

Abstract
Spurred by recent progress in medicinal chemistry, numerous lead compounds have sprung up in the past few years, although the majority are hindered by hydrophobicity, which greatly challenges druggability. In an effort to assess the potential of platinum (Pt) candidates, the nanosizing approach to alter the pharmacology of hydrophobic Pt(IV) prodrugs in discovery and development settings is described. The construction of a self-assembled nanoparticle (NP) platform, composed of amphiphilic lipid-polyethylene glycol (PEG) for effective delivery of Pt(IV) prodrugs capable of resisting thiol-mediated detoxification through a glutathione (GSH)-exhausting effect, offers a promising route to synergistically improving safety and efficacy. After a systematic screening, the optimized NPs (referred to as P6 NPs) exhibited small particle size (99.3 nm), high Pt loading (11.24%), reliable dynamic stability (~7 days), and rapid redox-triggered release (~80% in 3 days). Subsequent experiments on cells support the emergence of P6 NPs as a highly effective means of transporting a lethal dose of cargo across cytomembranes through macropinocytosis. Upon reduction by cytoplasmic reductants, particularly GSH, P6 NPs under disintegration released sufficient active Pt(II) metabolites, which covalently bound to target DNA and induced significant apoptosis. The PEGylation endowed P6 NPs with in vivo longevity and tumor specificity, which were essential to successfully inhibiting the growth of cisplatin-sensitive and -resistant xenograft tumors, while effectively alleviating toxic side-effects associated with cisplatin. P6 NPs are, therefore, promising for overcoming the bottleneck in the development of Pt drugs for oncotherapy.
Keywords: glutathione, nanoparticle, pharmacokinetics, pharmacodynamics, platinum(IV)
Graphical Abstract
The huge changes brought about by automated synthesis, combinatorial chemistry, and high-throughput screening have contributed to the production of a massive number of potent drug candidates as well as the introduction of more hydrophobic drugs into the pharmaceutical pipeline.1–4 In the discovery setting, it is important to recall that, as far as hydrophobic drugs are concerned, the likelihood of poor absorption or permeation hits occurs according to Lipinski’s rule of five.5,6 Unfortunately, the vast majority of drug substances under development exhibit poor solubility in water, which seriously hinders their clinical translation. Several strategies, such as complexation with cyclodextrins,7 conjugation to dendrimers,8 salification of ionizable drugs,9 cosolvency,10 and solubilization11 have demonstrated some value in overcoming the obstacle of solubility but have not always enhanced therapeutic benefit in drug treatment. This persistent problem is in part attributable to the suboptimal performance of the above formulations in parameters such as burst or stagnant release, specificity, and body clearance.
Over the past decade, advanced nanoscale system-mediated drug delivery has received tremendous attention for the treatment of a myriad of chronic diseases,12–23 most notably cancer, owing to the complexity of tumor biology, where untamed cell growth and protein transcription contribute to highly disorganized vasculature and congested extravascular environments (also known as the enhanced permeation and retention, or EPR, effect).24,25 With nanoparticle (NP) technology, it may be possible to (1) achieve delivery of poorly water-soluble candidates, (2) protect cargos from premature degradation or detoxification, (3) control pharmacokinetic and biodistribution profiles, (4) enrich drug delivery in a tissue- or cell-specific manner, and (5) improve intracellular penetration.26–29 As a result, a variety of NPs have been widely pursued to obtain safer and more-effective therapeutic modalities for hydrophobic drugs used in cancer treatment.30–34 Nevertheless, the current progress of NPs is still far from acceptable at the clinical due to various limitations. One of the more notable problems is that most reported NPs contain a substantial quantity of natural or synthetic materials that improve therapeutic performance at the cost of biosafety.
The attempt to devise platinum (Pt) drugs that surpass the antitumor potency of cisplatin has produced many candidates that display considerable promise in preclinical models; however, only a handful evoke the expected response in the human body.35,36 This failure may be associated with poor circulation and tumor nonselectivity as well as deactivation mechanisms that are mediated by thiol-containing species, e.g., glutathione (GSH) and metallothionein (MT), abundant in the cytoplasm.37,38 The irreversible thiol-induced alterations in the chemical structures of Pt candidates, particularly those of Pt(II) substances, render them ineffective in tumor therapy (through so-called detoxification).39 The aforementioned unfavorable pathways that sacrifice in vivo Pt pharmacology can be circumvented by adopting more-mature NP platforms or substituting more-inert Pt(IV) prodrugs.35 Recently, we demonstrated the synergistic effect of loading Pt drugs into poly(disulfide amide) NPs that, upon entry into cells, scavenged up-regulated GSH to reverse cisplatin resistance.40 Herein, the concept of self-assembled Pt(IV) prodrug NPs is proposed to address this issue by precisely tuning axial ligands from octahedral structures but not altering the pharmacophores that are ultimately released by reduction following cellular uptake.41,42
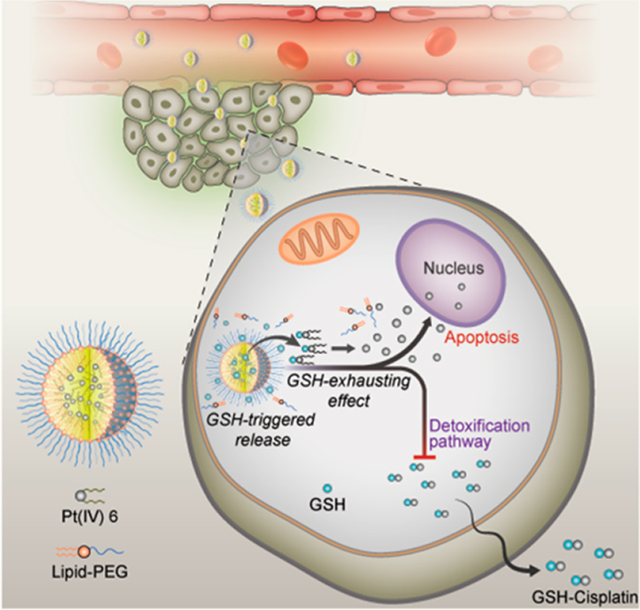
As proof of concept, we developed a series of Pt(IV) prodrugs with tunable hydrophobicity and sensitive redox behavior and designed a redox-responsive Pt(IV) prodrug platform that self-assembled with lipid-polyethylene glycol (lipid-PEG) to form Pt(IV) NPs. The process by which the nanoconstructs, P6 NPs (the optimal formulation after systematic optimization), function is depicted schematically in Figure 1. P6 NPs with desirable physicochemical properties were endocytosed through macropinocytosis with high efficiency. After that, the consumption of GSH resulted in the disassembly of NP structures and the untying of Pt centers tethered by superhydrophobic ligands, inducing significant mitochondrial control of apoptosis. This result could be attributed to the fact that the GSH-exhausting effect decreased the likelihood of thiol-mediated detoxification while simultaneously restoring the Pt sensitivity of tumor cells. PEG-functionalization methodology imparted P6 NPs with the ability to control their pharmacokinetics and biodistribution, which was verified by in vivo longer blood circulation and higher tumor accumulation. As a result, the PEGylated Pt(IV) 6 delivery platform achieved minimal off-target side-effects and full pharmacological outcomes, even inducing the reversal of cisplatin resistance.
Figure 1.

Schematic illustration of self-assembled Pt(IV) NPs for specific delivery of Pt drugs and effective suppression of cisplatin-resistant tumors. Redox-responsive P6 NPs were self-assembled with superhydrophobic Pt(IV) 6 and coated with amphiphilic lipid-PEG via nanoprecipitation. Benefiting from the extended blood circulation and selective tumor accumulation, P6 NPs could be endocytosed into tumor cells through macropinocytosis and then disintegrated by consumption of cytoplasmic thiol-containing species, especially GSH. The redox-triggered process contributed to the release of Pt(II) ions and their reduced probability of deactivation, which went on to rapidly diffuse into nuclei and covalently bind large amounts of DNA, ultimately resulting in the mitochondria-controlled apoptosis of cisplatin-resistant tumors.
RESULTS AND DISCUSSION
Self-Assembly and Characterization of P6 NPs
Pt(IV) prodrugs were prepared by oxidation of square-planar cisplatin, followed by the addition of hydrophobic ligands to the intermediates (Figure S1 and Table S1).41,43,44 The detailed synthesis and exhaustive characterization of those analogous octahedral prodrugs, namely cis,cis,trans-[Pt(NH3)2Cl2(OOC-(CH2)nCH3)2] (where n is 0, 2, 4, 6, 8, 10, 12, or 14), are presented in the Supporting Information (Figures S2–S9). The 1H nuclear magnetic resonance (NMR) spectra of Pt(IV) 1–8 all contained a typical resonance at approximately 7.00 ppm, consistent with protons on ammine ligands bound to a Pt(IV) center. For Pt(IV) 1–3, each addition of 2 methylene units to the carboxylate chain produced a well-resolved resonance in the aliphatic region that displayed the expected 1H–1H coupling pattern. As additional methylene linkers were added from Pt(IV) 3 to Pt(IV) 8, the overall intensity of the CH2 signal at approximately 1.40 ppm consistently increased. 13C NMR spectra all displayed resolved peaks for the expected number of carbon atoms despite nearly overlapping signals in some of the longer chains. Pt(IV) 1–5 have been described in investigations aimed at evaluating the effect of lipophilicity on biological activity through various quantitative structure–activity relationship (QSAR) approaches.45,46 It was found that the best QSAR model for the cytotoxicity of Pt(IV) prodrugs always contained a lipophilic descriptor, suggesting that the number of CH2 groups in the carboxylate chain increased lipophilicity as well as cytotoxicity. Pt(IV) 6–8, however, were rarely reported, mainly owing to their superhydrophobicity. For example, Pt(IV) 5 with decanoate ligands was readily soluble in the DMSO at concentrations up to 50 mg/mL at 25 °C, while for Pt(IV) 8, which had hexadecanoate ligands, a precipitate rapidly formed as the warm DMSO of equivalent concentration was cooled to room temperature.
Because Pt(IV) 1 –8 serve as prodrugs of cisplatin, reduction potentials are key pharmacological parameters to estimate their activities.47 Pt(IV) prodrugs with fairly high reduction potential do achieve fast reduction, followed by a series of severe side-effects, whereas prodrugs with relatively low reduction potential are not reduced quickly enough, even in tumors, and are excreted intact in significant amounts.48 Pt(IV) 1–8 prodrugs were investigated by cyclic voltammetry in dimethylformamide (DMF)/phosphate-buffered serum (PBS) solution at different pH values using KCl as the supporting electrolyte (Figures S10–S17). Electrochemical studies revealed the expected irreversible reduction maxima in cyclic voltammograms, corresponding to the loss of axial ligands, which resulted in a change of coordination geometry from octahedron to square-planar. Furthermore, reduction potentials of all Pt(IV) prodrugs were very similar, indicating that the increase in chain length did not significantly influence the electron density in Pt(IV) centers, identified with density functional theory calculations.46 However, as compared with reduction potentials extrapolated to 0 mV/s at pH 7.4, there were slightly positive shifts for all prodrugs at pH 6.0. For instance, the reduction potential of Pt(IV) 6 occurred −0.6183 V at pH 7.4 but −0.5581 V at pH 6.0. The more-facile reduction reflected the protonation of axial ligands, which dissociated upon the reduction of Pt atoms. This property might significantly affect the activity of Pt(IV) prodrugs in the low-pH environment of many solid tumors and inside endosomes.49,50
Self-assembled Pt(IV) NPs (designated as P1–8 NPs) were prepared by nanoprecipitation of the above Pt(IV) prodrugs with lipid-polyethylene glycol (PEG), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-3000] (ammonium salt) (denoted as DP 3000). Studies of the precise effect that varying the length of methylene linkers in Pt(IV) prodrugs had on the particle size and Pt loading identified Pt(IV) 6 as an ideal candidate for NP formulation (Table S2). When the carboxylate chain increased from ethyl to hexadecyl, the particle size of the corresponding NPs systematically increased from 45.6 to 158.7 nm, as well as Pt loading from 0.50% to 12.67%. The increased lipophilicity was beneficial for the self-assembly process of Pt(IV) prodrugs and the formation of hydrophobic NP cores during nanoprecipitation. Axial ligands with longer methylene spacers might also finely tune the ζ potential from electropositivity to electronegativity, due to the increasing density of the exterior PEG. Ultimately, P6 NPs were chosen for further evaluation owing to their small particle size (99.3 nm) and high Pt loading (11.24%) capacity (Figure 2a). Moreover, P6 NPs were proven colloidally stable in PBS over the course of 1 week (Figures 2b and S18).
Figure 2.

PEGylated Pt(IV) 6 delivery platform exhibition of redox-triggered degradation. (a) Histogram of particle-size distribution of P6 NPs obtained by DLS. (b) Particle-size and ζ-potential changes of P6 NPs monitored over the course of 1 week. (c, d) TEM morphologies of P6 NPs stored in (c) water or (d) 10 mM DTT at 25 °C for 24 h.
TEM was utilized to monitor disintegration of NPs in response to dithiothreitol (DTT) (Figure 2c,d). Unlike PBS, the incubation of P6 NPs with 10 mM DTT for 24 h contributed to the rapid degradation of spherical NPs into irregularly shaped debris, suggesting the redox-reactive disruption of nanostructures. Platinum released from NPs under 0, 1, or 10 mM DTT buffered with phosphate was measured by inductively coupled mass spectrometry (ICP-MS; Figure S19). A total of ~70% of the payload was liberated over the course of 48 h incubation of P6 NPs with 10 mM DTT. Meanwhile, ~30% was released following incubation in PBS containing 1 mM DTT and less than 15% was released in PBS alone, further supporting the redox-promoted discharge of payloads from P6 NPs.
Cytotoxicity of P6 NPs via Macropinocytosis and Redox-Triggered Disassembly
The extent of internalization was evaluated by treating A2780 and A2780cis cells with 50 or 100 μM cisplatin, P6 NPs, or Pt(IV) 6 solution (denoted P6 Soln) for 4 or 18 h (Figure 3a–d). ICP-MS and BCA protein assay confirmed that P6 NPs entered cells in both time- and concentration-dependent manners. Notably, P6 NPs were taken up by tumor cells much more effectively than cisplatin and P6 Soln. In addition, the cellular uptake of cisplatin by A2780cis cells sharply decreased in comparison to A2780 cells, whereas at least a 3-fold increase of P6 NPs could be achieved under the same conditions. Subsequently, to investigate the mechanism by which NPs were internalized, A2780 and A2780cis cells were co-incubated with various endocytic markers labeled with Alexa-Fluor@ 488 and P6 NPs using Dil as a fluorescent probe. P6 NPs co-localized with dextran, a fluid-phase marker known to enter cells via macropinocytosis but not with cholera toxin subunit B or transferrin, markers of caveolae- or clathrin-mediated endocytosis,51,52 as shown in Figures 4a and S20a. Moreover, one of the hallmarks of such intracellular trafficking was membrane ruffling and actin rearrangement, which was observed in A2780 and A2780cis cells within 15 min of NP application (Figures 4b and S20b).53 The effects of the macropinocytosis inhibitor, 5-N-ethyl-N-isoproamiloride (EIPA), and the actin polymerization inhibitor, cytochalasin D, on NP uptake were tested to further validate our findings.52 As shown in Figure 3e,f, both of the inhibitors dose-dependently lowered NP trafficking, indicating that the majority of P6 NPs likely entered tumor cells through macropinocytosis.
Figure 3.

Self-assembly of Pt(IV) 6 into NPs quantitatively maximized cellular uptake of Pt, which was easily interfered with by macropinocytosis inhibitors. (a–d) Pt uptake into (a, b) A2780 and (c, d) A2780cis cells after 4 and 18 h of exposure to cisplatin, P6 NPs, or P6 Soln at Pt concentrations of (a, c) 50 and (b, d) 100 μM. (e, f) Prior exposure of A2780 and A2780cis cells to (e) EIPA and (f) cytochalasin D reduced uptake of Dil-P6 NPs in a dose-dependent fashion.
Figure 4.

Confocal laser scanning microscopy utilization to visualize cytoplasmic delivery of P6 NPs. (a) Dil-P6 NPs were incubated with A2780cis cells in the presence of Alexa-Fluor 488-labeled markers of various endocytic pathways. NPs co-localized with dextran, a fluid-phase marker known to enter cells via macropinocytosis (white arrows). (b) Alexa-Fluor 488-labeled actin fibers revealed membrane ruffling and actin rearrangement (white arrows), hallmarks of uptake by macropinocytosis.
Intracellular disintegration of P6 NPs was then explored by Förster resonance energy transfer (FRET), a mechanism describing molecular distance-dependent energy transfer between a donor chromophore (Coumarin 6) and an acceptor chromophore (Nile red).54 Because the FRET pair is in the range of 1–10 nm, upon photoexcitation of Coumarin 6 at 410 nm, green fluorescence is induced (520 nm) and part of the emission is transferred to Nile red, resulting in red fluorescence (590 nm). However, complete separation of the chromophore pair severely attenuates the FRET effect, restoring green fluorescence and extinguishing red. In cisplatin-sensitive cells incubated with Coumarin 6 and Nile red-P6 NPs for 4 h, the chromophore pair undergoing efficient FRET gave off intense dual fluorescent signals, whereas in cisplatin-resistant cells, the FRET image was much weaker (Figure 5a). This difference might be attributed to up-regulated levels of thiol-containing species in A2780cis cells, which promoted the more-rapid separation of Coumarin 6 and Nile red. As the incubation time was prolonged to 18 h, only green fluorescence was captured in both cell lines, suggesting complete disassembly of the nanostructures. Nevertheless, once cells were pretreated with N-ethylmaleimide (NEM), a molecule inhibitor of GSH, persistent FRET could be easily observed, further demonstrating the interaction of intracellular thiol-containing species with the decomposition of NPs.55
Figure 5.

Rapid intracellular disintegration of P6 NPs accompanied by a GSH-exhausting effect. (a) A2780 and A2780cis cells incubated with Coumarin 6 and Nile red-P6 NPs for 4 and 18 h. To investigate the effect of GSH on NP disintegration, cells were also pretreated with NEM to consume cytosolic GSH. Images were taken under a 60× objective. (b, c) GSH and (d, e) GSSG percent of (b, d) A2780 and (c, e) A2780cis cells treated with cisplatin, P6 NPs, or P6 Soln at Pt concentrations of 0, 1.56, 6.25, or 25 μM.
Following uptake, Pt(IV) prodrugs are recovered to Pt(II) ions in the presence of abundant reductants in cellular milieu (namely, GSH, MT, and ascorbate). In the meantime, the competitively covalent binding between GSH and Pt(II) centers plays a crucial role in cellular detoxification for Pt chemotherapeutics. These GSH-consuming behaviors contribute to sequential transcription of γ-glutamylcysteine synthetase and γ-glutamyl transpeptidase, up-regulating GSH.37 Thus, intracellular reduced (GSH) and oxidized (GSSG) glutathione were assessed to investigate reduction of P6 NPs (Figure 5b–e). The significant increase in GSH percent was observed for both A2780 and A2780cis cells exposed to cisplatin, which was believed to be a consequence of restored GSH biosynthesis in response to GSH consumption during detoxification. Importantly, the increase in GSH percentage for A2780cis cells (from 3.76% to 10.92%) was more significant, while a slight increase (from 1.54% to 1.71%) was recorded for A2780 cells, demonstrating the much-more-sensitive GSH biosynthesis of cisplatin-resistant cells. Conversely, P6 NPs restrained the growth of GSH by greedily consuming intracellular GSH (from 1.56% to 0.48% for A2780 cells and from 3.80% to 1.23% for A2780cis cells). Additionally, unlike cisplatin’s limited effect on GSSG level (from 15.24% to 16.00% for A2780 cells and from 38.69% to 38.40% for A2780cis cells), the redox reaction induced by Pt(IV) prodrugs and GSH accounted for the formation of GSSG, accompanying its dramatically increased level (from 15.62% to 16.74% for A2780 cells and from 38.15% to 46.85% for A2780cis cells). Similar trends were evident for P6 Soln, albeit more moderate than P6 NPs, owing to lower uptake. In addition, intracellular GSH in cisplatin-resistant cells was twice as high as that in cisplatin-sensitive cells, which was identical with our postulation in FRET study and those reported previously.56
Subsequently, intracellular fate of Pt(II) metabolites after reduction was investigated. Figure S21 shows a snapshot of Pt distribution in each subcellular fraction of A2780 and A2780cis cells after treatment with Pt chemotherapeutics. Pt distribution of A2780cis cells treated with P6 NPs was observably higher than A2780 cells, while the opposite conclusion was drawn for cells undergoing cisplatin treatment, which was in good agreement with cellular uptake work. In addition, it was not surprising that cytoplasmic fractions were shown to have the largest Pt contents, accounting for more than 70% of the total Pt incorporated into tumor cells. This revealed that, despite the lipophilic character of Pt(IV) 6, it did not become trapped in the lipid bilayer.
The nuclear fractions are of special interest because they contain DNA, the ultimate target of Pt drugs. Compared with cells treated with cisplatin and P6 Soln, a larger proportion of Pt released from P6 NPs accumulated in the nucleus, indicative of more Pt-DNA adduct generation. This observation was further demonstrated by assessing the ability of cisplatin, P6 NPs, or P6 Soln to platinate nuclear DNA (Figure S22). The extent of DNA platination was determined to be 0.31 ng/mg for cisplatin, 7.32 ng/mg for P6 NPs, and 0.80 ng/mg for P6 Soln in the case of A2780 cells versus 0.22 ng/mg for cisplatin, 10.58 ng/mg for P6 NPs, and 1.18 ng/mg for P6 Soln in the case of A2780cis cells. For cells treated with NPs, the sharply increased amounts in Pt-DNA adduct might be driven by the Pt concentration gradient between the cytoplasm and nucleus and the avoidance of GSH-mediated detoxification.
Next, cytotoxicity in multiple cell lines, i.e., A2780, A2780cis, PC-3, MCF7, HCT116, A549, and H460 was extensively examined and their IC50 were calculated from the dose dependence of surviving cells after exposure to cisplatin, P6 NPs, or P6 Soln for 48 or 72 h (Figure 6a,b). Unlike cisplatin, which displayed limited toxicity against the cell lines evaluated (except A2780 and PC-3 cells), P6 NPs displayed the lowest IC50 values across the cells investigated. The significant increase in cytotoxicity of P6 NPs was mirrored by the dramatic increase in cellular uptake, suggesting that enhanced anti-proliferation could be attributed in large part to increased uptake. As expected, P6 Soln had modest potency in killing those cancer cells, with IC50 values lower than P6 NPs but higher than cisplatin. In vitro anticancer activity was also assessed by a live/dead cell assay (Figure S23). Live cells are distinguished by the presence of esterase activity, determined by intracellular enzymatic conversion of the non-fluorescent cell-permeant calcein AM to green-fluorescent calcein. Ethidium homodimer-1 enters dead cells with compromised membranes and undergoes an enhancement of red fluorescence upon binding to nucleic acids. A2780 and A2780cis cells treated with P6 NPs (the most potent member) for 24 h were mostly dead, while those that underwent cisplatin treatment had mostly survived. The observation in live/dead cell viability agrees with cytotoxicity data.
Figure 6.

P6 NPs enhancement of toxicity to various tumor types through the augmentation of apoptosis. (a, b) In vitro cytotoxicity of cells treated with cisplatin, P6 NPs, or P6 Soln for (a) 48 or (b) 72 h. (c, d) In vitro apoptosis of (c) A2780 and (d) A2780cis cells treated with cisplatin, P6 NPs, or P6 Soln for 24 h.
Using a dual-staining Annexin V/PI flow cytometry assay, apoptosis was studied in A2780 and A2780cis cells exposed to cisplatin, P6 NPs, or P6 Soln for 24 h (Figure 6c,d). Although both cells displayed dose-dependent apoptosis, a more-efficient Pt-induced apoptosis was found in A2780 cells, even at a low concentration of 1 μM. Consistently, P6 NPs promoted the largest population of A2780 and A2780cis cells to undergo early- and late-stage apoptosis. This result agrees well with the findings from our cytotoxicity experiment and may also be the result of the drastic difference in cellular uptake. Normally, apoptotic cells exhibit morphological changes with chromatin condensation and nuclear fragmentation, all of which can be identified by Hoechst staining. As shown in Figure S24, nuclei with homogeneous fluorescence were found in PBS- and DP 3000-treated A2780 and A2780cis cells; P6 NPs caused the most-severe fragmentation of nuclei, which had segmented into dense nuclear parts and been distributed into apoptotic bodies. Fluorescent images showed characteristic apoptotic changes following treatment of cells with P6 Soln but were much more moderate than those produced by P6 NPs.
Pharmacokinetics and Biodistribution
Although the number of exciting in vitro results is encouraging, in vivo application of the PEGylated Pt(IV) 6 delivery platform remained to be investigated. Therefore, pharmacokinetics was tested first; plots of Pt concentrations as a function of time are presented in Figure 7a. Cisplatin was eliminated from the blood circulation rapidly, whereas lipid-PEG or Tween 80 formulated Pt(IV) 6 produced much more stable and mild decline curves, suggesting the longer retention of superhydrophobic Pt (IV) prodrugs. Both a non-compartment model and a two-compartment model were used to study pharmacokinetic behaviors (Table S3). Non-compartmental methods estimate the exposure to a drug by integrating the area under the curve of a concentration—time graph and thus produce accurate results for bioequivalence evaluation. AUC0→inf and AUMC0→inf of P6 NPs and P6 Soln were all much higher than those of cisplatin, revealing enhanced bioavailability. CL of P6 NPs and P6 Soln decreased by ~70% versus cisplatin, confirming a significant retardation in clearance. The volume of distribution at steady state (Vss) of P6 NPs was about 1.8 times higher than that of P6 Soln, indicating greater tissue distribution. The Vss of cisplatin showed an anomalous increase to 0.79 L/kg, almost the same as P6 NPs. It might be speculated that fluid and blood retention caused by cisplatin-induced renal failure and cisplatin-plasmatic protein binding were the leading causes. MRT0→inf of P6 NPs and P6 Soln were remarkably extended compared to cisplatin, i.e., 4.3- and 2.2-fold, respectively, suggesting prolonged blood circulation. Classical compartmental methods use kinetic models to describe the concentration—time curve, and thus, they are based on a consideration of an organism as a number of related compartments. P6 NPs with mild distribution and elimination phases decreased A, α, B, and β. Rate constants (k10, k12, and k21) declined as well. Conversely, t1/2α and t1/2β both being more than two times larger than those of P6 NPs and P6 Soln greatly prolonged Pt’s presence in blood.
Figure 7.

Pharmacokinetics and biodistribution of P6 NPs. (a) Pharmacokinetics of cisplatin, P6 NPs, or P6 Soln in BALB/c mice (n = 3). (b–d) Biodistribution of (b) cisplatin, (c) P6 NPs, or (d) P6 Soln in A2780cis tumor-bearing athymic nude mice (n = 3). All data were normalized by weight or volume, i.e., microgram per gram or milligram per liter.
Next, A2780cis tumor-bearing athymic nude mice were given a single dose of cisplatin, P6 NPs, or P6 Soln to investigate Pt biodistribution (Figure 7b–d). After the intravenous injection of cisplatin, the liver showed the highest Pt concentration, with the kidney, plasma, spleen, tumor, lung, and heart following, suggesting severe hepatic and renal toxicity. Nevertheless, both P6 NPs and P6 Soln showed much lower Pt levels in the liver and kidney, the primary target organs of cisplatin, while Pt amounts in the mononuclear phagocyte system (MPS), i.e., the spleen and lung, were slightly enriched, especially in the case of P6 Soln. Unlike cisplatin and P6 Soln, Pt(IV) prodrugs encapsulated in NPs were released gradually over time, exposing the organs to a low-dose drug environment, whereas maximal Pt concentration was found in the tumor. This characteristic of P6 NPs could help avoid high peak drug levels and their severe toxicological effects on the organs, while maintaining strong pharmacological responses to tumors. Figure 7c also shows that over the course of 48 h following administration of P6 NPs, the second largest proportion of Pt is in the plasma, a favorable indication for the probability to promote more circulating Pt into target sites via the EPR effect. Prolonged drug residence in blood is a prerequisite for the realization of a nanostrategy for controlled systemic delivery of cisplatin in the form of a prodrug for cancer treatment.
To systemically validate biodistribution of the PEGylated Pt(IV) 6 delivery platform, near-infrared (NIR) imaging was employed to investigate trends of fluorescent signals from hydrosoluble (Cy5.5) or liposoluble (DID) probes in single or multiple tumor-bearing athymic nude mice (Figures 8, S25, and S26). After the intravenous injection of PBS to A2780cis tumor-bearing athymic nude mice, no fluorescence or a weak signal from background noise was captured during different time frames. Compared with free Cy5.5 and DID, both Cy5.5- or DID-P6 NPs exhibited completely different distribution behaviors, accumulating in tumors gradually. It appeared that the specific tumor enrichment of fluorescent signals from NPs was independent of dye species. Moreover, unlike DID, which was widely distributed throughout the bodies of multiple tumor-bearing athymic nude mice, DID-P6 NPs preferentially accumulated in A2780 (X), A2780cis (Y), and H460 (Z) xenograft tumors at the same time. Thus, it was concluded that tumor types may have a limited effect on the NPs’ ability to preferentially accumulate within tumor tissue. Ex vivo images showed that mice dosed with hydrosoluble Cy5.5 had probes mainly in the kidney (their point of excretion), then the lung, liver, tumor, spleen, plasma, and heart. However, liposoluble DID, which was metabolized by the liver, reduced probes in most tissues but had slightly improved content in the plasma. In contrast, in A2780cis tumor-bearing athymic nude mice treated with Cy5.5- or DID-loaded NPs, more signals were detected in the tumor and plasma, demonstrating good tumor accumulation of P6 NPs through the blood circulation and EPR effect. Besides, the plasma, organs, and tumors collected from multiple tumor-bearing athymic nude mice exhibited a fluorescence distribution pattern similar to those found in single tumor-bearing athymic nude mice. Additionally, these results correlated well with the good long circulation confirmed by pharmacokinetics.
Figure 8.

NIR images of dye or dye-loaded NPs in single or multiple tumor-bearing athymic nude mice. In vivo fluorescence images of mice captured using the Maestro 2 in vivo imaging system (n = 3).
Subsequently, the organs and tumors with immunofluorescence staining were utilized to validate in vivo disintegration of NPs. As displayed in Figure S27, P6 NPs effectively reduced probe distribution to off-target organs and primarily bypassed the MPS, resulting in much lower accumulation in the liver, spleen, and lung compared with free probes, suggesting a much milder side-effect profile. Meanwhile, at tumor sites, NPs readily extravasated microvessels, permeated the parenchyma, and disassembled inside cells, ultimately leaving red fluorescence across the entire microscopic field, indicating the potential for superior therapeutic performance.
Pharmacodynamics and Reversal Mechanism of Cisplatin Resistance
Next, antitumor evaluation and survival analysis were carried out using both A2780 and A2780cis xenograft models. After tumors had grown to ~100 mm3, animals were randomly assigned and injected once weekly with the following regimen: (I) PBS (control), (II) cisplatin at 4 mg Pt/kg, (III) P6 NPs at 4 mg Pt/kg, (IV) P6 Soln at 4 mg Pt/kg, or (v) DP 3000 (equal in weight to that of P6 NPs).
During in vivo antitumor evaluation, tumor volume and body weight were monitored at two-day intervals for 4 weeks (Figures 9a, 10a, and S28). The results showed that tumors in mice with subcutaneous A2780 cells initially grew fairly slowly, followed by exponential growth. Tumors in mice after the implantation of A2780cis cells, however, underwent unchecked growth throughout the observation period. Specifically, tumor volumes of PBS- and DP 3000-dosed groups increased sharply with time in both A2780 and A2780cis tumor-bearing athymic nude mice. Tumor growth during cisplatin treatment was well controlled in A2870 xenograft model but was largely unaffected in A2780cis xenograft model, confirming that cisplatin was unable to alter the natural progression of drug-resistant oophoroma. In comparison, all mice receiving P6 NPs developed tumors with average volumes no more than 250 mm3, suggesting that these tumors, including those developed in cisplatin-resistant cell lines, all grew at a decelerated rate. P6 Soln achieved similar biological responses but was less efficacious than the NP formulation. These results were further confirmed by full tumor images (Figures 9b and 10b). The tumor inhibition rates (TIRs) of Pt chemotherapeutics are listed in Table S4. Upon the termination of the regimen, for A2780 xenograft model, the TIR of 84.71% for cisplatin was comparable to 85.99% for NPs, slightly larger than 71.28% for P6 Soln. For the A2780cis xenograft model, however, the TIR for cisplatin declined drastically to 12.65% and was nearly the same as that of DP 3000 (9.42%), indicating that they were equally ineffective in cisplatin-resistant cancer therapy. In contrast, TIRs for P6 NPs (88.69%) and P6 Soln (72.82%) were well-maintained in xenograft tumors that were tolerant to their parent drugs. In addition, cisplatin-induced weight loss was observed at the dose studied for mice treated with P6 Soln, whereas those treated with P6 NPs under the same conditions had minimal changes in weight, implying a good prognosis for cancer therapy.
Figure 9.

P6 NPs mediation of an antitumor effect in a A2780 xenograft model. (a) Tumor growth curve of each mouse from different groups of A2780 tumor-bearing athymic nude mice during chemotherapy (n = 5). (b) The harvested A2780 xenograft tumors after systemic treatment captured using the Maestro 2 in vivo imaging system. (c) Western blot quantification of p53, Caspase 3, PARP, and cleaved PARP for A2780 xenograft tumors following treatment with PBS, cisplatin, P6 NPs, P6 Soln, or DP 3000.
Figure 10.

P6 NPs mediation of an antitumor effect in a A2780cis xenograft model. (a) Tumor growth curve of each mouse from different groups of A2780cis tumor-bearing athymic nude mice during chemotherapy (n = 5). (b) The harvested A2780cis xenograft tumors after systemic treatment captured using the Maestro 2 in vivo imaging system. (c) Western blot quantification of p53, Caspase 3, PARP, and cleaved PARP for A2780cis xenograft tumors following treatment with PBS, cisplatin, P6 NPs, P6 Soln, or DP 3000.
During in vivo survival analysis, deaths and physical performance were monitored during mimic cyclic chemotherapeutic dosing; time to natural death or euthanization was analyzed using Kaplan—Meier estimates. Of the mice subcutaneously challenged with A2780 cells, the median survival for PBS and DP 3000 treatment was 42 and 44 days, and administration of cisplatin and P6 Soln slightly increased this time to 50 and 54 days, respectively (Figure S29a). In stark contrast, P6 NPs maximally suppressed proliferation of A2780 xenograft tumors for up to 63 days, a significant improvement over both free parent drugs and prodrugs. Moreover, to determine whether the tumoricidal activity of P6 NPs could be demonstrated in a cisplatin-resistant xenograft model, the same process was conducted in A2780cis tumor-bearing athymic nude mice. As shown in Figure S29b, mice treated with PBS and DP 3000 all died from tumor burden between days 24 and 34 after the first injection (mean survival of 30 days in control mice and 32 days in mice receiving DP 3000). Importantly, mice receiving NP therapy benefitted greatly in terms of survival, with 80% still alive at the end of the experiment. Conversely, the cisplatin group hit 0% around day 30 (mean survival of 28 days), while P6 Soln-dosed mice survived longer but were all dead by day 63 (mean survival of 52 days). This suggests that the degree of growth inhibition observed with cisplatin is not adequate to have an impact on Pt-resistant tumors, whereas P6 NPs remain effective in both cases.
To elucidate the contribution of P6 NPs to tumor killing, a follow-up study aiming to uncover apoptotic pathways was performed in which Pt chemotherapeutics were intravenously administered for four doses. The comparative molecule expression (p53, Caspase 3, PARP, and cleaved PARP) in A2780 and A2780cis xenograft tumors was detected by Western blot. Cisplatin is known to exert its antitumor effect by disrupting DNA structure in nuclei through the formation of intra- and interstrand cross-links. Once activated in response to DNA damage induced by Pt-DNA adducts, the reinforced p53 destabilizes the Bcl-2 family balance between pro-apoptotic and antiapoptotic regulators.57 The imprisonment of antiapoptotic molecules and freeing of pro-apoptotic members go on to signal apoptosis to mitochondria. Following mitochondrial dysfunction, the apoptotic executor Caspase 3 is activated and thus promotes PARP cleavage-mediated cellular decomposition, ultimately leading to cell lysis and necrosis.58 As verified by blots and their quantification in Figures 9c, 10c, and S30, P6 NPs elicited the greatest p53 expression in both A2780 and A2780cis xenograft tumors. Thus, the caspase cascade was activated, in line with expectations. The dramatically upregulated caspase 3 started to catalyze the cleavage of PARP, yielding a minimal amount of intact PARP and a large fraction of cleaved PARP, supporting the proposed mitochondrial control of apoptosis. A similar trend was observed in tumors from mice treated with P6 Soln but a less-efficient effect than that elicited by NPs. In particular, cisplatin had an unsatisfactory pharmacological profile because although it successfully completed the mitochondrial signaling pathway in cisplatin-sensitive tumors, it was thoroughly blocked from initiating apoptosis in cisplatin-resistant tumors.
Current studies of cisplatin resistance demonstrate that tolerance is mediated through two broad mechanisms: an insufficient amount of Pt being able to reach DNA and a failure to achieve cell death after Pt-DNA adduct formation.39 As delineated earlier, the PEGylated Pt(IV) 6 delivery platform with longer blood circulation achieved higher tumor accumulation via the EPR effect. Following high efficiency uptake, up-regulated GSH in A2780cis cells promoted the rapid disintegration of P6 NPs and reduction of Pt(IV) prodrugs, leading to decreased GSH levels. This GSH-exhausting process was expected to minimize GSH-induced detoxification, providing an adequate source of Pt, increasing the likelihood of covalent binding between surviving Pt(II) ions and DNA bases in nuclei. Further, whether the other mechanism attributable to cisplatin resistance could be interfered with by P6 NPs was tested (Figures S31 and 32). After Pt-DNA adduct formation, cell death failed because of enhanced translesion DNA synthesis, aberrant apoptotic signals, or up-regulated nucleotide excision repair (NER).39 It is reported that DNA polymerases β and η are particularly important for bypassing Pt-DNA adducts and allowing translesion synthesis, thereby maintaining tumor proliferation. As a scaffold to recruit these polymerases involved in DNA replication,59,60 PCNA expression was investigated. Compared to free cisplatin and P6 Soln, administration of P6 NPs markedly reduced the percentage of proliferating PCNA-positive cells, revealing superior efficiency in inhibiting translesion synthesis of both tumor types. Moreover, drug resistance to cisplatin and other chemotherapeutics usually occurs via the loss of apoptotic signaling pathways mediated by various proteins such as p53, Bcl-2 family members, c-Jun N-terminal kinase (JNK), etc.61 As previously illustrated, once inside the nuclei, preferentially covalent binding between aqua cisplatin and N7 atoms contributes to intra- and interstrand cross-links on DNA. Being induced by this DNA damage, p53 promotes the transcription of pro-apoptotic BAX to neutralize antiapoptotic Bcl-2 on the mitochondrial membrane, which leads to mitochondria-controlled apoptosis. The most obvious upregulation of p53 in A2780 and A2780cis xenograft models after NP treatment has been demonstrated by Western blot analysis. P6 NPs here greatly increased BAX and decreased Bcl-2, reflecting the activation of apoptotic signals. Generally, cisplatin-resistant cells have been shown to increase DNA repair capacity in comparison to sensitive counterparts. The protein XPA appears to play a key role in NER at sites of damage as a scaffold for the repair protein ERCC1, to ensure that cisplatin lesions are appropriately removed from DNA.62,63 The evaluation of both A2780 and A2780cis tumors harvested from mice dosed with P6 NPs indicated the lowest ERCC1 and XPA levels, consistent with successful inhibition of tumor growth. Additionally, with particular attention focused through TUNEL assay, P6 NPs were proven to induce the most-extensive DNA fragmentation among all regimen, regardless of whether in A2780 or A2780cis tumors, confirming that NP treatment led to significant growth arrest and apoptosis in vivo.
Biosafety Evaluation
In spite of cisplatin’s ubiquitous application in oncology, numerous side effects have also been identified since its promising clinical trials. To investigate systemic toxicity, histopathology of organs and tumors collected from A2780 and A2780cis tumor-bearing athymic nude mice undergoing successive chemotherapy was performed with hematoxylin and eosin (H&E) staining (Figures S33 and 34). Pathological examination yielded no evidence of any obvious changes in organs tested from mice injected with PBS, P6 NPs, or DP 3000. In comparison, mice after cisplatin treatment presented important organ damage, such as liver injury with hyperemia, swelling, and vacuolar degeneration; incomplete spleen structure with diminished and shrunken white pulp; and collapsed alveoli with slight hemorrhage.64 The most serious symptoms induced by cisplatin were hyperemia, edema, and inflammatory infiltration of the kidney, accompanied by hydropic or ballooning degeneration of proximal tubular epithelial cells, cytoplasmic relaxation, and exudates from glomerular capsules, supporting its severe nephrotoxicity. Similar lesions were occasionally observed in organs harvested from mice dosed with P6 Soln. The antitumor effect of successive chemotherapy was also evaluated via pathological investigation of tumors. Upon the termination of the regimen, tumors harvested from PBS and DP 3000 groups displayed spherical or spindle cells with larger nuclei and abundant chromatin and karyosomes, signs of extensive proliferation. Notably, tumors exposed to P6 NPs displayed fragmented or shrunken cells, destruction of organelles, chromatin condensation, and pyknotic nuclei undergoing karyolysis or karyorrhexis, indicative of tumor necrosis.64 A2780 xenograft tumors treated with cisplatin presented similar necrosis, whereas vigorous proliferation was found in A2780 xenograft tumors, supporting the better curative effect of NPs over current cisplatin treatment. Necrosis elicited by P6 Soln in both tumor types was typically less severe than that produced by P6 NPs.
During the clinical progression of cisplatin, major side-effects stem from its cumulative and dose-dependent hepatotoxicity and nephrotoxicity.65 Thus, the blood biochemistry of ALT, AST, ALP, BUN, and Cre in plasma taken from A2780 and A2780cis tumor-bearing athymic nude mice was measured 24 h after the last injection (Figures S35 and 36). Cisplatin, owing to its harsh renal side-effects, significantly increased levels of BUN and Cre. Meanwhile, as a cytotoxic chemotherapeutic, multidose cisplatin led to the up-regulation of ALT, AST, and ALP, highlighting serious hepatotoxicity. Similarly, up-regulated levels of all the above indices were found in both xenograft models following repeated administration of P6 Soln. P6 NPs given at the same doses, conversely, had a negligible effect on all tested biomarkers compared with PBS or DP 3000, suggesting substantially less nephrotoxicity and hepatotoxicity. These results serve to reinforce the pathology work done in vivo, which described the role of multidose Pt drugs in regulating systemic toxicity.
As stated above, one of most serious complications of cisplatin regimen is acute renal failure (ARF), which has been increasingly appreciated with the involvement of IL-1β, IL-18, IL-6, and TNF-α.66 These cytokine levels in the kidney of healthy BALB/c mice were found 24 or 72 h after a single bolus injection of PBS, cisplatin, P6 NPs, P6 Soln, or DP 3000. As indicated in Figures S37 and 38, the up-regulation of renal IL-1β, IL-18, IL-6, and TNF-α following cisplatin treatment supported its notably injurious role in ARF. Treatment with P6 Soln after 24 h produced significant differences only in the concentration of IL-18, whereas mice undergoing P6 Soln after 72 h showed dramatic increases in all cytokines. However, IL-1β, IL-18, IL-6, and TNF-α in kidney homogenates from NP group were always within normal range, indicating that P6 NPs successfully mitigated the nephritic toxicity of Pt-based chemotherapy at the recommended dosage.
Subsequently, hemocompatibility was examined by incubation erythrocytes with cisplatin, P6 NPs, P6 Soln, DP 3000, and Tween 80 at gradient concentrations (Figure S39). Results indicated that free cisplatin, P6 NPs, and DP 3000 exhibited minimal hemolysis (<8%) across all tested concentrations, unlike Tween 80 controls (a commercial excipient intended for injectable use), suggesting that P6 NPs had better hemocompatibility. P6 Soln, which had the same excipient in an equal amount, displayed concentration-dependent hemolysis behavior similar to Tween 80.
CONCLUSIONS
A library of Pt(IV) prodrugs having various hydrophobic carboxylates and similar reduction potentials was prepared and formulated with amphiphilic lipid-PEG to obtain Pt(IV) NPs. Following systematic screening, the self-assembled P6 NPs with small particle size, high Pt loading, and reliable dynamic stability were subjected to in vitro and in vivo evaluation. Compared to free cisplatin with its short half-life and poor tumor selectivity (which was readily inactivated by cytoplasmic thiol-containing species, particularly GSH, at tumor sites), P6 NPs achieved longer blood circulation, leading to higher tumor accumulation through the EPR effect. Following cellular uptake through macropinocytosis, high levels of intracellular GSH promoted rapid disintegration of NPs and reduction of Pt(IV) prodrugs. As expected, this GSH-exhausting process was beneficial in minimizing thiol-mediated detoxification of recuperative Pt drugs, contributing to more-extensive covalent binding between Pt(II) ions and purine bases, ultimately allowing mitochondrial control of apoptosis. The nanostrategy of largely exhausting cytosolic GSH served a dual purpose: both fulfilling the conversion of parent drugs and protecting the cargos from detoxification, finally reactivating the pharmacology of Pt(IV) prodrugs. Therefore, these prepared P6 NPs can be used as promising and effective platforms for cancer therapy.
EXPERIMENTAL SECTION
Preparation of Pt(IV) NPs
The self-assembled Pt(IV) nanoparticles (NPs) coated with lipid-PEG in PBS were prepared as follows: 4.5 mg of Pt(IV) 6 and 10.5 mg of DP 3000 were dissolved in 0.3 mL of DMSO to form a homogeneous solution. Under vigorous stirring, the mixture was slowly added to 5.7 mL of water. The remaining free molecules and organic solvent were removed by washing with PBS (3 × 5.0 mL) using an Amicon Ultra-15 centrifugal filter (MWCO 100 kDa, Millipore). The retained P6 NPs were dispersed in 1.0 mL of PBS for further use. Dil-P6 NPs, Coumarin 6, and Nile red-P6 NPs, Cy5.5-P6 NPs, or DID-P6 NPs were prepared by mixing predetermined amounts of Pt(IV) 6, DP 3000, and dye (3 w/w% Dil, 0.1 w/w% Coumarin 6, and 1 w/w% Nile red, 3 w/w% Cy5.5, SE, or 3 w/w% DID) in DMSO, then following the nanoprecipitation procedure described above. P6 Soln was obtained by dissolving 2.0 mg of Pt(IV) 6 and 0.2 mL of Tween 80 in 0.4 mL of alcohol.
Endocytic Pathway
A2780 and A2780cis cells were seeded in 35 mm sterile glass bottom culture dishes (20 000 cells per dish) and incubated with 1 mL of RPMI-1640 medium containing 10% fetal bovine serum (FBS) for 24 h. Subsequently, cells were incubated with Dil-P6 NPs at 5 μg/mL. Alexa-Fluor 488-labeled cholera toxin subunit B, transferrin, or dextran (Thermo Fisher Scientific) was added during the final 15 min of NP incubation prior to nuclear staining with Hoechst 33342. Images were acquired with a Yokogawa Spinning Disk Confocal Microscope (Andor, UK). For examination of actin ruffling, cells were serum starved for 1 h, followed by the addition of P6 NPs for 15 min, fixed with 4% paraformaldehyde, permeated with 0.1% saponin, and stained with Alexa-Fluor 488-labeled phalloidin (Thermo Fisher Scientific,) and Hoechst 33342, respectively.
Endocytosis Interference by Inhibitors
A2780 and A2780cis cells were seeded in 6-well plates (50 000 cells per well) and incubated with 1 mL of RPMI-1640 medium containing 10% FBS for 24 h. After pre-incubation with 5-N-ethyl-N-isoproamiloride (EIPA) or cytochalasin D at different concentrations for 1 h, Dil-P6 NPs were added in the presence of inhibitors. A total of 2 hours later, cells were washed with PBS 3 times, collected, and analyzed via flow cytometry (FACSAriaTM III, BD).
NIR Imaging and IF Staining
The tumor-targeting capability of NPs was further investigated using a non-invasive near-infrared (NIR) optical imaging technique. A2780cis tumor-bearing athymic nude mice were randomly assigned to three groups (female, n = 3) and given an intravenous injection of (I) PBS, (II) Cy5.5, or (III) Cy5.5-P6 NPs at 1.0 mg Cy5.5/kg. A series of NIR images were collected from mice under isoflurane anesthesia using the Maestro 2 In Vivo imaging system (CRi Inc.). After that, mice were sacrificed, and their plasma, organs, and tumors were collected for ex vivo imaging. Organs and tumors were fixed with 4% paraformaldehyde, embedded in paraffin, and cut into sections for immunofluorescence (IF) staining. To image vasculatures, slices were heated at 60 °C for 1 h and then washed with xylene, ethanol, and PBS three times. After blocking with 10% FBS for 1.5 h, slices were incubated with rat anti-mouse CD31 antibodies (Abcam) at 4 °C for 1 h, washed with PBS/0.2% Triton X-100 three times, and then incubated with Alexa-Fluor 488-conjugated secondary antibodies (Goat anti-rat IgG, Abcam) for 1 h. Thereafter, slices were washed with PBS three times, stained with Hoechst 33342, and viewed under a CLSM.
A2780cis tumor-bearing athymic nude mice were randomly assigned to three groups (female, n = 3) and given an intravenous injection of (I) PBS, (II) DID, or (III) DID-P6 NPs at 1.0 mg DID/kg. Then, other procedures were the same as those described above.
A2780 (X), A2780cis (Y), and H460 (Z) tumor-bearing athymic nude mice were randomly assigned to 2 groups (female, n = 3) and given an intravenous injection of (I) DID or (II) DID-P6 NPs at 1.0 mg DID/kg. Then, all other procedures were the same as those described above.
In Vivo Antitumor Evaluation
A2780 and A2780cis tumor-bearing athymic nude mice were randomly assigned to 5 groups (female, n = 5) and intravenously injected with (I) PBS, (II) cisplatin, (III) P6 NPs, (IV) P6 Soln, or (V) DP 3000 at 4.0 mg Pt/kg. The amount of DP 3000 injected was equal in weight to that of P6 NPs. The mice were injected 4 times at 7-day intervals, with the first injection day designated day 0. Tumor volume was determined every 2 days with a caliper and calculated according to the equation V = a2 × b × 0.5, where a and b are the shortest and longest perpendicular diameters. Body weight was also recorded every 2 days to indicate systemic toxicity. After tumor monitoring, all mice were euthanized. Tumors were harvested, weighed, and imaged using the Maestro 2 In Vivo Imaging System. TIR was calculated according to the equation: TIR (%) = (m − mt)/m × 100, where m is the average weight of tumors in the PBS group, and mt is the average weight of tumors in the other groups.
IHC, TUNEL, and H&E
A2780 and A2780cis tumor-bearing athymic nude mice were randomly assigned to five groups (female, n = 5) and intravenously injected with (I) PBS, (II) cisplatin, (III) P6 NPs, (IV) P6 Soln, or (V) DP 3000 at 4.0 mg Pt/kg. The mice were injected four times at 7-day intervals. After that, mice were sacrificed, and their organs and tumors were quickly collected, immediately fixed with 4% paraformaldehyde, embedded in paraffin, and cut into sections.
Immunohistochemistry (IHC) of tumors was conducted to evaluate expression of proliferating cell nuclear antigen (PCNA), Bcl-2 associated X (BAX), B-cell lymphoma 2 (Bcl-2), excision repair cross-complementation group 1 (ERCC1), and Xeroderma pigmentosum A (XPA). Slices were incubated with corresponding primary antibodies (Cell Signaling or Abcam) and evaluated using the HRP/DAB Detection IHC Kit (Abcam) according to the manufacturer protocol.
The terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick-end labeling (TUNEL) of tumors was performed using an in situ Cell Death Detection Kit (Roche, Germany) according to the manufacturer protocol. All organs and tumors were stained with H&E before undergoing histopathological examination with an Upright Microscope (BX63, Olympus, Japan).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the U.S. National Institutes of Health grant no. HL127464, American Heart Association (AHA) 18CSA34080399, the David H. Koch-Prostate Cancer Foundation Program in Cancer Nanotherapeutics, China Scholarship Council grant no. 201507060015, National Natural Science Foundation of China grant no. 81730108, Key Project of Zhejiang province Ministry of Science and Technology grant no. 2015C03055, and Key Project of Hangzhou Ministry of Science and Technology grant nos. 20162013A07 and 20142013A63.
Footnotes
The authors declare the following competing financial interest(s): O.C.F. has financial interests in Selecta Bio-sciences, Tarveda Therapeutics, Placon Therapeutics, and Seer.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.8b06400.
Additional experimental details and spectral analysis results; figures showing synthetic routes, NMR results, cyclic voltammograms, polydispersity index change, Pt release profiles, fluorescence imaging, subcellular distribution, Pt-DNA adducts, cellular and nuclear staining, whole-body imaging, co-localization, tumor size and body weight changes, Kaplan—Meier survival curves, Western blot results, IHC and TUNEL results, H&E sections, serum levels, and hemolysis rates; tables showing Pt(IV) and corresponding NPs, NP characteristics, pharmacokinetics, and tumor-inhibition rates (PDF)
REFERENCES
- (1).Fahr A; Liu X Drug Delivery Strategies for Poorly Water- Soluble Drugs. Expert Opin. Drug Delivery 2007, 4, 403–416. [DOI] [PubMed] [Google Scholar]
- (2).Hird NW Automated Synthesis: New Tools for the Organic Chemist. Drug Discovery Today 1999, 4, 265–274. [DOI] [PubMed] [Google Scholar]
- (3).Corbett PT; Leclaire J; Vial L; West KR; Wietor JL; Sanders JK; Otto S Dynamic Combinatorial Chemistry. Chem. Rev 2006, 106, 3652–3711. [DOI] [PubMed] [Google Scholar]
- (4).Mayr LM; Bojanic D Novel Trends in High-Throughput Screening. Curr. Opin. Pharmacol 2009, 9, 580–588. [DOI] [PubMed] [Google Scholar]
- (5).Lipinski CA; Lombardo F; Dominy BW; Feeney PJ Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Delivery Rev 2001, 46, 3–26. [DOI] [PubMed] [Google Scholar]
- (6).Lipinski CA Drug-Like Properties and the Causes of Poor Solubility and Poor Permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [DOI] [PubMed] [Google Scholar]
- (7).Davis ME; Brewster ME Cyclodextrin-Based Pharmaceutics: Past, Present and Future. Nat. Rev. Drug Discovery 2004, 3, 1023–1035. [DOI] [PubMed] [Google Scholar]
- (8).Lee CC; MacKay JA; Frechet JM; Szoka FC Designing Dendrimers for Biological Applications. Nat. Biotechnol 2005, 23, 1517–1526. [DOI] [PubMed] [Google Scholar]
- (9).Serajuddin AT Salt Formation to Improve Drug Solubility. Adv. Drug Delivery Rev 2007, 59, 603–616. [DOI] [PubMed] [Google Scholar]
- (10).Akers MJ Excipient-Drug Interactions in Parenteral Formulations. J. Pharm. Sci 2002, 91, 2283–2300. [DOI] [PubMed] [Google Scholar]
- (11).Strickley RG Solubilizing Excipients in Oral and Injectable Formulations. Pharm. Res 2004, 21, 201–230. [DOI] [PubMed] [Google Scholar]
- (12).Wang C; Sun WJ; Ye YQ; Hu QY; Bomba HN; Gu Z In Situ Activation of Platelets with Checkpoint Inhibitors for Post-Surgical Cancer Immunotherapy. Nat. Biomed Eng 2017, 1, 1–10. [Google Scholar]
- (13).Zhang F; Ni Q; Jacobson O; Cheng S; Liao A; Wang Z; He Z; Yu G; Song J; Ma Y; Niu G; Zhang L; Zhu G; Chen X Polymeric Nanoparticles with a Glutathione-Sensitive Heterodimeric Multifunctional Prodrug for in Vivo Drug Monitoring and Synergistic Cancer Therapy. Angew. Chem. Int. Ed 2018, 57, 7066–7070. [DOI] [PubMed] [Google Scholar]
- (14).Kroll AV; Fang RH; Jiang Y; Zhou J; Wei X; Yu CL; Gao J; Luk BT; Dehaini D; Gao W; Zhang L Nanoparticulate Delivery of Cancer Cell Membrane Elicits Multiantigenic Antitumor Immunity. Adv. Mater 2017, 29, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Tong R; Chiang HH; Kohane DS Photoswitchable Nanoparticles for in Vivo Cancer Chemotherapy. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 19048–19053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Lee DY; Kim JY; Lee Y; Lee S; Miao W; Kim HS; Min JJ; Jon S Black Pigment Gallstone Inspired Platinum-Chelated Bilirubin Nanoparticles for Combined Photoacoustic Imaging and Photothermal Therapy of Cancers. Angew. Chem., Int. Ed 2017, 56, 13684–13688. [DOI] [PubMed] [Google Scholar]
- (17).Yin H; Song CQ; Dorkin JR; Zhu LJ; Li Y; Wu Q; Park A; Yang J; Suresh S; Bizhanova A; Gupta A; Bolukbasi MF; Walsh S; Bogorad RL; Gao G; Weng Z; Dong Y; Koteliansky V; Wolfe SA; Langer R; Xue W; Anderson DG Therapeutic Genome Editing by Combined Viral and Non-Viral Delivery of Crispr System Components in Vivo. Nat. Biotechnol 2016, 34, 328–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Sun W; Li S; Haupler B; Liu J; Jin S; Steffen W; Schubert US; Butt HJ; Liang XJ; Wu S An Amphiphilic Ruthenium Polymetallodrug for Combined Photodynamic Therapy and Photochemotherapy in Vivo. Adv. Mater 2017, 29, 1603702. [DOI] [PubMed] [Google Scholar]
- (19).Xu XD; Saw PE; Tao W; Li Y; Ji X; Bhasin S; Liu Y; Ayyash D; Rasmussen J; Huo M; Shi J; Farokhzad OC Ros- Responsive Polyprodrug Nanoparticles for Triggered Drug Delivery and Effective Cancer Therapy. Adv. Mater 2017, 29, 1700141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Xu XD; Cheng YJ; Wu J; Cheng H; Cheng SX; Zhuo RX; Zhang XZ Smart and Hyper-Fast Responsive Polyprodrug Nanoplatform for Targeted Cancer Therapy. Biomaterials 2016, 76, 238–249. [DOI] [PubMed] [Google Scholar]
- (21).Hu X; Hu J; Tian J; Ge Z; Zhang G; Luo K; Liu S Polyprodrug Amphiphiles: Hierarchical Assemblies for Shape-Regulated Cellular Internalization, Trafficking, and Drug Delivery. J. Am. Chem. Soc 2013, 135, 17617–17629. [DOI] [PubMed] [Google Scholar]
- (22).He S; Zhou D; Kuang H; Wu Y; Jing X; Huang Y Dextran-Platinum(Iv) Conjugate as Drug Carrier for Triggered Drug Release. J. Controlled Release 2015, 213, No. e96. [DOI] [PubMed] [Google Scholar]
- (23).Min Y; Mao CQ; Chen S; Ma G; Wang J; Liu Y Combating the Drug Resistance of Cisplatin Using a Platinum Prodrug Based Delivery System. Angew. Chem., Int. Ed 2012, 51, 6742–6747. [DOI] [PubMed] [Google Scholar]
- (24).Matsumura Y; Maeda H A New Concept for Macromolecular Therapeutics in Cancer Chemotherapy: Mechanism of Tumoritropic Accumulation of Proteins and the Antitumor Agent Smancs. Cancer Res. 1986, 46, 6387–6392. [PubMed] [Google Scholar]
- (25).Prabhakar U; Maeda H; Jain RK; Sevick-Muraca EM; Zamboni W; Farokhzad OC; Barry ST; Gabizon A; Grodzinski P; Blakey DC Challenges and Key Considerations of the Enhanced Permeability and Retention Effect for Nanomedicine Drug Delivery in Oncology. Cancer Res. 2013, 73, 2412–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Farokhzad OC; Langer R Impact of Nanotechnology on Drug Delivery. ACS Nano 2009, 3, 16–20. [DOI] [PubMed] [Google Scholar]
- (27).Peer D; Karp JM; Hong S; Farokhzad OC; Margalit R; Langer R Nanocarriers as an Emerging Platform for Cancer Therapy. Nat. Nanotechnol 2007, 2, 751–760. [DOI] [PubMed] [Google Scholar]
- (28).Shi J; Kantoff PW; Wooster R; Farokhzad OC Cancer Nanomedicine: Progress, Challenges and Opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Bertrand N; Wu J; Xu X; Kamaly N; Farokhzad OC Cancer Nanotechnology: The Impact of Passive and Active Targeting in the Era of Modern Cancer Biology. Adv. Drug Delivery Rev 2014, 66, 2–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Hu Q; Sun W; Qian C; Bomba HN; Xin H; Gu Z Relay Drug Delivery for Amplifying Targeting Signal and Enhancing Anticancer Efficacy. Adv. Mater 2017, 29, 1–8. [DOI] [PubMed] [Google Scholar]
- (31).Lin LS; Song J; Song L; Ke K; Liu Y; Zhou Z; Shen Z; Li J; Yang Z; Tang W; Niu G; Yang HH; Chen X Simultaneous Fenton-Like Ion Delivery and Glutathione Depletion by Mno2-Based Nanoagent to Enhance Chemodynamic Therapy. Angew. Chem., Int. Ed 2018, 57, 4902–4906. [DOI] [PubMed] [Google Scholar]
- (32).Zhang X; Angsantikul P; Ying M; Zhuang J; Zhang Q; Wei X; Jiang Y; Zhang Y; Dehaini D; Chen M; Chen Y; Gao W; Fang RH; Zhang L Remote Loading of Small-Molecule Therapeutics into Cholesterol-Enriched Cell-Membrane-Derived Vesicles. Angew. Chem., Int. Ed 2017, 56, 14075–14079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Lee H; Lee E; Kim DK; Jang NK; Jeong YY; Jon S Antibiofouling Polymer-Coated Superparamagnetic Iron Oxide Nanoparticles as Potential Magnetic Resonance Contrast Agents for in Vivo Cancer Imaging. J. Am. Chem. Soc 2006, 128, 7383–7389. [DOI] [PubMed] [Google Scholar]
- (34).Dong Y; Love KT; Dorkin JR; Sirirungruang S; Zhang Y; Chen D; Bogorad RL; Yin H; Chen Y; Vegas AJ; Alabi CA; Sahay G; Olejnik KT; Wang W; Schroeder A; Lytton-Jean AK; Siegwart DJ; Akinc A; Barnes C; Barros SA; Carioto M; Fitzgerald K; Hettinger J; Kumar V; Novobrantseva TI; Qin J; Querbes W; Koteliansky V; Langer R; Anderson DG Lipopeptide Nanoparticles for Potent and Selective Sirna Delivery in Rodents and Nonhuman Primates. Proc. Natl. Acad. Sci. U. S. A 2014, 111, 3955–3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Johnstone TC; Suntharalingam K; Lippard SJ The Next Generation of Platinum Drugs: Targeted Pt(Ii) Agents, Nanoparticle Delivery, and Pt(Iv) Prodrugs. Chem. Rev 2016, 116, 3436–3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Wong E; Giandomenico CM Current Status of Platinum-Based Antitumor Drugs. Chem. Rev 1999, 99, 2451–2466. [DOI] [PubMed] [Google Scholar]
- (37).Godwin AK; Meister A; O’Dwyer PJ; Huang CS; Hamilton TC; Anderson ME High Resistance to Cisplatin in Human Ovarian Cancer Cell Lines Is Associated with Marked Increase of Glutathione Synthesis. Proc. Natl. Acad. Sci. U. S. A 1992, 89, 3070–3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Kuppusamy P; Li H; Ilangovan G; Cardounel AJ; Zweier JL; Yamada K; Krishna MC; Mitchell JB Noninvasive Imaging of Tumor Redox Status and Its Modification by Tissue Glutathione Levels. Cancer Res. 2002, 62, 307–312. [PubMed] [Google Scholar]
- (39).Kelland L The Resurgence of Platinum-Based Cancer Chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [DOI] [PubMed] [Google Scholar]
- (40).Ling X; Chen X; Riddell IA; Tao W; Wang J; Hollett G; Lippard SJ; Farokhzad OC; Shi J; Wu J Glutathione-Scavenging Poly(Disulfide Amide) Nanoparticles for the Effective Delivery of Pt(Iv) Prodrugs and Reversal of Cisplatin Resistance. Nano Lett. 2018, 18, 4618–4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Johnstone TC; Lippard SJ The Effect of Ligand Lipophilicity on the Nanoparticle Encapsulation of Pt(Iv) Prodrugs. Inorg. Chem 2013, 52, 9915–9920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Zanellato I; Bonarrigo I; Colangelo D; Gabano E; Ravera M; Alessio M; Osella D Biological Activity of a Series of Cisplatin- Based Aliphatic Bis(Carboxylato) Pt(Iv) Prodrugs: How Long the Organic Chain Should Be? J. Inorg. Biochem 2014, 140, 219–227. [DOI] [PubMed] [Google Scholar]
- (43).Ling X; Shen Y; Sun RN; Zhang MZ; Li C; Mao JY; Xing J; Sun CM; Tu JS Tumor-Targeting Delivery of Hyaluronic Acid-Platinum(Iv) Nanoconjugate to Reduce Toxicity and Improve Survival. Polym. Chem 2015, 6, 1541–1552. [Google Scholar]
- (44).Ling X; Zhao CY; Huang LP; Wang QY; Tu JS; Shen Y; Sun CM Synthesis and Characterization of Hyaluronic Acid-Platinum(Iv) Nanoconjugate with Enhanced Antitumor Response and Reduced Adverse Effects. RSC Adv. 2015, 5, 81668–81681. [Google Scholar]
- (45).Gramatica P; Papa E; Luini M; Monti E; Gariboldi MB; Ravera M; Gabano E; Gaviglio L; Osella D Antiproliferative Pt(Iv) Complexes: Synthesis, Biological Activity, and Quantitative Structure-Activity Relationship Modeling. JBIC, J. Biol. Inorg. Chem 2010, 15, 1157–1169. [DOI] [PubMed] [Google Scholar]
- (46).Varbanov HP; Jakupec MA; Roller A; Jensen F; Galanski M; Keppler BK Theoretical Investigations and Density Functional Theory Based Quantitative Structure-Activity Relationships Model for Novel Cytotoxic Platinum(Iv) Complexes. J. Med. Chem 2013, 56, 330–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Choi S; Filotto C; Bisanzo M; Delaney S; Lagasee D; Whitworth JL; Jusko A; Li C; Wood NA; Willingham J; Schwenker A; Spaulding K Reduction and Anticancer Activity of Platinum(Iv) Complexes. Inorg. Chem 1998, 37, 2500–2504. [Google Scholar]
- (48).Reithofer MR; Bytzek AK; Valiahdi SM; Kowol CR; Groessl M; Hartinger CG; Jakupec MA; Galanski M; Keppler BK Tuning of Lipophilicity and Cytotoxic Potency by Structural Variation of Anticancer Platinum(Iv) Complexes. J. Inorg. Biochem 2011, 105, 46–51. [DOI] [PubMed] [Google Scholar]
- (49).Feazell RP; Nakayama-Ratchford N; Dai H; Lippard SJ Soluble Single-Walled Carbon Nanotubes as Longboat Delivery Systems for Platinum(Iv) Anticancer Drug Design. J. Am. Chem. Soc 2007, 129, 8438–8439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Lu Y; Aimetti AA; Langer R; Gu Z Bioresponsive Materials. Nat. Rev. Mater 2017, 2, 1–17. [Google Scholar]
- (51).Love KT; Mahon KP; Levins CG; Whitehead KA; Querbes W; Dorkin JR; Qin J; Cantley W; Qin LL; Racie T; Frank-Kamenetsky M; Yip KN; Alvarez R; Sah DW; de Fougerolles A; Fitzgerald K; Koteliansky V; Akinc A; Langer R; Anderson DG Lipid-Like Materials for Low-Dose, in Vivo Gene Silencing. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 1864–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Mercer J; Helenius A Vaccinia Virus Uses Macropinocytosis and Apoptotic Mimicry to Enter Host Cells. Science 2008, 320, 531–535. [DOI] [PubMed] [Google Scholar]
- (53).Araki N; Egami Y; Watanabe Y; Hatae T Phosphoinositide Metabolism During Membrane Ruffling and Macropinosome Formation in Egf-Stimulated A431 Cells. Exp. Cell Res 2007, 313, 1496–1507. [DOI] [PubMed] [Google Scholar]
- (54).Jares-Erijman EA; Jovin TM Fret Imaging. Nat. Biotechnol 2003, 21, 1387–1395. [DOI] [PubMed] [Google Scholar]
- (55).Yang J; Chen H; Vlahov IR; Cheng JX; Low PS Evaluation of Disulfide Reduction During Receptor-Mediated Endocytosis by Using Fret Imaging. Proc. Natl. Acad. Sci. U. S. A 2006, 103, 13872–13877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Lewis AD; Hayes JD; Wolf CR Glutathione and Glutathione-Dependent Enzymes in Ovarian Adenocarcinoma Cell Lines Derived from a Patient before and after the Onset of Drug Resistance: Intrinsic Differences and Cell Cycle Effects. Carcinogenesis 1988, 9, 1283–1287. [DOI] [PubMed] [Google Scholar]
- (57).Speidel D Transcription-Independent P53 Apoptosis: An Alternative Route to Death. Trends Cell Biol. 2010, 20, 14–24. [DOI] [PubMed] [Google Scholar]
- (58).Chalah A; Khosravi-Far R The Mitochondrial Death Pathway. Adv. Exp. Med. Biol 2008, 615, 25–45. [DOI] [PubMed] [Google Scholar]
- (59).Bassett E; Vaisman A; Tropea KA; McCall CM; Masutani C; Hanaoka F; Chaney SG Frameshifts and Deletions During in Vitro Translesion Synthesis Past Pt-DNA Adducts by DNA Polymerases Beta and Eta. DNA Repair 2002, 1, 1003–1016. [DOI] [PubMed] [Google Scholar]
- (60).Albertella MR; Green CM; Lehmann AR; O’Connor MJ A Role for Polymerase Eta in the Cellular Tolerance to Cisplatin-Induced Damage. Cancer Res. 2005, 65, 9799–9806. [DOI] [PubMed] [Google Scholar]
- (61).Gadducci A; Cosio S; Muraca S; Genazzani AR Molecular Mechanisms of Apoptosis and Chemosensitivity to Platinum and Paclitaxel in Ovarian Cancer: Biological Data and Clinical Implications. Eur. J. Gynaecol. Oncol 2002, 23, 390–396. [PubMed] [Google Scholar]
- (62).Mazzoni F; Cecere FL; Meoni G; Giuliani C; Boni L; Camerini A; Lucchesi S; Martella F; Amoroso D; Lucherini E; Torricelli F; Di Costanzo F Phase Ii Trial of Customized First Line Chemotherapy According to Ercc1 and Rrm1 Snps in Patients with Advanced Non-Small-Cell Lung Cancer. Lung Cancer 2013, 82, 288–293. [DOI] [PubMed] [Google Scholar]
- (63).Liu Y; Bernauer AM; Yingling CM; Belinsky SA Hif1alpha Regulated Expression of Xpa Contributes to Cisplatin Resistance in Lung Cancer. Carcinogenesis 2012, 33, 1187–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Shackelford C; Long G; Wolf J; Okerberg C; Herbert R Qualitative and Quantitative Analysis of Nonneoplastic Lesions in Toxicology Studies. Toxicol. Pathol 2002, 30, 93–96. [DOI] [PubMed] [Google Scholar]
- (65).Pezeshki Z; Khosravi A; Nekuei M; Khoshnood S; Zandi E; Eslamian M; Talebi A; Emami SN; Nematbakhsh M Time Course of Cisplatin-Induced Nephrotoxicity and Hepatotoxicity. J. Nephropathol 2017, 6, 163–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Faubel S; Lewis EC; Reznikov L; Ljubanovic D; Hoke TS; Somerset H; Oh DJ; Lu L; Klein CL; Dinarello CA; Edelstein CL Cisplatin-Induced Acute Renal Failure Is Associated with an Increase in the Cytokines Interleukin (Il)-1beta, Il-18, Il-6, and Neutrophil Infiltration in the Kidney. J. Pharmacol. Exp. Ther 2007, 322, 8–15. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.