

Abstract
Gold standard diagnosis of hematological cancers require complete white blood cell count, followed by flow cytometry with multiple CD markers, and cytology. It requires substantial time and specialized training. A dual-layer paper microfluidic chip was developed as a quicker, low-cost, and field-deployable alternative to detect ROR1+ (receptor tyrosine-like orphan receptor one) cancer cells from the undiluted and untreated buffy coat blood samples. The first capture layer consisted of a GF/D glass fiber substrate, preloaded with cancer specific anti-ROR1 conjugated fluorescent particles to its center for cancer cell capture and direct smartphone fluorescence imaging. The second flow layer was comprised of a grade 1 cellulose chromatography paper with wax-printed four channels for wicking and capillary flow-based detection. The flow velocity was used as measure of antigen concentration in the buffy coat sample. In this manner, intact cells and their antigens were separated and independently analyzed by both imaging and flow velocity analyses. A custom-made smartphone-based fluorescence microscope and automated image processing and particle counter software were developed to enumerate particles on paper, with the limit of detection of 1 cell/μL. Flow velocity analysis showed even greater sensitivity, with the limit of detection of 0.1 cells/μL in the first 6 s of assay. Comparison with capillary flow model revealed that great alignment with experimental data and greater correlation to viscosity than interfacial tension. Our proposed device is able to capture and on-chip image ROR1+ cancer cells within a complex sample matrix (buffy coat) while simultaneously quantifying cell concentration in a point-of-care manner.
Keywords: particle aggregation, capillary action, immunoagglutination, chronic lymphocytic leukemia (CLL), antibody receptor tyrosine kinase-like orphan receptor 1 (ROR1)
1. INTRODUCTION
Hematological cancers, cancers derived from the blood, are conventionally diagnosed in a manner that is very time consuming, requires expensive equipment, and demands for professionally trained personnel [1]. Such processes can be especially tiresome and invasive for patients suffering from chronic cancers that require months-worth of blood analysis. For example, chronic lymphocytic leukemia (CLL) diagnosis requires the presence of ≥ 5 × 109 B lymphocytes/L (= 106 cells/μL) in the peripheral blood, sustained for at least 3 months. The clonality of these B lymphocytes are then confirmed by demonstration of immunoglobulin light chain restriction via flow cytometry. Following, leukemia cells are also morphologically confirmed via a microscope blood smear to appear as small, mature lymphocytes with a narrow border of cytoplasm and a dense nucleus lacking discernable nucleoli with partially aggregated chromatin [2]. CLL is the most common form of leukemia in adults, accounting for 25% to 30% of all leukemia derivatives in the United States [3]. Globally, based off of data collected from 1990 to 2015, 2 out of 100,000 people (age-standardized) were reported to have CLL [4]. While a variety of treatment options are now available, CLL has a low response rate, and assessing the prognosis of patients remains difficult [5]. This project addresses the need to develop an inexpensive, rapid, and point-of-care (POC) method to identify, quantify, and detect hematological cancers, including CLL.
In recent years, various microfluidic POC assays have been developed for detection of hematologic cancer and CLL. Previously investigated microfluidic techniques include gene-specific amplification via on-chip quantitative reverse transcription polymerase chain reaction (qRT-PCR) [6,7], aptamer probe hybridization via electrochemical impedance spectroscopy [8], on-chip fluorescence microscopy of immunostained whole cells [9], cell sorting by magnetic trapping array and lectin affinity [10], and measurement of cell stiffness [11]. Although each technique has its advantage such as specificity, multiplexing capabilities, label-free, and single-cell sorting, there are also disadvantages. First, all microfluidic platforms mentioned utilize a custom silicone-based chip, whose fabrication process can be complex and expensive, requiring special equipment and cleanroom access. Secondly, the use of nucleic acid identification techniques such as PCR or apta-sensing requires pre-assay sample preparation (i.e. extraction via cell lysis, filtration, etc.) and expensive reagents with shortened storage life, making it undesirable for field and POC applications. Lastly, cell sorting techniques with on-chip immunofluorescence imaging requires lengthy procedures of cell fixation, immunostaining with multiple fluorophores, and access to benchtop fluorescence microscopy.
Use of paper microfluidic platforms with fluorescence sensing has been demonstrated for multiple chemical and biological applications. Advantages of paper microfluidics include: an inherent pump-free mechanism via capillary and wicking action, ease and low-cost of fabrication, disposability, and increased storage-life of reagents preloaded prior to performing assay. Fluorescent nanotechnologies such as quantum dots (QD), fluorescent micro/nanoparticles, and nanoclusters have consistently shown superior stability and high sensitivity in comparison to traditional fluorescent dyes [12–14]. Additionally, smartphone optical sensing was chosen due to its holistic capabilities of being portable, user-friendly interface, ability to connect and upload to a network, and on-board processing capability [15]. However, as paper is optically opaque and generates a large extent of back scattering noise (from its fibrous structure) as well as auto-fluorescence, such detection has always been considered challenging.
In this work, we developed a dual-layer paper microfluidic chip pre-loaded with antibody-conjugated, fluorescent microparticles. This platform consisted of the top capture layer, allowing efficient filtration of the buffy coat samples and capture of the cancer cells, and the bottom flow layer generating capillary flow through paper pores necessary for efficient mixing and subsequent antibody-antigen binding. Particle binding to the cancer cells captured on the top layer was quantified via smartphone-based on-chip microscopic imaging [16]. Particles were still aggregated by cell fragments and antigens in the bottom flow layer, causing changes in the capillary flow, which was also quantified via smartphone-based real-time monitoring of flow velocity [17]. In this manner, intact cells and their antigens were separated and independently analyzed. Through this dual detection, cancer cell concentration was evaluated in the undiluted buffy coat samples from healthy donors, dosed with known concentration of cancer cells. Buffy coat samples can easily be obtained by phlebotomy, although they still contain substantial amount of red blood cells and complex to be directly assayed in a handheld platform with satisfactory sensitivity. Through utilizing a novel image processing algorithm for microparticle immunoagglutination on paper demonstrated in [16], it is possible to make an extremely sensitive detection. Our method also involves a second mechanism of detection via flow velocity analysis, which has recently been demonstrated as an emerging alternative detection on paper microfluidic platforms [18].
The biorecognition element chosen for this assay is an antibody that has a high affinity to the receptor, tyrosine-like orphan receptor one (ROR1) protein. ROR1 has been a biomarker with increasing interest as a diagnostic tool for cancer, specifically for CLL and B lymphoma cells. The ROR1 antigen has been demonstrated in numerous studies, as a highly and specifically expressed biomarker for CLL disease expression with respect to normal B cells, normal tissues, and normal blood cells from healthy donors [19,20]. Other leukemia’s such as B cell acute lymphoblastic leukemia and Harry cell leukemia have also resulted in high expression of the ROR1 protein [21]. Unfortunately, ROR1 is not easily expressed in vitro leukemia cell culture [22,23]; a substitute model cell should preferably be tested for in vitro assays. In fact, ROR1 is a highly versatile biomarker and is found in other malignant cancers including lung, ovarian, and prostate cancer [24,25]. Therefore, for this proof-of-concept technology, we have used ROR1+ MDA-MB-231 breast cancer cells as the simulated target cells for this platform.
Overall, our unique method, which utilizes in situ imaging and flow-based washing, allows for fast and easy ROR1+ cancer cell detection to monitor the progression of the disease at the point-of-care. This novel assay is also far less expensive and requires much less specialized training than any of the current methods. These advantages successfully address the current need for simple ROR1+ cancer diagnostic and prognostic applications.
2. MATERIALS AND METHODS
2.1. Cell culture and suspension
Mammary gland/breast adenocarcinoma cell cultures (MDA-MB-231; HTB-26, ATCC; Manassas, VA, USA) were grown in HyClone Leibovitz L-15 media with 2.05 mM L-glutamine (SH30525.01; GE Healthcare; Maidstone, Kent, UK) supplemented with 10% (v/v) fetal bovine serum (FBS; 30–2021; ATCC; Manassas, VA, USA), 0.1% (v/v) of 50 mg/mL gentamycin sulfate solution (IB02030; IBI Scientific; Dubuque, IA, USA), and 0.2% (v/v) of 250 μg/mL Amphotericin B (GE Healthcare; Maidstone, Kent, UK). Cells were cultured at 37°C (HERAcell 150i; Cambridge Scientific; Watertown, MA, USA) in 0% CO2 until 80% confluent. After reaching 80% confluency, MDA-MB-231 cells were passaged following standard procedures [26]. Cells were resuspended, counted via a hemocytometer, diluted in media to 102 cells/μL, and stained for 15 min with NucBlue Live ReadyProbes Reagent (R37605; ThermoFisher Scientific; USA). Serial dilutions were made by pipette mixing (not by vortexing) to prevent cell fragmentation. Excess dye was removed by centrifugation at 1.3 g for 5 min and resuspended in prewarmed 1X DPBS, back to 102 cells/μL concentration.
2.2. Benchtop fluorescence imaging
An inverted benchtop fluorescence microscope (Eclipse TS100; Nikon Corp.; Tokyo, Japan) was used, equipped with fluorescence filters UV-2E/C and G-2A (A.G. Heinze, Lake Forest, CA, USA) and an imaging software (NIS Elements; Nikon Corp.). Greyscale images were also taken from each filter cube. Images were then superimposed with pseudo-colors: blue for NucBlue-stained cells and red for antibody-particles via ImageJ software (US National Institutes of Health; Bethesda, MD, USA).
2.3. Antibody conjugation to fluorescent microparticles
Rabbit polyclonal antibody to receptor-tyrosine-kinase-like orphan receptor 1 (anti-ROR1; OABF00363; Aviva Systems Biology; San Diego, CA, USA) was used for ROR1 expression on cancer cells. 1 M ethanolamine solution (398136; Sigma-Aldrich; St. Louis, MO, USA) was used to generate negative control signals with ROR1-positive cell cultures. Anti-ROR1 and ethanolamine were covalently conjugated to highly carboxylated, red, 1-μm diameter, fluorescent polystyrene particles (CAF-001UM; Magsphere Inc.; Pasadena, CA, USA) with reported 538 nm excitation and 584 nm emission wavelengths. Conjugation to particles were done by the following method: 1) particles were washed three times, first with 0.05% Triton-X 100 washing buffer, followed by deionized water with centrifugation of 8,100 g for 8 min in between each washing step; 2) washed particles were incubated with activation buffer [1 mg/mL 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDAC) in pH 6], 5 mM 2-(N-morpholino) ethanesulfonic acid (MES), and 1 M NaOH for 30 min on a rocker at room temperature; 3) 10 μL of 1 mg/mL ethanolamine or anti-ROR1 antibody solutions were added to activation buffer and particle mixture and placed on a rocker for 2 h at room temperature; 4) 1 M ethanolamine buffer was added to quench reaction and placed on a rocker for 30 min at room temperature; 5) antibody-particle or ethanolamine-particle suspensions were centrifuged and washed similar to step 1, but with added 5 min sonication in between resuspension and centrifugation. Particles were stored in deionized water in 3–8°C. This EDAC-based covalent conjugation of antibodies to carboxylated polystyrene particles has been well-documented to yield a uniform distribution of antibodies on the particle surface [27], since the carboxyl-containing co-monomers repel from each other and from hydrophobic main-monomer (styrene). In addition, a total of four batches of antibody-conjugated particles were used throughout this work to address the batch-to-batch variations.
2.4. Capture layer substrate selection - optimum cell retention and particle relief
The paper microfluidic chip is comprised of two layers: the first capture layer and the second flow layer. Pre- and post-washing steps were evaluated for two types of the capture layer’s ability to retain cells after washing and relieve unbound particles. NucBlue-stained cell suspension or anti-ROR1/ethanolamine conjugated fluorescent particle suspension was applied to 6 mm diameter mechanically-punched paper rounds of GF/D borosilicate microfiber filter (1823–047; Whatman GE Healthcare; Maidstone, Kent, UK) or G041 glass fiber conjugate pad sheets (Milipore Sigma, Billericia, MA, USA). After a 5 min waiting period, paper rounds were flipped and imaged using a fluorescence microscope with the addition of a drop of mineral oil to omit undesired scatter off glass fibers. The collected images were evaluated for pre-wash steps. Post-wash assay images were taken with the continuation of 100 μL of 0.05% Triton-X 100 (T8787, 398136; Sigma-Aldrich; St. Louis, MO, USA) washing buffer solution to paper rounds with a 1 in × 1 in (25.4 mm × 25.4 mm) grade 1 chromatography paper (Whatman Grade 1 Chr, GE Healthcare, Maidstone, Klent, UK) underneath. After a 5 min waiting period, the paper rounds were flipped and imaged with the addition of mineral oil.
2.5. Well plate assay procedure
6 × 104 MDA-MB-231 cells/mL were seeded into 96-well plates and incubated overnight. Cells were prepared for a specificity assay by washing with 1X DPBS twice, fixing with 4% paraformaldehyde for 15 min at room temperature, and washing again with 1X DPBS twice. Red fluorescent anti-ROR1 particles were then added and incubated for 30 min at room temperature. To remove unbound particles, a 0.05% Triton X-100 washing buffer was used to wash cells twice. 1X DPBS was then added to wells to prevent drying during imaging. As a control, wells with no cells were incubated in media overnight, followed by washing, fixing, and incubating with particles under the same conditions of the wells with cells. Four 400X fluorescent images were taken for each well, where each image represented one field-of-view (FOV). All particles were counted for each FOV and tallied for frequency in occurrence. Particles counts greater than 16 particles per FOV were omitted due to insufficient washing of unbound particles.
2.6. Paper chip design and fabrication
The microfluidic chip was comprised of two layers. The first layer (capture layer) was 6-mm diameter, mechanically punched rounds made out of GF/D or G041 glass fiber substrates, for preloading antibody-conjugated particles and initial capturing of whole cancer cells (Figure 1A). The second layer (flow layer) was designed using SolidWorks 2015 software (Dassault Systèmes, Vélizy-Villacoublay, France) and wax-printed (ColorQube 8580, Xerox, Norwalk, CT, USA) on grade 1 chromatography paper for wicking and capillary flow measurements along four channels. The width (2 mm) and length (5 mm) of the four channels on the second flow layer was selected from multiple iterations for a 10 μL sample to be distributed amongst the four channels evenly and to flow along the channel for measurable distances for the smartphone to take a video without magnification or zoom (Figure 1B). After the pattern was printed, chips were cut and melted at about 120°C for 5 min to allow for wax to flow into the paper fibers creating hydrophobic barriers throughout the depth of the paper.
Figure 1.
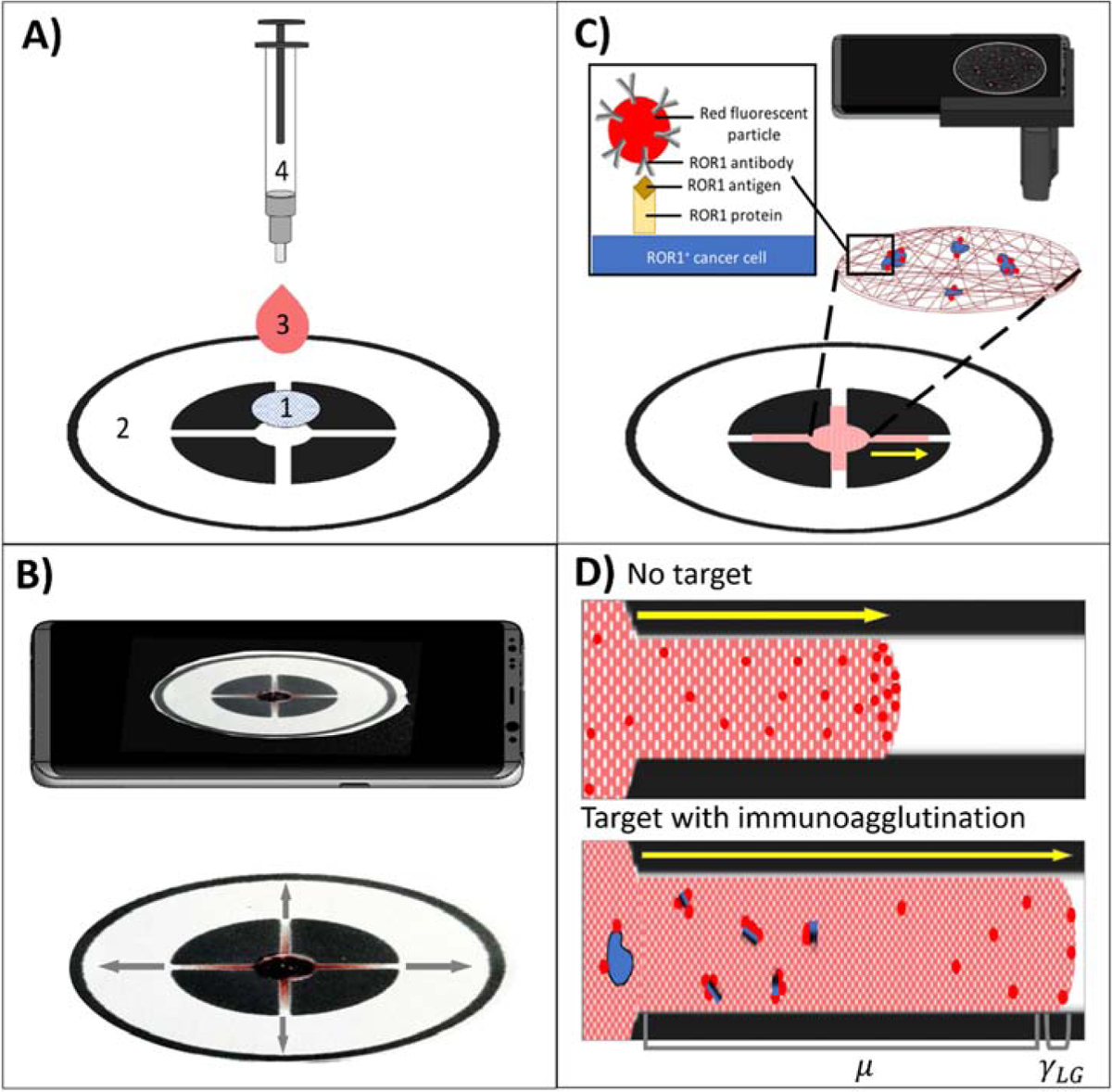
(A) Dual-layer paper chip assay procedure with 1) a capture layer (glass fiber), pre-loaded with red fluorescent, anti-ROR1 conjugated microparticles, 2) a flow layer (wax-printed chromatography paper), 3) a buffy coat sample with dosed cancer cells, and 4) a blunt-end needle tip syringe for uniform droplet application. (B) A smartphone captures a video for monitoring the flow velocity from the flow layer. (C) A smartphone-based fluorescence microscope images the capture layer for quantifying ROR1+ cancer cell. (D) Schematic of flow-based detection with no target (more particles at the flow front, increasing interfacial tension γLG) and with target (more particles not at the flow front due to immunoagglutination, increasing viscosity μ).
2.7. Paper chip assay procedure
10 μL of anti-ROR1 conjugated particles were preloaded to the center of the first capture layer of the paper microfluidic chip, followed by drying at room temperature, prior to its binding to the second flow layer. Cell suspensions of 102 cells/μL were serially diluted down to 0.1 cells/μL in 1X buffy coat (SER-BC-SDS, ZenBio, Research Triangle, NC, USA). 10 μL of dosed samples were applied to the center of the capture layer with a 1 mL syringe attached with an 18-gauge blunt needle tip (outer diameter = 1.27 mm) (Figure 1A). Flow along the four channels of the second flow layer of the paper chip was monitored via capturing 30 fps videos using a smartphone (Samsung Galaxy S8; Samsung, Suwon, South Korea), without separating the capture layer (Figure 1B). Once all channels have been saturated the video was ended and 3 steps of 30 μL washing buffer was added to the capture layer every 1 min. The capture layer was then separated and placed on a microscope stage for particle count analysis (Figure 1C).
2.8. Design and assembly of smartphone-based fluorescence microscope device
Cells captured on the capture layer were imaged with a custom-made smartphone fluorescence microscope. A commercially available smartphone microscope (AOMEKIE Cellphone Microscope Magnifier; 60X-100X zoom with a UV LED) was modified as follows: Firstly, the existing LED was replaced with a 525 nm green LED (MTE5052M3A-UBG, Marktech Optoelectronics, Thief River Falls, MN, USA) for fluorescence excitation of antibody-conjugated microparticles. Secondly, a 3D-printed attachment (an enclosure and a moveable stage) was constructed and added to the microscope to provide a dark enclosure and a moveable stage, made out of black acrylonitrile-butadiene-styrene (ABS) polymer using a 3D printer (Zortrax M200, Zortax Inc., Olsztyn, Poland). This attachment was designed using SolidWorks. Thirdly, the attachment was also designed to incorporate a 589 ± 2 nm optical bandpass filter (65–707, Edmund Optics, Barrington, NJ, USA) to capture fluorescence emission from the same particles.
2.9. Image processing algorithm - particle count
Both smartphone and benchtop microscope images were pre-processed and analyzed via MATLAB 2019a (MathWorks, Natick, Massachusetts, MA, USA). The pre-processing algorithm includes the use of a bandpass noise filter in the Fourier Space to enhance higher frequency signals and filter out low frequency signals caused by autofluorescence or undesirable light scatter. An optimal binary threshold of 0.6 was then applied, highlighting only the antibody-conjugated fluorescent particles on paper. A circular object function was then utilized to count and augment the measured diameters onto the pre–processed image. ImageJ 1.51m9 (National Institutes of Health, Bethesda, MD, USA) was also used to analyze benchtop microscope images for initial substrate optimization and specificity test.
2.10. Image processing algorithm - flow analysis
30 fps videos were parsed into 1 s time interval images. Images were processed and analyzed via MATLAB. Auto-contrast stretching [28] and chromatic adaptation function [29] were used for white balance as well as for the enhancement to better detect buffy coat color. Binary thresholds and fill-in functions were also used prior to measure lateral capillary flow distance from the edge of the first capture layer. A moving average was then applied to raw measurements. Capillary flow velocity was then determined as the slope of the average flow distance at 3 different time intervals: Δt1 Δt2, Δt3, and Δt4, which correspond to 2 to 6 s, 7 to 12 s, 13 to 22 s, and 23 to 32 s.
2.11. Viscosity measurements
The viscosity of each mixture containing particles and target cells in growth media (L15) was measured using oscillatory shear rheology on a Discovery Hybrid Rheometer 2 (TA Instruments, New Castle, DE, USA) with a 40 mm diameter and 0.5° cone geometry. Sample mixtures were pipetted and spread evenly on a 40 mm stage before lowering the gap height to 30 μm. A Peltier temperature-controlled stage maintained samples at 37°C during rheology measurements. A shear stress sweep was performed from 0.001 to 100 Pa at a constant 10 rad/s angular frequency. The viscosity of particles and cancer cell mixtures from 0 to 100 cells/μL was determined by the slope of the shear stress (τ)vs. shear strain rate (ẏ) plot [30,31].
2.12. Interfacial tension measurements
Interfacial tension (mN/m) was measured with an FTÅ200 contact angle/surface tension analyzer (First Ten Ångstroms, Portsmouth, VA, USA) using the pendant droplet method. Five 10 μL of anti-ROR1 particles with either 100, 10, 1, 0.1, or 0 (NTC) of MDA-MB-231 cells/μL were prepared. Particle-cell mixture were then extruded from an 18-gauge blunt needle (outer diameter = 1.27 mm) and measured.
3. RESULTS AND DISCUSSION
3.1. Selection of the capture layer substrate for optimum cell and particle retention
Two different types of capture layer were investigated to determine best ability to retain cells and relieve particles after the washing step. Figure 2A shows the average cells or cluster per field-of-view (FOV) for MDA-MB-231 breast cancer cells on G041 and GF/D glass fiber substrates. Average cells/cluster per FOV were 2.2 for pre-wash and remained the same for post-wash on G041. In comparison, average cells/cluster per FOV were 4.0 for pre-wash and 2.7 for post-wash with GF/D. More cells/clusters were observed with GF/D than G041 and washing was more effective with GF/D. Figure 2B illustrates capture layer’s ability to retain or relieve particles post-wash. More particles were captured with GF/D than G041, and more particles were retained after washing with GF/D than G041.
Figure 2.
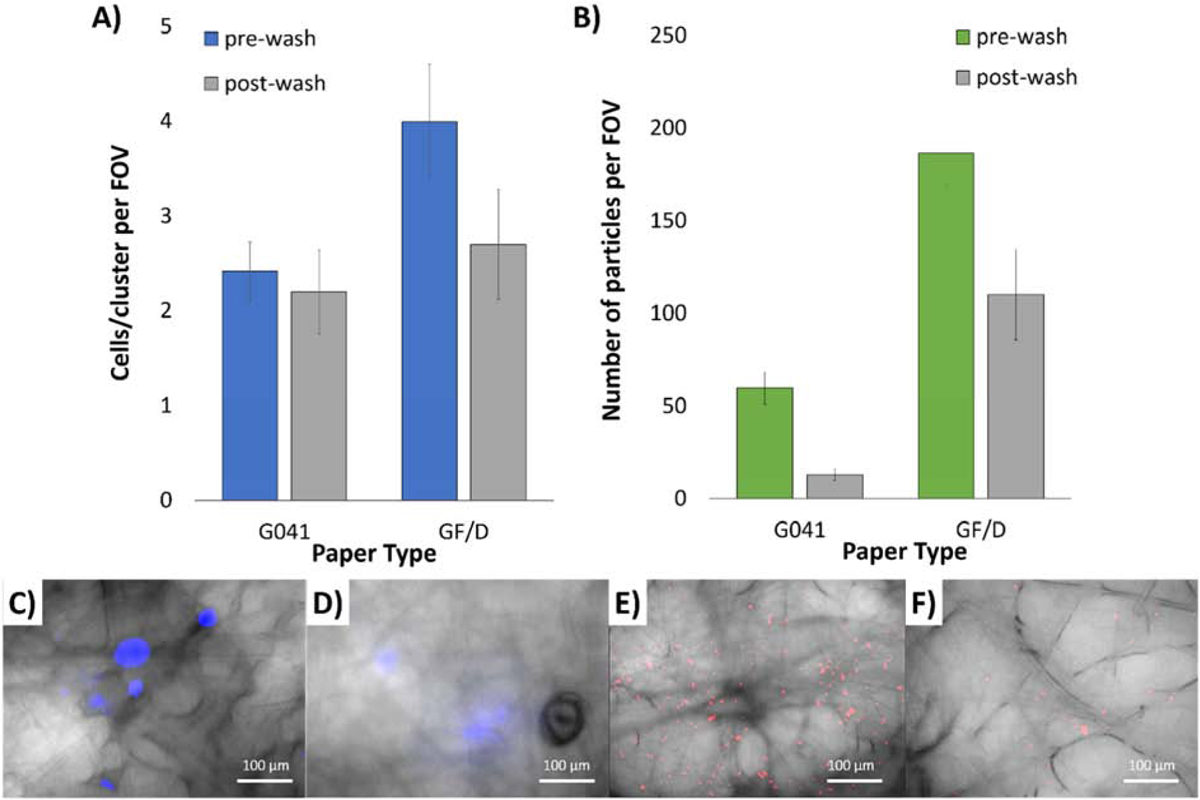
Selection of capture layer substrate for optimum cell retention comparing pre- and post-wash of (A) MDA-MB-231 cells and (B) BSA-conjugated particles, on G0401 and GF/D glass fiber substrates. Averages of 5 assays. Error bars represent standard errors. 400X microscopic overlaid images of (C) pre- and (D) post-wash NucBlue-stained MDA-MB-231 cells, as well as (E) pre- and (F) post-wash red fluorescent BSA-conjugated particles, both on GF/D glass fiber substrates.
In addition, we found that the GF/D produced less undesired background scatter over G041. G041 fibers acted as miniature optical fibers that transmitted and propagated the excitation light along the fiber itself causing difficulty in distinguishing the fluorescence from the NucBlue-stained cells or the fluorescent particles. While we did find that such background light scatter could be reduced when mineral oil was added. By adding mineral oil to the glass fiber substrate for fluorescence imaging we reduced the difference in refractive index between the glass fibers (n = 1.5) and the surrounding media (n = 1.47 for mineral oil vs. n = 1.00 for air). Unfortunately, it was not a significant enough reduction method in noise in comparison to the GF/D paper substrate. Taken together, GF/D was chosen as the better substrate for the capture layer.
3.2. Verification of specific binding of anti-ROR1 particles to cells via well-plate assays
MDA-MB-231 ROR1+ cells were seeded onto tissue culture well plates (TCP) and fixed with formaldehyde to run specificity tests of antibody-conjugated particles. Anti-ROR1 particles were added to wells with and without cells seeded, incubated, and washed. Supplementary Figure S1A depicts the frequency tally of randomly chosen FOVs in which the total number of particles were counted. As shown, anti-ROR1 particles had a higher frequency with higher number of particles in FOVs where MDA-MB-231 cells were present. The highest frequency count had 4 FOVs with 8 total particles, followed by 3 FOVs with 12 total particles. In comparison, areas with no cells had low frequency FOV counts of 1, however ranging from 0 to 12 total particles in FOV. Supplementary Figure S1B and S1C are FOVs in which anti-ROR1 is specifically binding to MDA-MB-231 cells. It is also observed that anti-ROR1 particles are also clustered in areas near, but with no MDA-MB-231. This suggests that although whole cells are not present, antigens may be responsible for particle binding and clustering, presumably due to the use of a surfactant-based washing buffer as well as the inherent mobility of cells during growth.
3.3. On-chip particle counting using a benchtop fluorescence microscope
A dual-layer paper microfluidic chip was designed and fabricated as shown in Figure 1A. The first capture layer is a mechanically punched, 6-mm diameter GF/D glass fiber where the cells are loaded and captured. GF/D was selected as the optimal paper substrate in capture and retention of cells and particles, as previously discussed. The second flow layer of cellulose chromatography paper allowed for wicking of applied sample and washing buffer. Four channels were designed and wax-printed to create hydrophobic barriers.
Fluorescence imaging was initially conducted for the first capture layer using a benchtop fluorescence microscope (Figure 3A). Particle area in FOV was evaluated in relation to varying cell concentration in a buffy coat matrix utilizing a custom MATLAB particle analyzer code (Figure 3B). Image pre-processing algorithm with Fourier bandpass noise filtering and binary thresholding was used prior to measuring particles in FOV (Figure 3C). The Fourier bandpass filtration mitigates low frequency and high frequency signals leaving an image in which an optimal frequency corresponding to the fluorescence light of the anti-ROR1 particles. This leaves an image of varying light intensities, in which a binary threshold is applied to result in fluorescence from the particles in a clear format to determine the total pixel area of captured particles. ROR1+ cells were serially diluted to 0.1 to 100 cells per/μL in buffy coat. 400X images were taken, processed, and measured for area of particles in FOV. The general trend was as cell concentration increased, particle area in FOV increased. With 100 cells/μL an average particle area of 2264 pixels in FOV was statistically different in comparison to no target control (NTC; no cancer cells) with the p value of 1×10−6. Following, 10 cells/mL was also statistically different with an average particle area of 1164 pixels in FOV with the p value of 0.007. Following, cell concentrations of 1 and 0.1 cells/μL with average particle areas of 132 and 206 pixels were statistically different from NTC with the p values of 0.023 and 0.001 (Figure 3D).
Figure 3.
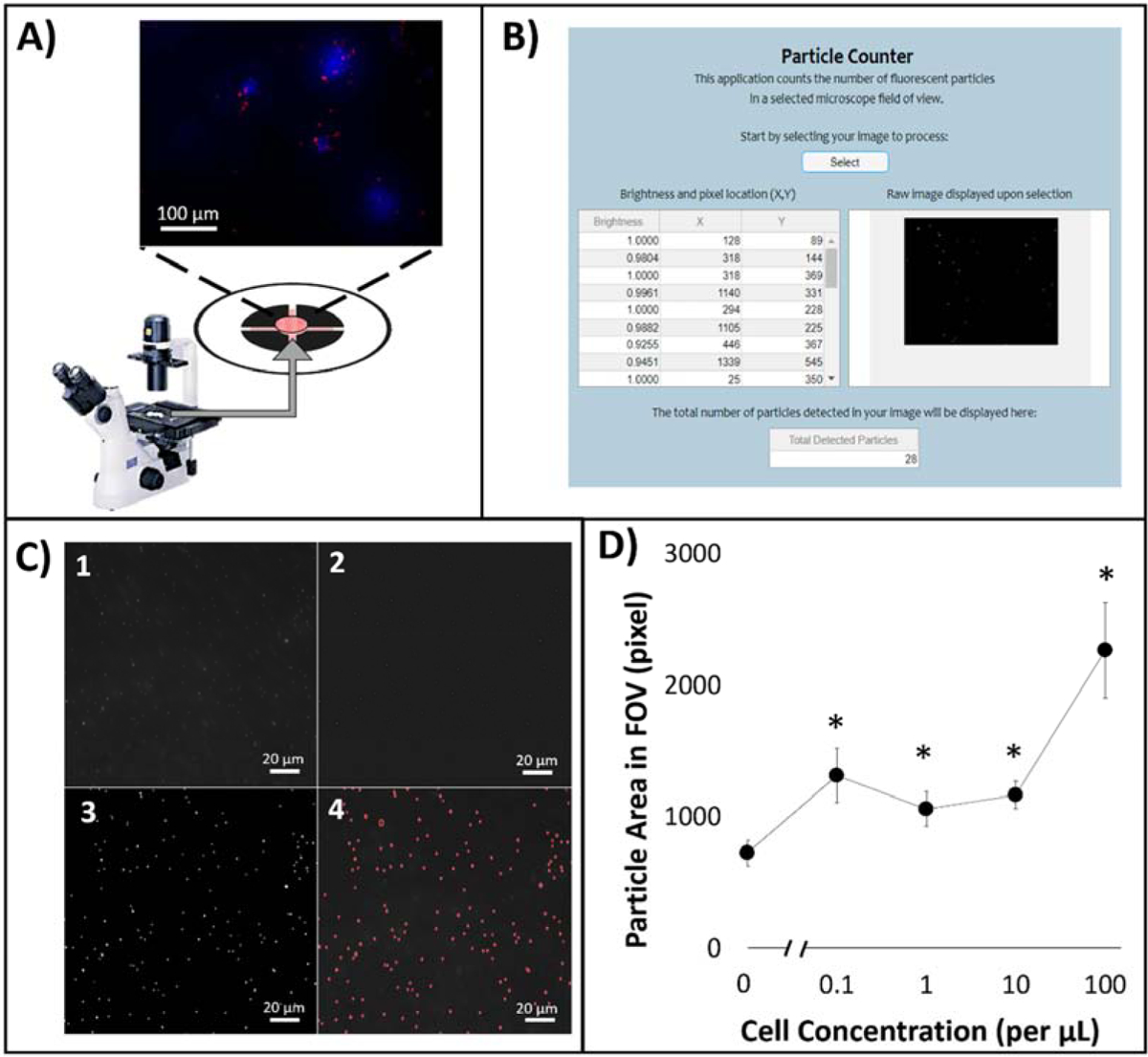
(A) On-chip particle counting analysis from the first capture layer of the dual-layer paper microfluidic chip using a benchtop fluorescence microscope. (B) MATLAB graphical user interface (GUI) for counting particle areas. (C) Raw and processed images: (1) raw 400X image, (2) Fourier bandpass filtered image with adjusted brightness and contrast, (3) image after grayscale normalization and global threshold binarization, and (4) final image with augmented particle detection. (D) Particle area in FOV in relation to dosed ROR1+ cell concentration from 0.1 to 100 cells/μL.
3.4. On-chip particle counting using a smartphone-based fluorescence microscope
The correlation between particles area in FOV and concentration of dosed ROR1+ cancer cells in buffy coat was demonstrated utilizing a smartphone-based fluorescence microscope, for its application in a more user-friendly and field-deployable manner. As shown in Figure 4A, a smartphone microscope was attached to the camera of a smartphone via a phone case and used to capture images of fluorescent anti-ROR1 particles on the second flow layer of the dual-layer paper microfluidic platform. To filter true signals from noise, especially from the GF/D glass fiber substrate, further physical light filtering and image digital processing were utilized. The smartphone attachment was comprised of three components: 1) 589 ± 2 nm optical bandpass filter that allows only the fluorescence emission from anti-ROR1 particles to the smartphone camera, 2) a commercially available smartphone microscope, replaced with a 525 nm LED, specific to the excitation wavelength to the anti-ROR1 particles, and 3) 3D printed enclosure and user-movable stage that housed the paper microfluidic chip with captured cells and particles. The optical bandpass filter mitigates weaker, non-specific fluorescence as well as reducing the amount of light. A Fourier bandpass filtering and binary thresholding algorithm (used for the benchtop fluorescence images) was also used to quantifying particles in FOV (Figure 4B). The MATLAB particle analyzer code and its GUI used for benchtop fluorescence microscopy (Figure 3B) were also used for smartphone-based fluorescence microscopy, using MATLAB Mobile (Supplementary Figure S2A). The operation procedure for final user is also illustrated in Supplementary Figure S2B.
Figure 4.
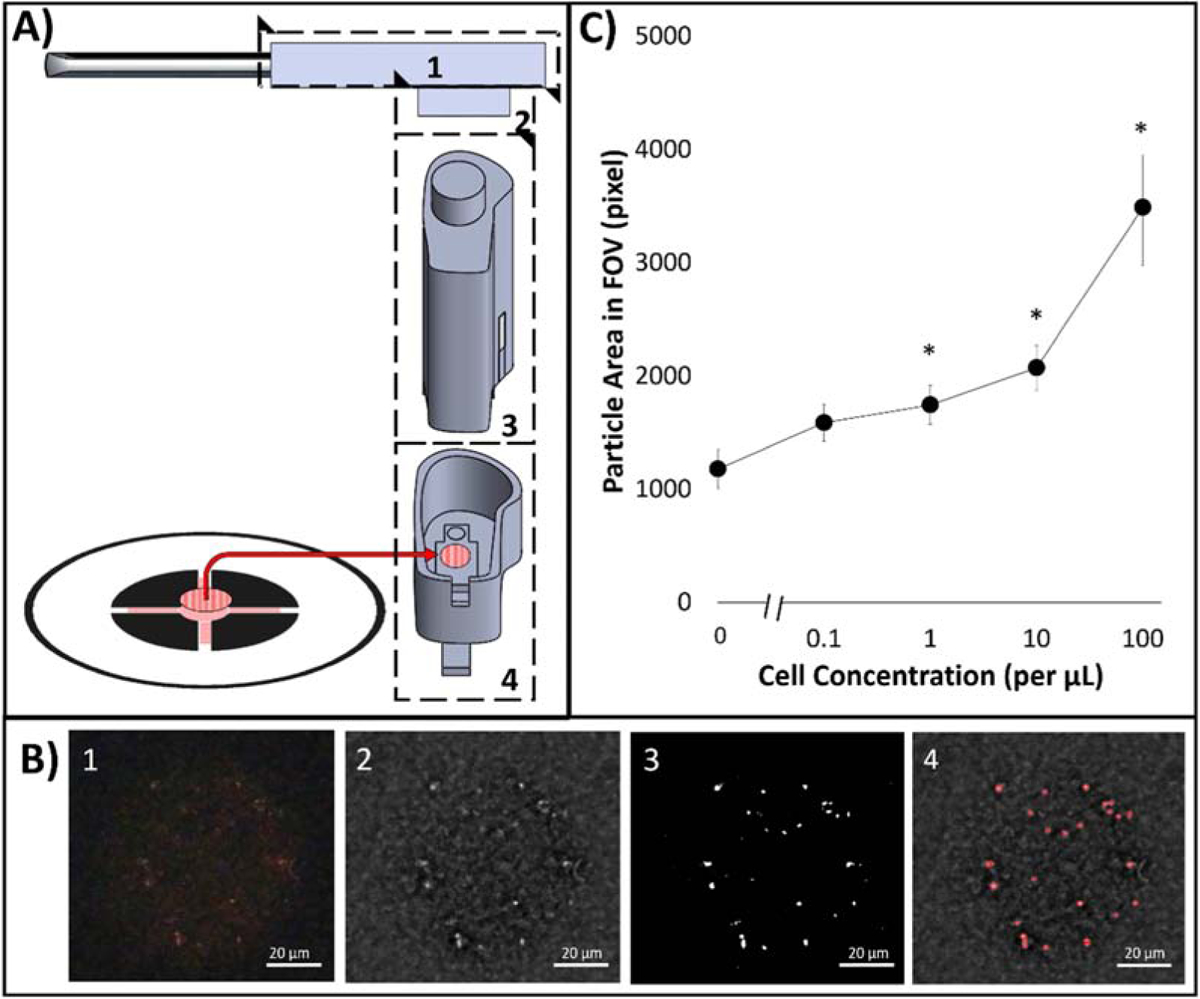
(A) On-chip particle counting analysis from the first capture layer of the dual-layer paper microfluidic chip using a smartphone-based fluorescence microscope. The smartphone-based fluorescence microscope is comprised of (1) a smartphone case (180 mm × 80 mm), (2) a 25-mm diameter, 589 ± 2 nm optical bandpass filter, (3) a commercial smartphone microscope attachment, replaced with a 525 nm LED, and (4) a 3D printed enclosure and a moveable stage with manual user lever. (B) Raw and processed smartphone images: (1) raw cropped 300X image, (2) Fourier bandpass noise filtered image, (3) image after grayscale normalization and global threshold binarization with threshold of 0.4, and (4) final image with augmented particle detection. (C) Particle areas in FOV in relation to dosed ROR1+ cell concentration from 0.1 to 100 cells/μL.
ROR1+ cells were serially diluted in buffy coat to obtain cell concentrations of 0.1, 1, 10, and 100 cells/μL. 200X images were taken via smartphone-based microscope, processed, and measured for particle area in FOV. A similar trend to the benchtop fluorescence microscope was observed. With increasing cell concentration, particle area in FOV present increased. At 100 cells/μL, an average particle area of 3487 pixels were measured in FOV and was statistically different in comparison to no cells (NTC). Similarly, 10 and 1 cells/μL concentration dosed in buffy coat were statistically different from NTC with average particle areas in FOV of 2071 and 1744 pixels. However, 0.1 cells/μL was not statistically different from NTC having an average particle area in FOV of 1179 pixels (Figure 4C).
3.5. Flow velocity analysis using a smartphone camera
The second flow layer of the dual-layer microfluidic chip was designed to have four channels to wick the sample solution from the center and to quantify antigens and cell fragments not captured in the first capture layer. A smartphone captured 30 fps videos during the assay, which were parsed into 1 s interval images, color enhanced, and measured for flow distance (Figure 5A). Antibody-conjugated particles are aggregated upon binding to targets, in this case, mostly antigens released from cells or possibly cell fragments, but not the whole cells (ranging from 5 to 10 μm) as they are mostly captured in the first capture layer and are too big to flow through the second flow layer (20 μm). Non-aggregated particles easily adsorb to the liquid-air interface (i.e. moving front) within paper fibers due to their relative hydrophobicity, and subsequently lowers the interfacial tension. Aggregated particles, however, diffuse to the liquid-air interface substantially slower, and the resulting flow velocities (flow distances) should be substantially different between non-aggregated and aggregated particles, i.e. between target non-presence and presence [14] (see Figure 1C for illustrated flow detection phenomenon). The presence and nature of blood components also affects the flow velocity (distance) [32].
Figure 5.
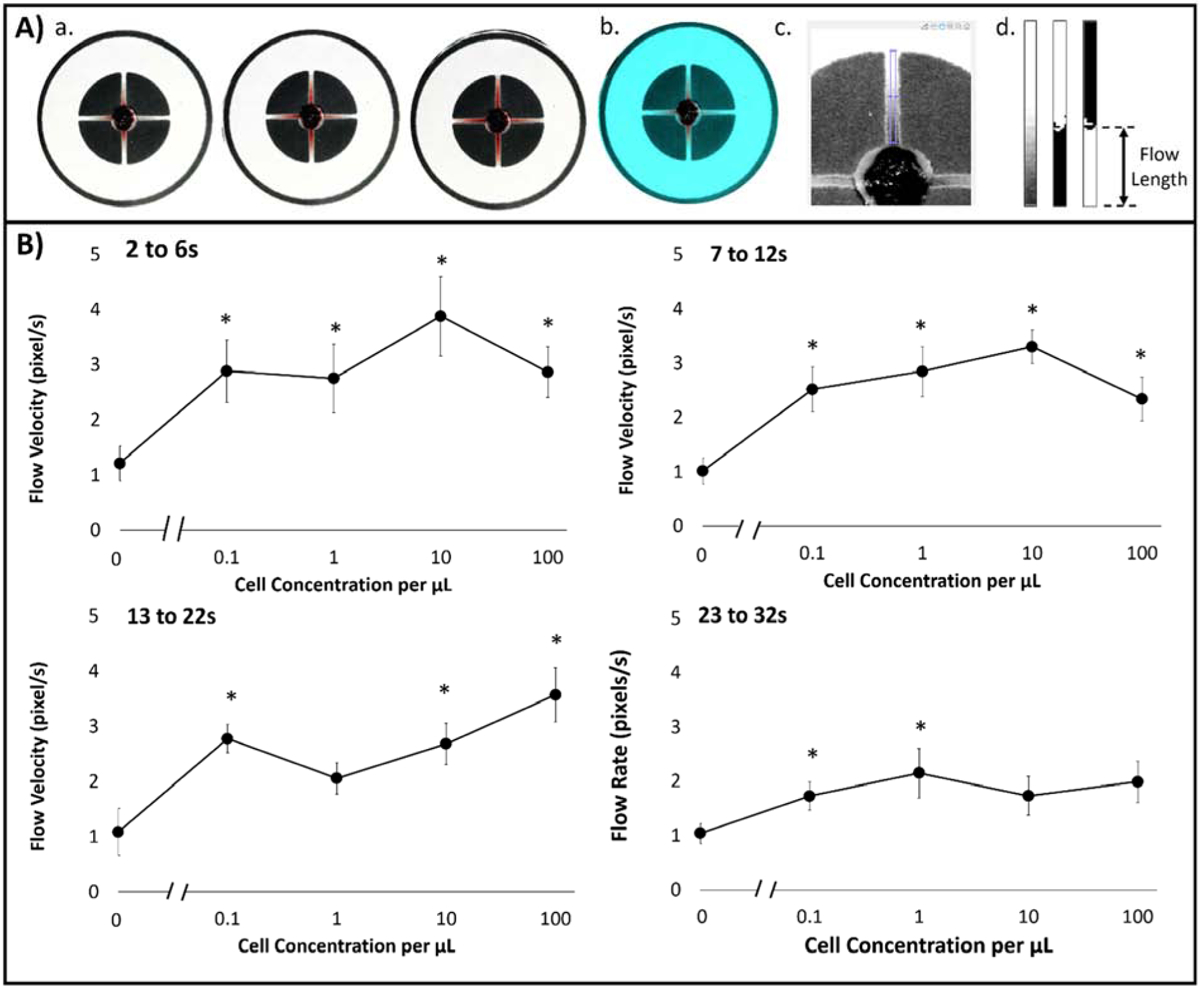
(A) Image processing algorithm for evaluating capillary flow velocities from smartphone-acquired videos: (1) Parsed 30 fps video to 1 s image shots; (2) Color balanced image to [150, 100, 100] RGB pixel intensities; (3) Extracted green channel image with user-designated channel crop tool; (4) Cropped, binary, and inverted images of channel of interest, from left to right. (B) Capillary flow velocities in relation to dosed buffy coat with cell concentration from 0 to 102 cells/μL at four different time intervals: Δt1, Δt2, Δt3, and Δt4, which correlated to 2 to 6 s, 7 to 12 s, 13 to 22 s, and 23 to 32 s, respectively.
The flow velocity in which applied sample flows from the loading zone was investigated, originating from spontaneous wicking effects of paper in correlation to cancer cell concentration present in buffy coat. Flow velocity was measured at four time-intervals: Δt1, Δt2, Δt3, and Δt4, which correlated to 2 to 6 s, 7 to 1 2s, 13 to 22 s, and 23 to 32 s, respectively (Figure 5B). Δt1 showed the greatest sensitivity and significant differences in flow velocities for all cell concentrations in comparison to NTC (no cancer cells) samples. Average flow velocities ranged from 2.75 pixels/s at 1 cell/μL and 3.88 pixels/s at 10 cells/μL. Δt2 and Δt3 showed significant differences in average flow velocities for all cell concentrations in comparison to NTC samples except for 1 cell/μL at Δt2. Average flow velocities ranged from 2.05 pixel/s and 3.57 pixels/s. Lastly, Δt4 displayed the least dynamic range and least significant difference in average flow velocity to NTC samples. Overall Δt3 time interval flow velocities in correspondence to dosed cancer cells in buffy coat showcased the best overall dynamic range, where 100 cells/μL (high concentration) delayed signal, was able to be detected. Therefore, Δt3 time interval flow velocities were used as the empirical comparison to a capillary fluid dynamic model.
3.6. Comparison with Lucas-Washburn capillary flow model
A common and widely used model for capillary flow through paper microfluidics is the Lucas-Washburn (L-W) model relating flow distance (l) over time (t) [33].
| (1) |
As demonstrated in [16], the changes in R (pore size) and ϑ (contact angle) were negligible compared to those in viscosity (μ) and interfacial tension (γLG) upon changing the target concentration and particle aggregation. Therefore, we assumed R and ϑ to be constant and measured μ and γLG of 0 to 102 cells/μL cell suspensions with anti-ROR1 particles as input parameters into the model. Relative viscosity (μ) was determined from shear stress (τ) versus shear rate (ẏ) curves (Supplementary Figure S3) via cone-plate rheology. It should be noted that 0.1 cells/ μL was mitigated from data set since number of cancer cells in sample volume is too low for accurate rheology measurements due to competitive adsorption to surface [30]. It showed an inverse relationship with increased cell concentration, with up to a 31% decrease in viscosity at 100 cells/μL (Figure 6A). Relative interfacial tension (γLG) measurements via pendant droplet analysis showed no significant trends with increased cell concentration. IFT differed from no cell samples by an increased 5% at the maximum concentration of 100 cells/μL (Figure 6B). Therefore, the most influential driving force for our system with regards to capillary flow-based detection is viscosity.
Figure 6.
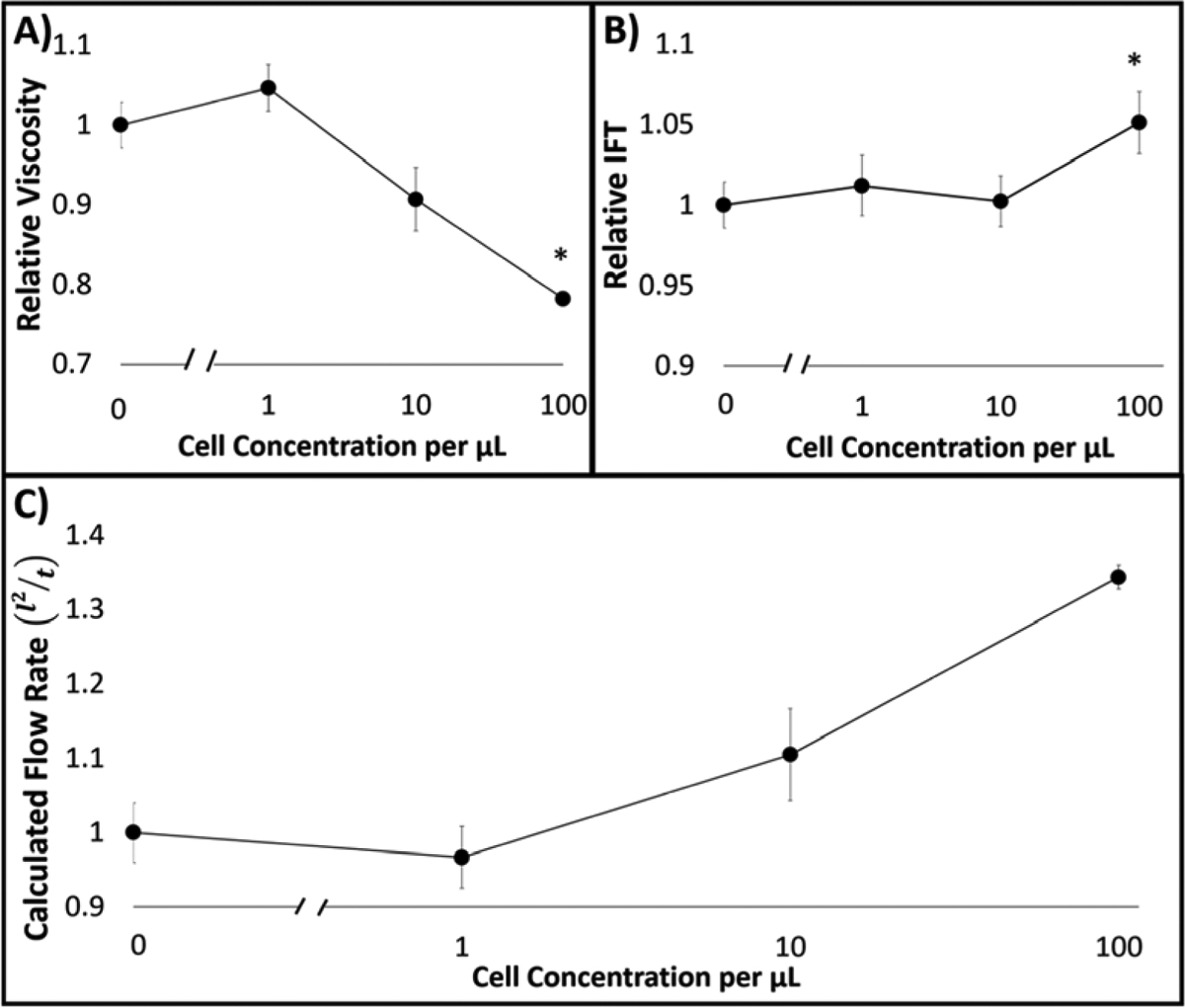
(A) Relative viscosity (μ) measurements via cone-plate rheology for 0 to 102 cells/μL cell suspensions with anti-ROR1 particles. (B) Relative interfacial tension (γLG) measurements via pendant drop analysis for the same. (C) Relative flow (l2/t) normalized to zero cell concentration, calculated with Lucas-Washburn capillary flow model using the parameters obtained from A and B.
Inputting measured viscosity and IFT values to the equation 1 and normalized to that of 0 cell/μL provided the relative flow rate (l2/t), where the constant R and ϑ are cancelled out. It resulted in an increasing flow as cell concentration increases with up to 34% at 100 cells/μL in comparison to no cell samples. Relaying back to the empirical data collected for flow velocities with varying cancer cell concentrations in comparison to the L-W modified model, similar trends were achieved (compare Figure 5B: Δt3 = 13 to 22 s and Figure 6C). This suggests that particle immunoagglutination due to free antigen presence decreases relative viscosity before the moving front contributing to less unbound particles at the interface, thus resulting in increased flow. In comparison, absence of particle immunoagglutination due to no target presence, allows more free particles to quickly move to the moving front increasing the relative flow along the channel (Figure 1C).
This viscosity contribution to the flow velocity-based assay may be affected by another factor: the viscosity variance of buffy coat blood samples among patients. In a case study [34], blood rheology parameters were investigated for red blood cells, white blood cells, and total cholesterol levels. Changes in blood viscosity were primarily due to red blood cell related factors, but not the white blood cell or cholesterol related factors. Even the variance in red blood cells was marginal when the shear rate is lower than 40 s−1. Therefore, patient variations can potentially be neglected when the buffy coat samples are tested.
4. CONCLUSION
In summary, we have developed a dual-layer paper microfluidic chip quantifying both whole cells captured on-chip through imaging the antibody conjugated fluorescent particles and antigen/cell fragment through monitoring capillary flow velocities. This proposed method was demonstrated for detecting ROR1+ cancer cells in a complex matrix of buffy coat samples that would be similar and relevant for clinical use. The similarity between these two assays also suggests that they can be used in a complementary manner in quantifying ROR1+ cancer cells. It is a quick and low-cost alternative to detect hematological cancer including chronic lymphocytic leukemia (CLL) in comparison to traditional, lengthy, and expensive cell counting methodologies and label-specific assays. MATLAB codes and GUIs were developed for on-chip image analysis as well as flow measurement. Direct imaging of particles found in the capture layer demonstrated the device’s ability to quantify cell concentration down to 1 cell/μL utilizing a smartphone based fluorescent microscope. Flow velocity measurements from parsed and color-enhanced smartphone videos showed statistical differences from no target samples as early as 6 s for the cell concentration down to 0.1 cells/μL. Flow measurements were validated and fitted to a Lucas-Washburn (L-W) model that was fed empirically collected relative viscosity and interfacial tension measurements. The L-W model fitting suggested that the flow detection method for our specific system, was driven by viscosity as most influential parameter contributing to increased flow with increased cell concentration. The overall procedure requires one pipetting for loading a sample, separation of a dual–layer paper microfluidic chip, and acquisition of a video and microscope images, with the assay time less than 10 minutes. This device and method can easily be translated and modified for clinical applications as well as other targets of interests such as other ROR1+ cancer cells (with high cell load), mammalian, bacterial, or plant cell types.
Supplementary Material
HIGHLIGHTS.
Dual layer paper microfluidic chip identifies ROR1+ cells directly from buffy coat blood samples
Cells are captured on the capture layer, while antigens flow through the flow layer
Smartphone quantified the cells by microscopic counting and the antigens by flow velocities
Capillary flow model identified viscosity as a major contributor but not interfacial tension
Detection limit is 0.1 cells/μL from 10 μL sample, without any dilution, concentration, or incubation
ACKNOWLEDGEMENTS
The authors would like to thank Ms. Katelyn Sosnowski for help and expertise in MATLAB flow analysis software Dr. Minkyu Kim for rheology instrumentation and lab access. R.Z. acknowledges W.L. Gore & Associates, Inc. for undergraduate research fellowship. A.G. acknowledges The University of Arizona’s Maximizing Access to Research Careers (MARC) program for undergraduate research fellowship, funded by U.S. National Institutes of Health (grant number T34GM008718). This work was supported by the Cardiovascular Biomedical Engineering Training Grant from U.S. National Institutes of Health (grant number T32HL007955).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
REFERENCES
- [1].Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Döhner H, Hillmen P, Keating MJ, Montserrat E, Rai KR, Kipps TJ 2008. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: A report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute - Working Group 1996 guidelines. Blood 111, 5446–5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Rozman C, Montserrat E 1995. Chronic lymphocytic leukemia. New Engl. J. Med 333, 1052–1057. [DOI] [PubMed] [Google Scholar]
- [3].Rai KR, Stilgenbauer S, Aster J 2019. Clinical features and diagnosis of chronic lymphocytic leukemia/small lymphocytic lymphoma - UpToDate.<https://www.uptodate.com/contents/clinical-features-and-diagnosis-of-chronic-lymphocytic-leukemia-small-lymphocytic-lymphoma> (accessed 07-Sep-2019).
- [4].Global Burden of Disease Cancer Collaboration. 2017. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: A systematic analysis for the global burden of disease study. JAMA Oncol 3, 524–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Rai KR, Sawitsky A, Cronkite EP, Chanana AD, Levy RN, Pasternack BS 1975. Clinical staging of chronic lymphocytic leukemia. Blood 46, 219–234.1139039 [Google Scholar]
- [6].Abruzzo LV, Lee KY, Fuller A, Silverman A, Keating MJ, Medeiros LJ, Coombes KR 2005. Validation of oligonucleotide microarray data using microfluidic low-density arrays: A new statistical method to normalize real-time RT-PCR data. BioTechniques 38, 785–792. [DOI] [PubMed] [Google Scholar]
- [7].Abruzzo LV, Barron LL, Anderson K, Newman RJ, Wierda WG, O’Brien S, Ferrajoli A, Luthra M, Talwalkar S, Luthra R, Jones D, Keating MJ, Coombes KR 2007. Identification and validation of biomarkers of IgVH mutation status in chronic lymphocytic leukemia using microfluidics quantitative real-time polymerase chain reaction technology. J. Mol. Diagn 9, 546–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ensafi AA, Amini M, Rezaei B, Talebi M 2016. A novel diagnostic biosensor for distinguishing immunoglobulin mutated and unmutated types of chronic lymphocytic leukemia. Biosens. Bioelectron 77, 409–415. [DOI] [PubMed] [Google Scholar]
- [9].Jackson JM, Taylor JB, Witek MA, Hunsucker SA, Waugh JP, Fedoriw Y, Shea TC, Soper SA, Armistead PM 2016. Microfluidics for the detection of minimal residual disease in acute myeloid leukemia patients using circulating leukemic cells selected from blood. Analyst 141, 640–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Vickers DAL, Hincapie M, Hancock WS, Murthy SK 2011. Lectin-mediated microfluidic capture and release of leukemic lymphocytes from whole blood. Biomed. Microdev 13, 565–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zheng Y, Wen J, Nguyen J, Cachia MA, Wang C, Sun Y 2015. Decreased deformability of lymphocytes in chronic lymphocytic leukemia. Sci. Rep 5, 7613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Qu X, Li M, Zhang H, Lin C, Wang F, Xiao M, Zhou Y, Shi J, Aldalbahi A, Pei H, Chen H, Li L 2017. Real-time continuous identification of greenhouse plant pathogens based on recyclable microfluidic bioassay system. ACS Appl. Mater. Interfaces 9, 31568–31575. [DOI] [PubMed] [Google Scholar]
- [13].Qu X, Xiao M, Li F, Lai W, Li L, Zhou Y, Lin C, Li Q, Ge Z, Wen Y, Pei H, Liu G 2018. Framework nucleic acid-mediated pull-down microRNA detection with hybridization chain reaction amplification. ACS Appl. Bio Mater 1, 859–864. [DOI] [PubMed] [Google Scholar]
- [14].Qi L, Xiao M, Wang X, Wang C, Wang L, Song S, Qu X, Li L, Shi J, Pei H 2017. DNA-encoded Raman-active anisotropic nanoparticles for microRNA detection. Anal. Chem 89, 9850–9856. [DOI] [PubMed] [Google Scholar]
- [15].Ulep T-H, Yoon J-Y 2018. Challenges in paper-based fluorogenic optical sensing with smartphones. Nano Converg 5, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chung S, Breshears LE, Perea S, Morrison CM, Betancourt WQ, Reynolds KA, Yoon J-Y 2019. Smartphone-based paper microfluidic particulometry of norovirus from environmental water samples at the single copy level. ACS Omega, 4, 11180–11188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Klug KE, Reynolds KA, Yoon J-Y 2018. A capillary flow dynamics-based sensing modality for direct environmental pathogen monitoring. Chem. Eur. J 24, 6025–6029. [DOI] [PubMed] [Google Scholar]
- [18].Chung S, Jennings CM, Yoon J-Y 2019. Distance vs. capillary flow dynamics-based detection methods on a microfluidic paper-based analytical device (μPAD). Chem. Eur. J 25, 13070–13077. [DOI] [PubMed] [Google Scholar]
- [19].Uhrmacher S, Schmidt C, Erdfelder F, Poll-Wolbeck SJ, Gehrke I, Hallek M, Kreuzer K-A 2011. Use of the receptor tyrosine kinase-like orphan receptor 1 (ROR1) as a diagnostic tool in chronic lymphocytic leukemia (CLL). Leukemia Res 35, 1360–1366. [DOI] [PubMed] [Google Scholar]
- [20].Aghebati-Maleki L, Shabani M, Baradaran B, Motallebnezhad M, Majidi J, Yousefi M 2017. Receptor tyrosine kinase-like orphan receptor 1 (ROR-1): An emerging target for diagnosis and therapy of chronic lymphocytic leukemia. Biomed. Pharmacother 88, 814–822. [DOI] [PubMed] [Google Scholar]
- [21].Daneshmanesh AH, Porwit A, Hojjat-Farsangi M, Jeddi-Tehrani M, Tamm KP, Grandér D, Lehmann S, Norin S, Shokri F, Rabbani H, Mellstedt H, Österborg A 2013. Orphan receptor tyrosine kinases ROR1 and ROR2 in hematological malignancies. Leukemia Lymphoma 54, 843–850. [DOI] [PubMed] [Google Scholar]
- [22].Yu J, Chen L, Hasan MK, Ghia EM, Zhang L, Wu R, Rassenti LZ, Widhopf GF, Shen Z, Briggs SP, Kipps TJ 2017. Wnt5a induces ROR1 to associate with 14–3-3ζ for enhanced chemotaxis and proliferation of chronic lymphocytic leukemia cells. Leukemia 31, 2608–2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Herlein E, Beckwith KA, Lozanski G, Chen TL, Towns WH, Johnson AJ, Lehman A, Ruppert AS, Bolon B, Andritsos L, Lozanski A, Rassenti L, Zhao W, Jarvinen TM, Senter L, Croce CM, Symer DE, Chapelle A Heerema NA, Byrd JC 2013. Characterization of a new chronic lymphocytic leukemia cell line for mechanistic in vitro and in vivo studies relevant to disease. PLoS ONE 8, e76607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhang S, Chen L, Wang-Rodriguez J, Zhang L, Cui B, Frankel W, Wu R, Kipps TJ 2012. The onco-embryonic antigen ROR1 is expressed by a variety of human cancers. Am. J. Pathol 181, 1903–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Yamaguchi T, Yanagisawa K, Sugiyama R, Hosono Y, Shimada Y, Arima C, Kato S, Tomida S, Suzuki M, Osada H, Takahashi T 2012. NKX2–1/TITF1/TTF-1-induced ROR1 is required to sustain EGFR survival signaling in lung adenocarcinoma. Cancer Cell 21, 348–361. [DOI] [PubMed] [Google Scholar]
- [26].Abcam, 2015. Cell Culture Guidelines. Abcam, Cambridge: <https://www.abcam.com/ps/pdf/protocols/cell_culture.pdf> (accessed 07-Sep-2019). [Google Scholar]
- [27].Molday RS, Dreyer WJ, Rembaum A, Yen SP 1975. New immunolatex spheres: visual markers of antigens on lymphocytes for scanning electron microscopy. J. Cell. Biol 64, 75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Roy D, 2009. AUTO CONTRAST - Automatic Adjustment of Contrast of Images, version 1.1.0.0. MathWorks, Natick: <https://www.mathworks.com/matlabcentral/fileexchange/10566-auto-contrast> (accessed 07-Sep-2019). [Google Scholar]
- [29].MathWorks, 2019. chromadapt - Adjust Color Balance of RGB Image with Chromatic Adaptation. <https://www.mathworks.com/help/images/ref/chromadapt.html> (accessed 07-Sep-2019).
- [30].Merrill EW 1969. Rheology of blood. Physiol. Rev 49, 863–888. [DOI] [PubMed] [Google Scholar]
- [31].Kim S, Cho YI, Jeon AH, Hogenauer B, Kensey KR, 2000. A new method for blood viscosity measurement. J. Non-Newtonian Fluid Mech 94, 47–56. [Google Scholar]
- [32].Sweeney RE, Nguyen V, Alouidor B, Budiman E, Wong RK, Yoon J-Y 2019. Flow rate and Raspberry Pi-based paper microfluidic blood coagulation assay device. IEEE Sens. J 19, 4743–4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Gasperino D, Baughman T, Hsieh HV, Bell D, Weigl BH 2018. Improving lateral flow assay performance using computational modeling. Annu. Rev. Anal. Chem 11, 219–244. [DOI] [PubMed] [Google Scholar]
- [34].Persson SU, Gustavsson CG, Larsson H, Persson S 1991. Studies on blood rheology in patients with primary pulmonary hypertension. Angiology 42, 836–842. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.