

Abstract
The chick embryo is an excellent model for studying eye morphogenesis, retinal cell fate determination, and retinotectal projections due to its accessibility and the available molecular tools. Avian replication-competent retroviruses allow efficient infection of proliferating cells and stable integration of the viral genome, including up to 2.3 kb of foreign cDNA, into the host chromosome. High-titer retroviruses are produced by transient transfection of avian DF-1 cells followed by centrifugation of the culture medium. Targeted infection of the optic vesicle, the lens vesicle, the retina and pigmented epithelium, the periocular mesenchyme, and the tectum can be performed at different developmental stages in ovo. In addition, retroviruses can be used to transduce genes of interest into various ocular tissue explants or cells in vitro. Virus-mediated gene expression can be detected within 12 h of infection. Therefore, avian replication-competent retroviruses serve as powerful tools to misexpress wild-type and mutant gene products and to study molecular mechanisms underlying vertebrate visual system development.
Keywords: Chick, Replication-competent retrovirus, RCAS, Eye, Retina, Gene, Misexpression, Development
1. Introduction
Among various vertebrate models used to conduct developmental studies, the chick embryo has the obvious advantages of being accessible throughout the entire period of embryogenesis and amenable to experimental manipulations such as tissue and cell transplantation. The chick eye is particularly suitable for studying eye morphogenesis, which occurs within the first 4 days of egg incubation [1], and retinal neurogenesis, which commences on embryonic day 3 (E3) and persists until E13, when cell proliferation ceases [2]. Accumulating evidence indicates that molecular mechanisms governing eye development are well conserved among vertebrate species such as the mouse and the chick. Although the technology to generate transgenic avian species has not yet been developed, other molecular approaches such as electroporation- and retrovirus-mediated gene transfer have been used successfully to perturb gene function and conduct mechanistic studies [3–11]. This article focuses on the production of avian replication-competent retroviruses and targeted infections of the developing chick visual system.
Retroviruses are naturally existing pathogens with specific host ranges. The proteins required for the retroviral life cycle are encoded by the viral RNA genome, which upon infection of host cells is replicated into DNA and randomly inserted into host chromosomes. For simple retroviruses, the proviral integration requires that the host cell undergoes the S phase of the cell cycle. Replication-competent retroviruses encode all necessary gene products for viral infection and replication, including POL for the reverse transcriptase, GAG for viral structural proteins, and ENV for the viral envelope protein. The host specificity of a retrovirus is determined by the ENV protein, which is responsible for mediating interactions between the viral particles and the host cell surface receptors.
Recombinant retroviral vectors are engineered to accommodate and express transgenes of interest. In chick, the most commonly used retroviral vectors are the RCAS vectors developed by Steven Hughes and colleagues [12,13]. The RCAS viruses are replication competent, i.e., encoding GAG, POL, and ENV proteins, and can be produced from an infected cell and spread to other cells. RCAS viral vectors are available in several envelope subtypes, some of which afford strain-specific infections, but none infect humans or other mammals [14]. The transcription of RCAS-encoded genes is initiated from the viral promoter located within the 5′ long terminal repeat and the transgene of interest is expressed through an alternatively spliced RNA product (Fig. 1). The avian RCAS viruses are efficient gene transfer tools and have been used successfully to misexpress (ectopically express or overexpress) wild-type or mutant proteins, including dominant-negative and constitutively active forms of gene products, in chick embryos. Compared to the electroporation method, retrovirus-mediated gene transfer is stable and long-lasting, thus allowing analysis of potential phenotypes throughout embryonic development from the time of infection until after hatching.
Fig. 1.

Schematic drawings of the genome and transcripts of the replication-competent retrovirus RCAS. The box indicates the proviral DNA inserted into the host chromosome. The long terminal repeats (LTRs) and the viral genes GAG, POL, and ENV are indicated. The cDNA of interest is designated “gene X” in the shaded box, flanked by the two ClaI restriction sites. The transcripts initiated from the 5′ LTR are shown as horizontal lines. The longest RNA encodes the GAG and POL proteins and also serves as the viral genome. The ENV protein and the transgene product “X” are translated from two alternatively spliced RNAs using a common 5′ splice donor (SD) site and two distinct splice acceptor (SA) sites.
2. Description of method
2.1. Construction of RCAS expression vectors
The maximum size of the transgene that can be successfully packaged and expressed by RCAS viruses is2.3 kb. A two-step cloning strategy (Fig. 2) is commonly used for RCAS virus construction [12]. First, the cDNA of interest is cloned into a “shuttle vector” plasmid (e.g., the ClaNco plasmid or Slax13) that contains a multiple cloning site flanked by two ClaI restriction sites [15]. It is important to eliminate the 5′ UTR and 3′ UTR, especially the polyadenylation sequence, from the cDNA insert, since they interfere with viral transcription and/or translation. It is also desirable to position the start codon (Met) of the cDNA directly within the NcoI site of the shuttle vector. This ensures that the transgene utilizes the translation initiation sequence located between the 5′ ClaI site and the NcoI site (Fig. 2). If desired, protein epitope tags can be incorporated into the cDNA, e.g., at the N- or the C-terminus, by PCR, for the detection of transgene expression from the viral vector. It is necessary to sequence any portion of the cDNA that has been altered during the cloning process to rule out unwanted mutations. Second, the cDNA insert is released from the shuttle vector by restriction digestion with the ClaI enzyme. The RCAS vectors contain a unique ClaI site, which allows the insertion of the cDNA of interest downstream of the viral ENV gene. Since the cDNA insert can ligate in either orientation into the RCAS vector, the directionality of the cDNA insert has to be determined. Clones containing the correct orientation, i.e., 5′-GAG–POL–ENV–cDNA X-3′, are selected and used for viral production.
Fig. 2.

Cloning strategy for constructing RCAS viral expression vectors. (A) The first step of viral vector construction involves inserting the cDNA (X) of interest into the multiple clone site (MCS) between the two ClaI sites, preferably fusing the start codon with the NcoI site of the ClaNco [12] or the Slax13 [15] shuttle vector plasmid. The sequence between the 5′ ClaI site and the NcoI site facilitates translation of the transgene. (B) The second step involves cloning the ClaI fragment containing the cDNA from the shuttle vector into the unique ClaI site in the RCAS viral vector in the correct orientation.
The most commonly used RCAS vectors encode the A or the B subclasses of viral ENV proteins. Both give a fairly broad range of tissue infection at early stages of chick embryonic development (Fig. 4), indicating that receptor distributions for either ENV protein are similar. Depending on experimental aims, users of the RCAS expression system can choose a RCAS vector encoding a specific ENV protein type, i.e., RCAS(A) or RCAS(B), and follow a similar cloning strategy to make the viral construct.
Fig. 4.
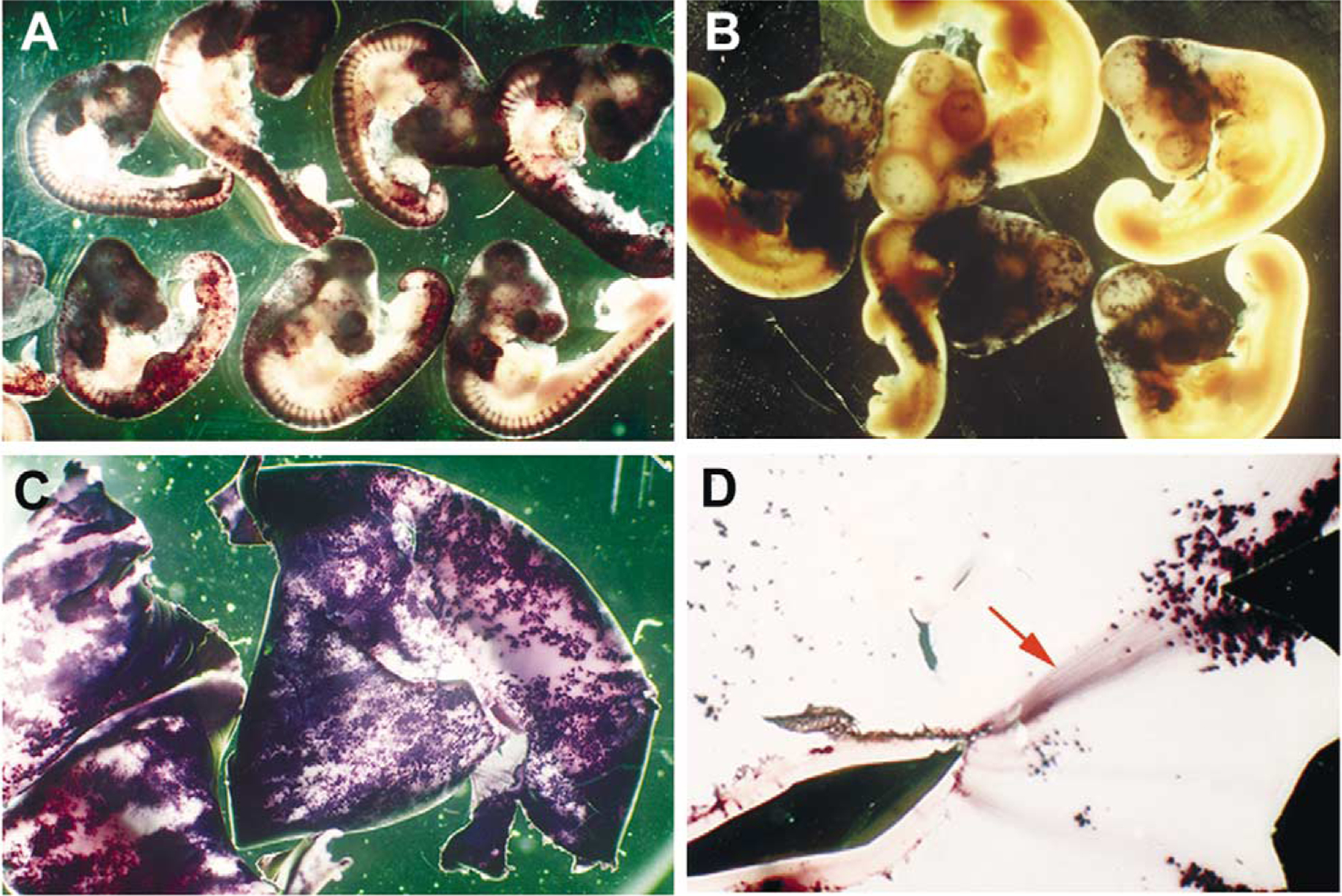
Whole-mount alkaline phosphatase histochemical staining of RCAS virus-infected embryos. All embryos were infected with viruses targeted toward the neural tube at stage 10. (A) Stage 20 embryos infected with RCAS(A) AP; note the extensive infection of the central nervous system and the dorsal root ganglia. (B) Stage 24 embryos infected with RCAS(B) AP. (C) Embryonic day 12 whole-mount retinas, showing extensive RCAS(A) AP viral infection. (D) An RCAS(A) AP-infected embryonic day 12 whole-mount retina, showing AP-positive retinal ganglion cell axons (red arrow).
2.2. Production of RCAS viral stocks
Transient transfection followed by viral propagation is used to generate high-titer RCAS viral stocks. The producer cell DF-1 is an avian fibroblast cell line established by Douglas N. Foster (University of Minnesota, St. Paul, MN) and is available through ATCC (No. CRL 12203). The day before transfection (16–20 h prior to), DF-1 cells cultured as a confluent monolayer in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal calf serum are dissociated by trypsin digestion and replated into 60-mm petri dishes at 1:5 dilution. Normally, transfection of a single 60-mm-diameter dish is sufficient for each virus. On the day of transfection, the DF-1 cells should reach 70% confluency. A lipid-based transfection method, e.g., LipofectAMINE (Gibco BRL, Gaithersburg, MD), is routinely used according to the manufacturer’s instructions. When transfected DF-1 cells reach confluency (2–3 days after transfection), the cells are split into five 10-cm petri dishes with 10 ml of DF-1 medium each and the incubation is continued until the cells reach confluency (3–4 days). This period of incubation ensures that all cells are infected by the retrovirus. To harvest the retrovirus, the culture medium containing viral particles is collected from the 10-cm dishes and the medium is frozen at −80 °C. The medium in each dish is replaced with 5 ml DMEM containing 1% FCS. Harvesting of viruses is continued every 12 or 24 h for up to 5 days.
Because RCAS viruses can easily infect uninfected proliferating avian cells, extreme caution is necessary during all stages of viral production and postproduction operations to avoid cross-contamination among different viruses and between viral-producing and uninfected cells. It is highly recommended that one handle a single viral producer at a time in the tissue culture hood, segregate all solutions and media, use pipette tips with filter plugs, and decontaminate surface areas, tools, and instruments during experimental operations.
2.3. Concentration of RCAS viral particles
For in vivo studies, it is often desirable to achieve high infection efficiency, thus high-titer viral stocks are necessary. Viral stocks stored frozen at −80 °C are stable for at least a year and can be further concentrated by centrifugation. Beckman SW28 Ultraclear centrifugation tubes (or equivalent) and the swing buckets are rinsed with 70% ethanol, and the tubes and buckets are dried in the tissue culture hood with the UV light on. Tubes containing viral stocks are thawed in a 37 °C water bath. As soon as the viral stock is thawed, the stocks of the same virus are pooled in a tissue culture hood and filtered through a 0.4-μm sterile filter unit outfitted with a prefilter, which reduces clogging by the cell debris. (Note: usage of 0.2-μm filters will result in loss of viral particles.) Media containing the virus is then subdivided into the sterile SW28 ultracentrifugation tubes at ~35–40 ml per tube. The loaded tubes are then centrifuged at 20,000 rpm (50,000g) for 2 h at 4 °C in the SW28 rotor. After centrifugation, the medium is decanted immediately from the tubes in a tissue culture hood, followed by aspiration of excess medium from the side of the tubes. The residual medium should be no more than ~100–200 μl and is retained at the bottom of each tube. The tubes are then placed back into the rotor buckets, capped, and rocked on ice for 1 h in a vertical position, which resuspends the viral particles gathered at the bottom of the tube. The tubes are then opened in a tissue culture hood, and viruses are further resuspended by gentle pipetting. (Note: creating air bubbles will disrupt viral particles and thus should be avoided). Concentrated viruses are dispensed as 10- to 20-μl aliquots into screw-cap vials and stored at −80 °C.
2.4. Determination of viral titers
The titer of RCAS viral stocks is commonly determined by immunocytochemical staining using antibodies that recognize the viral GAG protein. The day before titering, confluent DF-1 cells are split at 1:5 into 24- or 96-well dishes. The next day, a vial of virus is thawed and serial 10-fold dilutions are made in DMEM with 10% FCS. Diluted viruses are then added directly to the culture medium of the previously plated DF-1 cells, which should be at 70–80% confluency (e.g., for the 24-well dish, 200 μl diluted viral stock is added per well). For infection by RCAS(B) virus, Polybrene at 8 μg/ml is included in the culture medium. The cells are returned to incubation for 2–3 days until they reach confluency. The cells are fixed by 4% paraformaldehyde in PBS for 20 min at room temperature and then processed by standard immunostaining procedures using either the monoclonal anti-GAG antibody 3C2 (purchased from DSHB; [16]) or the rabbit anti-GAG antibody P27 (available from SPAFAS) followed by horseradish peroxidase (HRP) histochemical detection (Vector Laboratories). The numbers of viral protein-positive clones (clusters of two to eight cells) per well are counted under the microscope. Viral titer is defined as colony forming units per milliliter (cfu/ml) and can be calculated based on the observed number of clones for a given dilution. For example, if 4 clones are observed in a well infected with 200 μl of the viral stock at 107 dilution, the titer should be 4cfu × 107 × (1.0/0.2ml) = 2 × 108 cfu/ml.
In addition to immunocytochemistry against viral-encoded proteins, viral titers can also be determined by using virally expressed marker genes, such as the human placental alkaline phosphatase (AP). After the infected DF-1 cells reach confluency, they can be fixed and processed for AP histochemistry [17]. The viral production method described above routinely produces viral stocks with titers between 108 and 109 cfu/ml.
2.5. Infection of ocular tissues by RCAS viruses in ovo and in vitro
Retroviral stocks can be targeted toward different ocular tissues at distinct developmental stages. The infection of chick embryos in ovo typically requires a dissecting microscope, a micromanipulator, and a humidified chick incubator. For infection of the optic vesicle, stage 10 chick embryos and the following method are used on the bench top (not in a tissue culture hood) (Fig. 3).
Fig. 3.

Targeted injection of the developing visual system in ovo. A loaded glass pipette needle is targeted toward: (A) the optic vesicles of a stage 10 chick embryo, which are visualized against the black ink injected underneath the embryo; (B) the optic cup of a stage 17 chick embryo; and (C) the subretinal space (blue arrow) of a stage 17 chick embryo.
(a) A 50-μl glass syringe (No. 705; Hamilton, Reno, NV) is mounted on a hand-operated tilt-arm micromanipulator (No. 50132; Stoelting, Wood Dale, IL), which allows adjustments in three dimensions as well as of the angle and advancement of the injection needle. The syringe and an attached 18-gauge disposable hypodermal needle are filled with heavy mineral oil (also called heavy white oil, No. 400–5; Sigma). Next, a piece of 2-cm-long Tygon tubing is attached to the end of the needle (0.7 mm inner diameter, 2.4 mm outer diameter, 0.8 mm wall thickness; www.Tygon.com) and the tubing is filled with oil by depressing on the syringe plunger. With a rotating motion a pulled glass pipette needle (1.0 mm outer diameter, 0.75 mm inner diameter, 10–15 μm tip diameter; FHC, Bowdoinham, ME) is attached to the tubing. A minimum of 0.5 cm of the end of the pipette needle is inserted into the tubing. The entire needle is filled with oil by gently pushing or rotating the plunger of the loaded syringe, and care should be taken not to trap air bubbles.
(b) A vial of frozen viral stock is thawed on ice, mixed with 1/10 volume of 0.25% Fast green dye, and pipetted onto a small petri dish. For in ovo injection of RCAS(B)-based viruses, Polybrene at 80 μg/ml is added to the viral solution. The dish is placed under the dissecting microscope and the needle tip is positioned near the viral droplet in the petri dish by maneuvering the micromanipulator. The tip is lowered into the viral solution using the micromanipulator under the microscope. The viral solution is slowly drawn into the needle, while care is taken not to draw air into the hydraulic system. Loading is stopped when most of the viral solution is loaded into the needle. One should wait until the pressure equilibrates inside and outside the needle before taking the tip of the needle out of the viral solution. The tip of the needle is then submerged into a dish containing sterile PBS until injection.
(c) Fertilized chick eggs are incubated in a horizontal position with periodic rotations for 36 h at 37.5 °C. Before the egg is removed from the incubator, the top position of the egg is marked with a pen and the egg is kept top side up. The eggshell is then rinsed with 70% ethanol and air dried. A sharp needle is used to drill a small hole at the large end of the egg and 3–5 ml of albumin is withdrawn with a 5-ml syringe and a 20-gauge needle. The hole is then sealed with a piece of Scotch tape. Next, a piece of the eggshell (2–3 cm diameter) is removed from the top around the pen mark using a pair of curved scissors to expose the embryo. For better visualization, India ink (black artist ink) diluted fivefold with PBS can be injected below the embryo with a 1-ml syringe and 30-gauge needle.
(d) The tip of the microinjection needle is aimed at the midbrain and forebrain junction at a 45° angle and the viral stock is injected into the ventricle until blue-colored solution fills the anterior neural tube, including the optic vesicles. It may be necessary to directly target each optic vesicle to result in optimal filling due to leakage from the anterior neural pore and the constriction at the base of the optic vesicle at later stages. After withdrawal of the needle, its tip is placed back into PBS immediately and the window on the eggshell is sealed with transparent packing tape. The egg is then returned to a humidified incubator in a stationary position until harvest.
Retroviral injection can be targeted toward various ocular tissues at different developmental stages, thus permitting temporally and spatially regulated gene perturbation. For example, after the formation of the double-layered optic cup, viral stocks can be delivered into the subretinal space between stage 17 and stage 18 to allow infection of the neural retina and retinal pigmented epithelium (Fig. 3). In this case, an injection needle with an 8- to 10-μm tip is used because it allows more precise targeting of the narrow subretinal space[18]. For lens vesicle injection, viral stock can be injected into the newly formed lens vesicle between stage 14 and stage 16 [7]. In addition, it is possible to obtain a good infection of the periocular mesenchyme at stage 17 by injecting multiple times into the region surrounding the optic cup [19].
The RCAS retroviruses can also be used to deliver and express transgenes in tissue explants and primary cell cultures as long as cell proliferation continues in vitro. To perform in vitro infection, viral stock can be directly added to culture medium to result in a high rate of infection. For RCAS(B) virus-based infection, Polybrene at 8 μg/ml is included in the medium to facilitate infection.
2.6. Evaluation of viral infection rate and phenotypes
Virally infected animals can be harvested at various developmental stages that are of interest to the investigator. The rate of viral infection and subsequent spread in a given tissue can be examined by antiviral protein immunostaining or marker gene detection on whole-mount tissues or sections (Fig. 4). Virus-mediated gene expression can be determined by costaining with antiviral protein antibodies and antibodies against the transgene product. This method also permits the identification of individual virally infected cells to discern possible cell-autonomous effects of transgene misexpression.
Several features of the avian replication-competent retrovirus warrant consideration when interpreting the biological phenotypes caused by virus-mediated gene expression. First, proviral integration sites in individual infected cells are presumably different. This often causes different levels of transgene expression due to positional effects. Second, since infected cells can produce new viruses which can further infect neighboring cells, not all infected cells will express the transgene at the same time, depending on the time of infection. Third, compared to rapid gene expression mediated by in ovo electroporation (within 2 h of introducing DNA; [20]), retrovirus-mediated gene expression is more stable and longer lasting but with a delayed onset of expression. Finally, since RCAS viruses are replication competent, viral infection often spreads to other regions of the embryo instead of being restricted to the initial injection site; therefore it is essential to use appropriate controls, e.g., embryos similarly infected with the parental RCAS virus or the RCAS AP virus, in the analysis of potential phenotypes.
Acknowledgments
I thank Helen Wilmore for carefully reading the manuscript and for helpful suggestions. The work in my laboratory is supported by grants from the Research to Prevent Blindness Foundation, the March of Dimes Birth Defect Foundation, the Karl Kirchgessner Foundation, the Foundation Fighting Blindness, and the National Institutes of Health (EY12270).
References
- [1].Hamburger V, Hamilto HL, Dev. Dyn 195 (1992) 231–272. [DOI] [PubMed] [Google Scholar]
- [2].Spence SG, Robson JA, Neuroscience 32 (1989) 801–812. [DOI] [PubMed] [Google Scholar]
- [3].Austin CP, Feldman DE, Ida JA Jr., Cepko CL, Development 121 (1995) 3637–3650. [DOI] [PubMed] [Google Scholar]
- [4].Cheng H-J, Nakamoto M, Bergermann AD, Flanagan JG, Cell 82 (1995) 371–381. [DOI] [PubMed] [Google Scholar]
- [5].Koshiba-Takeuchi K, Takeuchi JK, Matsumoto K, Momose T, Uno K, Hoepker V, Ogura K, Takahashi N, Nakamura H, Yasuda K, Ogura T, Science 287 (2000) 134–137. [DOI] [PubMed] [Google Scholar]
- [6].Nakamoto M, Cheng HJ, Friedman GC, McLaughlin T, Hansen MJ, Yoon CH, O’Leary DD, Flanagan JG, Cell 86 (1996) 755–766. [DOI] [PubMed] [Google Scholar]
- [7].Ogino H, Yasuda K, Science 280 (1998) 115–118. [DOI] [PubMed] [Google Scholar]
- [8].Schulte D, Furukawa T, Peters MA, Kozak CA, Cepko CL, Neuron 24 (1999) 541–553. [DOI] [PubMed] [Google Scholar]
- [9].Yan R-T, Ma W-X, Wang S-Z, Proc. Natl. Acad. Sci. USA 98 (2001) 15014–15019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zhang XM, Yang XJ, Development 128 (2001) 943–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Watanabe Y, Nakamura H, Development 127 (2000) 1131–1140. [DOI] [PubMed] [Google Scholar]
- [12].Hughes SH, Greenhouse JJ, Petropoulos CJ, Sutrave P, J. Virol 61 (1987) 3004–3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Petropoulos CJ, Hughes SH, J. Virol 65 (1991) 3728–3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Morgan BA, Fekete DM, Methods Cell Biol. 51 (1996) 185–218. [DOI] [PubMed] [Google Scholar]
- [15].Riddle RD, Johnson RL, Laufer E, Tabin C, Cell 75 (1993) 1401–1416. [DOI] [PubMed] [Google Scholar]
- [16].Stoker AW, Bissell MJ, J. Gen. Virol 68 (1987) 2481–2485. [DOI] [PubMed] [Google Scholar]
- [17].Fields-Berry SC, Halliday AL, Cepko CL, Proc. Natl. Acad. Sci. USA 89 (1992) 693–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zhang XM, Yang XJ, Dev. Biol 233 (2001) 271–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hsieh YW, Zhang XM, Lin E, Oliver G, Yang XJ, Dev. Biol 248 (2002) 265–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Nakamura H, Funahashi J-I, Methods 24 (2001) 43–48. [DOI] [PubMed] [Google Scholar]