

Abstract
Motivation
Numerous sequencing studies, including transcriptomics of host-pathogen systems, sequencing of hybrid genomes, xenografts, mixed species systems, metagenomics and meta-transcriptomics, involve samples containing genetic material from divergent organisms. A crucial step in these studies is identifying from which organism each sequencing read originated, and the experimental design should be directed to minimize biases caused by cross-mapping of reads to incorrect source genomes. Additionally, pooling of sufficiently different genetic material into a single sequencing library could significantly reduce experimental costs but requires careful planning and assessment of the impact of cross-mapping. Having these applications in mind we designed Crossmapper, the first to our knowledge tool able to assess cross-mapping prior to sequencing, therefore allowing optimization of experimental design.
Results
Using any combination of reference genomes, Crossmapper performs read simulation and back-mapping of those reads to the pool of references, quantifies and reports the cross-mapping rates for each organism. Crossmapper performs these analyses with numerous user-specified parameters, including, among others, read length, read layout, coverage, mapping parameters, genomic or transcriptomic data. Additionally, it outputs the results in highly interactive and publication-ready reports. This allows the user to perform multiple comparisons at once and choose the experimental setup minimizing cross-mapping rates. Moreover, Crossmapper can be used for resource optimization in sequencing facilities by pooling different samples into one sequencing library.
Availability and implementation
Crossmapper is a command line tool implemented in Python 3.6 and available as a conda package, allowing effortless installation. The source code, detailed information and a step-by-step tutorial is available at our GitHub page https://github.com/Gabaldonlab/crossmapper.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
There are various biological problems addressed by next-generation sequencing (NGS) in which the samples contain genetic material from multiple species. These include, but are not limited to studies involving host-pathogen interaction (Westermann et al., 2017), symbiont-host or microbial interaction (Burns et al., 2017; González-Torres et al., 2015), metagenomics (Quince et al., 2017) or hybrid organisms (Metzger et al., 2017). A challenging step in these experimental setups is to assign each sequencing read to the corresponding source organism, which is usually done by mapping the reads to the set of reference genomes (Wolf et al., 2018). A similar strategy is applied in allele-specific expression (ASE) studies in the case of phased reference genomes (Yuan and Qin, 2012). Successful read separation depends on numerous factors, including mainly read length, read layout, similarity of sequenced genomes and different mapping parameters. Thus, if these parameters are not carefully planned, downstream analyses can be biased by cross-mapping of reads to non-corresponding references. For example, in a human-Salmonella interaction study it was observed that ∼1.44% of total reads map equally well (multi-mapped) to both reference genomes (Westermann and Vogel, 2018). While the amount of erroneously mapped reads can be low for highly divergent species, in metagenomics (Petersen et al., 2017) and ASE studies, erroneously mapped and multi-mapped reads constitute the majority of the data (Yuan and Qin, 2012). Despite the importance of sequencing design in aforementioned studies, today there are no computational tools to assist in their planning so that optimal results are obtained. To overcome this, we developed Crossmapper––a pipeline assessing, prior to sequencing, the potential rates of multi-mapping and erroneous mapping for various combinations of sequencing parameters and any number of reference sequences.
2 Workflow and implementation
Crossmapper proceeds as follows (Fig. 1A). It first takes as input any number of reference genomes and allows to simulate DNA and RNA reads in a wide range of experimental setups. This step is performed by wgsim (Li et al., 2009) with the possibility to define different parameters such as read length, error rates, outer distance, among others. Crossmapper allows to simulate many different sequencing configurations at once. The user can specify genome annotations to limit read simulations from specific parts of the genomic regions (i.e. for transcriptomic or exome sequencing studies).
Fig. 1.
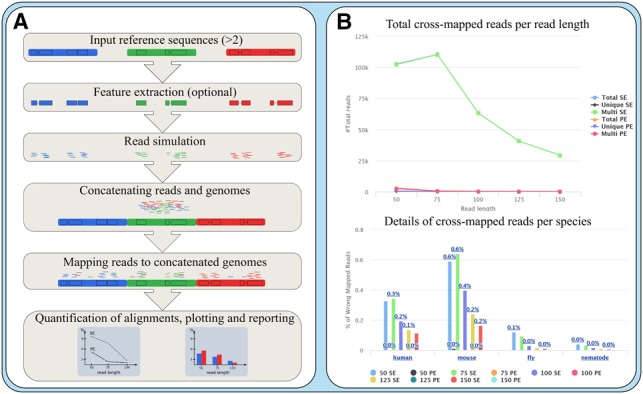
(A) The general workflow of Crossmapper (see main text for details). (B) An example of Crossmapper output
After read simulation, Crossmapper concatenates fastq files from different organisms and maps the reads back to a concatenated set of reference genomes. By default Crossmapper uses BWA-MEM (Li and Durbin, 2009), and STAR (Dobin et al., 2013) for mapping DNA and RNA data, respectively. However, we also implemented the –mapper-template option allowing to use any desired mapping software with custom parameters by supplying the configuration file to the Crossmapper (a documentation for creating a configuration file is given in the GitHub page). The final bam file for each read length and layout contains alignments of all simulated reads collectively mapped to all source reference genomes. Since simulated data preserve information regarding the source genome and exact location, Crossmapper can calculate the rate of multi-mapped and erroneously mapped reads for all source genomes. After the quantification step Crossmapper produces an extensive html report, which includes several interactive, publication-ready plots summarizing mapping rates, as well as tables with detailed mapping statistics for each experimental configuration. Based on this report users can decide the optimal experimental and mapping parameters prior to the actual sequencing. In addition, coordinates of cross-mapped reads are reported so these regions can be filtered, if necessary, in downstream analyses.
3 Usage case
Several examples of Crossmapper usage are available in the GitHub site of this tool. Here, we explain how to use of Crossmapper to optimize resources by pooling of genetic material of different organisms into a single sequencing library. Indeed, the cost of sequencing has dropped dramatically in the past decade (Goodwin et al., 2016) largely due to throughput increase. However, the costs for library preparation do not follow the same trend and often constitute a financial bottleneck. A simple pooling of genetic materials of different species into one library could save a substantial amount of resources, provided reads from different sources could effectively be separated computationally. This has to be carefully planned to avoid aforementioned biases in downstream analyses. Crossmapper can achieve this task in a single run. Below is an example of sequencing design optimization for pooling genetic material of widely analyzed organisms––human, mouse, fly and nematode––in a single library.
Command syntax
Crossmapper DNA -t 8 -gb -rlay both -g homo.fasta mus.fasta dros.fasta caeno.fasta -gn human mouse fly nematode -N 2500000 2500000 2500000 2500000 -rlen 50,75,100,125,150 -r 0.01
Lets Crossmapper to simulate 2.5 million DNA reads per organism at 50, 75, 100, 125 and 150 read lengths at both single- and paired-end layouts, map the data to the pool of reference genomes [obtained from Ensembl (Zerbino et al., 2018)] and report mapping rates for all sequencing configurations (Fig. 1B). Using Intel Xeon 3.5 GHz, 64 GB of RAM and 8 cores the analysis takes ∼11 h. In this case of very large reference genome (c.a. 6.5 GB) and 10 mapping jobs, the main bottleneck for the speed of the analysis is genome indexing and read mapping, which collectively takes ∼8 h.
The results of this analysis (Supplementary File S1) demonstrate that by pooling the DNA of the four species reads can be effectively separated by mapping. However, single-end sequencing produces relatively high rates of multi-mapping (maximum 4.95% and 6.06% for 150 and 50 bp, respectively) and erroneous mapping (maximum 0.16% and 0.52%, for 150 and 50 bp, respectively) which potentially can bias differential expression or variant calling analysis. On the other hand, paired-end sequencing with 75 bp reads significantly reduces multi- and erroneous-mapping (0.01% and 0%, respectively) rates. Thus, the pooling strategy with 2 x 75 bp reads can be the most efficient balance between accuracy and sequencing cost. Repeating this test with a higher number of reads (40, 30, 20 and 10 million reads for human, mouse, fly and nematode, respectively), showed similar rates of cross-mapping (Supplementary File S2), which indicates that low-coverage simulations are sufficient to properly estimate cross-mapping rates.
4 Conclusion
Crossmapper allows to design numerous types of NGS experiments that share a common feature of sequencing several organisms as one sample. Crossmapper is easy to install and use. It is highly customizable and outputs the results in intuitive, interactive and publication-ready reports. We believe that Crossmapper will benefit both research and industrial communities by helping to optimize sequencing strategies and available resources.
Funding
This work was supported by the Spanish Ministry of Economy, Industry and Competitiveness (MEIC) for the EMBL partnership and the grant ‘Centro de Excelencia Severo Ochoa’ SEV-2012-0208 cofounded by European Regional Development Fund (ERDF); from the CERCA Programme/Generalitat de Catalunya; from the Catalan Research Agency (AGAUR) SGR857 and grants from the European Union’s Horizon 2020 research and innovation programme under the grant agreement ERC-2016-724173 and the Marie Sklodowska-Curie grant agreement No H2020-MSCA-ITN-2014-642095. The group also receives support from a INB Grant (PT17/0009/0023–ISCIII-SGEFI/ERDF).
Conflict of Interest: none declared.
Supplementary Material
References
- Burns J.A. et al. (2017) Transcriptome analysis illuminates the nature of the intracellular interaction in a vertebrate-algal symbiosis. Elife, 6, 22054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A. et al. (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Torres P. et al. (2015) Interactions between closely related bacterial strains are revealed by deep transcriptome sequencing. Appl. Environ. Microbiol., 81, 8445–8456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin S. et al. (2016) Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet., 17, 333–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. et al. (2009) The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger B.P.H. et al. (2017) Evolutionary dynamics of regulatory changes underlying gene expression divergence among Saccharomyces species. Genome Biol. Evol., 9, 843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen T.N. et al. (2017) MGmapper: reference based mapping and taxonomy annotation of metagenomics sequence reads. PLoS One, 12, e0176469.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quince C. et al. (2017) Corrigendum: shotgun metagenomics, from sampling to analysis. Nat. Biotechnol., 35, 1211.. [DOI] [PubMed] [Google Scholar]
- Westermann A.J. et al. (2017) Resolving host–pathogen interactions by dual RNA-seq. PLoS Pathog., 13, e1006033.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann A.J., Vogel J. (2018) Host-pathogen transcriptomics by dual RNA-Seq. Methods Mol. Biol., 1737, 59–75. [DOI] [PubMed] [Google Scholar]
- Wolf T. et al. (2018) Two’s company: studying interspecies relationships with dual RNA-seq. Curr. Opin. Microbiol., 42, 7–12. [DOI] [PubMed] [Google Scholar]
- Yuan S., Qin Z. (2012) Read-mapping using personalized diploid reference genome for RNA sequencing data reduced bias for detecting allele-specific expression. IEEE Int. Conf. Bioinform. Biomed. Workshops, 2012, 718–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino D.R. et al. (2018) Ensembl 2018. Nucleic Acids Res., 46, D754–D761. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.