

Abstract
Human immunodeficiency virus (HIV) infection is now pandemic. Targeting HIV-1 reverse transcriptase (HIV-1 RT) has been considered one of the most successful targets for the development of anti-HIV treatment. Among the HIV-1 RT inhibitors, non-nucleoside reverse transcriptase inhibitors (NNRTIs) have gained a definitive place due to their unique antiviral potency, high specificity and low toxicity in antiretroviral combination therapies used to treat HIV. Till now, >50 structurally diverse classes of compounds have been reported as NNRTIs. Among them, six NNRTIs were approved for HIV-1 treatment, namely, nevirapine (NVP), delavirdine (DLV), efavirenz (EFV), etravirine (ETR), and rilpivirine (RPV), and doravirine (DOR). In this perspective, we focus on the six NNRTIs and lessons learned from their journey through development to clinical studies. It demonstrates the obligatory need of understanding the physicochemical and biological principles (lead optimization), resistance mutations, synthesis and clinical requirements for drugs.
Graphical Abstract
1. INTRODUCTION
Acquired immune deficiency syndrome (AIDS) is caused by human immunodeficiency virus (HIV) infection and is today considered one of the most highly reviewed antiviral drug targets.1–6 According to the Joint United Nations Program on HIV/AIDS (UNAIDS) report 2017 more than 36 million people are today living with HIV including 2.1 million children under the age of 15 years. Among these only ~18 million are on antiretroviral therapy.7 Although AIDS-associated death has been significantly reduced as per 2016 data (1 million) as compared to 2005 (1.9 million), the number of new infections unfortunately remains constant per year. One of the primary reasons for the reduction of AIDS-related mortality is the introduction of combination antiretroviral therapy (also called cART). However, most of the currently available drugs for clinical use are prone to development of drug resistance.8 Briefly, cART targets multiple points of the replication cycle and typically include two nucleoside reverse transcriptase inhibitors (NRTIs), and either a protease inhibitor (PI), a non-nucleoside reverse transcriptase inhibitor (NNRTI), or an integrase inhibitor (InSTI). 9–10
HIV virus enters the host cell and uses host cell machinery to replicate, which eventually leads to significant weakening of the host immune system. HIV-1 replication is a multi-step process, and each step is crucial for successful viral replication and therefore a potential target for anti-retroviral drugs. Briefly, the life cycle begins when it binds to a CD4+ receptor, which is present on the surface of a T-lymphocyte. In addition to binding to a CD4+ receptor, HIV must bind to one of the chemokine receptors such as CCR5 or CXCR4 in order to enter into a cell (Figure 1A). These receptors interact with viral envelope proteins such as glycoproteins gp120 and trans-membrane gp41 protein. When HIV-1 approaches the host cell surface, gp120 binds to the CD4+ receptor, and this further promotes binding with the co-receptor, which leads to a conformational change in gp120 which allows gp120 to unfold into the host cell membrane and folds back on itself to induce fusion of their membranes. Viral nucleocapsid enters the host cell and releases the RNA strands and key enzymes such as reverse transcriptase (RT), integrase (IN) and protease (PR). To replicate inside the host cell, the viral DNA polymerase uses the host RNA primer, especially tRNAlys3 in order to synthesize single strand viral DNA (−), subsequently, it is hybridized with a viral RNA template to form a RNA:DNA hybrid. Further, the RNase H (ribonuclease H) domain of reverse transcriptase (RT) removes the RNA strand from the hybrid and further assists the formation of a purine-rich sequence of HIV RNA (also called “polypurine tract” or PPT), which serves as a primer for the synthesis of viral DNA strand (+). RNase H plays a crucial role in the removal of the PPT portion after priming of (+) DNA synthesis (Figure 1B). The polymerase then continues to convert the remaining viral DNA into double-stranded viral DNA (dsDNA). Subsequently, IN takes the dsDNA into the nucleus and integrates viral DNA into the host cell genome. Activation of the cell induces the transcription of pro-viral DNA into viral messenger RNA. The viral messenger RNA then translocates into the cytoplasm where it is used as a blueprint to build longer blocks of the new virus. For instance, part of the viral mRNA is translated as polyproteins: gag (structural) pol (replication) the latter as a gag-pol fusion following a frame shift event. Protease enzyme (PR) processes some of the viral proteins and digestion of a longer viral protein into smaller individual core proteins; this step is crucial in order to create an infectious virus. At this stage of the process, viral genetic material and key enzymes are assembled with co-proteins and form a viral capsid, which leaves the host cell with envelope proteins at the surface of the host cell.
Figure 1.

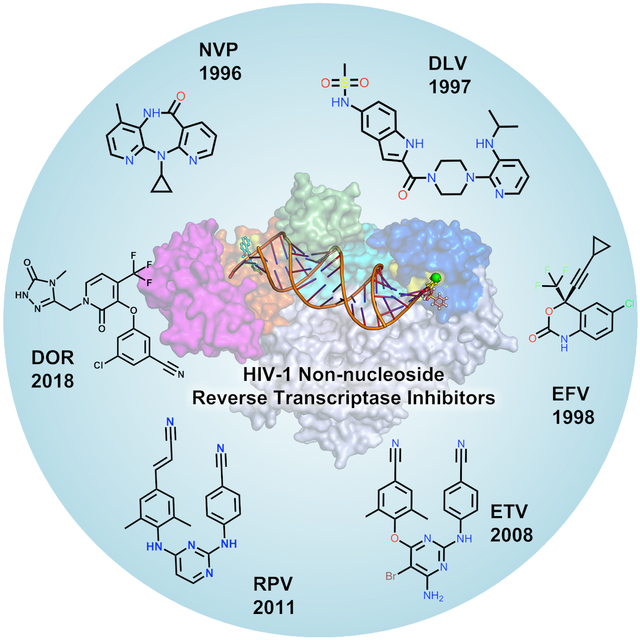
A) The overall architecture of HIV-1 is shown and B) Three-dimensional structure of HIV-1 RT (p66/p51) in complex with nucleic acids is highlighted in different colors. C) A brief timeline of HIV-1 drug development and in particular the drugs identified for RT are highlighted. The two-dimensional (2D) representation of approved NNRTI drugs is depicted.
Drugs that interfere at key replication steps will stop the replication of virus; 1) blocking of viral entry into the host cell with entry inhibitors, including fusion inhibitors (FIs); 2) inhibition of reverse transcriptase stops the further step in the replication process either by nucleoside reverse transcriptase inhibitors (NRTIs), non-nucleoside reverse transcriptase inhibitors (NNRTIs) or nucleotide analog reverse-transcriptase inhibitor (NtRTI); 3) inhibition of integration of viral DNA with host genetic material by blocking integrase activity (InSTI); 4) protease inhibitor (PI) that blocks further viral maturation. 1, 5, 11–12
1.1. Reverse Transcriptase (RT) Inhibitors
Reverse transcriptase (RT) is an RNA-dependent DNA polymerase that utilizes a strand of RNA to synthesize double-stranded viral DNA.13 On the basis of the binding site and chemical class of compounds, RT inhibitors are classified into NRTIs and NNRTIs.14–15 Figure 1C shows a brief milestone in the development of HIV-1 drugs and the important RT inhibitors are highlighted. The list of currently approved NNRTIs with their key parameters is shown in Table 1. The NRTIs are initially phosphorylated at the deoxyribose moiety in the host cell by cellular kinases, subsequently, deoxynucleotide triphosphate (i.e. NNRT-triphosphates) is incorporated into a growing strand of DNA acting as ‘chain-terminators’ in the RT active site (Figure 2A). The NNRTIs are also called allosteric inhibitors, which bind to a hydrophobic pocket distal to the active site within RT. The allosteric site-binding pocket is also termed the non-nucleoside inhibitor-binding pocket (NNIBP), which influences the geometry of the RT active site and interferes with viral DNA synthesis.
Table 1.
The approved RT-targeting drugs
| Generic | Brand | Manufacturer | FDA Approved date | Recommended dose (mg) | Serum half-life (h) |
|---|---|---|---|---|---|
| Nevirapine | Viramune | Boehringer Ingelheim | 21 June, 1996 | 200 b.i.d | 25–30 |
| Delavirdine* | Rescriptor | Upjohn, Pharmacia, Pfizer | 4 April, 1997 | 400 q.d | 2–11 |
| Efavirenz | Sustiva | Bristol-Myers Squibb | 17 September, 1998 | 600 q.d | 40–55 |
| Etravirine | Intelence | Tibotec, Johnson and Johnson | 18 January 2008 | 200 b.i.d | 41 |
| Rilprivine | Edurant | Tibotec, Johnson and Johnson | 20 May, 2011 | 25 q.d | 50 |
| Doravirine | Pifeltro | Merck | 30 August, 2018 | 100 q.d | 11–15 |
The approved drugs generic name, brand name, manufacture, date of approval, recommended daily dosage in mg and serum half-life of the drugs in hours are provided.
Discontinued by FDA. q.d: once a day, b.i.d: two times a day.
Figure 2.

(A) Ligand binding pockets of HIV-1 RT are shown with nevirapine (grey) and zidovudine (green) and the key structural domains are highlighted including non- nucleoside inhibitor-binding pocket (NNIBP) (see text for detail). (B) The NNIBP of HIV-1 RT including important secondary structural domains and amino acid residues in the pocket.
Overall, the NNIBP is largely hydrophobic and comprises amino acids L100, V106, T107, V108, V179, Y181, Y188, G190, F227, W229, L234, and Y318 that can interact through their side chains (cf. Figure 2B).16 The pocket also includes a few non-hydrophobic residues including K103, K101 and P236. The size of the NNIBP greatly varies depending on the position of the β10 strand (residues 232–234), β11 strand (residues 239–241) and the P236 hairpin loop of RT (Figure 2B). A small NNIBP includes a hydrogen bond between the carbonyl oxygen of P236 and the nitrogen of K103. Larger NNIBPs do not include such a hydrogen bond and instead provide an expanded volume for NNRTI binding.17–19
1.2. Mechanism of resistance
The allosteric binding site is not crucial to RT function and is not directly involved in substrate binding or viral DNA synthesis. As a consequence, point mutations can occur within the NNIBP that hinder NNRTI binding to NNIBP but do not interfere with RT`s role in viral DNA synthesis. A rapid selection of drug-resistant HIV-1 strains in cell culture as well as in HIV-1 infected patients has been observed with NNRTIs.20–21 Primary and secondary resistance-associated mutations occur within or near the NNIBP and reduce the efficacy of NNRTIs.22
Because of the rapid development of resistance, NNRTIs cannot be applied as monotherapy in the management of HIV23 and the major NNRTI resistance mutations include K103N/S, V106A/M, Y181C/I/V, Y188L/C/H, and G190A/S/E.24–26 The minor mutations usually occur in combination with one of the major NNRTI resistance mutations and include L100I, K101P, P225H, F227L, M230L, and K238T. In addition, double mutants also frequently occur in the RT and these include K103N/Y181C and K103N/L100I.
The first-generation NNRTIs including nevirapine (NVP), and efavirenz (EFV) show a significant loss of activity with single-point mutations such as Y181C, P236L and K103N, and the second-generation NNRTIs etravirine (ETR) and rilpivirine (RPV) show loss of activity against mutants such as K101E, E138K and Y181V. As shown in Figure 3, the effectiveness of NNRTIs has been limited by the rapid emergence of drug-resistance from new viral strains, thus the development of novel NNRTIs with more potency, improved resistance profiles and less toxicity is required. The reported X-ray crystal structures with the approved NNRTI drugs are listed in Supplementary Table S1.
Figure 3.

Venn diagram showing the most common clinically significant NNRTI-resistance mutations published for NNRTI approved drugs.27
Due to the combined availability of supercomputing facilities and sophisticated computational chemistry and biophysical approaches, computer-aided drug design plays an important role in designing novel and effective drug candidates for HIV-1.2, 28–30 For instance, in silico approaches such as virtual screening, quantitative structure-activity relationship (QSAR) and pharmacophore modeling are often used for identification and optimization of lead compounds. Recently, artificial intelligence (AI) approaches such as neural network (NN) etc. have also expanded to drug discovery projects particularly in pharmaceutical and data analytics companies31. AI approaches are particularly demanding to mine and find relationship patterns from the massive chemical data and protein sequence information towards the development of novel drug candidates. On the other hand, learning from known drugs would also help to improve the discovery and design process. Such learning could potentially be used to design new medicinal chemistry approaches that possibly could make new connections between the data. Thus, the scientific perspectives of these six NNRTIs from development to clinic will be very valuable information in the development of next-generation NNRTIs. In this perspective, we have summarized the most important drug development processes which include lead identification, lead optimization, pharmacokinetic optimization, synthetic procedures, clinical evaluation studies, etc. In addition, detailed information on protein-ligand interactions and mutations has been included to explain the mechanism of resistance of NNRTIs.
2. DISCOVERY AND DEVELOPMENT OF NEVIRAPINE (NVP)
Nevirapine (also known as BI-RG-587) was the first NNRTI drug emerged as a selective non-competitive inhibitor/NNRTI drug developed from the tricyclic pyridobenzo- and dipyridodiazepinones series by Boehringer Ingelheim Pharmaceuticals, Inc.1–3 In 1990, compound BI-RG-587 was identified from high-throughput screening (HTS) of a large collection of the company’s internal compound database. Among the screened compounds, compound pyrido[2,3-b][1,4]benzodiazepinones (1) was shown as a weak HIV-1 RT enzyme inhibitor with an IC50 of 6 μM, and structurally similar isomeric compounds such as pyrido[2,3-b][1,5]benzodiazepinones (2), dipyrido[3,2-b:2’,3’-e][1,4]diazepinones (3) and dibenzo[b,e][1,4]diazepinones (4)32 compounds were also screened against HIV-1 RT.
2.1. Lead optimization
Molecular hybridization and bioisosterism are two classical concepts that were primarily used in the lead optimization. In the case of NVP development, both concepts were successfully applied. For instance, compounds 3 and 4 are hybridization products of 1 and 2. For synthetic purposes, compound 2 was chosen as a lead structure and further structural modifications were introduced. The N-alkylated (5 and 11 positions) derivative of compound 2 6,11-dihydro-11-ethyl-6-methyl-5H-pyrido[2,3-b][1,5]benzodiazepinone (5a) was developed and found to be active against HIV-1 RT with an IC50 value of 350 nM. Later, compound 3 was optimized from compound 2 by N-alkylation and this led to the identification of a potent dipyrido compound (5b). Although dipyrido compound 5b showed a strong inhibition in HIV-1 RT, because of the fast metabolism through N-dealkylation (N5 methyl and N11-ethyl) by CYP450, the activity of 5b was drastically reduced. Based on the knowledge from SAR study and metabolism profiling, the methyl group was moved from the N5 to the C4 position at the aromatic ring and yielded a metabolically stable compound (6) with improved potency (compared to 5b).33–34It is worth noting that moving a methyl group from the N5 position to the aromatic ring helped to show a favorable interaction with Y181 at the NNIBP site and improved the overall binding affinity towards HIV-1 RT. Moreover, substitution of the 4-methyl group in the aromatic ring significantly reduced the N-deethylation at the 11th position because CYP450 oxidation takes place at 4-methyl group and the rate of 4-methyl hydroxylation is very slow as compared to N-dealkylation (cf. Figure 4).
Figure 4.

The key steps and intermediate compounds in the lead optimization process of NVP. The possible sites of CYP450 metabolism are indicated with a yellow sphere.
From the detailed SAR studies, it was found that the N11-position is key for HIV-1 RT inhibition, for instance, smaller alkyl groups such as methyl, ethyl (7) or cyclopropyl (8) groups improve the binding affinity and pharmacokinetic properties. Later, these compounds were chosen for detailed preclinical studies.
2.2. Metabolism of NVP
NVP undergoes an extensive CYP450 oxidative metabolism which primarily occurs in liver microsomes at the C-2, C-3 C-4 methyl and C-8 positions (by CYP3A4 and 2B6) resulting in five major metabolites: 2-Hydroxy-nevirapine, 3-Hydroxy-nevirapine, 4-Hydroxymethyl-nevirapine, 4-carboxy-nevirapine and 8-Hydroxy-nevirapine (cf. Figure 4).35–38 Hydroxylated primary metabolites undergo glucuronidation by UGT enzymes. Because of low protein binding 90% of NVP is immediately absorbed.
2.3. RT-NVP interactions
Figure 5 demonstrates key interactions for NVP within the NNIBP of HIV-1. It occupies the hydrophobic pocket made up of L100, V106, V179, Y181, Y188, G190, F227, W229, L234 and Y318. Hydrophobic and stacking interactions have been observed between the two pyrimidine rings and the alkyl substitutions of NVP and L100, V106 and the aromatic side chains of Y188 and Y181.
Figure 5.

(A) Binding mode of NVP (green) with important amino acid residues in the binding pocket of HIV-1 RT (PDB code: 3V81) and (B) schematic representation of ligand pharmacophore models is encircled in red.
2.4. Resistance of NVP
Resistance to NVP has resulted from one or more of the amino acid substitutions K101E, G190A/S, K103N, V106A/M, V108I, Y188C/L, A98G, F227L, and M230L.22 Among these, K103N, Y181C and G190A substitutions appeared frequently.39–40 Based on the three-dimensional structure of NVP in mutant RT compared to wild-type RT, the interactions between the two-pyrimidine rings of NVP and the aromatic side chains of Y181 and Y188 could explain the significant reduction in inhibition when Y181C and Y188C mutations emerge.41 The mutations G190A and L100I sterically hinder the binding of NVP to NNIBP.42–43 The mutants G190A and K103N possibly affect the flexibility of the β-strand and this diminishes the overall binding affinity NNRTIs.42,44 NVP is a fairly rigid molecule with no linker fragments that could allow for flexibility. Due to the rigidity of its structure, this molecule cannot adapt to point mutations within the NNIBP and resistance develops rapidly to NVP.39
2.5. Clinical trials of NVP
NVP was approved for use in HIV-1 infected adults on the basis of two clinical trials namely BI 1090 and BI 1046 (INCAS Trial, a randomized trial conducted in Italy, the Netherlands, Canada, and Australia). Trial BI 1090 was a double-blind, placebo-controlled, randomized study initiated in 1995.45 2249 HIV-1 infected patients with CD4+ cells <200 cells/mm3 were randomized into a NVP + lamivudine (3TC) + background therapy (BT) arm or a 3TC + BT arm. The patient’s median CD4+ count at baseline was 96 cells/mm3 and HIV-RNA baseline was 4.58 log10 copies/mL. 89% of patients were treatment experienced. The percentage of responders (HIV-1 RNA <50 copies/mL) at week 48 were 18% for the NVP arm and 2% for the placebo arm.
BI 1046 (INCAS trial) was a placebo-controlled, double-blind, randomized 3-arm clinical trial comparing nevirapine (NVP) + zidovudine (AZT) + didanosine (ddI), NVP + AZT, and AZT + ddI.46 151 HIV-1 infected patients with CD4+ counts between 200–600 cells/mm3 entered into the study. The patients had a median baseline CD4+ count of 376 cells/mm3, and baseline RNA was 4.41 log10copies/mL. Patients with HIV-1 viral loads <400 copies/mL at 48 weeks were 45%, 19%, and 0% for the NVP + AZT + ddI arm, AZT + ddI arm, and NVP + AZT arm respectively.
NVP is available as 100, 200, and 400 mg tablets and a 50 mg/5mL oral suspension. It can be taken with or without food. The standard adult dose is 200 mg (1 tablet) daily for the first two weeks of treatment and 200 mg, twice daily, thereafter. NVP is no longer recommended as first-line or salvage therapy in US treatment guidelines. Severe rash and hepatotoxicity have been reported as adverse events associated with NVP.47,48 It is also characterized by a low barrier to resistance, where the emergence of one mutation compromising NVP efficacy can compromise future treatment with other NNRTIs.49
2.6. Commercial synthesis of NVP
Synthesis of NVP (and its derivatives) was first reported by Merluzzi et al. and Grozinger et al. starting from 2-chloro-3-nitro-4-methylpyridine (9) and 2-chloronicotinoyl chloride (11).37, 50 Compound 9 was used as a precursor for building 2-chloro-3-amino-4-methylpyridine (10) by hydrogenation over 5% Rh/C. Precursors 10 and 11 were reacted together in solvent system 1,4-dioxane, cyclohexane and pyridine to obtain 12 in 65% yield followed by the reaction with cyclopropyl amine in xylene at 110 °C to give 13 in 83% yield. Compound 13, on cyclization under basic conditions (NaH in diglyme) at 160 °C, afforded NVP (8) in 67% yield (Scheme 1).
Scheme 1:

Synthesis of NVP (8) by Merluzzi et al.50 and Grozinger et al.37
As described above in Scheme 1, compound 9 was the main block for the synthesis of NVP. In earlier reports51 on synthesis on NVP, 9 was prepared from 14 due to complications in separation of two regioisomers (1: 13.2) formed during the reaction; production of 9 on a bigger scale was a bottle-neck. Therefore, a subsequent advancement was reported in which 10 was synthesized from 17 which was commercially accessible in bulk quantities. To get rid of hydroxyl groups in compound 17 it was subjected to halogenation by POCl3 at 110 °C – 140 °C and subsequently dehalogenated by reduction over Pd-C. In order to convert the nitrile group into an amine, 19 was partially hydrolyzed by Amberlite IRA-400 and subsequently subjected to Hofmann reaction to give 21 with 75% yield. Halogenation of 21 by Cl2/HCl given 10 in 84% yield (Scheme 2).34, 51–54
Scheme 2:

Synthesis of key building blocks required for NVP (8)
Alternatively, Grozinger et al.36 attempted a synthesis of NVP from 18 with an idea that the 6-chloro group can be dehalogenated in the last step and keeping it might help in cyclization. Compound 18 was hydrolyzed using H2SO4 and subjected to Hofmann reaction to form 23 in 92–93% yield. Similar to 10 in Scheme 1, 23 was also reacted with 11 to obtain 24 (80% yield) and followed by reaction with cyclopropylamine to give 22 in 84% yield. Cyclisation of compound 22 by NaH and dehalogenation over Pd-C/H2 gave NVP (13) in 72% yield. The main shortcoming of this method was insolubility issues with 23 when attempting to produce NVP (8) at bulk scale (Scheme 3). 37, 55
Scheme 3:

Synthesis of NVP (8) proposed by Grozinger et al.36
Although the above methods (Scheme 2 and 3) are best known for synthesis of NVP, the described methods had a few drawbacks; cyclopropyl amine was more likely to vaporize in the process of synthesizing 13 and the process needed to be done in a sealed vessel in order to avoid the vaporization. However, 13 was not very stable at high temperature (>145 °C) and prone to explode at such high temperature. Thus, these methods were improved again by reacting cyclopropyl amine with 24 at 77 to 100 °C to achieve 25 followed by basic hydrolysis and activation of the carbonyl group by SOCl2. The resultant product reacted with 10 to form 13. Ring closure of 13 was achieved by sodium bis(trimethylsilyl)amide (NaHMDS) in THF to obtain the product, NVP (8) (Scheme 4).56
Scheme 4:

Recent synthetic procedure developed for NVP (8)
However, because this method uses expensive reagents including NaHMDS, the method was modified again by facilitating the SNAr at 80 °C by adding KI in the transformation from 12 to 13 (Scheme 1). Likewise, the reaction of 13 to 8 (Scheme 4) was also optimized at 130 °C.57
3. DISCOVERY AND DEVELOPMENT OF DELAVIRDINE (DLV)
Delavirdine (DLV) is the second NNRTI drug candidate approved by the FDA for clinical use in 1997, and can be one of the components of cART. At the time of its introduction, DLV was recommended as first-line therapy in combination with other NRTIs.58 DLV shows greater ligand flexibility in comparison with NVP and other NNRTIs, but is less effective against various mutants.
3.1. Lead optimization
The initial hit molecule of DLV mesylate (also known as PNU-90152) was first identified in 1992 by Upjohn scientists from a chemical library of 1500 structurally diverse compounds and shows very similar structural features tobis(heteroaryl)piperazine (BHAP).59 During computational filtration followed by in vitro screening of HIV-1 RT inhibition, several compounds showed weak to moderate inhibition. Among these compounds pyridylpiperazine derivative (PNU-80493E) was identified with an IC50 of 20 μM (ED50 = 2 μM), which is weakly potent than AZT (0.07 μM), but PNU-80493E showed better selectivity for polymerase α and δ as compared to AZT. In addition, PNU-80493E also had a better cytotherapeutic ratio (CC50/ED50) as compared to AZT. Other compounds that possessed phenylpiperazines moieties showed less potency compared to PNU-80493E. Therefore, this compound was chosen as the lead compound for further optimization.60
The lead compound was further optimized through SAR studies and before introducing an indole group in the lead molecule, the linker between piperazine and the tri-substituted phenyl ring was optimized. Although a carbonyl spacer group did not increase the binding affinity as compared to an alkyl linker, introducing a hydrogen bond acceptor (carbonyl group) greatly improved the binding affinity with HIV-1 RT as seen in the crystal structure (cf. Figure 6 and Figure 7A). The lead compound U-85199E was further modified by introducing an indole moiety in the place of the phenyl ring which led to several potent compounds.
Figure 6.

The key steps, techniques and intermediate compounds in the lead optimization process of DLV. The possible sites of CYP450 metabolism are indicated with a yellow sphere.
Figure 7.

(A) The 2D ligand-interaction diagram of RT and DLV (PDB ID: 1KLM). The important amino acids residues in the binding pocket are shown. (B) The 3D representation of DLV indicating the conformational flexibility by specifying the number of rotatable bonds.
Further, Romero et al. 61 explored anti-HIV potency of indol-2-yl substituted arylpiperazines, in particular positions X and Y were extensively optimized in wild-type and mutants e.g. P236L and Y181C (cf. Figure 6). Additionally, the flexibility of the molecules also increased to accommodate in the binding site of RT (Figure 7B). In total, six possible clinical candidates (including U-85199E) against the HIV RT enzyme were chosen and metabolic stability was optimized at the 5-position of indole and N-alkyl group. Compounds substituted with 5-methoxy and 5-fluoro congeners (U-87201, U88352 and U88353) at indole led to less metabolic deactivation via hydroxylation. Likewise, N-alkyl substituents such as bulky N-isopropylamine and N-teritarybutyl amine created the sterically hindered region to reduce the rate of N-dealkylation metabolic conversion. These structural modifications resulted in superior oral bioavailability in laboratory animals followed by good antiviral potency.
3.2. Pharmacokinetic studies of DLV
Based on pharmacokinetic studies of six compounds (cf. Figure 6), PNU-87201E (atevirdine mesylate salt form) was chosen as the first-generation drug candidate which showed better physical and metabolic stability.62 The total plasma clearance of atevirdine mesylate was found to be approximately two times slower compared to unsubstituted indole analogs such as U-85961 and U-88204.62In addition, PNU-87201E demonstrated good oral bioavailability in animals (50 ±20% in female beagle dogs; 62 ± 20% in male rats) and drug concentrations in serum greatly exceeded those required for in vitro antiviral activity (Cmax= 16–33 μM, Tmax= 1 h, in female beagle dogs; Cmax = 4.6–17.4 μM, Tmax= 0.5–1 h, in male rats. Moreover, in preclinical studies atevirdine mesylate (U-87201E) proved to have a good margin of safety upon multiple dosing.63 Further, the metabolic stability due to indole substituent, central spacer and pyridine heterocyclic was also studied. Based on the toxicokinetic studies three compounds showed better profiles, e.g., 5-substituted indole PNU-90152, the 6-substituted indole PNU-89388, and the (alkylamino)piperidine PNU-90328. Among these three analogs, only PNU-90152 inhibited HIV-1 replication in cell culture and exhibited the highest serum concentrations as well as good tolerability.64–65
3.3. Metabolism of DLV
DLV is primarily metabolized in the liver by cytochrome P450 3A4. The key metabolic process includes N-dealkylation, hydroxylation of N-dealkyldelavirdine, amidic hydrolysis and hydroxylation at the pyridine ring (cf. Figure 6, Supplementary Figure S1). From metabolic studies in mouse, rat, dog, monkey, rabbit, and human, N-desisopropyldelavirdine is the major metabolite which undergoes hydroxylation either at C-4’ or C-6’ position of the pyridine ring and subsequently conjugates with sulfate. In addition, DLV is also directly hydroxylated at the C-6’ pyridine ring and forms hydroxylated 6’-pyridinoldelavirdine as major metabolite. A minor portion of DLV is hydrolyzed at the amide bond by indole carboxylic acid and N-isopropylpyridinepiperazine.66–67 All metabolites are inactive and primarily excreted renally.
3.4. RT-DLV interactions
The greater size of DLV allows interaction with more residues in the NNIBP as compared to NVP (cf. Figure 6).68 Compared to NVP, DLV introduces new interactions, which include hydrogen bonding between a carbonyl spacer group to K102 and K103 and hydrophobic interactions with P236. The piperazine ring as a linker molecule allows interactions with V106 and the indole ring of DLV forms hydrogen bonding to the main chain of K103 and a hydrophobic interaction with P236. The isopropylamino and pyridine group of DLV are positioned in close proximity for aromatic interactions with Y181, Y188 and W229.
3.5. Resistance of DLV
The common resistance mutations for DLV include L100I, K103N, and Y181C, and also P236L.54, 64 Although DLV makes extensive hydrophobic contacts with the P236 side-chain via its indole ring in the NNRTI binding pocket, the mutation P236L significantly reduces the potency of DLV. The possible reason could be the extreme molecular flexibility and in particular at the carbonyl and sulfonamide groups (terminal), which reduce the binding affinity of the indole ring towards P236L mutation. Furthermore a study by Romero et al.61 showed that removal of the terminal sulfonamide group (i.e. un-substituted indole at R3 and R4 position) increases its potency against P236L mutants but significantly reduced its effectiveness against Y181C mutant 69 and showed very poor pharmacokinetic properties.
Although the P236L mutation confers resistance against DLV, it increases the effectiveness of other NNRTIs. While DLV is also selective for P236L mutation, only 6% of DLV- resistant HIV isolates from patients receiving DLV monotherapy contain the P236L mutation, whereas 80% include the K103N mutation.70–71 It has been suggested that the P236L mutation confers a replication defect in comparison to wild-type that accounts for a predominance of the K103N mutant compared to P236L. Interestingly, P236L confers resistance to DLV, but it does not decrease susceptibility to other NNRTIs, and therefore failing therapy with DLV does not result in NNRTI cross-resistance.59 DLV is unique in its capacity to induce a G190A and G190E mutation that demonstrates enhanced inhibition of RT function.72–73 The increased susceptibility to RT inhibition by DLV is explained based on G190E RT being less processive compared to G190A RT or wild-type HIV-1 RT with regard to both polymerase and RNase H function.
3.6. Clinical trials of DLV
DLV was approved on the basis of two clinical trials; study 21 part II and study 13C.74 Both studies were double-blind, placebo-controlled, randomized trials evaluating DLV in HIV-1 infected patients.75–77 Both trials enrolled patients who were treatment-naïve or treatment-experienced. Study 21 part II compared DLV + AZT + 3TC, DLV + AZT, and AZT + 3TC in 373 HIV-1 infected patients. Intent to treat analysis of patients with viral loads <400 copies/mL at 54 weeks was 45%, 2%, and 14% for the DLV + AZT + 3TC arm, DLV + AZT arm, and AZT + 3TC arm, respectively. Study 13C randomized 345 HIV-1 infected patients into 2 arms. The first arm was DLV + AZT + either ddI, 3TC, or zalcitabine (ddC). The comparator arm was AZT + either ddI, 3TC, or ddC. The proportion of patients with viral loads <400 copies/mL at 54 weeks was 29% for the DLV arm and 10% for the non-DLV containing arm.
DLV is rarely used today and is not recommended in the U.S. Department of Health and Human Services (DHHS) or International Antiviral Society (IAS) HIV treatment guidelines due to high pill burden, dosing schedule, and drug interactions.78 The most common adverse event is severe rash, which occurred in 18% of patients in clinical trials.74, 79
DLV and NVP were studied during a time before the utility of cART was determined. Regimens were suboptimal by today’s standards; the efficacy of a three-drug regimen (anchor + NRTI backbone) had not been established. As such, many of the clinical trials described were not differentiated by treatment-naive or treatment-experienced patients. Indeed, many patients during this time had previously been treated with monotherapy AZT and harbored NRTI resistance mutations prior to enrolment.
3.7. Commercial synthesis of DLV
Synthesis of DLV was initially reported by Romero et al.63–64 Piperazine (28) was reacted with 2-chloro-3-nitro pyridine (29) and followed by protection of amine as t-butyloxycarbamate to obtain 30 (96% yield). The nitro group in 30 was reduced by hydrogenation on Pd/C and subsequently condensed with acetone to form imine, which was reduced by sodium cyanoborohydride to form 31 (91% yield). The t-butylcarbamate group in 31 was deprotected by TFA and reacted with 32 using peptide coupling agents 1,1’-carbonyldiimidazole/N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (CDI/EDC) to obtain 33 (74% yield). The nitro group in 33 was reduced by hydrogenation on Pd/C and reacted with mesityl chloride in the presence of pyridine as a base to obtain DLV (34) (Scheme 5).
Scheme 5:

Synthesis of DLV by Romero et al.63
Although the above method was very efficient in preparation of a number of DLV derivatives, it had a few drawbacks concerning bulk scale production. For instance, Pd/C was responsible for increasing the cost of production and purification of side products generated by CDI/EDC at bulk scale was problematic. These issues were solved by replacing Pd/C with a low-cost Raney Ni and coupling agents CDI/EDC was also replaced by pre-activation of the carboxylic group using SOCl2 (Scheme 6).80–81 Compound 36 was obtained from 35 by ester hydrolysis using potassium hydroxide in methanol followed by reduction of nitro group using Raney Ni (an alternative to Pd-C) in >90% yield. Mesylation of 36 was done by mesityl chloride and sodium carbonate to obtain 37 (83% yield). Compound 37 was coupled to 39 by pre-activation of carboxylic group with thionyl chloride to give DLV (34) in 63% yield (Scheme 6).
Scheme 6:

Modified synthetic procedure of DLV (34)
Despite this method providing a reasonable yield in each step, 38 was moisture sensitive and it was difficult to maintain dry conditions at bulk scale. Recently, another method has been reported in which 40 was reduced using Fe powder and a cheaper alternative to Pd-C/Raney Ni to obtain 41. This was followed by mesylation using mesityl chloride/pyridine to attain 42 in 89% yield. Then, 42 was reacted with 39 at 60 °C to give DLV (34) in 63% yield (Scheme 7).82
Scheme 7:

Recent synthetic procedure developed for DLV (34)
4. Discovery and Development of Efavirenz (EFV)
The third approved NNRTI drug for the treatment of HIV-1 infection is efavirenz (EFV, DMP 266, L-743,726) which belongs to the series of benzoxazinones. This drug exhibited potent inhibition against wild-type HIV-1 RT enzyme (Ki = 2.93 nM) as well as a panel of NNRTI-resistant single RT mutant viruses with 95% inhibitory concentrations of <1.5 μM.83
4.1. Lead Optimization
The initial prototype for the development of EFV was initiated with a benzothiadiazine compound NSC287474, which showed potent activity against a variety of HIV-1 strains.84 In the development process of EFV, the bioisosteric replacement strategy was utilized to obtain the lead compound L-608,788.85
The combined introduction of acetylene and cyclopropyl improved the activity but decreased the oral bioavailability. A variety of aryl groups were explored at the acetylene group and identified cyclopropylarylethynylquinazolin-2(1H)-ones) a racemic mixture (IC50 = 23.2 nM) enhanced the potency against HIV-RT and also improved oral bioavailability.86–88 Furthermore, the obtained (−)-4(S) enantiomer was found to be the active enantiomer (IC50 = 12nM) and the other one (+)-4(S) identified was inactive (IC50 = 9300 nM)89. Initial structural modification of quinazolines with alkylbenzoxazepinones, benzothiadiazepines decreased the activity and 1,4-dihydro-2H-3,1-benzoxazin-2-ones (L-743, 726 or efavirenz, EFV) was found to be a potent NNRTI. Due to possible oxidative metabolism of N-methyl or NH- groups at position 3, further optimized with bioisosteres. Subsequent studies with different substituents (see Figure 8) on the benzoxazinone ring explain why bulky substituents at position 6 were not allowed due to a decrease in activity. Attempts to replace the cyclopropylacetlylene group at the C4 position with small heterocyclic (furan, pyridine) or alkoxy groups (saturated or unsaturated) yielded only equivalent or less potent molecules in comparison to EFV. Later studies demonstrated that the CF3 group is essential and improves the potency by reducing pKa and forming strong hydrogen bond interactions with the enzyme.90 Later, molecular modeling studies predicted different interactions for the molecules replacing the benzoxazinone with indolin-2-one ring which improves the activity.91
Figure 8.

The lead compounds and important compounds synthesized and tested in the process of EFV lead optimization
4.2. Metabolism of EFV
EFV undergoes cytochrome P450 oxidative metabolism and primarily occurs in liver microsomes at C-7 and C-8 methyl positions resulting in two major metabolites: 7- and 8-hydroxy-efavirenz, respectively (Supplementary Figure S3). The minor 7-hydroxy-efavirenz metabolite undergoes additional metabolic conversion to 8,14-dihydroxy-efavirenz. The hydroxyl metabolites are excreted in urine in the form of glucuronide conjugates. However, in rat 8-hydroxy-efavirenz is identified as a major metabolite which conjugates with sulfur. The presence of a triple bond was expected to be a potential risk in formation of reactive metabolites.92–93 In particular, the metabolites identified from rats, monkey and humans revealed that rats produce a unique glutathione adduct and cause nephrotoxicity.94 Due to lack of specific glutathione S-transferase (GST) and related enzymes in human and monkey might result in the absence of glutathione adducts. Detailed studies revealed that decreasing the cycloproponal metabolite or inhibiting the γ-glutamyltranspeptidase reduces the nephrotoxicity in rats.
4.3. RT-EFV interaction
The benzoxazin-2-one ring is bound between the side chains of L100 and V106, and makes edge-on contacts with Y318 and V179. Hydrogen bonds are observed between the benzoxazin-2-one NH and carbonyl group and the backbone carbonyl oxygen and backbone amine of K101. The nitrogen of the benzoxazin-2-one ring of EFV is also located close to the side chain of K103 allowing for a van der Waals interaction. The cyclopropyl–propynyl group is situated in a pocket flanked by the aromatic side chains of Y181, Y188, F227 and W229.
4.4. Resistance of EFV
EFV has a different resistance profile compared to NVP and DLV. Resistance resulting in reduced susceptibility in cell culture to EFV has nonetheless been observed. EFV has a somewhat greater barrier to resistance compared to NVP and DLV and is considered a second-generation NNRTI.41 The propynyl–cyclopropyl group of EFV is smaller than the aromatic pyridine ring of NVP.44 Consequently, interactions with mutable Y181 and Y188 sidechains are more limited compared to NVP. Thus, the Y181C and Y188C mutations do not have as much of an effect on EFV binding compared to NVP.44 This is consistent with the three-dimensional structure of EFV with Y181C RT. K103N substitution also caused a decrease in susceptibility to EFV, although the decrease in affinity due to the K103N mutation was less for EFV compared to NVP.83, 95–96 A hydrogen bond was observed for EFV, but not for NVP, with the backbone carbonyl of K103.25 The ability of the smaller EFV molecule to rearrange in the mutated K103N NNIBP compared to the bulkier, rigid structure of NVP could explain the different binding affinities.44 The G190A and L100I mutations sterically hinder binding to EFV, similar to NVP. Resistance to EFV has also resulted from one or more of the amino acid substitutions L100I, K101P, G190A/S, V106M, Y188L, G190A/S/E, F227L, and M230L that sterically hinder binding to EFV similar to NVP.26,97
4.5. Clinical trials of EFV
EFV was the third NNRTI approved by the FDA. It gained approval for use in HIV-1 infected adults in 1998 on the basis of two clinical trials. Study 006 enrolled 1266 HIV-1 infected patients into an open-label trial who were randomized to either an EFV + AZT + 3TC arm, an EFV + indinavir (IDV) arm, or an IDV + AZT + 3TC arm. Median CD4+ count at baseline was 345 cells/mm3; median baseline HIV-1 RNA was 4.8 log10 copies/mL. Virologic response (viral load <400 copies/mL) at 48 weeks was 70% for the EFV + AZT + 3TC arm, 53% for the EFV + IDV arm, and 48% for the IDV + AZT + 3TC arm.98 This result demonstrated the increased efficacy of EFV + 2 NRTIs compared to IDV + 2 NRTIs or IDV + EFV. The combination of EFV + 2 NRTIs was shown as a potent combination of antiretroviral agents.
AIDS Clinical Trials Group (ACTG) 364 enrolled 196 NRTI-experienced patients into a double-blind, randomized, placebo-controlled study. Patients were randomized to 1 of 3 arms; EFV + nelfinavir (NFV) + NRTIs; EFV + NRTIs; or NFV + NRTIs. All patients were assigned new NRTIs based on their past NRTI exposure. Baseline CD4+ cell count was 389 cells/mm3; baseline HIV-1 RNA was 8130 copies/mL. At 48 weeks, the percentage of patients with viral loads <500 copies/mL was 78% for the EFV + NFV + NRTIs, 63% for the EFV + NRTIs, and 40% for NFV + NRTIs.98 This study demonstrated that in treatment-experienced patients the combination of EFV + NFV + 2 NRTIs was the most effective in suppressing viral loads compared to only 1 NNRTI or PI + 2 NRTIs.
EFV was the most widely used in treatment-naive patients among the three NNRTIs approved in the late 1990’s. Its most common formulation is a fixed-dose combination with tenofovir disproxil fumarate (TDF)/emtricitabine (FTC), making it the first one pill once a day tablet. The standard dose of EFV, either alone or in combination, is 600 mg per day. It is currently recommended as an alternative regimen in the DHHS guidelines based on its widespread use and virologic efficacy; however, patients on EFV-based regimens have exhibited relatively high rates of neuropsychiatric/central nervous system side effects, affecting overall tolerability.99
4.6. Commercial synthesis of EFV
Synthesis of EFV was first reported by Young et al.100 and subsequent advancements were made over the years.101–103 Reports from Pierce et al. (from Merck and duPont)104 and Dai et al. (Lonza Ltd.)105 represent reasonable approaches for the synthesis of EFV at bulk scale. The scientists from Lonza’s Ltd. made advancements by using 1,4-dichlorobenzene because of the necessity of amine moiety to be protected by a protecting group if present at the beginning and using 1,4-dichlorobenzene may shorten the route by avoiding amine protection. Considering this learning, Correia et al. discovered a new route to EFV by flow synthesis.106
Pierce et al.104 started the synthesis from 4-chloro aniline (43) and in order to introduce the trifluoroacetyl group meta to chloro, amino group was protected as t-butyl carbamate and subsequently reacted with n-BuLi/ethyl trifluoroacetate. The deprotection of t-butyl carbamate in 45 by HCl-CH3COOH gave 46, which was purified by crystallization at 5 °C and isolated in 84% yield. For further reactions 46 was needed in neutralized form; therefore, it was stirred with NaOAc in MTBE to provide 47. p-Methoxybenzylation of 47 was done by p-methoxy benzyl alcohol, an inexpensive and less toxic alternative to the previous reports103 to form 48 (90% yield). Compound 48 was enantioselectively alkynylated using 49 in presence of ligand 50 in THF-toluene-hexane mixture at −50 °C and gave 51 in 91–93% yield and >99.5% enantiomeric excess (ee). Cyclization of 51 by COCl2/TEA in 95% yield followed by p-methoxy benzyl deprotection by ceric ammonium nitrate gave EFV (53) in 76% yield. However, p-methoxy benzyl deprotection and ceric ammonium nitrate were responsible for the generation of side products like p-methoxybenzaldehyde and some cerium salts. Therefore, an alternative method was explored in which 51 was reacted with DDQ in toluene to give 54a/54b followed by reaction with NaOMe-MeOH which gave amino alcohol 55 (94% yield) and the side product p-methoxybenzaldehyde was converted into p-methoxy benzyl alcohol by reduction with NaBH4. Ring closure of 55 was achieved by phosgene to obtain EFV (53) in 95% yield (>99.5% purity, >99.5% enantiomeric excess or ee after crystallization with THF-heptane), alternative attempts of ring closure of 55 by methyl carbamate only give EFV in 83% yield and using p-nitrophenylcarbamate achieved EFV in 94% yield after recrystallization (Scheme 8).
Scheme 8:

Synthesis steps of EFV developed by Pierce et al.104
Although the procedure from Merck and duPont provide good yields (<90%) in the majority of steps, Chen et al.107 reported an alternative procedure in which protection of anilinic amine can be avoided by using Me2Zn and 57 in the presence of additives (2,2-dimethyl propionicacid, 2,2,2-trichloroethanol) and reacted with 56 at −10 °C to form 55 in 95.2% yield (99.8% purity, 99.3% ee) (Scheme 9). Subsequent advancement in the reaction scheme was made by Dai et al. from Lonza in which protection/deprotection of anilinic moiety was bypassed by starting the synthesis from 1,4-dichlorobenzene.105
Scheme 9:

Advancements in the synthesis of EFV by Chen et al.107
5. DISCOVERY AND DEVELOPMENT OF ETRAVIRINE (ETR) AND RILPIVIRINE (RPV)
5.1. Lead optimization
The development of the diarylpyrimidine (DAPY) series as NNRTIs was started in 1987 by Janssen Pharmaceuticals in collaboration with the Rega Institute for Medical Research. The search was initiated by screening 600 compounds on the MT-4 cell-based anti-HIV assay. Among those screened, 4,5,6,7-tetrahydro-5-methylimidazo[4,5,1-jk][1,4]benzodiazepin-2(1H)-one (TIBO, R14458) was recognized as first NNRTI which inhibited HIV-1 replication at a moderate level.108–109 Subsequently, the hydrophobic ring was broken and analogs of α-anilinophenylacetamide (α-APA) were synthesized and named loviride.110 These two scaffolds exhibited excellent potency against wild-type HIV-1 but had lesser potency against the common NNRTI-resistant mutants.111 Later in the same year, further steps were taken by increasing the space between the two ring fragments and cyclization of the linker which resulted in a new class of diaryltriazine (DATA, R106168) and revealed high activity against wild-type and mutant strains, in particular the G190A mutant, but reduced potency against the double mutants L100I/K103N112. Consequently, the central triazine ring replaced the bioisostere pyrimidone and continued the development of new prototype diarylpyrimidine (DAPY) analogue of TMC120 (Dapivirine, DPV or R147681) to improve the solubility, bioavailability, pharmacokinetics, and potency against HIV-1 double mutations.
In collaboration with Tibotec Pharmaceuticals Ltd. and the International Partnership for Microbicides (IPM), DPV was developed as a vaginal microbicide to prevent/decrease the rate of sexual transmission of HIV-1.113 The unsubstituted pyrimidine ring of dapivirine was further optimized with a bromine atom introduced at the 5-position and amine group at the 6-position which led to the identification of etravirine (ETR).72, 114 ETR shows high potency against wild-type and mutant strains include L100I, K103N, V106A, Y181C, Y188L, L100I/K103N, and K103N/Y181C.115–116 Further, the help of medicinal chemists, crystallographers, and computational approaches resulted in the discovery of the cyanovinyl DAPY derivative called as TMC278 (rilpivirine, RPV).117 The extended cyanovinyl group of RPV makes the compound highly active with an IC50 <5 nM against both wild-type and many clinically relevant mutants by interacting directly with the highly conserved W229 residue of RT. The development of ETR and RPV took about 20 years (1987 to 2001) from TIBO to DAPY series by long-term optimization (Figure 11). These two drugs are very active against mutant strains through diverse modes of interaction within the highly flexible NNRTI binding site of RT through molecular flexibility and conformational interconversion for i.e. “wiggling” and “jiggling” models.117–118 In addition, Ren et al have proposed alternative binding models using benzophenone derivatives such as “up- and down” position models.119
Figure 11.

Key lead optimization process of ETR and RPV
5.2. Metabolism of ETR
The elimination half-life of ETR is 30 to 40 hours which is fairly long due to the high protein bound (99.9%) affinity (mainly to the albumin protein and α1-acid glycoprotein). The metabolism of ETR is largely catalyzed by cytochrome P450 subtypes 3A, 2C9 and 2C19. The key hydroxylation process at the dimethylbenzonitrile moiety causes two forms of monohydroxy-etravirine as major metabolites (Supplementary Figure S3). Later the metabolites were further hydrolyzed to form dihydroxy-etravirine. Subsequently, the metabolites undergo glucuronide conjugation by means of uridinediphosphateglucuronosyltransferase (UGT).120–121
5.3. Metabolism of RPV
RPV is metabolized by cytochrome P450 3A4 and 3A5. The key metabolic process is hydroxylation at the dimethylphenylacrylonitrile (Supplementary Figure S4). A total of seven different metabolites were identified using tandem mass spectral analysis. Among these, four form as oxygenated RPV metabolites with two mono- (M1, M2) and hydroxy-RPV (M3, M4) each. The remaining three conjugate form metabolites as N-linked and O-linked glucuronidations (M5-M7) by UGT subtypes 1A1 and 1A4.122
5.4. RT-ETR interaction
ETR binds to NNIBP in a horseshoe conformation20–21, 123–125 and demonstrates single-digit nanomolar potencies against both wild-type and mutant HIV-1 isolates. The pyrimidine scaffold of ETR is positioned between L100 and V179.72, 118 The primary amine and bromo substituents point toward an opening in the pocket between K101 on the p66 subunit and E138 of the p51 subunit. A hydrogen bond is observed between the N1 pyrimidine nitrogen and the carbonyl oxygen from the K101 main chain. A hydrogen bond is also observed between the primary amine and carbonyl group of the E138 sidechain. The benzonitrile substitution occupies a sub-pocket comprised of V106, P225, P236, F227, Y318, H235, P236 and L234. K103 is located closely to the benzonitrile group and is possibly involved in van der Waals interactions. The dimethylcyanophenyl substituent is positioned in a sub-pocket lined with the aromatic side chains which include Y188, W229, and F227. The positioning of the cyano groups of the benzonitrile and dimethylcyanophenyl substitutions on either side of F227 results in the horseshoe shape observed for ETR within the NNIBP.
5.5. RT-RPV interaction
RPV occupies a similar position as ETR in the NNIBP118 and a horseshoe conformation is observed between the two cyano substitutions. Two hydrogen bonds are observed to bind to the main chain atoms of K101. The first is between the pyrimidine N and the backbone NH of K101 and the second between the aniline NH and the backbone carbonyl of K101. In the absence of a primary amino substitution on the pyrimidine scaffold, no interactions are observed with the E138 side chain within NNIBP. The benzonitrile substitution occupies a subpocket comprising of V106, P225, P236, F227, Y318, H235, P236 and L234. The (E)-3-(3,5- dimethylphenyl)acrylonitrile occupies the subpocket made up of W229, Y188, F227 and L234. The cyanovinyl group interacts closely with W229 and F227.
5.6. Resistance of ETR and RPV
ETR retains activity against several RT resistant variants with K103N, Y181C, Y188L, and G190A mutations.126 ETR is a highly potent and flexible molecule that can adapt to the steric interference caused by G190A and Y181C mutations (cf. Figure 13A).127 Also fewer interactions were observed with the residues Y181 and Y188 of NNIBP, allowing for greater tolerance of the Y181C and Y188L mutations compared to NVP and EFV.
Figure 13.

Schematic representation of (A) ETR bound to K103N mutant (PDB ID: 3MED) and (B) RPV bound to K103N/L100I double mutant (PDB ID: 2ZE2) in the NNIBP of RT.
Additionally, ETR retains activity against the K103N mutants owing to interactions with main chain residues 100–103.128 The mutated asparagine side chain is located below ETR and the horseshoe conformation maintains binding interaction within NNIBP in the K103N RT mutant. A decrease in susceptibility was observed for ETR in clinical trials for RT mutants with amino acid substitutions at L100I, Y181I/V, or G190S/E.129,126 Cross-resistance to NVP, DLV, and EFV is likely after virologic failure with an ETR containing regimen.
RPV has activity against resistance-associated mutations seen for earlier NNRTIs. Similar to ETR, RPV has retained activity against several RT resistant variants with K103N, Y181C, Y188L and G190A mutations126 and form interactions with Y181 and Y188 which allows greater tolerance towards Y181C and Y188L mutants in comparison to NVP and EFV.130
Owing to the flexibility of its structure, the conformation adopted by RPV in the L100I/K103N double mutant RT differs significantly from the binding mode in wild-type RT to accommodate for changes in the binding pocket caused by the K103N/L100I mutation. Similarly, for RPV bound to the K103N/Y181C double mutant RT, loss of the aromatic ring interaction caused by the Y181C mutation is compensated by interactions between the cyanovinyl group of RPV and the aromatic side chain of Y183.
A slight repositioning of the amino acid chains allows for complementarity between the NNIBP and RPV in wild-type and mutant forms of HIV-1 RT. However, K101E and K101P mutations have emerged that impair NNRTI binding and cause a significant reduction in efficacy up to 243-fold to RPV and >50-fold to EFV.
However, a distinct resistance profile with new amino acid substitutions have emerged.131 A comparison of the potency of EFV and RPV in a single infectivity assay is shown in Table 2.132
Table 2:
Potency values obtained for EFV and RPV using a single infectivity assay in wild-type and mutant RTs.132
| Reverse Transcriptase | EFV (EC50, nM) | RPV (EC50, nM) |
|---|---|---|
| wild-type | 15 | 13 |
| K103N | 806 | 13 |
| K101P | 870 | 1142 |
| Y181C | 41 | 51 |
A decreased susceptibility to RPV has been observed for L100I, K101E/P, Y181I/V, E138K/A/Q/G, V179I/L, Y188L, H221Y, F227C/L and M230L.25 The K101P mutations cause a significant reduction in efficacy for RPV compared to EFV (Table 2). The K101P mutation has been suggested to enhance misincorporation producing an RT enzyme that is more likely to generate new mutations in the viral genome.132 The K101E mutation also confers high resistance to RPV.133 The E138A/G/K/Q/R mutation in combination with the M184I mutation significantly decreases susceptibility to RPV and ETR.24, 134 The E138K substitution was suggested to unfavorably impact the dissociation/association equilibrium for the RPV in complex with RT thereby reducing RPV-mediated RT inhibition. A combination of E138K and M184I showed a 4-fold reduction in susceptibility to RPV compared with 2-fold for the single mutant E138K.135–136 Cross-resistance to EFV, ETR and NVP is expected to lead virological failure and development of RPV resistance.
As shown in Table 2 above, the Y181C mutation caused a slight decrease in efficacy for RPV and EFV in Y181C RT compared to wild-type RT. The Y181I and Y181V substitutions also conferred 15- and 12-fold decreased susceptibility to RPV.137
5.7. Clinical trials of ETR
ETR was approved for use by the FDA in 2008 and is the fourth NNRTI approved for the management of HIV-1 infection. The clinical efficacy results from the phase III Demonstrate Undetectable viral load in patients Experienced with ARV Therapy (DUET) 1 and 2 trials.138 The trials were identical and the pooled results established efficacy in treatment experienced HIV-1 infected patients.139 These patients had prior exposure to EFV, DLV, NVP and enfuvirtide (T-20). All patients had at least one NNRTI mutation and three or more protease inhibitor mutations. Patients were randomized to an ETR + background regimen or a placebo + background regimen arm. Median baseline CD4+ count was 99 cells/mm3 in the ETR arm and 109 in the placebo arm. Median baseline plasma viral load was 4.8 log10 copies/mL. Virologic response at 48 weeks, defined as viral load <50 copies/mL, was achieved by 60% of patients in the ETR + background regimen and 38% in the placebo arm.
ETR is unique among the approved NNRTIs in that it is indicated specifically for use in treatment-experienced patients. It has not been studied in treatment-naive patients with a phase III clinical program. Its resistance profile represents an increased genetic barrier to resistance and virologic efficacy against many common NNRTIs such as Y181C and G190S.140 The recommended daily dose is 200 mg twice daily per day and it is available in 25 mg, 100 mg and 200 mg tablets. It must be taken with a meal in order to increase systemic exposure. DHHS guidelines do not recommend ETR as part of an initial regimen for treatment-naïve patients, citing a lack of data in this population.138
5.8. Clinical trials of RPV
The FDA approved RPV in 2011, making it the fifth NNRTI approved for use in HIV-1 disease management. It has been approved for use in treatment-naive adults on the basis of the ECHO and THRIVE phase 3 registration trials.141 In ECHO, the arms consisted of RPV + TDF)/FTC and EFV + TDF/FTC. In THRIVE, the arms consisted of RPV vs EFV each with investigator-selected NRTI combinations of either TDF/FTC, AZT + 3TC or abacavir (ABC) + 3TC. Randomization of patients was baseline viral load-dependent, with 48–54% of patients harboring viral loads <100,000 copies/mL.
Pooled data from both trials reports 76% of patients in the RPV arm suppressed viral load to <50 copies/mL at week 96 vs. 77% of patients in the EFV arm.142 RPV is available in a number of different combinations; in addition to a standalone 25 mg tablet, it has been coformulated with TDF/FTC into one tablet that must be taken once a day. It has also been coformulated with tenofovir alfenamide fumarate (TAF)/FTC into a one pill once a day regimen. RPV is one half of the first two-drug combination approved for use as maintenance therapy in HIV-1 infected patients; it is coformulated with the integrase inhibitor dolutegravir (DTG). This is likewise one pill once a day regimen. The recommended daily dose of RPV in 25 mg and must be taken with a meal. Systemic exposure was found to be 40% lower under fasting conditions. RPV is recommended as an alternative regimen for treatment-naïve patients in the U.S. Department of Health and Human Services (DHHS) guidelines based on its virologic efficacy and lower rates of CNS adverse events, lipid effects, and rash compared to EFV. However, RPV is not recommended in patients with HIV-1 RNA viral loads of > 100,000 copies/mL or a CD4+ count < 200 cells/mm3 based on the lower rates of virologic suppression in patients with these characteristics during the phase III clinical trial program.
5.9. Commercial synthesis of ETR
Synthesis of ETR was first reported by De Corte et al.113, 143 in which 4-cyanophenyl guanidine (58) and diethyl malonate were reacted together in the presence of sodium ethoxide to obtain pyrimidinediol (59) in 76% yield and followed by halogenation with POCl3 at 80–90 °C to form 60 (86% yield). The bromo group in 60 was introduced by the reaction with Br2 to give 61 (78% yield).
Compound 61 reacted with 62 in the solvent system NMP, 1,4-dioxane to obtain 63 (45% yield) (Scheme 10) followed by reaction with NH3 in isopropanol to give ETR ( 64) in 41% yield.
Scheme 10:

Initial synthetic procedure for ETR proposed by De Corte et al.113, 143
Considering the lower yield in the last two steps and longer reaction time (4 days) in the last step, further advancements were made by Joshi et al. (Scheme 11).144 It was speculated that poor yield (41%) in amination step in the preparation of 64 was due to the bromide group adjacent to the reaction site; therefore, attempts were made to do bromination after the amination step.97 Compound 65 reacts with 66 in the presence of DIEA in 1,4-dioxane at 70 °C to form 67 with an 80% yield and then, 67 reacts with 68 in the presence of NaH/t-BuOK to give 69a as the main product and 69b as a side product. However, the formation of 69b was lowered by cooling down the reaction to −15 °C. Compound 69 on reaction with aq. NH3 at 120–125 °C in 8–12 h gave 70 with 2–3% of side product formed because of 69b in the reaction mixture. Side products were removed by crystallization using dioxane to give 70 in 82% yield which on bromination by liquid Br2 in CH2Cl2 at 0–5 °C gave ETR (64) in 85% yield.
Scheme 11:

Modified synthesis of ETR (64) by Joshi et al.144
To improve overall yield and reaction time, subsequent advancements were made by Fier et al. by using their recently developed chemistry on SNAr reaction by AgF2 (Scheme 12).145 The initial building block 72 was made by SNAr reaction between 71 and 66 using K2CO3 in DMF to obtain 72 in 81% yield. Compound 72 on reaction with 68 at 165 °C and t-butyloxycarbamate protection resulted in 73 in 88% yield. Fluorination of 73 with AgF2 followed by reaction with NH3/i-PrOH at 80 °C and t-butyloxycarbamate deprotection gave ETR (64) in 56% yield.
Scheme 12:

Modified synthesis scheme of ETR (64) by Fier et al.145
5.10. Commercial synthesis of RPV
RPV was invented by Janssen Pharmaceuticals and Tibotec.117, 146–148 The two key building blocks 77 and 83 (Scheme 13) were the main requirements for the synthesis of RPV.117, 146–149 Compound 77 was prepared from thiouracil (74), which was commercially available in bulk scale. Thiouracil (74) was alkylated by MeI/NaOH at 60 °C to form 75 in 90% yield followed by reaction with 68 at 150 °C resulted 76 in 70% yield. The halogenation of 76 was done with POCl3 to obtain 77 in 80% yield.117, 146–149 Compound 83, another key intermediate, was prepared in four steps from 3,5-dimethyl-4-amino-benzaldehyde (78). The amino group in 78 was protected as dimethylformamidine using N,N-dimethylformamide dimethyl acetal (79) to form 80 which was subsequently reacted with n-BuLi to give 81. Compound 81 on Wadsworth–Emmons reaction with (diethoxyphosphinyl)acetonitrile followed by dimethylformamide protection by ZnCl2 yielded 83. The compunds 76 and 83 were reacted together at 150 °C to obtain RPV (84).
Scheme 13:

Synthetic procedure developed for RPV (84) by Janssen Pharmaceuticals
In another route for RPV, ethyl 4-amino-3,5- dimethylbenzoate (85) was reacted with 76 at 150 °C to form 86, which on reduction with LiAlH4 followed by oxidation with MnO2 resulted in the aldehyde derivative 87. Likewise, in the preparation of 81, Wadsworth–Emmons reaction with (diethoxyphosphinyl)acetonitrile through a rather complex system 87 also yielded RPV (84) (Scheme 14).
Scheme 14:

Modified synthetic process of RPV (84)
Later it was reported that 88 and 77 can also react without using any precursor for aldehyde moiety or any additional protection on aldehyde group to obtain 89 which was followed by Wadsworth–Emmons reaction with (diethoxyphosphinyl)acetonitrile to provide RPV (84) (Scheme 15).
Scheme 15:

Alternative synthetic procedure developed for RPV (84)
Recently various advancements were made to make RPV (84) by the introduction of an acrylonitrile moiety through Heck reaction (Scheme 16). Compounds 90 and 77 were reacted at 150 °C to form 91 which was reacted with acrylonitrile using Pd(OAc)2/P(o-tol)3 to obtain RPV (84) (E:Z 96:4).
Scheme 16:

Synthesis of RPV (84) by Heck reaction 148
Advancement was made for the production of RPV at bulk scale using Heck reaction. The iodo analogue (93) was used instead of the bromo analogue in the Heck reaction and use of expensive ligands in Heck reaction was avoided by the use of Pd/C 0.5%, although this method provided a mixture of E: Z isomers (83:83a 80:20); they could be converted to major E isomer(E:Z 98:2) by treatment with 6N HCl to give 83 in 70% yield (Scheme 17).148
Scheme 17:

Synthesis of block 83 by Heck reaction
5.11. RPV-based long acting antiviral agents
Recently, ViiV Healthcare developed two new long-acting injectable nanosuspensions of antiretrovirals, cabotegravir (INSTIs) and RPV (NNRTI) for the treatment of chronic HIV infection. The crystalline nanoparticle formulations have two different drugs with different inhibition mechanisms. The LATTE-2 (Long Acting Antiretroviral Treatment Enabling) study was conducted to evaluate the long-acting HIV-1 viral suppression and comparison of the antiviral activity, tolerability, and safety of the two intramuscular dosing regimens of cabotegravir and RPV compared to oral cabotegravir plus abacavir–lamivudine through 96 weeks.150–151 This combination regimen was found to be well-tolerated, with no adverse reactions and few withdrawals due to adverse effects were observed. Moreover, this combination is administered once every 4 or 8 weeks and it maintains viral suppression in about 90% of people who started therapy with an undetectable viral load.152
6. OTHER NNRTIs IN CLINICAL DEVELOPMENT
6.1. Elsulfavirine
Elsulfavirine, initially developed by Viriom, is a prodrug of VM1500A (cf. Figure 14) that has been approved (June 2017) in Russia for use in treatment-naive HIV-1 infected patients. The drug exhibited in vitro antiviral potency with an EC50 value of 1.2 nM, and serum-adjusted EC50value of 13.8 nM against the wt HIV RT and a broad range of mutant clinical HIV isolates as well as no cross-resistance with other NNRTIs. The orally formulated prodrug Elsulfavirine is transformed into VM1500A, is rapidly taken up into the red blood cells (RBCs), and gets accumulated reversibly through binding to RBC carbonic anhydrase. Hence, RBCs act as a reservoir that slowly releases the drug back to plasma, and from plasma to the peripheral blood mononuclear cell, the target cells. This pre-clinical phenomenon may provide more advantages than long-acting oral and parenteral formulation development as a result of infrequent dosing, improving patients’ compliance and likely improving the long-term treatment and prevention of HIV/AIDS.153–154
Figure 14.

2D chemical structures of NNRTIs in clinical development.
The approval of elsulfavirine came after a phase IIb, double-blind, randomized study comparing elsulfavirine vs EFV with TDF/FTC. 120 patients were enrolled. Baseline CD4+ counts were similar between the 2 arms; elsulfavirine - 349 cells/mm3 and EFV – 379 cells/mm3. Baseline HIV viral load was 4.7 log10 copies/mL. At 48 weeks, a modified intent to treat analysis showed 81% of patients in the elsulfavirine arm had suppressed to <50 copies/mL compared to 73.7% of patients in the EFV arm. Elsulfavirine has not been approved for use outside Russia.155
The recommended dose of elsulfavirine is 20 mg once daily in addition to an NRTI backbone (TDF/FTC). Patients administered elsulfavirine reported fewer CNS and skin adverse events when compared to the EFV group. The long half-life of VM1500A has prompted a preclinical study of long-acting injectable formulations as well as an oral, once-weekly formulation.155
6.2. Doravirine
Merck has developed a next-generation NNRTI, doravirine, which commenced by intergrating a tetrazolethioacetanilides scaffold (94) (Figure 15) and related acetanilide para-sulfonamide series from GW-8248 (95) to form a novel class of diaryl ether NNRTIs (96). In this new class of diarylether derivatives the benzophenone moiety in 95 has been replaced by a 1,3-phenoxy fragment.156–158 The diaryl class of the compound showed only moderate inhibition of wt HIV RT, and clinically relevant mutants K103N and Y181C. However, the SAR of this class is interesting and showed that remarkable enhancement in potency occurred with low nanomolar inhibition against the wild type HIV RT (IC50 = 0.1 nM), K103N (IC50 = 0.2 nM) and Y181C (IC50 = 0.3 nM) enzymes by the insertion of a chlorine atom at the 3-position of ring B followed by mono- and di-substitution of ring A that resulted in highly potent inhibitors (97).159
Figure 15.

Schematic representation doravirine design and lead optimization process.
The diaryl ether phenoxyacetanilides undergoes rapid in vivo clearance due to metabolic hydrolytic reactions carried out at the anilide group of the molecule. Hence, a wide-range of heterocyclic amide isosteres were substituted in the acetanilide moiety and the resulting indazole derivative (98) showed an excellent enzymatic potency towards wt HIV RT (IC50 = 1.1 nM), clinically relevant mutants (K103N, IC50 1.1 nM and Y181C IC50 2.6 nM), and improved in vivo metabolic stability. Further replacement of indazole by 6-amino-7-azaindazole found an optimal pre-clinical candidate (99) and was labeled MK-4965, which exhibited improved bioavailability with in vivo clearance in rats and beagle dogs and low potential for drug-drug interactions.160
Further developments were initiated to increase the human pharmacokinetic properties by Roche in which the side chains were modified by the elimination of oxygen atom. Exploration of heteroaryl replacements resulted in forming a 2-pyridone derivative 100 and found promising potency values against both wt HIV RT (IC50 2.5nM) and mutants K103N (IC50 3.7 nM) and Y181C (IC50 3.1 nM).161–162 Optimization with small lipophilic substituents like halogens, alkyl groups, fluorinated alkyl groups and thioethers were performed at the 4-position of the 2-pyridone core. The 4-trifluoromethyl analog 101, showed improved plasma stability and excellent wtHIV-1 RT and double mutant (K103N/Y183C) enzyme inhibition. Due to highly ordered intermolecular H-bonding in the pyrazolopyridine side chain it has poor solubility and oral absorption (15% and 9% of bioavailability rats and dogs, respectively).163 The isosteric replacement of pyrazolopyridine with methyltriazolinone moiety 102 proved to have a significantly better pharmacokinetic profile along with high potency against the clinically relevant single and double mutants when compared to standard NNRTIs efavirenz and rilpivirine (cf. Figure 15). Later compound 102 was selected for pre-clinical development, and assigned as MK-1439 followed by USAN name doravirine.164
Doravirine (DOR) has recently been approved by the FDA (July 2018) for the treatment of ART-naive HIV-1 infected adults (cf. Figure 14). 100 mg of doravirine, in combination with TDF/FTC, has been evaluated against ritonavir-boosted darunavir (DRV/r) and EFV in the phase III trials DRIVE-FORWARD and DRIVE-AHEAD, respectively. DRIVE-FORWARD study compared DOR to once-daily ritonavir-boosted darunavir (DRV+r), each administered in combination with TDF/FTC or abacavir (ABC)/3TC, in treatment-naïve adults. DRIVE-AHEAD study compared DOR/3TC/TDF to EFV/FTC/TDF in treatment-naive adults.
In DRIVE-FORWARD, 769 HIV-1 infected patients were enrolled and randomized to either the DOR or DRV/r arm. Doravirine was non-inferior to DRV/r with 84% of participants in the DOR arm vs. 80% in the DRV/r arm with HIV-1 RNA <50 copies/mL at week 48.165Both regimens were well-tolerated and safe. In DRIVE-AHEAD; doravirine was non-inferior to EFV with 84% of participants in the DOR arm vs. 81% in the EFV arm with HIV-1 RNA <50 copies/mL at week 48. Fixed dose combination of doravirine/lamivudine/TDF is non-inferior to EFV/emtricitabine/TDF in treatment-naive adults with HIV-1 infection: week 48 results of Phase 3 DRIVE-AHEAD study.166 There were fewer discontinuations due to reasons other than virologic failure in the DOR arm with lower rates of neuropsychiatric AEs. Based on these phase 3 trials, doravirine has been shown to be non-inferior to both EFV and boosted darunavir. Doravirine is currently being evaluated in treatment-switch studies.
7. CONCLUDING REMARKS
7.1. Structural features of NNRTIs for binding within NNIBP
Key functional groups and molecular conformations that are found among NNRTIs include a core scaffold that binds within the NNIBP and orients its pendant substitutions for interactions with the NNIBP.85, 167–168 Varied scaffolds have been tolerated including a diazepin-6-one in NVP, a piperazine in DLV, a benzoxazine in EFV, and a pyrimidine in ETR and RPV. The choice of scaffold has been suggested to control the flexibility of the overall NNRTI and its ability to reposition itself in wild-type and mutant HIV-1 RT. The core scaffold is substituted with planar, aromatic or heterocyclic rings that allow for hydrophobic/π-π stacking interactions with the side chains of residues Y181, Y188, W229, F227, V106, P236, L100, L234, and Y318. Substitutions on the aromatic rings include halogens, nitrile and alkyl groups that influence the potency of HIV-1 RT inhibition. The core scaffold is further substituted with moieties that are able to form hydrogen bonds with the backbone of K101, K103, and P236. Linker fragments between the pendant aromatic and heteroaromatic rings and the core scaffold may be present to allow for flexibility and the ability to adopt multiple conformations within the NNIBP. The NNRTIs are largely hydrophobic and include polar substitutions to improve pharmacokinetic profiles and provide an optimal balance of hydrophilicity and hydrophobicity for an oral route of administration. The currently available NNRTI drugs fall within the drug-like chemical space. The calculated physicochemical properties of the approved NNRTI drugs are provided in Table 3.
Table 3:
The calculated physicochemical properties of approved NNRTI drugs.
| Generic | Properties |
|||||
|---|---|---|---|---|---|---|
| MW | PSA | cLogP | HBA | HBD | NrotB | |
| Nevirapine | 266.30 | 58.12 | 2.05 | 3 | 1 | 1 |
| Delavirdine | 456.56 | 118.81 | −0.06 | 4 | 3 | 6 |
| Efavirenz | 315.67 | 38.33 | 3.68 | 2 | 1 | 2 |
| Etravirine | 435.28 | 120.64 | 6.11 | 5 | 2 | 5 |
| Rilprivine | 366.42 | 97.42 | 5.64 | 4 | 2 | 6 |
| Doravirine | 425.74 | 104.42 | 3.47 | 6 | 1 | 5 |
The calculated physicochemical properties of the approved drugs are provided. The values are calculated from Canvas tool (Canvas, Schrödinger, LLC, New York, NY, 2016).
Although approved NNRTIs share several common structural features as outlined above, they also display important differences.123–125 The NNRTIs differ in their size and flexibility (cf. Table 3), and also in the conformation they adopt within NNIBP and preference for binding to small or large NNIBP. These differences translate to varied potency and resistance profiles.
7.2. Non-nucleoside RT inhibitors (NNRTIs)
The NNRTIs are classified into three generations depending on their discovery and resistance profile. A greater barrier to the development of resistance is observed for the third-generation NNRTIs compared to earlier generations, although the efficacy of all approved NNRTIs can be compromised by 1–2 mutations.20–21 First-generation NNRTIs relied on π-stacking interactions to Y181 and Y188. The replacement of the tyrosine residue by a non-aromatic cysteine residue prevented this interaction and resistance developed to these molecules.85, 124, 167–168 New NNRTIs reduced interactions with Y181 and Y188, and expanded to incorporate interactions with W229 which is less prone to mutation. The newer NNRTIs also incorporated a central hydrogen bond to the backbone carbonyl of K101.
Resistance to first-generation NNRTIs has also developed rapidly due to the rigidity of their structures that prevent them from adapting to mutations in the NNIBP. The newer generation is more flexible and may be accommodated within a mutated NNIBP based on drug molecule movements within the NNIBP and slight side-chain rearrangements. Thus, newer generation NNRTIs are less prone to the development of resistance.
The first-generation NNRTI include NVP and DLV that possess rigid structures and adopt a “butterfly-like” conformation in NNIBP.68, 169 EFV is a second-generation NNRTI and is more flexible compared to first-generation NNRTIs with a slightly improved resistance profile.44 Third-generation NNRTIs include the diarylpyrimidine derivatives, ETR and RPV that are more flexible, and adopt a horseshoe conformation within NNIBP.118 The horseshoe conformation allows them to reposition themselves within NNIBP despite small changes within the NNIPB due to point mutations. The third-generation NNRTIs, ETR, RPV and DOR are effective in wild-type and K103N HIV-1 RT. Repositioning within the NNIBP allows for the accommodation of third-generation NNRTIs within mutation-modified NNIBP.85, 124, 167–168
The NNRTIs are well absorbed orally and are available as tablets and as suspensions for oral use as immediate-release or extended-release formulations.170 Since resistance develops rapidly to monotherapy, they are typically used in combination with other antiretroviral agents, for instance, cART.8 Additionally, EFV, RPV, and DOR are available in single tablet combination regimens with two NRTIs, TDF/FTC, as Atripla, Complera, and Delstrigo, respectively.171–172 Single tablet combination regimens provide the advantages of cART such as lowered dose due to additive or synergistic inhibition among the individual drugs, and a greater barrier to the development of resistance.39 Additionally, single tablet combination regimens improve patient adherence and reduce pill burden.173
7.3. NNRTIs as preexposure prophylaxis (PrEP) agent
NNRTIs can be used to prevent HIV infection in a number of situations. Single-dose NVP can prevent mother-to-child transmission when administered to both mother and baby.174 This approach has been adopted in low-income countries as a cost-efficient and simple way to reduce the incidence of mother-to-child-transmission.175
DPV has been studied as pre-exposure prophylaxis (PREP) specifically for women as part of an intravaginal ring. This DPV gel-containing ring was studied in 2 phase III, randomized, placebo, double-blind trials, MTM-020/ASPIRE and RING.176–177 These studies evaluated the efficacy of monthly intravaginal rings in African women. Both studies demonstrated marked reductions in HIV incidence, ranging from 27–37%. Interestingly, the studies differed over the effect of participant age on rate of protection. Baeten et al. showed that women older than 21 years of age had a higher protection rate compared to those 21 and younger (56% vs. −21%).176 Nel et al. reported no significant differences between age groups (hazard ratio 0.63 over 21 vs 0.85 21 and younger).177 These studies are ongoing with open-label extensions; full results are expected in 2019.178
RPV, in a long-acting (LA) formulation, has been studied as a PrEP agent. This formulation is a nanosuspension of RPV-containing nanoparticles that is administered as an intramuscular (IM) injection.179 The phase I study MWRI-01 shoed that the majority of study participants reported injection site reactions, though no severe (grade 3 or 4) adverse events were attributed to RPV.180
HPTN 076, a phase II, double-blind, randomized clinical trial enrolled 136 sexually active HIV-negative women to assess the safety of the RPV LA IM injection vs. placebo.181–182 The participants were started on QD oral pills for 4 weeks to determine tolerability before moving to the IM phase. There was no difference in safety outcomes between placebo and RPV arms, with 79% and 84% of participants reporting side effects, respectively. Weight loss was the most commonly reported side effect. All but 2% of participants who received all 6 scheduled injections had RPV concentrations above protein-adjusted IC90 concentrations after week 4, indicating optimal adherence led to consistent therapeutic levels of drug over the course of the study.
The NNRTI class has a number of potential candidates for PREP in various stages of development. Though the HIV armamentarium is vast there is currently only 1 approved regimen for PrEP; tenofovir disproxil fumarate/emtricitabine as an oral pill, once-daily. The NNRTI candidates represent new PrEP modalities that warrant further research in an effort to increase preventative options for individuals at high risk of contracting HIV.
8. MEDICINAL CHEMISTRY PERSPECTIVE
a). Conformational flexibility
From several biochemical studies, it was revealed that various domains in the RT structure are flexible including NNRTI and RNase H, but the flexibility of RT is reduced when a ligand binds at the pocket. This is primarily due to the fact that inhibitors restrict the movement of the “thumb” region.183 Flexibility is a key for not only stronger binding but also sufficiently flexible for confronting the mutations in the NNIPB. There is an excellent correlation between the number of rotatable bonds (nRotB) with binding affinity and ability to retain activity against possible mutations among these drugs (excluding DLV).2 It is evident that compounds having more freedom of rotation in the binding site are able to show better activity versus RT mutants, for instance, NVP, a fairly rigid molecule (nRotB = 1), has shown low activity against V106A, Y188C, Y1991I, Y188V, L100I, K101P, Y181I and Y181C mutations (bold letter refers residues that show mutations by all NNRTI drugs) (cf. Figure 3, Table 3).
On the other hand, RPV (nRotB = 5) showed significantly improved activity profile to various mutants; due to the rotatable dihedral angle around the cyanovinyl group and NH-linked aniline (central ring). Particularly, the cyanovinyl group showed a favorable binding to the hydrophobic tunnel which is formed by Y188, F227, W229, L234 residues and nitrogen atoms of diarylpyrimidine ring form a hydrogen bond with both wild-type and mutants e.g K101P and K103N (cf. Figure 16).
Figure 16.

Overlay of X-ray crystal conformations and binding mode of NVP (A) and RPV (B) is shown. Important residues are highlighted.
b). Physicochemical properties
The NNRTI drugs are mostly lipophilic and predominantly unionized at physiologic pH.170 They are highly plasma protein bound with > 95% plasma protein binding for DLV, EFV, ETR and RPV (cf. Table 1).39, 97, 129, 137, 184NVP is approximately 60% plasma protein bound and has a long half-life. The NNRTIs primarily undergo metabolism via cytochrome P450 oxidative metabolism, primarily via CYP3A4 and CYP2B6. Most NNRTIs are also inhibitors of CYP2C9 and inducers of CYP3A4. The metabolic profile, the half-life of NNRTI and adverse effect profile are important considerations when considering NNRTIs as part of antiretroviral regimens.
c). Fluorine-a privileged atom in NNRTI design
Fluorine is a key atom in drug designing strategies due to its impact in improving metabolic stability modulating physicochemical properties, and binding affinity towards targets.185–187 Fluorine also highly influences antiviral activity, evidenced in the design of DAPY analogs. The effect of the presence of fluorine substitution has been well explained in the DAPY series, e.g. non-fluorinated (Figure 16A, EC50=5.5 nM) showed five times less activity as compared to fluorinated compounds (EC50 = 1.0 nM) in both wild-type and mutants.188–189
In general, several fluorinated NNRTIs compounds have been reported, and this includes the approved drug EFV. A representative fluorinated-NNRTI binding interaction is shown in Figure 10.In many of X-ray crystal structures of NNRTI-bound to RT complex, fluorinated compounds did not directly interact with residues but mainly modulate the conformational preference of the whole molecule due to high electronegativity (Figure 18). Moreover, the fluorine atom showed non-bonded hydrophobic interactions particularly with V106, V179, L234, and W229 residues. It was also observed that because of its strong electron-withdrawing nature, introduction of the fluorine atom in the aromatic ring significantly increases the binding affinity.
Figure 10.

A) The binding mode of EFV in the binding pocket of HIV-1 RT (PDB ID: 1JKH) with important amino acids in the pocket and B) the 2D ligand-interaction diagram of EFV is shown.
Figure 18.

The X-ray crystal structures of HIV-1 RT in complex with fluorine-containing NNRTIs doravirine (PDB ID: 4NCG, IC50 0.011 μM, colored green); and KRV-2110 (PDB ID: 3LAK, IC50 0.0034 μM, colored pink)
In addition to fluorine, the other halogens including bromine in ETR contribute to the electrostatic-type interactions. This was also proven with another halogen, i.e the chlorine atom in indolylarylsulfone derivatives influences the antiviral activity (Figure 16B).190 Thus, in modern drug design introduction of halogens has to be taken into account.
d). Clinical prospectives
NNRTI development and clinical evaluation has continued in a number of areas including, but not limited to: prevention, as part of cART, and as part of dual-therapy regimens.191–192 DPV has been evaluated for use in HIV prevention in phase III clinical trials in Africa.176–177 These trials evaluated DPV as part of a monthly vaginal ring that provided controlled release of the compound at the site of viral transmission. Adherence to oral therapy and vaginal gels was low in this population, necessitating an alternative prevention methodology that did not rely on daily oral adherence or dosing around coital situations.193–194 DPV reduced HIV incidence by 27% & 31% compared to placebo groups in 2 separate trials.176–177 The efficacy showed in these trials warrants further studies in alternative prevention strategies.
The use of NNRTIs in conjunction with 2 NRTIs as cART has and is still being studied clinically. Current clinical trials evaluating cART regimens that include DOR or elsulfavirine have shown efficacy in suppressing HIV below the limit of detection in treatment-naïve patients. Clinical trials examining the efficacy and safety of 2-drug regimens have also been performed with the aim of reducing overall patient exposure to NRTIs. The combination of the integrase inhibitor DTG and RPV represents the first FDA-approved 2-drug treatment for the maintenance of HIV suppression.195 This regimen was shown to be non-inferior to a 3-drug regimen in maintaining suppression of viral load at 48 weeks. All patients in the study had undetectable viral loads for 6 months prior to enrolment in the study. At 48 weeks, both groups exhibited a viral suppression rate of 95% in the intent-to-treat analysis.195 The results from these studies demonstrate the value of continuing research into the NNRTI class, with potential benefits to a number of treatment modalities.
9. Lessons Learned from NNRTI drugs
Two classical approaches for the lead optimization i.e. molecular hybridization and bioisosteric replacement are the key for NNRTI drug development.
N-alkyl substituents create the sterically hindered region, which reduces the rate of N-dealkylation metabolic conversion (cf. NVPlead optimization).
When the molecule is rigid and focuses more on positional adaptability, the molecule cannot adapt to point mutations within the NNIBP and resistance develops rapidly to the molecule.
Introduction of a flexible linker increases the solubility but is also prone to compound promiscuity (cf. DLV lead optimization).
Substitution with a CF3 group reduces the pKa value of the drug and improves the potency against HIV-1 inhibitory activity (cf. lead optimization of EFV).
The presence of triple bond in a molecule was expected to be a potential risk for reactive metabolism and found inactive against HIV-1 (cf. lead optimization of EFV).
Introduction of a cyanovinyl group makes the compound highly active against both wildtype and mutated HIV-1 RTs(cf. lead optimization of ETR and RPV).
Introduction of fluorine or halogen atoms in the aromatic ring significantly increases the binding affinity (cf. lead optimization of ETR and EFV).
Most of NNRTI drugs were synthesized by efficient synthetic procedures, which provided an excellent opportunity to explore as many derivatives as needed in developing the lead molecule for improved inhibitory activity.
HIV-1 can develop resistance mutations to all currently available NNRTIs. Further development of newer compounds with unique and better resistance profiles remains a priority.
Future design perspective
As discussed above there are various factors to be considered for next generation NNRTI development.2, 196–197 From a medicinal chemistry point of view these can be summarized as (a) improving conformational flexibility of the compound by maintaining its positional adaptability,2, 72, 198–199 (b) forming extensive hydrogen bond interactions with the main chain of the protein to avoid resistance towards mutations,200–201 (c) targeting the highly conserved residues such as W229 for stronger interactions or with the adjacent binding site,202–204 (d) involving additional interaction with other atoms, including halogen or nitrile groups,187 (e)improving physiochemical properties by introducing solubilizing groups into entrance of the NNIBP or utilizing prodrug approaches.205 For development of novel NNRTIs, new technologies with orthogonal HTS methods will provide a strong basis for rapid screening of large chemical libraries.206 Additionally, computational approaches including QSAR,167 virtual screening207, prediction of pharmacologically important properties, and the estimation of binding affinities208 will certainly contribute to the rapid development of future anti-viral drugs against HIV infection.
Supplementary Material
Figure 12.

The 2D protein-ligand interaction diagram for (A) ETR (PDB ID: 3MEC) and (B) RPV (PDB ID: 2ZD1) and the important amino acids in the NNIBP of RT.
Figure 17.

(A). Inhibition of DAPY series in wild-type and (B) Inhibition of indolylarylsulfone derivatives influenced by the introduction of halogen atom.
ACKNOWLEGMENT
V.M acknowledges SERB-DST (Science & Engineering Research Board, Government of India) for a Young Scientist Fellowship (YSS/2015/001938). S.N.B thanks NIH R01-AI129745 and MH062261 for financial support. Z.P acknowledges Young Scholars Program of Shandong University (YSPSDU No. 2016WLJH32), the Fundamental Research Funds of Shandong University (No. 2017JC006), L.X thanks Key Project of NSFC for International Cooperation (No. 81420108027) and the Key research and development project of Shandong Province (No. 2017CXGC1401). The authors like to thank European Molecular Biology laboratory (EMBL), for providing the evaluation license of LigPlot+. We are indebted to Dr. Vasanthanathan Poongavanam for his valuable discussion during the design of this manuscript, and preparation of figures, also thankful to Dr. Mohit Tyagi, Uppsala University, Sweden for his rigorous proof read of synthetic schemes for the manuscript.
ABBREVIATIONS USED
- 2D
two-dimensional
- AIDS
acquired immune deficiency syndrome
- HIV
human immuno deficiency virus
- RT
reverse transcriptase
- IN
integrase
- PR
protease
- NNIBP
non-nucleoside inhibitor-binding pocket
- PI
protease inhibitor
- INSTI
integrase inhibitor
- NNRTIs
non-nucleoside reverse transcriptase inhibitors
- NRTIs
nucleoside reverse transcriptase inhibitors
- FDA
food and drug administration
- AZT
zidovudine
- NVP
nevirapine
- DLV
delavirdine
- EFV
efavirenz
- ETR
etravirine
- RPV
rilpivirine
- DPV
dapivirine
- UNAIDS
the joint united nations programme on HIV/AIDS
- cART
combination antiretroviral therapy
- gp
glycoproteins
- QSAR
quantitative structure-activity relationship
- AI
artificial intelligence
- NN
neural network
- q.d.
once a day
- b.i.d.
two times a day
- TDF
tenofovirdisoproxilfumarate
- FTC
emtricitabine
- TAF
tenofoviralafenamidefumarate
- 3TC
lamivudine
- BT
background therapy
- ddI
didanosine
- NaHMDS
sodium bis(trimethylsilyl)amide
- HTS
high-throughput screening
Biography
Vigneshwaran Namasivayam is a senior research scientist at Pharmaceutical Institute, University of Bonn, Germany (Since 2010) and involved in the field of cheminformatics, computational chemistry, data analysis and molecular modeling. He gained his doctoral degree under the supervision of Prof. Dr. Hans-Jörg Hofmann from Leipzig University, Germany (2009). He carried out his postdoctoral research at the Technical University of Munich, Germany (2010). Prior to his doctoral studies in Germany, he worked as a Research Executive (2004–2006) at Orchid Chemical and Pharmaceutical Limited, Chennai, India.
Murugesan Vanangamudi received his Ph.D (2013) from CSIR-Central Drug Research Institute (CDRI), Lucknow. Since 2013, he has been working as Associate Professor at the Department of Medicinal and Pharmaceutical Chemistry, Sree Vidyanikethan College of Pharmacy, Tirupathi, India. His research work included rational drug design and synthesis of novel small molecules against HIV-1 reverse transcriptase.
Victor G. Kramer obtained an M.S. degree in Applied Immunology & Molecular Biology under the mentorship of Drs. Ah-kau Ng and Trebor Lawton at the University of Southern Maine. He then spent 4 years in the SHIV laboratory of Dr. Ruth Ruprecht at Dana-Farber Cancer Institute in Boston, MA. He obtained his Ph.D in Experimental Medicine under the mentorship of Dr. Mark Wainberg at the McGill University AIDS Center and Lady Davis Institute in Montreal, Quebec, Canada. In 2014, he joined Abbvie and worked for their HIV franchise. He now works for Merck Canada Inc. in the Medical Affairs department and focuses on HIV and hepatitis C therapeutics.
Sonali Kurup secured a PhD in medicinal chemistry from Duquesne University in 2009 and completed her postdoctoral training at the University of Michigan in 2011. Following her training, Dr. Kurup worked at the College of Pharmacy at Roosevelt University as Assistant Professor of Medicinal Chemistry (2011–2017) and Associate Professor of Medicinal Chemistry (2017–2018). She has recently joined the College of Pharmacy at Ferris State University as Associate Professor of Medicinal Chemistry. Her research focuses on developing novel molecules against resistant pathways for non-small cell lung cancer.
Peng Zhan obtained his B.S. degree from Shandong University, China, in 2005. Then he earned his M.S. and Ph.D degrees in Medicinal Chemistry from Shandong University in 2008 and 2010, respectively. He subsequently joined the research group of Professor Xinyong Liu as a Lecturer (2010–2012). From 2012 to 2014, he worked as a Postdoctoral Fellow funded by JSPS (Japan Society for the Promotion of Science) in the Graduate School of Medical Science, Kyoto Prefectural University of Medicine, Japan. He is now working as an Associate Professor in the laboratory of Professor Xinyong Liu. His research interests involve the discovery of novel antiviral, anticancer, and neurodegenerative disease-related agents based on rational drug design and combinatorial chemistry approaches.
Xinyong Liu received his B.S. and M.S. degrees from School of Pharmaceutical Sciences, Shandong University, China, in 1984 and in 1991, respectively. From 1997 to 1999 he worked at Instituto de Quimica Medica (CSIC) in Spain as a Senior Visiting Scholar. He obtained his Ph.D. from Shandong University in 2004. He is currently a Full Professor of the Institute of Medicinal Chemistry, Shandong University. His research interests include rational drug design and synthesis and antiviral activity evaluation of a variety of molecules that interact with specific enzymes and receptors in the viral life cycle (HIV, HBV, HCV, and FluV). Other ongoing programs include molecular modification and structure-activity relationships study of natural products used to treat cardiovascular diseases, and drug delivery research using PEGylated small-molecular agents.
Jacob Kongsted completed his Ph.D. in Theoretical and Computational Chemistry at the University of Copenhagen in 2005, followed by post-doctoral training at Lunds University (Sweden) and University of Aarhus (Denmark). In 2009 he moved to University of Southern Denmark as an Assistant Professor and from 2015 he is Full Professor of computational quantum chemistry at the University of Southern Denmark. Kongsted has a broad research interest in theoretical and computational chemistry and has especially contributed to this field by development of novel quantum chemistry embedding methods as well as application of these computational models to systems of biophysical and biochemical interest. Recent research use quantum chemistry methods for rational design of light-induced biological functional materials and advanced drug design.
Siddappa N. Byrareddy is an Associate Professor, in the Department of Pharmacology and Experimental Neurosciences, University of Nebraska Medical Center, Omaha. He obtained a Ph.D. from the National Institute of Mental Health and Neurosciences, Bangalore, India and completed postdoctoral training at the Dana-Farber Cancer Institute/Harvard Medical School, Boston, USA. Dr. Byrareddy has a strong background in molecular retrovirology, lentiviral evolution, drugs of abuse and mucosal virus transmission studies with 12 years of research experience, with over 80 peer-reviewed publications in the field. His laboratory research focuses on the development of effective AIDS vaccine/HIV Cure strategies and therapeutics to control HIV infections.
Footnotes
CONFLICT OF INTEREST
Victor Kramer is an employee in the medical affairs department of Merck Canada Inc. Kirkland, QC, Canada and previously worked at the McGill AIDS Center/Lady Davis Institute from 2009–2015.
REFERENCES
- 1.Arts EJ; Hazuda DJ HIV-1 antiretroviral drug therapy. Cold Spring Harbor Perspect. Med 2012, 2 (4), a007161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Poongavanam V; Namasivayam V; Vanangamudi M; Al Shamaileh H; Veedu RN; Kihlberg J; Murugan NA Integrative approaches in HIV-1 non-nucleoside reverse transcriptase inhibitor design. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2018, 8 (1), e1328. [Google Scholar]
- 3.Zhan P; Pannecouque C; De Clercq E; Liu X Anti-HIV drug discovery and development: Current innovations and future trends. J. Med. Chem. 2016, 59 (7), 2849–78. [DOI] [PubMed] [Google Scholar]
- 4.Mehellou Y; De Clercq E Twenty-six years of anti-HIV drug discovery: where do we stand and where do we go? J. Med. Chem. 2010, 53 (2), 521–538. [DOI] [PubMed] [Google Scholar]
- 5.Flexner C HIV drug development: the next 25 years. Nat. Rev. Drug Discovery 2007, 6 (12), 959–966. [DOI] [PubMed] [Google Scholar]
- 6.Zdanowicz MM The pharmacology of HIV drug resistance. Am. J. Pharm. Educ. 2006, 70 (5), 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.UNAIDS data 2017. http://www.unaids.org/sites/default/files/media_asset/20170720_Data_book_2017_en.pdf (accessed Feb 10, 2018).
- 8.Life expectancy of individuals on combination antiretroviral therapy in high-income countries: a collaborative analysis of 14 cohort studies. The Lancet 2008, 372 (9635), 293–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shafer RW; Vuitton DA Highly active antiretroviral therapy (HAART) for the treatment of infection with human immunodeficiency virus type 1. Biomed. Pharmacother. 1999, 53 (2), 73–86. [DOI] [PubMed] [Google Scholar]
- 10.Brechtl JR; Breitbart W; Galietta M; Krivo S; Rosenfeld B The use of highly active antiretroviral therapy (HAART) in patients with advanced HIV infection. J. Pain Symptom Manage. 21 (1), 41–51. [DOI] [PubMed] [Google Scholar]
- 11.Meanwell NA; Krystal MR; Nowicka-Sans B; Langley DR; Conlon DA; Eastgate MD; Grasela DM; Timmins P; Wang T; Kadow JF Inhibitors of HIV-1 attachment: the discovery and development of temsavir and its prodrug fostemsavir. J. Med. Chem. 2018, 61 (1), 62–80. [DOI] [PubMed] [Google Scholar]
- 12.De Clercq E Strategies in the design of antiviral drugs. Nat. Rev. Drug Discovery 2002, 1 (1), 13–25. [DOI] [PubMed] [Google Scholar]
- 13.De Clercq E The role of non-nucleoside reverse transcriptase inhibitors (NNRTIs) in the therapy of HIV-1 infection. Antiviral Res. 1998, 38 (3), 153–179. [DOI] [PubMed] [Google Scholar]
- 14.De Clercq E Non-nucleoside reverse transcriptase inhibitors (NNRTIs): past, present, and future. Chem. Biodiversity 2004, 1 (1), 44–64. [DOI] [PubMed] [Google Scholar]
- 15.De Clercq E; Li G Approved antiviral drugs over the past 50 years. Clin. Microbiol. Rev. 2016, 29 (3), 695–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schafer W; Friebe WG; Leinert H; Mertens A; Poll T; von der Saal W; Zilch H; Nuber B; Ziegler ML Non-nucleoside inhibitors of HIV-1 reverse transcriptase: molecular modeling and X-ray structure investigations. J. Med. Chem. 1993, 36 (6), 726–732. [DOI] [PubMed] [Google Scholar]
- 17.Hsiou Y; Ding J; Das K; Clark AD Jr.; Hughes SH; Arnold E Structure of unliganded HIV-1 reverse transcriptase at 2.7 A resolution: implications of conformational changes for polymerization and inhibition mechanisms. Structure 1996, 4 (7), 853–860. [DOI] [PubMed] [Google Scholar]
- 18.Kohlstaedt LA; Wang J; Friedman JM; Rice PA; Steitz TA Crystal structure at 3.5 A resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science 1992, 256 (5065), 1783–1790. [DOI] [PubMed] [Google Scholar]
- 19.Bauman JD; Das K; Ho WC; Baweja M; Himmel DM; Clark AD Jr.; Oren DA; Boyer PL; Hughes SH; Shatkin AJ; Arnold E Crystal engineering of HIV-1 reverse transcriptase for structure-based drug design. Nucleic Acids Res. 2008, 36 (15), 5083–5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Das K; Arnold E HIV-1 reverse transcriptase and antiviral drug resistance. Part 2. Curr. Opin. Virol. 2013, 3 (2), 119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Das K; Arnold E HIV-1 reverse transcriptase and antiviral drug resistance. Part 1. Curr. Opin. Virol. 2013, 3 (2), 111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shafer RW; Schapiro JM HIV-1 drug resistance mutations: an updated framework for the second decade of HAART. AIDS Rev. 2008, 10 (2), 67–84. [PMC free article] [PubMed] [Google Scholar]
- 23.AIDS guidelines. https://aidsinfo.nih.gov/guidelines (accessed February 22, 2018).
- 24.Sluis-Cremer N The emerging profile of cross-resistance among the nonnucleoside HIV-1 reverse transcriptase inhibitors. Viruses 2014, 6 (8), 2960–2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wensing AM; Calvez V; Gunthard HF; Johnson VA; Paredes R; Pillay D; Shafer RW; Richman DD 2014 Update of the drug resistance mutations in HIV-1. Top. Antivir. Med. 2014, 22 (3), 642–650. [PMC free article] [PubMed] [Google Scholar]
- 26.Melikian GL; Rhee SY; Varghese V; Porter D; White K; Taylor J; Towner W; Troia P; Burack J; Dejesus E; Robbins GK; Razzeca K; Kagan R; Liu TF; Fessel WJ; Israelski D; Shafer RW Non-nucleoside reverse transcriptase inhibitor (NNRTI) cross-resistance: implications for preclinical evaluation of novel NNRTIs and clinical genotypic resistance testing. J. Antimicrob. Chemother. 2014, 69 (1), 12–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.HIV Drug resistance database. https://hivdb.stanford.edu/dr-summary/resistance-notes/NNRTI/ (accessed April 10, 2018).
- 28.Smith RH Jr.; Jorgensen WL; Tirado-Rives J; Lamb ML; Janssen PA; Michejda CJ; Kroeger Smith MB Prediction of binding affinities for TIBO inhibitors of HIV-1 reverse transcriptase using monte carlo simulations in a linear response method. J. Med. Chem 1998, 41 (26), 5272–5286. [DOI] [PubMed] [Google Scholar]
- 29.Jorgensen WL; Ruiz-Caro J; Tirado-Rives J; Basavapathruni A; Anderson KS; Hamilton AD Computer-aided design of non-nucleoside inhibitors of HIV-1 reverse transcriptase. Bioorg. Med. Chem. Lett. 2006, 16 (3), 663–667. [DOI] [PubMed] [Google Scholar]
- 30.Ragno R; Mai A; Sbardella G; Artico M; Massa S; Musiu C; Mura M; Marturana F; Cadeddu A; La Colla P Computer-aided design, synthesis, and anti-HIV-1 activity in vitro of 2-alkylamino-6-[1-(2,6-difluorophenyl)alkyl]-3,4-dihydro-5-alkylpyrimidin-4(3H)-o nes as novel potent non-nucleoside reverse transcriptase inhibitors, also active against the Y181C variant. J. Med. Chem. 2004, 47 (4), 928–934. [DOI] [PubMed] [Google Scholar]
- 31.Bender A; Brown N Special issue: Cheminformatics in drug discovery. ChemMedChem 2018, 13 (6), 467–469. [DOI] [PubMed] [Google Scholar]
- 32.Carmine AA; Brogden RN Pirenzepine. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic efficacy in peptic ulcer disease and other allied diseases. Drugs 1985, 30 (2), 85–126. [DOI] [PubMed] [Google Scholar]
- 33.Hargrave KD; Proudfoot JR; Grozinger KG; Cullen E; Kapadia SR; Patel UR; Fuchs VU; Mauldin SC; Vitous J; Behnke ML; Klunder JM; Pal K; Skiles JW; McNeil DW; Rose JM; Chow GC; Skoog MT; Wu JC; Schmidt G; Engel WW; Eberlein WG; Saboe TD; Campbell SJ; Rosenthal AS; Adams J Novel non-nucleoside inhibitors of HIV-1 reverse transcriptase. 1. Tricyclic pyridobenzo- and dipyridodiazepinones. J. Med. Chem. 1991, 34 (7), 2231–2241. [DOI] [PubMed] [Google Scholar]
- 34.Grozinger K; Proudfoot J; Hargrave K, Discovery and development of nevirapine In Drug Discovery Development, John Wiley & Sons, Inc.: 2006; pp 353–363. [Google Scholar]
- 35.Fan-Havard P; Liu Z; Chou M; Ling Y; Barrail-Tran A; Haas DW; Taburet A-M; the ASG Pharmacokinetics of phase I nevirapine metabolites following a single dose and at steady state. Antimicrob. Agents Chemother. 2013, 57 (5), 2154–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grozinger KG; Byrne DP; Nummy LJ; Ridges MD; Salvagno A Synthesis of five nevirapine metabolites. J. Heterocycl. Chem. 2000, 37 (2), 229–239. [Google Scholar]
- 37.Grozinger KG; Fuchs V; Hargrave KD; Mauldin S; Vitous J; Campbell S; Adams J Synthesis of nevirapine and its major metabolite. J. Heterocycl. Chem. 1995, 32 (1), 259–263. [Google Scholar]
- 38.Riska P; Lamson M; MacGregor T; Sabo J; Hattox S; Pav J; Keirns J Disposition and biotransformation of the antiretroviral drug nevirapine in humans. Drug Metab. Dispos. 1999, 27 (8), 895. [PubMed] [Google Scholar]
- 39.Nevirapine FDA package. https://www.fda.gov/ (accessed May 6, 2018).
- 40.Wang Y; Xing H; Liao L; Wang Z; Su B; Zhao Q; Feng Y; Ma P; Liu J; Wu J; Ruan Y; Shao Y The development of drug resistance mutations K103N Y181C and G190A in long term Nevirapine-containing antiviral therapy. AIDS Res: Ther: 2014, 11, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ren J; Nichols C; Bird L; Chamberlain P; Weaver K; Short S; Stuart DI; Stammers DK Structural mechanisms of drug resistance for mutations at codons 181 and 188 in HIV-1 reverse transcriptase and the improved resilience of second generation non-nucleoside inhibitors. J. Mol. Biol. 2001, 312 (4), 795–805. [DOI] [PubMed] [Google Scholar]
- 42.Kleim JP; Bender R; Kirsch R; Meichsner C; Paessens A; Riess G Mutational analysis of residue 190 of human immunodeficiency virus type 1 reverse transcriptase. Virology 1994, 200 (2), 696–701. [DOI] [PubMed] [Google Scholar]
- 43.Ren J; Nichols CE; Chamberlain PP; Weaver KL; Short SA; Stammers DK Crystal structures of HIV-1 reverse transcriptases mutated at codons 100, 106 and 108 and mechanisms of resistance to non-nucleoside inhibitors. J. Mol. Biol. 2004, 336 (3), 569–578. [DOI] [PubMed] [Google Scholar]
- 44.Ren J; Milton J; Weaver KL; Short SA; Stuart DI; Stammers DK Structural basis for the resilience of efavirenz (DMP-266) to drug resistance mutations in HIV-1 reverse transcriptase. Structure 2000, 8 (10), 1089–1094. [DOI] [PubMed] [Google Scholar]
- 45.Lange JM Efficacy and durability of nevirapine in antiretroviral drug naive patients. J. Acquired Immune Defic. Syndr. 2003, 34 Suppl 1, S40–52. [DOI] [PubMed] [Google Scholar]
- 46.Montaner JS; Reiss P; Cooper D; Vella S; Harris M; Conway B; Wainberg MA; Smith D; Robinson P; Hall D; Myers M; Lange JM A randomized, double-blind trial comparing combinations of nevirapine, didanosine, and zidovudine for HIV-infected patients: the INCAS Trial. Italy, The Netherlands, Canada and Australia Study. Jama 1998, 279 (12), 930–937. [DOI] [PubMed] [Google Scholar]
- 47.Baylor MS; Johann-Liang R Hepatotoxicity associated with nevirapine use. J. Acquired Immune Defic. Syndr. 2004, 35 (5), 538–539. [DOI] [PubMed] [Google Scholar]
- 48.Sanne I; Mommeja-Marin H; Hinkle J; Bartlett JA; Lederman MM; Maartens G; Wakeford C; Shaw A; Quinn J; Gish RG; Rousseau F Severe hepatotoxicity associated with nevirapine use in HIV-infected subjects. J. Infect. Dis. 2005, 191 (6), 825–829. [DOI] [PubMed] [Google Scholar]
- 49.de Bethune MP Non-nucleoside reverse transcriptase inhibitors (NNRTIs), their discovery, development, and use in the treatment of HIV-1 infection: a review of the last 20 years (1989–2009). Antiviral Res. 2010, 85 (1), 75–90. [DOI] [PubMed] [Google Scholar]
- 50.Merluzzi VJ; Hargrave KD; Labadia M; Grozinger K; Skoog M; Wu JC; Shih CK; Eckner K; Hattox S; Adams J Inhibition of HIV-1 replication by a nonnucleoside reverse transcriptase inhibitor. Science 1990, 250 (4986), 1411–1413. [DOI] [PubMed] [Google Scholar]
- 51.Burton AG; Halls PJ; Katritzky AR The selective 3- and 5-nitration of 2-pyridones. Tetrahedron Lett. 1971, 12 (24), 2211–2212. [Google Scholar]
- 52.Bobbitt JM; Scola DA Synthesis of isoquinoline alkaloids. II. The synthesis and reactions of 4-methyl-3-pyridinecarboxaldehyde and other 4-methyl-3-substituted pyridines1,2. J. Org. Chem. 1960, 25 (4), 560–564. [Google Scholar]
- 53.Grozinger KG; Hargrave KD; Adams J Method for the Preparation of 3-amino-2-chloro-4-methylpyridines. US5668287 A, 1997.
- 54.Johnson DS; Lie JJ, The Art of Drug Synthesis. Wiley-Interscience: Ljubljana, 2007. [Google Scholar]
- 55.Grozinger KG; Hargrave KD; Adams J Method for the Preparation of 5,11-dihydro-6h-dipyrido [3,2-b:2’,3’-e][1,4]diazepines. US5571912, 1996.
- 56.Boswell RF; Gupton BF; Lo YS Improved Method for Making Nevirapine. CN1280293C, 2006.
- 57.Zhiquan Z; Laisheng Z; Xiujuan W; Fei X Methods for preparation of nevirapine and intermediate thereof. CN104774204A, 2015.
- 58.Scott LJ; Perry CM Delavirdine. Drugs 2000, 60 (6), 1411–1444. [DOI] [PubMed] [Google Scholar]
- 59.Dueweke TJ; Poppe SM; Romero DL; Swaney SM; So AG; Downey KM; Althaus IW; Reusser F; Busso M; Resnick L U-90152, a potent inhibitor of human immunodeficiency virus type 1 replication. Antimicrob. Agents Chemother. 1993, 37 (5), 1127–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Romero DL; Busso M; Tan CK; Reusser F; Palmer JR; Poppe SM; Aristoff PA; Downey KM; So AG; Resnick L Nonnucleoside reverse transcriptase inhibitors that potently and specifically block human immunodeficiency virus type 1 replication. Proc. Natl. Acad. Sci. U. S. A. 1991, 88 (19), 8806–8810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Romero DL; Olmsted RA; Poel TJ; Morge RA; Biles C; Keiser BJ; Kopta LA; Friis JM; Hosley JD; Stefanski KJ; Wishka DG; Evans DB; Morris J; Stehle RG; Sharma SK; Yagi Y; Voorman RL; Adams WJ; Tarpley WG; Thomas RC Targeting delavirdine/atevirdine resistant HIV-1: identification of (alkylamino)piperidine-containing bis(heteroaryl)piperazines as broad spectrum HIV-1 reverse transcriptase inhibitors. J. Med. Chem. 1996, 39 (19), 3769–3789. [DOI] [PubMed] [Google Scholar]
- 62.Adams WJ; Aristoff PA; Jensen RK; Morozowich W; Romero DL; Schinzer WC; Tarpley WG; Thomas RC, Discovery and development of the BHAP nonnucleoside reverse transcriptase inhibitor delavirdine mesylate In Integration of Pharmaceutical Discovery and Development: Case Histories, Borchardt RT; Freidinger RM; Sawyer TK; Smith PL, Eds. Springer; US: Boston, MA, 1998; pp 285–312. [DOI] [PubMed] [Google Scholar]
- 63.Romero DL; Morge RA; Biles C; Berrios-Pena N; May PD; Palmer JR; Johnson PD; Smith HW; Busso M; Tan CK; Voorman RL; Reusser F; W. AI; Downey KW; So AG; Resnik L; Tarpley WG; Aristoff PA Discovery, synthesis, and bioactivity of bis(heteroaryl)piperazines. 1. A novel class of non-nucleoside HIV-1 reverse transcriptase inhibitors. J. Med. Chem. 1994, 37 (7), 999–1014. [DOI] [PubMed] [Google Scholar]
- 64.Romero DL; Morge RA; Genin MJ; Biles C; Busso M; Resnick L; Althaus IW; Reusser F; Thomas RC; Tarpley WG Bis(heteroaryl)piperazine (BHAP) reverse transcriptase inhibitors: structure-activity relationships of novel substituted indole analogues and the identification of 1-[(5-methanesulfonamido-1H-indol-2-yl)-carbonyl]-4-[3- [(1-methylethyl)amino]-pyridinyl]piperazine monomethanesulfonate (U-90152S), a second-generation clinical candidate. J. Med. Chem. 1993, 36 (10), 1505–1508. [DOI] [PubMed] [Google Scholar]
- 65.Adams WJ; Aristoff PA; Jensen RK; Morozowich W; Romero DL; Schinzer WC; Tarpley WG; Thomas RC Discovery and development of the BHAP nonnucleoside reverse transcriptase inhibitor delavirdine mesylate. Pharm. Biotechnol. 1998, 11, 285–312. [DOI] [PubMed] [Google Scholar]
- 66.Voorman RL; Maio SM; Hauer MJ; Sanders PE; Payne NA; Ackland MJ Metabolism of delavirdine, a human immunodeficiency virus type-1 reverse transcriptase inhibitor, by microsomal cytochrome P450 in humans, rats, and other species: probable involvement of CYP2D6 and CYP3A. Drug Metab. Dispos. 1998, 26 (7), 631–639. [PubMed] [Google Scholar]
- 67.Voorman RL; Maio SM; Payne NA; Zhao Z; Koeplinger KA; Wang X Microsomal metabolism of delavirdine: evidence for mechanism-based inactivation of human cytochrome P450 3A. J. Pharmacol. Exp. Ther. 1998, 287 (1), 381–388. [PubMed] [Google Scholar]
- 68.Esnouf RM; Ren J; Hopkins AL; Ross CK; Jones EY; Stammers DK; Stuart DI Unique features in the structure of the complex between HIV-1 reverse transcriptase and the bis(heteroaryl)piperazine (BHAP) U-90152 explain resistance mutations for this nonnucleoside inhibitor. Proc. Natl. Acad. Sci. U. S. A. 1997, 94 (8), 3984–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Genin MJ; Poel TJ; Yagi Y; Biles C; Althaus I; Keiser BJ; Kopta LA; Friis JM; Reusser F; Adams WJ; Olmsted RA; Voorman RL; Thomas RC; Romero DL Synthesis and bioactivity of novel bis(heteroaryl)piperazine (BHAP) reverse transcriptase inhibitors: structure-activity relationships and increased metabolic stability of novel substituted pyridine analogs. J. Med. Chem. 1996, 39 (26), 5267–5275. [DOI] [PubMed] [Google Scholar]
- 70.Demeter LM; Shafer RW; Meehan PM; Holden-Wiltse J; Fischl MA; Freimuth WW; Para MF; Reichman RC Delavirdine susceptibilities and associated reverse transcriptase mutations in human immunodeficiency virus type 1 isolates from patients in a phase I/II trial of delavirdine monotherapy (ACTG 260). Antimicrob. Agents Chemother. 2000, 44 (3), 794–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gerondelis P; Archer RH; Palaniappan C; Reichman RC; Fay PJ; Bambara RA; Demeter LM The P236L delavirdine-resistant human immunodeficiency virus type 1 mutant is replication defective and demonstrates alterations in both RNA 5’-end- and DNA 3’-end-directed RNase H activities. J. Virol. 1999, 73 (7), 5803–5813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Das K; Clark AD Jr.; Lewi PJ; Heeres J; De Jonge MR; Koymans LM; Vinkers HM; Daeyaert F; Ludovici DW; Kukla MJ; De Corte B; Kavash RW; Ho CY; Ye H; Lichtenstein MA; Andries K; Pauwels R; De Bethune MP; Boyer PL; Clark P; Hughes SH; Janssen PA; Arnold E Roles of conformational and positional adaptability in structure-based design of TMC125-R165335 (etravirine) and related non-nucleoside reverse transcriptase inhibitors that are highly potent and effective against wild-type and drug-resistant HIV-1 variants. J. Med. Chem. 2004, 47 (10), 2550–2560. [DOI] [PubMed] [Google Scholar]
- 73.Uhlmann EJ; Tebas P; Storch GA; Powderly WG; Lie YS; Whitcomb JM; Hellmann NS; Arens MQ Effects of the G190A substitution of HIV reverse transcriptase on phenotypic susceptibility of patient isolates to delavirdine. J. Clin. Virol. 2004, 31 (3), 198–203. [DOI] [PubMed] [Google Scholar]
- 74.Scott LJ; Perry CM Delavirdine: a review of its use in HIV infection. Drugs 2000, 60 (6), 1411–1444. [DOI] [PubMed] [Google Scholar]
- 75.Davey RT Jr.; Chaitt DG; Reed GF; Freimuth WW; Herpin BR; Metcalf JA; Eastman PS; Falloon J; Kovacs JA; Polis MA; Walker RE; Masur H; Boyle J; Coleman S; Cox SR; Wathen L; Daenzer CL; Lane HC Randomized, controlled phase I/II, trial of combination therapy with delavirdine (U-90152S) and conventional nucleosides in human immunodeficiency virus type 1-infected patients. Antimicrob. Agents Chemother. 1996, 40 (7), 1657–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Been-Tiktak AM; Boucher CA; Brun-Vezinet F; Joly V; Mulder JW; Jost J; Cooper DA; Moroni M; Gatell JM; Staszewski S; Colebunders R; Stewart GJ; Hawkins DA; Johnson MA; Parkin JM; Kennedy DH; Hoy JF; Borleffs JC Efficacy and safety of combination therapy with delavirdine and zidovudine: a European/Australian phase II trial. Int. J. Antimicrob. Agents 1999, 11 (1), 13–21. [DOI] [PubMed] [Google Scholar]
- 77.Friedland GH; Pollard R; Griffith B; Hughes M; Morse G; Bassett R; Freimuth W; Demeter L; Connick E; Nevin T; Hirsch M; Fischl M Efficacy and safety of delavirdine mesylate with zidovudine and didanosine compared with two-drug combinations of these agents in persons with HIV disease with CD4 counts of 100 to 500 cells/mm3 (ACTG 261). ACTG 261 Team. J. Acquired Immune Defic. Syndr. 1999, 21 (4), 281–292. [DOI] [PubMed] [Google Scholar]
- 78.Current and resolved drug shortages and discontinuations reported to FDA. https://www.accessdata.fda.gov/scripts/drugshortages/dsp_ActiveIngredientDetails.cfm?AI=Delavirdine%20Mesylate%20(RESCRIPTOR)%20Tablets&st=d (accessed May 6, 2018).
- 79.Chou R; Fu R; Huffman LH; Korthuis PT Initial highly-active antiretroviral therapy with a protease inhibitor versus a non-nucleoside reverse transcriptase inhibitor: discrepancies between direct and indirect meta-analyses. Lancet 2006, 368 (9546), 1503–1515. [DOI] [PubMed] [Google Scholar]
- 80.Wenjie S; Xuhua X; Weiyong M Preparing Method of 1-(5-nitro-1H-indole-2-carbonyl)-4-{3-[(1-methylethyl)amino]-2-pyridine}. CN101016295A, 2007.
- 81.Ensi W; Jing G; Shengxiu N; Nan Z; Liping J Synthesis Method of Delavirdine. CN102675284 A, 2012.
- 82.Min G Synthesis Method of anti-AIDS Medicine Delavirdine. CN102633778A, 2012.
- 83.Young SD; Britcher SF; Tran LO; Payne LS; Lumma WC; Lyle TA; Huff JR; Anderson PS; Olsen DB; Carroll SS L-743, 726 (DMP-266): a novel, highly potent nonnucleoside inhibitor of the human immunodeficiency virus type 1 reverse transcriptase. Antimicrob. Agents Chemother. 1995, 39 (12), 2602–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Buckheit RW; Fliakas-Boltz V; Decker WD; Roberson JL; Pyle CA; White EL; Bowdon BJ; McMahon JB; Boyd MR; Bader JP; Nickell DG; Barth H; Antonucci TK Biological and biochemical anti-HIV activity of the benzothiadiazine class of nonnucleoside reverse transcriptase inhibitors. Antiviral Res. 1994, 25 (1), 43–56. [DOI] [PubMed] [Google Scholar]
- 85.Zhan P; Chen X; Li D; Fang Z; De Clercq E; Liu X HIV-1 NNRTIs: structural diversity, pharmacophore similarity, and implications for drug design. Med. Res. Rev. 2013, 33 Suppl 1, E1–72. [DOI] [PubMed] [Google Scholar]
- 86.Britcher SF; Goldman ME; Huff JR; Lumma WC; Lyle TA; Payne LS; Quesada ML; Sanders WM; Tucker TJ; Young SD Quinazoline Derivatives as Inhibitors of HIV Reverse Transcriptase. WO1993004047A1, 1993.
- 87.Houpis IN; Molina A; Douglas AW; Xavier L; Lynch J; Volante RP; Reider PJ Synthesis of a new generation reverse transcriptase inhibitor via the BCl3/GaCl3-induced condensation of anilines with nitriles (sugasawa reaction). Tetrahedron Lett. 1994, 35 (37), 6811–6814. [Google Scholar]
- 88.Tucker TJ; Lyle TA; Wiscount CM; Britcher SF; Young SD; Sanders WM; Lumma WC; Goldman ME; O’Brien JA Synthesis of a series of 4-(Arylethynyl)-6-chloro-4-cyclopropyl-3,4-dihydroquinazolin-2(1H)-ones as novel non-nucleoside HIV-1 Reverse transcriptase inhibitors. J. Med. Chem. 1994, 37 (15), 2437–2444. [DOI] [PubMed] [Google Scholar]
- 89.Huffman MA; Yasuda N; DeCamp AE; Grabowski EJJ Lithium alkoxides of cinchona alkaloids as chiral controllers for enantioselective acetylide addition to cyclic N-acyl ketimines. J. Org. Chem. 1995, 60 (6), 1590–1594. [Google Scholar]
- 90.Purser S; Moore PR; Swallow S; Gouverneur V Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37 (2), 320–330. [DOI] [PubMed] [Google Scholar]
- 91.Nubia B; Kover WB; Vera B; Monica MB; Nelilma CR; Maria LGA; Wagner W Design, synthesis and pharmacological evaluation of HIV-1 reverse transcriptase inhibition of new indolin-2-Ones. Med. Chem. 2007, 3 (6), 533–542. [DOI] [PubMed] [Google Scholar]
- 92.Ogburn ET; Jones DR; Masters AR; Xu C; Guo Y; Desta Z Efavirenz primary and secondary metabolism in vitro and in vivo: identification of novel metabolic pathways and cytochrome P450 2A6 as the principal catalyst of efavirenz 7-hydroxylation. Drug Metab. Dispos. 2010, 38 (7), 1218–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ward BA; Gorski JC; Jones DR; Hall SD; Flockhart DA; Desta Z The cytochrome P450 2B6 (CYP2B6) is the main catalyst of efavirenz primary and secondary metabolism: implication for HIV/AIDS therapy and utility of efavirenz as a substrate marker of CYP2B6 catalytic activity. J. Pharmacol. Exp. Ther. 2003, 306 (1), 287–300. [DOI] [PubMed] [Google Scholar]
- 94.Mutlib AE; Gerson RJ; Meunier PC; Haley PJ; Chen H; Gan LS; Davies MH; Gemzik B; Christ DD; Krahn DF; Markwalder JA; Seitz SP; Robertson RT; Miwa GT The Species-Dependent Metabolism of Efavirenz Produces a Nephrotoxic Glutathione Conjugate in Rats. Toxicol. Appl. Pharmacol. 2000, 169 (1), 102–113. [DOI] [PubMed] [Google Scholar]
- 95.Lindberg J; Sigurdsson S; Lowgren S; Andersson HO; Sahlberg C; Noreen R; Fridborg K; Zhang H; Unge T Structural basis for the inhibitory efficacy of efavirenz (DMP-266), MSC194 and PNU142721 towards the HIV-1 RT K103N mutant. Eur. J. Biochem. 2002, 269 (6), 1670–1677. [DOI] [PubMed] [Google Scholar]
- 96.Hsiou Y; Ding J; Das K; Clark AD Jr.; Boyer PL; Lewi P; Janssen PA; Kleim JP; Rosner M; Hughes SH; Arnold E The Lys103Asn mutation of HIV-1 RT: a novel mechanism of drug resistance. J. Mol. Biol. 2001, 309 (2), 437–445. [DOI] [PubMed] [Google Scholar]
- 97.Efavirenz FDA package. https://www.fda.gov/ (accessed May 6, 2018).
- 98.Staszewski S; Morales-Ramirez J; Tashima KT; Rachlis A; Skiest D; Stanford J; Stryker R; Johnson P; Labriola DF; Farina D; Manion DJ; Ruiz NM Efavirenz plus zidovudine and lamivudine, efavirenz plus indinavir, and indinavir plus zidovudine and lamivudine in the treatment of HIV-1 infection in adults. Study 006 Team. N. Engl. J. Med. 1999, 341 (25), 1865–1873. [DOI] [PubMed] [Google Scholar]
- 99.https://aidsinfo.nih.gov/contentfiles/lvguidelines/adultandadolescentgl.pdf (accessed May 14, 2018).
- 100.Young SD; Britcher SF; Payne LS; Tran LO; Lumma WC Benzoxazinones as Inhibitors of HIV Reverse Transcriptase. US5519021A, 1996.
- 101.Radesca LA; Lo† YS; Moore JR; Pierce ME Synthesis of HIV-1 reverse transcriptase inhibitor DMP 266. Synth. Commun. 1997, 27 (24), 4373–4384. [Google Scholar]
- 102.Thompson A; Corley EG; Huntington MF; Grabowski EJJ; Remenar JF; Collum DB Lithium ephedrate-mediated addition of a lithium acetylide to a ketone: Solution structures and relative reactivities of mixed aggregates underlying the high enantioselectivities. J. Am. Chem. Soc. 1998, 120 (9), 2028–2038. [Google Scholar]
- 103.Thompson AS; Corley EG; Huntington MF; Grabowski EJJ Use of an ephedrine alkoxide to mediate enantioselective addition of an acetylide to a prochiral ketone: asymmetric synthesis of the reverse transcriptase inhibitor L-743,726. Tetrahedron Lett. 1995, 36 (49), 8937–8940. [Google Scholar]
- 104.Pierce ME; Parsons RL; Radesca LA; Lo YS; Silverman S; Moore JR; Islam Q; Choudhury A; Fortunak JMD; Nguyen D; Luo C; Morgan SJ; Davis WP; Confalone PN; Chen C.-y.; Tillyer RD; Frey L; Tan L; Xu F; Zhao D; Thompson AS; Corley EG; Grabowski EJJ; Reamer R; Reider PJ Practical asymmetric synthesis of efavirenz (DMP 266), an HIV-1 reverse transcriptase inhibitor. J. Org. Chem. 1998, 63 (23), 8536–8543. [Google Scholar]
- 105.Dai D; Long X; Luo B; Kulesza A; Reichwagen J; Guo Y Process for Preparation of Efavirenz by Cyclisation. WO2012097510A1, 2012.
- 106.Correia CA; Gilmore K; McQuade DT; Seeberger PH A concise flow synthesis of efavirenz. Angew. Chem., Int. Ed. 2015, 54 (16), 4945–4948. [DOI] [PubMed] [Google Scholar]
- 107.Chen CY; Tillyer RD; Tan L Efficient Enantioselective Addition Reaction using an Organozinc Reagent. WO1998051676A1, 1998.
- 108.Ding J; Das K; Moereels H; Koymans L; Andries K; Janssen PA; Hughes SH; Arnold E Structure of HIV-1 RT/TIBO R 86183 complex reveals similarity in the binding of diverse nonnucleoside inhibitors. Nat. Struct. Biol. 1995, 2 (5), 407–415. [DOI] [PubMed] [Google Scholar]
- 109.Pauwels R; Andries K; Desmyter J; Schols D; Kukla MJ; Breslin HJ; Raeymaeckers A; Gelder JV; Woestenborghs R; Heykants J; Schellekens K; Janssen MAC; Clercq ED; Janssen PAJ Potent and selective inhibition of HIV-1 replication in vitro by a novel series of TIBO derivatives. Nature 1990, 343, 470. [DOI] [PubMed] [Google Scholar]
- 110.Ding J; Das K; Tantillo C; Zhang W; Clark JAD; Jessen S; Lu X; Hsiou Y; Jacobo-Molina A; Andries K; Pauwels R; Moereels H; Koymans L; Janssen PAJ; Smith JRH; Koepke MK; Michejda CJ; Hughes SH; Arnold E Structure of HIV-1 reverse transcriptase in a complex with the non-nucleoside inhibitor α-APA R 95845 at 2.8 å resolution. Structure 1995, 3 (4), 365–379. [DOI] [PubMed] [Google Scholar]
- 111.Pauwels R; Andries K; Debyser Z; Van Daele P; Schols D; Stoffels P; De Vreese K; Woestenborghs R; Vandamme AM; Janssen CG Potent and highly selective human immunodeficiency virus type 1 (HIV-1) inhibition by a series of alpha-anilinophenylacetamide derivatives targeted at HIV-1 reverse transcriptase. Proc. Natl. Acad. Sci. U. S. A. 1993, 90 (5), 1711–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ludovici DW; Kavash RW; Kukla MJ; Ho CY; Ye H; De Corte BL; Andries K; de Bethune MP; Azijn H; Pauwels R; Moereels HE; Heeres J; Koymans LM; de Jonge MR; Van Aken KJ; Daeyaert FF; Lewi PJ; Das K; Arnold E; Janssen PA Evolution of anti-HIV drug candidates. Part 2: Diaryltriazine (DATA) analogues. Bioorg. Med. Chem. Lett. 2001, 11 (17), 2229–2234. [DOI] [PubMed] [Google Scholar]
- 113.Ludovici DW; De Corte BL; Kukla MJ; Ye H; Ho CY; Lichtenstein MA; Kavash RW; Andries K; de Bethune MP; Azijn H; Pauwels R; Lewi PJ; Heeres J; Koymans LM; de Jonge MR; Van Aken KJ; Daeyaert FF; Das K; Arnold E; Janssen PA Evolution of anti-HIV drug candidates. Part 3: Diarylpyrimidine (DAPY) analogues. Bioorg. Med. Chem. Lett. 2001, 11 (17), 2235–2239. [DOI] [PubMed] [Google Scholar]
- 114.Andries K; Azijn H; Thielemans T; Ludovici D; Kukla M; Heeres J; Janssen P; De Corte B; Vingerhoets J; Pauwels R; de Bethune MP TMC125, a novel next-generation nonnucleoside reverse transcriptase inhibitor active against nonnucleoside reverse transcriptase inhibitor-resistant human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 2004, 48 (12), 4680–4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Deeks ED; Keating GM Etravirine. Drugs 2008, 68 (16), 2357–2372. [DOI] [PubMed] [Google Scholar]
- 116.Elsayed RK; Caldwell DJ Etravirine: a novel nonnucleoside reverse transcriptase inhibitor for managing human immunodeficiency virus infection. Am. J. Health-Syst. Pharm. 2010, 67 (3), 193–205. [DOI] [PubMed] [Google Scholar]
- 117.Janssen PAJ; Lewi PJ; Arnold E; Daeyaert F; de Jonge M; Heeres J; Koymans L; Vinkers M; Guillemont J; Pasquier E; Kukla M; Ludovici D; Andries K; de Béthune M-P; Pauwels R; Das K; Clark AD; Frenkel YV; Hughes SH; Medaer B; De Knaep F; Bohets H; De Clerck F; Lampo A; Williams P; Stoffels P In search of a novel anti-HIV Drug: multidisciplinary coordination in the discovery of 4-[[4-[[4-[(1E)-2-Cyanoethenyl]-2,6-dimethylphenyl]amino]-2- pyrimidinyl]amino]benzonitrile (R278474, Rilpivirine). J. Med. Chem. 2005, 48 (6), 1901–1909. [DOI] [PubMed] [Google Scholar]
- 118.Lansdon EB; Brendza KM; Hung M; Wang R; Mukund S; Jin D; Birkus G; Kutty N; Liu X Crystal structures of HIV-1 reverse transcriptase with etravirine (TMC125) and rilpivirine (TMC278): implications for drug design. J. Med. Chem. 2010, 53 (10), 4295–4429. [DOI] [PubMed] [Google Scholar]
- 119.Ren J; Chamberlain PP; Stamp A; Short SA; Weaver KL; Romines KR; Hazen R; Freeman A; Ferris RG; Andrews CW; Boone L; Chan JH; Stammers DK Structural basis for the improved drug resistance profile of new generation benzophenone non-nucleoside HIV-1 reverse transcriptase inhibitors. J Med Chem 2008, 51 (16), 5000–8. [DOI] [PubMed] [Google Scholar]
- 120.Scholler-Gyure M; Kakuda TN; Raoof A; De Smedt G; Hoetelmans RM Clinical pharmacokinetics and pharmacodynamics of etravirine. Clin. Pharmacokinet. 2009, 48 (9), 561–574. [DOI] [PubMed] [Google Scholar]
- 121.Yanakakis LJ; Bumpus NN Biotransformation of the antiretroviral drug etravirine: metabolite identification, reaction phenotyping, and characterization of autoinduction of cytochrome P450-dependent metabolism. Drug Metab. Dispos. 2012, 40 (4), 803–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lade JM; Avery LB; Bumpus NN Human biotransformation of the nonnucleoside reverse transcriptase inhibitor rilpivirine and a cross-species metabolism comparison. Antimicrob. Agents Chemother. 2013, 57 (10), 5067–5079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Alcaro S; Alteri C; Artese A; Ceccherini-Silberstein F; Costa G; Ortuso F; Parrotta L; Perno CF; Svicher V Molecular and structural aspects of clinically relevant mutations related to the approved non-nucleoside inhibitors of HIV-1 reverse transcriptase. Drug Resist. Updates 2011, 14 (3), 141–149. [DOI] [PubMed] [Google Scholar]
- 124.Frey KM; Puleo DE; Spasov KA; Bollini M; Jorgensen WL; Anderson KS Structure-based evaluation of non-nucleoside inhibitors with improved potency and solubility that target HIV reverse transcriptase variants. J. Med. Chem. 2015, 58 (6), 2737–2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Paris KA; Haq O; Felts AK; Das K; Arnold E; Levy RM Conformational landscape of the human immunodeficiency virus type 1 reverse transcriptase non-nucleoside inhibitor binding pocket: lessons for inhibitor design from a cluster analysis of many crystal structures. J. Med. Chem. 2009, 52 (20), 6413–6420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Basson AE; Rhee SY; Parry CM; El-Khatib Z; Charalambous S; De Oliveira T; Pillay D; Hoffmann C; Katzenstein D; Shafer RW; Morris L Impact of drug resistance-associated amino acid changes in HIV-1 subtype C on susceptibility to newer nonnucleoside reverse transcriptase inhibitors. Antimicrob. Agents Chemother. 2015, 59 (2), 960–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Xu H; Quan Y; Brenner BG; Bar-Magen T; Oliveira M; Schader SM; Wainberg MA Human immunodeficiency virus type 1 recombinant reverse transcriptase enzymes containing the G190A and Y181C resistance mutations remain sensitive to etravirine. Antimicrob. Agents Chemother. 2009, 53 (11), 4667–4672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sharma M; Saravolatz LD Rilpivirine: a new non-nucleoside reverse transcriptase inhibitor. J. Antimicrob. Chemother. 2013, 68 (2), 250–256. [DOI] [PubMed] [Google Scholar]
- 129.Etravirine FDA package. https://www.fda.gov/ (accessed May 6, 2018).
- 130.Johnson BC; Pauly GT; Rai G; Patel D; Bauman JD; Baker HL; Das K; Schneider JP; Maloney DJ; Arnold E; Thomas CJ; Hughes SH A comparison of the ability of rilpivirine (TMC278) and selected analogues to inhibit clinically relevant HIV-1 reverse transcriptase mutants. Retrovirology 2012, 9, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ripamonti D; Bombana E; Rizzi M Rilpivirine: drug profile of a second-generation non-nucleoside reverse transcriptase HIV-inhibitor. Expert Rev. Anti-Infect. Ther. 2014, 12 (1), 13–29. [DOI] [PubMed] [Google Scholar]
- 132.Gray WT; Frey KM; Laskey SB; Mislak AC; Spasov KA; Lee WG; Bollini M; Siliciano RF; Jorgensen WL; Anderson KS Potent inhibitors active against HIV reverse transcriptase with K101P, a mutation conferring rilpivirine resistance. ACS Med. Chem. Lett. 2015, 6 (10), 1075–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Xu HT; Colby-Germinario SP; Huang W; Oliveira M; Han Y; Quan Y; Petropoulos CJ; Wainberg MA Role of the K101E substitution in HIV-1 reverse transcriptase in resistance to rilpivirine and other nonnucleoside reverse transcriptase inhibitors. Antimicrob. Agents Chemother. 2013, 57 (11), 5649–5657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Singh K; Marchand B; Rai DK; Sharma B; Michailidis E; Ryan EM; Matzek KB; Leslie MD; Hagedorn AN; Li Z; Norden PR; Hachiya A; Parniak MA; Xu HT; Wainberg MA; Sarafianos SG Biochemical mechanism of HIV-1 resistance to rilpivirine. J. Biol. Chem. 2012, 287 (45), 38110–3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wei L; Wang HL; Huang L; Chen CH; Morris-Natschke SL; Lee KH; Xie L Drug-like property-driven optimization of 4-substituted 1,5-diarylanilines as potent HIV-1 non-nucleoside reverse transcriptase inhibitors against rilpivirine-resistant mutant virus. Bioorg. Med. Chem. Lett. 2017, 27 (12), 2788–2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kulkarni R; Babaoglu K; Lansdon EB; Rimsky L; Van Eygen V; Picchio G; Svarovskaia E; Miller MD; White KL The HIV-1 reverse transcriptase M184I mutation enhances the E138K-associated resistance to rilpivirine and decreases viral fitness. J. Acquired Immune Defic. Syndr. 2012, 59 (1), 47–54. [DOI] [PubMed] [Google Scholar]
- 137.Rilpivirine FDA package. https://www.fda.gov/ (accessed May 6, 2018).
- 138.Etravirine prescribing information. https://aidsinfo.nih.gov/drugs/398/etravirine/16/professional (accessed May 14, 2018).
- 139.Katlama C; Haubrich R; Lalezari J; Lazzarin A; Madruga JV; Molina JM; Schechter M; Peeters M; Picchio G; Vingerhoets J; Woodfall B; De Smedt G Efficacy and safety of etravirine in treatment-experienced, HIV-1 patients: pooled 48 week analysis of two randomized, controlled trials. Aids 2009, 23 (17), 2289–2300. [DOI] [PubMed] [Google Scholar]
- 140.de Bethune MP Non-nucleoside reverse transcriptase inhibitors (NNRTIs), their discovery, development, and use in the treatment of HIV-1 infection: a review of the last 20 years (1989–2009). Antiviral Res 2010, 85 (1), 75–90. [DOI] [PubMed] [Google Scholar]
- 141.Rilpivirine prescribing information. https://aidsinfo.nih.gov/drugs/426/rilpivirine/19/professional (accessed May 14, 2018).
- 142.Cohen CJ; Molina JM; Cahn P; Clotet B; Fourie J; Grinsztejn B; Wu H; Johnson MA; Saag M; Supparatpinyo K; Crauwels H; Lefebvre E; Rimsky LT; Vanveggel S; Williams P; Boven K Efficacy and safety of rilpivirine (TMC278) versus efavirenz at 48 weeks in treatment-naive HIV-1-infected patients: pooled results from the phase 3 double-blind randomized ECHO and THRIVE Trials. J. Acquired Immune Defic. Syndr. 2012, 60 (1), 33–42. [DOI] [PubMed] [Google Scholar]
- 143.Corte D; De Jonge MR; Heeres J; Ho CY; Janssen PAJ; Kavash RW; Koymans LMH; Kukla MJ; Ludovici DW; Van Aken KJA HIV Replication Inhibiting Pyrimidines. US6878717B2, 2005.
- 144.Joshi S; Maikap GC; Titirmare S; Chaudhari A; Gurjar MK An improved synthesis of etravirine. Org. Process Res. Dev. 2010, 14 (3), 657–660. [Google Scholar]
- 145.Fier PS; Hartwig JF Synthesis and late-stage functionalization of complex molecules through C–H fluorination and nucleophilic aromatic substitution. J. Am. Chem. Soc. 2014, 136 (28), 10139–10147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Andries KJLM; De CB; DE JMR; Heeres J; Ho CY; Janssen MAC; Janssen PAJ; Koymans LMH; Kukla MJ; Ludovici DW HIV Inhibiting Pyrimidine Derivatives. WO1999050250A1, 1999.
- 147.Guillemont J; Pasquier E; Palandjian P; Vernier D; Gaurrand S; Lewi PJ; Heeres J; de Jonge MR; Koymans LM; Daeyaert FF; Vinkers MH; Arnold E; Das K; Pauwels R; Andries K; de Bethune MP; Bettens E; Hertogs K; Wigerinck P; Timmerman P; Janssen PA Synthesis of novel diarylpyrimidine analogues and their antiviral activity against human immunodeficiency virus type 1. J. Med. Chem. 2005, 48 (6), 2072–2079. [DOI] [PubMed] [Google Scholar]
- 148.Schils DPR; Stappers AE Process for Preparing 4-[(1,6-dihydro-6-oxo-2-pyrimidinyl)amino]benzonitrile. US20080171878A1, 2008.
- 149.Ding HX; Liu KK; Sakya SM; Flick AC; O’Donnell CJ Synthetic approaches to the 2011 new drugs. Bioorg. Med. Chem. 2013, 21 (11), 2795–2825. [DOI] [PubMed] [Google Scholar]
- 150.Margolis DA; Boffito M Long-acting antiviral agents for HIV treatment. Curr. Opin. HIV AIDS 2015, 10 (4), 246–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Margolis DA; Gonzalez-Garcia J; Stellbrink HJ; Eron JJ; Yazdanpanah Y; Podzamczer D; Lutz T; Angel JB; Richmond GJ; Clotet B; Gutierrez F; Sloan L; Clair MS; Murray M; Ford SL; Mrus J; Patel P; Crauwels H; Griffith SK; Sutton KC; Dorey D; Smith KY; Williams PE; Spreen WR Long-acting intramuscular cabotegravir and rilpivirine in adults with HIV-1 infection (LATTE-2): 96-week results of a randomised, open-label, phase 2b, non-inferiority trial. Lancet 2017, 390 (10101), 1499–1510. [DOI] [PubMed] [Google Scholar]
- 152.Kerrigan D; Mantsios A; Gorgolas M; Montes ML; Pulido F; Brinson C; deVente J; Richmond GJ; Beckham SW; Hammond P; Margolis D; Murray M Experiences with long acting injectable ART: A qualitative study among PLHIV participating in a Phase II study of cabotegravir + rilpivirine (LATTE-2) in the United States and Spain. PLoS One 2018, 13 (1), e0190487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Murphy R; Kravchenko AV; Orlova-Morozova E; Nagimova F; Kozirev O; Shimonava T; Deulina M; Vostokova N; Zozulya O; Bichko V, Elsulfavirine as compared to Efavirenz in combination with TDF/FTC: 48-week study. In Conference on Retroviruses and Opportunistic Infections (CROI), Seattle, Washington, 2017; p Poster 452LB. [Google Scholar]
- 154.Rai MA; Pannek S; Fichtenbaum CJ Emerging reverse transcriptase inhibitors for HIV-1 infection. Expert Opin. Emerging Drugs 2018, 23 (2), 149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Al-Salama ZT Elsulfavirine: First global approval. Drugs 2017, 77 (16), 1811–1816. [DOI] [PubMed] [Google Scholar]
- 156.Muraglia E; Kinzel OD; Laufer R; Miller MD; Moyer G; Munshi V; Orvieto F; Palumbi MC; Pescatore G; Rowley M; Williams PD; Summa V Tetrazole thioacetanilides: potent non-nucleoside inhibitors of WT HIV reverse transcriptase and its K103N mutant. Bioorg. Med. Chem. Lett. 2006, 16 (10), 2748–2752. [DOI] [PubMed] [Google Scholar]
- 157.De La Rosa M; Kim HW; Gunic E; Jenket C; Boyle U; Koh YH; Korboukh I; Allan M; Zhang W; Chen H; Xu W; Nilar S; Yao N; Hamatake R; Lang SA; Hong Z; Zhang Z; Girardet JL Tri-substituted triazoles as potent non-nucleoside inhibitors of the HIV-1 reverse transcriptase. Bioorg. Med. Chem. Lett. 2006, 16 (17), 4444–4449. [DOI] [PubMed] [Google Scholar]
- 158.Romines KR; Freeman GA; Schaller LT; Cowan JR; Gonzales SS; Tidwell JH; Andrews CW 3rd; Stammers DK; Hazen RJ; Ferris RG; Short SA; Chan JH; Boone LR Structure-activity relationship studies of novel benzophenones leading to the discovery of a potent, next generation HIV nonnucleoside reverse transcriptase inhibitor. J. Med. Chem. 2006, 49 (2), 727–739. [DOI] [PubMed] [Google Scholar]
- 159.Tucker TJ; Saggar S; Sisko JT; Tynebor RM; Williams TM; Felock PJ; Flynn JA; Lai MT; Liang Y; McGaughey G; Liu M; Miller M; Moyer G; Munshi V; Perlow-Poehnelt R; Prasad S; Sanchez R; Torrent M; Vacca JP; Wan BL; Yan Y The design and synthesis of diaryl ether second generation HIV-1 non-nucleoside reverse transcriptase inhibitors (NNRTIs) with enhanced potency versus key clinical mutations. Bioorg. Med. Chem. Lett. 2008, 18 (9), 2959–2966. [DOI] [PubMed] [Google Scholar]
- 160.Tucker TJ; Sisko JT; Tynebor RM; Williams TM; Felock PJ; Flynn JA; Lai MT; Liang Y; McGaughey G; Liu M; Miller M; Moyer G; Munshi V; Perlow-Poehnelt R; Prasad S; Reid JC; Sanchez R; Torrent M; Vacca JP; Wan BL; Yan Y Discovery of 3-{5-[(6-amino-1H-pyrazolo[3,4-b]pyridine-3-yl)methoxy]-2-chlorophenoxy}−5-chloro benzonitrile (MK-4965): a potent, orally bioavailable HIV-1 non-nucleoside reverse transcriptase inhibitor with improved potency against key mutant viruses. J. Med. Chem. 2008, 51 (20), 6503–6511. [DOI] [PubMed] [Google Scholar]
- 161.Gomez R; Jolly SJ; Williams T; Vacca JP; Torrent M; McGaughey G; Lai MT; Felock P; Munshi V; Distefano D; Flynn J; Miller M; Yan Y; Reid J; Sanchez R; Liang Y; Paton B; Wan BL; Anthony N Design and synthesis of conformationally constrained inhibitors of non-nucleoside reverse transcriptase. J. Med. Chem. 2011, 54 (22), 7920–7933. [DOI] [PubMed] [Google Scholar]
- 162.Sweeney ZK; Harris SF; Arora SF; Javanbakht H; Li Y; Fretland J; Davidson JP; Billedeau JR; Gleason SK; Hirschfeld D; Kennedy-Smith JJ; Mirzadegan T; Roetz R; Smith M; Sperry S; Suh JM; Wu J; Tsing S; Villasenor AG; Paul A; Su G; Heilek G; Hang JQ; Zhou AS; Jernelius JA; Zhang FJ; Klumpp K Design of annulated pyrazoles as inhibitors of HIV-1 reverse transcriptase. J. Med. Chem. 2008, 51 (23), 7449–7458. [DOI] [PubMed] [Google Scholar]
- 163.Gomez R; Jolly S; Williams T; Tucker T; Tynebor R; Vacca J; McGaughey G; Lai MT; Felock P; Munshi V; DeStefano D; Touch S; Miller M; Yan Y; Sanchez R; Liang Y; Paton B; Wan BL; Anthony N Design and synthesis of pyridone inhibitors of non-nucleoside reverse transcriptase. Bioorg. Med. Chem. Lett. 2011, 21 (24), 7344–7350. [DOI] [PubMed] [Google Scholar]
- 164.Cote B; Burch JD; Asante-Appiah E; Bayly C; Bedard L; Blouin M; Campeau LC; Cauchon E; Chan M; Chefson A; Coulombe N; Cromlish W; Debnath S; Deschenes D; Dupont-Gaudet K; Falgueyret JP; Forget R; Gagne S; Gauvreau D; Girardin M; Guiral S; Langlois E; Li CS; Nguyen N; Papp R; Plamondon S; Roy A; Roy S; Seliniotakis R; St-Onge M; Ouellet S; Tawa P; Truchon JF; Vacca J; Wrona M; Yan Y; Ducharme Y Discovery of MK-1439, an orally bioavailable non-nucleoside reverse transcriptase inhibitor potent against a wide range of resistant mutant HIV viruses. Bioorg. Med. Chem. Lett. 2014, 24 (3), 917–922. [DOI] [PubMed] [Google Scholar]
- 165.Molina JM; Squires K; Sax PE; Cahn P; Lombaard J; DeJesus E; Lai MT; Xu X; Rodgers A; Lupinacci L; Kumar S; Sklar P; Nguyen BY; Hanna GJ; Hwang C Doravirine versus ritonavir-boosted darunavir in antiretroviral-naive adults with HIV-1 (DRIVE-FORWARD): 48-week results of a randomised, double-blind, phase 3, non-inferiority trial. Lancet HIV 2018, 5 (5), e211–e220. [DOI] [PubMed] [Google Scholar]
- 166.In Fixed dose combination of doravirine/lamivudine/TDF is non-inferior to efavirenz/emtricitabine/TDF in treatment-naïve adults with HIV-1 infection: week 48 results of the Phase 3 DRIVE-AHEAD study, 9th IAS Conference on HIV Science, Paris, France, 23–26 July 2017; Paris, France, 2017. [Google Scholar]
- 167.Vanangamudi M; Poongavanam V; Namasivayam V HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors: SAR and Lead Optimization Using CoMFA and CoMSIA Studies (1995–2016). Curr. Med. Chem. 2017, 24 (34), 3774–3812. [DOI] [PubMed] [Google Scholar]
- 168.Li D; Zhan P; De Clercq E; Liu X Strategies for the design of HIV-1 non-nucleoside reverse transcriptase inhibitors: lessons from the development of seven representative paradigms. J. Med. Chem. 2012, 55 (8), 3595–613. [DOI] [PubMed] [Google Scholar]
- 169.Das K; Martinez SE; Bauman JD; Arnold E HIV-1 reverse transcriptase complex with DNA and nevirapine reveals non-nucleoside inhibition mechanism. Nat. Struct. Mol. Biol. 2012, 19 (2), 253–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Usach I; Melis V; Peris JE Non-nucleoside reverse transcriptase inhibitors: a review on pharmacokinetics, pharmacodynamics, safety and tolerability. J. Int. AIDS Soc. 2013, 16, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Atripla FDA package. https://www.fda.gov/ (accessed May 6, 2018).
- 172.Odefsey FDA package. https://www.fda.gov/ (accessed May 6, 2018).
- 173.Collier R Reducing the “pill burden”. Can. Med. Assoc. J. 2012, 184 (2), E117–E118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Guay LA; Musoke P; Fleming T; Bagenda D; Allen M; Nakabiito C; Sherman J; Bakaki P; Ducar C; Deseyve M; Emel L; Mirochnick M; Fowler MG; Mofenson L; Miotti P; Dransfield K; Bray D; Mmiro F; Jackson JB Intrapartum and neonatal single-dose nevirapine compared with zidovudine for prevention of mother-to-child transmission of HIV-1 in Kampala, Uganda: HIVNET 012 randomised trial. Lancet 1999, 354 (9181), 795–802. [DOI] [PubMed] [Google Scholar]
- 175.Ayouba A; Tene G; Cunin P; Foupouapouognigni Y; Menu E; Kfutwah A; Thonnon J; Scarlatti G; Monny-Lobe M; Eteki N; Kouanfack C; Tardy M; Leke R; Nkam M; Nlend AE; Barre-Sinoussi F; Martin PM; Nerrienet E Low rate of mother-to-child transmission of HIV-1 after nevirapine intervention in a pilot public health program in Yaounde, Cameroon. J. Acquired Immune Defic. Syndr. 2003, 34 (3), 274–280. [DOI] [PubMed] [Google Scholar]
- 176.Baeten JM; Palanee-Phillips T; Brown ER; Schwartz K; Soto-Torres LE; Govender V; Mgodi NM; Matovu Kiweewa F; Nair G; Mhlanga F; Siva S; Bekker LG; Jeenarain N; Gaffoor Z; Martinson F; Makanani B; Pather A; Naidoo L; Husnik M; Richardson BA; Parikh UM; Mellors JW; Marzinke MA; Hendrix CW; van der Straten A; Ramjee G; Chirenje ZM; Nakabiito C; Taha TE; Jones J; Mayo A; Scheckter R; Berthiaume J; Livant E; Jacobson C; Ndase P; White R; Patterson K; Germuga D; Galaska B; Bunge K; Singh D; Szydlo DW; Montgomery ET; Mensch BS; Torjesen K; Grossman CI; Chakhtoura N; Nel A; Rosenberg Z; McGowan I; Hillier S Use of a vaginal ring containing dapivirine for HIV-1 prevention in women. N. Engl. J. Med. 2016, 375 (22), 2121–2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Nel A; van Niekerk N; Kapiga S; Bekker LG; Gama C; Gill K; Kamali A; Kotze P; Louw C; Mabude Z; Miti N; Kusemererwa S; Tempelman H; Carstens H; Devlin B; Isaacs M; Malherbe M; Mans W; Nuttall J; Russell M; Ntshele S; Smit M; Solai L; Spence P; Steytler J; Windle K; Borremans M; Resseler S; Van Roey J; Parys W; Vangeneugden T; Van Baelen B; Rosenberg Z Safety and efficacy of a dapivirine vaginal ring for HIV prevention in women. N. Engl. J. Med. 2016, 375 (22), 2133–2143. [DOI] [PubMed] [Google Scholar]
- 178.Buchbinder SP; Liu AY CROI 2018: Epidemic Trends and Advances in HIV Prevention. Top. Antivir. Med. 2018, 26 (1), 1–16. [PMC free article] [PubMed] [Google Scholar]
- 179.Kovarova M; Council OD; Date AA; Long JM; Nochi T; Belshan M; Shibata A; Vincent H; Baker CE; Thayer WO; Kraus G; Lachaud-Durand S; Williams P; Destache CJ; Garcia JV Nanoformulations of rilpivirine for topical pericoital and systemic coitus-independent administration efficiently prevent HIV transmission. PLoS Pathog 2015, 11 (8), e1005075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.McGowan I; Dezzutti CS; Siegel A; Engstrom J; Nikiforov A; Duffill K; Shetler C; Richardson-Harman N; Abebe K; Back D; Else L; Egan D; Khoo S; Egan JE; Stall R; Williams PE; Rehman KK; Adler A; Brand RM; Chen B; Achilles S; Cranston RD Long-acting rilpivirine as potential pre-exposure prophylaxis for HIV-1 prevention (the MWRI-01 study): an open-label, phase 1, compartmental, pharmacokinetic and pharmacodynamic assessment. Lancet HIV 2016, 3 (12), e569–e578. [DOI] [PubMed] [Google Scholar]
- 181.In HPTN 076: TMC278 LA Safe, Tolerable and Acceptable for HIV Pre-Exposure Prophylaxis, Conference on Retroviruses and Opportunistic Infections (CROI), Seattle, Washington, Seattle, Washington, 2017. [Google Scholar]
- 182.HPTN 076. https://www.hptn.org/research/studies/hptn076 (accessed August 16, 2018).
- 183.Seckler JM; Barkley MD; Wintrode PL Allosteric Suppression of HIV-1 Reverse Transcriptase Structural Dynamics upon Inhibitor Binding. Biophys. J. 2011, 100 (1), 144–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Delavirdine FDA package. https://www.fda.gov/ (accessed May 6, 2018).
- 185.Hans-Joachim B; David B; Stefanie B; Manfred K; Bernd K; Klaus M; Ulrike OS; Martin S Fluorine in medicinal chemistry. ChemBioChem 2004, 5 (5), 637–643. [DOI] [PubMed] [Google Scholar]
- 186.Meanwell NA Fluorine and fluorinated motifs in the design and application of bioisosteres for drug design. J. Med. Chem. 2018, 61 (14), 5822–5880. [DOI] [PubMed] [Google Scholar]
- 187.Sharaf NG; Ishima R; Gronenborn AM Conformational plasticity of the NNRTI-binding pocket in HIV-1 reverse transcriptase: A fluorine nuclear magnetic resonance study. Biochemistry 2016, 55 (28), 3864–3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Huang B; Kang D; Yang J; Zhan P; Liu X Novel diarylpyrimidines and diaryltriazines as potent HIV-1 NNRTIs with dramatically improved solubility: a patent evaluation of US20140378443A1. Expert Opin Ther Pat 2016, 26 (2), 281–289. [DOI] [PubMed] [Google Scholar]
- 189.Meng Q; Liu N; Huang B; Zhan P; Liu X Novel fluorine-containing DAPY derivatives as potent HIV-1 NNRTIs: a patent evaluation of WO2014072419. Expert Opin Ther Pat 2015, 25 (12), 1477–1486. [DOI] [PubMed] [Google Scholar]
- 190.La Regina G; Coluccia A; Piscitelli F; Bergamini A; Sinistro A; Cavazza A; Maga G; Samuele A; Zanoli S; Novellino E; Artico M; Silvestri R Indolyl aryl sulfones as HIV-1 non-nucleoside reverse transcriptase inhibitors: role of two halogen atoms at the indole ring in developing new analogues with improved antiviral activity. J. Med. Chem. 2007, 50 (20), 5034–5038. [DOI] [PubMed] [Google Scholar]
- 191.Li D; Zhan P; De Clercq E; Liu X Strategies for the design of HIV-1 non-nucleoside reverse transcriptase inhibitors: lessons from the development of seven representative paradigms. J. Med. Chem. 2012, 55 (8), 3595–3613. [DOI] [PubMed] [Google Scholar]
- 192.Zhan P; Pannecouque C; De Clercq E; Liu X Anti-HIV drug discovery and development: Current innovations and future trends. J. Med. Chem. 2016, 59 (7), 2849–2878. [DOI] [PubMed] [Google Scholar]
- 193.Marrazzo JM; Ramjee G; Richardson BA; Gomez K; Mgodi N; Nair G; Palanee T; Nakabiito C; van der Straten A; Noguchi L; Hendrix CW; Dai JY; Ganesh S; Mkhize B; Taljaard M; Parikh UM; Piper J; Masse B; Grossman C; Rooney J; Schwartz JL; Watts H; Marzinke MA; Hillier SL; McGowan IM; Chirenje ZM Tenofovir-based preexposure prophylaxis for HIV infection among African women. N. Engl. J. Med. 2015, 372 (6), 509–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Van Damme L; Corneli A; Ahmed K; Agot K; Lombaard J; Kapiga S; Malahleha M; Owino F; Manongi R; Onyango J; Temu L; Monedi MC; Mak’Oketch P; Makanda M; Reblin I; Makatu SE; Saylor L; Kiernan H; Kirkendale S; Wong C; Grant R; Kashuba A; Nanda K; Mandala J; Fransen K; Deese J; Crucitti T; Mastro TD; Taylor D Preexposure prophylaxis for HIV infection among African women. N. Engl. J. Med. 2012, 367 (5), 411–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Llibre JM; Hung CC; Brinson C; Castelli F; Girard PM; Kahl LP; Blair EA; Angelis K; Wynne B; Vandermeulen K; Underwood M; Smith K; Gartland M; Aboud M Efficacy, safety, and tolerability of dolutegravir-rilpivirine for the maintenance of virological suppression in adults with HIV-1: phase 3, randomised, non-inferiority SWORD-1 and SWORD-2 studies. Lancet 2018, 391 (10123), 839–849. [DOI] [PubMed] [Google Scholar]
- 196.Sluis-Cremer N Future of nonnucleoside reverse transcriptase inhibitors. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (4), 637–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Calmy A; Tarr PE Will NNRTIs be driving forward again? Lancet HIV 2018, 5 (5), e200–e201. [DOI] [PubMed] [Google Scholar]
- 198.Mislak AC; Frey KM; Bollini M; Jorgensen WL; Anderson KS A mechanistic and structural investigation of modified derivatives of the diaryltriazine class of NNRTIs targeting HIV-1 reverse transcriptase. Biochim. Biophys. Acta. 2014, 1840 (7), 2203–2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Bruccoleri A Positional adaptability in the design of mutation-resistant nonnucleoside HIV-1 reverse transcriptase inhibitors: a supramolecular perspective. AIDS Res. Hum. Retroviruses 2013, 29 (1), 4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Zhan P; Liu X; Li Z; Pannecouque C; De Clercq E Design strategies of novel NNRTIs to overcome drug resistance. Curr. Med. Chem. 2009, 16 (29), 3903–3917. [DOI] [PubMed] [Google Scholar]
- 201.Lai M-T; Munshi V; Lu M; Feng M; Hrin-Solt R; McKenna PM; Hazuda DJ; Miller MD Mechanistic Study of Common Non-Nucleoside Reverse Transcriptase Inhibitor-Resistant Mutations with K103N and Y181C Substitutions. Viruses 2016, 8 (10), 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Huo Z; Zhang H; Kang D; Zhou Z; Wu G; Desta S; Zuo X; Wang Z; Jing L; Ding X; Daelemans D; De Clercq E; Pannecouque C; Zhan P; Liu X Discovery of novel diarylpyrimidine derivatives as potent HIV-1 NNRTIs targeting the “NNRTI adjacent” binding site. ACS Med. Chem. Lett. 2018, 9 (4), 334–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Kang D; Ding X; Wu G; Huo Z; Zhou Z; Zhao T; Feng D; Wang Z; Tian Y; Daelemans D; De Clercq E; Pannecouque C; Zhan P; Liu X Discovery of Thiophene[3,2-d]pyrimidine Derivatives as Potent HIV-1 NNRTIs Targeting the Tolerant Region I of NNIBP. ACS Med. Chem. Lett. 2017, 8 (11), 1188–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.La Regina G; Coluccia A; Silvestri R Looking for an active conformation of the future HIV type-1 non-nucleoside reverse transcriptase inhibitors. Antiviral Chem. Chemother. 2010, 20 (6), 213–237. [DOI] [PubMed] [Google Scholar]
- 205.Kang D; Wang Z; Zhang H; Wu G; Zhao T; Zhou Z; Huo Z; Huang B; Feng D; Ding X; Zhang J; Zuo X; Jing L; Luo W; Guma S; Daelemans D; Clercq E; Pannecouque C; Zhan P; Liu X Further exploring solvent-exposed tolerant regions of allosteric binding pocket for novel HIV-1 NNRTIs discovery. ACS Med. Chem. Lett. 2018, 9 (4), 370–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Kudalkar SN; Beloor J; Quijano E; Spasov KA; Lee W-G; Cisneros JA; Saltzman WM; Kumar P; Jorgensen WL; Anderson KS From in silico hit to long-acting late-stage preclinical candidate to combat HIV-1 infection. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (4), E802–E811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Poongavanam V; Corona A; Steinmann C; Scipione L; Grandi N; Pandolfi F; Di Santo R; Costi R; Esposito F; Tramontano E; Kongsted J Structure-guided approach identifies a novel class of HIV-1 ribonuclease H inhibitors: binding mode insights through magnesium complexation and site-directed mutagenesis studies. MedChemComm 2018, 9 (3), 562–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Barreiro G; Guimaraes CR; Tubert-Brohman I; Lyons TM; Tirado-Rives J; Jorgensen WL Search for non-nucleoside inhibitors of HIV-1 reverse transcriptase using chemical similarity, molecular docking, and MM-GB/SA scoring. J. Chem. Inf. Model 2007, 47 (6), 2416–2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.