APPsw-tauvlw mice overexpressing human mutant APP [73,74]. Finally, brain infusion of a specific GSK3 inhibitor SB216763 confirmed the beneficial effect of the reduction of GSK3-β activity in an AD model [75].
Although GSK-3 is a highly conserved kinase only a few GSK-3 inhibitors (synthetic and non-synthetic) previously tested in the preclinical phase have reached clinical trials. Additionally, although some encouraging results in animal models, the fact that GSK3-β is ubiquitously expressed and involved in several key cellular biological processes have raised many concerns and significant toxicological challenges for long-term treatment studies in both pre-clinical and clinical phase [76,77]. The first candidate for phase I of clinical trial as GSK3-β inhibitor was AZD2558 [78]. This highly selective GSK3-β inhibitor successfully reduced tau phosphorylation and gliosis in vitro and in vivo. However, the severity of the off target effects associated with its administration in vivo prevented this drug from being tested for chronic treatment of AD. A second attempt was made with AZD1080 previously validated for its capacity of reducing tau phosphorylation in vitro and in pre-clinic studies [78]. Unfortunately, chronic administration of this drug was also associated with significant sides effects and the phase I study was discontinued [78]. So far, only one GSK-3 inhibitor made it to phase II clinical trial for the treatment of AD and progressive supranuclear palsy, Tideglusib [79–81]. Tideglusib has been tested in two small Phase II clinical trials in patients with mild to moderate AD, which showed good tolerability but no significant clinical improvements [77].
Pilot clinical trials have been conducted with lithium chloride in patients with a clinical diagnosis of mild cognitive impairment and AD. Lithium chloride, an FDA approved drug for the treatment of bipolar disorders [82], is a weak and unspecific GSK3-β inhibitor. At therapeutic doses, it can reduce GSK3-β activity by approximately 25% with no adverse effects [82]. Small clinical studies with this agent have shown some positive results including improved cognitive performance and reduction in tau phosphorylation [77] suggesting that lithium might hold potential as disease modifying therapy for AD.
Conclusions
In the past years, GSK3-β has been recognized as an important key player in the AD pathogenesis. Since the initial discovery of GSK3-β overexpression and overactivity in the CNS of AD patients, the role of this kinase has been extensively investigated in connection to tau and Aβ pathophysiology. As one of the main tau kinases, GSK3-β promotes tau hyperphosphorylation and NFTs formation. Strong evidence has also pointed out that increased activity of GSK3-β can induce Aβ formation through transcriptional regulation of BACE1 gene expression and through its regulatory function on PS1. Given the existence of sex differences in AD incidence, some studies have attempted to address the question on whether GSK3β regulation is sexually dimorphic but all the current data available so far are not sufficient to conclude any significant gender-related effect on the activity of this kinase that could be relevant to the onset of development of AD pathology. In addition, recently GSK3-β has been implicated in the modulation of synaptic plasticity, memory, neurogenesis and inflammation making GSK3-β an excellent candidate as disease modifying agent for AD. However, current clinical research has failed so far to identify an effective and safe inhibitor of this kinase. GSK3-β inhibition comes with many challenges related to its ubiquitous expression and multiple regulatory cellular functions. Future possible strategies aiming at bypassing the potentially serious off target effects should be to specifically direct GSK3-β inhibition in the CNS and in the cells of interest. Therefore, given the promising preclinical studies, more research needs to be devoted to the development of selective and organ specific inhibitors that efficiently and safely regulate GSK3-β activity in the CNS as a viable therapeutic approach to prevent or halt AD.
Highlights.
GSK3-β kinase increases with age and in AD pathology.
GSK3-β dysregulation modulates Aβ production and accumulation in AD.
Hyperactive GSK3-β promotes phosphorylation and formation of toxic tau species.
GSK3-β activation is pro-inflammatory and impairs LTP, neurogenesis and memory.
GSK3-β represents a good therapeutic target against AD.
Acknowledgements
Domenico Praticò is the Scott Richards North Star Charitable Foundation Chair for Alzheimer’s Research. The work from the author’s laboratory presented in this article has been supported in part by grants from the Pennsylvania Department of Health, PA-CURE (SAP#4100083099) and the National Institute on Health (NIA, AG051684).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Reference
- [1].2019. Alzheimer’s Disease Facts and Figures Report.
- [2].U.S. Census Bureau, Population Projections. [Google Scholar]
- [3].Dos Santos Picanco LC, Ozela PF, de Fatima de Brito Brito M, Pinheiro AA, Padilha EC, Braga FS, de Paula da Silva CHT, Dos Santos CBR, Rosa JMC, da Silva Hage-Melim L. (2018). Alzheimer’s Disease: A Review from the Pathophysiology to Diagnosis, New Perspectives for Pharmacological Treatment. Curr Med Chem. 25(26):3141–3159. doi: 10.2174/0929867323666161213101126. [DOI] [PubMed] [Google Scholar]
- [4].Bagyinszky E, Youn YC, An SS, Kim S. (2014). The genetics of Alzheimer’s disease. Clin Interv Aging. 9:535–51. doi: 10.2147/CIA.S51571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Woodgett JR. (1990). Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 9(8):2431–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lee SJ, Chung YH, Joo KM, Lim HC, Jeon GS, Kim D, Lee WB, Kim YS, Cha CI. (2006). Age-related changes in glycogen synthase kinase 3beta (GSK3beta) immunoreactivity in the central nervous system of rats. Neurosci Lett. 409(2):134–9. doi: 10.1016/j.neulet.2006.09.026. [DOI] [PubMed] [Google Scholar]
- [7].Leroy K, Yilmaz Z, Brion JP. (2007). Increased level of active GSK-3beta in Alzheimer’s disease and accumulation in argyrophilic grains and in neurons at different stages of neurofibrillary degeneration. Neuropathol Appl Neurobiol. 33(1):43–55. doi: 10.1111/j.1365-2990.2006.00795. [DOI] [PubMed] [Google Scholar]
- [8].Llorens-Martín M, Jurado J, Hernández F, Avila J. (2014). GSK-3β, a pivotal kinase in Alzheimer disease. Front Mol Neurosci. 7:46. doi: 10.3389/fnmol.2014.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hu S, Begum AN, Jones MR, Oh MS, Beech WK, Beech BH, Yang F, Chen P, Ubeda OJ, Kim PC, Davies P, Ma Q, Cole GM, Frautschy SA. (2009). GSK3 inhibitors show benefits in an Alzheimer’s disease (AD) model of neurodegeneration but adverse effects in control animals. Neurobiol Dis. 33(2):193–206. doi: 10.1016/j.nbd.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Embi N, Rylatt DB, Cohen P. (1980). Glycogen Synthase Kinase-3 from Rabbit Skeletal Muscle: Separation from Cyclic-AMP-Dependent Protein Kinase and Phosphorylase Kinase. European Journal of biochemistry. 107(2), 519–527. [PubMed] [Google Scholar]
- [11].Frame S & Cohen P. (2001). GSK3 takes center stage more than 20 years after its discovery. Biochemical Journal. 359(1), 1–16. doi: 10.1042/0264-6021:3590001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Woodgett JR (1990). Molecular cloning and expression of glycogen synthase kinase‐3/factor A. The EMBO journal. 9(8), 2431–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Soutar MP, Kim WY, Williamson R, Peggie M, Hastie CJ, McLauchlan H, Snider WD, Gordon-Weeks PR, Sutherland C. (2010), Evidence that glycogen synthase kinase-3 isoforms have distinct substrate preference in the brain. Journal of Neurochemistry, 115: 974–983. doi: 10.1111/j.1471-4159.2010.06988.x. [DOI] [PubMed] [Google Scholar]
- [14].Hoeflich KP1, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR. (2000). Requirement for glycogen synthase kinase-3β in cell survival and NF-κB activation. Nature. 406(6791), 86. doi: 10.1038/35017574. [DOI] [PubMed] [Google Scholar]
- [15].Kaidanovich-Beilin O, Lipina TV, Takao K, van Eede M, Hattori S, Laliberté C, Khan M, Okamoto K, Chambers JW, Fletcher PJ, MacAulay K, Doble BW, Henkelman M, Miyakawa T, Roder J, Woodgett JR. (2009). Abnormalities in brain structure and behavior in GSK-3alpha mutant mice. Molecular Brain. 2(1), 35. doi: 10.1186/1756-6606-2-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ly PT1, Wu Y, Zou H, Wang R, Zhou W, Kinoshita A, Zhang M, Yang Y, Cai F, Woodgett J, Song W. (2013). Inhibition of GSK3β-mediated BACE1 expression reduces Alzheimer-associated phenotypes. The Journal of Clinical Investigation. 123(1), 224–235. doi: 10.1172/JCI64516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhang W, Yang J, Liu Y, Chen X, Yu T, Jia J, & Liu C (2009). PR55α, a regulatory subunit of PP2A, specifically regulates PP2A-mediated β-catenin dephosphorylation. Journal of Biological Chemistry, 284(34), 22649–22656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ishiguro K1, Shiratsuchi A, Sato S, Omori A, Arioka M, Kobayashi S, Uchida T, Imahori K (1993). Glycogen synthase kinase 3β is identical to tau protein kinase I generating several epitopes of paired helical filaments. FEBS letters, 325(3), 167–172. [DOI] [PubMed] [Google Scholar]
- [19].Takashima A, Honda T, Yasutake K, Michel G, Murayama O, Murayama M, Ishiguro K, Yamaguchi H (1998). Activation of tau protein kinase I/glycogen synthase kinase-3β by amyloid β peptide (25–35) enhances phosphorylation of tau in hippocampal neurons. Neuroscience Research, 31(4), 317–323. [DOI] [PubMed] [Google Scholar]
- [20].Phiel CJ, Wilson CA, Lee VMY, Klein PS. (2003). GSK-3α regulates production of Alzheimer’s disease amyloid-β peptides. Nature. 423(6938), 435. doi: 10.1038/nature01640. [DOI] [PubMed] [Google Scholar]
- [21].Llorens-Martín M, Jurado J, Hernández F, Avila J. (2014). GSK-3β, a pivotal kinase in Alzheimer disease. Front Mol Neurosci. 7:46. doi: 10.3389/fnmol.2014.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Koike H, Tomioka S, Sorimachi H, Saido TC, Maruyama K, Okuyama A, Fujisawa-Sehara A, Ohno S, Suzuki K, Ishiura S. (1999). Membrane-anchored metalloprotease MDC9 has an alpha-secretase activity responsible for processing the amyloid precursor protein. Biochem J. 1999 October 15;343 Pt 2(Pt 2):371–5. [PMC free article] [PubMed] [Google Scholar]
- [23].Cai Z, Zhao Y, Zhao B. (2012). Roles of glycogen synthase kinase 3 in Alzheimer’s disease. Current Alzheimer Research. 9(7), 864–879. doi: 10.2174/156720512802455386. [DOI] [PubMed] [Google Scholar]
- [24].Uemura K, Kuzuya A, Shimozono Y, Aoyagi N, Ando K, Shimohama S, Kinoshita A. (2007). GSK3beta activity modifies the localization and function of presenilin 1. J Biol Chem. 282(21):15823–32. [DOI] [PubMed] [Google Scholar]
- [25].Chen CH, Zhou W, Liu S, Deng Y, Cai F, Tone M, Tone Y, Tong Y, Song W. (2012). Increased NF-κB signalling up-regulates BACE1 expression and its therapeutic potential in Alzheimer’s disease. Int J Neuropsychopharmacol. 15(1):77–90. doi: 10.1017/S1461145711000149. [DOI] [PubMed] [Google Scholar]
- [26].Sun X, Sato S, Murayama O, Murayama M, Park JM, Yamaguchi H, Takashima A. (2002). Lithium inhibits amyloid secretion in COS7 cells transfected with amyloid precursor protein C100. Neurosci lett. 321(1–2), 61–64. doi: 10.1016/s0304-3940(01)02583-6. [DOI] [PubMed] [Google Scholar]
- [27].Luo Y, Bolon B, Kahn S, Bennett BD, Babu-Khan S, Denis P, Fan W, Kha H, Zhang J, Gong Y, Martin L, Louis JC, Yan Q, Richards WG, Citron M, Vassar R. (2001). Mice deficient in BACE1, the Alzheimer’s β-secretase, have normal phenotype and abolished β-amyloid generation. Nature Neuroscience, 4(3), 231. doi: 10.1016/s0304-3940(01)02583-6. [DOI] [PubMed] [Google Scholar]
- [28].Jolivalt CG, Lee CA, Beiswenger KK, Smith JL, Orlov M, Torrance MA, Masliah E (2008). Defective insulin signaling pathway and increased glycogen synthase kinase-3 activity in the brain of diabetic mice: parallels with Alzheimer’s disease and correction by insulin. J Neurosci Res. 86(15):3265–74. doi: 10.1002/jnr.21787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kurochkin IV, Goto S. (1994). Alzheimer’s β-amyloid peptide specifically interacts with and is degraded by insulin degrading enzyme. FEBS letters, 345(1), 33–37. [DOI] [PubMed] [Google Scholar]
- [30].Qiu WQ, Walsh DM, Ye Z, Vekrellis K, Zhang J, Podlisny MB, Rosner MR, Safavi A, Hersh LB, Selkoe DJ. (1998). Insulin-degrading enzyme regulates extracellular levels of amyloid β-protein by degradation. Journal of Biological Chemistry, 273(49), 32730–32738. [DOI] [PubMed] [Google Scholar]
- [31].Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, Eckman CB, Tanzi RE, Selkoe DJ, Guenette S (2003). Insulin-degrading enzyme regulates the levels of insulin, amyloid β-protein, and the β-amyloid precursor protein intracellular domain in vivo. Proceedings of the National Academy of Sciences, 100(7), 4162–4167. doi: 10.1073/pnas.0230450100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Magdesian MH1, Carvalho MM, Mendes FA, Saraiva LM, Juliano MA, Juliano L, Garcia-Abreu J, Ferreira ST. (2008). Amyloid-β binds to the extracellular cysteine-rich domain of Frizzled and inhibits Wnt/β-catenin signaling. Journal of Biological Chemistry, 283(14), 9359–9368. doi: 10.1074/jbc.M707108200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hernández F, Gómez de Barreda E, Fuster-Matanzo A, Lucas JJ, Avila J. (2010). GSK3: a possible link between beta amyloid peptide and tau protein. Experimental Neurology, 223(2), 322–325. doi: 10.1016/j.expneurol.2009.09.011. [DOI] [PubMed] [Google Scholar]
- [34].Gabbouj S, Ryhänen S, Marttinen M, Wittrahm R, Takalo M, Kemppainen S, Martiskainen H, Tanila H, Haapasalo A, Hiltunen M, Natunen T. (2019). Altered insulin signaling in Alzheimer’s disease brain–special emphasis on PI3K-Akt pathway. Frontiers in Neuroscience, 13, 629. doi: 10.3389/fnins.2019.00629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hong M, Lee VMY. (1997). Insulin and insulin-like growth factor-1 regulate tau phosphorylation in cultured human neurons. Journal of Biological Chemistry, 272(31), 19547–19553. [DOI] [PubMed] [Google Scholar]
- [36].Jackson GR, Wiedau-Pazos M, Sang TK, Wagle N, Brown CA, Massachi S, Geschwind DH (2002). Human wild-type tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila. Neuron. 34(4), 509–519. doi: 10.1016/s0896-6273(02)00706-7. [DOI] [PubMed] [Google Scholar]
- [37].Pérez M, Hernández F, Gómez-Ramos A, Smith M, Perry G, Avila J. (2002). Formation of aberrant phosphotau fibrillar polymers in neural cultured cells. European Journal of Biochemistry. 269(5), 1484–1489. doi: 10.1046/j.1432-1033.2002.02794.x. [DOI] [PubMed] [Google Scholar]
- [38].Ryves WJ, Harwood AJ. (2001). Lithium inhibits glycogen synthase kinase-3 by competition for magnesium. Biochemical and Biophysical Research Communications. 280(3), 720–725. doi: 10.1006/bbrc.2000.4169. [DOI] [PubMed] [Google Scholar]
- [39].Avila J, Hernández F. (2007). GSK-3 inhibitors for Alzheimer’s disease. Expert Review of Neurotherapeutics. 7(11), 1527–1533. doi: 10.1586/14737175.7.11.1527. [DOI] [PubMed] [Google Scholar]
- [40].Pérez M, Hernández F, Lim F, Díaz-Nido J, Avila J. (2003). Chronic lithium treatment decreases mutant tau protein aggregation in a transgenic mouse model. Journal of Alzheimer Dis. 5(4), 301–308. doi: 10.3233/jad-2003-5405. [DOI] [PubMed] [Google Scholar]
- [41].Goodenough S, Schleusner D, Pietrzik C, Skutella T, Behl C. (2005). Glycogen synthase kinase 3β links neuroprotection by 17β-estradiol to key Alzheimer processes. Neuroscience, 132(3), 581–589. doi: 10.1016/j.neuroscience.2004.12.029. [DOI] [PubMed] [Google Scholar]
- [42].Preece P, Virley DJ, Costandi M, Coombes R, Moss SJ, Mudge AW, Jazin E, Cairn NJ. (2003). β-Secretase (BACE) and GSK-3 mRNA levels in Alzheimer’s disease. Molecular Brain Research, 116(1–2), 155–158. doi: 10.1016/s0169-328x(03)00233-x. [DOI] [PubMed] [Google Scholar]
- [43].Giese KP. (2009). GSK-3: a key player in neurodegeneration and memory. IUBMB Life. 61(5):516–21. doi: 10.1002/iub.187. [DOI] [PubMed] [Google Scholar]
- [44].Peineau S, Taghibiglou C, Bradley C, Wong TP, Liu L, Lu J, Lo E, Wu D, Saule E, Bouschet T, Matthews P, Isaac JT, Bortolotto ZA, Wang YT, Collingridge GL. (2007). LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron. 53(5):703–17. doi: 10.1016/j.neuron.2007.01.029. [DOI] [PubMed] [Google Scholar]
- [45].Hooper C, Markevich V, Plattner F, Killick R, Schofield E, Engel T, Hernandez F, Anderton B, Rosenblum K, Bliss T, Cooke SF, Avila J, Lucas JJ, Giese KP, Stephenson J, Lovestone S. (2007). Glycogen synthase kinase-3 inhibition is integral to long-term potentiation. Eur J Neurosci. 25(1):81–6. doi: 10.1111/j.1460-9568.2006.05245.x. [DOI] [PubMed] [Google Scholar]
- [46].Hui J, Zhang J, Pu M, Zhou X, Dong L, Mao X, Shi G, Zou J, Wu J, Jiang D, Xi G. (2018). Modulation of GSK-3β/β-Catenin Signaling Contributes to Learning and Memory Impairment in a Rat Model of Depression. Int J Neuropsychopharmacol. 21(9):858–870. doi: 10.1093/ijnp/pyy040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Gumbiner BM.(1996). Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell 84:345–357. [DOI] [PubMed] [Google Scholar]
- [48].Murase S, Mosser E, Schuman EM.(2002). Depolarization drives beta-catenin into neuronal spines promoting changes in synaptic structure and function. Neuron 35:91–105. doi: 10.1016/s0896-6273(02)00764-x. [DOI] [PubMed] [Google Scholar]
- [49].Maguschak KA, Ressler KJ. (2008). β-catenin is required for memory consolidation. Nature Neuroscience. 11, pages1319–1326. doi: 10.1038/nn.2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Nelson WJ. (2008). Regulation of cell-cell adhesion by the cadherin-catenin complex. Biochem Soc Trans. 36(Pt 2):149–55. doi: 10.1042/BST0360149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Vallée A, Lecarpentier Y. (2016). Alzheimer Disease: Crosstalk between the Canonical Wnt/Beta-Catenin Pathway and PPARs Alpha and Gamma. Front Neurosci. 10:459. doi: 10.3389/fnins.2016.00459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Ding Y, Qiao A, Fan GH. (2010). Indirubin-3’-monoxime rescues spatial memory deficits and attenuates β-amyloid-associated neuropathology in a mouse model of Alzheimer’s disease. Neurobiol Dis. 39:156–168. doi: 10.1016/j.nbd.2010.03.022. [DOI] [PubMed] [Google Scholar]
- [53].Onishi T, Iwashita H, Uno Y, Kunitomo J, Saitoh M, Kimura E, Fujita H, Uchiyama N, Kori M, Takizawa M. (2011). A novel glycogen synthase kinase-3 inhibitor 2-methyl-5-(3-{4-[(S)-methylsulfinyl]phenyl}−1-benzofuran-5-yl)-1,3,4-oxadiazole decreases tau phosphorylation and ameliorates cognitive deficits in a transgenic model of Alzheimer’s disease. J Neurochem. 119:1330–1340. doi: 10.1111/j.1471-4159.2011.07532.x [DOI] [PubMed] [Google Scholar]
- [54].Avrahami L, Farfara D, Shaham-Kol M, Vassar R, Frenkel D, Eldar-Finkelman H. (2013). Inhibition of glycogen synthase kinase-3 ameliorates β-amyloid pathology and restores lysosomal acidification and mammalian target of rapamycin activity in the Alzheimer disease mouse model: in vivo and in vitro studies. J Biol Chem. 288:1295–1306. doi: 10.1074/jbc.M112.409250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Sirerol-Piquer M, Gomez-Ramos P, Hernández F, Perez M, Morán MA, Fuster-Matanzo A, Lucas JJ, Avila J, García-Verdugo JM. (2011). GSK3β overexpression induces neuronal death and a depletion of the neurogenic niches in the dentate gyrus. Hippocampus. 21(8):910–22. doi: 10.1002/hipo.20805. [DOI] [PubMed] [Google Scholar]
- [56].Zhao C, Deng W, Gage FH. (2008). Mechanisms and functional implications of adult neurogenesis. M. Cell 132(4):645–60. doi: 10.1016/j.cell.2008.01.033. [DOI] [PubMed] [Google Scholar]
- [57].Moreno-Jiménez EP, Flor-García M, Terreros-Roncal J, Rábano A, Cafini F, Pallas-Bazarra N, Ávila J, Llorens-Martín M. (2019). Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat Med. 25(4):554–560. doi: 10.1038/s41591-019-0375-9. [DOI] [PubMed] [Google Scholar]
- [58].Hernandez F, Lucas JJ, Avila J. (2013). GSK3 and tau: two convergence points in Alzheimer’s disease. J Alzheimer’s Dis. 33 Suppl 1:S141–4. doi: 10.3233/JAD-2012-129025. [DOI] [PubMed] [Google Scholar]
- [59].Morales-Garcia JA, Luna-Medina R, Alonso-Gil S, Sanz-Sancristobal M, Palomo V, Gil C, Santos A, Martinez A, Perez-Castillo A. (2012). Glycogen synthase kinase 3 inhibition promotes adult hippocampal neurogenesis in vitro and in vivo. ACS Chem Neurosci. 3(11):963–71. doi: 10.1021/cn300110c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Hardy JA, Higgins GA. (1992). Alzheimer’s disease: the amyloid cascade hypothesis,” Science, 256(5054):184–185. [DOI] [PubMed] [Google Scholar]
- [61].Crouch PJ, Harding SME, White AR, Camakaris J, Bush AI, Masters CL, (2008). Mechanisms of Aβ mediated neurodegeneration in Alzheimer’s disease. International Journal of Biochemistry Cell Biology. 40(2):181–198. doi: 10.1016/j.biocel.2007.07.013. [DOI] [PubMed] [Google Scholar]
- [62].(Michelucci A, Heurtaux T, Grandbarbe L, Morga E, Heuschling P. Michelucci A, Heurtaux T, Grandbarbe L, Morga E, and Heuschling P, 2009). Characterization of the microglial phenotype under specific pro-inflammatory and anti-inflammatory conditions: Effects of oligomeric and fibrillar amyloid-beta. J Neuroimmunol. 210(1–2):3–12. doi: 10.1016/j.jneuroim.2009.02.003. [DOI] [PubMed] [Google Scholar]
- [63].Heneka MT, O’Banion MK, Terwel D, Kummer MP. (2010). Neuroinflammatory processes in Alzheimer’s disease. J Neural Transm (Vienna). 117(8):919–47. doi: 10.1007/s00702-010-0438-z. [DOI] [PubMed] [Google Scholar]
- [64].Eikelenboom P, van Gool WA. (2004). Neuroinflammatory perspectives on the two faces of Alzheimer’s disease. J Neural Transm (Vienna). 111(3):281–94. doi: 10.1007/s00702-003-0055-1. [DOI] [PubMed] [Google Scholar]
- [65].Koistinaho J, Malm T, Goldsteins G. (2011). Glycogen synthase kinase-3β: a mediator of inflammation in Alzheimer’s disease? Int J Alzheimers Dis. 2011:129753. doi: 10.4061/2011/129753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Beurel E, Jope RS. (2008). Differential regulation of STAT family members by glycogen synthase kinase-3. J Biol Chem. 283(32):21934–44. doi: 10.1074/jbc.M802481200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Yuskaitis CJ, Jope RS. (2009). Glycogen synthase kinase-3 regulates microglial migration, inflammation, and inflammation-induced neurotoxicity. Cell Signal. 21(2):264–73. doi: 10.1016/j.cellsig.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Perez M, Hernandez F, Lim F, Diaz-Nido J, Avila J. (2003). Chronic lithium treatment decreases mutant tau protein aggregation in a transgenic mouse model. J. Alzheimer’s Dis. 5, 301–308. doi: 10.3233/jad-2003-5405. [DOI] [PubMed] [Google Scholar]
- [69].Phiel CJ, Wilson CA, Lee VM, Klein PS. (2003). GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature 423, 435–439. doi: 10.1038/nature01640. [DOI] [PubMed] [Google Scholar]
- [70].Noble W, Planel E, Zehr C, Olm V, Meyerson J, Suleman F, Gaynor K, Wang L, LaFrancois J, Feinstein B, Burns M, Krishnamurthy P, Wen Y, Bhat R, Lewis J, Dickson D, Duff K. (2005). Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc. Natl. Acad. Sci. U.S.A. 102, 6990–6995. doi: 10.1073/pnas.0500466102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Nakashima H, Ishihara T, Suguimoto P, Yokota O, Oshima E, Kugo A, Terada S, Hamamura T, Trojanowski JQ, Lee VM, Kuroda S. (2005). Chronic lithium treatment decreases tau lesions by promoting ubiquitination in a mouse model of tauopathies. Acta Neuropathol. 110, 547–556. doi: 10.1007/s00401-005-1087-4. [DOI] [PubMed] [Google Scholar]
- [72].Caccamo A, Oddo S, Tran LX, LaFerla FM. (2007). Lithium reduces tau phosphorylation but not A beta or working memory deficits in a transgenic model with both plaques and tangles. Am. J. Pathol 170, 1669–1675. doi: 10.2353/ajpath.2007.061178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Serenó L, Coma M, Rodríguez M, Sánchez-Ferrer P, Sánchez MB, Gich I, Agulló JM, Pérez M, Avila J, Guardia-Laguarta C, Clarimón J, Lleó A, Gómez-Isla T. (2009). A novel GSK-3beta inhibitor reduces Alzheimer’s pathology and rescues neuronal loss in vivo. Neurobiol Dis. 35(3):359–67. doi: 10.1016/j.nbd.2009.05.025 [DOI] [PubMed] [Google Scholar]
- [74].Wang H, Huang S, Yan K, Fang X, Abussaud A, Martinez A, Sun HS, Feng ZP. (2016). Tideglusib, a chemical inhibitor of GSK3β, attenuates hypoxic-ischemic brain injury in neonatal mice. Biochim Biophys Acta. 1860(10):2076–85. doi: 10.1016/j.bbagen.2016.06.027. [DOI] [PubMed] [Google Scholar]
- [75].Hu S, Begum AN, Jones MR, Oh MS, Beech WK, Beech BH, Yang F, Chen P, Ubeda OJ, Kim PC, Davies P, Ma Q, Cole GM, Frautschy SA. (2009). GSK3 inhibitors show benefits in an Alzheimer’s disease (AD) model of neurodegeneration but adverse effects in control animals. Neurobiol Dis. 33(2):193–206. doi: 10.1016/j.nbd.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Medina M, Garrido JJ, Wandosell FG. (2011). Modulation of GSK-3 as a Therapeutic Strategy on Tau Pathologies. Front Mol Neurosci. 4:24. doi: 10.3389/fnmol.2011.00024. eCollection 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Congdon EE, Sigurdsson EM. (2018). Tau-targeting therapies for Alzheimer disease. Nat Rev Neurol. 14(7):399–415. doi: 10.1038/s41582-018-0013-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Bhat RV, Andersson U, Andersson S, Knerr L, Bauer U, Sundgren-Andersson AK. (2018). The Conundrum of GSK3 Inhibitors: Is it the Dawn of a New Beginning? J Alzheimer’s Dis. 64(s1):S547–S554. doi: 10.3233/JAD-179934. [DOI] [PubMed] [Google Scholar]
- [79].del Ser T, Steinwachs KC, Gertz HJ, Andrés MV, Gómez-Carrillo B, Medina M, Vericat JA, Redondo P, Fleet D, León T. (2012).Treatment of Alzheimer’s Disease with the GSK-3 Inhibitor Tideglusib: A Pilot Study. J Alzheimer’s Dis. 33(1):205–15. doi: 10.3233/JAD-2012-120805. [DOI] [PubMed] [Google Scholar]
- [80].Lovestone S, Boada M, Dubois B, Hüll M, Rinne JO, Huppertz HJ, Calero M, Andrés MV, Gómez-Carrillo B, León T, del Ser T, ARGO investigators. (2015). A phase II trial of tideglusib in Alzheimer’s disease. J Alzheimer’s Dis. 45(1):75–88. doi: 10.3233/JAD-141959. [DOI] [PubMed] [Google Scholar]
- [81].Höglinger GU, Huppertz HJ, Wagenpfeil S, Andrés MV, Belloch V, León T, Del Ser T, TAUROS MRI Investigators. (2014). Tideglusib reduces progression of brain atrophy in progressive supranuclear palsy in a randomized trial. Mov Disord. 29(4):479–87. doi: 10.1002/mds.25815. [DOI] [PubMed] [Google Scholar]
- [82].Machado-Vieira R, Manji HK, Zarate CA Jr. (2009). The role of lithium in the treatment of bipolar disorder: convergent evidence for neurotrophic effects as a unifying hypothesis. Bipolar Disord. 11(2):92–109. doi: 10.1111/j.1399-5618.2009.00714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]