

Abstract
Recent crystal structures of multiple G protein-coupled receptors (GPCRs) have revealed a highly conserved intracellular pocket that can be used to modulate these receptors from the inside. This novel intracellular site partially overlaps with the G protein and β-arrestin binding site, providing a new manner of pharmacological intervention. Here we provide an update of the architecture and function of the intracellular region of GPCRs, until now portrayed as the signaling domain. We review the available evidence on the presence of intracellular binding sites among chemokine receptors and other class A GPCRs, as well as different strategies to target it, including small molecules, pepducins and nanobodies. Finally, the potential advantages of intracellular (allosteric) ligands over orthosteric ligands are also discussed.
Keywords: G protein-coupled receptors, Allosteric modulation, Antagonism, Intracellular binding site, Small molecules
Multiple binding sites to target a GPCR
G protein-coupled receptors (GPCRs, see Glossary) comprise one of the largest families of drug targets, with approximately 34% of the currently marketed drugs targeting this receptor class [1]. As lack of efficacy continues to be the main reason of failure in Phase II and Phase III clinical trials [2], novel approaches to successfully target these receptors are still necessary. As it is apparent from most GPCR crystal structures reported so far, small molecules often occupy a binding site exposed to the extracellular solvent—the so-called orthosteric binding site which is used by endogenous ligands [3] (Figure 1A). However, targeting GPCRs has proved to be quite challenging, especially when drugs need to compete with a high (local) concentration of the endogenous ligand, as is the case of targeting chemokine receptors during inflammatory conditions [4]. Hence, the development of allosteric modulators (Box 1) that bind to spatially distinct binding sites [5] has emerged as a promising approach to improve not only drug efficacy, but also selectivity and safety [6–8]. A variety of different allosteric binding sites have already been identified in GPCRs, most of them close to the orthosteric binding site; yet, unexpected ligand binding sites have recently been found in crystal structures of class A and class B GPCRs [5]. In this regard, the recent crystal structures of CC chemokine receptor 2 (CCR2) [9], CC chemokine receptor 9 (CCR9) [10], and β2-adrenergic receptor (β2AR) [11] have for the first time revealed a spatially conserved intracellular binding site for small molecules in class A GPCRs (Figure 1A), providing a new avenue to inhibit or modulate these receptors in different pathologies.
Figure 1. Novel allosteric binding site in class A GPCRs.
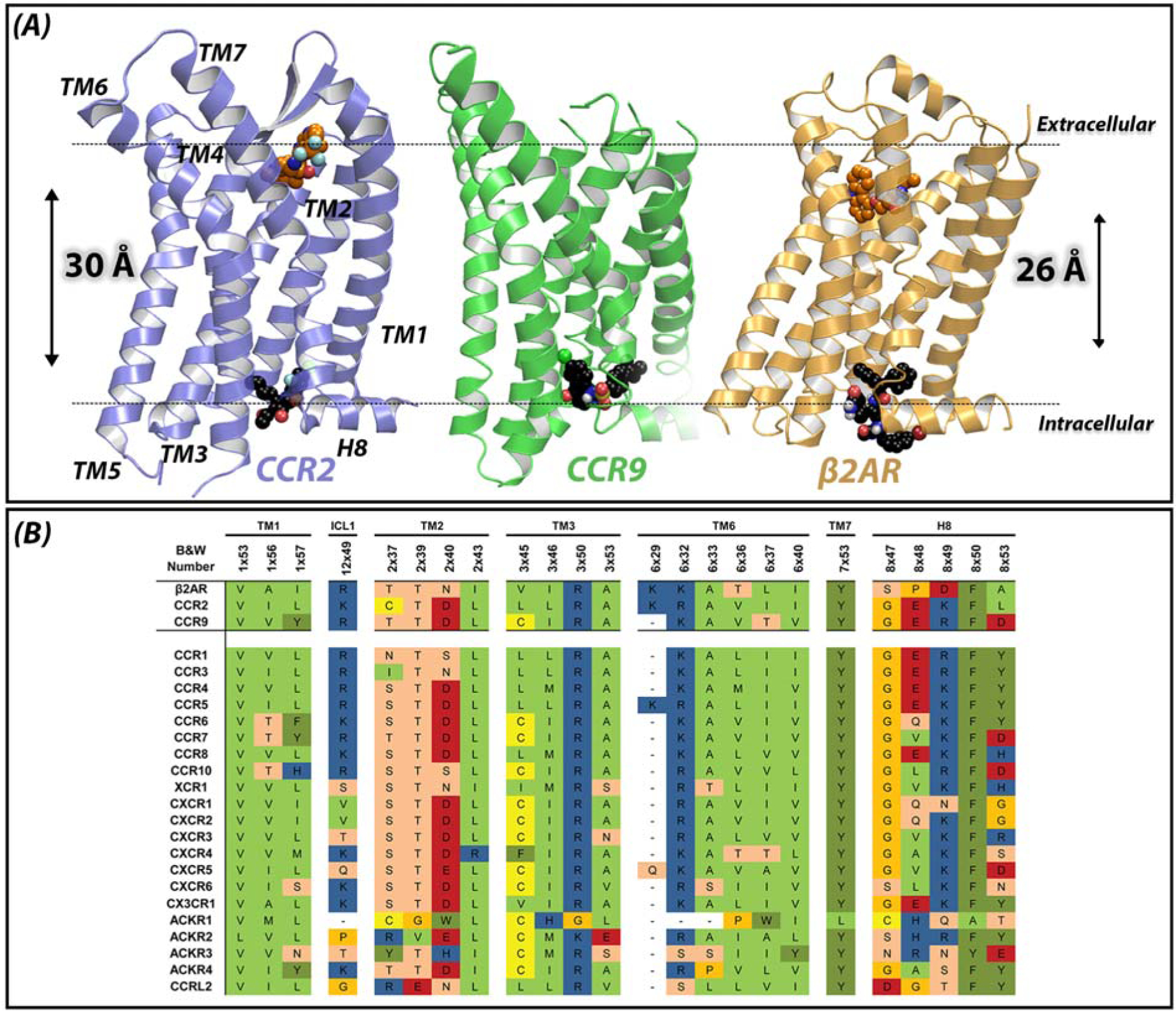
(A) Endogenous ligands bind close to the extracellular region of GPCRs, in the so-called orthosteric binding site. Most of the co-crystallized small molecules also bind in this extracellular region, such as BMS-681 in CCR2 and carazolol in β2AR. Recently, the crystal structures of CCR2 (purple, PDB 5T1A), CCR9 (green, PDB 5LWE) and β2AR (yellow, PDB 4XT1) have revealed an allosteric solvent-exposed binding site, located in the intracellular region of GPCRs, around 30 Å away from the orthosteric binding site. This novel binding site challenges the traditional view of the upper 7TM region of GPCRs as ligand binding domain and the intracellular region as signaling domain only. As shown in the structures, this intracellular binding site can also be targeted by small molecules such as CCR2-RA-[R] in CCR2, vercirnon in CCR9 and 15-PA in β2AR. Dotted lines represent the plane of the membrane. (B) Sequence conservation among chemokine receptors and β2AR, based on the GPCR database (GPCRdb, http://www.gpcrdb.org). Residues shown are residues involved in the intracellular binding site of CCR2, CCR9 and β2AR (upper three rows). Some of these residues have also been found to be important for ligand binding to other class A GPCRs, as well as for G protein and β-arrestin binding.
Box 1. Allosteric modulation in GPCRs.
G protein-coupled receptors (GPCRs) are considered natural allosteric proteins, as the site of interaction of the endogenous ligand—the orthosteric binding site—differs from the site of the signaling effectors, such as G proteins and β-arrestins [74]. In addition to the orthosteric site, GPCRs possess a variety of topologically distinct allosteric binding sites where ligands can bind [5]. When allosteric modulators bind, they modulate the activity of orthosteric ligands by inducing conformational changes in the receptor.
Orthosteric ligands are competitive and thus, they replace the endogenous ligand resulting in a single pharmacological state. In contrast, by modulating the activity of another ligand, allosteric ligands have the potential for fine-tuning a receptor response, maximizing the efficacy in some therapeutic contexts [6, 7], and/or minimizing the potential side effects and other liabilities [6, 8]. Depending on their effect, allosteric modulators can be divided in [6, 7, 75]:
Positive allosteric modulators (PAMs): Enhance the affinity and/or efficacy of the endogenous or orthosteric ligand.
Negative allosteric modulators (NAMs): Decrease the affinity and/or efficacy of the endogenous or orthosteric ligand.
Ago-PAMs: PAMs with some inherent level of agonist activity on their own
Silent allosteric modulators (SAMs): Have no effect on the affinity or efficacy of the endogenous or orthosteric ligand. Their presence may lead to for instance enhanced thermostability of the receptor and increased signaling lifetime.
Some key pharmacological properties of allosteric modulators are:
Insurmountability: The ability of allosteric ligands to cause a decrease in the potency and/or efficacy of the endogenous agonist, even when the endogenous ligand is present at high concentrations.
Selectivity: Generally, allosteric binding sites show less evolutionary pressure leading to a less-conserved amino acid sequence and thus, higher ligand selectivity that the orthosteric binding site. If an allosteric site is highly conserved, selectivity can be achieved via optimization of cooperativity with the orthosteric ligand or by targeting specific non-conserved amino acids.
Saturability or ceiling effect: The limit of the pharmacological effect produced by the allosteric ligand due to saturation of the effect after full occupancy of the allosteric site.
Probe-dependence: Both the magnitude and direction of the allosteric effect achieved by the allosteric modulator are dependent on the orthosteric ligand used as a “probe”.
Biased signaling: The ability of a ligand to preferentially stabilize a conformation that leads to the selective activation of a signaling pathway.
Intracellular region of GPCRs: Beyond signaling
In general, GPCRs share a similar structure consisting of three different domains (Figure 1A): the extracellular domain that includes three extracellular loops (ECLs) and the N terminus, which vary in length and structure depending on the GPCR subfamily [12]; the transmembrane (TM) domain that comprises seven TM helices; and the intracellular domain that includes three intracellular loops (ICLs), an amphipathic helix (H8) and the C terminus [3]. Traditionally, the upper TM section and the extracellular domain have been considered to encompass the ligand binding domain. In contrast, the lower TM section and the intracellular domain have been considered to be the signaling domain [3, 13]. Structurally, the intracellular domain is more highly conserved and flexible than the extracellular region containing the orthosteric binding site [3, 13], which is probably related to a common mechanism of receptor activation and G protein-coupling [14]. In this regard, analysis of several active- and inactive-state crystal structures has revealed a conserved rearrangement of residue contacts near the G protein-binding site, involving residues 3×46 in TM3, 6×37 in TM6, and 7×53 from the highly conserved NPxxY motif located in TM7 (residues according to structure-based Ballesteros-Weinstein numbering [15]) [14]. In addition, this region is also involved in the coupling and selective recognition of different G proteins [16, 17] and other signaling proteins such as β-arrestin [18], which can lead to a multitude of different signaling pathways upon activation of a GPCR. Recently, the traditional view of a separate ligand binding and signaling domain has been challenged as more evidence suggests that the intracellular domain of GPCRs can also be bound by ligands and thus be used for receptor modulation (Figure 1) [5, 9–11].
A common intracellular binding site in class A GPCRs
Among GPCRs there is now mutational, pharmacological and structural evidence of ligand binding sites located at their intracellular interface. This evidence is particularly extensive in the case of chemokine receptors (Box 2); thus, before extending to other class A GPCRs, we will first review the evidence available for chemokine receptors.
Box 2. Chemokine Receptors.
Chemokine receptors represent one of the largest subfamilies within class A G protein-coupled receptors (GPCRs). So far, 23 chemokine receptors have been identified that can be activated by more than 45 chemokine ligands (IUPHAR/BPS Guide to Pharmacology, http://www.guidetopharmacology.org, accessed on 04–12-2017). Chemokines and chemokine receptors are subdivided in four different families, according to the number and arrangement of conserved cysteine residues in the N-terminus of the chemokine ligands: C, with only one conserved cysteine present; CC, CXC and CX3C, with zero, one and three extra residues between two conserved cysteine residues. [76].
Both chemokines and chemokine receptors comprise the so-called chemokine system, which plays an important role in the migration and positioning of immune cells in homeostatic or pathological conditions [77]. According to their immune function, chemokine receptors can be classified as homeostatic, or dual inflammatory/homeostatic [78]. The chemokine system is a complex, seemingly redundant system in which one chemokine ligand is able to activate multiple chemokine receptors, and one chemokine receptor can be activated by multiple chemokine ligands. Yet, evidence suggests it is a highly fine-tuned system as it is tightly regulated by specific spatial and temporal control of chemokine expression [79, 80].
Dysregulation of this complex system has been implicated in a variety of inflammatory and immune diseases, including arthritis, diabetes, inflammatory bowel disease and cancer [81]. Three drugs targeting chemokine receptors have already gained market approval: maraviroc, a small-molecule targeting CCR5; plerixafor, a small-molecule targeting CXCR4; and mogamulizumab, an anti-CCR4 antibody [76].
Intracellular binding site at Chemokine Receptors
The recent X-ray structure of CCR2 in complex with an orthosteric antagonist and the negative allosteric modulator (NAM, Box 1) CCR2-RA-[R] (PDB 5T1A) [9], and of CCR9 in complex with the NAM vercirnon (PDB 5LWE) [10] (Figure 2) have provided structural confirmation of such intracellular binding site in chemokine receptors. The two structures report an overlapping solvent-exposed binding site in the intracellular domain of these receptors, located more than 30 Å from the orthosteric binding site and enclosed by the intracellular ends of TM1 – TM3, TM6, TM7 and H8 (Figure 1A) [9, 10]. The NAMs bind this intracellular pocket where they interact with several conserved amino acid residues (Figure 1B).
Figure 2. Chemical structures of selected intracellular small molecule ligands for different class A GPCRs.
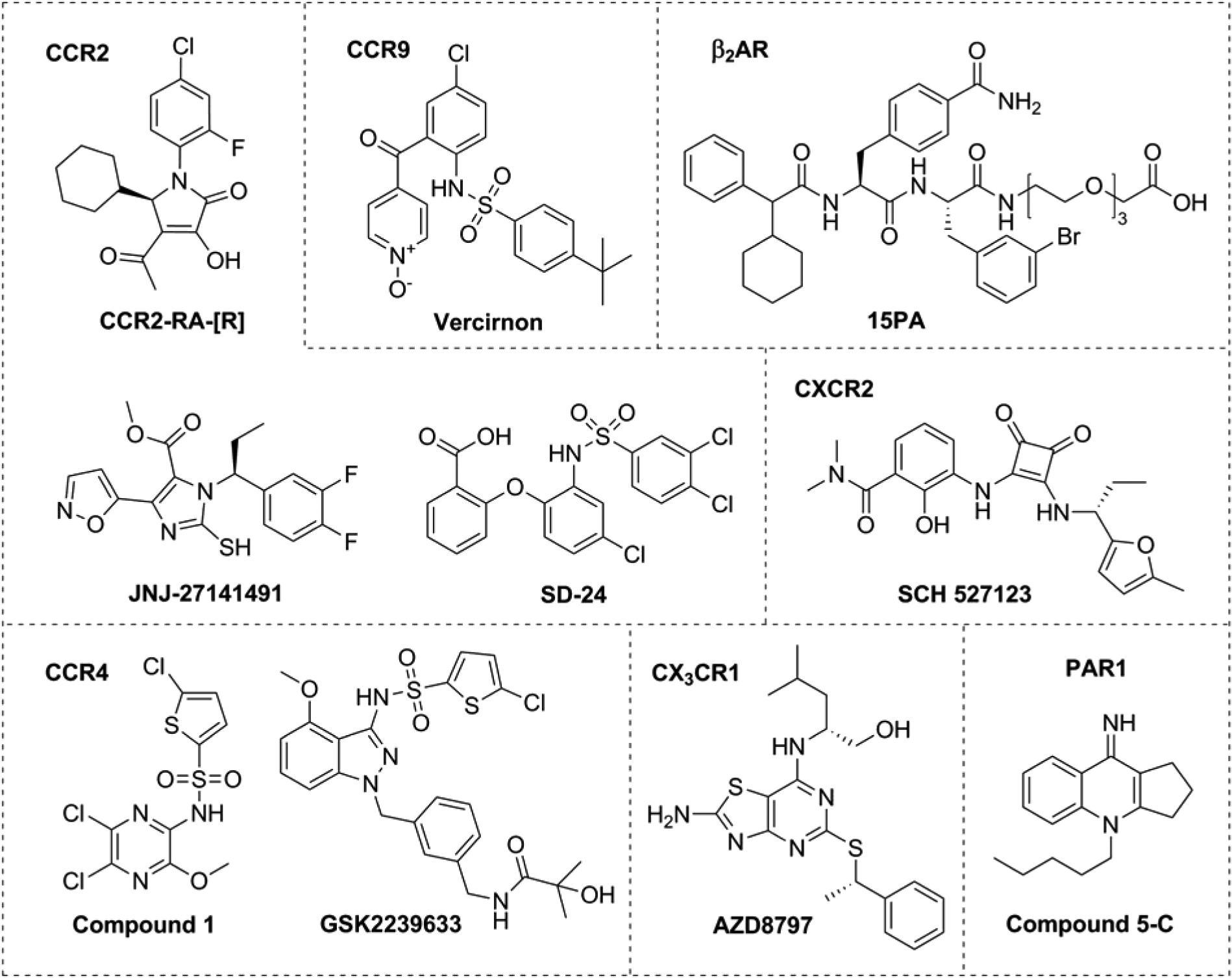
Upper row shows the chemical structures of cocrystallized intracellular ligands with their corresponding receptor: CCR2-RA-[R] with CCR2, Vercirnon with CCR9, and 15PA with β2AR. Vercirnon, SCH 527123 and GSK2239633 are examples of intracellular ligands that have progressed to clinical trials.
Interestingly, before these crystal structures were solved, intracellular ligand binding sites had already been suggested for chemokine receptors. In 2008, a putative intracellular binding site for small-molecule compounds had been identified in CCR4, CCR5, CXCR1 and CXCR2 [19, 20]. Functional data from these studies suggested that a series of compounds required intracellular access in order to exert their activity. Specifically, for CCR4 it was shown that several compounds similar to compound 1 (Figure 2) exhibited a lack of correlation in their potencies when measured in membrane or cellular assays. However, after permeabilization of the cells with saponin the potencies became comparable in both assays [19]. In CXCR2, the loss of cellular potency seemed to be dependent on the lipophilicity (logD) of the compounds. Lower lipophilicities resulted in a greater loss of potency, indicating that these compounds needed a certain level of lipophilicity to cross the cell membrane and reach the intracellular binding site [20]. A subsequent chimeric approach, with CCR4-CCR5 or CXCR1-CXCR2 chimeras, led to the suggestion that the C terminus was part of the binding site for these molecules [19, 20]. In CXCR2, this intracellular binding site was further mapped with help of homology modeling and mutational studies, which resulted in the identification of several C terminal residues as part of this allosteric binding site, including D842×40, T832×39, A2496×33, Y3147×53, and K3208×49 (Figure 1B) [20, 21]. Thus, these studies in CCR4 and CXCR2 provided the first biochemical evidence of the existence of such binding sites.
Using a similar approach, a homologous binding site was discovered in CCR2, where small molecules such as CCR2-RA-[R], JNJ-27141491 and SD-24 can bind (Figure 2) [22, 23]. Similar key residues were identified, including V2446×36, K3118×49, Y3057×53 and F3128×50 (Figure 1B) [22], which have now been confirmed by the X-ray structure [9]. A similar binding site has also been suggested in CX3CR1 after pharmacological characterization of compound AZD8797 (Figure 2), a non-competitive inhibitor of CX3CR1 with structural similarity to known CXCR2 intracellular ligands [24]. In addition, several pepducins derived from ICL1 of CXCR4 have been shown to interact selectively with CXCR4 in a non-competitive manner [25, 26]. Specifically, CXCR4 pepducin ATI-2341 has been predicted to interact with most of the residues located in ICL1 – ICL3 [27], indicating that this receptor can also be targeted from the intracellular side. Finally, the structure of the viral chemokine receptor US28 in complex with the chemokine ligand CX3CL1 and the nanobody Nb7 shows that Nb7 binds in a similar subpocket composed by the intracellular ends of TM3, TM5, TM6 and H8. Moreover, Nb7 interacts with several residues also involved in the binding of small-molecules or pepducins in human chemokine receptors, or in interactions with signaling proteins [28, 29].
Intracellular binding site at other class A GPCRs
This conserved intracellular binding site is not limited to chemokine receptors, as evidence for this site has been found in other class A GPCRs. In this regard, the crystal structure of β2AR (PDB 5X7D) has been solved with the small-molecule ligand 15PA (Figure 2), a polyethylene glycol- carboxylic acid derivative of compound 15 [30], co-crystalized at the intracellular interface [11]. Compound 15PA binds in a pocket formed by the intracellular ends of TM1, TM2, TM6, TM7, H8 and ICL1, where it interacts with key residues also identified in CCR2 and CCR9 [11] (Figure 3). Moreover, this binding pocket partially overlaps with the binding site of nanobodies Nb60 and Nb80 in β2AR [31, 32], Fab2838 in Adenosine A2A receptor (A2AAR) [33], Nb9–8 in M2R [34] and Nb39 in the μ-opioid receptor (μOR) [35]. Previous to these crystal structures, different computational tools predicted intracellular binding pockets in rhodopsin and M2R [36, 37]. Molecular docking studies and virtual screening identified several rhodopsin inhibitors that bind at the interface between the GPCR and G protein [38, 39], in an intracellular pocket resembling that identified in chemokine receptors. More evidence for a generalized intracellular pocket comes from the proteinase activated receptor 1 (PAR1), where a series of small molecules such as compound 5-C (Figure 2) and ICL3-derived pepducins were shown to interact with residues located in TM7 and H8 [40, 41]. Similar ICL-derived pepducins have also been developed for PAR2 [42], PAR4 [43], sphingosine-1-phosphate receptor 3 (S1P3) [44] and formylpeptide receptors 1 and 2 (FPR1 and FPR2) [45]. Taken together, there is mounting evidence for the presence of a spatially conserved intracellular pocket, not only in chemokine receptors but among several class A GPCRs.
Figure 3. Overview of structural features of the intracellular binding site.
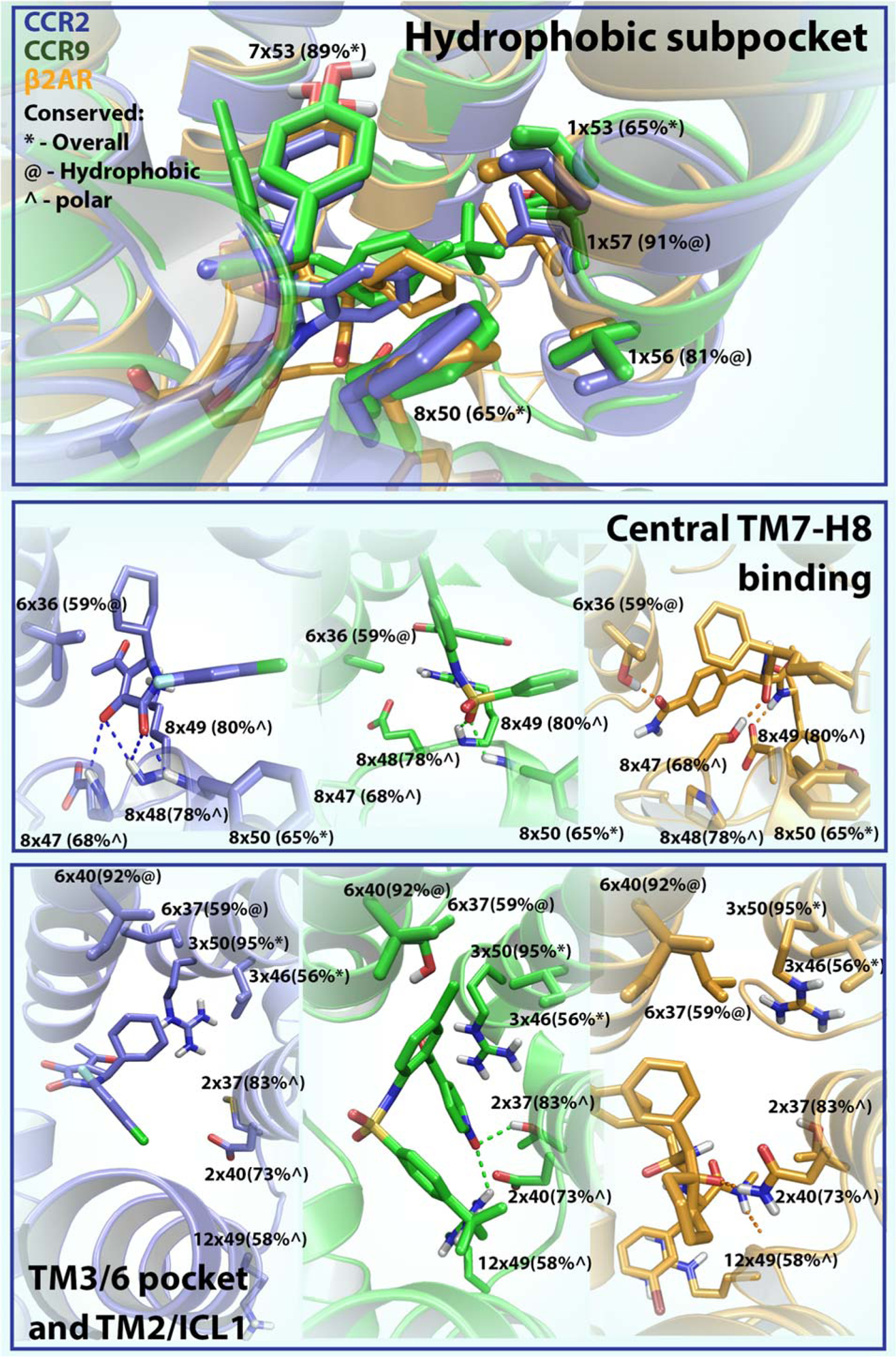
Common features in intracellular ligand binding derived from the crystal structures of CCR2 (PDB 5T1A), CCR9 (PDB 5LWE) and β2AR (PDB 4XT1). Residues are numbered using structure-based Ballesteros-Weinstein numbers [15]. Residue conservation among all class A GPCRs is shown in the following way; residues that are overall conserved (identical) in class A (>50%) are shown first (*); for residues that are not conserved we show how conserved they are in terms of polarity (^) or hydrophobicity (@). The three different boxes represent three different sections of the intracellular binding sites, in the upper panel all receptors are superimposed while in the lower two boxes the receptors are shown separately. CCR2 is colored blue, CCR9 is colored green and β2AR is colored orange.
Structural features of the intracellular binding site
The recent X-ray structures of CCR2 [9], CCR9 [10] and β2AR [11] are providing structural information on the features that determine binding and selectivity in this intracellular binding site (Figure 3, Key Figure). Moreover, these structures provide new opportunities for the application of structure-based drug design (SBDD) methods, such as virtual screening campaigns, which might allow the identification and/or optimization of novel intracellular ligands for these or other homologous receptors [5, 28]. Below, features of this site are discussed in terms of three component parts: a hydrophobic subpocket above H8, a central TM7-H8 binding region, and a region formed by TM3/6 and TM2/ICL1.
Hydrophobic subpocket
All ligands share a highly conserved hydrophobic subpocket above H8. Three highly conserved residues amongst class A GPCRs form the basis of this pocket: V1×53 (65% conserved), Y7×53 (89% conserved) and F8×50 (65% conserved) (Figure 3, upper panel). While there is only some evidence for the role of V1×53 in activation [46], numerous publications have shown the role of the latter two residues in signaling and intracellular ligand binding at different GPCRs [14, 47, 48]. In terms of hydrophobicity, residues 1×56 and 1×57 are also highly conserved (Figure 3, upper panel). However, in CCR9 Y1×57 adopts an orientation that further opens up the pocket, allowing the large 4-tert-butyl substituent of the ligand to reach deeper into this pocket, indicating a role in conferring ligand selectivity.
Central TM7-H8 binding region
The central part of the pocket consists of the kink between H8 and TM7, formed by either P8×48 (β2AR) or G8×47 (chemokine receptors). This subpocket includes residues 8×47 to 8×49, which are conserved in terms of polarity, and residue 6×36 (Figure 3, central panel). For chemokine receptors this kink allows ligands to interact with the backbone of residues K/R8×49 and F8×50. In CCR2, the specific conformation of this subpocket allows the ligand to bind closer to H8, where the negatively charged oxygen of the ligand is also able to interact with the backbone of E8×48. In β2AR, P8×48 forces S8×47 inwards, allowing it to interact with the oxygen of the amide in the ligand, while a second interaction is formed between the nitrogen of another amide and D8×49. Noteworthy is position 6×36 which is not strongly conserved (59% in terms of hydrophobicity) among GPCRs. This residue is key for ligand binding in both β2AR and CCR2: in β2AR, T6×36 forms a hydrogen bond with an amide of 15PA; in CCR2, V6×36 makes a hydrophobic interaction with the cyclohexyl substituent of the ligand. However, different effects have been reported upon mutation of this residue. While the mutation V6×36A abolished ligand binding in CCR2 [22], it increased the stability of CCR9, facilitating its crystallization. In CXCR4, a T6×36P mutation abolished signaling [49], whereas M6×36T made the delta opioid receptor a constitutively active mutant (CAM) [50]. Finally, in the Adenosine A2B receptor (A2BAR) this residue acts as a determinant for G protein selectivity [51], indicating that this position might be crucial for target selectivity of intracellular ligands as well.
Region formed by TM3/6 and TM2/ICL1
The largest differences are observed in this region of the binding site; residues found in TM3 include R3×50 from the highly conserved DRY motif, and residue 6×40 conserved in terms of hydrophobicity (Figure 3, lower panel). Residue 6×37 seems to be important for selectivity, as exemplified by T6×37 in CCR9 that allows the chloro substituent of vercirnon to go deeper into this pocket. Interestingly, mutation of this residue has been implicated in altered signaling [51] and improved stability of A2AAR to facilitate crystallization [52]. Polar residues found at the TM2/ICL1 interface interact with both the CCR9 and β2AR ligand. For example, R12×49 (ICL1) forms a cation-pi interaction with the β2AR ligand while in CCR9 it interacts with both D2×40 and the nitro group of the ligand. However, these polar residues do not interact with the CCR2 ligand, indicating a different binding mode.
Strategies for intracellular modulation
In general, three main strategies have been used to target the intracellular side of GPCRs so far: small molecules, pepducins and nanobodies or “intrabodies”.
Small molecules
Small molecules currently account for the majority of drug types in clinical trials targeting GPCRs [1]. Although most of these small molecules are presumed to be orthosteric, the number of confirmed allosteric modulators targeting GPCRs is increasing in clinical trials[1]. In this regard, several intracellular small molecules have already been identified for a number of GPCRs, but few of these have progressed to clinical trials and none has made it to the market.
The largest number of small-molecule intracellular ligands reported so far target chemokine receptors, including CCR2 [9, 22], CCR4 [19, 53, 54], CCR9 [10], CXCR1 and CXCR2 [20, 21]. These intracellular ligands share similar chemical features such as the presence of acidic groups acting as hydrogen-bond acceptors when interacting with the target (Figure 2). A good balance of hydrophobic and polar residues make this binding site highly druggable, as described in a previous section [9]. However, intracellular ligands must cross the cellular membrane in order to exert their effect; therefore attention must be paid to the overall physicochemical properties of these intracellular small molecules, such as lipophilicity and molecular weight to ensure good permeability. Most of these intracellular ligands have been found using a traditional medicinal-chemistry approach. However, in the case of the β2AR, the co-crystallized compound 15PA (Figure 2) was derived from a novel β2AR NAM (compound 15) identified in a screening campaign using DNA-encoded small-molecule libraries, suggesting a novel approach to discover intracellular modulators in GPCRs [30].
One of the suggested intracellular ligands, the CCR2 antagonist CCX140-B from Chemocentryx (structure undisclosed) [55], has recently demonstrated positive results in a Phase II clinical trial in patients with type 2 diabetes and diabetic nephropathy [56]. The CCR9 intracellular antagonist, vercirnon (Figure 2) [10], also showed promising results in Phase II clinical trials in patients with Crohn’s disease [57]; however, it did not demonstrate clinical efficacy in the last Phase III study [58]. In case of CCR4, GlaxoSmithKline (GSK) has identified more than three different chemical scaffolds for intracellular antagonists—termed “site 2” antagonists by GSK [54]. Yet, only one of these ligands, GSK2239633 (Figure 2), progressed to Phase I clinical trials, before failing due to lack of efficacy [59]. Development of CXCR1-CXCR2 intracellular ligands such as SCH 527123 (Figure 2) has also resulted in several clinical trials for the treatment of chronic obstructive pulmonary disease (COPD) and asthma [60]. Although none of these ligands has been approved yet, this strategy has led to several clinical studies that might ultimately lead to a new marketed therapeutic agent.
Pepducins and Nanobodies
Another strategy for intracellular targeting of GPCRs is the use of pepducins, peptides derived from the ICLs of the target receptor, or nanobodies. As the use and pharmacology of several pepducins [61, 62] and nanobodies [63, 64] have been recently reviewed elsewhere, we will only briefly discuss them here. The pepducin approach has been explored with several GPCRs, including CXCR1, CXCR2 [65], CXCR4 [26, 66], PAR1 [41] and β2AR [67]. Although in many cases pepducins have been employed as pharmacological tools, several in vitro and in vivo preclinical studies support the role of pepducins as therapeutic agents [62]. In the case of PAR1, a recent clinical trial involving pepducin PZ-128 demonstrated positive results in patients with coronary artery disease [68]. Finally, the intracellular domain can also be targeted with intracellular nanobodies or “intrabodies”, as exemplified by US28 [29], β2AR [16, 31, 32, 69], A2AAR [33], M2R [34] and μOR [35]. Although most of these intrabodies have been used to aid GPCR crystallization and understand receptor function, their therapeutic potential has also been highlighted [70].
Advantages and therapeutic implications of intracellular ligands
As a consequence of their ability to bind to distinct sites on a GPCR, intracellular allosteric modulators can have unique properties compared to compounds that target the (orthosteric) binding site of endogenous ligands [8]. Some of these key properties include the modulation of affinity and/or efficacy of orthosteric ligands, improved selectivity, polypharmacology, or biased signaling (Box 1, Figure 4).
Figure 4. Potential advantages of intracellular allosteric modulators.
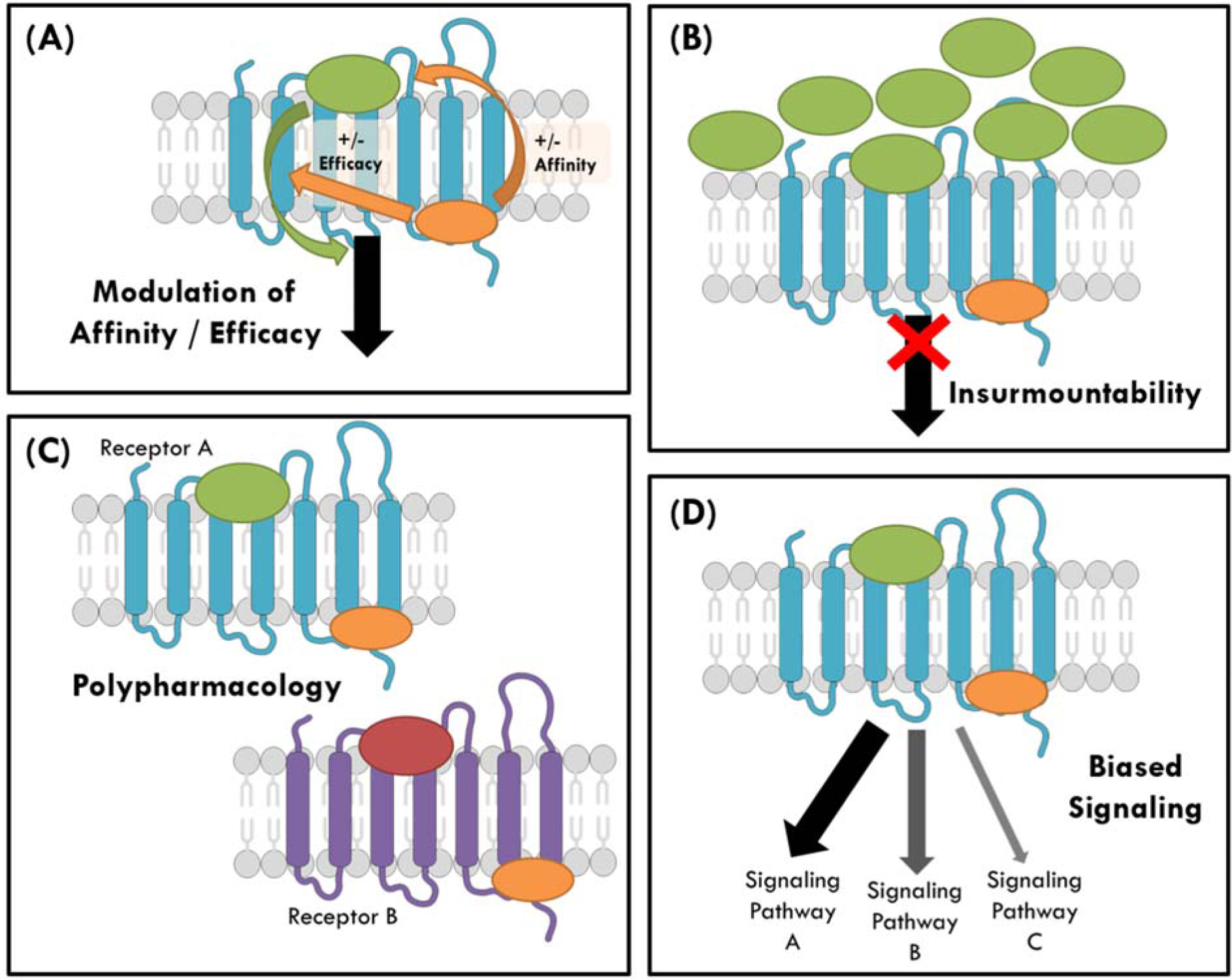
(A) Intracellular allosteric modulators (small molecules, pepducins or intrabodies, shown in orange) have the potential to positively or negatively modulate the affinity and/or the efficacy of the endogenous ligand (shown in green or red) or any orthosteric ligand. The ultimate response depends on the level of positive or negative cooperativity between the two ligands. (B) Intracellular ligands can display insurmountability, as they can inhibit the receptor (shown in blue) even when high concentrations of endogenous ligand are present. (C) A highly-conserved intracellular binding site provides the possibility of designing intracellular ligands that bind and exert their effect in multiple receptors (receptor A in blue and receptor B in purple). These pharmacological ligands, as opposed to selective ligands, might be advantageous in diseases where more than one receptor is involved. (D) Intracellular ligands can also promote biased signaling, by preferentially modulating one signaling pathway over another upon activation by the endogenous ligand. For instance, they can stabilize G protein signaling over β-arrestin signaling. Source of cellular biology illustrations: Servier Medical Art by Servier, available from https://smart.servier.com/.
Modulation of affinity and efficacy of orthosteric ligands
In β2AR, two allosteric intrabodies, a NAM and a PAM, were able to modulate the affinity of the orthosteric agonist isoprenaline by more that 15,000-fold, an unexpectedly large dynamic range (Figure 4A) [32]. Although both intrabodies insert into the pocket where the G protein binds, they modulate the functional state of the receptor differently by engaging with other residues within the binding pocket. The impact of the PAM and NAM intrabodies on a panel of orthosteric ligands of different efficacies was also shown to be consistent with the presence of multiple receptor states. The concept of more than two functional states—inactive and active—may allow for finer control of functional responses than previously thought. In addition, the demonstrated ability of allosteric ligands to differentially modulate the activity of distinct orthosteric ligands (referred to as probe dependence) has important implications regarding the selectivity of drugs for receptors that are activated by multiple ligands, as is the case for chemokine receptors. Intracellular NAMs of CCR2 and CCR9 are thought to function by directly inhibiting the interaction with intracellular signaling proteins, while at the same time blocking the outward motion of TM6 and the upward motion of TM3, required for receptor activation [9, 10]. By preventing G protein coupling and stabilizing an inactive state, they also presumably reduce the affinity of the endogenous agonists. In addition, intracellular NAMs inhibit the receptor in an insurmountable manner (Figure 4B). As previously demonstrated in CCR2, CCR2-RA-[R] was able to decrease the maximum effect of the endogenous chemokine CCL2, even at the highest CCL2 concentration tested [23]. Another advantage of allosteric over orthosteric inhibitors is their saturability or the so-called “ceiling effect”, which limits the allosteric activity to a certain level, despite further increments in the dose of the modulator [6–8]. Whether compounds targeting this site can be appropriately designed with the right level of saturability will become clear with more intracellular compounds in clinical studies.
Selectivity vs. Polypharmacology
As this intracellular binding site is likely present in most chemokine receptors, it may be a useful site for simultaneously blocking multiple chemokine receptors in disease contexts where polypharmacology has been deemed useful (Figure 4C). This may hold true in multiple sclerosis or rheumatoid arthritis where multiple chemokine receptors have been found to play a role [71]. As allosteric modulators, pepducins may prove useful for polypharmacology because they are derived from the intracellular loops of GPCRs, which often display a high degree of sequence similarity amongst related receptors [61]. For example, the pepducin P4pal-10 was shown to inhibit diverse Gq-coupled receptors without affecting β2AR (Gs) or CXCR4 (Gi) signaling [72]. Its broad spectrum inhibition profile was exploited to investigate the effect of blocking Gq-mediated signaling from a number of receptors for the treatment of asthma, which involves multiple GPCRs. On the other hand, intracellular allosteric antagonists exhibiting >100-fold selectivity for CXCR2 over CXCR1 have been discovered indicating that selectivity can also be achieved in this binding site [20].
Biased signaling
Pepducins have also been shown to promote biased signaling of GPCRs. Biased signaling tends to involve preferential activation of G protein-dependent over G protein-independent signaling (e.g., via β-arrestin)or vice versa (Figure 4D). AT1–2341 is a pepducin derived from the ICL1 of CXCR4 that promotes specific Gi-mediated signaling without G13-coupling or β-arrestin recruitment [66]. Similarly pepducin ICL3–9 derived from ICL3 of β2AR showed Gs-biased signaling [67], which may be advantageous for the treatment of asthma by limiting β-arrestin-mediated desensitization and potential tachyphylaxis from chronic use of β-agonists [73].
Concluding Remarks and Future Perspectives
There is now ample evidence from mutational, computational, and structural studies in class A GPCRs for novel allosteric binding pockets, located in close proximity to the G protein or β-arrestin binding site. This highly-conserved solvent-exposed intracellular pocket can be used to inhibit or modulate the receptor in an allosteric manner. Intracellular receptor modulation is not limited to small molecules, as intracellular pepducins and intrabodies have also been used to modulate GPCRs from the inside. These intracellular agents bring new pharmacological opportunities, but also new challenges including optimization of their selectivity profile, and their permeability properties to allow access to the inside of the cell and ultimately to cross the intestinal wall. These and other key issues have been summarized in the Outstanding Questions. Although none of these intracellular agents is yet on the market, promising (pre)clinical results have been already reported, pointing to their clinical potential. Importantly, the recent crystal structures in complex with these ligands provide a detailed view of the intracellular pockets, allowing for a better understanding and a rational design of novel intracellular ligands to target these and other GPCRs in a wide variety of diseases.
Glossary
- Affinity
Parameter that describes how strong a ligand binds to its target
- Allosteric binding site
A binding site nonoverlapping and topographically distinct from the orthosteric binding site
- Allosteric modulator
Any ligand that binds to an allosteric binding site, from which they can modulate the activity of orthosteric ligands
- Constitutively active mutant (CAM)
Mutation that leads to a permanent and agonist-independent active state of the GPCR, compared with the wild-type receptor
- Efficacy
Parameter that describes the degree of effect or response achieved by a specific ligand upon binding to its target
- G protein-coupled receptors (GPCRs)
Family of transmembrane proteins that transduce a variety of extracellular signals into intracellular responses via G protein-dependent or -independent signaling pathways. In vertebrates, GPCRs are divided in four different classes or subfamilies: class A (rhodopsin-like), class B (secretin), class C (metabotropic glutamate), and class F (frizzled/smoothened)
- Intrabodies
Intracellularly-expressed antibody fragments aimed at intracellular targets
- Nanobodies
Recombinant small size antibody (12 – 15 kDa), containing a variable-domain fragment derived from camelid heavy-chain antibodies
- Orthosteric binding site
The binding site recognized and used by the endogenous ligand for a corresponding receptor
- Orthosteric ligand
Any ligand that binds to the orthosteric binding site of the receptor. Orthosteric ligands include the endogenous ligands and non-endogenous agonists, antagonists, or inverse agonists
- Pepducins
Lipidated peptides derived from the intracellular loops or the C terminus of GPCRs, which specifically target their cognate receptor by acting as allosteric agonists or antagonists
- Polypharmacology
The ability of a ligand to (purposely and) effectively bind to several targets
- Potency
Parameter that describes the activity of a drug by defining how much of a ligand (concentration) is needed to produce a half-maximal effect
- Structure-based Ballesteros-Weinstein numbering
Numbering system for amino acid residues, which takes into account structural information to correct for bulges and constrictions. In this numbering scheme the first number denotes the transmembrane domain and the second number denotes the residue position in relation to the most conserved amino acid, the latter always in position 50. E.g. Y7×53 indicates that this tyrosine in located in TM7, in position 53. This numbering system is currently used by the GPCR database (GPCRdb http://www.gpcrdb.org) [15]
Footnotes
Disclaimer Statement
The authors declare that they have no conflicts of interest.
References
- 1.Hauser AS et al. (2017) Trends in GPCR drug discovery: new agents, targets and indications. Nature Reviews Drug Discovery. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harrison RK (2016) Phase II and phase III failures: 2013–2015. Nat Rev Drug Discov 15 (12), 817–818. [DOI] [PubMed] [Google Scholar]
- 3.Venkatakrishnan AJ et al. (2013) Molecular signatures of G-protein-coupled receptors. Nature 494 (7436), 185–94. [DOI] [PubMed] [Google Scholar]
- 4.Koelink PJ et al. (2012) Targeting chemokine receptors in chronic inflammatory diseases: an extensive review. Pharmacol Ther 133 (1), 1–18. [DOI] [PubMed] [Google Scholar]
- 5.Congreve M et al. (2017) Applying Structure-Based Drug Design Approaches to Allosteric Modulators of GPCRs. Trends Pharmacol Sci 38 (9), 837–847. [DOI] [PubMed] [Google Scholar]
- 6.Foster DJ and Conn PJ (2017) Allosteric Modulation of GPCRs: New Insights and Potential Utility for Treatment of Schizophrenia and Other CNS Disorders. Neuron 94 (3), 431–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Christopoulos A (2014) Advances in G protein-coupled receptor allostery: from function to structure. Mol Pharmacol 86 (5), 463–78. [DOI] [PubMed] [Google Scholar]
- 8.Kenakin TP (2012) Biased signalling and allosteric machines: new vistas and challenges for drug discovery. Br J Pharmacol 165 (6), 1659–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zheng Y et al. (2016) Structure of CC chemokine receptor 2 with orthosteric and allosteric antagonists. Nature 540 (7633), 458–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oswald C et al. (2016) Intracellular allosteric antagonism of the CCR9 receptor. Nature 540 (7633), 462–465. [DOI] [PubMed] [Google Scholar]
- 11.Liu X et al. (2017) Mechanism of intracellular allosteric β2AR antagonist revealed by X-ray crystal structure. Nature 548 (7668), 480–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lagerstrom MC and Schioth HB (2008) Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat Rev Drug Discov 7 (4), 339–57. [DOI] [PubMed] [Google Scholar]
- 13.Katritch V et al. (2012) Diversity and modularity of G protein-coupled receptor structures. Trends in Pharmacological Sciences 33 (1), 17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Venkatakrishnan AJ et al. (2016) Diverse activation pathways in class A GPCRs converge near the G-protein-coupling region. Nature 536 (7617), 484–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Isberg V et al. (2015) Generic GPCR residue numbers – aligning topology maps while minding the gaps. Trends in Pharmacological Sciences 36 (1), 22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rasmussen SGF et al. (2011) Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477 (7366), 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moreira IS (2014) Structural features of the G-protein/GPCR interactions. Biochimica et Biophysica Acta (BBA) - General Subjects 1840 (1), 16–33. [DOI] [PubMed] [Google Scholar]
- 18.Kang Y et al. (2015) Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature 523 (7562), 561–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andrews G et al. (2008) An intracellular allosteric site for a specific class of antagonists of the CC chemokine G protein-coupled receptors CCR4 and CCR5. Mol Pharmacol 73 (3), 855–67. [DOI] [PubMed] [Google Scholar]
- 20.Nicholls DJ et al. (2008) Identification of a putative intracellular allosteric antagonist binding-site in the CXC chemokine receptors 1 and 2. Mol Pharmacol 74 (5), 1193–202. [DOI] [PubMed] [Google Scholar]
- 21.Salchow K et al. (2010) A common intracellular allosteric binding site for antagonists of the CXCR2 receptor. Br J Pharmacol 159 (7), 1429–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zweemer AJ et al. (2014) Discovery and mapping of an intracellular antagonist binding site at the chemokine receptor CCR2. Mol Pharmacol 86 (4), 358–68. [DOI] [PubMed] [Google Scholar]
- 23.Zweemer AJ et al. (2013) Multiple binding sites for small-molecule antagonists at the CC chemokine receptor 2. Mol Pharmacol 84 (4), 551–61. [DOI] [PubMed] [Google Scholar]
- 24.Cederblad L et al. (2016) AZD8797 is an allosteric non-competitive modulator of the human CX3CR1 receptor. Biochem J 473 (5), 641–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Janz JM et al. (2011) Direct interaction between an allosteric agonist pepducin and the chemokine receptor CXCR4. J Am Chem Soc 133 (40), 15878–81. [DOI] [PubMed] [Google Scholar]
- 26.Tchernychev B et al. (2010) Discovery of a CXCR4 agonist pepducin that mobilizes bone marrow hematopoietic cells. Proc Natl Acad Sci U S A 107 (51), 22255–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Planesas JM et al. (2015) Studying the binding interactions of allosteric agonists and antagonists of the CXCR4 receptor. J Mol Graph Model 60, 1–14. [DOI] [PubMed] [Google Scholar]
- 28.Arimont M et al. (2017) Structural Analysis of Chemokine Receptor-Ligand Interactions. J Med Chem. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burg JS et al. (2015) Structural basis for chemokine recognition and activation of a viral G protein-coupled receptor. Science 347 (6226), 1113–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ahn S et al. (2017) Allosteric “beta-blocker” isolated from a DNA-encoded small molecule library. Proc Natl Acad Sci U S A 114 (7), 1708–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rasmussen SG et al. (2011) Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature 469 (7329), 175–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Staus DP et al. (2016) Allosteric nanobodies reveal the dynamic range and diverse mechanisms of G-protein-coupled receptor activation. Nature 535 (7612), 448–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hino T et al. (2012) G-protein-coupled receptor inactivation by an allosteric inverse-agonist antibody. Nature 482 (7384), 237–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kruse AC et al. (2013) Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 504 (7478), 101–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang W et al. (2015) Structural insights into μ-opioid receptor activation. Nature 524 (7565), 315–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miao Y et al. (2014) Mapping of allosteric druggable sites in activation-associated conformers of the M2 muscarinic receptor. Chem Biol Drug Des 83 (2), 237–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yanamala N and Klein-Seetharaman J (2010) Allosteric Modulation of G Protein Coupled Receptors by Cytoplasmic, Transmembrane and Extracellular Ligands. Pharmaceuticals (Basel) 3 (10), 3324–3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taylor CM et al. (2008) Modulating G-protein coupled receptor/G-protein signal transduction by small molecules suggested by virtual screening. J Med Chem 51 (17), 5297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yanamala N et al. (2009) pH-dependent interaction of rhodopsin with cyanidin-3-glucoside. 1. Structural aspects. Photochem Photobiol 85 (2), 454–62. [DOI] [PubMed] [Google Scholar]
- 40.Dowal L et al. (2011) Identification of an antithrombotic allosteric modulator that acts through helix 8 of PAR1. Proc Natl Acad Sci U S A 108 (7), 2951–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang P et al. (2015) Allosteric Activation of a G Protein-coupled Receptor with Cell-penetrating Receptor Mimetics. J Biol Chem 290 (25), 15785–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sevigny LM et al. (2011) Interdicting protease-activated receptor-2-driven inflammation with cell-penetrating pepducins. Proc Natl Acad Sci U S A 108 (20), 8491–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Covic L et al. (2002) Pepducin-based intervention of thrombin-receptor signaling and systemic platelet activation. Nat Med 8 (10), 1161–5. [DOI] [PubMed] [Google Scholar]
- 44.Severino B et al. (2013) Identification of a pepducin acting as S1P3 receptor antagonist. J Pept Sci 19 (11), 717–24. [DOI] [PubMed] [Google Scholar]
- 45.Winther M et al. (2014) Antibacterial activity of pepducins, allosterical modulators of formyl peptide receptor signaling. Antimicrob Agents Chemother 58 (5), 2985–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Naville D et al. (1996) Demonstration by transfection studies that mutations in the adrenocorticotropin receptor gene are one cause of the hereditary syndrome of glucocorticoid deficiency. J Clin Endocrinol Metab 81 (4), 1442–8. [DOI] [PubMed] [Google Scholar]
- 47.Chen A et al. (2001) Constitutive Activation of A3 Adenosine Receptors by Site-Directed Mutagenesis. Biochemical and Biophysical Research Communications 284 (3), 596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sun Y et al. (2016) Actinin-1 binds to the C-terminus of A2B adenosine receptor (A2BAR) and enhances A2BAR cell-surface expression. Biochemical Journal 473 (14), 2179–2186. [DOI] [PubMed] [Google Scholar]
- 49.Wu B et al. (2010) Structures of the CXCR4 Chemokine GPCR with Small-Molecule and Cyclic Peptide Antagonists. Science 330 (6007), 1066–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tryoen-Tóth P et al. (2005) Inverse Agonism and Neutral Antagonism at Wild-Type and Constitutively Active Mutant Delta Opioid Receptors. Journal of Pharmacology and Experimental Therapeutics 313 (1), 410–421. [DOI] [PubMed] [Google Scholar]
- 51.Liu R et al. (2014) A yeast screening method to decipher the interaction between the adenosine A2B receptor and the C-terminus of different G protein α-subunits. Purinergic Signalling 10 (3), 441–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Doré Andrew S. et al. (2011) Structure of the Adenosine A2A Receptor in Complex with ZM241385 and the Xanthines XAC and Caffeine. Structure 19 (9), 1283–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Slack RJ et al. (2013) Antagonism of human CC-chemokine receptor 4 can be achieved through three distinct binding sites on the receptor. Pharmacol Res Perspect 1 (2), e00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miah AH et al. (2017) Identification of pyrazolopyrimidine arylsulfonamides as CC- chemokine receptor 4 (CCR4) antagonists. Bioorg Med Chem 25 (20), 5327–5340. [DOI] [PubMed] [Google Scholar]
- 55.Carter PH (2013) Progress in the discovery of CC chemokine receptor 2 antagonists, 2009 – 2012. Expert Opinion on Therapeutic Patents 23 (5), 549–568. [DOI] [PubMed] [Google Scholar]
- 56.de Zeeuw D et al. (2015) The effect of CCR2 inhibitor CCX140-B on residual albuminuria in patients with type 2 diabetes and nephropathy: a randomised trial. Lancet Diabetes Endocrinol 3 (9), 687–96. [DOI] [PubMed] [Google Scholar]
- 57.Keshav S et al. (2013) A randomized controlled trial of the efficacy and safety of CCX282- B, an orally-administered blocker of chemokine receptor CCR9, for patients with Crohn’s disease. PLoS One 8 (3), e60094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Feagan B et al. (2015) Randomised clinical trial: vercirnon, an oral CCR9 antagonist, vs. placebo as induction therapy in active Crohn’s disease. Alimentary pharmacology & therapeutics 42 (10), 1170–1181. [DOI] [PubMed] [Google Scholar]
- 59.Cahn A et al. (2013) Safety, tolerability, pharmacokinetics and pharmacodynamics of GSK2239633, a CC-chemokine receptor 4 antagonist, in healthy male subjects: results from an open-label and from a randomised study. BMC Pharmacology and Toxicology 14 (1), 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dwyer MP and Yu Y (2014) CXCR2 modulators: a patent review (2009–2013). Expert opinion on therapeutic patents 24 (5), 519–534. [DOI] [PubMed] [Google Scholar]
- 61.Carr R 3rd and Benovic JL (2016) From biased signalling to polypharmacology: unlocking unique intracellular signalling using pepducins. Biochem Soc Trans 44 (2), 555–61. [DOI] [PubMed] [Google Scholar]
- 62.Zhang P et al. (2015) Pepducins and other lipidated peptides as mechanistic probes and therapeutics. Cell-Penetrating Peptides: Methods and Protocols, 191–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mujic-Delic A et al. (2014) GPCR-targeting nanobodies: attractive research tools, diagnostics, and therapeutics. Trends Pharmacol Sci 35 (5), 247–55. [DOI] [PubMed] [Google Scholar]
- 64.Manglik A et al. (2017) Nanobodies to Study G Protein-Coupled Receptor Structure and Function. Annu Rev Pharmacol Toxicol 57, 19–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kaneider NC et al. (2005) Reversing systemic inflammatory response syndrome with chemokine receptor pepducins. Nat Med 11 (6), 661–5. [DOI] [PubMed] [Google Scholar]
- 66.Quoyer J et al. (2013) Pepducin targeting the C-X-C chemokine receptor type 4 acts as a biased agonist favoring activation of the inhibitory G protein. Proc Natl Acad Sci U S A 110 (52), E5088–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Carr R 3rd et al. (2014) Development and characterization of pepducins as Gs-biased allosteric agonists. J Biol Chem 289 (52), 35668–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gurbel PA et al. (2015) Cell-penetrating pepducin therapy targeting PAR1 in subjects with coronary artery disease. Arteriosclerosis, thrombosis, and vascular biology, ATVBAHA. 115.306777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Staus DP et al. (2014) Regulation of beta2-adrenergic receptor function by conformationally selective single-domain intrabodies. Mol Pharmacol 85 (3), 472–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shukla AK (2014) Biasing GPCR signaling from inside. Sci Signal 7 (310), pe3. [DOI] [PubMed] [Google Scholar]
- 71.Horuk R (2009) Chemokine receptor antagonists: overcoming developmental hurdles. Nat Rev Drug Discov 8. [DOI] [PubMed] [Google Scholar]
- 72.Carr R 3rd et al. (2016) Interdicting Gq Activation in Airway Disease by Receptor-Dependent and Receptor-Independent Mechanisms. Mol Pharmacol 89 (1), 94–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Whalen EJ et al. (2011) Therapeutic potential of beta-arrestin- and G protein-biased agonists. Trends Mol Med 17 (3), 126–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bartuzi D et al. (2017) Signaling within Allosteric Machines: Signal Transmission Pathways Inside G Protein-Coupled Receptors. Molecules 22 (7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Müller CE et al. (2012) Allosteric modulators of rhodopsin-like G protein-coupled receptors: Opportunities in drug development. Pharmacology & Therapeutics 135 (3), 292–315. [DOI] [PubMed] [Google Scholar]
- 76.Bachelerie F et al. (2014) International Union of Basic and Clinical Pharmacology. LXXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol Rev 66 (1), 1–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Griffith JW et al. (2014) Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol 32, 659–702. [DOI] [PubMed] [Google Scholar]
- 78.Lopez-Cotarelo P et al. (2017) Beyond Chemoattraction: Multifunctionality of Chemokine Receptors in Leukocytes. Trends Immunol. [DOI] [PubMed] [Google Scholar]
- 79.Schall TJ and Proudfoot AE (2011) Overcoming hurdles in developing successful drugs targeting chemokine receptors. Nat Rev Immunol 11 (5), 355–63. [DOI] [PubMed] [Google Scholar]
- 80.Zweemer AJ et al. (2014) Bias in chemokine receptor signalling. Trends Immunol 35 (6), 243–52. [DOI] [PubMed] [Google Scholar]
- 81.Viola A and Luster AD (2008) Chemokines and their receptors: drug targets in immunity and inflammation. Annu Rev Pharmacol Toxicol 48, 171–97. [DOI] [PubMed] [Google Scholar]