

SUMMARY
Neuronal migration, axon fasciculation, and axon guidance need to be closely coordinated for neural circuit assembly. Spinal motor neurons (MNs) face unique challenges during development because their cell bodies reside within the central nervous system (CNS) and their axons project to various targets in the body periphery. The molecular mechanisms that contain MN somata within the spinal cord while allowing their axons to exit the CNS and navigate to their final destinations remain incompletely understood. We find that the MN cell surface protein TAG-1 anchors MN cell bodies in the spinal cord to prevent their emigration, mediates motor axon fasciculation during CNS exit, and guides motor axons past dorsal root ganglia. TAG-1 executes these varied functions in MN development independently of one another. Our results identify TAG-1 as a key multifunctional regulator of MN wiring that coordinates neuronal migration, axon fasciculation, and axon guidance.
Graphical Abstract

In Brief
Suter et al. demonstrate that the motor neuron cell surface molecule TAG-1 confines motor neurons to the central nervous system, promotes motor axon fasciculation, and steers motor axons past inappropriate targets. This study highlights how a single cell adhesion molecule coordinates multiple steps in neuronal wiring through partially divergent mechanisms.
INTRODUCTION
Nervous system wiring during development involves a carefully orchestrated sequence of events, including neuronal migration and axon extension. The migrating neuronal cell body is pulled by a leading process (Marín et al., 2010), but, once properly localized, the soma remains in place while the nascent axon projects to its target. Axons are guided by attractive and repulsive cues that activate receptors on their growth cones (Kolodkin and Tessier-Lavigne, 2011); pathfinding is further facilitated by fasciculation, mediated by axon-axon interactions via cell adhesion molecules (CAMs) or channeling via surround repulsion, as late-extending axons can rely on guidance decisions made by pioneer axons (Raper and Mason, 2010; Wang and Marquardt, 2013). Despite their central importance to neural circuit assembly, the cellular and molecular mechanisms that control and cross-coordinate neuronal migration, axon guidance, and axon fasciculation are not completely understood.
Motor neurons (MNs) in the hindbrain and spinal cord face unique challenges during wiring, as their cell bodies reside in the central nervous system (CNS), but their axons project into the body periphery. In the developing spinal cord, postmitotic MNs migrate from their birthplace in the ventricular zone to settle in the ventral horn. Motor axons subsequently leave the neural tube through motor exit points (MEPs) and fasciculate to form segmentally repeated ventral roots (VRs) (Figure 1A) (Bonanomi and Pfaff, 2010). Previous work indicates that MN cell bodies need to be actively prevented from following their axons into the periphery. For instance, ablation of VR-associated boundary cap cells or the repulsive cue Sema6A, which is expressed by these cells, results in MN emigration from the spinal cord (Bron et al., 2007; Mauti et al., 2007; Vermeren et al., 2003). Additionally, radial glia endfeet have been implicated in sealing off MEPs from within, preventing MNs from exiting the neural tube (Lee and Song, 2013). Still, the precise mechanisms that confine MN somata to the CNS while allowing motor axon exit remain largely unexplored.
Figure 1. TAG-1 Is Expressed on Motor Neuron Cell Bodies and Axons during Early Stages of Differentiation.
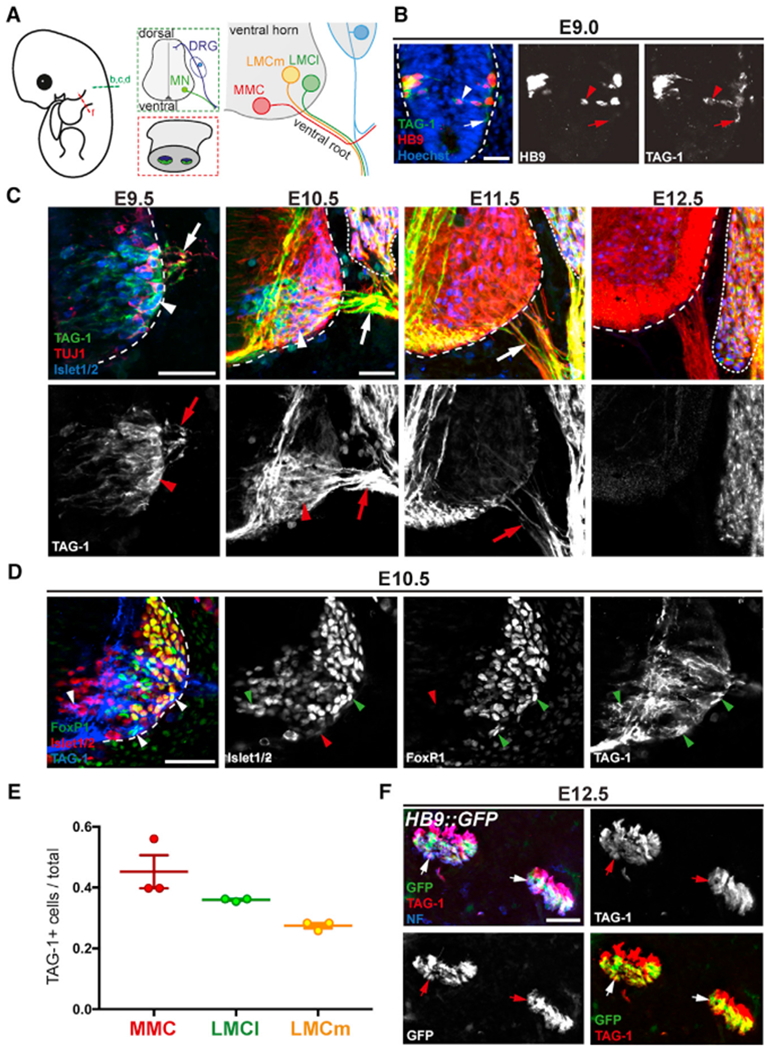
(A) Schematic depicting transverse sections of mouse embryonic spinal cord (green) and limb bud (red), as shown in (B–D) and (F). Right panel shows organization of motor columns in brachial spinal cord ventral horn.
(B and C) Time course of TAG-1 expression in brachial, ventral spinal cord (dashed outlines), visualized using an antibody against TAG-1 and other markers. (B) At E9.0, TAG-1 is expressed on all HB9+ MNs (arrowhead) and their growth cones (arrow). Hoechst stain labels cell nuclei. (C) At E9.5 and 10.5, Islet1/2+ MNs express TAG-1 on their cell bodies (arrowheads). Motor axons in VRs (arrows) express TAG-1 between E9.5 and E11.5. TUJ1 antibody against class III β-tubulin labels all axons, and Islet1/2 immunostaining labels MNs in the spinal cord and sensory neurons in the DRG (dotted line).
(D) E10.5 brachial spinal cord was stained with antibodies against TAG-1, FoxP1, and Islet1/2 to detect TAG-1 in MMC, LMCl, and LMCm neurons (see STAR Methods). TAG-1 is expressed by neurons belonging to all motor columns (arrowheads; in single-channel images, green indicates positive and red indicates negative for that marker).
(E) Quantification of TAG-1+ neurons in MMC, LMCl, and LMCm shows TAG-1 expression across motor columns (n = 3 embryos).
(F) Limb bud transverse sections from E12.5 HB9::GFP embryos were stained with antibodies against TAG-1 and the panaxonal marker neurofilament (NF). TAG-1 is present on NF+/GFP+ motor axons (arrows), as well as sensory axons (NF+ only).
Scale bars: 25 μm in (B); 50 μm in (C), (D), and (F). Error bars indicate SEM.
Adding to the complexity of MN development, MNs are grossly segregated into motor columns, and distinct genetic programs endow these different MN populations with specific guidance receptor profiles, which direct their axons to various peripheral targets via series of successive choice points (Bonanomi and Pfaff, 2010). At brachial level, the medial motor column (MMC) sends axons to epaxial muscles, and the lateral motor column (LMC) innervates the limb; the LMC is further partitioned into lateral (LMCl) and medial (LMCm) subdivisions, which send their axons into the dorsal and ventral halves of the limb bud, respectively (Bonanomi and Pfaff, 2010; Stifani, 2014). MMC and LMC axons initially co-extend in VRs but segregate just ventral to dorsal root ganglia (DRGs), where sensory axons join motor projections and follow them toward peripheral targets (Figure 1A) (Bonanomi, 2019; Stifani, 2014; Wang et al., 2011). While previous work has identified multiple motor column-specific axon guidance mechanisms (Bonanomi and Pfaff, 2010; Stifani, 2014), the underpinnings of the diverse guidance decisions made by differentmotoraxon populations are still not completely understood.
Here, we identified transient axonal glycoprotein type-1 (TAG-1) as an essential regulator of MN containment within the spinal cord, motor axon fasciculation, and LMC axon guidance. TAG-1 is a glycosylphosphatidylinositol-linked CAM of the Contactin family that is transiently expressed by a variety of neuronal populations during neural circuit assembly (Karagogeos et al., 1991; Masuda, 2017; Wolfer et al., 1994; Yamamoto et al., 1986). Previous in vitro experiments and limited genetic analysis in vivo have demonstrated that TAG-1 participates in homophilic interactions and binds multiple other CAMs to regulate axon growth, guidance, and fasciculation (Baeriswyl and Stoeckli, 2008; Furley et al., 1990; Masuda, 2017; Ruegg et al., 1989). Additionally, TAG-1 is an indirect mediator of repulsive Semaphorin signaling in DRG neurons, and positively regulates axonal response to other, as-yet-unidentified, diffusible repellants (Dang et al., 2012; Law et al., 2008; Masuda et al., 2000, 2003). The now 33-year-old discovery that TAG-1 is transiently expressed by MNs during early stages of development suggested that it regulates MN wiring (Yamamoto et al., 1986), but its function in MNs had since remained elusive.
Using a combination of mouse genetics, in vivo phenotype analysis, and in vitro assays, we demonstrate that TAG-1 plays an integral role in multiple aspects of MN development. In particular, TAG-1 expression by MNs anchors their cell bodies in the ventral horn, preventing their emigration from the spinal cord. Additionally, TAG-1 mediates axon-axon interactions essential for motor axon fasciculation within VRs, and it is required in MNs to guide LMC, but not MMC, axons past DRGs. These three functions of TAG-1 are independent from one another and appear to involve partially divergent molecular mechanisms. Our results identify TAG-1 as a key multifunctional regulator of MN wiring that controls both pan-MN and motor column-specific aspects of motor circuit assembly.
RESULTS
TAG-1 Is Transiently Expressed on Migrating Motor Neuron Cell Bodies and Localizes to Distal Motor Axons during Neurite Extension
In mice, TAG-1 was first reported to be expressed by spinal MNs and their axons exclusively at embryonic day 10.0 (E10.0), based on immunohistochemistry (IHC) using a monoclonal TAG-1 antibody (Yamamoto et al., 1986). A new, polyclonal antibody against TAG-1 provides increased sensitivity (Xu et al., 2014), and we found that it strongly labels commissural axons, as well as DRG neurons and their axons; unlike the monoclonal antibody, it also labels spinal cord VRs of E11.5 wild-type, but not TAG-1 mutant mice (Figure S1A), confirming its specificity and improved sensitivity. We used this antibody to reevaluate the time window of TAG-1 expression in developing MNs via IHC on transverse sections of mouse embryonic brachial spinal cord and limb bud (Figure 1A). We found that TAG-1 is first expressed by newly differentiated MNs on their cell bodies and leading processes at E9.0 as they migrate from the ventricular zone to the presumptive ventral horn (Figure 1B). At E9.5, when the first motor axons exit the spinal cord and begin to form VRs, TAG-1 continues to be expressed on all MN somata (Figure 1C). Cell body expression becomes restricted to a subset of MNs at E10.5 and is abolished by E11.5 (Figure 1C). We examined TAG-1 expression on E10.5 MN cell bodies more closely by co-labeling with antibodies against motor column-specific markers and found that subsets of brachial MMC, LMCl, and LMCm neurons are TAG-1-positive (TAG-1+) (Figures 1D and 1E), indicating that gradual loss of TAG-1 is not restricted to any particular motor column(s). Broad expression of TAG-1 across the entire MN population was also observed in cervical, thoracic, and lumbar E10.5 spinal cord, as well as in the hindbrain (Figure S1B; data not shown). When we examined TAG-1 expression on motor axons, we found that TAG-1 is present on all VR axons at E9.5 and E10.5, expressed on a subset at E11.5, and lost from VRs by E12.5 (Figure 1C). However, TAG-1 IHC on limb buds of E12.5 embryos carrying the MN-specific HB9::GFP transgene revealed that distal motor axons still express TAG-1 (Figure 1F). Together, these data demonstrate that TAG-1 is transiently present on MN cell bodies as they migrate and settle in the ventral horn, and that it localizes to the distal segment of motor axons as they exit the neural tube through VRs and extend into the body periphery.
TAG-1 Prevents Motor Neuron Emigration from the Spinal Cord
The early expression of TAG-1 on MN somata is consistent with the idea that it regulates MN positioning. To test this possibility, we examined MN cell bodies in mice lacking TAG-1 (Poliak et al., 2003) by IHC. We found that, at brachial level, a subset (≈3.5%) of MNs fail to be contained within the spinal cord in E11.5 TAG-1−/− embryos; these MNs are instead aberrantly positioned outside of the neural tube within motor axon tracts (Figure 2A). In some cases, MNs have migrated more than 550 μm away from the MEP, e.g., into the limb plexus (data not shown). Ectopic MNs are not observed in TAG-1−/− embryos (Figure 2B), even though TAG-1 protein levels in these heterozygous animals are reduced by about 50% compared to wild type (Figures S1C and S1D). Time course analysis revealed that MNs in TAG-1−/− embryos are normally positioned within the ventral horn at E9.5, have migrated out of the spinal cord at E10.5, and persist in the periphery through at least E13.5 (Figure 2C); however, no ectopic MNs are present at postnatal day 0 (P0), suggesting that these mispositioned neurons are eliminated over time (data not shown). We found that, at brachial level, MNs belonging to all motor columns (MMC, LMCl, and LMCm) emigrate from the TAG-1−/− spinal cord (Figures 2D and 2E). Compared to wild type, the total number of MNs within the brachial TAG-1−/− spinal cord is slightly but not significantly decreased across all motor columns (Figure 2F). When we examined MN positioning in E11.5 wild-type and TAG-1−/− mice at hindbrain, cervical, thoracic, and lumbar levels, we consistently observed a significant increase in ectopic MNs in TAG-1 mutant mice (Figure S2A). In the spinal cord, this defect arises predominately from the emigration of non-MMC MNs; MMC MN emigration is significantly increased at brachial level, appears elevated at cervical and lumbar levels without reaching statistical significance, and does not appear to be a driver of the phenotype at thoracic level (Figure S2A). Overall motor column segregation appears normal in TAG-1 mutant mice, except adjacent to MEPs where MMC and LMC neurons intermingle on their way out of the spinal cord (Figures S2B and S2C). Thus, TAG-1 is required to prevent MNs from exiting the spinal cord, irrespective of motor column identity and without controlling motor column organization.
Figure 2. Motor Neurons Leave the Spinal Cord in TAG-1−/− Mice.
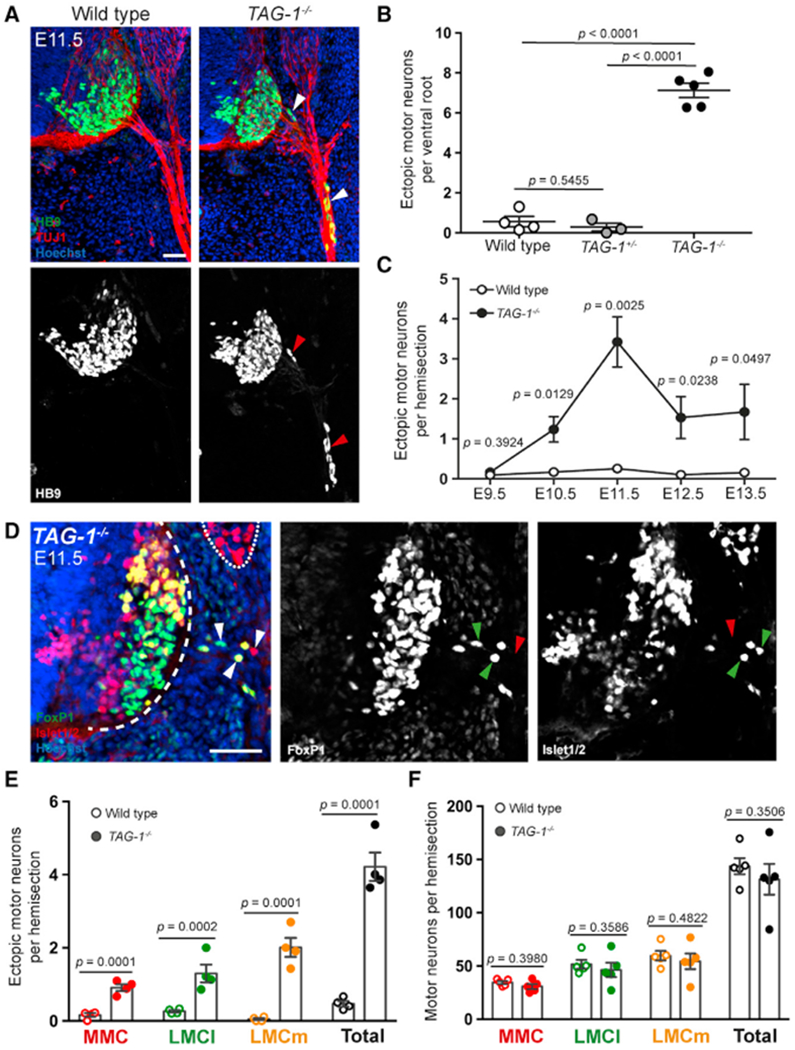
(A) Brachial spinal cord transverse sections of E11.5 wild-type and TAG-1−/− embryos were labeled with antibodies against HB9 and TUJ1, and Hoechst stain. MN cell bodies are found within axon tracts outside of the spinal cord in TAG-1−/− embryos (arrowheads), but not wild-type littermates.
(B) Quantification of ectopic MNs per VR shows significantly increased MN emigration in E11.5 TAG-1−/− embryos compared to wild-type and TAG-1+/− littermates (n = 3–5 embryos per genotype).
(C) Quantification of ectopic MNs from spinal cord hemisections in wild-type and TAG-1−/− embryos over time shows significantly increased MN emigration in TAG-1−/− mice between E10.5 and E13.5 (n = 4–8 embryos per age per genotype).
(D) E11.5 TAG-1−/− spinal cord sections were stained for FoxP1, Islet1/2, and nuclei. MMC, LMCl, and LMCm MNs are present outside of the spinal cord (arrowheads; in single-channel images, green indicates positive and red indicates negative for that marker). The dashed line indicates edge of spinal cord, and the dotted line indicates DRG.
(E and F) Quantification of MNs outside (E) and inside the spinal cord (F) of wild-type and TAG-1−/− embryos shows significant MN emigration in TAG-1−/− mice across motor columns but no significant change of MN numbers in the spinal cord (n = 4–5 embryos per genotype).
Scale bars: 50 μm. Error bars indicate SEM.
TAG-1 Expression by Motor Neurons Anchors Them within the Spinal Cord
Previous work suggests that radial glia endfeet at MEPs prevent MN emigration from the ventral horn (Lee and Song, 2013). Additionally, MEP-associated boundary cap cells are required to contain MNs within the spinal cord (Bron et al., 2007; Mauti et al., 2007; Vermeren et al., 2003). Hence, we addressed the possibility that MNs leave the spinal cord in TAG-1 mutant mice due to defects in the organization of these cell types. Using IHC, we observed normal morphology of radial glia and their endfeet in tight association with MEPs in E11.5 TAG-1 embryos (Figure S2D). We also examined the localization of boundary cap cells via in situ hybridization for Krox20, which labels these cells (Schneider-Maunoury et al., 1993; Wilkinson et al., 1989). We found that boundary cap cells are associated with VRs at MEPs in TAG-1−/−; HB9::GFP embryos, just as in HB9::GFP control littermates (Figures S2E and S2F), and their number per VR does not differ between these two genotypes (HB9::GFP, 18.20 ± 1.934; TAG-1−/−; HB9::GFP, 18.75 ± 0.6439; p = 0.7058; n = 3 embryos per genotype). Collectively, these results indicate that ectopic MN migration in TAG-1−/− mice is not a consequence of defects in radial glia endfeet organization or boundary cap cell clustering at MEPs.
To determine whether TAG-1 is required cell autonomously in MNs to prevent their exit from the spinal cord, we generated a TAG-1 conditional knockout allele (TAG-1cKO) (Figures S3A and S3B). We first validated this allele by deleting TAG-1 in the germline using E2A::Cre mice (Laksoetal., 1996). Mice homozygous for the Cre-deleted version of the conditional allele (TAG-1Δ) exhibited a dramatic reduction or complete loss of TAG-1 protein, as determined by IHC (Figures S3C and S3D) or western blot (Figures S3E and S3F), respectively, indicating that the allele is null or near null upon Cre-mediated deletion. For MN-specific Cre expression, we used Olig2Cre mice (Dessaud et al., 2007). By crossing to a ROSA26LSL-tdTom reporter line that expresses tdTomato in a Cre-dependent manner (Madisen et al., 2010), we confirmed that, as early as E9.5, Olig2Cre drives Cre activity in 100% of MNs (Figures S4A and S4B). We then generated Olig2Cre/+; TAG-1cKO/cKO mice to delete TAG-1 in MNs. Using IHC, we found that TAG-1 protein is lost from MNs and VRs in Olig2Cre/+; TAG-1cKO/cKO embryos, but not TAG-1cKO/cKO littermates, at both E9.5 (Figures S4C and S4D) and E11.5, while commissural axons and DRG neurons/axons retain normal TAG-1 levels (Figures S3G and S3H). Thus, TAG-1 is selectively and efficiently ablated from developing MNs in Olig2Cre/+; TAG-1cKO/cKO mice. When we assessed MN positioning in Olig2Cre/+; TAG-1cKO/cKO; HB9::GFP mice, we observed ectopic MNs outside of the spinal cord, with the severity of this phenotype being indistinguishable from the complete TAG-1 knockout (Figures 3A and 3B; compare to Figure 2B). These data indicate that TAG-1 is specifically required in MNs to prevent their emigration from the neural tube.
Figure 3. TAG-1 in Motor Neurons Prevents Emigration from the Ventral Horn.
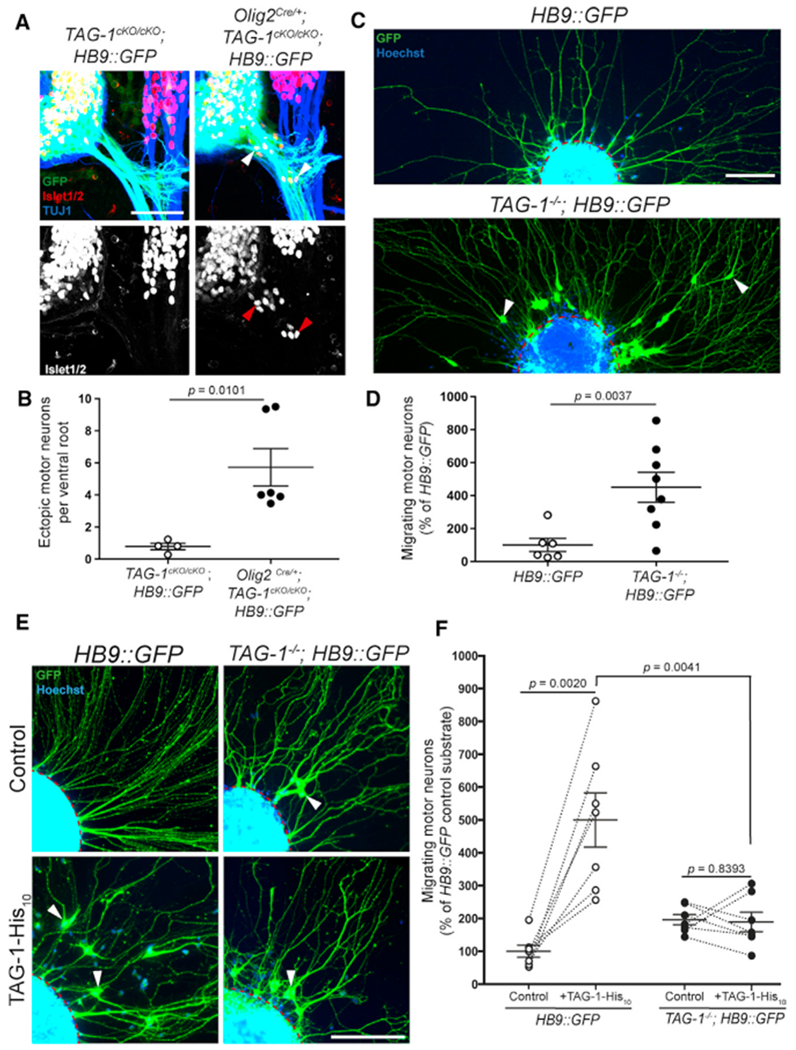
(A) Vibratome spinal cord sections from E11.5 TAG-1cKO/cKO; HB9::GFP and Olig2Cre/+; TAG-1cKO/cKO; HB9::GFP embryos were stained for Islet1/2 and TUJ1. MNs are found within VRs of Olig2Cre/+; TAG-1cKO/cKO; HB9::GFP embryos (arrowheads), but not TAG-1cKO/cKO; HB9::GFP littermates.
(B) Quantification of ectopic MNs per VR shows significantly increased MN emigration in Olig2Cre/+; TAG-1cKO/cKO; HB9::GFP embryos (n = 4–6 embryos per genotype).
(C) Ventral spinal cord explants from E11.5 HB9::GFP and TAG-1−/−; HB9::GFP embryos were cultured and stained for GFP and Hoechst. GFP+ MNs migrate out of TAG-1−/−; HB9::GFP explants (arrowheads). The red dashed lines indicate the edge of explants.
(D) Quantification of MNs outside of explants shows significantly more MNs migrating out of TAG-1−/−; HB9::GFP explants compared to HB9::GFP controls (n = 6–8 embryos per genotype).
(E) Ventral spinal cord explants from E11.5 HB9::GFP and TAG-1−/−; HB9::GFP embryos were cultured on either control substrate or recombinant TAG-1 (TAG-1-His10)-coated substrate and stained for GFP and Hoechst. MNs emigrate from TAG-1−/−; HB9::GFP explants on both substrates, as well as from HB9::GFP explants on TAG-1 (arrowheads). The red dashed lines indicate the edge of explants.
(F) Quantification of MN migration outside of explants shows significantly more MNs migrating out of HB9::GFP explants on TAG-1 substrate compared to control substrate or TAG-1−/−; HB9::GFP explants on TAG-1 (n = 7 embryos per genotype).
Scale bars: 50 μm in (A); 100 μm in (C) and (E). Error bars indicate SEM.
We also deleted TAG-1 using a different Cre-expressing line, Olig2IRES-Cre (Zawadzka et al., 2010), which results in stochastic Cre-mediated recombination in about 40% (39.17 ± 0.33%; n = 3 embryos) of MNs (Figures S4E and S4F), and found that ectopic MN migration does not occur in Olig2IRES-Cre/+; TAG-1cKO/cKO; HB9::GFP embryos (data not shown). Thus, expression of TAG-1 in the majority (≈60%) of MNs can compensate for loss of TAG-1 from individual MNs and prevent their exit from the spinal cord, suggesting that TAG-1 does not act strictly cell autonomously in MN migration.
Boundary cap cell-derived Sema6A has been implicated as a repulsive signal that confines MN cell bodies to the spinal cord via Neuropilin-2 (Nrp2) (Bron et al., 2007). Because TAG-1 can positively regulate Semaphorin signaling (Dang et al., 2012; Law et al., 2008), we investigated the possibility that TAG-1 prevents MN emigration from the spinal cord via the Sema6A-Nrp2 pathway. We analyzed E11.5 Sema6A−/− mice and found that MNs exit the spinal cord at both forelimb and hindlimb level; however, at the brachial level, this phenotype is less severe than in TAG-1 mutants (Figures S5A and S5B; compare to Figure 2B), and it is more pronounced at the lumbar level (Figures S5A and S5C), unlike the TAG-1 knockout (Figure S2A). Furthermore, when we deleted Nrp2 in MNs of Olig2Cre/+; Nrp2cKO/cKO; HB9::GFP mice, we observed no increase in MN emigration compared to littermate controls (Figures S5D–S5F). Together, these results argue against disrupted Sema6A signaling as main driver of MN emigration in mice lacking TAG-1.
Previous work supports the idea that TAG-1 is required for responsiveness to repulsive guidance molecules other than Semaphorins (Law et al., 2008; Masuda et al., 2000, 2003, 2004), leaving open the possibility that MN emigration in TAG-1−/− mice is caused by loss of MN repulsion from other cues in the spinal cord periphery. To test this idea, we studied MN migration in cultures of spinal cord ventral horn explants, which eliminates interactions with tissues outside the neural tube. Cultured E11.5 MNs express TAG-1 on their cell bodies and axons (Figure S6A), allowing investigation of TAG-1 function in vitro. When we cultured ventral spinal cord explants from E11.5 HB9::GFP and TAG-1−/−; HB9::GFP embryos, we found that an increased number of MN cell bodies migrate out of explants from mice lacking TAG-1 compared to littermate control explants (Figures 3C, 3D, and S6B). This demonstrates that TAG-1 prevents MN emigration from the ventral horn through spinal cord-intrinsic mechanisms, not via interactions with the spinal cord periphery.
To determine whether TAG-1 activates a signal that restricts MN migration or instead anchors MNs in the spinal cord by mediating adhesion, we cultured ventral spinal cord explants on a substrate containing recombinant TAG-1. We found that MN emigration from HB9::GFP explants is drastically increased on TAG-1 compared to control substrate (Figures 3E and 3F), indicating that ectopic TAG-1 can compete with endogenous TAG-1 as an adhesive substrate for MNs. In contrast, MNs from TAG-1−/−; HB9::GFP explants do not exhibit increased migration on a TAG-1 substrate, and their rate of migration is below that of HB9::GFP MNs cultured on TAG-1 (Figures 3E and 3F). This result demonstrates that MN-derived TAG-1 is required for the pro-migratory effect of ectopic TAG-1. Together, these findings support the idea that TAG-1 expression on MNs anchors them within the spinal cord by mediating homophilic adhesion.
TAG-1 Controls Ventral Root Thickness
Expression of TAG-1 in spinal cord VRs suggests that it controls motor axon behavior during exit from the neural tube. To test this idea, we examined VRs in E11.5 TAG-1−/−; HB9::GFP embryos. We found that VRs are expanded in mice lacking TAG-1 (Figure 4A), resulting in a nearly 50% increase in root diameter compared to TAG-1 heterozygous and wild-type littermates (Figure 4B). Time course analysis of this phenotype in whole, optically cleared embryos (Figure S7A) revealed that VR diameter in TAG-1−/− embryos is already increased at E9.5 and remains elevated through E12.5; at E13.5, wild-type roots reach the same size as in TAG-1 knockout mice (Figure 4C). VRs are also expanded in the rostro-caudal dimension and frequently fail to completely separate from one another in E11.5 TAG-1−/− embryos; the magnitude of this effect is similarly diminished by E13.5 (Figures S7B and S7C). We observed no correlation between VR diameter and the number of ectopic MNs per root in TAG-1−/− mice (Figure 4D). Combined with the fact that VR expansion in TAG-1 mutants is detectable a full day before presence of ectopic MNs, this argues against root expansion being a secondary consequence of MN emigration. Thus, TAG-1 negatively regulates VR size, independent from its function in containing MN cell bodies within the spinal cord.
Figure 4. Ventral Roots Are Expanded in TAG-1−/− Mice.

(A) Brachial spinal cord transverse sections from E11.5 HB9::GFP and TAG-1−/−; HB9::GFP embryos. GFP+ VR diameter (lines) is increased in TAG-1−/−; HB9::GFP mice.
(B) Quantification of VR diameter shows significant expansion in E11.5 TAG-1−/−; HB9::GFP embryos compared to HB9::GFP and TAG-1+/−; HB9::GFP littermates (n = 3–6 embryos per genotype).
(C) Time course analysis of VR diameter in wild-type and TAG-1−/− embryos using iDISCO visualization of NF (see STAR Methods) shows significant root expansion between E9.5 and E12.5 (n = 4–5 embryos per genotype per age).
(D) Correlation analysis of ectopic MN number per VR and root diameter in E11.5 TAG-1−/−; HB9::GFP embryos (analyzed in Figures 2B and 4B) reveals no significant relationship between these parameters (n = 74 VRs from 5 embryos).
Scale bar: 50 μm. Error bars indicate SEM.
Motor Neuron-Derived TAG-1 Promotes Motor Axon Fasciculation Independent of Interactions with Peripheral Tissues
TAG-1 can promote axon fasciculation in multiple neuronal populations (Buttiglione et al., 1998; Masuda et al., 2000; Ruegg et al., 1989; Stoeckli and Landmesser, 1995; Stoeckli et al., 1991). However, we first considered the possibility that VR expansion in TAG-1−/− mice does not result from motor axon defasciculation and tested two alternative hypotheses—that an increased number of Schwann cells or presence of additional axons within VRs cause the observed phenotype. Using IHC for the neural crest marker Sox10, we found comparable numbers of Schwann cell precursors in VRs of E11.5 wild-type and TAG-1−/− embryos (Figures S8A and S8B). MNs are generated in equal numbers in wild-type and TAG-1−/− embryos (Figures S8C, S8D, and 2F), arguing against the presence of additional motor axons in TAG-1−/− VRs. To determine whether neuronal populations other than MNs aberrantly project axons into TAG-1−/− VRs, we conducted DiI tracing experiments (Figure S8E). Anterograde tracing from dorsal spinal cord labeled contralaterally and ipsilaterally projecting axons and showed that neither of these axonal populations project into VRs (Figure S8F) of TAG-1−/− and wild-type embryos. Conversely, retrograde tracing from VRs exclusively labeled neurons in the portion of the ventral horn occupied by MNs (Figure S8G). Taken together, these data suggest that neither excess Schwann cells nor extraneous axons cause VR expansion in mice lacking TAG-1; rather, they support the idea that the phenotype reflects a requirement for TAG-1 in motor axon fasciculation.
To determine whether TAG-1 is required in MNs to control VR size, we analyzed E11.5 Olig2Cre/+; TAG-1cKO/cKO; HB9::GFP embryos. We found that root diameter is increased by about 50% (Figures 5A and 5B), and rostro-caudal VR separation is impaired (Figure S7D), similar to the complete TAG-1 knockout. Stochastic deletion of TAG-1 using Olig2IRES-Cre causes a slight (≈13%) increase in VR diameter (at E11.5, TAG-1cKO/cKO; HB9::GFP, 26.36 ± 0.95 μm; Olig2IRES-Cre/+; TAG-1cKO/cKO; HB9::GFP, 30.11 ± 0.77 μm; p = 0.022; n = 4 embryos per genotype). These data indicate that TAG-1 acts cell-autonomously in MNs to regulate VR size.
Figure 5. TAG-1 in Motor Neurons Promotes Axonal Fasciculation.

(A) Transverse sections of E11.5 TAG-1cKO/cKO; HB9::GFP and Olig2Cre/+; TAG-1cKO/cKO; HB9::GFP brachial spinal cord. GFP+ VR diameter (lines) is increased in Olig2Cre/+; TAG-1cKO/cKO; HB9::GFP mice.
(B) Quantification of VR diameter shows significant expansion in Olig2Cre/+; TAG-1cKO/cKO; HB9::GFP embryos compared to TAG-1cKO/cKO; HB9::GFP (n = 4–6 embryos per genotype).
(C) Ventral spinal cord explants from E11.5 HB9::GFP and TAG-1−/−; HB9::GFP embryos were cultured and labeled with GFP antibody and Hoechst stain. The dashed box indicates area of magnified inset. GFP+ motor axons from HB9::GFP explants form thicker fascicles (red arrowhead) than axons from TAG-1−/−; HB9::GFP explants. The white arrowheads indicate precociously migrating MNs, and the red dashed lines indicate the edge of the explants.
(D) Quantification of motor axon bundle diameter shows significantly increased numbers of small motor axon fascicles and a decrease of larger motor axon fascicles in TAG-1−/−; HB9::GFP explants compared to controls (n = 7–8 experiments per genotype).
(E) Ventral spinal cord explants from E11.5 HB9::GFP embryos were cultured on either control substrate or recombinant TAG-1(TAG-1-His10)-coated substrate and labeled with GFP antibody and Hoechst stain. The dashed box indicates area of magnified inset. Motor axons form more thick fascicles (red arrowhead) on control substrate than on TAG-1. The white arrowheads indicate precociously migrating MNs, and the red dashed lines indicate the edge of the explants.
(F) Quantification of motor axon bundle diameter from HB9::GFP explants shows a shift from midrange diameter (2.5–5 μm) bundles to smaller (0–2.5 μm) bundles when cultured on a TAG-1-coated substrate versus control substrate (n = 7 embryos per genotype).
Scale bars: 50 μm in (A); 100 μm in (C) and (E); 10 μm in (C) and (E) insets. Error bars indicate SEM.
To investigate whether TAG-1 controls motor axon fasciculation, we examined axon growth from ventral spinal cord explants in vitro. We found that motor axons emerge from E11.5 HB9::GFP explants as a mix of individual axons and fascicles of various sizes, while TAG-1−/−; HB9::GFP motor axons form fewer thick bundles and remain less fasciculated (Figures 5C and 5D). Thus, TAG-1 expression by MNs promotes motor axon fasciculation, and this function does not require interactions with tissues outside of the spinal cord.
To distinguish between a signaling and an adhesion mechanism for TAG-1’s function in motor axon fasciculation, we cultured ventral spinal cord explants on a TAG-1 substrate. We found that fasciculation of axons from HB9::GFP explants is slightly impaired on a TAG-1-coated surface compared to control (Figures 5E and 5F), indicating that ectopic TAG-1 competes with axonal TAG-1 as the preferred substrate for motor axon growth. Substrate-bound TAG-1 did not affect axon fasciculation from TAG-1−/−; HB9::GFP explants (data not shown), indicating that endogenous TAG-1 on motor axons is required for the adhesive effect of ectopic TAG-1. Together, these results suggest that TAG-1 homophilic binding mediates axon-axon adhesion to promote motor axon fasciculation.
TAG-1 Prevents LMC Motor Axons from Invading DRGs
TAG-1 is expressed on motor axons as they navigate to their targets in the periphery, raising the possibility that it controls motor axon guidance. To test this idea, we examined motor axon projections in TAG-1 mutant mice. We did not observe motor axon tract expansion distal to VRs (data not shown). However, in E11.5 TAG-1−/−; HB9::GFP embryos, some motoraxons deviate from their normal trajectory at the point where VRs coalesce with sensory axons and instead misproject into DRGs; in most instances, these axons reenter the spinal cord at the dorsal root entry zone (DREZ) after traversing the entire DRG (Figure 6A). Motor axons do not enter DRGs in HB9::GFP littermates, but the DRG invasion phenotype is present in TAG-1 heterozygous mutant animals, with a severity of about 50% relative to homozygous knockouts (Figures 6A and 6B), indicating that this defect is sensitive to gene dosage. Analysis of whole, optically cleared E12.5 TAG-1−/−; HB9::GFP embryos revealed motor axon misprojections at all rostro-caudal levels except the most anterior cervical spinal cord (data not shown); after reentering the neural tube, motor axons join the dorsal funiculus and extend longitudinally over multiple spinal cord segments (Figure 6C). DRG invasion by motor axons in TAG-1−/−; HB9::GFP embryos is not observed before E11.5 but persists through at least E13.5; even at birth, a significant number of ectopic motor axon projections remain within DRGs (Figures S9A–S9D). The delayed onset, increased duration, exclusion from the most rostral spinal cord, and gene dosage sensitivity of this TAG-1 mutant motor axon guidance defect distinguish it from the MN emigration and VR expansion phenotypes. Together, these data indicate that TAG-1 guides motor axons past DRGs, independent from its functions in MN cell body anchoring and axon fasciculation.
Figure 6. LMC Motor Axons Misproject through DRGs in TAG-1−/− Mice.
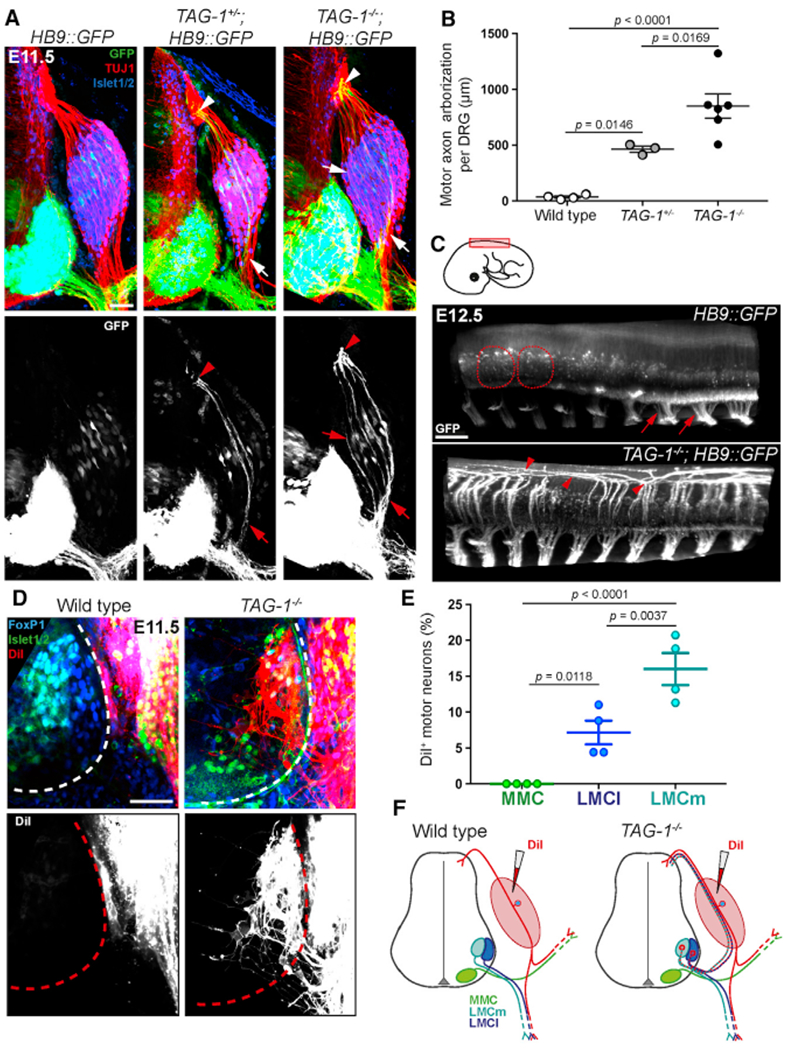
(A) E11.5 brachial spinal cord transverse sections from HB9::GFP, TAG-1+/−; HB9::GFP and TAG-1−/−; HB9::GFP embryos were stained for TUJ1 and Islet1/2. GFP+ motor axons in TAG-1+/−; HB9::GFP and TAG-1−/−; HB9::GFP mice invade the DRG (arrows) and re-enter the spinal cord at the DREZ (arrowheads).
(B) Quantification of GFP+ motor axon arborization within DRGs shows significant DRG invasion in TAG-1+/−; HB9::GFP and TAG-1−/−; HB9::GFP embryos, but not HB9::GFP littermates (n = 3–6 embryos per genotype).
(C) Lateral views of iDISCO-cleared E12.5 HB9::GFP and TAG-1−/−; HB9::GFP embryos at brachial and thoracic level (schematic shows location and orientation; cropped to one-half of spinal cord) show GFP+ motor axons projecting through DRGs and re-entering the spinal cord along the rostro-caudal axis in TAG-1−/−; HB9::GFP mice. The dashed outlines indicate DRGs, and the arrows point to VRs. The arrowheads in TAG-1−/−; HB9::GFP panel highlight motor axons that project longitudinally in the dorsal spinal cord after re-entering through the DREZ.
(D) Spinal cord transverse sections taken after DiI injections into DRGs (schematized in F) of E11.5 wild-type and TAG-1−/− embryos were labeled with antibodies against FoxP1 and Islet1/2. Retrogradely traced neurons are found in the TAG-1−/−, but not wild-type, spinal cord ventral horn (dashed outline) and are FoxP1+.
(E) Quantification of DiI+ neurons in different motor columns of TAG-1−/− embryos shows that only LMC (both LMCl and LMCm) neurons misproject axons into DRGs (n = 4 embryos).
(F) Schematic showing DiI injection strategy for retrograde tracing of misprojecting MNs and the observed labeling in wild-type and TAG-1−/− embryos.
Scale bars: 50 μm in (A) and (D); 200 μm in (C). Error bars indicate SEM.
Next, we sought to elucidate whether axonal misprojections into DRGs originate from MNs across different motor column identities. We injected E11.5 brachial-level DRGs with DiI and determined the motor column identity of retrogradely traced MNs in the ventral horn by IHC (Figure 6F). As expected, no DiI-labeled MNs were observed in wild-type embryos (Figure 6D). In TAG-1−/− embryos, we found that retrogradely traced MNs are exclusively of LMC (LMCl and LMCm) identity (Figures 6D–6F), accounting for about 12% of all MNs in this motor column. When we examined motor innervation of limb buds in E12.5 TAG-1−/−; HB9::GFP embryos and HB9::GFP littermates, we observed thinned motor axon projections in mice lacking TAG-1 (Figure S9E), consistent with a partial failure of LMC axons to reach the limb. Thus, TAG-1 is specifically required for LMC axon guidance past DRGs and into the target field.
TAG-1 Functions in Motor Neurons to Guide Motor Axons Past DRGs
To determine whether TAG-1 acts cell autonomously to prevent motor axons from misprojecting into DRGs, we deleted TAG-1 specifically in MNs. We found that, in E11.5 Olig2Cre/+; TAG-1cKO/cKO; HB9::GFP mice but not TAG-1cKO/cKO; HB9::GFP littermates, motor axons invade DRGs (Figures 7A and 7C), indicating that MN-derived TAG-1 is required for axon guidance. We also performed TAG-1 deletion in a subset of MNs using the Olig2IRES-Cre line and marked the targeted neurons and their axons via the ROSA26LSL-tdTom allele, which is a more sensitive label for individual motor axons than the HB9::GFP transgene (Figure 7B). We observed GFP+ motor axon invasion of DRGs in Olig2IRES-Cre/+; TAG-1cKO/cKO; ROSA26LSL-tdTom/+; HB9::GFP embryos (Figures 7B and 7C), demonstrating that stochastic deletion of TAG-1 is sufficient to elicit the axon guidance defect. While most of the misprojecting GFP+ motor axons are also tdTomato+, a small fraction appears negative for tdTomato (Figures 7D and 7E), supporting the idea that the TAG-1 mutant DRG invasion phenotype is mostly cell autonomous but that misrouted TAG-1-deficient motor axons can occasionally pull wild-type axons into the DRG with them. In both conditional knockout lines, the extent of DRG invasion by motor axons is lower than in the complete knockout and more closely resembles the TAG-1 heterozygous null (Figure 7C; compare to Figure 6B). Together, these results indicate that TAG-1 in MNs helps guide motor axons past DRGs, but they also suggest that sources of TAG-1 other than MNs, such as sensory neurons, might contribute to this function.
Figure 7. TAG-1 Is Required in Motor Neurons to Guide Their Axons Past DRGs.

(A) E11.5 TAG-1cKO/cKO; HB9::GFP and Olig2Cre/+; TAG-1cKO/cKO; HB9::GFP brachial spinal cord transverse sections were stained for TUJ1 and Islet1/2. GFP+ motor axons invade DRGs (arrow) in Olig2Cre/+; TAG-1cKO/cKO; HB9::GFP mice and re-enter the spinal cord at the DREZ (arrowhead).
(B) Spinal cord sections from E11.5 Olig2IRES-Cre/+; TAG-1cKO/cKO; ROSA26LSL-tdTom/+; HB9::GFP embryos were stained with antibodies against Islet1/2 and RFP to amplify tdTomato (tdTom) signal. TdTom+/GFP+ motor axons invade the DRG (arrows; in single-channel images, green indicates positive, and red indicates negative for that marker).
(C) Quantification of GFP+ motor axon arborization within DRGs shows significant DRG invasion in Olig2Cre/+; TAG-1cKO/cKO; HB9::GFP and Olig2IRES-Cre/+; TAG-1cKO/cKO; ROSA26LSL-tdTom/+; HB9::GFP embryos but not TAG-1cKO/cKO; HB9::GFP mice (n = 4–6 embryos per genotype). The dashed line indicates that Olig2IRES-Cre/+; TAG-1cKO/cKO; ROSA26LSL-tdTom/+; HB9::GFP embryos were not littermates of the other two genotypes.
(D) High-magnification view of GFP+ motor axons in DRGs of Olig2IRES-Cre/+; TAG-1cKO/cKO; ROSA26LSL-tdTom/+; HB9::GFP embryos. TdTom−/GFP+ motor axons are found defasciculating from tdTom+/GFP+ axon bundles (arrowheads; in single-channel images, green indicates positive and red indicates negative for that marker).
(E) Quantification of motor axon arborization within DRGs of Olig2IRES-Cre/+; TAG-1cKO/cKO; ROSA26LSL-tdTom/+; HB9::GFP embryos, broken down by GFP and tdTom labeling, shows that a minor subset of GFP+ motor axons are tdTom− (n = 4 embryos).
Scale bars: 50 μm in (A) and (B); 10 μm in (D). Error bars indicate SEM.
DISCUSSION
During development, MNs project axons to peripheral targets while their cell bodies remain in the CNS. We demonstrate that MN-expressed TAG-1 anchors MN somata in the spinal cord to prevent their emigration, mediates motor axon fasciculation during CNS exit, and helps guide LMC axons to their target field. We provide evidence that these multiple functions of TAG-1 during different stages of spinal MN wiring are independent from each other. These results identify TAG-1 as a multifunctional regulator of MN development that coordinates neuronal migration, axon fasciculation, and axon guidance, likely through at least partially separate molecular mechanisms.
TAG-1 Anchors Motor Neuron Cell Bodies in the Spinal Cord
To be in position for receiving appropriate presynaptic inputs, MNs must be prevented from following their axons into the periphery. Sema6A-Nrp2 and Netrin5-DCC signaling from boundary cap cells to MNs have been implicated as important contributors to this process (Bron et al., 2007; Garrett et al., 2016; Mauti et al., 2007; Vermeren et al., 2003). Moreover, radial glia endfeet have been proposed to form a barrier for migrating MNs at MEPs, as Reelin mutant mice exhibit disorganization of spinal cord radial glia and MN mispositioning outside the neural tube (Lee and Song, 2013). With few, incompletely understood exceptions (Arber et al., 1999; Kim et al., 2017; Lee et al., 2009; Thaler et al., 2004), additional mechanisms that help contain MNs within the spinal cord have remained elusive.
We find that TAG-1 is expressed on and required in migrating MNs, across motor column identities, to prevent their exit from the spinal cord in vivo. MN emigration in mice lacking TAG-1 is more severe than in Sema6A, Nrp2, and Netrin5 mutant mice, indicating that TAG-1 does not control MN positioning solely by mediating either Sema6A-Nrp2 or Netrin5-DCC signaling. Furthermore, the TAG-1 mutant phenotype is also stronger than what is observed in mice with disorganized radial glia end-feet, and detailed phenotype analysis of TAG-1 knockout mice demonstrates that TAG-1 does not prevent MN emigration by regulating radial glia or boundary cap cell organization at MEPs. In vitro analysis shows that TAG-1 anchors MNs in the spinal cord independently from any interactions with peripheral tissues and indicates that TAG-1 on MN somata tethers MNs in the ventral horn through homophilic adhesion, consistent with TAG-1’s ability to engage in homo- and heterophilic adhesive interactions (Felsenfeld et al., 1994; Rader et al., 1993). Sparse TAG-1 deletion in about 40% of MNs shows that TAG-1 does not act in a strictly cell-autonomous manner and suggests that TAG-1-mediated adhesion between the majority of MNs can prevent spinal cord exit of individual TAG-1-deficient MNs, possibly via other CAMs or by physically trapping these MNs. Combined with previous findings, our results highlight the multiplicity of both spinal cord-intrinsic and -extrinsic mechanisms that collaborate in containing MNs within the spinal cord (Suter and Jaworski, 2019).
Previously, TAG-1 has been implicated in facilitating facial MN migration in the zebrafish hindbrain (Gurung et al., 2018). This contrasts with our finding that TAG-1 restricts, rather than promotes, migration and aberrant spinal cord exit of MNs in the mouse hindbrain. How TAG-1 might act as an either pro- or anti-migratory factor in different contexts or organisms remains to be determined.
TAG-1 Mediates Axon-Axon Interactions for Motor Axon Fasciculation
Motor axons are tightly fasciculated en route to their target fields. Various mechanisms promote motor axon bundling during different stages of growth, including surround repulsion by Semaphores, autocrine Slit-Robo signaling, and ALCAM-mediated axon-axon adhesion (Huber et al., 2005; Huettl et al., 2011; Jaworski and Tessier-Lavigne, 2012; Moret et al., 2007; Weiner et al., 2004). Additional molecules are likely to contribute to motor axon fasciculation.
Nascent motor axons express TAG-1, and we find that global or MN-specific TAG-1 deletion causes disorganization and expansion of VRs, but not of more distal motor axon tracts. Detailed TAG-1−/− phenotype analysis provides multiple lines of evidence that this defect is not a consequence of additional cells or axons in VRs. In vitro results directly demonstrate TAG-1-dependent motor axon fasciculation without signaling from tissues in the spinal cord periphery and indicate that TAG-1 homophilic binding mediates axon-axon adhesion. Collectively, these findings support the idea that TAG-1 on motor axons controls VR size by promoting motor axon interactions and fasciculation, consistent with TAG-1’s function in mediating fasciculation in multiple other neuronal populations (Buttiglione et al., 1998; Kunz et al., 1998; Masuda et al., 2000; Ruegg et al., 1989; Stoeckli and Landmesser, 1995; Stoeckli et al., 1991). It remains to be determined why TAG-1-dependent fasciculation is specifically required during early motor axon growth and VR formation, but not at later stages.
TAG-1 Guides LMC Axons Past DRGs
To reach their final destinations, motor axons avoid inappropriate targets along the way. Multiple studies indicate that motor axons need to be actively prevented from projecting into DRGs after exiting the spinal cord, as subsets of motor axons invade DRGs in EphA3/4 double knockouts, EfnB1−/− mice, CNS-specific FGFR1 knockouts, and Robo1/2 double mutants (Gallarda et al., 2008; Kim et al., 2019; Luxey et al., 2013; Shirasaki et al., 2006). Additionally, overexpression of Sema3A in MNs causes motor axon misprojection into DRGs (Moret et al., 2007). However, the precise mechanisms that promote or prevent motor axon entry into DRGs have remained elusive.
We find that motor axons express TAG-1 as they navigate through the periphery and that, in mice lacking TAG-1, they misproject through DRGs and reenter the spinal cord at the DREZ. The developmental timing, MN subtype specificity, and TAG-1 protein level sensitivity of this phenotype indicate that it is not a secondary consequence of impaired MN anchoring or motor axon fasciculation. At brachial level, only LMC, but not MMC, axons enter DRGs in TAG-1 mutant mice, demonstrating that TAG-1 specifically guides LMC axons past DRGs. However, presence of the motor axon projection defect along most of the rostro-caudal axis suggests that TAG-1 guides additional, likely non-MMC motor axons at other spinal cord levels. Because EphA and FGFR1 signaling prevent MMC, not LMC, axons from entering DRGs (Gallarda et al., 2008; Shirasaki et al., 2006), our results indicate that TAG-1 is not a mediator of these pathways in motor axon guidance. TAG-1 can facilitate Semaphorin signaling, and previous work implicates class III Semaphorins in repelling motor axons from DRGs (Moret et al., 2007), but motor axons do not invade DRGs in mice lacking the Semaphorin receptors Nrp1, Nrp2, PlexinA3, or PlexinA4 (this study; unpublished data). This argues against a requirement for Semaphorin repulsion in guiding motor axons past DRGs. TAG-1 also promotes axonal responses to other, as-yet-unidentified, repellants (Law et al., 2008; Masuda et al., 2000, 2003, 2004), leaving open the possibility that TAG-1-dependent repulsion from these cues prevents motor axons from entering DRGs. Analysis of Robo1/2 mutants revealed sporadic motor axon misprojections into DRGs with much lower phenotype severity compared to TAG-1 knockouts (unpublished data), indicating that TAG-1 does not guide motor axons by mediating Slit repulsion. The precise molecular mechanism for TAG-1-dependent motor axon guidance remains elusive, but MN-specific deletion of TAG-1 demonstrates that TAG-1 expression on motor axons is important for this function. Our results underscore the necessity of multiple signals that prevent motor axon entry into DRGs and identify a LMC-specific mechanism. Differences in the timing of axon extension or the existence of redundant mechanisms in MMCs (such as EphA-mediated repulsion) might explain why LMC axons require TAG-1 to avoid DRGs while MMC axons do not, despite both populations expressing TAG-1. Because MN-specific TAG-1 deletion does not fully reproduce the TAG-1−/− axon guidance phenotype, additional sources of TAG-1 likely contribute to its guidance function. DRG sensory neurons express high levels of TAG-1 and are therefore a strong candidate, but further investigation is needed to determine whether, and how, MN- and DRG-derived TAG-1 collaborate to prevent motor axons from entering DRGs.
TAG-1 Multifunctionality Coordinates Sequential Steps in Neuronal Wiring
Our results show that TAG-1 mediates MN cell body anchoring, motor axon fasciculation, and motor axon guidance, and we provide strong evidence that these three functions of TAG-1 are not interdependent. Of note, both MN soma anchoring and motor axon bundling appear to rely on homophilic adhesion via TAG-1, yet the severity of axon defasciculation after loss of TAG-1 exceeds that of cell body overmigration. The lack of correlation between these two phenotypes underscores their independence from one another, but it also raises the question which factors render individual MNs vulnerable to either of the defects. Partially redundant adhesive interactions between MN cell bodies, mediated by CAMs other than TAG-1, might well explain why only a subset of MNs is susceptible to spinal cord exit in TAG-1−/− mice. Furthermore, our time course analysis of TAG-1 expression in MNs shows that TAG-1 is first present on cell bodies and gradually redistributes to nascent axons. As axons continue to extend, TAG-1 localizes exclusively to their distal segments, consistent with the previous in vitro observation of selective TAG-1 delivery to growth cones (Vogt et al., 1996). This dynamic, sequential targeting to neuronal somata and axons might allow TAG-1 to independently mediate its multiple functions during sequential stages of MN wiring by engaging distinct molecular pathways or by acting in different cellular compartments.
Motor Neuron Wiring Defects after Loss of TAG-1 Are Partially Corrected during Later Development
Despite their defects in MN migration and wiring, mice lacking TAG-1 are postnatally viable (Fukamauchi et al., 2001; Poliak et al., 2003), although they have been reported to exhibit gait abnormalities and impaired performance on a rotarod (Savvaki et al., 2008, 2010). This suggests that motor circuitry in adult TAG-1 mice is largely normal, with limited functional consequences. Our results show that the early VR expansion phenotype in TAG-1 mutants disappears by E13.5, since VR diameter in wild-type mice gradually increases to match the knockout. It is possible that, during this E9.5-E13.5 time period, newly emerging axons in TAG-1 mice project through gaps between less fasciculated, earlier projecting axons, and ventral root diameter therefore remains essentially constant. Compensatory upregulation of other CAMs that can drive motor axon fasciculation is an alternative explanation for recovery of the mutant phenotype. As for the TAG-1−/− MN emigration and motor axon guidance defects, ectopic MNs are no longer detected at birth, and the DRG invasion phenotype is strongly diminished, yet not fully abolished by P0. This suggests that mispositioned MNs undergo apoptosis and misprojecting motor axons are largely pruned over time, likely due to lack of trophic support. Collectively, these results highlight the plasticity of developing motor circuits and their ability to correct defects in early MN wiring. This plasticity is at least partly afforded by the overproduction of MNs during development and the subsequent removal of neurons that fail to successfully integrate into functional circuits.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Alexander Jaworski (alexander_jaworski@brown.edu). All unique reagents, including mouse lines, generated in this study are available from the lead contact.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All experimental procedures had institutional approval through Brown University’s Institutional Animal Care and Use Committee and followed the guidelines provided by the National Institutes of Health. Mouse lines were maintained in a CD-1 (TAG-1−/−; HB9::GFP) or mixed CD-1 / C56bl6 background (all other lines). For timed pregnancies, the day of vaginal plug was defined as E0.5, and littermate embryos of either sex were used for all experiments. Spinal cord sizes did not differ significantly between littermates of different genotypes (data not shown), indicating comparable overall development.
Mouse strains used were: TAG-1cKO, TAG-1−/− (MGI: 2677610, official nomenclature Cntn2tm1Furl) (Poliak et al., 2003), E2A::Cre (MGI: 2137691, official nomenclature Tg(EIIa-cre)C5379Lmgd) (Lakso et al., 1996), Olig2Cre/+(MGI: 3774124, official nomenclature Olig2tm1(cre)Tmj) (Dessaud et al., 2007), Olig2IRES-Cre/+ (MGI: 4461156, official nomenclature Olig2tm1.1(cre)Wdr) (Zawadzka et al., 2010), HB9::GFP (MGI: 3056906, official nomenclature Tg(Hlxb9-GFP)1Tmj) (Wichterle et al., 2002), ROSA26LSL-tdTom/+ (MGI: 3809523, official nomenclature Gt(ROSA)26Sortm9(CAG-tdTomato)Hze)(Madisen etal., 2010), Sema6A(MGI: 2156202, official nomenclature Sema6aGt(KST069)Byg) (Leighton et al., 2001), Nrp2cKO/cKO (MGI: 3712029, official nomenclature Nrp2tm1.1Mom) (Walz et al., 2002), Nrp1cKO/cKO(MGI: 3512101, official nomenclature Nrp1tm2Ddg) (Gu et al., 2003), PlexinA3−/− (MGI: 2386961, official nomenclature Plxna3tm1Matl) (Cheng et al., 2001), PlexinA4−/− (MGI: 3579185, official nomenclature Plxna4tm1Matl) (Yaron et al., 2005), and FLPeR (MGI: 2429412, official nomenclature Gt(ROSA)26Sortm1(FLP1)Dym) (Farley et al., 2000). These mouse lines were genotyped as previously reported.
Cell lines
CV-1 in Origin with SV40 genes (COS-7) cell lines (ATCC, Catalog #CRL-1651) were grown as previously described (Jaworski and Tessier-Lavigne, 2012) at 37°C with 5% CO2.
In vitro primary cell cultures
Primary motor neurons from E11.5 embryos of both sexes were dissected in L-15 media (GIBCO) with 5% horse serum. Neurons were placed in 8-chamber glass slides, which had been pre-coated with 1 mg/ml poly-D-Lysine (PDL; Sigma, #P6407) in water and 50 ng/ml laminin (Millipore, #CC095) in PBS (control conditions). For TAG-1 substrate conditions, 50 mg/ml recombinant TAG-1 protein (TAG-1-His10; R&D Systems, #1714-CN) was added to 50 ng/ml laminin with PBS and incubated overnight at 4°C prior to explant dissection. Neurons were cultured for 40–44 h in MN growth medium (10 ng/ml BDNF (Cell Science, #CRB600B), 50 ng/mL GDNF (Sigma, #SRP3200), 2% B-27, penicillin/streptomycin/glutamine (both Invitrogen), 0.5% glucose, and 0.5% methylcellulose in Neurobasal medium) at 37°C with 5% CO2.
METHOD DETAILS
Generation of TAG-1cKO mouse
The TAG-1cKO allele was generated using a targeting construct that was assembled via a combination of gene synthesis (Blue Heron Biotechnology, Inc) and Red/ET recombineering (GeneBridges). Part of a BAC clone containing the TAG-1 locus (CHORI, RP23-69J15) was retrieved and modified by inserting LoxP sites into non-conserved intronic regions 553bp upstream and 530bp downstream of TAG-1 exon 2. The 3′ LoxP site was preceded by a Frt-flanked neomycin (Neo) cassette (Figure S3). The targeting vector was introduced into 129/B6 hybrid embryonic stem (ES) cells and G418-resistant colonies were screened via Southern blot using probes located outside of the vector’s homology arms. The 5′- and 3′-probes were generated by PCR amplification from isolated BAC DNA (primer sequences: 5′ probe, TTAAAGGTTGACCGTAGATCTCTCTGT and GGAGGTTCAGGGGTTCTGGTT; 3′-probe, CAACATCACCTGGCTGACACA and TTTATAATGGGTCAGGCCTATCTGG). Probes were 32P-radiolabeled using the Prime-It II Random Primer Labeling Kit (Agilent). Correctly targeted ES cell clones were injected into B6 blastocysts at the Brown University Transgenic Services Laboratory to obtain chimeric TAG-1cKO-Neo/+ mice. The Neo cassette was then deleted by crossing chimeras to FLPeR mice, which express FLPe recombinase from the ROSA26 locus (Farley et al., 2000), to generate TAG-1cKO/+ mice.
The TAG-1Δ allele was generated by crossing TAG-1cKO/cKO mice with E2A::Cre mice, which express Cre recombinase in the germline (Lakso et al., 1996). Deletion of TAG-1 in TAG-1 and TAG-1Δ/Δ mice was confirmed by various methods (Figures S1, S3, and S4). Normal expression of the CAM Neurofascin, which is encoded by a genomic locus adjacent to TAG-1 (Hadas et al., 2013), in these mouse lines was confirmed by IHC on E11.5 brachial spinal cord sections (data not shown). For genotyping, TAG-1 alleles were detected by PCR (primer sequences: Fwd1 - CATGTGGTGGGAGTGTGGCTG, Rev1 - ACATCTCTCTTCCTACTTTTCTTCCCA TAG, and Rev2 - AACTCTCCAGATGTTGGATCCCTT) from genomic DNA, yielding band sizes of 212, 246, and 173 base pairs (bps) for the TAG-1+, TAG-1cKO, and TAG-1Δ alleles, respectively.
Immunohistochemistry
All spinal cord sections were collected from brachial level unless specified otherwise. Unless indicated otherwise, all incubations were conducted at room temperature (RT). Immunohistochemistry (IHC) on cryosections were performed as previously reported (Jaworski et al., 2010).
Embryos for vibratome sectioning were fixed overnight at 4°C in 4% paraformaldehyde (PFA) in PBS and washed three times for 10 min in PBS. Embryos were then embedded in 2% agarose in PBS and sectioned in PBS at 100 μm, and P0 animals were embedded in 3.5% agarose in PBS and sectioned at 150 μm. Free-floating vibratome sections were incubated in blocking buffer (2.5% bovine serum albumin (BSA), 0.1% Triton X-100 in PBS for embryonic sections and 4% BSA, 0.5% Triton X-100 in PBS for P0 sections) for 3—4 h). Primary antibodies diluted in blocking buffer were added to sections and incubated at 4°C overnight. After three 20-min washes with blocking buffer, sections were incubated with secondary antibodies diluted in blocking buffer overnight at 4°C. Three washes with blocking buffer for 20 min and one rinse with PBS were completed prior to mounting sections under Fluoromount-G for imaging.
Whole embryo IHC and clearing was performed via iDISCO. Embryos were fixed in 4% PFA in PBS overnight at 4°C and washed three times for 5 min with PBS. Embryos were then processed, as previously described, using the iDISCO IHC and methanol protocol (Renier et al., 2014).
Explants and dissociated MN cultures were fixed for 2 h with 4% PFA in PBS and washed three times for 5 min in PBS. The tissue was then stained using the IHC protocol for cryosections with solutions added directly to the 8-well glass chamber slides.
Mouse antibodies raised against NF (DHSB, #2H3, 1:500 (cryosections) or 1:200 (iDISCO)), TAG-1 (DSHB, #4D7, 1:200), and Islet1/2 (DHSB, #39.4D5, 1:200) were used, as well as goat antibodies raised against TAG-1 (R&D Systems, #AF4439, 1:200) and Sox10 (Santa Cruz, #sc-17342,1:200). All TAG-1 immunolabeling was performed using the polyclonal R&D Systems antibody unless specifically stated otherwise. Additionally, rabbit antibodies raised against TUJ1 (BioLegend, #802001, 1:500 and Covance #MrB-435P, 1:1000), HB9 (described previously; Thaler et al., 1999; 1:10000), Neurofascin (described previously; Sherman et al., 2005; 1:2000), Lhx3 (Abcam, #ab14555, 1:400), FoxP1 (Abcam, #ab16645, 1:500), Olig2 (Millipore, #AB9610, 1:500) and RFP (Rockland, #600-401-379, 1:500) and a chick antibody against GFP (Abcam, #ab13970, 1:200) were used. Secondary antibodies were: Alexa488-conjugated donkey anti-goat, Alexa488-conjugated donkey anti-rabbit, Alexa488-conjugated donkey anti-mouse, Alexa488-conjugated goat anti-chicken, Alexa594-conjugated donkey anti-goat, Alexa594-conjugated donkey anti-rabbit, Alexa594-conjugated donkey anti-mouse, Alexa647-conjugated donkey anti-goat, Alexa647-conjugated donkey anti-rabbit, Alexa647-conjugated donkey anti-mouse, Alexa647-conjugated rabbit anti-goat, Alexa647-conjugated goat anti-chicken, Alexa555-conjugated goat anti-mouse, and Alexa488-conjugated goat anti-mouse IgM (all Invitrogen, 1:200). Alexa488 donkey anti-chicken (Jackson ImmunoResearch, 1:200) was also used. Hoechst 33342 (Invitrogen; 1:1000) was added with secondary antibodies.
Imaging
Cryosections were imaged on a Nikon Eclipse Ti inverted microscope with Andor CCD camera or an Olympus FV3000 confocal laser scanning microscope. Vibratome sections were imaged using either a Zeiss LSM 810 confocal microscope, Zeiss LSM 510 meta confocal microscope, or Olympus FV3000 confocal microscope. Embryos cleared with iDISCO were either imaged on a Zeiss LSM 510 meta confocal laser scanning microscope with a 10x/0.45 Plan Apochromat objective or a LaVision Biotec light sheet microscope (Ultramicroscope II) equipped with a 2x/0.5 objective lens (MVPLAPO 2x) with 6mm working distance dipping cap, a sCMOS camera (Andor Neo), and Imspector Microscope controller software.
Assignment of motor column identity
To differentiate between motor columns at brachial level, spinal cord sections were immunolabeled for the transcription factors Islet1/2 and FoxP1. MMC MNs are immunoreactive only for Islet1/2; the LMC contains LMCl neurons, which are labeled by the FoxP1 antibody and exhibit weak Islet1/2 immunoreactivity, and LMCm neurons, which are strongly co-labeled by both the FoxP1 and Islet1/2 antibodies (Francius and Clotman, 2014; Stifani, 2014). For cervical and thoracic levels, spinal cord sections were immunolabeled for the transcription factors Islet1/2 and Lhx3. MMC MNs are immunoreactive for Islet1/2 and Lhx3, while all non-MMC MNs (hypaxial motor column, preganglionic motor column, phrenic motor column, and spinal accessory column) only exhibit Islet1/2 immunoreactivity (Stifani, 2014).
DiI tracing
Dil (Invitrogen, #V22888) was injected into either the DRG (Figure 6F) or the dorsal spinal cord (Figure S8E) of intact, fixed embryos (see IHC section for fixation method) using a micropipette and incubated in PBS for 24 h in the dark. Embryos were then vibratome-sectioned (see IHC methods). For DiI tracing from VRs (Figure S8E), fixed embryos were first embedded in 2% agarose and vibra-tome-sectioned at 150 μm. DiI was then injected into VRs using a micropipette, and free-floating sections were incubated in PBS for 24 h in the dark. Sections were then fixed for 10 min with 4% PFA in PBS and washed three times for 5 min with PBS before proceeding with the vibratome section IHC protocol (see above). This protocol was modified by replacing Triton X-100 with 0.1% Tween-20 in all blocking buffers, and no detergent was used in any wash buffers.
Fluorescent in situ hybridization
Embryos were fixed with 4% PFA in PBS and cryosectioned at 20 μm under RNase-free conditions. Slides were then processed according to the ACD RNAscope fresh frozen tissue pretreatment and fluorescent multiplex assay manual. The Mm-Egr2 probe (ACD Bio-techne, #407871), which has been described previously (Atlan et al., 2018; Martinez-Moreno et al., 2017), was used. IHC on cryosections (see above) was performed following completion of the RNAscope protocol.
Cell culture
COS-7 cells were grown as previously described (Jaworski and Tessier-Lavigne, 2012). The mouse TAG-1 coding sequence under control of a combined CMV/b-actin enhancer/promoter (or empty vector as negative control) were introduced into cells using Viofectin (Viogene, #VFT1001), following the manufacturer-recommended protocol.
Western blot analysis
For western blot analysis of embryonic tissue, the spinal column area (spinal cord, DRGs, and surrounding tissue) was dissected, lysed, and homogenized. COS cells were lysed and homogenized 24 h after transfection. Lysis/homogenization was performed on ice, and all subsequent incubation steps were conducted at RT unless otherwise specified. Protein concentration was determined by BCA protein assay (Pierce, #23335), and equal amounts of protein, falling within the linear range of western blot detection, were separated using 4%–12% Bis-Tris polyacrylamide gels (Bio-Rad, #3450124) and blotted onto Immobilon-P PVDF membranes (Millipore, #IVPH00010). Membranes were rinsed three times in TBST (20mM Tris base, 150mM NaCl, 0.1% Tween 20, pH 7.5) and cut between the 50-kD and 75-kD protein ladder bands (Kaleidoscope protein ladder, Bio-Rad, #1610375) to allow for the separate detection of TAG-1 (≈135 kD) and β-actin (≈42 kD). Membranes were blocked in 5% non-fat dry milk in TBST before being incubated with primary antibody in blocking buffer overnight at 4°C. Membranes were then incubated with HRP-conjugated secondary antibodies for 2 h, quickly rinsed three times with TBST, and then washed three times for 5 min with TBST. Blots were developed using SuperSignal™ West Dura Extended Duration Substrate (Pierce, #32106) for 1 min and imaged on an Azure C series digital imager. Primary antibodies used were goat anti-TAG-1 (R&D system, #AF4439, 1:25) and rabbit anti-β-actin (Novus, #NB600-503, 1:200). Secondary antibodies used were HRP-conjugated donkey anti-goat (Jackson ImmunoResearch, #705-035-003, 1:2500) and HRP-conjugated goat anti-rabbit (Jackson ImmunoResearch, #111-035-144,1:1500). Protein band intensities were quantified using ImageJ. TAG-1 band intensities were normalized to β-actin loading controls.
Ventral spinal cord explant and neuron culture
E11.5 embryos were harvested in L-15 media (GIBCO) with 5% horse serum. The Royal Blue NightSea system (Electron Microscopy Sciences, #SFA-LFS-RB) was used to identify HB9::GFP+ embryos for dissection. To generate ventral spinal cord explants, open-book preparations of E11.5 mouse brachial and cervical spinal cords were used. Under NightSea fluorescent illumination, the ventral third of the spinal cord, containing GFP+ MNs but excluding the floor plate, was collected and cut into approximately square explants. Ventral spinal cord explants were plated in 8-chamber glass slides, which had been pre-coated with 1 mg/ml poly-D-Lysine (PDL; Sigma, #P6407) in water and 50 ng/ml laminin (Millipore, #CC095) in PBS (control conditions). For TAG-1 substrate conditions, 50 μg/ml recombinant TAG-1 protein (TAG-1-His10; R&D Systems, #1714-CN) was added to 50 ng/ml laminin with PBS and incubated overnight at 4°C prior to explant dissection. Explants were cultured for 40-44 h in MN growth medium (10 ng/ml BDNF (Cell Science, #CRB600B), 50 ng/mL GDNF (Sigma, #SRP3200), 2% B-27, penicillin/streptomycin/glutamine (both Invitrogen), 0.5% glucose, and 0.5% methylcellulose in Neurobasal medium) at 37°C before processing for IHC (see above).
Dissociated MN cultures were generated by dissecting out the ventral spinal cord from an open book preparation (see above). Dissociation into single-cell suspension was conducted as described before (Langlois et al., 2010). Cells were plated at low density on PDL- and laminin-coated 8-well chamber slides and cultured for 24 h in MN growth medium before processing for IHC.
QUANTIFICATION AND STATISTICAL ANALYSIS
TAG-1 expression in motor neuron subtypes
To determine TAG-1 expression across motor columns, the number of TAG-1-immunolabeled MNs belonging to different brachial motor columns (see above; MMC (Islet1/2+), LMCl (FoxP1+), LMCm (Islet1/2+/FoxP1+)) was determined and expressed as a fraction of the total number of MNs within these columns. These quantifications were carried out on spinal cord cryosections.
Ectopic motor neurons
MNs were considered ectopic if the nucleus had broken the edge of the spinal cord at the MEP or left the spinal cord completely. To quantify the number of ectopic MNs per VR, 100 μm vibratome sections from embryos carrying the HB9::GFP transgene were stained for Islet1/2. The total number of Islet1/2+/GFP+ MNs within the VR and more distal motor nerve segment was counted. Time course analysis of ectopic MNs was performed using cryosections stained for HB9. The total number of HB9+ MNs outside the spinal cord per hemisection with a visible VR were counted. To quantify the number of ectopic MNs per motor column, cryosections were stained for motor column markers (see above), and ectopic neurons were counted per VR-containing hemisection.
Motor column mixing
To quantify motor column spatial organization, ImageJ software was used to create ROIs outlining the MMC and LMC within each hemisection, as defined by FoxP1 and Islet1/2 staining (see above). The perimeters of the LMC (ROILMC) and the MMC (ROIMMC) encompassed each column’s total area and was defined by vertices placed on the somas of the most peripheral cells within each column. A third ROI, which encompassed the area of overlap between the two columns, was then generated (ROIoverlap). All defined ROIs were convex polygons (interior angles < 180°). The areas of the three ROIs (ALMC, AMMC, and Aoverlap) were then measured using ImageJ. The amount of overlap between the LMC and MMC was expressed as Aoverlap/(ALMC + AMMC − Aoverlap).
Ventral root diameter
VR diameter quantification in 100 μm vibratome sections from embryos carrying the HB9::GFP transgene was conducted using ImageJ. Maximum-intensity projections were generated from confocal z stacks containing entire VRs. The diameter of the root was measured perpendicular to the VR at a distance of 30 μm from the MEP. All GFP+ axons were included in the measurement (Figures 4A and 5A), and VR diameter was expressed as a percentage of the average for control littermates. For the time course analysis of VR diameter, maximum-intensity projections of z stacks from NF-stained, iDISCO-cleared embryos were generated and used to measure each VR maximum diameter in the transverse plane using ImageJ (Figure S7A). Measurements were taken at different distances from the spinal cord depending on age: E9.5 – 20 μm, E10.5 – 30 μm, E11.5 – 30 μm, E12.5 – 35 μm, and E13.5 – 40 μm. Quantification of VR diameter from longitudinal cryosections was performed as previously reported (Figure S7B) (Roffers-Agarwal and Gammill, 2009).
Motor axon DRG invasion
To quantify motor axon invasion of DRGs, maximum-intensity projections of z stacks collected from 100 μm vibratome sections of embryos and 150 μm vibratome sections from P0 animals carrying the HB9::GFP transgene were generated. Using the ImageJ plugin NeuronJ, the total summed length of all GFP+ motor axons was quantified per DRG. Measurements were made from the point where motor axons left the main VR and covered the entire arbor within the DRG. In Olig2IRES-Cre/+; TAG-1cKO/cKO; ROSA26LSL-tdTom/+; HB9::GFP embryos, DRG invasion was quantified based on GFP+, tdTom+, and GFP+/tdTom+ motor axons.
DiI retrograde tracing
Following DiI injection into DRGs and IHC staining of vibratome sections, DiI+ MNs were counted and assigned motor column identity (see above). Individual optical sections from z stacks were used to unambiguously identify cells positive for both DiI and motor column markers. The number of DiI+ MNs in different motor columns was expressed as a fraction of the total number of MNs within these columns.
In vitro migration and axon fasciculation
Quantification of MN migration in vitro was completed in ImageJ using cell counter and measurement tools. The total number of GFP+/Hoechst+ MNs that had fully emigrated from their explant (defined by solid Hoechst+ area) was counted. The explant diameter was measured (in μm) and used to normalize the number of migrating neurons to the size of the explant. Quantification of in vitro motor axon fasciculation was performed by measuring the diameter of all GFP+ fascicles at a distance of 100 μm away from the edge of the explant. These measurements were made using the automated measurement tool in the NIS Elements software, and correct diameter measurements were confirmed by eye. We defined five bins (0-2.5 μm, 2.5-5 μm, 5-7.5 μm, 7.5-10 μm, and 10+ μm) and calculated the total number of fascicles that fell into each bin. These counts were then normalized to the total number of GFP+ fascicles for the entire experiment, so that the number of bundles within each bin was expressed as a percentage of total fascicle number.
Motor neurons with Cre activity
To determine the proportion of MNs with Cre activity, tdTom+ (and therefore Cre-expressing) cells in E9.5 Olig2-labeled or E11.5 Islet1/2-labeled cryosections from Olig2IRES-Cre/+; ROSA26LSL-tdTom/+ and Olig2Cre/+; ROSA26LSL-tdTom/+ embryos were counted using ImageJ cell counter. The numbers of Olig2+/tdTom+ or Islet1/2+/tdTom+ neurons were expressed as fractions of the overall total number of Olig2+ or Islet1/2+ cells, respectively.
TAG-1 immunofluorescence analysis
Quantification of TAG-1 fluorescence intensity was performed using Nikon NIS Elements software. All cryosections were stained in parallel and imaged using identical, sub-saturating exposure times for the TAG-1, TUJ1, and Hoechst channels. For each hemisection, 3-5 ROIs were drawn into each anatomical structure based on TUJ1 staining (TAG-1 channel was hidden to prevent selection bias). The amount of summed fluorescence signal for each of the TUJ1 and TAG-1 channels in each ROI was measured in arbitrary units (a.u.). Background fluorescence was measured by placing ROIs within the (TAG-1-and TUJ1-negative) progenitor zone of each section analyzed. Fluorescence measurements for TAG-1 and TUJ1 from each ROI within a given anatomical structure were averaged across the hemisection, resulting in one fluorescence measurement per protein per anatomical structure, from which background fluorescence was then subtracted. TAG-1 fluorescence intensity was then normalized to TUJ1 fluorescence intensity.
Motor neuron, Schwann cell, and boundary cap cell count
HB9+ MNs within the spinal cord were counted per cryosection and normalized to total spinal cord cross-section area. Schwann cell quantification was conducted by staining cryosections for Sox10. All Sox10+ cells within VRs were counted, and TUJ1 staining was used to determine the VR area, which was traced and measured using ImageJ. The total number of Sox10+ Schwann cells was normalized to VR area. Quantification of boundary cap cells at MEPs was performed on cryosections after Krox20 in situ hybridization and Sox10 IHC. VR-associated, dual Sox10+/Krox20+ cells were counted per VR-containing hemisection.
Statistical analysis
A minimum of 3 embryos (biological replicates, n) were examined for all descriptive statistics (exact n for each analysis is indicated in figure legends or in the Results section). For all tissue section analyses, quantification was performed on hemisections, with the number of hemisections per n ranging from 6 to 27. For in vitro analyses, a total of 4-22 explants per embryos were analyzed. Values from multiple hemisections or explants per embryo were averaged, generating a single mean value per n. The averages and SEMs across biological replicates within each experimental group were calculated and displayed graphically or stated in the Results section. Analysis of in vitro experiments using control versus TAG-1 substrate were analyzed using paired two-tailed t tests when comparing within the same genotype. All remaining pairwise comparisons between groups were performed using unpaired, two-tailed t tests with p values indicated in figures or the Results section. Multiple comparisons were performed by one-way ANOVA, followed by Holm’s post hoc test (α = 0.05), with p values shown in figures. All data were blinded prior to analysis to prevent scoring bias.
DATA AND CODE AVAILABILITY
This study did not generate any unique datasets or code.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| mouse-anti-Neurofilament (1:200) | DHSB | Cat#2H3; RRID:AB_531793 |
| mouse-anti-Islet1/2 (1:200) | DHSB | Cat#39.4D5 |
| mouse-anti-TAG-1 (1:200) | DHSB | Cat#4D7; RRID: AB_531775 |
| goat polyclonal-anti-TAG-1 (1:200) | R&D Systems | Cat#AF4439; RRID: AB_2044647 |
| goat-anti-Sox10 (1:200) | Santa Cruz | Cat#sc-17342; RRID: AB_2195374 |
| rabbit-anti-TUJ1 (1:500) | Biolegend | Cat#802001; RRID: AB_2313773 |
| rabbit-anti-TUJ1 (1:1000) | Covance | Cat#MrB-435P; RRID: AB_663339 |
| rabbit-anti-HB9 (1:10000) | gift from Samuel L. Pfaff | Thaler et. al., 1999 |
| rabbit-anti-Neurofascin (1:2000) | kind gift from Peter Brophy | Sherman et al., 2005 |
| rabbit-anti-Lhx3 (1:400) | Abcam | Cat#ab14555; RRID: AB_301332 |
| rabbit-anti-FoxP1 (1:500) | Abcam | Cat#ab16645; RRID: AB_732428 |
| rabbit-anti-Olig2 (1:500) | Millipore | Cat#AB9610 |
| rabbit-anti-RFP (1:500) | Rockland | Cat#600-401-379; RRID: AB_2209751 |
| chick-anti-GFP (1:200) | Abcam | Cat#ab13970; RRID: AB_300798 |
| Alexa488-conjugated donkey anti-goat (1:200) | Invitrogen | Cat#A11055 |
| Alexa488-conjugated donkey anti-rabbit (1:200) | Invitrogen | Cat#A21206 |
| Alexa488-conjugated donkey anti-mouse (1:200) | Invitrogen | Cat#A21202 |
| Alexa488-conjugated goat anti-chicken (1:200) | Invitrogen | Cat#A11039 |
| Alexa594-conjugated donkey anti-goat (1:200) | Invitrogen | Cat#A11058 |
| Alexa594-conjugated donkey anti-rabbit (1:200) | Invitrogen | Cat#A21207 |
| Alexa594-conjugated donkey anti-mouse (1:200) | Invitrogen | Cat#A21203 |
| Alexa647-conjugated donkey anti-goat (1:200) | Invitrogen | Cat#A21447 |
| Alexa647-conjugated donkey anti-rabbit (1:200) | Invitrogen | Cat#A31573 |
| Alexa647-conjugated rabbit anti-goat (1:200) | Invitrogen | Cat#A27018 |
| Alexa647-conjugated goat anti-chicken (1:200) | Invitrogen | Cat#A21449 |
| Alexa555-conjugated goat anti-mouse (1:200) | Invitrogen | Cat#A28180 |
| Alexa488-conjugated goat anti-mouse IgM (1:200) | Invitrogen | Cat#A21042 |
| Alexa488 donkey anti-chicken (1:500) | Jackson ImmunoResearch | Cat#703-545-155; RRID: AB_2340375 |
| Hoechst 33342 (1:1000) | Invitrogen | Cat#H3570 |
| donkey-anti-goat-HRP (1:2500) | Jackson ImmunoResearch | Cat#705-035-003; RRID: AB_2340390 |
| donkey-anti-rabbit-HRP (1:1500) | Jackson ImmunoResearch | Cat#711-005-152; RRID: AB_2340585 |
| rabbit-anti-Beta Actin (1:200) | Novus | Cat#NB600-503; RRID:AB_1216153 |
| Bacterial and Virus Strains | ||
| TAG-1 Locus | CHORI | RP23-69J15 |
| DH5-alpha | ThermoFisher | Cat#18265017 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| recombinant TAG-1 protein (TAG-1-His10) | R&D Systems | Cat#1714-CN; Lot #:OCX031803A |
| poly-D-Lysine (PDL) | Sigma | Cat#P6407 |
| Laminin | Millipore | Cat#CC095 |
| Bovine Serum Albumin (BSA) | Sigma | Cat#A2153 |
| DiI | Invitrogen | Cat#V22888 |
| GDNF | Sigma | Cat#SRP3200 |
| BDNF | Cell Science | Cat#CRB600B |
| Critical Commercial Assays | ||
| Prime-It II Random Primer Labeling Kit | Agilent | Cat#300385 |
| RNAscope® Intro Pack for Multiplex Fluorescent Reagent Kit- Fixed Frozen-Rn | ACD Bio-techne | Cat#323134 |
| Viofectin | Viogene | Cat#VFT1001 |
| BCA protein assay | Pierce | Cat#23335 |
| Experimental Models: Cell Lines | ||
| COS-7 Cell line | ATCC | Cat# CRL-1651 |
| Experimental Models: Organisms/Strains | ||
| mouse: TAG-1cKO/cKO | this article | N/A |
| mouse: TAG-1−/−: Cntn2tm1Furl | Poliak et al., 2003 | MGI: 2677610 |
| mouse: E2A::Cre: Tg(EIIa-cre)C5379Lmgd | Lakso et al., 1996 | MGI: 2137691 |
| mouse: Olig2Cre : Olig2tm1(cre)Tmj | Dessaud et al., 2007 | MGI: 3774124 |
| mouse: Olig2IRES-Cre: Olig2tm1.1(cre)Wdr | Zawadzka et al., 2010 | MGI: 4461156 |
| mouse: HB9::GFP: Tg(Hlxb9-GFP)1Tmj | Wichterle et al., 2002 | MGI: 3056906 |
| mouse: ROSA26LSL-tdTom: Gt(ROSA)26Sortm9(CAG-tdTomato)Hze | Madisen et al., 2010 | MGI: 3809523 |
| mouse: Sema6A−/− : Sema6aGt(KST069)Byg | Leighton et al., 2001 | MGI: 2156202 |
| mouse: Nrp2cKO/cKO : Nrp2tm1.1Mom | Walz et al., 2002 | MGI: 3712029 |
| mouse: Nrp1cKO/cKO: Nrp1tm2Ddg | Gu et al., 2003 | MGI: 3512101 |
| mouse: PlexinA3−/−: Plxna3tm1Matl | Cheng et al., 2001 | MGI: 2386961 |
| mouse: PlexinA4−/− : Plxna4tm1Matl | Yaron et al., 2005 | MGI: 3579185 |
| mouse: FLPeR : Gt(ROSA)26Sortm1(FLP1)Dym | Farley et al., 2000 | MGI: 2429412 |
| Oligonucleotides | ||
| Mm-Egr2 probe | ACD Bio-techne | Cat#407871 |
| Recombinant DNA | ||
| pCAGGs-Cntn2 | This article | N/A |
| Software and Algorithms | ||
| Prism 7 for Mac OS X | Graphpad | https://download.cnet.com/GraphPad-Prism/3000-2053_4-8453.html |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Illustrator CC | Adobe | https://www.adobe.com/jp/products/illustrator.html |
| Photoshop CC | Adobe | https://www.adobe.com/products/photoshop.html |
| NIS Elements | Nikon | https://www.microscope.healthcare.nikon.com/products/software/nis-elements |
| Imaris | Oxford Instruments | https://imaris.oxinst.com/downloads |
Highlights.
TAG-1 expression during motor neuron development shifts from cell bodies to axons
TAG-1 is required to anchor motor neuron cell bodies in the central nervous system
Motor axons fail to fasciculate and exhibit guidance defects in TAG-1−/− mice
TAG-1 fulfills its multiple functions independently of one another
ACKNOWLEDGMENTS
We thank Yonghong Zhou and Tess Puopolo for technical assistance and the Brown University Mouse Transgenic and Gene Targeting Facility for assistance in generation of the TAG-1cKO mouse line. We are grateful to Alex Kolodkin for sharing Nrp1, Nrp2, and Sema6A mutant mouse lines; Ben Novitch and Jeremy Dasen for sending us Olig2Cre mice; and Peter Brophy for sharing an antibody against Neurofascin. We are also thankful to members of the Jaworski laboratory for thoughtful comments on the manuscript. This work was supported by the National Institutes of Health (F31 NS098643 and T32 GM077995 to T.A.C.S.S., RI-INBRE P20 GM103430 to A.J. and K.P.D., and P30 GM1034310 to the Brown Transgenic Facility); the Rhode Island Foundation (Medical Research Grant 20154259 to A.J.); a New Frontiers Award from the Rhode Island Neuroscience Consortium (to A.J.); and funding from Brown University.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.12.085.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Arber S, Han B, Mendelsohn M, Smith M, Jessell TM, and Sockanathan S (1999). Requirement for the homeobox gene Hb9 in the consolidation of motor neuron identity. Neuron 23, 659–674. [DOI] [PubMed] [Google Scholar]
- Atlan G, Terem A, Peretz-Rivlin N, Sehrawat K, Gonzales BJ, Pozner G, Tasaka GI, Goll Y, Refaeli R, Zviran O, et al. (2018). The claustrum supports resilience to distraction. Curr. Biol. 28, 2752–2762.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeriswyl T, and Stoeckli ET (2008). Axonin-1/TAG-1 is required for path finding of granule cell axons in the developing cerebellum. Neural Dev. 3, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonanomi D (2019). Axon pathfinding for locomotion. Semin. Cell Dev. Biol. 85, 26–35. [DOI] [PubMed] [Google Scholar]
- Bonanomi D, and Pfaff SL (2010). Motor axon pathfinding. Cold Spring Harb. Perspect. Biol. 2, a001735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bron R, Vermeren M, Kokot N, Andrews W, Little GE, Mitchell KJ, and Cohen J (2007). Boundary cap cells constrain spinal motor neuron somal migration at motor exit points by a semaphorin-plexin mechanism. Neural Dev. 2, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttiglione M, Revest JM, Pavlou O, Karagogeos D, Furley A, Rougon G, and Faivre-Sarrailh C (1998). A functional interaction between the neuronal adhesion molecules TAG-1 and F3 modulates neurite outgrowth and fasciculation of cerebellar granule cells. J. Neurosci. 18, 6853–6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HJ, Bagri A, Yaron A, Stein E, Pleasure SJ, and Tessier-Lavigne M (2001). Plexin-A3 mediates semaphorin signaling and regulates the development of hippocampal axonal projections. Neuron 32, 249–263. [DOI] [PubMed] [Google Scholar]
- Dang P, Smythe E, and Furley AJW (2012). TAG1 regulates the endocytic trafficking and signaling of the semaphorin3A receptor complex. J. Neurosci. 32, 10370–10382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessaud E, Yang LL, Hill K, Cox B, Ulloa F, Ribeiro A, Mynett A, Novitch BG, and Briscoe J (2007). Interpretation of the sonic hedgehog morphogen gradient by a temporal adaptation mechanism. Nature 450, 717–720. [DOI] [PubMed] [Google Scholar]
- Farley FW, Soriano P, Steffen LS, and Dymecki SM (2000). Widespread recombinase expression using FLPeR (flipper) mice. Genesis 28, 106–110. [PubMed] [Google Scholar]
- Felsenfeld DP, Hynes MA, Skoler KM, Furley AJ, and Jessell TM (1994). TAG-1 can mediate homophilic binding, but neurite outgrowth on TAG-1 requiresan L1-like molecule and beta 1 integrins. Neuron 12, 675–690. [DOI] [PubMed] [Google Scholar]
- Francius C, and Clotman F (2014). Generating spinal motor neuron diversity: a long quest for neuronal identity. Cell. Mol. Life Sci. 71, 813–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukamauchi F, Aihara O, Wang YJ, Akasaka K,Takeda Y, Horie M, Kawano H, Sudo K, Asano M, Watanabe K, and Iwakura Y (2001). TAG-1-deficient mice have marked elevation of adenosine A1 receptors in the hippocampus. Biochem. Biophys. Res. Commun. 281, 220–226. [DOI] [PubMed] [Google Scholar]
- Furley AJ, Morton SB, Manalo D, Karagogeos D, Dodd J, and Jessell TM (1990). The axonal glycoprotein TAG-1 is an immunoglobulin superfamily member with neurite outgrowth-promoting activity. Cell 61, 157–170. [DOI] [PubMed] [Google Scholar]
- Gallarda BW, Bonanomi D, Müller D, Brown A, Alaynick WA, Andrews SE, Lemke G, Pfaff SL, and Marquardt T (2008). Segregation of axial motor and sensory pathways via heterotypic trans-axonal signaling. Science 320, 233–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett AM, Jucius TJ, Sigaud LP, Tang FL, Xiong WC, Ackerman SL, and Burgess RW (2016). Analysis of expression pattern and genetic deletion of netrin5 in the developing mouse. Front. Mol. Neurosci. 9, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu C, Rodriguez ER, Reimert DV, Shu T, Fritzsch B, Richards LJ, Kolodkin AL, and Ginty DD (2003). Neuropilin-1 conveys semaphorin and VEGF signaling during neural and cardiovascular development. Dev. Cell 5, 45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurung S, Asante E, Hummel D, Williams A, Feldman-Schultz O, Halloran MC, Sittaramane V, and Chandrasekhar A (2018). Distinct roles for the cell adhesion molecule Contactin2 in the development and function of neural circuits in zebrafish. Mech. Dev. 152, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadas Y, Nitzan N, Furley AJW, Kozlov SV, and Klar A (2013). Distinct cis regulatory elements govern the expression of TAG1 in embryonic sensory ganglia and spinal cord. PLoS ONE 8, e57960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber AB, Kania A, Tran TS, Gu C, De Marco Garcia N, Lieberam I, Johnson D, Jessell TM, Ginty DD, and Kolodkin AL (2005). Distinct roles for secreted semaphorin signaling in spinal motor axon guidance. Neuron 48, 949–964. [DOI] [PubMed] [Google Scholar]
- Huettl RE, Soellner H, Bianchi E, Novitch BG, and Huber AB (2011). Npn-1 contributes to axon-axon interactions that differentially control sensory and motor innervation of the limb. PLoS Biol. 9, e1001020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaworski A, and Tessier-Lavigne M (2012). Autocrine/juxtaparacrine regulation of axon fasciculation by Slit-Robo signaling. Nat. Neurosci. 15, 367–369. [DOI] [PubMed] [Google Scholar]
- Jaworski A, Long H, and Tessier-Lavigne M (2010). Collaborative and specialized functions of Robo1 and Robo2 in spinal commissural axon guidance. J. Neurosci. 30, 9445–9453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karagogeos D, Morton SB, Casano F, Dodd J, and Jessell TM (1991). Developmental expression of the axonal glycoprotein TAG-1: differential regulation by central and peripheral neurons in vitro. Development 112, 51–67. [DOI] [PubMed] [Google Scholar]
- Kim M, Fontelonga TM, Lee CH, Barnum SJ, and Mastick GS (2017). Motor axons are guided to exit points in the spinal cord by Slit and Netrin signals. Dev. Biol. 432, 178–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Bjorke B, and Mastick GS (2019). Motor neuron migration and positioning mechanisms: new roles for guidance cues. Semin. Cell Dev. Biol. 85, 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodkin AL, and Tessier-Lavigne M (2011). Mechanisms and molecules of neuronal wiring: a primer. Cold Spring Harb. Perspect. Biol. 3, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunz S, Spirig M, Ginsburg C, Buchstaller A, Berger P, Lanz R, Rader C, Vogt L, Kunz B, and Sonderegger P (1998). Neurite fasciculation mediated by complexes of axonin-1 and Ng cell adhesion molecule. J. Cell Biol. 143, 1673–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakso M, Pichel JG, Gorman JR, Sauer B, Okamoto Y, Lee E, Alt FW, and Westphal H (1996). Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc. Natl. Acad. Sci. USA 93, 5860–5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlois SD, Morin S, Yam PT, and Charron F (2010). Dissection and culture of commissural neurons from embryonic spinal cord. J. Vis. Exp. 39, 2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law CO, Kirby RJ, Aghamohammadzadeh S, and Furley AJW (2008). The neural adhesion molecule TAG-1 modulates responses of sensory axons to diffusible guidance signals. Development 135, 2361–2371. [DOI] [PubMed] [Google Scholar]
- Lee H, and Song MR (2013). The structural role of radial glial endfeet in confining spinal motor neuron somata is controlled by the Reelin and Notch pathways. Exp. Neurol. 249, 83–94. [DOI] [PubMed] [Google Scholar]
- Lee S, Lee B, Lee JW, and Lee SK (2009). Retinoid signaling and neurogenin2 function are coupled for the specification of spinal motor neurons through a chromatin modifier CBP. Neuron 62, 641–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leighton PA, Mitchell KJ, Goodrich LV, Lu X, Pinson K, Scherz P, Skarnes WC, and Tessier-Lavigne M (2001). Defining brain wiring patterns and mechanisms through gene trapping in mice. Nature 410, 174–179. [DOI] [PubMed] [Google Scholar]
- Luxey M, Jungas T, Laussu J, Audouard C, Garces A, and Davy A (2013). Eph:ephrin-B1 forward signaling controls fasciculation of sensory and motor axons. Dev. Biol. 383, 264–274. [DOI] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin O, Valiente M, Ge X, and Tsai LH (2010). Guiding neuronal cell migrations. Cold Spring Harb. Perspect. Biol. 2, a001834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Moreno M, O’Shea TM, Zepecki JP, Olaru A, Ness JK, Langer R, and Tapinos N (2017). Regulation of peripheral myelination through transcriptional buffering of Egr2 by an antisense long non-coding RNA. Cell Rep. 20, 1950–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda T (2017). Contactin-2/TAG-1, active on the front line for three decades. Cell Adhes. Migr. 11, 524–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda T, Okado N, and Shiga T (2000). The involvement of axonin-1/SC2 in mediating notochord-derived chemorepulsive activities for dorsal root ganglion neurites. Dev. Biol. 224, 112–121. [DOI] [PubMed] [Google Scholar]
- Masuda T, Tsuji H, Taniguchi M, Yagi T, Tessier-Lavigne M, Fujisawa H, Okado N, and Shiga T (2003). Differential non-target-derived repulsive signals play a critical role in shaping initial axonal growth of dorsal root ganglion neurons. Dev. Biol. 254, 289–302. [DOI] [PubMed] [Google Scholar]
- Masuda T, Fukamauchi F, Takeda Y, Fujisawa H, Watanabe K, Okado N, and Shiga T (2004). Developmental regulation of notochord-derived repulsion for dorsal root ganglion axons. Mol. Cell. Neurosci. 25, 217–227. [DOI] [PubMed] [Google Scholar]
- Mauti O, Domanitskaya E, Andermatt I, Sadhu R, and Stoeckli ET (2007). Semaphorin6A acts as a gate keeper between the central and the peripheral nervous system. Neural Dev. 2, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moret F, Renaudot C, Bozon M, and Castellani V (2007). Semaphorin and neuropilin co-expression in motoneurons sets axon sensitivity to environmental semaphorin sources during motor axon pathfinding. Development 134, 4491–4501. [DOI] [PubMed] [Google Scholar]
- Poliak S, Salomon D, Elhanany H, Sabanay H, Kiernan B, Pevny L, Stewart CL, Xu X, Chiu S-Y, Shrager P, et al. (2003). Juxtaparanodal clustering of Shaker-like K+ channels in myelinated axons depends on Caspr2 and TAG-1. J. Cell Biol. 162, 1149–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rader C, Stoeckli ET, Ziegler U, Osterwalder T, Kunz B, and Sonderegger P (1993). Cell-cell adhesion by homophilic interaction of the neuronal recognition molecule axonin-1. Eur. J. Biochem. 215, 133–141. [DOI] [PubMed] [Google Scholar]
- Raper J, and Mason C (2010). Cellular strategies of axonal pathfinding. Cold Spring Harb. Perspect. Biol. 2, a001933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renier N, Wu Z, Simon DJ, Yang J, Ariel P, and Tessier-Lavigne M (2014). iDISCO: a simple, rapid method to immunolabel large tissue samples for volume imaging. Cell 159, 896–910. [DOI] [PubMed] [Google Scholar]
- Roffers-Agarwal J, and Gammill LS (2009). Neuropilin receptors guide distinct phases of sensory and motor neuronal segmentation. Development 136, 1879–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruegg MA, Stoeckli ET, Lanz RB, Streit P, and Sonderegger P (1989). A homologue of the axonally secreted protein axonin-1 is an integral membrane protein of nerve fiber tracts involved in neurite fasciculation. J. Cell Biol. 709, 2363–2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savvaki M, Panagiotaropoulos T, Stamatakis A, Sargiannidou I, Karatzioula P, Watanabe K, Stylianopoulou F, Karagogeos D, and Kleopa KA (2008). Impairment of learning and memory in TAG-1 deficient mice associated with shorter CNS internodes and disrupted juxtaparanodes. Mol. Cell. Neurosci. 39, 478–90. [DOI] [PubMed] [Google Scholar]
- Savvaki M, Theodorakis K, Zoupi L, Stamatakis A, Tivodar S, Kyriacou K, Stylianopoulou F, and Karagogeos D (2010).The expression of TAG-1 in glial cells is sufficient for the formation of the juxtaparanodal complex and the phenotypic rescue of tag-1 homozygous mutants in the CNS. J. Neurosci. 30, 13943–13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider-Maunoury S, Topilko P, Seitandou T, Levi G, Cohen-Tannoudji M, Pournin S, Babinet C, and Charnay P (1993). Disruption of Krox-20 results in alteration of rhombomeres 3 and 5 in the developing hindbrain. Cell 75, 1199–1214. [DOI] [PubMed] [Google Scholar]
- Sherman DL, Tait S, Melrose S, Johnson R, Zonta B, Court FA, Macklin WB, Meek S, Smith AJH, Cottrell DF, and Brophy PJ (2005). Neurofascins are required to establish axonal domains for saltatory conduction. Neuron 48, 737–742. [DOI] [PubMed] [Google Scholar]
- Shirasaki R, Lewcock JW, Lettieri K, and Pfaff SL (2006). FGF as a target-derived chemoattractant for developing motor axons genetically programmed by the LIM code. Neuron 50, 841–853. [DOI] [PubMed] [Google Scholar]
- Stifani N (2014). Motor neurons and the generation of spinal motor neuron diversity. Front. Cell. Neurosci. 8, 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeckli ET, and Landmesser LT (1995). Axonin-1, Nr-CAM, and Ng-CAM play different roles in the in vivo guidance of chick commissural neurons. Neuron 14, 1165–1179. [DOI] [PubMed] [Google Scholar]
- Stoeckli ET, Kuhn TB, Duc CO, Ruegg MA, and Sonderegger P (1991). The axonally secreted protein axonin-1 is a potent substratum for neurite growth. J. Cell Biol. 112, 449–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter TACS, and Jaworski A (2019). Cell migration and axon guidance at the border between central and peripheral nervous system. Science 365, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaler J, Harrison K, Sharma K, Lettieri K, Kehrl J, and Pfaff SL (1999). Active suppression of interneuron programs within developing motor neurons revealed by analysis of homeodomain factor HB9. Neuron 23, 675–687. [DOI] [PubMed] [Google Scholar]
- Thaler JP, Koo SJ, Kania A, Lettieri K, Andrews S, Cox C, Jessell TM, and Pfaff SL (2004). A postmitotic role for Isl-class LIM homeodomain proteins in the assignment of visceral spinal motor neuron identity. Neuron 41, 337–350. [DOI] [PubMed] [Google Scholar]
- Vermeren M, Maro GS, Bron R, McGonnell IM, Charnay P, Topilko P, and Cohen J (2003). Integrity of developing spinal motor columns is regulated by neural crest derivatives at motor exit points. Neuron 37, 403–415. [DOI] [PubMed] [Google Scholar]
- Vogt L, Giger RJ, Ziegler U, Kunz B, Buchstaller A, Kaplitt MG, Rosenfeld MR, Pfaff DW, Verhaagen J, and Sonderegger P; Hermens WTJMC (1996). Continuous renewal of the axonal pathway sensor apparatus by insertion of new sensor molecules into the growth cone membrane. Curr. Biol. 6, 1153–1158. [DOI] [PubMed] [Google Scholar]
- Walz A, Rodriguez I, and Mombaerts P (2002). Aberrant sensory innervation of the olfactory bulb in neuropilin-2 mutant mice. J. Neurosci. 22, 4025–4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, and Marquardt T (2013). What axons tell each other: axon-axon signaling in nerve and circuit assembly. Curr. Opin. Neurobiol. 23, 974–982. [DOI] [PubMed] [Google Scholar]
- Wang L, Klein R, Zheng B, and Marquardt T (2011). Anatomical coupling of sensory and motor nerve trajectory via axon tracking. Neuron 71, 263–277. [DOI] [PubMed] [Google Scholar]
- Weiner JA, Koo SJ, Nicolas S, Fraboulet S, Pfaff SL, Pourquié O, and Sanes JR (2004). Axon fasciculation defects and retinal dysplasias in mice lacking the immunoglobulin superfamily adhesion molecule BEN/ALCAM/SC1. Mol. Cell. Neurosci. 27, 59–69. [DOI] [PubMed] [Google Scholar]
- Wichterle H, Lieberam I, Porter JA, and Jessell TM (2002). Directed differentiation of embryonic stem cells into motor neurons. Cell 110, 385–397. [DOI] [PubMed] [Google Scholar]
- Wilkinson DG, Bhatt S, Chavrier P, Bravo R, and Charnay P (1989). Segment-specific expression of a zinc-finger gene in the developing nervous system of the mouse. Nature 337, 461–464. [DOI] [PubMed] [Google Scholar]
- Wolfer DP, Henehan-Beatty A, Stoeckli ET, Sonderegger P, and Lipp HP (1994). Distribution of TAG-1/axonin-1 in fibre tracts and migratory streams of the developing mouse nervous system. J. Comp. Neurol. 345, 1–32. [DOI] [PubMed] [Google Scholar]
- Xu K, Wu Z, Renier N, Antipenko A, Tzvetkova-Robev D, Xu Y, Minchenko M, Nardi-Dei V, Rajashankar KR, Himanen J, et al. (2014). Neural migration. Structures of netrin-1 bound to two receptors provide insight into its axon guidance mechanism. Science 344, 1275–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Boyer AM, Crandall JE, Edwards M, and Tanaka H (1986). Distribution of stage-specific neurite-associated proteins in the developing murine nervous system recognized by a monoclonal antibody. J. Neurosci. 6, 3576–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaron A, Huang PH, Cheng HJ, and Tessier-Lavigne M (2005). Differential requirement for Plexin-A3 and -A4 in mediating responses of sensory and sympathetic neurons to distinct class 3 Semaphorins. Neuron 45, 513–523. [DOI] [PubMed] [Google Scholar]
- Zawadzka M, Rivers LE, Fancy SPJ, Zhao C, Tripathi R, Jamen F, Young K, Goncharevich A, Pohl H, Rizzi M, et al. (2010). CNS-resident glial progenitor/stem cells produce Schwann cells as well as oligodendrocytes during repair of CNS demyelination. Cell Stem Cell 6, 578–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.