

Abstract
Campylobacter is the major bacterial agent of human gastroenteritis worldwide and represents a crucial global public health burden. Species differentiation of C. jejuni and C. coli and phylogenetic analysis is challenged by inter-species horizontal gene transfer. Routine real-time PCR on more than 4000 C. jejuni and C. coli field strains identified isolates with ambiguous PCR results for species differentiation, in particular, from the isolation source eggs. K-mer analysis of whole genome sequencing data indicated the presence of C. coli hybrid strains with huge amounts of C. jejuni introgression. Recombination events were distributed over the whole chromosome. MLST typing was impaired, since C. jejuni sequences were also found in six of the seven housekeeping genes. cgMLST suggested that the strains were phylogenetically unrelated. Intriguingly, the strains shared a stress response set of C. jejuni variant genes, with proposed roles in oxidative, osmotic and general stress defence, chromosome maintenance and repair, membrane transport, cell wall and capsular biosynthesis and chemotaxis. The results have practical impact on routine typing and on the understanding of the functional adaption to harsh environments, enabling successful spreading and persistence of Campylobacter.
Subject terms: Bacterial genomics, Food microbiology, Pathogens
Introduction
Since 2005, Campylobacter is the major zoonotic agent in the European Union, causing 250,161 confirmed campylobacteriosis cases in 20171. Around one third of the cases can be directly attributed to handling, preparation and consumption of broiler meat2. Measures for Campylobacter reduction focus on virulence mechanisms and persistence factors, enabling the pathogen to successfully circulate within the food chain.
Typing of Campylobacter by species differentiation methods and by multi-locus sequence typing (MLST) has become key tools for diagnostics and source attribution. Specific gene targets have proven stable and were, therefore, chosen for this purpose. Two of commonly used species differentiation markers3–5 are mapA, a fitness factor in chicken colonization6 and ceuE playing a role in iron acquisition7. For MLST, central enzymatic functions, which are conserved in the genome were defined8 and are commonly used for phylogenetic analysis.
It was shown that high level of interspecies transfer of genetic material can occur between C. jejuni and C. coli9. Adaptation to hosts can modulate the gene pool and allele variants and was suggested to be of more relevance than geographical location10.
Here we identified extensive interspecies gene transfer from C. jejuni to C. coli, impairing species differentiation and MLST analysis. Whole genome sequencing revealed that these hybrid strains shared C. jejuni gene variants, involved in stress response. Since the hybrids had predominantly been isolated from egg shells, we suggest that gene variations due to C. jejuni sequence introgression might have been a consequence of selection of survivors in a harsh environment.
Results
Isolates had been collected from food and animal matrices during routine sampling or zoonosis monitoring by the Federal State Laboratories between January 2016 and December 2018 according to ISO 1027211. The isolates were analysed by real-time PCR in the German National Reference Laboratory for Campylobacter4,5. The target for C. jejuni is a fragment of mapA, coding for an outer membrane protein. The C. coli specific target ceuE encodes the enterochelin uptake substrate-binding protein, involved in iron acquisition3. Out of 4.335 C. jejuni and C. coli isolates, 31 delivered ambiguous PCR results (0.72%). Ambiguous PCR results were defined by either amplification of both specific targets for C. jejuni and C. coli with similar Ct values (Cj/Cc mix, 28/31) or by no amplification at all (none, 3/31). A subsequent gel-based multiplex PCR12, targeting the hipO gene of C. jejuni and the glyA gene of C. coli, indicated that all of these isolates belonged to the species C. coli.
The 31 strains with ambiguous real-time PCR signal had been isolated from poultry meat, turkey cecum or skin and eggs. The Federal State Laboratories either did not report any species (9/31), correctly identified C. coli (20/31) or in one case falsely reported C. jejuni (1/31). Surprisingly, when we compared the number of strains with ambiguous real-time PCR result with the total number of isolates analysed during the same time of collection, proportionally the highest percentage of strains with ambiguous qPCR results was derived from eggs (Table 1), although the total number of analysed eggs was low.
Table 1.
Distribution of Campylobacter isolation sources of isolates with ambiguous PCR results.
| Isolation source* | # total isolates investigated (Cj and Cc) | # isolates with ambiguous PCR | % of isolates with ambiguous PCR relative to Cj and Cc | # total Cc isolates | % of isolates with ambiguous PCR relative to Cc |
|---|---|---|---|---|---|
| eggs | 39 | 5 | 12.8 | 11 | 45.5 |
| duck meat | 63 | 1 | 1.6 | 20 | 5 |
| chicken meat | 1245 | 7 | 0.6 | 281 | 2.5 |
| turkey meat | 351 | 5 | 1.4 | 95 | 5.3 |
| turkey cecum/skin | 777 | 13 | 1.7 | 414 | 3.1 |
Cj, C. jejuni; Cc, C. coli; *isolates were obtained in the years 2016–2018; isolation source are displayed, from which isolates with ambiguous PCR results were obtained. Further 1860 isolates from other sources did not result in an ambiguous PCR result and were omitted from the table.
We characterized these isolates by whole genome sequence analysis. In addition, further C. coli isolates from previous years (n = 26, 2009–2015) were included. As the prevalence of strains with ambiguous qPCR was highest from eggs, we included those from eggs and additional isolates from laying hens, chicken meat and pig feces.
We performed a k-mer based analysis using the KmerFinder 3.1 (CGE, DTU, Denmark)13–15. For a typical C. coli it is expected that the k-mers match to different C. coli reference genomes. However, as expected from the real-time PCR results, the k-mers of the input C. coli sequences with ambiguous PCR results also exhibited C. jejuni genomic content. Different percentages of k-mers matching to C. jejuni reference genomes were observed, ranging from 0 (undetectable) to 15.5% (Fig. 1). Also correctly PCR-identified C. coli exhibited various amounts of C. jejuni content (Fig. 1, red squares). From the latter, those with the highest C. jejuni content (>10%) were exclusively from eggs, two others with C. jejuni content between 4.4–5.3% were from chicken meat and turkey cecum. These isolates apparently harboured an extended amount of C. jejuni sequences but maintained C. coli sequences at mapA and ceuE. In total, 29 C. coli isolates with a k-mer percentage of more than 10% C. jejuni content (Fig. 1, categorized C. coli with high C. jejuni content, named “hybrids”) were identified. “Half hybrid” strains were defined as harbouring <10% C. jejuni sequences but displaying an ambiguous qPCR result. According to MALDI-TOF analysis, all hybrid and half hybrid strains belonged to C. coli with a score of ≥ 2.000, which was previously validated as indicative for correct species identification of Campylobacter spp.16–18. In order to clarify genetic relationship of “hybrid” and “half hybrid” strains within the Campylobacter population we performed an average nucleotide identity (ANI) analysis using the tool FastANI19. Population and ANI studies of recent years showed that organisms sharing at least 95% ANI among themselves are defined to be of the same species20,21. Results of the ANI analysis were visualized in Fig. 2A and reveal that “hybrid“ strains form a separate cluster but still share an ANI of 96.95% with C. coli isolates, proving them to be part of the C. coli population. In comparison, the ANI in the C. jejuni population ranges from 97% to 100% and shares 87.92% ANI with the “hybrid” isolates. “Half hybrid” strains are closely related with C. coli (98.96% ANI) and cannot be separated by ANI. A very similar observation can be made while looking at the core genome phylogeny in Fig. 2B based on Roary analysis using a sequence identity of at least 80%. The “half hybrid” isolates are found among the original C. coli population, whereas the hybrid strains form a separated, but still closely related group nearby C. coli. In general, both analyses show that the diversity/identity between C. coli including “hybrids” is similar than the diversity within the C. jejuni population, confirming that the hybrid strains indeed belong to the species C. coli.
Figure 1.
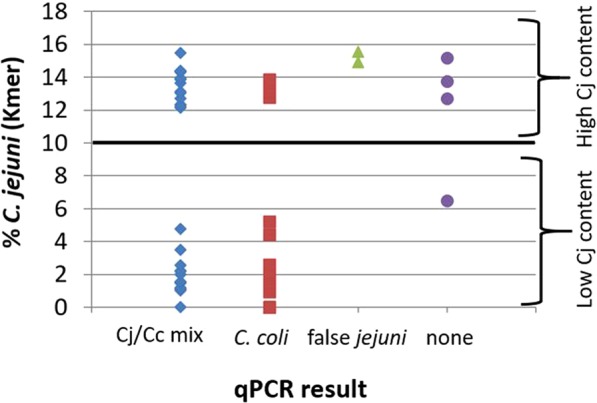
Percentage of C. jejuni genome content in C. coli isolates detected by k-mer analysis, categorized by qPCR result using mapA/ceuE as targets. Cj/Cc mix, both targets for C. jejuni and C. coli were amplified; C. coli, C. coli was correctly detected; false jejuni, C. coli was falsely detected as C. jejuni.; none, none of the targets was amplified.
Figure 2.

Relatedness of C. jejuni (turquoise), C. coli (pink), hybrid strains (purple) and half hybrid strains (mustard) according to ANI (A) and core genome analysis using Roary (B). (A) heatmap visualization of ANI values across all isolates. Hybrid strains form a separate cluster but still share ~97% ANI with C. coli. Half hybrid isolates are spread across the C. coli population. (B) phylogeny of the Campylobacter core genomes based on Roary analysis. The branch length between C. coli, including hybrid and half hybrid strains and C. jejuni has been shortened for better visualization.
We further asked whether the hybrid strains were of clonal origin and disseminated upon the/multiple horizontal C. jejuni gene transfer events had occurred. For this purpose, the MLST type was analysed based on the seven housekeeping genes (8, PubMLST.org). As visualized in Fig. 3 the isolates belonged to different sequence types, from which only two could be attributed to any known clonal complex (both ST-1150 complex), implicating that none of the isolates belonged to the CC-828 complex, which is the most abundant clonal complex of C. coli. More specific sequence analysis showed that these housekeeping genes were also affected by C. jejuni sequence introgression (see below). Thus, MLST typing apparently has its limitations in those Campylobacter with substantial horizontal gene transfer activity.
Figure 3.

The 29 C. coli with at least 10% C. jejuni genome content are not related and isolated from different locations and at distinct timepoints. Minimum spanning tree of cgMLST analysis, based on 1343 core genes defined previously22. Circles represent C. coli isolates; numbers illustrate allele differences between nearest neighbours; isolation matrix is indicated as text; colour code for MLST ST-type as depicted in the legend. Unknown, new ST-type; no group assigned, at least one allele of the 7 housekeeping genes is new. New alleles and ST-types were submitted to PuMLST and numbers are shown in Supplementary Table S1.
Analysis via Ridom Seqsphere+ software using allele-based cgMLST22 of 1343 genes indicated that the 29 C. coli hybrids were in majority unrelated. The median number of allele differences between the nearest neighbour was 343 (¼ of all analysed genes, Fig. 3). Thus, most of the strains displayed a phylogenetically diverse origin. This was substantiated by different isolation dates, ranging from 2009 to 2018 and different isolation locations from six federal states of Germany. Taken together, these data indicate that horizontal gene transfer from C. jejuni independently occurred in the C. coli hybrids.
Where did C. jejuni introgression take place
An in-house k-mer analysis was performed. The C. coli sequences with >10% of C. jejuni introgression were split into 16-mers or 31-mers, which were compared against a set of 95 complete C. coli genomes obtained from the NCBI database and 18 further C. coli strains without PCR ambiguity, for which whole genome sequencing was performed in the laboratory (Supplementary Table S1). We excluded three genomes from the NCBI database, which were apparently C. jejuni strains, falsely annotated as C. coli (GCA_001292485.1, GCA_001292205.1 and GCA_001292265.1). K-mers with direct matching were subtracted from the k-mer pool and the residual k-mers were compared against 152 complete C. jejuni genomes from the NCBI database and 3 additional sequences from the BfR strain collection (Supplementary Table S1). Those k-mers, which matched sequences present in at least 95% of the C. jejuni and maximal 5% of the C. coli genomes, were mapped against the reference C. jejuni strain NCTC 11168. Example k-mer mappings of a “hybrid” (>10% C. jejuni introgression) and a “half hybrid” strain (<10% C. jejuni introgression but with ambiguous qPCR result) against the NCTC 11168 reference sequence are depicted in Fig. 4. The recombination events of C. jejuni sequences in C. coli appear to be distributed all over the chromosome.
Figure 4.

Cc/Cj hybrid strains show recombination events of C. jejuni sequences throughout the chromosome. k-mer mapping of C. jejuni sequences from two example strains to reference sequence of C. jejuni NCTC 11168 (in light yellow, visualized linearly from left (1st base) to right (1,64 Mb)), visualized with Geneious Prime 2019.2.1. Upper panel, “half hybrid” BfR-CA-15281 (<10% C. jejuni introgression); middle panel, “hybrid” BfR-CA-14731 (>10% C. jejuni introgression); lower panel, cumulative plot of recombination events in 29 “hybrid” strains indicate common locations of C. jejuni sequence introgression. Black bars, k-mer matching at the indicated location in the C. jejuni genome.
An analysis of the recombination size was performed with a minimal assumed recombination event size of 100 bp and various maximal gaps between events of 100–500 bp between k-mer matchings. As expected, the recombination size was increased with increasing size of maximal gaps. However, the overall median size of recombination events ranged between 297 and 512 bp and the maximal event was between 11.4 and 11.8 kb detected in strain BfR-CA-08318. This might hint at the potential of C. coli to incorporate large regions of more than 10 kb within one recombination event but that most of them were below 1 kb. The number of detected recombination events per strain ranged in median between 218 and 230 events. Note that our analysis might underestimate the number and the size due to the fact that only k-mers with exact and unique matches to the reference C. jejuni NCTC 11168 and to 95% of all C. jejuni strains included in the study were considered. A cumulative plot of recombination events (from gap size analysis of maximal 100 bp) in each strain sorted by the chromosomal location of the reference sequence is depicted in Fig. 4, indicating that common recombination events occurred in multiple C. coli hybrid strains and that the overall C. jejuni content in these strains was similar as analysed by k-mer analysis via the KmerFinder 3.1 of CGE.
What has happened at mapA and ceuE loci
In order to find out, why qPCR results led to ambiguous and even false results, k-mer mapping of these isolates to C. jejuni NCTC 11168 was visualized with Geneious Prime. The k-mer analysis revealed different patterns of C. jejuni introgression in C. coli mapA and ceuE genes and their gene context (Fig. 5). In mapA either several small or large recombination events, covering also the adjacent 3′ genes Cj1028c and gyrA and the 5′ upstream located lepA, were identified. In the false positive C. jejuni isolates, the ceuE locus exhibited a mosaic allele structure with the 5′ start of ceuE displaying a typical C. coli sequence and the 3′ end matching C. jejuni sequences. In one strain with no amplification of any of both targets (“none”), the complete ceuE, including adjacent ceuD and ceuC and 3′ downstream located tRNA and Cj1356c were exchanged by C. jejuni sequences. Thus, although introgression of C. jejuni sequences into C. coli mapA locus was more frequently observed, the ceuE locus can also partially or fully be introgressed by C. jejuni DNA in contrast to previous observations23.
Figure 5.

Strains with ambiguous qPCR result display various C. jejuni recombination events at the mapA and ceuE locus. In blue, coverage of k-mers to the reference sequence of C. jejuni NCTC 11168 of example strains with ambiguous qPCR results. Cj/Cc mix, integration of C. jejuni sequences at the mapA locus; none, integration of C. jejuni sequences in the ceuE locus; false C. jejuni, integration of C. jejuni sequences in both targets.
For a further analysis we looked at the annealing sites of the oligos used for mapA and ceuE amplification. A multiple alignment of the genes, displaying C. jejuni sequence content as identified by k-mer analysis, was done using MegAlign Pro 14 (Supplementary Fig. S1). As expected from the real-time PCR result, those strains from which both targets mapA and ceuE were amplified, exhibited a sequence at mapA, which is typical for C. jejuni but maintained a typical C. coli ceuE allele. Those four strains with no real-time PCR signal at all (“none” strains), had a C. coli allele at mapA but either a complete C. jejuni sequence of ceuE (BfR-CA-15489) or a mosaic gene as indicated in Fig. 5. Besides, this analysis corroborated the idea that independent recombination events led to similar outcome of the PCR result, since the sequence of the annealing sites of oligos and probes at the mapA locus was different in the strains with ambiguous PCR results and overall sequence of mapA and ceuE varied within PCR categories.
Are the strains with ambiguous qPCR result typable by other PCR assays
There are various other PCR assays published for species differentiation of C. jejuni and C. coli with and without detection of further Campylobacter spp12,24–29. Thus, the whole-genome sequences of the strains leading to ambiguous species differentiation with mapA/ceuE targets3,5 and the correctly identified C. coli hybrids with high C. jejuni content were further assessed in silico (Supplementary Fig. S1). First, a second multiplex PCR targeting mapA/ceuE was evaluated25, leading to 56% ambiguous or false results. The sequence data revealed that also cpn60 detection24 would lead to false species identification of all C. coli hybrid strains but would identify C. coli correctly in the “half hybrids”, in which mapA/ceuE were no reliable targets. Besides, strain BfR-CA-17110 harboured a C. jejuni sequence in the target cadF28,29, leading to false C. jejuni identification of this C. coli strain. All other targets hipO, glyA, lpxA and ccoN displayed either no or low C. jejuni introgression. If C. jejuni sequences were detected within the genes, the annealing sites of the PCR oligos were not affected, thus, a correct output of the PCR is expected.
As mentioned above, MLST typing based on the seven housekeeping genes aspA, glyA, gltA, glnA, tkt, uncA, pgm was impaired in the C. coli hybrid strains with high content of C. jejuni sequence introgression (Fig. 3 and Supplementary Table S1). In particular, aspA and tkt contained C. jejuni sequences in all hybrid strains for at least 38 or 26% gene coverage, respectively. pgm displayed C. jejuni sequences in 24 of the 29 hybrid strains between 13 and 29% of the gene length. Low amount of C. jejuni introgression up to 16% were found in gltA, glnA and uncA in 28 strains. glyA was the most “stable”, since all strains harboured a classical C. coli allele or displayed just one C. jejuni k-mer match, which was not significant.
Which genes/locations were exchanged by C. jejuni sequences in the C. coli hybrid strains
Considering the typing results with some genes overrepresented for C. jejuni introgression (e. g. cpn60 or aspA) and the observations of distinct recombination regions, the question arises, whether C. jejuni sequence exchange had common patterns in the 29 C. coli hybrid strains. 346 genes were covered by k-mers in at least 20% of the gene length in minimally one of the high content C. coli hybrid genomes. They were depicted as heatmap by the R package pheatmap v.1.0.12. (Supplementary Fig. S2). For better visualization (Fig. 6), the 194 genes were filtered to 50% of gene length exchange by C. jejuni sequences in minimally one of the hybrid strains. As shown above, recombination events, i. e. genes with C. jejuni content, were distributed throughout the chromosome. Intriguingly, similar genes were exchanged in multiple C. coli hybrid strains with similar extent of C. jejuni sequence exchange (same coloured lines in Fig. 6). Therefore, we looked at those genes, which were exchanged for at least 20% gene length coverage in at least 25 of the 29 strains and identified 104 genes, identified with both k-mer analysis (16 bp or 31 bp) and additional 8 and 14 genes with 16 bp and 31 bp k-mer analysis, respectively (Supplementary Table S2). Gene categories were annotated by EggNOG 4.5.130 and, after integration of additional information on Campylobacter homologues, summarized in functional categories (Supplementary Table S2). Assuming that C. jejuni sequence introgression into C. coli strains was random, we simulated 300 recombination events in 800 core genes of 30 strains by using a Python script (available at https://gitlab.com/microbial_genomics/relative-kmer-project). As expected, and statistically proven significant (p < 0.05 by Chi2 test), the random distribution of genes recombined in multiple strains was distinct from the real distribution of observations in the hybrid strains (Supplementary Fig. S4). This supports our hypothesis of a selective process on the gene set affected by C. jejuni introgression in the hybrid strains.
Figure 6.

Cc/Cj hybrid strains share common genes with C. jejuni sequence content, which are distributed over the chromosome. Visualization of genes with at least 50% k-mer coverage of C. jejuni sequences identified in at least one of the 29 Cc/Cj hybrid strains (with >10% C. jejuni introgression). X-axis, strains; y-axis, genes. Left, heatmap sorted according to gene location in the reference C. jejuni sequence; right, heatmap sorted according to genes with high number of strains and high C. jejuni sequence exchange. Colours indicate coverage of gene length by C. jejuni sequence specific k-mer (16 bp) matches in % as detailed in the figure. Example genes with high coverage in the majority of strains are indicated. Below heatmaps, isolation source of the strains: c, chicken meat; tc, turkey cecum; tm, turkey meat and eggs.
Almost half of all identified genes encode proteins involved in oxidative stress response (katA, Cj1386, mrsB, canB, Cj0833c hydA, hydA2, nadD, nuoA, nuoB, nuoC, Cj0081), stress response in general (clpA, htrB, htrA, cpn10, cpn60), DNA metabolism and repair (purF, pyrG, thyX, rarA, recJ, ung, ribA, guaB, dut), chemotaxis and flagellar motor switch (cheA, cheV, cheW, fliY), signal transduction (Cj1110c, Cj1227c, Cj1258), membrane transporters (crcB, cj0832c, ktrA, ktrB, Cj1257c, Cj1687), cell wall and capsule biosynthesis (kpsS, kpsE, kpsF, kpsD, kpsT, murE) and metK encoding a S-adenosylmethionine transferase, involved in providing the substrate for methylation reactions. This suggests that C. jejuni sequence recombination in the C. coli hybrid genes was not random but might modulate the fitness of the C. coli hybrid strains, selected for survival in a harsh environment. Intriguingly, an American isolate, C. coli RM4661, from turkey carcass origin (NZ_CP007181.1) was identified as a Cc/Cj-hybrid strain, sharing 106 of the 126 C. jejuni introgressed genes revealed in the majority of our hybrid strains (Supplementary Table S2). We propose that this strain underwent a similar selection procedure, which corroborates our hypothesis of independent functional adaptation upon selection in a harsh environment.
Which C. jejuni sequence exchange leads to amino acid exchange in the protein and might represent a functional adaptation in C. coli
We checked whether the gene variants of the C. coli hybrid strains lead to protein variants different from C. coli proteins. Since C. jejuni and C. coli proteins differ in average by nearly 40 amino acids31, it was expected that most of the observed C. jejuni introgression covering at least 20% of the gene length leads to changes in protein sequence. BLAST analysis (https://blast.ncbi.nlm.nih.gov) was performed on a subset of the above-mentioned identified gene translations and in all of the cases amino acid exchanges were detected in the hybrid variants as compared to C. coli typical protein sequence. It remains to be investigated in future studies, how these variations impact protein function with respect to C. coli survival capacity under stress conditions.
Discussion
The German National Reference Laboratory for Campylobacter has access to a large collection of representative isolates from Germany. With this set of isolates in hand we were able to identify multiple strains with ambiguous species differentiation, in particular, isolated from eggs but also from poultry meat and turkey cecum. Further isolates from eggs showed that from this isolation source nearly half of all C. coli displayed an extended amount of C. jejuni sequences incorporated in the genome.
A study comparing C. coli clade 1 (ST-828 and ST-1150) from agriculture with nonagricultural unintrogressed C. coli clade 2 and 3 demonstrated the potential of incorporation of substantial C. jejuni sequences in clade 1 C. coli31. The authors identified 26 C. jejuni genes present in C. coli clade 1 but absent in clade 2 and 3. Our analysis focussed on C. coli hybrid strains as a fraction of clade 1 and deciphers ongoing extended C. jejuni introgression in these strains. As expected, we only have an overlap of 2 genes (Cj0555 and htrB) out of the identified 26 with the study of Sheppard et al.31, since we compared our hybrid strains against 113 C. coli sequences (mostly clade 1), including sequences from the NCBI database. This supports the notion that the C. jejuni recombination events found in this study represent a further development of C. coli strains. Since the C. coli hybrids were predominantly isolated from eggs, this supports the notion that the identified C. jejuni sequence incorporations might be a consequence of functional adaptation to survival in a harsh environment. Campylobacter is transmitted on egg shells via fecal contamination. On the shell, the bacterium encounters oxidative stress but also dryness and, thus, osmotic stress as well as nutrient and cold stress. Usually after 5–6 days, Campylobacter are no longer cultivatable from faeces32,33.
Adaptation to harsh environment might explain shared C. jejuni recombinations in C. coli hybrids
The hybrid strains carried gene variants of C. jejuni or mosaic genes involved in oxidative stress response, such as katalase (katA) and Cj1386, which was shown to encode an atypical hemin-binding protein, mediating the trafficking of hemin to katalase34. Katalase is one of the key enzymes for protection against oxidative stress by cleaving peroxide to water and oxygen. mrsB (cj1112c) encoding a methionine sulphoxide reductase, was shown to protect C. jejuni against oxidative and nitrosative stress35. Furthermore, canB displayed C. jejuni sequences in the hybrid strains, encoding carbonic anhydrase, an enzyme important for growth at low CO2 concentrations36. A further oxidoreductase (Cj0833c) and genes encoding for the Ni/Fe hydrogenase small subunit hydA (Cj1267c) and hydA2 (Cj1399c) as well as nadD (Cj1404) involved in the synthesis of the redox cofactor NAD + were found to harbour C. jejuni sequences. Furthermore, nuoA, nuoB, nuoC implicated in transfer of electrons in the respiration chain and Cj0081, encoding the cyanide-resistant CioAB, which is proposed to lower oxygen levels and maintain microaerobic conditions37, were identified to bear C. jejuni sequences in the hybrid strains. The htrB gene encoding a lipid A acyltransferase was proposed to play a role in regulation of cell responses to environmental harsh conditions, such as acid, heat, oxidative and osmotic stress38. As mentioned above, also htrA, which encodes a protease and chaperone activity with roles in virulence and oxidative stress defence39–41 was among the genes with C. jejuni sequence detected in all hybrid strains. Besides cgb, encoding a single-domain haemoglobin, was suggested to protect Campylobacter against nitric oxide and nitrosative stress42.
Interestingly, also genes implicated in general stress response as the clpA ATPase and the chaperone genes cpn10 and cpn60 were affected by C. jejuni introgression. The latter cpn60 (groEL) also serves as target for species differentiation24, inevitably leading to false species identification of the hybrid strains. Among the genes with C. jejuni introgression in the hybrid strains were several with roles in DNA metabolism and repair, such as purF, pyrG, thyX, rarA, recJ, ung, ribA, guaB and dut. Moreover, motility-associated genes, like the chemotaxis genes cheA, cheV, cheW and fliY, encoding a flagellar motor switch protein, displayed C. jejuni sequences.
In addition, our list of genes with C. jejuni content in the hybrid strains, also contained genes implicated in cell wall (murE) and capsule biosynthesis (kpsS, kpsE, kpsF, kpsD, kpsT). Consistently, in C. jejuni strains enhanced biofilm formation capacity, which might also be associated with enhanced survival under oxidative stress, was attributed to genes implicated in oxidative stress defence, motility, cell wall and capsular biosynthesis43.
We suggest that modification of genes by recombination of C. jejuni sequences in a common set of genes in most of the hybrid strains, might reflect selection of survivors from harsh environments.
Practical implications for diagnostics
The hybrid strains can elude molecular typing, such as species differentiation using the mapA/ceuE targets and MLST. It was previously found that mapA/ceuE targets might lead to ambiguous qPCR results in six identified strains out of a data collection of around 1700 sequences23. In our study we identified in total 37 strains (21 “hybrid” strains and 16 “half hybrid” strains), which were not identifiable in the qPCR using mapA/ceuE targets, including two isolates, which were falsely identified as C. jejuni. All “hybrid” strains failed to be typed using the cpn60 target24 and one “half-hybrid” would be incorrectly typed as C. jejuni using the cadF target28,29.
C. jejuni sequence introgression into aspA and adjacent regions (including Cj0081) was previously detected in two C. coli strains from turkey44. Our data showed that MLST as phylogenetic assay has limitations, since C. jejuni sequences were found in six of the seven housekeeping genes in the majority of “hybrid” strains. Hence, standardized typing methods should consider perturbations due to extended recombination activity in Campylobacter. Thus, it is recommendable to include multiple independent species differentiation methods as future molecular annex to ISO 10272-1/2:2017 and to be aware of phylogenetic bias in source attribution analysis.
Conclusions and further aspects
There are various studies dealing with the differential survival of C. jejuni and C. coli under different environmental and host conditions. It has to be noted, that stress survival of the microaerobic Campylobacter is one of the major and still enigmatic topics in order to explain the pathogens widespread dissemination. C. jejuni was shown to survive longer in liver juice45. Aerotolerant C. jejuni strains were identified46 but also aerotolerant C. coli isolates were highly prevalent in other studies47. Survival in harsh environments might be a result of various factors and also dependent on the specific genomic background. In aerotolerant C. coli point mutations were detected in other genes, not obviously implicated in oxidative stress response48. Thus, it remains elusive, how Campylobacter species modulate their gene pool in order to adapt to changing environments. However, the identification of hybrid strains, mainly selected from a harsh environment, exhibiting an extended amount of C. jejuni sequences in a common gene set, shows the enormous potential of Campylobacter for extensive genetic exchange for fitness enhancement.
Materials and Methods
Strains and growth conditions
C. jejuni and C. coli field strains were isolated from different food matrices and animal samples by the Federal State Laboratories according to ISO 1027211. At the National Reference Laboratory, isolates were cultured on Columbia agar (Oxoid, Germany) supplemented with 5% sheep blood (Oxoid, Germany) (ColbA) or passaged through Bolton broth and subcultured on mCCDA in case strains still exhibited non-Campylobacter background flora. Incubation was performed for 48 h under microaerobic conditions (5% O2, 10% CO2, rest N2) at 42 °C. Strains were stored at −80 °C using the cryobank system (Mast Diagnostica GmbH, Germany). For DNA extraction strains from −80 °C stocks were grown on ColbA for 24 h under microaerobic conditions at 42 °C and once subcultured for another 24 ± 4 h prior to use.
Species differentiation by PCR
DNA of the strains was extracted by resuspension of the cell pellet in 5% Chelex 100 resin (Bio-Rad Laboratories GmbH, Germany), followed by incubation for 15 min at 95 °C and subsequent centrifugation. The supernatant was used for PCR analysis. For detection and species identification a real-time PCR method, targeting either a C. jejuni specific fragment of the mapA gene, a C. coli specific fragment of the ceuE gene or a C. lari specific fragment of the glyA gene was performed4,5. In case of ambiguity of the results, a second gel-based multiplex-PCR was applied, targeting specific fragments of the hipO gene for C. jejuni, the glyA gene for C. coli and C. upsaliensis, the cpn60 for C. lari, the sapB2 gene of C. fetus and a Campylobacterales specific fragment of the 23 S rRNA gene12.
Matrix-assisted laser desorption/ionization (MALDI-TOF) analysis
Colony material of a 24 h ColbA plate was spotted onto the target plate (MSP 96 target polished steel (MicroScout Target) plate; Bruker Daltonik, Germany). After air drying the spots were overlaid with 1 µl of saturated α-cyano-4-hydroxy-cinnamic acid matrix solution (200 mg in 2.5% trifluoroacetic acid/50% acetonitrile) and dried completely. MALDI-TOF MS analysis was performed using MALDI-TOF Microflex LT (Bruker Daltonics, Germany) using a range of 2,000–20,000 m/z (mass to charge ratio) following the calibration with Bacterial Test Standard (Bruker Daltonics, Germany). For each spectrum 240 laser shots were summed up in 40 shot steps, but at least 80 shots per raster spot from different positions within the sample were acquired by the AutoXecute method using the software FlexAnalysis 3.4. The spectra were compared with the MBT Compass Library, Revision F (Bruker Daltonics, Germany). Each identification obtains a score value. The identification at the species level with a score ≥ 2.000 was considered correct16–18.
Whole genome sequence analysis
Campylobacter strains grown on ColbA for 24 h under microaerobic atmosphere at 42 °C were harvested and DNA was extracted using the PureLink Genomic DNA Mini Kit (Thermo Fisher Scientific, USA). The quality of the DNA was evaluated by spectral analysis (NanoDrop Spectrophotometer, Thermo Fisher Scientific, USA) and the concentration was fluorimetrically quantified by Qubit 3.0 Fluorometer (dsDNA HS Assay Kit 0.2–100 ng; Thermo Fisher Scientific, USA). DNA libraries were prepared using the Nextera XT DNA Library Prep kit or the Nextera DNA Flex Library Prep Kit according to the manufacturer’s instructions (Illumina, San Diego, USA). Quality of the libraries was assessed by gel analysis or on a fragment analyser 3408 (Advanced Analytical Technologies Inc., USA). Paired-end sequencing was performed on the Illumina MiSeq (2×301 cycles) or the NextSeq (2×151 cycles) platform using the MiSeq v3 (600 cycles) reagent kit or the NextSeq 500/550 Mid Output kit v2.5 (300 cycles), respectively. The sequences were published within the BioProject No. PRJNA595957, BioSample No. SAMN13577876-SAMN13577920, SRA accession No. SRR10698060-SRR10698104 at NCBI sequence read archive (SRA). New MLST alleles and MLST-ST types were uploaded to PubMLST.
Sequence analysis
Sequences were analyzed by Ridom Seqsphere+ v. 6.0.0 (2019–04) (Ridom, Muenster, Germany) using the cgMLST scheme of 1343 gene targets previously proposed22, with 98% required identity and 98% required percentage of coverage to one of the known alleles (allele library status June 2018). Quality trimming was performed in a window of 20 bp with Phred score 30. The obtained average coverage (processed, unassembled) was >75-fold. Raw reads were de-novo assembled via SPAdes 3.11.149 with careful option, which performs a mismatch correction. The number of assembled contigs was between 31 and 130, the total size of the assemblies ranged from 1.65 to 1.92 Mb. At least 95% “good targets” were found for cgMLST-based analysis using the previously proposed cgMLST scheme22. Average nucleotide identity (ANI) analysis was done using the tool FastANI19. Core genome phylogeny was calculated using Roary v.3.12.050 with a sequence identity of at least 80%. This resulted in 800 core genes that were used to build a phylogenetic tree with RAxML v.8.2.1051 (100 bootstraps). Finally, the phylogenetic tree was adjusted for recombination sites using ClonalFrameML v.1.1152.
For prescreening of sequences for C. jejuni introgression, assembled contigs were analyzed on the web-based KmerFinder 3.1 (Center for Genomic Epidemiology, DTU, Denmark)13–15, which splits the assembly contigs into overlapping 16-mers and searches for homology matches in sequenced bacterial organisms, filtered on coding sequences (CDS; starting with ATG). The percentage of k-mers matching to distinct reference genomes was received as output data. An in-depth k-mer analysis was performed using an in-house pipeline. For this purpose the assemblies from Ridom Seqsphere+ after SPAdes assembly were used. A k-mer based databases from assembled and closed genomes from C. jejuni and C. coli (Supplementary Table S1) were built by kmc v.353 with a k-mer size of 16 bases or 31 bases. In order to identify C. jejuni specific genes, the database of C. jejuni includes k-mers present in at least 95% of the genomes whereas the C. coli database contains k-mers present in at least 5% of the genomes. K-mers from mixed isolates were subsequently counted and compared against the databases with kmc_tools. K-mers present in C. jejuni database and absent in C. coli genomes were mapped against a closed reference strain NCTC 11168 (NC_002163.1) of C. jejuni by bowtie254 and corresponding genes were identified with BEDTools v2.27.155. Since read mappers are optimized for longer sequences, k-mers of size 16 bp were only considered for exact and unique matches for further downstream analysis. Since k-mers of size 31 bp are more specific, mapping results were not filtered at this step.
Genes covered by k-mers in at least 20% or 50% of the gene length and in one of the high content C. coli hybrid genome were visualized as heatmap by the R package pheatmap v.1.0.12. K-mers matched in C. coli hybrid genomes (as.bam files) were visualized in Geneious v.2019.2.1 using C. jejuni strain NCTC 11168 (NC_002163.1) as reference. Source code and scripts used to perform those steps are freely available at https://gitlab.com/microbial_genomics/relative-kmer-project.
Supplementary information
Acknowledgements
J.G. was financed by the Federal Ministry of Education and Research (BMBF) within the PAC-CAMPY project IP3/01KI1725B, whereas L.E. was financed by IP10/01KI1725F. The study was supported by the 1322–646 SFB project of BfR. The funders had no influence on the design, data collection, analysis and conclusions of the study. We thank Nicole vom Ort from the BfR section Diagnostics, Pathogen Characterization, Parasites in Food and all members of the National Reference Laboratory for excellent technical assistance. We are grateful for the continuous collaboration with the Federal State Laboratories. We thank Dag Harmsen and Joerg Rothgaenger for implementation of the cgMLST scheme22 into the Ridom Seqsphere+ software. The authors declare that they have no conflict of interest.
Author contributions
J.G. performed experiments, analysed data and co-wrote the manuscript; L.E. designed the k-mer analysis, analysed data, co-wrote the manuscript; M.K., performed experiments; F.H., analysed data; M.B., C.D., B.M. and T.S. provided critical advice in experimental design and edited the paper; K.S. designed the study, analysed data and wrote the manuscript. All authors reviewed the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Julia C. Golz and Lennard Epping.
Supplementary information
is available for this paper at 10.1038/s41598-020-60320-y.
References
- 1.ECDC. European Centre for Disease Prevention and Control. Campylobacteriosis. In: ECDC. Annual epidemiological report for 2017. Stockholm: ECDC; 201.
- 2.EFSA. Scientific Opinion on Campylobacter in broiler meat production: control options and performance objectives and/or targets at different stages of the food chain. EFSA Journal 20119(4):2105. [141 pp.], 10.2903/j.efsa.2011.2105 (2011).
- 3.Best EL, Powell EJ, Swift C, Grant KA, Frost JA. Applicability of a rapid duplex real-time PCR assay for speciation of Campylobacter jejuni and Campylobacter coli directly from culture plates. FEMS Microbiol. Lett. 2003;229:237–241. doi: 10.1016/S0378-1097(03)00845-0. [DOI] [PubMed] [Google Scholar]
- 4.LFGB. German Food and Feed Code, § 64,L-06.32-1, Detection of Campylobacter spp. in minced meat – real-time PCR methods, Federal Office of Consumer Protection and Food Safety (BVL), Beuth GmbH, Germany. (2013).
- 5.Mayr AM, et al. Rapid detection and differentiation of Campylobacter jejuni, Campylobacter coli, and Campylobacter lari in food, using multiplex real-time PCR. J. Food Prot. 2010;73:241–250. doi: 10.4315/0362-028X-73.2.241. [DOI] [PubMed] [Google Scholar]
- 6.Johnson JG, Livny J, Dirita VJ. High-throughput sequencing of Campylobacter jejuni insertion mutant libraries reveals mapA as a fitness factor for chicken colonization. J. Bacteriol. 2014;196:1958–1967. doi: 10.1128/JB.01395-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller CE, Williams PH, Ketley JM. Pumping iron: mechanisms for iron uptake by Campylobacter. Microbiology. 2009;155:3157–3165. doi: 10.1099/mic.0.032425-0. [DOI] [PubMed] [Google Scholar]
- 8.Dingle KE, et al. Multilocus sequence typing system for Campylobacter jejuni. J. Clin. Microbiol. 2001;39:14–23. doi: 10.1128/JCM.39.1.14-23.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sheppard SK, McCarthy ND, Falush D, Maiden MC. Convergence of Campylobacter species: implications for bacterial evolution. Sci. 2008;320:237–239. doi: 10.1126/science.1155532. [DOI] [PubMed] [Google Scholar]
- 10.Sheppard SK, Maiden MC. The evolution of Campylobacter jejuni and Campylobacter coli. Cold Spring Harb. Perspect. Biol. 2015;7:a018119. doi: 10.1101/cshperspect.a018119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.ISO. 10272-1:2006 revised 2017, Microbiology of the food chain - Horizontal method for detection and enumeration of Campylobacter spp. - Part 1: Detection method. [DOI] [PubMed]
- 12.Wang G, et al. Colony multiplex PCR assay for identification and differentiation of Campylobacter jejuni, C. coli, C. lari, C. upsaliensis, and C. fetus subsp. fetus. J. Clin. Microbiol. 2002;40:4744–4747. doi: 10.1128/jcm.40.12.4744-4747.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clausen P, Aarestrup FM, Lund O. Rapid and precise alignment of raw reads against redundant databases with KMA. Bmc Bioinforma. 2018;19:307. doi: 10.1186/s12859-018-2336-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hasman H, et al. Rapid whole-genome sequencing for detection and characterization of microorganisms directly from clinical samples. J. Clin. Microbiol. 2014;52:139–146. doi: 10.1128/JCM.02452-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Larsen MV, et al. Benchmarking of methods for genomic taxonomy. J. Clin. Microbiol. 2014;52:1529–1539. doi: 10.1128/JCM.02981-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bessede E, Solecki O, Sifre E, Labadi L, Megraud F. Identification of Campylobacter species and related organisms by matrix assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometry. Clin. Microbiol. Infect. 2011;17:1735–1739. doi: 10.1111/j.1469-0691.2011.03468.x. [DOI] [PubMed] [Google Scholar]
- 17.Emele MF, et al. Proteotyping as alternate typing method to differentiate Campylobacter coli clades. Sci. Rep. 2019;9:4244. doi: 10.1038/s41598-019-40842-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsieh YH, et al. Application of MALDI-TOF MS systems in the rapid identification of Campylobacter spp. of public health importance. J. AOAC Int. 2018;101:761–768. doi: 10.5740/jaoacint.17-0266. [DOI] [PubMed] [Google Scholar]
- 19.Jain C, Rodriguez RL, Phillippy AM, Konstantinidis KT, Aluru S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018;9:5114. doi: 10.1038/s41467-018-07641-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Figueras, M. J., Beaz-Hidalgo, R., Hossain, M. J. & Liles, M. R. Taxonomic affiliation of new genomes should be verified using average nucleotide identity and multilocus phylogenetic analysis. Genome Announc2, 10.1128/genomeA.00927-14 (2014). [DOI] [PMC free article] [PubMed]
- 21.Goris J, et al. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007;57:81–91. doi: 10.1099/ijs.0.64483-0. [DOI] [PubMed] [Google Scholar]
- 22.Cody AJ, Bray JE, Jolley KA, McCarthy ND, Maiden MCJ. Core genome multilocus sequence typing scheme for stable, comparative analyses of Campylobacter jejuni and C. coli human disease isolates. J. Clin. Microbiol. 2017;55:2086–2097. doi: 10.1128/JCM.00080-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jansen van Rensburg MJ, Swift C, Cody AJ, Jenkins C, Maiden MC. Exploiting bacterial whole-genome sequencing data for evaluation of diagnostic assays: Campylobacter species identification as a case study. J. Clin. Microbiol. 2016;54:2882–2890. doi: 10.1128/JCM.01522-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chaban B, Musil KM, Himsworth CG, Hill JE. Development of cpn60-based real-time quantitative PCR assays for the detection of 14 Campylobacter species and application to screening of canine fecal samples. Appl. Env. Microbiol. 2009;75:3055–3061. doi: 10.1128/AEM.00101-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Denis M, et al. Development of a m-PCR assay for simultaneous identification of Campylobacter jejuni and C. coli. Lett. Appl. Microbiol. 1999;29:406–410. doi: 10.1046/j.1472-765x.1999.00658.x. [DOI] [PubMed] [Google Scholar]
- 26.Klena JD, et al. Differentiation of Campylobacter coli, Campylobacter jejuni, Campylobacter lari, and Campylobacter upsaliensis by a multiplex PCR developed from the nucleotide sequence of the lipid A gene lpxA. J. Clin. Microbiol. 2004;42:5549–5557. doi: 10.1128/JCM.42.12.5549-5557.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.LaGier MJ, Joseph LA, Passaretti TV, Musser KA, Cirino NM. A real-time multiplexed PCR assay for rapid detection and differentiation of Campylobacter jejuni and Campylobacter coli. Mol. Cell Probes. 2004;18:275–282. doi: 10.1016/j.mcp.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 28.Shams S, Bakhshi B, Tohidi Moghadam T. In silico analysis of the cadF gene and development of a duplex polymerase chain reaction for species-specific identification of Campylobacter jejuni and Campylobacter coli. Jundishapur J. Microbiol. 2016;9:e29645. doi: 10.5812/jjm.29645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Toplak N, Kovac M, Piskernik S, Mozina SS, Jersek B. Detection and quantification of Campylobacter jejuni and Campylobacter coli using real-time multiplex PCR. J. Appl. Microbiol. 2012;112:752–764. doi: 10.1111/j.1365-2672.2012.05235.x. [DOI] [PubMed] [Google Scholar]
- 30.Huerta-Cepas J, et al. eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016;44:D286–293. doi: 10.1093/nar/gkv1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sheppard SK, et al. Progressive genome-wide introgression in agricultural Campylobacter coli. Mol. Ecol. 2013;22:1051–1064. doi: 10.1111/mec.12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ahmed MF, Schulz J, Hartung J. Survival of Campylobacter jejuni in naturally and artificially contaminated laying hen feces. Poult. Sci. 2013;92:364–369. doi: 10.3382/ps.2012-02496. [DOI] [PubMed] [Google Scholar]
- 33.Bui XT, Wolff A, Madsen M, Bang DD. Reverse transcriptase real-time PCR for detection and quantification of viable Campylobacter jejuni directly from poultry faecal samples. Res. Microbiol. 2012;163:64–72. doi: 10.1016/j.resmic.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 34.Flint A, Stintzi A. Cj1386, an atypical hemin-binding protein, mediates hemin trafficking to KatA in Campylobacter jejuni. J. Bacteriol. 2015;197:1002–1011. doi: 10.1128/JB.02346-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Atack JM, Kelly DJ. Contribution of the stereospecific methionine sulphoxide reductases MsrA and MsrB to oxidative and nitrosative stress resistance in the food-borne pathogen Campylobacter jejuni. Microbiology. 2008;154:2219–2230. doi: 10.1099/mic.0.2008/019711-0. [DOI] [PubMed] [Google Scholar]
- 36.Al-Haideri H, White MA, Kelly DJ. Major contribution of the type II beta carbonic anhydrase CanB (Cj0237) to the capnophilic growth phenotype of Campylobacter jejuni. Env. Microbiol. 2016;18:721–735. doi: 10.1111/1462-2920.13092. [DOI] [PubMed] [Google Scholar]
- 37.Jackson RJ, et al. Oxygen reactivity of both respiratory oxidases in Campylobacter jejuni: the cydAB genes encode a cyanide-resistant, low-affinity oxidase that is not of the cytochrome bd type. J. Bacteriol. 2007;189:1604–1615. doi: 10.1128/JB.00897-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Phongsisay V, Perera VN, Fry BN. Expression of the htrB gene is essential for responsiveness of Salmonella typhimurium and Campylobacter jejuni to harsh environments. Microbiology. 2007;153:254–262. doi: 10.1099/mic.0.29230-0. [DOI] [PubMed] [Google Scholar]
- 39.Baek KT, Vegge CS, Skorko-Glonek J, Brondsted L. Different contributions of HtrA protease and chaperone activities to Campylobacter jejuni stress tolerance and physiology. Appl. Env. Microbiol. 2011;77:57–66. doi: 10.1128/AEM.01603-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boehm M, et al. Rapid paracellular transmigration of Campylobacter jejuni across polarized epithelial cells without affecting TER: role of proteolytic-active HtrA cleaving E-cadherin but not fibronectin. Gut Pathog. 2012;4:3. doi: 10.1186/1757-4749-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boehm M, Lind J, Backert S, Tegtmeyer N. Campylobacter jejuni serine protease HtrA plays an important role in heat tolerance, oxygen resistance, host cell adhesion, invasion, and transmigration. Eur. J. Microbiol. Immunol. 2015;5:68–80. doi: 10.1556/EUJMI-D-15-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Elvers KT, Wu G, Gilberthorpe NJ, Poole RK, Park SF. Role of an inducible single-domain hemoglobin in mediating resistance to nitric oxide and nitrosative stress in Campylobacter jejuni and Campylobacter coli. J. Bacteriol. 2004;186:5332–5341. doi: 10.1128/JB.186.16.5332-5341.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pascoe B, et al. Enhanced biofilm formation and multi-host transmission evolve from divergent genetic backgrounds in Campylobacter jejuni. Env. Microbiol. 2015;17:4779–4789. doi: 10.1111/1462-2920.13051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chan K, Elhanafi D, Kathariou S. Genomic evidence for interspecies acquisition of chromosomal DNA from Campylobacter jejuni by Campylobacter coli strains of a turkey-associated clonal group (cluster II) Foodborne Pathog. Dis. 2008;5:387–398. doi: 10.1089/fpd.2008.0113. [DOI] [PubMed] [Google Scholar]
- 45.Karki AB, Wells H, Fakhr MK. Retail liver juices enhance the survivability of Campylobacter jejuni and Campylobacter coli at low temperatures. Sci. Rep. 2019;9:2733. doi: 10.1038/s41598-018-35820-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oh E, McMullen L, Jeon B. High prevalence of hyper-aerotolerant Campylobacter jejuni in retail poultry with potential implication in human infection. Front. Microbiol. 2015;6:1263. doi: 10.3389/fmicb.2015.01263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Karki AB, Marasini D, Oakey CK, Mar K, Fakhr MK. Campylobacter coli from retail liver and meat products is more aerotolerant than Campylobacter jejuni. Front. Microbiol. 2018;9:2951. doi: 10.3389/fmicb.2018.02951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.O’Kane PM, Connerton IF. Characterisation of aerotolerant forms of a robust chicken colonizing Campylobacter coli. Front. Microbiol. 2017;8:513. doi: 10.3389/fmicb.2017.00513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bankevich A, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Page AJ, et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinforma. 2015;31:3691–3693. doi: 10.1093/bioinformatics/btv421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinforma. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Didelot X, Wilson DJ. ClonalFrameML: efficient inference of recombination in whole bacterial genomes. PLoS Comput. Biol. 2015;11:e1004041. doi: 10.1371/journal.pcbi.1004041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kokot M, Dlugosz M, Deorowicz S. KMC 3: counting and manipulating k-mer statistics. Bioinforma. 2017;33:2759–2761. doi: 10.1093/bioinformatics/btx304. [DOI] [PubMed] [Google Scholar]
- 54.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinforma. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.