

SUMMARY
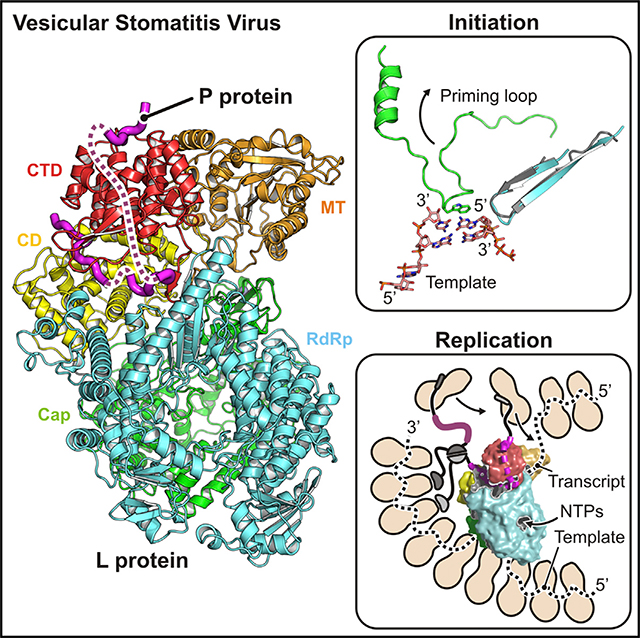
The large (L) proteins of non-segmented, negativestrand RNA viruses are multifunctional enzymes that produce capped, methylated, and polyadenylated mRNA and replicate the viral genome. A phosphoprotein (P), required for efficient RNA-dependent RNA polymerization from the viral ribonucleoprotein (RNP) template, regulates the function and conformation of the L protein. We report the structure of vesicular stomatitis virus L in complex with its P cofactor determined by electron cryomicroscopy at 3.0 Å resolution, enabling us to visualize bound segments of P. The contacts of three P segments with multiple L domains show how P induces a closed, compact, initiationcompetent conformation. Binding of P to L positions its N-terminal domain adjacent to a putative RNA exit channel for efficient encapsidation of newly synthesized genomes with the nucleoprotein and orients its C-terminal domain to interact with an RNP template. The model shows that a conserved tryptophan in the priming loop can support the initiating 5′ nucleotide.
In Brief
Jenni et al. describe a 3.0 Å resolution cryo-EM structure of vesicular stomatitis virus L protein, bound with its P-protein cofactor, suggesting molecular features of RNA-synthesis initiation, transcript capping, and replication-product encapsidation.
Graphical Abstract
INTRODUCTION
The large (L) protein encoded by the genomes of nonsegmented, negative-sense (NNS) RNA viruses carries out all the various catalytic steps associated with transcription and replication. A virally encoded phosphoprotein (P) is an essential cofactor, both for the incorporation of L into virions and for the regulation of replication and transcription. In addition to its RNA-dependent RNA polymerase (RdRp) activity, L caps and methylates the 5′ ends of transcripts.
The vesicular stomatitis virus (VSV) genome encodes an untranslated (and uncapped) 5′ leader sequence and five proteins, in the order of N, P, M, G, and L. The template for transcription is a full-length ribonucleoprotein (RNP)—that is, a genome-sense RNA fully coated with protein N. Each N subunit accommodates nine nucleotides of RNA in a groove along the waist of an elongated, two-lobe protein. Transcription of successive genes (with about 70% efficiency) initiates upon the termination and polyadenylation of the upstream transcript, produced by stuttering on a U7 sequence at the end of each gene (Iverson and Rose, 1981).
We described five years ago the structure of a VSV L-P complex from a cryo-electron microscopy (EM) reconstruction at 3.8 Å resolution (Liang et al., 2015). The multifunctional L protein—a single, 2109-amino-acid-long polypeptide chain— folds into three catalytic and two structural domains (Figure 1A; Data S1). The N-terminal RdRp domain and a capping (Cap) domain, which follows in primary sequence and has polyribonucleotidyl transferase (PRNTase) activity, form the core of the structure (Figure 1B). Negative-stain EM images had shown previously that in the absence of P, the three remaining domains—the connector domain (CD), the methyltransferase (MT), and the C-terminal domain (CTD)—have no fixed position with respect to the RdRp-Cap core structure, and the full molecular model from the cryo-EM showed particularly long linker segments between the Cap domain and CD and between the CD and MT (Liang et al., 2015; Rahmeh et al., 2010).
Figure 1. Structure of the VSV L Protein in Complex with the Phosphoprotein.

(A) Linear domain organization of VSV L and P. Domain boundary numbers are shown. L-protein domains are colored as follows: RNA-dependent RNA polymerase domain (RdRp), cyan; capping domain (Cap), green; connector domain (CD), yellow; methyltransferase (MT), orange; C-terminal domain (CTD), red. Catalytic residues (CRs) are shown above. CRI–VI: conserved regions in L proteins of NNS RNA viruses. The L-protein scheme is adapted from Liang et al. (2015). P-protein N-terminal (PNTD), oligomerization (POD), and C-terminal (PCDT) domains are in gray; its L protein-binding domain (PL) is in magenta.
(B) Cryo-EM structure of the L-P complex determined at 3.0 Å resolution. Full-length L was incubated with a P fragment comprising residues 35–106. Domains colored as in (A). Dashed lines show flexible PL segments, connecting the three L-bound segments.
(C) L-protein-binding motif 1 of P shown as sticks (carbon, magenta; nitrogen, blue; oxygen, red). The density map (EMD- 20614, B sharpened map) is shown as gray mesh around P (carving radius =2.0 Å ).
(D) L-protein-binding motif 2 of P, colored and with density map shown as described in (C). Absence of density for almost all acidic amino acid side chains (Asp and Glu, indicated by asterisks) is characteristic of cryo-EM density maps.
(E) L-protein-binding motif 3 of P, colored and with density map shown as described in (C) and (D).
See also Figures S2 and S3 and Table S1.
The polymerase catalytic site faces a cavity at the center of the RdRp domain (Liang et al., 2015). Channels between this cavity and the surface of the molecule allow entry and exit of the template RNA strand and entry of nucleoside triphosphates (NTPs). A loop from the Cap domain, projecting into the RdRp catalytic cavity, appears to be a priming loop to support the initiating nucleotide. Analogous loops are present in other viral RdRps that do not initiate on a polynucleotide primer (Lu et al., 2008; Tao et al., 2002). The priming loop must shift out of the way to allow elongation, as seen in the transition from initiation to elongation in the reovirus polymerase (Tao et al., 2002). In VSV L, reconfiguration of the priming loop would allow the leading end of the elongating product strand to move into a cavity in the Cap domain that includes its active-site residues.
The capping reaction catalyzed by the L proteins of NNS RNA viruses proceeds through intermediates different from those of the mammalian-cell capping process (and different from the related and well-characterized capping mechanism of double-strand RNA viruses). Instead of an attack by the 5′ end of the RNA on a guanosine monophosphate (GMP) covalently attached to a lysine side chain, the monophosphoylated 5′ end of the transcript attaches covalently to a histidine side chain; resolution of this intermediate through attack by guanosine diphosphate (GDP) or guanosine triphosphate (GTP) results in a guanylated 5′ terminus. The Cap domain is thus a PRNTase; 2′-O methylation and 7-N methylation, reactions carried out by the single active site on the MT domain, complete the modification.
Our previous L-P reconstruction resulted in a few regions of uncertain chain trace and did not clearly identify features corresponding to P (Liang et al., 2015). It was clear, however, that the binding of a fragment covering P residues 35–106, the sequence between the conserved N-terminal segment (PNTD) and the oligomerization domain (POD) (Figure 1A; Data S1), induced a “closed” structure of L in which the three terminal domains docked in well-defined positions onto the RdRp-Cap domain (Figure 1B). We interpreted the observed conformation as a preinitiation complex. Poor density in the region between the active sites of the RdRp and Cap domains prevented analysis of the coupling of transcription and capping.
We have now extended the resolution of the VSV L-P structure to 3.0 Å, from new images recorded on a Titan Krios microscope. The new reconstruction has allowed us to trace three segments of P bound to L and to correct a few small errors in the previous model. The molecular L-P interactions explain how P binding leads to a closed structure. Improved definition of several parts of the Cap domain and a clearer trace of the connections into and out of the connector domain have allowed us to identify a channel leading from the polymerase active site to a potential genome and anti-genome exit site at a position appropriate for acquisition of N, held close to that site by interaction with the N-terminal segment of P. Direct deposition of N onto the emerging RNA could afford prompt encapsidation and maximal protection. Better definition of the Cap domain active-site cavity also allows us to consider possible conformational changes that accompany formation of the covalent 5’-monophosphate-RNA intermediate and its transfer to GDP.
RESULTS AND DISCUSSION
Structure of VSV L-P from Cryo-EM Reconstruction at 3.0 Å Resolution
We prepared the VSV L-P complex for cryo-EM imaging as described previously (Liang et al., 2015). We expressed full-length VSV L in insect cells and a P fragment (residues 35–106) in bacteria. We mixed L and P in vitro and purified the complex by gel filtration. We recorded images (Figures S1A and S1B) and carried out the image processing steps as described in the Method Details. The final map used for the modeling and interpretation reported here (Figure 1C) had an overall resolution of 3.0 Å, as determined by the spatial frequency at which the Fourier shell correlation (FSC) between half maps dropped below 0.143 (Figure S1C). The data quality allowed us not only to improve the overall resolution of the reconstruction, but also to classify out a more homogeneous set of particle images, yielding a density map in which all domains were equally well resolved.
We could place our previous model (PDB: 5A22) into the density without any major adjustments. We traced connections into and out of the connector domain more clearly than with the previous, lower-resolution reconstruction; corrected a local chain trace error associated with these connections; corrected a few errors in assigning sequence register to segments poorly defined in the previous map; and adjusted many side-chain rotamers, which were generally well defined in the new map. Data collection, model refinement, and validation statistics are in Table S1. The positions of various differences between the updated and the original coordinates are shown in Figures S2 and S3.
Interactions of the P Subunit with L
The most important new feature was interpretable density for parts of P (Figure S4), enabling us to model three ordered segments from residues 49 to 56, 82 to 89, and 94 to 105 (Figures 2A and 2B). The first segment binds in a shallow pocket on the outward-facing surface of the CTD. The interactions are mostly hydrophobic around a conserved tyrosine residue PTyr53 that also hydrogen bonds to the side chain of LAsp1981 (Figure 2C). P then wraps around the CTD with a stretch of flexible residues that we can detect in low-pass-filtered density maps (Figure S4B). The second segment binds between the CTD and the RdRp and interacts with the C-terminal arm as it inserts along the edge of the RdRp. This P segment appears to interact with hydrogen bonds that involve main-chain atoms, either the amino nitrogen or carbonyl oxygen. These interactions, which do not directly depend on side-chain residues, explain the relaxed conservation of residues in segment 2. Binding is further stabilized by hydrophobic contacts of PVal84 and PPhe87 and a salt bridge between PGlu85 and the CTD residue LLys2022 (Figure 2D). The third segment lies in the groove between the CTD and the connector. We count two salt bridges and two hydrogen bonds at this binding site. One hydrogen bond, between PGlu99 and LGly1911, connects to the CTD. The PTyr95 side chain stacks between LArg1419 and LTyr1438. A conserved PVal102 lies a hydrophobic L pocket, and a conserved PPhe104 stacks on LPro1535 (Figure 2E). The CTD is thus an adaptor domain of L for its association with P, which then stabilizes the closed conformation by reinforcing interdomain contacts.
Figure 2. Molecular Details of Phosphoprotein Interactions with the L Protein.

(A) Multiple-sequence alignment for P proteins of different NNS viruses. For sequence accession identifiers, see Data S1. The alignment is shown only for VSV P residues included in the expression construct. Magenta solid bars beneath the sequences are residues modeled in our structure; magenta dashed lines are residues included in the expression construct. Binding motifs 1, 2, and 3 are labeled.
(B) Overview of how P binds L. P is shown as a magenta tube; L is colored as in Figure 1. Motif 1 binds on the outer surface of the CTD. Motif 2 binds between the CTD and the RdRp and also interacts with the L-protein C-terminal arm. Motif 3 interacts with the CD and the CTD.
(C) Molecular interactions with L of P segment 1. The L backbone is in ribbon representation and colored as in Figure 1. Interacting residues (main-chain and/or side-chain atoms) are shown as gray sticks; P residues are shown as magenta sticks; hydrogen bonds and salt bridges are shown as yellow dashed lines.
(D) Molecular interactions with L of P segment 2.
(E) Molecular interactions with L of P segment 3.
See also Figure S4.
VSV P dimerizes through a domain (POD) that extends from residue 109 to residue 170 (Figure 1; Data S1). We analyzed, by projection matching with the present structure (Data S2), images of the negatively stained L-P dimers described in much earlier work (Figure 3A) (Rahmeh et al., 2010). We found that the mean distance between residues 105 on the P chains of the two matched complexes (60 Å ) (Figure 3B) agreed well with that expected from the known structure of the dimerization domain (Ding et al., 2006), allowing for an extension of the short segment from 105 to 109 on each of the dimerized complexes (Figure 3C). The agreement validates the polarity of the P-chain trace and the assignment of residues in segment 3.
Figure 3. Characterization of P Binding and Function.

(A) Projection matching of VSV L-P dimer negative-stain class averages. Top, representative negative-stain class average (Rahmeh et al., 2010). Middle, projection image of our reconstruction obtained from two aligned complexes. Scale bar, 100 Å. Bottom, two aligned L-P complexes, in surface representation. The last modeled P residue (PThr105) in each complex is a large magenta sphere. See Data S2 for details of the projection-matching analysis.
B) Statistical analysis of the distance between the C-terminal residues (PThr105) of P-binding motif 3 in the negative-stain class averages of VSV L-P dimers. For each of the 20 class averages, the distance obtained from the projection-matching analysis is shown as a magenta dot. The average distance is shown as a black dot.
(C) Structural model of the P-protein oligomerization domain (POD). The dimer structure is from PDB: 2FQM (residues 111–171) (Ding et al., 2006); residues 105–110 were modeled in extended conformation. The distance between PThr105 residues is 59 Å.
(D) Cell-based assay using an EGFP reporter as a readout for L activity when RNA synthesis is reconstituted with the various P mutants.
(E) Plaque assay with VSV-EGFP-P_wt and VSVEGFP-P_Y53F. GFP signal was monitored 24 h post-infection.
(F) Reconstituted RNA synthesis in vitro on naked RNA (left) and nucleocapsid-RNA template (right).
(G) Model for replication of RNP by VSV L-P. N is in beige and drawn to approximate scale. Threading the template into the polymerase active site appears to require a separation of RNA from at least 3 N molecules. P binding stabilizes the closed conformation of L, leads to processive polymerization, and positions the N-terminal domain (PNTD) close to a putative product exit channel, facilitating the transfer of N from N0-P to emerging RNA. The C-terminal domain of P (PCTD) interacts with the RNP; it may guide the polymerase along the template. P dimerization through the oligomerization domain (POD) would lead to additional RNP and N0-P interactions.
See also Data S2.
In segment 1, PTyr53 fits into a pocket on the outward-facing surface of the CTD. For a series of mutations at position 53, we found greatly decreased RNA synthesis in cells transfected with L, P, and N and infected with particles expressing a reporter template (Figure 3D). When incorporated into virions, the mutations resulted in no growth (Y53A and Y53D) or substantially attenuated growth (Y53F) (Figure 3E). They did not, however, affect in vitro transcription by L-P (Figure 3F). Anchoring of the P N terminus at the apex of the CTD (Figure 3G) thus contributes to RNA synthesis during viral infection but does not influence RNA polymerase activity directly. The most plausible interpretation of these results is that the defect is in replication, perhaps because of a failure to deliver N to the nascent RNA.
The interaction of rabies virus (RABV) P with RABV L is similar the VSV P-L interaction described here (Figure S4C), with contacts that anchor the CD, MT, and CTD and stabilize a closed conformation of L (Horwitz et al., 2019). The PL sequences for the two viruses have diverged substantially, however, and the specific molecular interactions differ. Recently determined structures of the respiratory syncytial virus (RSV) (Gilman et al., 2019) and human metapneumovirus (HMPV) (Pan et al., 2019) L-P complexes show that the tetrameric P proteins of those viruses contact only the RdRp and Cap domains of L (Figure S4C) and that those interactions do not fix the positions of the remaining domains.
Priming, Initiation, and Elongation
The loop between residues 1152 and 1173 in the Cap domain projects into the catalytic cavity of the RdRp and appears to be a priming loop to support the nucleotide that initiates polymerization. As shown in Figure 4A, superposition of the catalytic residues of L on corresponding residues of the reovirus λ3 initiation complex (Tao et al., 2002) (Table S2) places the initiating nucleotide so that it stacks directly on a prominent tryptophan (LTrp1167) at the tip of the VSV L priming loop. Mutation of this tryptophan to alanine severely restricts the initiation at the end of the genome or antigenome; mutation to tyrosine or phenylalanine has a smaller effect (Ogino et al., 2019). Mutation of the homologous tryptophan in RABV L also compromises the end initiation, but it appears not to affect the internal initiation of transcripts, and it does not prevent capping (Ogino et al., 2019).
Figure 4. Model for RNA Synthesis by the VSV L-P Complex.

(A) Model of an initiation complex. The priming loop of VSV L Cap is in green; the RdRp hairpin harboring polymerase active-site residues GDN is in cyan. Template and transcript nucleotides are from the crystal structure of the reovirus λ3 polymerase initiation complex (ReoV) (PDB: 1N1H) (Tao et al., 2002) superposed on the VSV L-P structure. The first phosphodiester bond of the transcript is formed between the nucleotide in the priming position and the incoming nucleotide. LTrp1167 supports the priming nucleotide by a stacking interaction with the base.
(B) Model of an elongation complex. To allow the passage of the growing template-transcript RNA duplex, shown here from the superposed structure of transcribing rotavirus VP1 polymerase (RotaV) (PDB: 6OJ6) (Jenni et al., 2019), the priming loop may retract to a position similar to that seen in the structure of the HMPV polymerase (PDB: 6U5O).
(C) Tunnels in VSV L-P. Structure is cut open to expose central cavities. RNA template and transcript strands are from the rotavirus structure as in (B). The template-entry channel is at the bottom. The template-exit channel is at the rear, bottom left. Nucleotide substrates enter the catalytic site laterally from the right (shown by an arrow). Catalytic sites with conserved residues for polymerization (RdRp), capping (Cap), and methylation (MT) are show by colored spheres. The tunnel was probed after omitting priming loop residues 1152–1173. Priming loop retraction into the space between the Cap domain and the CD (asterisk) opens a continuous path for the transcript RNA (dashed line).
See also Table S2.
Further polymerization requires retraction of the priming loop or, potentially, displacement of the entire Cap domain. The HMPV (Pan et al., 2019) and RSV (Gilman et al., 2019) polymerase structures show a fully retracted priming loop with no substantial displacement of the rest of the Cap domain. The priming loops of those two enzymes and the catalytic cavities of their Cap domains are similar, and we can show by superposition that the VSV priming loop can also retract without any major rearrangements in the rest of the Cap domain (Figure 4B). Moreover, when we model the VSV L priming loop based on the HMPV L protein structure, we find that a twisting tunnel connects the RdRp catalytic site with an opening where the CTD, RdRp, and MT all meet (Figure 4C). We suggest that this tunnel might be the exit pathway for uncapped products (i.e., the full-length antigenome and genome during replication and the uncapped leader during transcription), which bypass the active sites of the Cap and MT domains.
The N-terminal segment of P (PNTD) binds a single subunit of N, forming the so-called N0-P complex. The likely function of this complex is to deliver N to the nascent genome and antigenome as they emerge from the polymerase. About 15 residues connect the N-binding element (residues 6–34) with the segment of P docked onto the outer face of the CTD. The N0 passenger could therefore lie close to the proposed exit site for uncapped products. A possible explanation for the defect in Y53 mutants is the release of the docked N-terminal region of P from its site on the CTD, causing inefficient delivery of N to the replication products. This defect would not affect transcription.
At the other end of the P polypeptide chain, a small C-terminal domain (PCTD) interacts with the RNP. The binding site is at the C-terminal lobe of the N proteins (Green and Luo, 2009). Together with a presumptive interaction of the RNP with L at the template entry site, the contact with P could facilitate the passage of the template strand through the enzyme. We have proposed previously that the subunits of N (probably three) that must separate from the RNA template as it threads through the polymerase do not dissociate from each other and instead rebind the RNA as polymerization proceeds. The N-N interaction creates a chain-like array, with an N-terminal arm and a C-proximal loop contacting the two neighboring subunits in the chain (Ge et al., 2010; Green et al., 2006). Interaction with the C terminus of L-bound P could then ensure the correct reassociation of this putative chain of N subunits with exiting RNA (see Figure 3G).
Transcription of all products except the uncapped leader requires a different sequence of events. One possibility is that the immediately upstream transcript leaves, and the priming loop snaps back into the position seen in our current structure. Internal initiation does not appear to depend on LTrp1167, however; one possibility is that the 3′ end of the departing transcript, still base paired with a few residues of the oligo-U tract at the 3′ end of the upstream gene, supports the initiating NTP instead. The interaction would need to account for the intergenic GA sequence on the template. Further polymerization would then displace the upstream transcript (Shuman, 1997).
Acquisition of the guanylate cap occurs when the transcript length has reached at least 31 nucleotides (Tekes et al., 2011). If we suppose that the formation of the covalent intermediate with LHis1227 occurs before the addition of the 31st nucleotide to the transcript, then approximately 30 nucleotides of transcript would need to fit into the combined active-site cavities of the RdRp and Cap domain. The volume of the Cap active-site cavity, marked with an asterisk in Figure 4C, is 1.2 × 104 Å3. Assuming a volume occupied per Dalton (Da) of RNA (Vm) of 2.1 to 4.6 Å3 and an average mass of 340 Da per nucleotide (Speir and Johnson, 2012), the interior of the Cap domain could accommodate between 8 and 17 transcript nucleotides, in addition to the ~10 nucleotides still base paired with the template in the RdRp catalytic cavity. These relatively generous estimates suggest that the addition of the guanylate to the 5′ end of the transcript requires an opening of the multi-domain assembly from the closed conformation present in our structure. A plausible transition would be the release of the CD, MT, and CTD from their docked positions, as indeed occurs in the absence of P. If that transition also enabled a guanine nucleotide to access the RNA-protein covalent linkage, release of the capped 5′ end would allow it to diffuse efficiently into the MT active site, since both the transcript (still anchored to the RdRp at its growing, 3′ end) and the MT (linked to RdRp-Cap through the CD) cannot move far from each other.
The VSV L-P structure described here illustrates how the VSV P cofactor regulates L function by locking the domain organization into a single, pre-initiation configuration. It allows an explicit comparison with three other NNS viral L-P complexes shown in Figure S4C. In the VSV and RABV L-P complexes, an extended segment of P contacts several domains, thereby fixing the relative positions of the RdRp-Cap and CD-MT-CTD modules; the oligomerization domain of P does not participate in the contact with L. In the pneumovirus complexes, the oligomerization domain of P interacts directly with L, and the extended segment binds only the RdRp, allowing the three C-terminal domains to move freely about the linkers that connect them. A more complete analysis will require structures with RNP templates, such as transcribing intermediates, and studies of viral L proteins in complex with inhibitors.
STAR ★ METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for reagents may be directed to Stephen C. Harrison (harrison@crystal.harvard.edu). All unique reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement. Requests for reagents can be addressed to spjwhelan@wustl.edu.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Expression plasmids contained cDNA sequences from vesicular stomatitis virus (VSV) Indiana strain. Recombinant VSV-L protein was overexpressed in Sf21 insect cells. VSV-P protein (residues 35–106) was overexpressed in Escherichia coli Rosetta BL21 (DE3) cells (Novagen) (Liang et al., 2015).
METHOD DETAILS
Cryo-EM Structure Determination
The VSV L-P protein complex was prepared for structural analysis as previously described (Liang et al., 2015). Briefly, 3 μL of protein at ~0.35 mg/mL were applied to a C-flat grid (CF-1.2/1.3–4C, Protochips) that had been glow discharged at 40 mA for 30 s. Grids were plunge-frozen with an Vitrobot Mark II (FEI Company) after blotting for 3 s at 4°C and 85% relative humidity. 3307 movies of vitrified protein solution were collected using SerialEM (Mastronarde, 2005) on a Titan Krios electron microscope (ThermoFisher) at 300 kV and 59,000 nominal magnification, on a K2 Summit detector (Gatan, Inc.) operated in counting mode, resulting in a calibrated pixel size of 0.485 Å. Each movie contained 50 frames, collected in a 10 s exposure at 2 electrons/Å 2/frame. Most micrographs showed well-dispersed particles (Figure S1A). Movies were aligned and exposure-filtered using Unblur (Grant and Grigorieff, 2015). 3235 movies were selected for further processing based on the frame motion traces, excluding movies with traces suggesting large and zig-zag motion. The contrast transfer function (CTF) was determined using CTFFIND4 (Rohou and Grigorieff, 2015). CTF fringes on many micrographs were fitted to better than 3.0 Å resolution (Figure S1B). 188,004 particles were picked with IMAGIC (van Heel et al., 1996), command PICK_M_ALL. Particles were windowed into 400×400 pixel images and aligned using cisTEM (Grant et al., 2018) against the pixel size-adjusted 2015 map (EMD-6337) at an initial resolution of 8.0 Å, then refined at successively higher resolution, up to a resolution limit of 5.5 Å. Refinement was continued with 3D classification of two classes, at increasing resolution up to 4.0 Å, resulting in two reconstructions at 3.6 and 3.2 Å resolution (69,312 and 118,692 particles, respectively). Particles assigned to the 3.2-Å reconstruction were further refined at 4.0-Å resolution, and classified again into two classes with reconstructions at 3.8 and 3.1 Å resolution (34,713 and 83,979 particles, respectively). The goal of our classification was to obtain the highest possible resolution (globally). The discarded classes had poorer resolution throughout, not just in a particular domain. Further refinement of the best class at 4.0 Å resolution resulted in the final 3.0 Å reconstruction. The reconstruction was sharpened using cisTEM’s sharpening tool, amplifying signal at high resolution with a B factor-equivalent of 75 Å2.
Model Building and Refinement
We used the program O (Jones et al., 1991) to adjust our previously determined VSV L protein structure (PDB-ID 5A22, determined at 3.8 Å resolution) (Liang et al., 2015) to fit the 3.0 Å resolution cryo-EM map. We modeled the P protein residues de novo. We refined coordinates and B factors with phenix.real_space_refine (version 1.16–3549) (Afonine et al., 2018). In addition to the standard chemical and grouped B factor restraints, we used secondary structure, rotamer, and Ramachandran restraints (weight = 1.0, nonbonded_weight = 500, rama_weight = 6.0). We used phenix.mtriage (Afonine, 2017) and MolProbity (Chen et al., 2010) to calculate the validation results shown in Figure S1C and Table S1. The final VSV L-P model comprises L protein residues 35–1211, 1217– 1332, 1339–1590, and 1596–2109; P protein residues 49–56, 82–89, and 94–105; and two zinc atoms. Improved model regions between updated (PDB-ID 6U1X) and original (PDB-ID 5A22) coordinates (Figures S2 and S3) are as follows. RdRp residues: 61– 64 (loop remodeled, sequence register shift of 60–64), 414–430 (sequence register shift of 414–430), 587–593 (loop remodeled, sequence register shift of 586–593). Cap residues: 958 (loop adjusted), 1035–1046 (sequence register shift of 1035–1046), 1072– 1075 (loop remodeled), 1100 (loop adjusted), 1122–1125 (loop remodeled, sequence register shift of 1124–1126), 1164–1170 (loop remodeled, sequence register shift of 1161–1170), 1195–1232 (sequence register shift of 1195–1227), 1262 (loop adjusted), 1314 (loop adjusted), 1321–1332 (region adjusted). CD residues: 1380–1395 (region remodeled, sequence register shift of 1379– 1396), 1498 (loop adjusted), 1535–1539 (loop remodeled, sequence register shift of 1535–1538). MT residues: 1718 (loop adjusted), 1806–1851 (sequence register shift of 1805–1841). CTD residues: 1952–1955 (loop remodeled, sequence register shift of 1950– 1955), 2006–2008 (shift in β hairpin tip), 2066 (loop remodeled), 2086–2094 (loop remodeled, sequence register shift of 2084– 2094), 2109 (Cα in main chain density instead side chain density of 2108).
Projection Matching of Negative-Stain Class Averages
We extracted 20 negative-stain class averages of L-P dimers from a screenshot of a previously published figure (Figure S4C in Rahmeh et al. (2010)) using RELION-3 (Zivanov et al., 2018). From those dimer images we manually extracted 40 subparticles, one of each protomer, and masked them with a soft spherical mask. We obtained rotation angles for reference projections of our reconstruction with healpy (http://healpy.readthedocs.io ), the Python implementation of the HEALPix library of routines for equal area sampling of a sphere (Górski et al., 2005). Because the L-P complex has no symmetry and we did not allow mirroring of reference projections in the subsequent alignment step, we sampled angles on the entire sphere. We aligned the subparticle images to the reference projections with e2simmx.py from EMAN2 (Bell et al., 2016). Alignments were scored by calculating a cross-correlation coefficient (CCC). Because information in the negative-stain class averages is restricted to low resolution, reference projections from very different angles resulted in similar CCCs, and the “correct” solution was not always the top scoring projection. We therefore looked at the top five alignment peaks, which we located with the CCP4 program PEAKMAX (Winn et al., 2011), and then manually selected for further analysis the one whose reference projections most closely matched the particle image as judged by eye (Data S2). To calculate the distance between the P protein C termini of the two promoters, we transformed the model coordinates according to the determined alignment parameters. The analysis results for all class averages can be found in Data S2.
Functional Assays
Cells
BSR-T7 cells (a kind gift from K. Conzelmann) (Buchholz et al., 1999) were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Corning Inc., 10–013-CV) containing 10% fetal bovine serum (FBS; Tissue Culture Biologicals, TCB 101) at 37C and 5% CO2.
Plasmids
For mammalian expression, plasmids expressing Y53 mutants were generated by site-directed mutagenesis using the Q5 polymerase (New England Biolabs) on the parental P expressing plasmid (pMB-NS) (Pattnaik and Wertz, 1990) with dedicated primers (for Y53A: TAGGCCCTCTGCGTTTCAGGCAGC and GTATGCTCTTCCACTCCG, for Y53D: TAGGCCCTCTGATTTTCAGGC and GTATGCTCTTCCACTCCG, for Y53F: CATACTAGGCCCTCTTTTTTTCAGGCAGC and AGAGGGCCTAGTATGCTC, for deltaY53: TTTCAGGCAGCAGATGATTC and AGAGGGCCTAGTATGCTC). For bacterial expression and purification, fusion proteins made of a 6 histidine tag followed by the maltose-binding protein (MBP), the human rhinovirus type 14 (HRV) 3C protease cleavage site and the P protein, were cloned into a pET16b plasmid. The MBP tag was added to increase P solubility. A PCR product containing the 6xHis-MBP fragment was amplified from pET16b-MBP-MACV-Z (Kranzusch and Whelan, 2011) using AAGGAGATATACA TATGGCTCACCATCACCATCACC and CTGGAACAGTACTTCCAAACCAGAACCCTCGATCCCGAGGTTG, and one containing the HRV_3C-P fragment was amplified from pET16b-P (Rahmeh et al., 2012) using GAAGTACTGTTCCAGGGTCCTATGGATAATCT CACAAAAGTTC and GTTAGCAGCCGGATCCA. Both were inserted into pET16b-MBP-MACV-Z digested with NdeI and BamHI using the Infusion kit (Takara) to form pET16–6xHis-MBP-3Cc-P. Y53 mutations were introduced in pET16–6xHis-MBP-3Cc-P by site-directed mutagenesis with primers described above. To rescue viruses expressing P_Y53 mutants, Y53 mutations were introduced into pVSV1(+)-eGFP backbone (Whelan et al., 2000).
Protein purification
L and P proteins were purified as previously described (Rahmeh et al., 2012). MBP-P was purified from Rosetta cells (EMD Millipore) after induction with 0.3 mM IPTG overnight at 18°C. Cells were then pelleted and resuspended with 5 mL cold lysis buffer per gram of pellet (50 mM HEPES (pH 7.4), 100 mM NaCl, 5% glycerol, 5 mM imidazole, 5 mM 2-mercaptoethanol and 1X protease inhibitor cocktail [cOmpleteTM, Roche Cat. # 4693116001]). After sonication, 40 mL cell lysate were clarified by centrifugation at 20,000 × g for 20 min, and the supernatant was incubated with 2 mL of Ni-NTA beads (QIAGEN) overnight at 4°C. Beads were washed three times with lysis buffer (without protease inhibitor), and MBP-P proteins were eluted in 5 mL elution buffer (50 mM HEPES (pH 7.4), 100 mM NaCl, 5% glycerol, 5 mM imidazole and 5 mM 2-mercaptoethanol). The eluate was concentrated with a centrifugal filter (Amicon) and passed through a Superdex S200 size exclusion chromatography column (GE Healthcare) in storage buffer (20 mM HEPES (pH7.4), 100 mM NaCl, 5% glycerol and 5 mM 2-mercaptoethanol). MBP-P proteins eluted in a single peak and were kept at −80°C in storage buffer.
Virus
Attempts to rescue viruses expressing P_Y53 mutants, were performed as previously described (Whelan et al., 1995) using pVSV1(+)-eGFP-P_Y53x plasmids. VSV-eGFP-P_Y53F was the only mutant virus forming plaques. It was amplified and titered on BSR-T7 cells.
N-RNA purification
Nucleocapsid template was purified essentially as described previously (Ongrádi et al., 1985). To limit background activity due to residual P bound to N-RNA, nucleocapsids were extracted from a recombinant VSV expressing a monomeric P protein whose oligomerization domain has been deleted (VSV-PΔOD). Briefly, 10 mg of gradient purified virions were disrupted on ice for 1 h in 10 mL of virion disruption buffer (VDB: 20 mM Tris-HCl (pH 7.4), 0.1% Triton X-100, 5% glycerol, 1 mM EDTA, 2 mM dithioerythritol and 600 mM NH4Cl). Lysed virions were centrifuged at 240,000 × g for 3.5 h through a glycerol step gradient of 0.25 mL each of 40, 45, and 50% glycerol in VDB. Pellets were resuspended overnight in 0.5 mL of 10 mM Tris-HCl (pH 7.4), 100 mM NaCl, 1 mM EDTA and 1 mM DTT, and disrupted again on ice for 1 h after mixing with 0.5 mL of 2X high salt VDB (1X HSVDB: 20 mM Tris-HCl (pH 7.4), 0.1% Triton X-100, 5% glycerol, 1 mM EDTA, 2 mM dithioerythritol and 1.5 M NH4Cl). Nucleocapsids were isolated by banding on a CsCl gradient. In a 5 mL tube, 1 mL of disrupted virions was added at the top of a step gradient of 2 mL of each 20 and 40% (wt/wt) CsCl in HSVDB. After centrifugation at 190,000 × g for 3 h, N-RNA were recovered by side puncture and diluted fivefold in NaCl-Tris-EDTA-DTT buffer (NTED: 10 mM Tris-HCl (pH 7.4), 100 mM NaCl, 1 mM EDTA and 2 mM DTT). N-RNA was then centrifuged through a 0.5 mL cushion of 50% glycerol in NTED buffer, resuspended overnight in NTE buffer and stored in aliquots at −80°C.
Production of minigenome particles
Production of transcription competent minigenome particles was as described previously with minor modifications (Pattnaik and Wertz, 1990; Wertz et al., 1994). BSR-T7 cells were plated in a 60mm dish and infected the next day with a vaccinia virus expressing T7 polymerase (vTF7–3) (Fuerst et al., 1986) at multiplicity of infection (MOI) 3 for 1 h in Dulbeco’s Phosphate Buffered Saline liquid (DPBS; Sigma Cat# 59300C). Cells were then transfected using Lipofectamine 2000 with plasmids expressing N (5.5 μg), P (1.6 μg), M (1.25 μg), G (2.5 μg), L (0.5 μg) and a minigenome containing an eGFP reporter gene (7.5 μg). Four hours later, the medium was replaced with 3 mL DMEM containing 2% FBS. The cell supernatant containing the DI particles (DI stock) was harvested 2 days after transfection.
Cell-based gene expression assay
BSR-T7 cells were plated in a 96-well plate and infected the next day with a vaccinia virus expressing T7 polymerase (vTF7–3) at MOI 3 for 1 h in DPBS. Cells were then transfected with plasmids expressing N (91 ng), P (26 ng) and L (7.5 ng) using Lipofectamine 2000. Four hours later, cells in each well were infected with 3 μL DI stock in 30 μL DMEM for 1 h. Two days after transfection, GFP signal was measured using a Typhoon FLA 9500 scanner (GE Healthcare).
In vitro transcription assays
RNA synthesis assay on naked RNA was described previously (Morin et al., 2012). Briefly, purified L (100 nM) was incubated with purified P or MBP-P (250 nM) and a 19nt-long RNA corresponding to the first nucleotides of the genomic promoter (1.4 μM). Reactions were performed in 10 μL at 30°C for 5 h in presence of radiolabeled GTP (32P-αGTP, PerkinElmer) and analyzed on a 20% polyacrylamide/7 M urea gel. The gel was exposed overnight to a phosphor screen (GE Healthcare), which was scanned on a Typhoon FLA 9500 scanner. The transcription assay on encapsidated template was carried out as described elsewhere (Li et al., 2008) by incubating 1 μg of purified L protein with or without 0.5 μg of P or 1.2 μg of MBP-P proteins and 5 μg of N-RNA purified from VSV-PΔOD virions. Reactions were performed in 100 μL in the presence of 1 mM ATP, 0.5 mM CTP, GTP, and UTP, 15 μCi of radiolabeled GTP (32P-αGTP, PerkinElmer), 30% vol/vol rabbit reticulocyte lysate (Promega), 0.05 μg/μL actinomycin D (Sigma) in transcription buffer (30 mM Tris-HCl pH 8.0, 33 mM NH4HCl, 50 mM KCl, 4.5 mM Mg(OAC)2, 1 mM DTT, 0.2 mM spermidine and 0.05% Triton X-100) at 30°C for 5 h. RNA was extracted with Trizol reagent (Invitrogen Cat# 15596018) following manufacturer’s protocol, boiled at 100°C for 1 min, incubated on ice for 2 min, mixed with a 1.33 X loading buffer (33.3 mM citrate pH 3, 8 M urea, 20% sucrose and 0.001% bromophenol blue) and analyzed on a 25 mM citrate pH 3, 1.75% agarose, 6 M urea gel run for 18 h at 4°C and 180 V. Gels were fixed (in 30% methanol and 10% acetic acid), dried, and exposed overnight to a phosphor screen (GE Healthcare), which was scanned on a Typhoon FLA 9500 scanner.
Superposition of Atomic Models
To compare the VSV L-P structure to other viral polymerases, we used the program LSQMAN (Kleywegt and Jones, 1997) for superposition, except for the structure of the reovirus λ3 polymerase initiation complex, for which we also used the program O (Jones et al., 1991) to focus the alignment on the RdRp active site. Superposition statistics are summarized in Table S2.
Cavity Volume Calculation
We used the program VOIDOO (Kleywegt and Jones, 1994) to calculate the volume of the Cap active site cavity (Figure 4C, marked by an asterisk). The radius of the probe sphere was 1.4 Å. The calculated volume corresponds to the probe-occupied volume. We closed off the cavity of interest at the constrictions leading to the RdRp and MT active sites cavities, respectively, with dummy atoms.
Figure Preparation
We used MAFFT to calculate the amino acid multiple sequence alignments (Katoh et al., 2002). We prepared the figures with PyMol (The PyMOL Molecular Graphics System, Version 2.1 Schrödinger, LLC), Pov-Ray (www.povray.org), matplotlib (Hunter, 2007), and ESPript (Robert and Gouet, 2014). Secondary structure elements were assigned using DSSP (Kabsch and Sander, 1983).
QUANTIFICATION AND STATISTICAL ANALYSIS
In the plot of Figure 3B, the box extends from the lower to upper quartile values of the data. The line is at the median. The whiskers extending from the box show the range of the data. In Figure 3D, data are represented as mean ± SEM.
DATA AND CODE AVAILABILITY
The accession number for the EM 3-D reconstruction maps reported here is EMDB: 20614; the accession number for the atomic coordinates is PDB: 6U1X.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| Escherichia coli Rosetta BL21 (DE3) | MilliporeSigma | Cat#70954 |
| Vaccinia virus expressing T7 polymerase (vTF7-3) | Fuerst et al., 1986 | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Q5® High-Fidelity DNA Polymerase | New England Biolabs | Cat#M0491S |
| Protease inhibitor cocktail (cOmpleteTM) | Roche | Cat#4693116001 |
| Trizol reagent | Thermo Fisher Scientific | Cat#15596018 |
| Deposited Data | ||
| Atomic coordinates, VSV L-P structure | Protein Data Bank | PDB-ID: 6U1X |
| Cryo-EM density map, VSV L-P structure | Electron Microscopy Data Bank | EMD-20614 |
| Experimental Models: Organisms/Strains | ||
| Spodoptera frugiperda Sf21 cells | Thermo Fisher Scientific | Cat#B82501 |
| BSR-T7 cells | Buchholz et al., 1999 | N/A |
| Oligonucleotides | ||
| Primers for Y53A mutation: Forward: TAGGCCCTCTGCGTTTCAGGCAGC; Reverse: GTATGCTCTTCCACTCCG | This paper | N/A |
| Primers for Y53D mutation: Forward: TAGGCCCTCTGATTTTCAGGC; Reverse: GTATGCTCTTCCACTCCG | This paper | N/A |
| Primers for Y53F mutation: Forward: CATACTAGGCCCTCTTTTTTTCAGGCAGC; Reverse: AGAGGGCCTAGTATGCTC | This paper | N/A |
| Primers for deltaY53 mutation: Forward: TTTCAGGCAGCAGATGATTC; Reverse: AGAGGGCCTAGTATGCTC | This paper | N/A |
| Primers for MBP-tagging of the P protein: Forward: AAGGAGATATACATATGGCTCACCATCACCATCACC; Reverse: CTGGAACAGTACTTCCAAACCAGAACCCTCGATCCCGAGGTTG | This paper | N/A |
| Primers for P protein: Forward: GAAGTACTGTTCCAGGGTCCTATGGATAATCTCACAAAAGTTC; Reverse: GTTAGCAGCCGGATCCA | This paper | N/A |
| Recombinant DNA | ||
| pFastBac-Dual-GFP-L | Liang et al., 2015 | N/A |
| plasmid-P(35–106) | Liang et al., 2015 | N/A |
| pMB-NS | Pattnaik and Wertz, 1990 | N/A |
| pET16b-MBP-MACV-Z | Kranzusch and Whelan, 2011 | N/A |
| pET16b-P | Rahmeh et al., 2012 | N/A |
| pET16-6xHis-MBP-3Cc-P | This paper | N/A |
| pVSV1(+)-eGFP | Whelan et al., 2000 | N/A |
| Software and Algorithms | ||
| SerialEM | Mastronarde, 2005 | https://bio3d.colorado.edu/SerialEM/ |
| Unblur | Grant and Grigorieff, 2015 | https://cistem.org/ctffind4 |
| CTFFIND4 | Rohou and Grigorieff, 2015 | https://cistem.org |
| IMAGIC | van Heel et al., 1996 | https://www.imagescience.de |
| cisTEM | Grant et al., 2018 | https://cistem.org |
| O | Jones et al., 1991 | http://xray.bmc.uu.se/alwyn/TAJ/Home.html |
| phenix.real_space_refine | Afonine et al., 2018 | https://www.phenix-online.org |
| phenix.mtriage | Afonine, 2017 | https://www.phenix-online.org |
| MolProbity | Chen et al., 2010 | http://molprobity.biochem.duke.edu |
| RELION-3 | Zivanov et al., 2018 | https://www3.mrc-lmb.cam.ac.uk/relion/index.php/Main_Page |
| healpy, HEALPix | Górski et al., 2005 | http://healpy.readthedocs.io |
| EMAN2 | Bell et al., 2016 | https://blake.bcm.edu/emanwiki/EMAN2 |
| CCP4 | Winn et al., 2011 | https://www.ccp4.ac.uk/ |
| LSQMAN | Kleywegt and Jones, 1997 | http://xray.bmc.uu.se/usf |
| VOIDOO | Kleywegt and Jones, 1994 | http://xray.bmc.uu.se/usf |
| MAFFT | Katoh et al., 2002 | https://mafft.cbrc.jp/alignment/software/ |
| PyMol | Schrödinger, LLC | https://pymolwiki.org/index.php/Main_Page |
| Pov-Ray | N/A | https://www.povray.org |
| matplotlib | Hunter, 2007 | https://matplotlib.org |
| ESPript | Robert and Gouet, 2014 | http://espript.ibcp.fr |
| Other | ||
| C-flat grids (CF-1.2/1.3-4C) | Protochips | Cat#CF-1.2/1.3-4C |
Highlights.
Cryo-EM structure of VSV L protein with bound P cofactor at 3.0 Å resolution
P interacts with multiple domains of L to lock it in an initiation competent state
Conserved tryptophan in priming loop supports transcription-initiating nucleotide
L structure shows potential RNA exit channel for full-length replication product
ACKNOWLEDGMENTS
We acknowledge support from NIH grant R37 AI059371 to S.P.J.W. N.G. and S.C.H. are investigators in the Howard Hughes Medical Institute.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.12.024.
REFERENCES
- Afonine PV (2017). phenix.mtriage: a tool for analysis and validation of cryo-EM 3D reconstructions. Comput. Crystallogr. Newsl 8, 25. [Google Scholar]
- Afonine PV, Poon BK, Read RJ, Sobolev OV, Terwilliger TC, Urzhumtsev A, and Adams PD (2018). Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D Struct. Biol 74, 531–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell JM, Chen M, Baldwin PR, and Ludtke SJ (2016). High resolution single particle refinement in EMAN2.1. Methods 100, 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchholz UJ, Finke S, and Conzelmann KK (1999). Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J. Virol 73, 251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H, Green TJ, Lu S, and Luo M (2006). Crystal structure of the oligomerization domain of the phosphoprotein of vesicular stomatitis virus. J. Virol 80, 2808–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuerst TR, Niles EG, Studier FW, and Moss B (1986). Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc. Natl. Acad. Sci. USA 83, 8122–8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge P, Tsao J, Schein S, Green TJ, Luo M, and Zhou ZH (2010). Cryo-EM model of the bullet-shaped vesicular stomatitis virus. Science 327, 689–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman MSA, Liu C, Fung A, Behera I, Jordan P, Rigaux P, Ysebaert N, Tcherniuk S, Sourimant J, Eléouët JF, et al. (2019). Structure of the Respiratory Syncytial Virus Polymerase Complex. Cell 179, 193–204.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Górski KM, Hivon E, Banday AJ, Wandelt BD, Hansen FK, Reinecke M, and Bartelmann M (2005). HEALPix: A Framework for High-Resolution Discretization and Fast Analysis of Data Distributed on the Sphere. Astrophys. J 622, 759–771. [Google Scholar]
- Grant T, and Grigorieff N (2015). Measuring the optimal exposure for single particle cryo-EM using a 2.6 Å reconstruction of rotavirus VP6. eLife 4, e06980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant T, Rohou A, and Grigorieff N (2018). cisTEM, user-friendly software for single-particle image processing. eLife 7, e35383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green TJ, and Luo M (2009). Structure of the vesicular stomatitis virus nucleocapsid in complex with the nucleocapsid-binding domain of the small polymerase cofactor, P. Proc. Natl. Acad. Sci. USA 106, 11713–11718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green TJ, Zhang X, Wertz GW, and Luo M (2006). Structure of the vesicular stomatitis virus nucleoprotein-RNA complex. Science 313, 357–360. [DOI] [PubMed] [Google Scholar]
- Horwitz JA, Jenni S, Harrison SC, and Whelan SPJ (2019). Structure of a rabies virus polymerase complex from electron cryo-microscopy. bioRxiv. 10.1101/794073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter JD (2007). Matplotlib: A 2D graphics environment. Comput. Sci. Eng 9, 90–95. [Google Scholar]
- Iverson LE, and Rose JK (1981). Localized attenuation and discontinuous synthesis during vesicular stomatitis virus transcription. Cell 23, 477–484. [DOI] [PubMed] [Google Scholar]
- Jenni S, Salgado EN, Herrmann T, Li Z, Grant T, Grigorieff N, Trapani S, Estrozi LF, and Harrison SC (2019). In situ Structure of Rotavirus VP1 RNA-Dependent RNA Polymerase. J. Mol. Biol 431, 3124–3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, and Kjeldgaard M (1991). Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47, 110–119. [DOI] [PubMed] [Google Scholar]
- Kabsch W, and Sander C (1983). Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K, and Miyata T (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleywegt GJ, and Jones TA (1994). Detection, delineation, measurement and display of cavities in macromolecular structures. Acta Crystallogr. D Biol. Crystallogr 50, 178–185. [DOI] [PubMed] [Google Scholar]
- Kleywegt GJ, and Jones TA (1997). Detecting folding motifs and similarities in protein structures. Methods Enzymol. 277, 525–545. [DOI] [PubMed] [Google Scholar]
- Kranzusch PJ, and Whelan SP (2011). Arenavirus Z protein controls viral RNA synthesis by locking a polymerase-promoter complex. Proc. Natl. Acad. Sci. USA 108, 19743–19748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Rahmeh A, Morelli M, and Whelan SP (2008). A conserved motif in region v of the large polymerase proteins of nonsegmented negative-sense RNA viruses that is essential for mRNA capping. J. Virol 82, 775–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang B, Li Z, Jenni S, Rahmeh AA, Morin BM, Grant T, Grigorieff N, Harrison SC, and Whelan SPJ (2015). Structure of the L Protein of Vesicular Stomatitis Virus from Electron Cryomicroscopy. Cell 162, 314–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, McDonald SM, Tortorici MA, Tao YJ, Vasquez-Del Carpio R, Nibert ML, Patton JT, and Harrison SC (2008). Mechanism for coordinated RNA packaging and genome replication by rotavirus polymerase VP1. Structure 16, 1678–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde DN (2005). Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol 152, 36–51. [DOI] [PubMed] [Google Scholar]
- Morin B, Rahmeh AA, and Whelan SP (2012). Mechanism of RNA synthesis initiation by the vesicular stomatitis virus polymerase. EMBO J. 31, 1320–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogino M, Gupta N, Green TJ, and Ogino T (2019). A dual-functional priming-capping loop of rhabdoviral RNA polymerases directs terminal de novo initiation and capping intermediate formation. Nucleic Acids Res. 47, 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ongrádi J, Cunningham C, and Szilágyi JF (1985). The role of polypeptides L and NS in the transcription process of vesicular stomatitis virus New Jersey using the temperature-sensitive mutant tsE1. J. Gen. Virol 66, 1011–1023. [DOI] [PubMed] [Google Scholar]
- Pan J, Qian X, Lattmann S, El Sahili A, Yeo TH, Jia H, Cressey T, Ludeke B, Noton S, Kolocsay M, Fearns R, and Lescar J (2019). Structure of the human metapneumovirus polymerase phosphoprotein complex. Nature, Published online November 7, 2019. 10.10.38/s41586-019-1759-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattnaik AK, and Wertz GW (1990). Replication and amplification of defective interfering particle RNAs of vesicular stomatitis virus in cells expressing viral proteins from vectors containing cloned cDNAs. J. Virol 64, 2948–2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahmeh AA, Schenk AD, Danek EI, Kranzusch PJ, Liang B, Walz T, and Whelan SP (2010). Molecular architecture of the vesicular stomatitis virus RNA polymerase. Proc. Natl. Acad. Sci. USA 107, 20075–20080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahmeh AA, Morin B, Schenk AD, Liang B, Heinrich BS, Brusic V, Walz T, and Whelan SP (2012). Critical phosphoprotein elements that regulate polymerase architecture and function in vesicular stomatitis virus. Proc. Natl. Acad. Sci. USA 109, 14628–14633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert X, and Gouet P (2014). Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohou A, and Grigorieff N (2015). CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol 192, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuman S (1997). A proposed mechanism of mRNA synthesis and capping by vesicular stomatitis virus. Virology 227, 1–6. [DOI] [PubMed] [Google Scholar]
- Speir JA, and Johnson JE (2012). Nucleic acid packaging in viruses. Curr. Opin. Struct. Biol 22, 65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y, Farsetta DL, Nibert ML, and Harrison SC (2002). RNA synthesis in a cage–structural studies of reovirus polymerase lambda3. Cell 111, 733–745. [DOI] [PubMed] [Google Scholar]
- Tekes G, Rahmeh AA, and Whelan SP (2011). A freeze frame view of vesicular stomatitis virus transcription defines a minimal length of RNA for 5′ processing. PLoS Pathog. 7, e1002073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Heel M, Harauz G, Orlova EV, Schmidt R, and Schatz M (1996). A new generation of the IMAGIC image processing system. J. Struct. Biol 116, 17–24. [DOI] [PubMed] [Google Scholar]
- Wertz GW, Whelan S, LeGrone A, and Ball LA (1994). Extent of terminal complementarity modulates the balance between transcription and replication of vesicular stomatitis virus RNA. Proc. Natl. Acad. Sci. USA 91, 8587–8591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan SP, Ball LA, Barr JN, and Wertz GT (1995). Efficient recovery of infectious vesicular stomatitis virus entirely from cDNA clones. Proc. Natl. Acad. Sci. USA 92, 8388–8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan SP, Barr JN, and Wertz GW (2000). Identification of a minimal size requirement for termination of vesicular stomatitis virus mRNA: implications for the mechanism of transcription. J. Virol 74, 8268–8276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, et al. (2011). Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, and Scheres SH (2018). New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the EM 3-D reconstruction maps reported here is EMDB: 20614; the accession number for the atomic coordinates is PDB: 6U1X.