

Abstract
Objectives
Primary open‐angle glaucoma (POAG) is a neurodegenerative disorder leading to a gradual vision loss caused by progressive damage to the optic nerve. Immunological processes are proposed to be involved in POAG pathogenesis. Altered serological autoantibody levels have been frequently reported, but complete analyses of the natural autoantibodies with respect to disease‐related alterations are scarce. Here, we provide an explorative analysis of pathways and biological processes that may involve naturally immunogenic proteins and highlight POAG‐specific alterations.
Methods
Mass spectrometry‐based antibody‐mediated identification of autoantigens (MS‐AMIDA) was carried out in healthy and glaucomatous trabecular meshwork (TM) cell lines, using antibody pools purified from serum samples of 30 POAG patients and 30 non‐glaucomatous subjects. Selected antigens were validated by protein microarray (n = 120). Bioinformatic assessment of identified autoantigens, including Gene Ontology (GO) enrichment analysis and protein–protein interaction networks, was applied.
Results
Overall, we identified 106 potential autoantigens [false discovery rate (FDR) < 0.01], from which we considered 66 as physiological targets of natural autoantibodies. Twenty‐one autoantigens appeared to be related to POAG. Bioinformatic analysis revealed that the platelet‐derived growth factor receptor beta (PDGFRB) pathway involved in TM fibrosis was particularly rich in POAG‐related antigens. Antibodies to threonine‐tRNA ligase (TARS), component 1 Q subcomponent‐binding protein (C1QBP) and paraneoplastic antigen Ma2 (PNMA2) showed significantly (P < 0.05) higher levels in POAG patients as validated by protein microarray.
Conclusion
This study provides new insights into autoimmunity in health and glaucoma. Bioinformatic analysis of POAG‐related autoantigens showed a strong association with the PDGFRB pathway and also increased levels of PNMA2, TARS, and C1QBP autoantibodies in the serum of POAG patients as potential glaucoma biomarkers.
Keywords: autoantigen, biomarker, glaucoma, immunoproteomics, natural autoantibodies, trabecular meshwork
In this study, we identified 66 potential antigens in the trabecular meshwork (TM) as targets of natural autoimmunity. Of these antigens, 21 can be assigned a connection to primary open‐angle glaucoma (POAG). The platelet‐derived growth factor receptor beta (PDGFRB) pathway involved in TM fibrosis is especially represented by POAG‐related antigens.

Introduction
Primary open‐angle glaucoma (POAG) is an optic neuropathy generally characterised by an elevated intraocular pressure (IOP) above 21 mmHg,1 apoptosis of retinal ganglion cells and gradual loss of vision.2 In addition to old age,3 genetic predisposition4, 5 and decreased central corneal thickness,6 a high IOP is the major risk factor for POAG,7 but alone it is not sufficient for its onset. An elevated IOP in POAG is caused by an increased outflow resistance of the aqueous humour, triggered by pathological changes in the trabecular meshwork (TM). Outflow resistance in the TM is primarily caused by the cells and extracellular matrix of the juxtacanalicular region that is subject to constant remodelling to maintain aqueous humour efflux. In the glaucomatous eye, signalling pathways regulating extracellular matrix deposition, the actin cytoskeleton and cell–cell/cell–matrix connections are dysregulated.8, 9 Transforming growth factor β levels in aqueous humour are increased, leading to a rise in its downstream effector, connective tissue growth factor. This matricellular protein promotes the excessive accumulation of extracellular matrix proteins in the extracellular space, causing the TM tissue to be denser and thereby decreasing outflow.10
Furthermore, cross‐linking of the actin cytoskeleton in the TM increases. This limits the ability of the tissue to actively respond to changes in IOP by actomyosin‐mediated contraction of TM cells. The β‐integrin‐induced formation of cross‐linked actin networks increases the overall rigidity of the tissue and exacerbates the resistance in the TM outflow pathway.11 The stiffness of the tissue also accumulates with age,12 elevating the chances of drainage failure. The TM in glaucoma is about 20 times more rigid than in healthy tissue,13 reflecting enormous changes in TM biomechanics during the course of the disease. However, in addition, another form of open‐angle glaucoma without increased pathological IOP exists. This particular subgroup is known as normal‐tension glaucoma and comprises between 20% and 61% of all open‐angle glaucomas.14 This also suggests that other pathomechanisms are involved in the generation of glaucomatous damage to the optic nerve. In the case of normal‐tension glaucoma, a weakness of the lamina cribrosa has been proposed, making the retinal ganglion cells more prone to mechanical damage, even under physiological IOP.15 The optic nerve leaves the posterior eye through this mesh‐like tissue which has similar properties to the TM. Therefore, as the LC provides mechanical support to the optic nerve, compression and distortion of this structure may be involved in the damage to retinal ganglion cell axons in glaucoma.16, 17
Another factor that has been forwarded by previous studies is a possible involvement of immunological mechanisms. Several research groups already found altered levels of autoantibodies (AAbs) against diverse ocular antigens.18, 19, 20, 21, 22, 23 Interestingly, not only increased but also downregulated AAb reactivities have been identified,24, 25, 26 suggesting a complex alteration of the natural autoantibody repertoire. Many scientists have abandoned the paradigm of Paul Ehrlich's ‘horror autotoxicus’, which links AAbs only to pathological effects. The physiological immunome most likely fulfils more tasks than a direct defence against invading pathogens as part of humoral immunity. Instead, the immune system is also involved in homeostasis and clearance of cellular debris.27 The impairment of these functions can likely favor the development or progression of diseases.
Up to now, the human immunome has not been well explored. In this study, we looked at antigens in TM cells and their serological AAbs, as well as their function, to identify pathways involved in the emergence of autoantigens. Furthermore, we searched for hints of any altered immunogenicity of these proteins in POAG and validated an increase in AAbs against of paraneoplastic antigen Ma2 (PNMA2), threonine‐tRNA ligase (TARS) and complement component 1 Q subcomponent‐binding protein (C1QBP) by antigen microarray.
Results
Identification of autoantigens in human TM cells using MS‐AMIDA
Mass spectrometry‐based antibody‐mediated identification of autoantigens (MS‐AMIDA) was used to detect serological AAbs to antigens in human TM cells. The workflow comprises the isolation of IgGs from human non‐glaucomatous serum (for characteristics, see Table 1, MS‐AMIDA CTRL group) and their cross‐linking to protein G‐coated magnetic beads. These beads were subsequently incubated with TM cell lysates from immortalised human cell lines. The putative autoantigens bind to the immobilised antibodies, and after on‐bead tryptic digestion, they were identified by high‐resolution MS. The IgG repertoire of non‐glaucomatous subjects against antigens from healthy (HTM) and glaucomatous TM (GTM) cells was tested. Also, negative controls, without IgG, and a mock control were included, which was incubated only with lysis buffer instead of cell lysate. An overview of the experimental design is depicted in Table 2.
Table 1.
Characteristics of the study population
| POAG | CTRL | |
|---|---|---|
| MS‐AMIDA | ||
| Samples (n) | 30 (3 pools) | 30 (3 pools) |
| Sex (m/f) | 17/13 | 15/15 |
| Mean age (±SD) | 63.93 ± 9.13 | 64.5 ± 9.82 |
| Eye surgery | None | None |
| Autoimmune disease | No reported | No reported |
| Other eye disease | Cataract (9 of 30) | Cataract (7 of 30) |
| Microarray validation | ||
| Samples (n) | 120 | 120 |
| Sex (m/f) | 65/55 | 65/55 |
| Mean age (±SD) | 67.25 ± 10.95 | 67.17 ± 11.89 |
Table 2.
Experimental design for the initial MS‐AMIDA profiling
| ID | Sample name | IgG source | Protein source | Comment |
|---|---|---|---|---|
| NC1 | Negative control 1 | None | Healthy TM cells | Unspecific binding to beads |
| NC2 | Negative control 2 | None | Glaucomatous TM cells | Unspecific binding to beads |
| MA | Mock A | POAG | Lysis buffer | Mock control; 3 replicates |
| MB | Mock B | CTRL | Lysis buffer | Mock control; 3 replicates |
| PH | POAG/HTM | POAG | Healthy TM cells | 3 replicates |
| PG | POAG/GTM | POAG | Glaucomatous TM cells | 3 replicates |
| CH | CTRL/HTM | CTRL | Healthy TM cells | 3 replicates |
| CG | CTRL/GTM | CTRL | Glaucomatous TM cells | 3 replicates |
With this approach, we identified 157 proteins in all groups, which were reproducibly found in all three replicates of at least one experimental group, with high confidence [false discovery rate (FDR) < 0.01]. Fifty‐one of these proteins were also found in the negative control and mock control groups and were excluded from further analysis. We considered the remaining 106 proteins as potential autoantigens (Supplementary table 1). To elucidate the natural autoantibody repertoire detectable with this method, the control experimental group, incubated with HTM cell lysates (‘CH’), was evaluated with special interest. In this group, 66 antigens were identified (Table 3). The analysis of the intensity‐based absolute quantification (iBAQ) values revealed that 12 antigens contribute to 67.3% of all identified proteins (Figure 1). The most abundant antigens were histone H4 with 14.4% and histone H2B type 1‐L (9.7%), followed by the histone H4‐binding partner protein SET with 8.2%. Other antigens among the top 10 most abundant were proteins from the tubulin beta chain (TBB6 6.5% and TBB4B 5.9%) and 40S ribosomal proteins (RS16 4.6%, RS2 3.3%, RS24 2.6%). Forty of the (auto)‐antibody‐captured proteins showed relative low abundance with < 1%. A complete list with all iBAQ values can be found in Table 3.
Table 3.
Autoantigens identified in HTM cells, captured by autoantibodies of CTRL sera (Group ID: ‘CH’)
| No. | Entry name (*_HUMAN) | UniProt accession | Protein name | Sequence coverage (%) | No. of peptides | No. of unique peptides | iBAQ (%) |
|---|---|---|---|---|---|---|---|
| 1 | FLNA | P21333 | Filamin‐A | 11 | 20 | 16 | 1.24 |
| 2 | TBB6 | Q9BUF5 | Tubulin beta‐6 chain | 46.6 | 15 | 6 | 6.51 |
| 3 | SYEP | P07814 | Bifunctional glutamate/proline‐tRNA ligase | 11 | 12 | 12 | 1.15 |
| 4 | SET | Q01105 | Protein SET | 19.3 | 3 | 3 | 8.23 |
| 5 | TBB4B | P68371 | Tubulin beta‐4B chain | 51.7 | 17 | 1 | 5.88 |
| 6 | SYIC | P41252 | Isoleucine‐tRNA ligase, cytoplasmic | 12.1 | 12 | 12 | 0.99 |
| 7 | FAS | P49327 | Fatty acid synthase | 4.8 | 7 | 7 | 0.44 |
| 8 | TERA | P55072 | Transitional endoplasmic reticulum ATPase | 12.8 | 6 | 6 | 1.14 |
| 9 | H4 | P62805 | Histone H4 | 35.9 | 4 | 4 | 14.39 |
| 10 | XRCC5 | P13010 | X‐ray repair cross‐complementing protein 5 | 23.8 | 8 | 8 | 2.28 |
| 11 | RS2 | P15880 | 40S ribosomal protein S2 | 18.8 | 4 | 4 | 3.09 |
| 12 | H2B1L | Q99880 | Histone H2B type 1‐L | 19 | 2 | 2 | 9.69 |
| 13 | HNRPF | P52597 | Heterogeneous nuclear ribonucleoprotein F | 11.3 | 3 | 2 | 4.65 |
| 14 | RS16 | P62249 | 40S ribosomal protein S16 | 14.4 | 2 | 2 | 4.6 |
| 15 | SYTC | P26639 | Threonine‐tRNA ligase, cytoplasmic | 11.1 | 7 | 7 | 1.77 |
| 16 | PLPHP | O94903 | Pyridoxal phosphate homeostasis protein | 17.5 | 3 | 3 | 1.81 |
| 17 | HS90B | P08238 | Heat shock protein HSP 90‐beta | 20.9 | 10 | 7 | 1.41 |
| 18 | ATPA | P25705 | ATP synthase subunit alpha, mitochondrial | 15.9 | 5 | 5 | 1.56 |
| 19 | SERPH | P50454 | Serpin H1 | 14.1 | 4 | 4 | 1.6 |
| 20 | NPM | P06748 | Nucleophosmin | 12.6 | 3 | 3 | 3.29 |
| 21 | MCM7 | P33993 | DNA replication licensing factor MCM7 | 7.4 | 4 | 4 | 0.55 |
| 22 | CCD47 | Q96A33 | Coiled‐coil domain‐containing protein 47 | 14.3 | 4 | 4 | 2.03 |
| 23 | ADT2 | P05141 | ADP/ATP translocase 2 | 14.4 | 4 | 2 | 1.69 |
| 24 | SYRC | P54136 | Arginine‐tRNA ligase, cytoplasmic | 17.7 | 10 | 10 | 0.74 |
| 25 | IF4A1 | P60842 | Eukaryotic initiation factor 4A‐I | 27.8 | 6 | 6 | 1.69 |
| 26 | IQGA1 | P46940 | Ras GTPase‐activating‐like protein IQGAP1 | 2.8 | 2 | 2 | 0.1 |
| 27 | HNRPK | P61978 | Heterogeneous nuclear ribonucleoprotein K | 7.8 | 3 | 3 | 1.01 |
| 28 | EIF3L | Q9Y262 | Eukaryotic translation initiation factor 3 subunit L | 3 | 1 | 1 | 0.54 |
| 29 | SYK | Q15046 | Lysine‐tRNA ligase | 8.9 | 4 | 4 | 0.5 |
| 30 | SYDC | P14868 | Aspartate‐tRNA ligase, cytoplasmic | 7.8 | 4 | 4 | 0.66 |
| 31 | SRP68 | Q9UHB9 | Signal recognition particle subunit SRP68 | 12.1 | 5 | 5 | 0.51 |
| 32 | TBA1A | Q71U36 | Tubulin alpha‐1A chain | 46.1 | 17 | 1 | 1.07 |
| 33 | RS24 | P62847 | 40S ribosomal protein S24 | 9 | 1 | 1 | 2.63 |
| 34 | RL15 | P61313 | 60S ribosomal protein L15 | 5.9 | 1 | 1 | 1.3 |
| 35 | EIF3E | P60228 | Eukaryotic translation initiation factor 3 subunit E | 2.9 | 1 | 1 | 0.45 |
| 36 | EIF3F | O00303 | Eukaryotic translation initiation factor 3 subunit F | 5.3 | 1 | 1 | 0.76 |
| 37 | SYQ | P47897 | Glutamine‐tRNA ligase | 9 | 5 | 5 | 0.36 |
| 38 | TCPZ | P40227 | T‐complex protein 1 subunit zeta | 7.2 | 2 | 2 | 0.47 |
| 39 | MYO1C | O00159 | Unconventional myosin‐Ic | 1.2 | 1 | 1 | 0.05 |
| 40 | EIF3A | Q14152 | Eukaryotic translation initiation factor 3 subunit A | 3.5 | 4 | 4 | 0.16 |
| 41 | DEST | P60981 | Destrin | 10.3 | 1 | 1 | 1.63 |
| 42 | EIF3B | P55884 | Eukaryotic translation initiation factor 3 subunit B | 3.4 | 1 | 1 | 0.26 |
| 43 | SYMC | P56192 | Methionine‐tRNA ligase, cytoplasmic | 1.2 | 1 | 1 | 0.33 |
| 44 | NSUN2 | Q08J23 | tRNA (cytosine(34)‐C(5))‐methyltransferase | 3.9 | 2 | 2 | 0.43 |
| 45 | COPG1 | Q9Y678 | Coatomer subunit gamma‐1 | 4.9 | 2 | 2 | 0.2 |
| 46 | MCM3 | P25205 | DNA replication licensing factor MCM3 | 2.7 | 2 | 2 | 0.13 |
| 47 | PSMD2 | Q13200 | 26S proteasome non‐ATPase regulatory subunit 2 | 4.5 | 2 | 2 | 0.12 |
| 48 | OLA1 | Q9NTK5 | Obg‐like ATPase 1 | 3.8 | 1 | 1 | 0.19 |
| 49 | RS5 | P46782 | 40S ribosomal protein S5 | 13.7 | 1 | 1 | 0.9 |
| 50 | ACTN1 | P12814 | Alpha‐actinin‐1 | 3.8 | 3 | 3 | 0.27 |
| 51 | PYRG1 | P17812 | CTP synthase 1 | 2.7 | 1 | 1 | 0.19 |
| 52 | PPIL4 | Q8WUA2 | Peptidyl‐prolyl cis‐trans isomerase‐like 4 | 8.7 | 2 | 2 | 0.34 |
| 53 | NU160 | Q12769 | Nuclear pore complex protein Nup160 | 1.3 | 1 | 1 | 0.06 |
| 54 | P5CS | P54886 | Delta‐1‐pyrroline‐5‐carboxylate synthase | 4.5 | 2 | 2 | 0.14 |
| 55 | TKT | P29401 | Transketolase | 6.9 | 2 | 2 | 0.21 |
| 56 | DCTN1 | Q14203 | Dynactin subunit 1 | 3.7 | 3 | 3 | 0.08 |
| 57 | CLH1 | Q00610 | Clathrin heavy chain 1 | 0.5 | 1 | 1 | 0.07 |
| 58 | PRP8 | Q6P2Q9 | Pre‐mRNA‐processing‐splicing factor 8 | 0.9 | 1 | 1 | 0.03 |
| 59 | DX39A | O00148 | ATP‐dependent RNA helicase DDX39A | 4.9 | 2 | 2 | 0.31 |
| 60 | TIM50 | Q3ZCQ8 | Mitochondrial import inner membrane translocase subunit TIM50 | 2.8 | 1 | 1 | 0.4 |
| 61 | CPSF7 | Q8N684 | Cleavage and polyadenylation specificity factor subunit 7 | 5.3 | 1 | 1 | 0.25 |
| 62 | SPT5H | O00267 | Transcription elongation factor SPT5 | 1.7 | 1 | 1 | 0.07 |
| 63 | IMB1 | Q14974 | Importin subunit beta‐1 | 1 | 1 | 1 | 0.06 |
| 64 | RT35 | P82673 | 28S ribosomal protein S35, mitochondrial | 3.1 | 1 | 1 | 0.17 |
| 65 | TGFI1 | O43294 | Transforming growth factor beta‐1‐induced transcript 1 protein | 4.3 | 1 | 1 | 0.11 |
| 66 | DDX46 | Q7L014 | Probable ATP‐dependent RNA helicase DDX46 | 1 | 1 | 1 | 0.05 |
Proteins were reproducibly identified in all three replicates. MaxQuant iBAQ values were used to calculate protein abundance.
Figure 1.
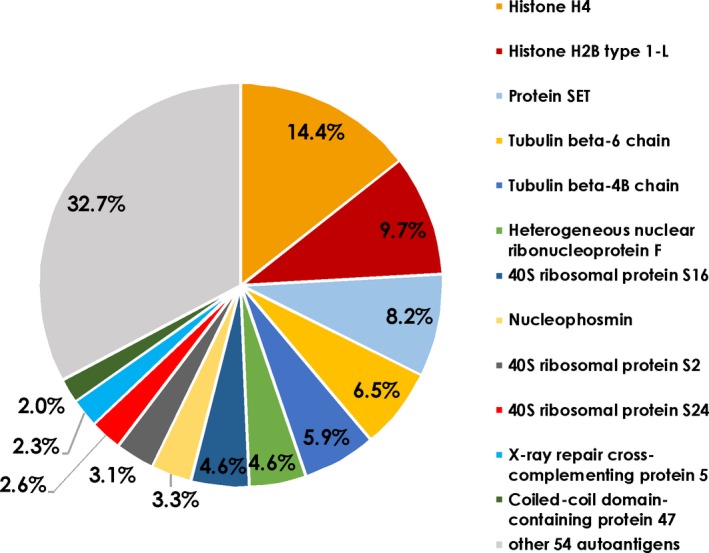
iBAQ values of captured autoantigens using CTRL serum and HTM cells. The iBAQ values are proportional to the molar quantity of the respective antigen measured by mass spectrometry. These proportions represent the abundance of the associated autoantibodies in natural autoimmunity. The diagram shows the distribution of different autoantigens from healthy TM cells captured by antibodies in the control sera (n = 3). The percentage of total peptide intensities is shown.
Characterisation of natural autoantigens by GO enrichment and pathway analysis
Gene Ontology (GO) enrichment was analysed using DAVID (https://david.ncifcrf.gov/) to get an overview of the characteristics of the identified targets of the natural AAbs in the CH experiment group. Figure 2 shows significantly enriched GO terms among the autoantigens for biological process (A), cellular component (B) and molecular function (C).
Figure 2.
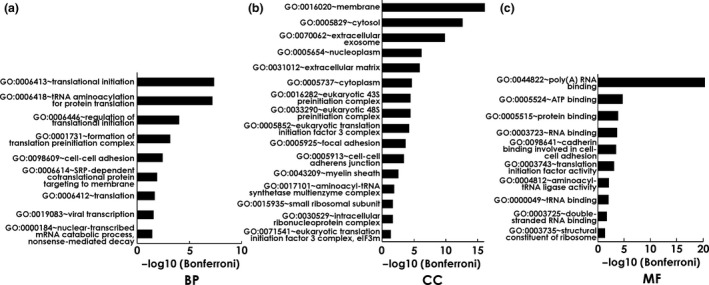
GO enrichment analysis of autoantigens from HTM captured by autoantibodies in CTRL sera (n = 3), representing natural autoimmunity. Negative log10‐transformed P‐values of significantly enriched GO terms (P < 0.05 with Bonferroni correction) are shown in (a) biological process (BP), (b) cellular component (CC) and (c) molecular function (MF).
The analysis showed that the antigens are profoundly involved in several processes associated with translation‐ and RNA‐related processes. Furthermore, the detected immunogenic proteins are involved in virus‐associated processes, as well as cell–cell adhesion. Highlighting the molecular functions of the autoantigens, nearly all of them showed the ability to bind poly (A) or other forms of RNA. Also, the binding of cadherin in cell–cell adhesion represented a frequent function among the autoantigens captured by antibodies from the control sera. We also found marked enrichment of the immunogenic proteins in the cellular components membrane, cytosol and extracellular exosomes.
Identification of POAG‐related autoantigens
The involvement of immunological processes has been frequently shown for several neurodegenerative diseases and for the ocular neuropathy, POAG. To identify antigens that are related to POAG, we analysed autoantibody abundance in the sera of POAG patients and a non‐glaucomatous control group (Table 1). To this end, the proteins identified by MS‐AMIDA were quantified using MaxQuant label‐free quantification (LFQ) intensities. The experimental groups CH and PH (Figure 3a) and also CG and PG (Figure 3b) were analysed for significant differences in autoantibody levels. These were tested using Student's t‐test with permutation‐based FDR (< 0.01) to adjust for multiple testing. The test, however, revealed no significant alterations. Principal component analysis revealed that there is a higher association of the autoantibody levels with the TM cell line used than with the antibody source (Figure 3c). Component 1 and Component 2 explain 68.11% of the variance in the data (see also scree plot in Supplementary figure 1). The influence of single autoantibody levels on the components can be evaluated by observing the component coefficients in Supplementary table 2. Component 1 is mainly influenced by EIF5A1, H4, C1QBP and PNMA2, whereas Component 2 is mainly influenced by NUCL, NSUN2, HS90B and ACTN1. Hierarchical clustering of these antigens based on Euclidean distances (Figure 4a) supports the findings from the principal component analysis (Figure 3c). The experimental groups cluster together based on the specific TM cells, rather than the serum source. The antigens showed two clusters with 11 antigens having lower levels in the glaucomatous TM cells and 27 with increased levels. Therefore, we also examined the differences in the captured autoantigens between GTM and HTM cells. Here, 38 (auto)‐antibody‐captured proteins were found at significantly different levels (Supplementary table 3). We wanted to use a larger sample size to analyse whether different autoantibody serum levels caused these observed changes. To select suitable candidates for further validation, antigens were not only analysed for significant hits but also their fold change (Figure 4b). Only antigens with an FDR < 0.01 and a log2 fold change of > 2 were considered eligible. A total of 21 autoantigens passed these criteria (Table 4). From the subset of the 21 eligible autoantigens, nine targets were chosen for further validation by a protein microarray analysis.
Figure 3.
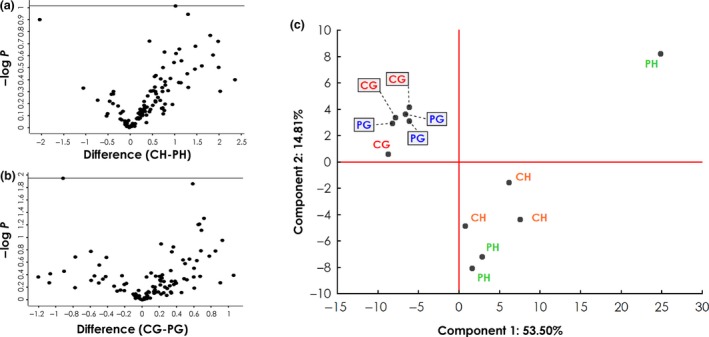
Analysis of autoantigen abundance in different experiment groups (IDs: PH, CH, PG, CG). Detailed information about the experimental groups can be found in Table 2. (a, b) Volcano plots showing negative log10‐transformed P‐values (t‐test with permutation‐based FDR to correct for multiple testing; y‐axis) against the differences of the means (log2; x‐axis) of two groups. (a) Comparison of antigens from healthy TM cell lysates captured by antibodies from control (CH; n = 3) and POAG sera (PH; n = 3). (b) Comparison of antigens from glaucomatous TM cell lysates captured by antibodies from control (CG; n = 3) and POAG sera (PG; n = 3). (c) Principal component analysis, including all four experiment groups, reveals that most of the variance between the groups can be attributed to the cell line used rather than the antibody source.
Figure 4.
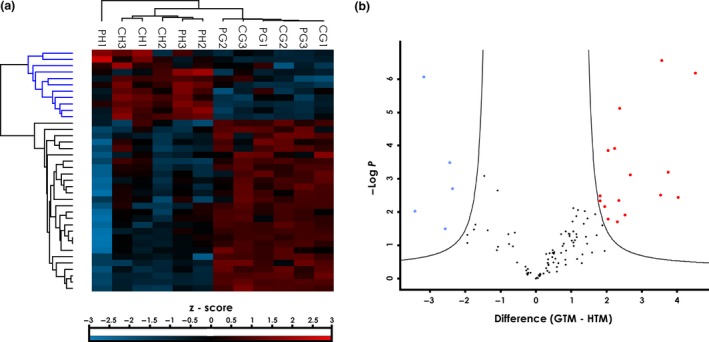
Statistical analysis of captured autoantigens from HTM and GTM cells. (a) Hierarchical clustering of antigens in single samples with significantly different abundance between the cell lines HTM and GTM (38 proteins; t‐test with permutation‐based FDR < 0.01). Clustering is based on z‐score‐transformed LFQ intensities. (b) Volcano plot for candidate selection. Highlighted dots represent proteins with significant differences between cell lines and a log2 fold change of 2 or more, to ensure relevant effect size. Blue dots represent antigens with lower levels and red dots antigens with higher levels in the GTM samples than HTM samples.
Table 4.
Candidate biomarkers with P‐value < 0.05 and log2 fold change of > 2 (log2 differences of the means)
| UniProt accession | Entry name | Gene | Protein | −Log(P‐value) | Difference (GTM – HTM) |
|---|---|---|---|---|---|
| P62805 | H4_HUMAN | HIST1H4A | Histone H4 | 2.028 | −3.428 |
| Q08J23 | NSUN2_HUMAN | NSUN2 | tRNA (cytosine(34)‐C(5))‐methyltransferase | 6.063 | −3.172 |
| P13010 | XRCC5_HUMAN | XRCC5 | X‐ray repair cross‐complementing protein 5 | 1.496 | −2.564 |
| P12814 | ACTN1_HUMAN | ACTN1 | Alpha‐actinin‐1 | 3.484 | −2.437 |
| P26639 | SYTC_HUMAN | TARS | Threonine‐tRNA ligase, cytoplasmic | 2.706 | −2.362 |
| Q6UVK1 | CSPG4_HUMAN | CSPG4 | Chondroitin sulphate proteoglycan 4 | 2.490 | 1.816 |
| Q02878 | RL6_HUMAN | RPL6 | 60S ribosomal protein L6 | 2.340 | 1.819 |
| Q14566 | MCM6_HUMAN | MCM6 | DNA replication licensing factor MCM6 | 2.164 | 1.950 |
| Q96AB3 | ISOC2_HUMAN | ISOC2 | Isochorismatase domain‐containing protein 2 | 3.856 | 2.043 |
| Q9NTK5 | OLA1_HUMAN | OLA1 | Obg‐like ATPase 1 | 1.786 | 2.053 |
| Q00005 | 2ABB_HUMAN | PPP2R2B | Serine/threonine‐protein phosphatase 2A 55 kDa regulatory subunit B beta isoform | 3.909 | 2.226 |
| P10809 | CH60_HUMAN | HSPD1 | 60 kDa heat shock protein, mitochondrial | 1.705 | 2.305 |
| P42704 | LPPRC_HUMAN | LRPPRC | Leucine‐rich PPR motif‐containing protein, mitochondrial | 2.342 | 2.350 |
| O00159 | MYO1C_HUMAN | MYO1C | Unconventional myosin‐Ic | 5.121 | 2.368 |
| P63104 | 1433Z_HUMAN | YWHAZ | 14‐3‐3 protein zeta/delta | 1.907 | 2.527 |
| P46940 | IQGA1_HUMAN | IQGAP1 | Ras GTPase‐activating‐like protein IQGAP1 | 3.117 | 2.680 |
| Q07021 | C1QBP_HUMAN | C1QBP | Complement component 1 Q subcomponent‐binding protein, mitochondrial | 2.513 | 3.536 |
| Q9ULC4 | MCTS1_HUMAN | MCTS1 | Malignant T‐cell‐amplified sequence 1 | 6.556 | 3.560 |
| P27708 | PYR1_HUMAN | CAD | CAD protein | 3.204 | 3.754 |
| P63241 | IF5A1_HUMAN | EIF5A | Eukaryotic translation initiation factor 5A‐1 | 2.437 | 4.028 |
| Q9UL42 | PNMA2_HUMAN | PNMA2 | Paraneoplastic antigen Ma2 | 6.185 | 4.521 |
Characterisation of POAG‐related autoantigens
To investigate in which biological processes these possibly POAG‐related autoantigens are involved, we used DAVID's functional annotation clusters and also Metascape enrichment and pathway analysis. DAVID identified six functional clusters among 19 of the 21 targets analysed using GO terms, UniProt (UP) keywords and UP sequence features (seq feature) (Figure 5). These clusters comprise cell–cell adhesion and junctions (cluster 1), nucleotide and ATP‐binding (cluster 2), cytoplasmic proteins with different isoforms (cluster 3), mitochondria‐associated proteins (cluster 4), DNA‐binding proteins (cluster 5) and transcription (cluster 6). The analysis with Metascape revealed eight enriched GO and Kyoto Encyclopaedia of Genes and Genomes (KEGG) terms (Figure 6a and b). The target antigens were enriched in translation, mRNA transport, cell junction assembly, positive regulation of apoptotic process and establishment of protein localisation to membrane, as well as viral carcinogenesis pathways, G2/M checkpoints and the platelet‐derived growth factor receptor beta (PDGFRB) pathway. The protein–protein interaction (PPI) network shows only antigens that are known to interact with at least one other of the identified targets (Figure 6c). This analysis showed that 15 of the 21 targets are connected in the PPI network. Of these, the Metascape molecular complex detection (MCODE) algorithm identified one densely connected network component, consisting of the proteins CAD, HSPD1, YWHAZ, OLA1 and EIF5A. The independent enrichment analysis for this MCODE component revealed an enrichment of the five antigens in translation, peptide biosynthetic process and the PDGFRB pathway (Figure 6d).
Figure 5.

DAVID functional annotation clusters of potentially POAG‐related autoantigens. Nineteen out of the 21 included proteins showed an enrichment in at least one cluster. Enrichment scores: cluster 1 (2.54), cluster 2 (1.7), cluster 3 (1.59), cluster 4 (1.32), cluster 5 (1.02) and cluster 6 (0.39). The analysis includes Gene Ontology terms (GO terms), UniProt keywords (UP keywords) and UniProt sequence features (UP seq feature). Green indicates enrichment in the cluster for the respective protein.
Figure 6.
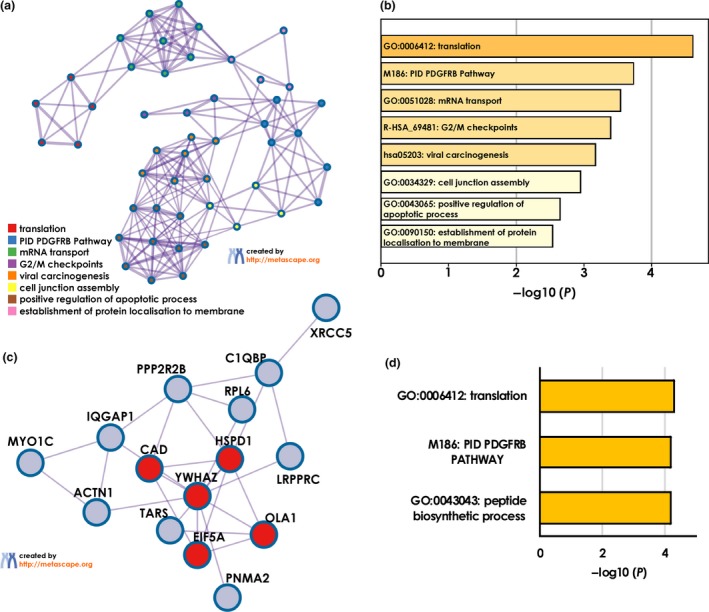
Metascape analysis of POAG‐related autoantigens. (a) Network of enriched GO and KEGG terms coloured by cluster. (b) Enriched GO and KEGG terms from (a) with corresponding P‐values. (c) Protein–protein interaction network with densely connected components (red) identified by the MCODE algorithm. (d) Enrichment analysis for MCODE cluster only.
Validation of selected targets by protein microarray
We chose alpha‐actinin‐1 (ACTN1), serine/threonine‐protein phosphatase 2A 55 kDa regulatory subunit B beta isoform (PPP2R2B), mitochondrial complement component 1 Q subcomponent‐binding protein (C1QBP), malignant T‐cell‐amplified sequence 1 (MCTS1), paraneoplastic antigen Ma2 (PNMA2), Ras GTPase‐activating‐like protein IQGAP1 (IQGAP1), unconventional myosin‐1c (MYO1C) and cytoplasmic threonine‐tRNA ligase (TARS) to be analysed by protein microarray. Even though it is not a new target, heat shock protein 60 (HSP60) has been included as a well‐defined glaucoma‐related antigen to serve as a positive control. The targets were obtained as recombinant proteins and were used to prepare customised microarray slides in our laboratory. Slides were hybridised with sera from 120 POAG patients and 120 non‐glaucomatous controls (Table 1, microarray validation). With this method, the samples were analysed for their antigen‐autoantibody reactions. Incubation with a fluorescent dye‐labelled anti‐human IgG antibody yielded signal intensities that were proportional to the concentration of the respective autoantibody. After data pre‐processing and normalisation, statistical tests and analysis were conducted. The results of the Mann–Whitney U‐test are listed in Table 5. This approach identified significant increased autoantibody levels to PNMA2, TARS, C1QBP and HSPD1 (Figure 7). Signals representing the binding of ACTN1, IQGAP1 and MYO1C AAbs were at the detection limit of this method and therefore could not be quantitatively evaluated.
Table 5.
Results of microarray validation
| Rank Sum POAG | Rank Sum CTRL | U | Z | P‐value | Z adjusted | P‐value | |
|---|---|---|---|---|---|---|---|
| PPP2R2B AAbs | 14 078 | 14 842 | 6818 | −0.710 | 0.477 | −0.712 | 0.477 |
| TARS Aabs | 16 741 | 12 179 | 4919 | 4.242 | 0.000 | 4.258 | < 0.001 |
| C1QBP Aabs | 15 714 | 13 206 | 5946 | 2.332 | 0.020 | 2.333 | 0.020 |
| PNMA2 Aabs | 16 032 | 12 888 | 5628 | 2.923 | 0.003 | 2.930 | 0.003 |
| MCTS1 Aabs | 15 015 | 13 905 | 6645 | 1.032 | 0.302 | 1.034 | 0.301 |
| HSPD1 Aabs | 16 793 | 12 127 | 4867 | 4.338 | 0.000 | 4.350 | < 0.001 |
Comparison of autoantibody levels in POAG and CTRL samples (n = 120) assessed by a two‐sided Mann–Whitney U‐test.
Figure 7.
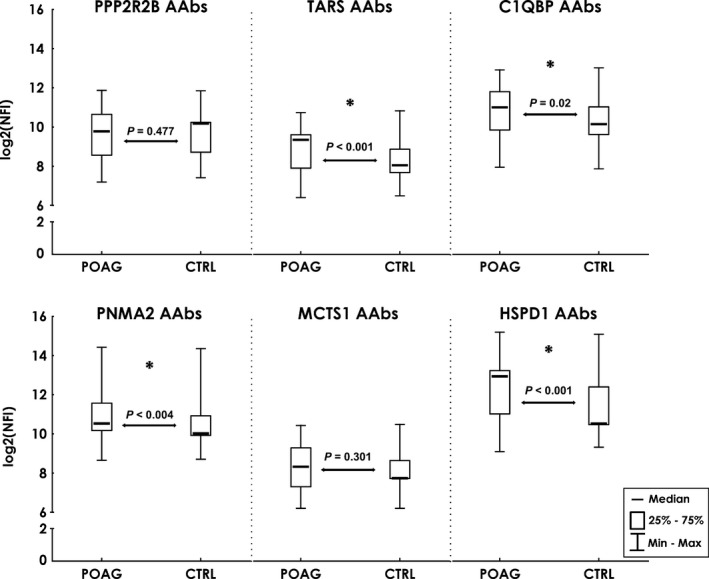
Results of the microarray validation. Comparison of autoantibody levels in POAG (n = 120) and CTRL (n = 120) serum. Median log2‐transformed normalised fluorescence intensities. Median intensities with 25–75% percentiles (box) and min/max values (whiskers). P‐values from a Mann–Whitney U‐test. Asterisks mark significant differences at P < 0.05.
Correlation with disease‐related clinical parameters
We wanted to examine further whether the detected increase in PNMA2, TARS, C1QBP and HSPD1 AAbs showed any relationship to clinical hallmarks of POAG pathogenesis. Calculation of Spearman's rank‐order correlation coefficients was used to detect correlations of the AAbs with cup disc ratio (CDR), visual field defect (‘mean deviation’; MD) and IOP (Table 6). The correlation coefficients reveal a weak negative correlation of PNMA2 and HSPD1 AAbs with CDR (R = −0.29 and R = −0.28), a moderate negative correlation of HSPD1 AAbs with visual field defect (R = −0.42) and a weak positive correlation between TARS AAbs and visual field defect (R = 0.34). None of the AAbs showed a relation to IOP. To investigate whether these AAbs show significant alterations in other systemic or eye diseases, we divided the control group into the categories, other eye disease (n = 39), systemic disease (n = 18), other eye + systemic disease (n = 40) and healthy subjects (n = 23). Kruskal–Wallis ANOVA for multiple group comparison revealed no significant difference (P > 0.05) for PNMA2, TARS, C1QBP and HSPD1 AAbs among these groups.
Table 6.
Spearman rank correlations of autoantibodies in POAG patients and glaucoma‐related clinical parameters: cup disc ratio (CDR); visual field defects (‘mean deviation’, MD); intraocular pressure (IOP). Spearman correlation coefficients and corresponding P‐values.
| Clinical parameter | Autoantibody | Valid N | Spearman R | t (N‐2) | P‐value |
|---|---|---|---|---|---|
| CDR | C1QBP | 82 | 0.082 | 0.732 | 0.466 |
| PNMA2 | 82 | −0.286 | −2.673 | 0.009 | |
| TARS | 82 | 0.098 | 0.884 | 0.379 | |
| HSPD1 | 82 | −0.284 | −2.648 | 0.010 | |
| MD | C1QBP | 55 | 0.113 | 0.826 | 0.413 |
| PNMA2 | 55 | −0.155 | −1.140 | 0.260 | |
| TARS | 55 | 0.335 | 2.586 | 0.012 | |
| HSPD1 | 55 | −0.421 | −3.376 | 0.001 | |
| IOP | C1QBP | 114 | −0.120 | −1.279 | 0.203 |
| PNMA2 | 114 | 0.091 | 0.964 | 0.337 | |
| TARS | 114 | 0.085 | 0.905 | 0.367 | |
| HSPD1 | 114 | 0.037 | 0.396 | 0.693 |
Evaluation of diagnostic potential
The establishment of reliable biomarkers for the objective diagnosis of POAG is still a crucial task in ophthalmic research. Therefore, we wanted to evaluate whether validated glaucoma‐related AAbs to PNMA2, TARS, C1QBP and HSPD1 hold the potential to serve as biomarker candidates. For this purpose, we applied a random forest algorithm to the microarray data set to seek a potential classification of POAG patients and non‐glaucomatous controls. The data set was randomly divided into a training (n = 165) and a test set (n = 75). The resulting random forest model was able to correctly classify 30 out of 38 POAG patients and 33 out of 37 controls (Figure 8). This translates to a sensitivity of 79% at 89% specificity with an overall accuracy of 84%.
Figure 8.

Random forest classification. Prediction is plotted using test samples not used for training (n = 75) only. Bars represent the number of predicted (POAG in green; CTRL in red) and observed cases. The random forest algorithm classified POAG samples and CTRL samples with a sensitivity of 79%, a specificity of 89% and 84% accuracy.
Discussion
Antibodies to self‐antigens are mostly associated with immune disorders such as systemic lupus erythematosus28, 29 or Hashimoto's disease,30 but are also frequently found in conditions that are not considered autoimmune. Antibodies to tumor antigens are present in the blood of cancer patients,31 and AAbs are even found in neurological disorders such as Alzheimer's32 or Parkinson's disease.33 In recent years, evidence has increased that AAbs are not only involved in pathology, but are also a part of physiologic processes. An important goal for the better understanding of the role of immunological processes in health and disease is to link the natural AAbs to specific biological processes and signalling pathways. Also, the formation of autoantigens in relation to diseases has not been elucidated conclusively. So far, several causes for proteins to become immunogenic have been discussed. These comprise the hypothesis of molecular mimicry,34 but alterations in post‐translational modification, protein overexpression, alternative splicing and the generation of neo‐epitopes in the course of apoptosis have also been proposed.35, 36, 37, 38 These proposals also infer that differences in circulating autoantibody abundances might reflect molecular changes. However, most identified autoantigens have not been studied for the underlying reason for their immunogenicity. Although the possible involvement of the immune system in glaucoma pathogenesis has been investigated in many studies, the qualitative characterisation of the involved antigens and AAbs is incomplete. The specific conditions under which an antigen can lead to a modified autoimmunity are also unclear. There is a further need to understand the nature and function of the natural autoantibody repertoire as a whole. That will also significantly help to understand the changes that occur under different disease states. In this study, we investigated the autoantibody repertoire against TM proteins as part of natural autoimmunity as well as POAG‐related alterations. This exploratory study also aims to stimulate the formation of hypotheses that would help to explain the properties of the natural immunome and the implications of autoimmunity in glaucoma.
We analysed autoantigens in healthy and glaucomatous TM cells that were captured by serological antibodies of POAG and non‐glaucomatous controls using MS‐AMIDA. At first, we wanted to identify targets of the naturally occurring AAbs and link them to biological processes and signalling pathways. To this end, we performed GO enrichment analysis with the 66 proteins captured by antibodies isolated from non‐glaucomatous sera. The GO analysis involved the evaluation of three different categories, ‘biological process’, ‘molecular function’ and ‘cellular component’. This analysis showed that there is a strong association of the antigens targeted by naturally occurring AAbs with translational processes as part of protein biosynthesis. Interestingly, several enzymes of the family of tRNA ligases were identified as immunogenic proteins. Many proteins representing subunits of the aminoacyl‐tRNA synthetase multienzyme complex were also enriched in different cellular components. However, the AAbs also directly targeted epitopes of the 40S ribosomal subunit. This was also reflected by the strong enrichment of RNA‐binding properties in the identified antigens.
Furthermore, the antigens were highly enriched in the cellular components, ‘membrane’ and ‘cytosol’. They were, however, also located in ‘extracellular exosomes’ and the ‘nucleoplasm’. That the autoantigens are also involved in intranuclear processes was also reflected by their enrichment in poly (A)‐RNA‐binding properties and nuclear‐transcribed mRNA processing in the control mechanism of nonsense‐mediated decay. Another study on the overall immunogenicity of autoantigens could also show a frequent enrichment of autoantigens with a function of RNA and protein binding and also a strong association with ribosomes and spliceosomes.39 The abundance of antigens associated with viral transcription‐related processes may be because the cell lines used in this experiment were transformed by simian virus 4040 that could be responsible for changes in cellular transcription proteins.
The most abundant antigens identified were nuclear proteins. At 23.3%, histone proteins were the most common, followed by the histone‐binding protein SET. Anti‐histone antibodies have been linked to the autoimmune disease systemic lupus erythematosus,28 but our results indicate that they might also be involved in natural autoimmunity. AAbs to ribonucleoproteins are associated with a spectrum of rheumatic diseases.41 It is presumed that RNA‐binding proteins can activate B cells and thereby enable AAb production by interaction of the RNA with TLR‐7 or TLR‐8.42 Since we have also found these antibodies in individuals who are not suffering from an autoimmune disease (according to anamnesis), there is also the probability that anti‐ribonucleoprotein antibodies are also part of the natural autoantibody repertoire. This also suggests that other criteria would need to be met for these AAbs to become pathogenic.
The second goal of this study was to investigate possible alterations in the immunoproteome in POAG patients. To this end, we compared the binding of serological antibodies, purified from POAG patients and non‐glaucomatous controls, to proteins derived from GTM and HTM cells. The autoantigens, identified and quantified by LFQ, showed more pronounced alterations in the groups with a different protein source (GTM vs HTM) than in the CTRL vs POAG sera groups. Antibody reactivity seems to be more influenced by possibly disease‐related antigen properties than by mere abundance of the antibodies. This could indicate that not only the antibodyome is changed during disease, but that natural AAbs bind with higher affinity to antigens with disease‐related alterations. Our data do not provide further insights into the nature of these alterations, but the occurrence of neo‐epitopes, specific altered post‐translational modifications or truncations are possible changes that could enhance the target binding of physiological AAbs. These explanations need to be evaluated through additional research, so the implications we can make from these results are merely theoretical. A hypothesis that needs further validation by subsequent experiments is that not only quantitative but also qualitative differences between natural and disease‐related AAbs may play a role. Hints that support such assumptions come from other studies that show the disease‐specific accumulation of distinct complementarity determining region peptides (CDR peptides) of the highly variable region of AAbs in glaucoma and other diseases.43, 44, 45 From these findings, it seems likely that not only alterations of the antigen but also of the AAbs themselves can lead to the onset of autoimmune processes. This raises an interesting question for future studies that aim to analyse whether alterations of either antibody or target protein can influence the binding properties. Also, the possible ability of these alterations to promote undesirable effects that may accompany or even accelerate pathological processes needs to be investigated.
The PPI network and pathway enrichment analysis of the POAG‐related autoantigens using Metascape revealed that especially those proteins involved in translation and the PDGFRB pathway show altered immunogenicity. Also, the five connected proteins, HSPD1, CAD, YWHAZ, EIF5A and OLA1, as identified by MCODE, are strongly enriched in these molecular processes. PDGF signalling has been linked to fibrotic diseases and is implicated in the proliferation of cells with myofibroblast‐like properties.46 TM cells also show similar characteristics to contractile myofibroblasts that express alpha‐smooth muscle actin (a‐SMA).47 Fibrosis also involves an accumulation of ECM, which was also observed in the glaucomatous TM.48 The hypothesis of an involvement of fibrotic processes in POAG‐related modulations of TM that could be mediated by increased levels of transforming growth factor β2 (TGFβ2) in the aqueous humour has already been discussed.49, 50 The MCODE cluster of AAbs potentially reflects POAG‐specific alterations of the PDGFRB pathway. However, a causative or synergetic effect of these AAbs in mediating autoimmune‐driven fibrosis in glaucomatous TM cannot be excluded. Thus, an investigation of the effects of AAbs on TM cells seems to be required. In a previous study from our group, a retinal ganglion cell line was incubated with serum or serological antibodies alone, derived from POAG patients or non‐glaucomatous controls.51 In this experiment, no changes in cell viability were observed. However, a change in protein expression towards a pro‐apoptotic state occurred when incubated with POAG serum or isolated POAG IgG, but not after incubation with control serum. We assume that the antibodies would exert similar effects on TM cells. Nonetheless, this would be an interesting subject for further studies, especially as the interaction of autoantibodies with targets in the PDGFR pathway of TM cells could give new insights into their role in glaucoma pathogenesis.
In other studies, which examined autoantibodies in glaucoma, but also in other neurodegenerative diseases such as Alzheimer's or Parkinson's disease, a disease‐specific change in the levels of various serum autoantibodies could be shown. We presume that an altered interplay of antigens and autoantibodies underlies both qualitative and quantitative changes that must be taken into account. Therefore, we wanted to analyse whether the antibody levels to the antigens identified here also show a change in abundance that can be detected by a more sensitive method. To analyse the relative abundance of the serological AAbs to selected targets in glaucomatous and non‐glaucomatous subjects, we used a protein microarray approach as a high‐throughput method. Even though specific attributes of the AAbs and their respective antigens seem to play a significant role in the characteristic of their interaction, many previous studies have also shown that disease‐related alterations of natural autoimmunity can also be related to the levels of the serological AAbs. The evaluation of these quantitative differences is especially valuable in disease diagnostics, as they are easier to measure in clinical tests. We used recombinant human proteins, produced with a wheat germ expression system, generating proteins without post‐translational modifications, to achieve a standardised experiment set‐up. The analysis of sera from 120 POAG and 120 non‐glaucomatous subjects revealed an increase of anti‐C1QBP, anti‐TARS, anti‐PNMA2 and anti‐HSPD1 AAbs in the POAG group. While antibodies to HSPD1 (mitochondrial 60 kDa heat shock protein) have frequently been observed in association with glaucoma,18, 52, 53, 54 AAbs to C1QBP, TARS and PNMA2 have not yet been described in the disease context.
Component 1 Q subcomponent‐binding protein (C1QBP) is a multifunctional protein and putative receptor for component 1Q. This function leads to the inhibition of the C1 complex and thereby to suppression of complement activation. It is also involved in the regulation of mRNA splicing via its RNA‐binding capacity. Our results showed upregulated levels of anti‐C1QBP antibodies in the serum of POAG patients. Although AAbs to C1QBP have not yet been described in glaucoma, activation of the complement system has been discovered in the retinal ganglion cells of glaucomatous eyes.55 C1QBP AAbs could presumably prevent the inhibition of C1 complex formation by blocking the C1QBP effector sites.
Our study also showed increased levels of autoantibodies to threonyl‐tRNA synthetase (TARS). TARS is an enzyme functioning as the ligase of threonine to its respective tRNA. Neither TARS AAbs nor the antigen itself has been investigated in the context of glaucoma. It cannot be clearly stated what causes the formation of these AAbs and whether they are involved in disease‐related tissue damage. Antibodies to different other tRNA synthetases are often found in patients with inflammatory myositis, especially Jo‐1 AAbs.56 Although these myositis‐specific AAbs are far better characterised, researchers did not propose the reason for their occurrence, or the implications for the disease.57 The characterisation of the origin and the properties of the TARS AAbs remains a task for further studies.
The paraneoplastic antigen 2 (PNMA2) is known to be linked to paraneoplastic disorders and is also related to neurodegenerative processes. AAbs are frequently found in cancer patients, as tumor cells are suspected of producing abnormal amounts of different members of the PNMA family.58 These proteins are also naturally expressed by neuronal cells. The presence of this protein in TM cells is conceivable, since this tissue is likely to have originated from the neural crest.59 Also, other PNMA proteins could be identified in porcine TM.60 It is assumed that AAbs to PNMA2 are produced as a consequence of protein overexpression in cancer cells. But it cannot be excluded that other properties of the antigen are responsible for its immunogenicity as well. The PNMA2 protein seems to be prone to an autoimmune reaction, which could also be relevant to other neurodegenerative diseases such as glaucoma. However, the AAbs do not seem to drive the progression of paraneoplastic neurologic disease, since their depletion did not result in relief of the symptoms, nor can the injection of the AAbs induce the disease.61 It could be an interesting topic for further studies to investigate the specific role of PNMA family members in the pathogenesis of glaucoma.
The identified AAbs seem to be associated with POAG but are mostly not exclusive to glaucoma. This is also true for several glaucoma‐related AAbs previously identified. For example, antibodies to HSP60, vimentin and annexin A5 have not only been reported in association with glaucoma,18, 23, 25, 52, 54 but are also frequently found in the sera of cancer patients.62, 63 Also, the antigens are not TM‐specific. Therefore, they might not be specific enough to serve as biomarker candidates. Although the application of random forest algorithms in this study enabled classification of POAG patients with a sensitivity of 79% at 89% specificity, a translation of these finding to be used in diagnostics might not be feasible and therefore needs additional evaluation. As observed with other autoantibody disease markers, variability in AAb levels in the population can be very high. Biomarkers often lack specificity since the putative disease markers are also widespread among subjects without the respective disease.64 The establishment of reliable biomarkers requires further knowledge about their roles in glaucoma and their distribution in the general population.
If the occurring autoantibodies are an epiphenomenon of the pathological changes in the TM, alterations in the involved proteins would necessarily occur beforehand. It has been shown that pathways, especially those regulating extracellular matrix deposition, the actin cytoskeleton and cell–cell/cell–matrix connections, are altered in glaucomatous TM. Several factors are thought to play a role in these changes, including increased expression of TGFβ. It seems quite possible that a single occurrence of elevated IOP could lead to proteomic alterations and consequently altered autoantibody profiles. In a previous study, our group was able to highlight changes in autoantibody levels after acute angle‐closure glaucoma attack, an event accompanied by a sudden increase of IOP and probable substantial damage to the optic nerve, in patients without previous glaucoma history.65
Although our study provides new insights into the properties and POAG‐related changes of the natural autoantibody repertoire, some limitations shall be pointed out. The proteomic analysis involved in the MS‐AMIDA approach requires a large amount of protein sample material. The availability of human TM tissue samples is limited, and primary cell cultures are also restricted in the amount of material they can yield. For the purpose of our explorative study, we used the most practical TM cell lines. These, however, have the disadvantage of being transformed by simian virus 40 that alters the cellular transcription machinery. Also, the donors of the TM cells were not the same age (GTM cells from a 74‐year‐old POAG patient and HTM cells from an 18‐year‐old donor). This makes further evaluation of our findings using more appropriate TM protein sources mandatory.
Conclusion
Overall, we could provide new insights into the natural AAb repertoire and highlight pathways which appear to harbour immunogenic proteins. PPI and pathway analysis of POAG‐related autoantigens showed a strong association with the PDGFRB pathway, among others. This pathway is assumed to play an essential role in TM fibrosis, and the related AAbs could play an active role in the pathology or not. Lastly, we could confirm the presence of increased levels of AAbs against PNMA2, TARS and C1QBP in the serum of POAG patients that deserve to be further analysed as potential glaucoma biomarkers.
Methods
Sera
The blood samples in this study were collected in accordance with the Declaration of Helsinki on biomedical research involving human subjects. Written informed consent was obtained from each subject. The ethics committee of the Landesärztekammer Rheinland‐Pfalz approved the usage of the samples (Vote: 827.228.11 (7770)). All subjects included in this study received an ophthalmic examination at the Department of Ophthalmology of the University Medical Center in Mainz, Germany. POAG patients were diagnosed according to the guidelines of the European Glaucoma Society,66 based on elevated IOP, visual field defects and optic nerve cupping. The control group consists of age‐ and sex‐matched non‐glaucomatous subjects. In the discovery phase using MS‐AMIDA, 30 serum samples of POAG patients and 30 non‐glaucomatous controls were analysed. Subjects with other eye diseases (except cataract), known autoimmune diseases (as evaluated from anamnesis) and previous eye surgeries were excluded to reduce the possible effects of other conditions on the serum antibody levels. With these exclusion criteria, we wanted to exclude bias from immunologic reactions that are not glaucoma‐related or might be a cause of the mechanical disruption of the ocular tissue caused by surgical intervention. This might lead to an exchange of intracellular proteins with blood or aqueous humour that otherwise would not be in contact under physiological or disease‐related conditions and could influence the conclusions drawn from the experiments. In the large‐scale microarray validation, 120 POAG samples were compared to 120 non‐glaucomatous, age‐ and sex‐matched control samples. Here, the exclusion criteria were abandoned, except for other glaucoma types in the control group. The characteristics of the study population can be found in Table 1.
IgG isolation
Mass spectrometry‐based antibody‐mediated identification of autoantigens was conducted using 30 POAG and 30 control serum samples (Table 2). Samples were pooled, creating three sample pools (10 samples/pool) for each group. IgG was isolated from 2 mL of pooled serum samples by affinity purification, using the NAb Protein G Spin Kit (Thermo Scientific, Rockford, IL, USA), according to the manufacturer's instructions.
Cell culture
As the source for TM proteins, the immortalised human TM cell line HTM5 and the glaucomatous TM cell line GTM3 were used. Both cell lines were a kind gift of Professor Abbot F. Clark and colleagues [North Texas Eye Research Institute (NTERI), Fort Worth, TX, USA]. The cell lines are derived from human TM as described elsewhere.40, 67 Cells were grown in DMEM low glucose medium (Sigma‐Aldrich, St. Louis, MO, USA), supplemented with 10% foetal calf serum (Gibco, Life Technologies, Carlsbad, USA), 1% penicillin/streptomycin solution (Sigma‐Aldrich, St. Louis, MO, USA) and 1% 200 mm L‐alanyl‐L‐glutamine (Biochrom, Berlin, Germany). The TM cells were cultured in an incubator at 37°C and 5% CO2. Media were changed every 2–3 days. On confluence, cells were collected using cell scrapers. Cell pellets were snap‐frozen in liquid nitrogen and stored at −20°C until further use.
Cell lysis
Cell pellets were incubated for 30 min on ice with lysis buffer, modified after Alhamdani et al. 68 [20 mm HEPES pH7.9, 1 mm MgCl2, 5 mm EDTA, 1 mm PMSF, 0.5% Triton X‐100, 0.5% NP40, 0.25% ASB‐14, 0.25% CHAPS (Carl Roth, Karlsruhe, Germany), 0.5% Protease Inhibitor Cocktail, 0.5% Phosphatase Inhibitor Cocktail (reagents obtained from Sigma‐Aldrich, Steinheim, Germany, unless otherwise stated)] with occasional vortexing. Additionally, cells were mechanically disrupted using an ultrasonic probe. The cells were sonicated in short burst, with occasionally cooling of the samples on ice. After an additional incubation for 15 min on ice, the cells were centrifuged at 20 000 g and 4°C. Afterwards, the supernatant was collected, and protein concentration was determined using a BCA assay kit (Thermo Scientific, Frankfurt, Germany).
Immunoprecipitation
For the immunoprecipitation (IP) of autoantigens from TM lysates, isolated IgG from serum was cross‐linked to magnetic Sepharose beads with Protein G as ligand (Protein G Mag Sepharose; GE Healthcare, Freiburg, Germany). The IP was carried out using a modified protocol, based on instructions from the manufacturer. One hundred microlitres of bead slurry was incubated with 2 mg of isolated IgG for 1 h at 4°C on a rotation incubator. Unbound IgG solution was removed, and beads were washed with TBS [Tris (Carl Roth)‐buffered saline; pH 7.5]. For the chemical cross‐linking of the antibodies, beads were first equilibrated in triethanolamine solution [TEA (Sigma‐Aldrich, Steinheim, Germany); 200 mm; pH 8.9] before incubation with dimethyl pimelimidate dihydrochloride (Sigma‐Aldrich, Steinheim, Germany; 50 mm in 200 mm TEA; pH 8.9) for 30 min with slow rotation at room temperature. Beads were washed with TEA solution following 30‐min incubation in 100 mm ethanolamine (Sigma‐Aldrich, Steinheim, Germany; pH 8.9). The beads were washed once with elution buffer [0.1 m Glycine (AppliChem, Darmstadt, Germany)‐HCl (Carl Roth), 2 m urea (Carl Roth); pH 2.9] and two times with TBS, to reduce unspecific binding. The beads covalently bound to the antibodies were incubated overnight with 2 mg TM protein (1 mg mL−1) at 4°C on a rotation incubator. After removing unbound TM proteins, the beads were washed three times with TBS and then four times with 100 mm ammonium bicarbonate solution (ABC; Sigma‐Aldrich, Steinheim, Germany).
On‐bead digestion
For the preparation of the samples prior to MS analysis, on‐bead tryptic digestion of the precipitated proteins was used, as described in Ref.69 To this end, 30 µL of trypsin solution (Promega, Madison, WI, USA; diluted in 100 mm ABC) was directly added to the beads following incubation for 15 min at room temperature with occasional vortexing. After overnight incubation at 37°C, supernatant was collected and stored at 4°C. Another 30 µL of trypsin solution was added to the beads before incubation for additional 4 h at 37°C. The supernatant was separated from the magnetic beads, and both digests were pooled. Formic acid (Merck, Darmstadt, Germany) was added to a final concentration of 5%. The samples were then dried in a vacuum concentrator. Prior to MS analysis, samples were diluted in 0.1% trifluoroacetic acid (TFA; Merck) in HPLC‐grade water (AppliChem) and purified using SOLAµ SPE plates (HRP 2 mg mL−1 96‐well plate; Thermo Scientific, Rockford, IL, USA) using the manufacturer's protocol with slight modifications.70 In brief, the plate was activated with 150 µL acetonitrile (ACN; AppliChem) and equilibrated with 0.1% TFA solution. Samples were loaded three consecutive times on the plates, followed by two washing steps with 0.1% TFA. Peptides were eluted twice with 25 µL 60% ACN. All samples were lyophilised by vacuum centrifugation (SpeedVac, Thermo Scientific, Waltham, MA, USA) and stored at −20°C until MS analysis.
LC‐ESI‐MS/MS
Following on‐bead tryptic digestion, the samples were analysed using an LC‐ESI‐MS/MS system (LTQ Orbitrap XL; Thermo Scientific, Rockford, IL, USA).60, 71 The samples were first solubilised in 10 µL of 0.1% TFA. The LC system consisted of a 30 × 0.5 mm BioBasic C18 column (Thermo Scientific, Rockford, IL, USA) and a Rheos Allegro pump (Thermo Scientific, Rockford, IL, USA). A PAL HTC autosampler (CTC Analytics, Zwingen, Switzerland) was used to inject 6 µL of the samples into the system, followed by a solvent gradient. The gradient was run for 120 min per sample: 0–40% solvent B (0–40 min), 40–80% solvent B (40–80 min), 80–100% solvent B (80–100 min), 100–80% solvent B (100–110 min), 80% solvent B (110–120 min) (solvent A: LC‐MS‐grade water (AppliChem) + 0.1% (v/v) formic acid; solvent B: LC‐MS grade ACN + 0.1% (v/v) formic acid). The LC system was coupled to an electrospray ionisation (ESI)–LTQ–Orbitrap XL MS (Thermo Scientific, Bremen, Germany) for the acquisition of the mass spectra data. The system was operated in a data‐dependent mode of acquisition to switch between Orbitrap‐MS and LTQ‐MS/MS acquisition automatically. The detection range was set to 300–2000 m/z with a resolution of 30 000. Parameters for dynamic exclusion were set to a repeat count of 1, a repeat duration of 30 s, with an exclusion list size of 50 and exclusion duration of 90 s. Collision‐induced dissociation (CID) fragmentation was used to isolate the five most intense precursor ions for fragmentation in the LTQ. Activation time was set to 30 ms with a repeat count of 10. Normalised collision energy (NCE) was set to 35%.
Mass spectrometry spectra were analysed using MaxQuant (version 1.5.3.30; Max Planck Institute of Biochemistry, Martinsried, Germany). The spectra were searched against UniProt reviewed protein database (homo sapiens; 11.02.2019). Mass tolerance was set to ±20 ppm for precursor ions and ±0.5 Da for fragmentation. Carbamidomethylation of cysteine was set as fixed modification, N‐terminal acetylation and oxidation of methionine as variable modifications. Trypsin was chosen as the digestive enzyme, and a maximum of two missed cleavages were allowed. Identification was based on a minimum peptide length of 7 with a FDR < 0.01. Proteins were quantified according to their peptide intensities using MaxQuant label‐free quantification (LFQ).
Antigen microarray analysis
Selected autoantigen candidates were purchased as recombinant proteins with no post‐translational modifications (wheat germ expression system). All antigens are listed in Supplementary table 4. The arrays were produced in our laboratory using a non‐contact array printer (SciFLEXARRAYER S3, Scienion, Berlin, Germany). The selected antigens were spotted in triplicate onto nitrocellulose‐covered glass slides (AVID Oncyte, 16 Pad NC slides, Grace Bio‐Labs, Bend, OR, USA). A human IgG mix (Sigma‐Aldrich, St. Louis, MO, USA) and PBS (Life Technologies, Paisley, UK) were included as positive and negative control spots. The spotting procedure was carried out in a humidity chamber at 60% humidity. After the array spotting, slides were allowed to dry on the spotter platform overnight. Array hybridisation was performed using 16‐well incubation chambers (ProPlate Multiwell chambers, Grace Bio‐Labs). All incubation steps were carried out on an orbital shaker at 4°C. At first, the arrays were incubated for 1 h with a blocking buffer (Super G, Grace Bio‐Labs) to reduce background signals. Then, the blocking buffer was discarded and the residual buffer was removed by washing the slides with phosphate‐buffered saline containing 0.5% Tween‐20 (PBST; Sigma‐Aldrich, Steinheim, Germany) three times. Afterwards, the arrays were incubated with 100 µL diluted serum samples (1:250 in PBS) overnight. PBS‐only negative controls were included on each slide. Next, slides were again washed three times with PBST followed by incubation with an anti‐human antibody conjugated with a fluorophore (Alexa Fluor® 647 AffiniPure Goat Anti‐Human IgG, Fcγ fragment‐specific, 109‐605‐008, Jackson ImmunoResearch, West Grove, PA, USA) as secondary antibody at a 1:500 dilution in PBS for 1 h. After this step, the arrays were washed twice with PBST and twice with ultrapure water. The slides were subsequently dried for 2 min in a vacuum centrifuge concentrator.
Array images were acquired as 16‐bit TIF files using a high‐resolution confocal laser scanner (428 Array Scanner, Affymetrix, Santa Clara, CA, USA). The image analysis software Imagene (Imagene 5.5, BioDiscovery Inc., Los Angeles, CA, USA) was used to quantify spot intensities. Poor‐quality spots were manually flagged and removed from the analysis.
Microarray data pre‐procession
Net signal intensities were calculated by subtraction of local background intensity. Signals reaching negative values after background subtraction were treated as missing data. Signals derived from the negative control included on each slide were subtracted from each spot to take unspecific binding of the secondary detection antibody into account. Intensities from the triplicate spots were averaged, resulting in one mean fluorescence intensity. All signals were then normalised to the IgG control spots included on each subarray by median centring to reduce intra‐slide variability and batch effects. Therefore, IgG median signal intensities were divided by the overall IgG signal median to attain a normalisation factor for each subarray. All further analyses are based on these normalised fluorescence intensities (NFI). To ensure robustness of the data set and reduce the influence of outliers, values below the 5th and above the 95th percentile were set as missing data. Targets with more than 25% missing data overall were not included in the statistical analyses. Missing data of targets with < 25% missing values were imputed using the k‐nearest neighbour (KNN) algorithm.
Statistical analysis
The statistical analysis of the MS data was performed with Perseus (version 1.6.2.3; Max Planck Institute of Biochemistry). Before statistical analysis, proteins were filtered, and potential contaminants, reverse hits and proteins only identified by site were excluded. Also, proteins needed to be identified in all three replicates in at least one group. Proteins that were also identified in any of the negative controls were excluded as well. Missing data have been imputed from normal distribution using the inbuilt algorithm to enable statistical testing. Differences in protein abundance were assessed with the Student's t‐test using a permutation‐based FDR of < 0.01 as a threshold for statistical significance to correct for multiple testing. Principal component analysis (PCA) was used to show the influence of the protein source on the data variability. Selected target proteins (based on significant altered levels in HTM and GTM samples) were displayed on a heat map with hierarchical clustering based on Euclidean distance using z‐score‐transformed LFQ intensities. For PCA and clustering analysis, missing data were replaced by random values drawn from a normal distribution.
Microarray statistics were calculated using Statistica (Statistica 13, StatSoft, Tulsa, OK, USA). Microarray data were evaluated for normality using the Shapiro–Wilk test. The data did not follow a normal distribution (P < 0.05), and so, the non‐parametric Mann–Whitney U‐test was used for hypothesis testing. Spearman's rank correlation coefficients were used to evaluate the correlations of autoantibody levels and clinical parameters. Kruskal–Wallis non‐parametric ANOVA was used for multi‐group comparisons. A P‐value of < 0.05 was considered statistically significant. A random forest classification algorithm was used to evaluate the diagnostic potential of the identified, disease‐related AAbs. Here, 165 cases were used as the training set and 75 samples as the test set.
Pathway and enrichment analysis
Gene Ontology enrichment analysis was done using DAVID (http://david.abcc.ncifcrf.gov/home.jsp).72, 73 GO terms with a P‐value < 0.05 after Bonferroni adjustment were considered as significantly enriched. Additionally, DAVID functional annotation clusters were used to characterise putative POAG‐related autoantigens. The analysis was carried out with whole homo sapiens proteome as background. Metascape (http://metascape.org)74 was used for further pathway and enrichment analysis of identified autoantigens. Also, Metascape's molecular complex detection (MCODE) algorithm was used to detect connected network components.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
Acknowledgments
This work was supported by the ‘German Federal Ministry of Education and Research’ (Bundesministerium für Bildung und Forschung); KMU‐innovativ: Medizintechnik (FKZ: 13GW0193B). We thank Professor Abbot F. Clark and colleagues [North Texas Eye Research Institute (NTERI), Fort Worth, TX, USA] for providing the TM cell lines. We also thank Dr Peter Taylor for proofreading the manuscript.
References
- 1. Ju Z, Liu W, Roebuck PL et al Development of a robust classifier for quality control of reverse‐phase protein arrays. Bioinformatics 2015; 31: 912–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Weinreb RN, Khaw PT. Primary open‐angle glaucoma. Lancet 2004; 363: 1711–1720. [DOI] [PubMed] [Google Scholar]
- 3. Mukesh BN, McCarty CA, Rait JL et al Five‐year incidence of open‐angle glaucoma: the visual impairment project. Ophthalmology 2002; 109: 1047–1051. [DOI] [PubMed] [Google Scholar]
- 4. Tielsch JM, Sommer A, Katz J et al Racial variations in the prevalence of primary open‐angle glaucoma. The Baltimore Eye Survey. JAMA 1991; 266: 369–374. [PubMed] [Google Scholar]
- 5. Wolfs RC, Klaver CC, Ramrattan RS et al Genetic risk of primary open‐angle glaucoma. Population‐based familial aggregation study. Arch Ophthalmol 1998; 116: 1640–1645. [DOI] [PubMed] [Google Scholar]
- 6. Francis BA, Varma R, Chopra V et al Intraocular pressure, central corneal thickness, and prevalence of open‐angle glaucoma: the Los Angeles Latino Eye Study. Am J Ophthalmol 2008; 146: 741–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. AGIS‐Investigators . The Advanced Glaucoma Intervention Study (AGIS): 7. The relationship between control of intraocular pressure and visual field deterioration. The AGIS Investigators. Am J Ophthalmol 2000; 130: 429–440. [DOI] [PubMed] [Google Scholar]
- 8. Vranka JA, Kelley MJ, Acott TS et al Extracellular matrix in the trabecular meshwork: intraocular pressure regulation and dysregulation in glaucoma. Exp Eye Res 2015; 133: 112–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Junglas B, Kuespert S, Seleem AA et al Connective tissue growth factor causes glaucoma by modifying the actin cytoskeleton of the trabecular meshwork. Am J Pathol 2012; 180: 2386–2403. [DOI] [PubMed] [Google Scholar]
- 10. Braunger BM, Fuchshofer R, Tamm ER. The aqueous humor outflow pathways in glaucoma: a unifying concept of disease mechanisms and causative treatment. Eur J Pharm Biopharm 2015; 95: 173–181. [DOI] [PubMed] [Google Scholar]
- 11. Bermudez JY, Montecchi‐Palmer M, Mao W et al Cross‐linked actin networks (CLANs) in glaucoma. Exp Eye Res 2017; 159: 16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Morgan JT, Raghunathan VK, Chang YR et al The intrinsic stiffness of human trabecular meshwork cells increases with senescence. Oncotarget 2015; 6: 15362–15374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Last JA, Pan T, Ding Y et al Elastic modulus determination of normal and glaucomatous human trabecular meshwork. Invest Ophthalmol Vis Sci 2011; 52: 2147–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gutteridge IF. Normal tension glaucoma: diagnostic features and comparisons with primary open angle glaucoma. Clin Exp Optom 2000; 83: 161–172. [DOI] [PubMed] [Google Scholar]
- 15. Zhavoronkov A, Izumchenko E, Kanherkar RR et al Pro‐fibrotic pathway activation in trabecular meshwork and lamina cribrosa is the main driving force of glaucoma. Cell Cycle 2016; 15: 1643–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Quigley HA, Hohman RM, Addicks EM et al Morphologic changes in the lamina cribrosa correlated with neural loss in open‐angle glaucoma. Am J Ophthalmol 1983; 95: 673–691. [DOI] [PubMed] [Google Scholar]
- 17. Sigal IA, Flanagan JG, Tertinegg I et al Predicted extension, compression and shearing of optic nerve head tissues. Exp Eye Res 2007; 85: 312–322. [DOI] [PubMed] [Google Scholar]
- 18. Wax MB, Barrett DA, Pestronk A. Increased incidence of paraproteinemia and autoantibodies in patients with normal‐pressure glaucoma. Am J Ophthalmol 1994; 117: 561–568. [DOI] [PubMed] [Google Scholar]
- 19. Tezel G, Seigel GM, Wax MB. Autoantibodies to small heat shock proteins in glaucoma. Invest Ophthalmol Vis Sci 1998; 39: 2277–2287. [PubMed] [Google Scholar]
- 20. Maruyama I, Ohguro H, Ikeda Y. Retinal ganglion cells recognized by serum autoantibody against gamma‐enolase found in glaucoma patients. Invest Ophthalmol Vis Sci 2000; 41: 1657–1665. [PubMed] [Google Scholar]
- 21. Ikeda Y, Maruyama I, Nakazawa M et al Clinical significance of serum antibody against neuron‐specific enolase in glaucoma patients. Jpn J Ophthalmol 2002; 46: 13–17. [DOI] [PubMed] [Google Scholar]
- 22. Grus FH, Joachim SC, Hoffmann EM et al Complex autoantibody repertoires in patients with glaucoma. Mol Vis 2004; 10: 132–137. [PubMed] [Google Scholar]
- 23. Boehm N, Wolters D, Thiel U et al New insights into autoantibody profiles from immune privileged sites in the eye: a glaucoma study. Brain Behav Immun 2012; 26: 96–102. [DOI] [PubMed] [Google Scholar]
- 24. Grus FH, Joachim SC, Bruns K et al Serum autoantibodies to alpha‐fodrin are present in glaucoma patients from Germany and the United States. Invest Ophthalmol Vis Sci 2006; 47: 968–976. [DOI] [PubMed] [Google Scholar]
- 25. Joachim SC, Bruns K, Lackner KJ et al Antibodies to α B‐crystallin, vimentin, and heat shock protein 70 in aqueous humor of patients with normal tension glaucoma and IgG antibody patterns against retinal antigen in aqueous humor. Curr Eye Res 2007; 32: 501–509. [DOI] [PubMed] [Google Scholar]
- 26. Joachim SC, Reichelt J, Berneiser S et al Sera of glaucoma patients show autoantibodies against myelin basic protein and complex autoantibody profiles against human optic nerve antigens. Graefes Arch Clin Exp Ophthalmol 2008; 246: 573–580. [DOI] [PubMed] [Google Scholar]
- 27. Nagele EP, Han M, Acharya NK et al Natural IgG autoantibodies are abundant and ubiquitous in human sera, and their number is influenced by age, gender, and disease. PLoS One 2013; 8: e60726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pradhan VD, Patwardhan MM, Ghosh K. Anti‐nucleosome antibodies as a disease marker in systemic lupus erythematosus and its correlation with disease activity and other autoantibodies. Indian J Dermatol Venereol Leprol 2010; 76: 145–149. [DOI] [PubMed] [Google Scholar]
- 29. Hahn BH. Antibodies to DNA. N Engl J Med 1998; 338: 1359–1368. [DOI] [PubMed] [Google Scholar]
- 30. Caturegli P, De Remigis A, Rose NR. Hashimoto thyroiditis: clinical and diagnostic criteria. Autoimmun Rev 2014; 13: 391–397. [DOI] [PubMed] [Google Scholar]
- 31. Zaenker P, Gray ES, Ziman MR. Autoantibody production in cancer‐the humoral immune response toward autologous antigens in cancer patients. Autoimmun Rev 2016; 15: 477–483. [DOI] [PubMed] [Google Scholar]
- 32. Wu J, Li L. Autoantibodies in Alzheimer's disease: potential biomarkers, pathogenic roles, and therapeutic implications. J Biomed Res 2016; 30: 361–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. De Virgilio A, Greco A, Fabbrini G et al Parkinson's disease: autoimmunity and neuroinflammation. Autoimmun Rev 2016; 15: 1005–1011. [DOI] [PubMed] [Google Scholar]
- 34. Getts MT, Miller SD. 99th Dahlem conference on infection, inflammation and chronic inflammatory disorders: triggering of autoimmune diseases by infections. Clin Exp Immunol 2010; 160: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Elkon K, Casali P. Nature and functions of autoantibodies. Nat Clin Pract Rheumatol 2008; 4: 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nissim A, Winyard PG, Corrigall V et al Generation of neoantigenic epitopes after posttranslational modification of type II collagen by factors present within the inflamed joint. Arthritis Rheum 2005; 52: 3829–3838. [DOI] [PubMed] [Google Scholar]
- 37. Anderton SM. Post‐translational modifications of self antigens: implications for autoimmunity. Curr Opin Immunol 2004; 16: 753–758. [DOI] [PubMed] [Google Scholar]
- 38. Ng B, Yang F, Huston DP et al Increased noncanonical splicing of autoantigen transcripts provides the structural basis for expression of untolerized epitopes. J Allergy Clin Immunol 2004; 114: 1463–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Backes C, Ludwig N, Leidinger P et al Immunogenicity of autoantigens. BMC Genom 2011; 12: 340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pang IH, Shade DL, Clark AF et al Preliminary characterization of a transformed cell strain derived from human trabecular meshwork. Curr Eye Res 1994; 13: 51–63. [DOI] [PubMed] [Google Scholar]
- 41. von Muhlen CA, Tan EM. Autoantibodies in the diagnosis of systemic rheumatic diseases. Semin Arthritis Rheum 1995; 24: 323–358. [DOI] [PubMed] [Google Scholar]
- 42. Murphy K, Weaver C. Janeway's Immunobiology, 9th Edition 2017:1–904. [Google Scholar]
- 43. Schmelter C, Perumal N, Funke S et al Peptides of the variable IgG domain as potential biomarker candidates in primary open‐angle glaucoma (POAG). Hum Mol Genet 2017; 26: 4451–4464. [DOI] [PubMed] [Google Scholar]
- 44. Singh V, Stoop MP, Stingl C et al Cerebrospinal‐fluid‐derived immunoglobulin G of different multiple sclerosis patients shares mutated sequences in complementarity determining regions. Mol Cell Proteomics 2013; 12: 3924–3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. de Costa D, Broodman I, Calame W et al Peptides from the variable region of specific antibodies are shared among lung cancer patients. PLoS One 2014; 9: e96029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Andrae J, Gallini R, Betsholtz C. Role of platelet‐derived growth factors in physiology and medicine. Genes Dev 2008; 22: 1276–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. de Kater AW, Shahsafaei A, Epstein DL. Localization of smooth muscle and nonmuscle actin isoforms in the human aqueous outflow pathway. Invest Ophthalmol Vis Sci 1992; 33: 424–429. [PubMed] [Google Scholar]
- 48. Tektas OY, Lutjen‐Drecoll E. Structural changes of the trabecular meshwork in different kinds of glaucoma. Exp Eye Res 2009; 88: 769–775. [DOI] [PubMed] [Google Scholar]
- 49. Kalouche G, Beguier F, Bakria M et al Activation of prostaglandin FP and EP2 receptors differently modulates myofibroblast transition in a model of adult primary human trabecular meshwork cells. Invest Ophthalmol Vis Sci 2016; 57: 1816–1825. [DOI] [PubMed] [Google Scholar]
- 50. Tellios N, Belrose JC, Tokarewicz AC et al TGF‐β induces phosphorylation of phosphatase and tensin homolog: implications for fibrosis of the trabecular meshwork tissue in glaucoma. Sci Rep 2017; 7: 812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bell K, Funke S, Pfeiffer N et al Serum and antibodies of glaucoma patients lead to changes in the proteome, especially cell regulatory proteins, in retinal cells. PLoS One 2012; 7: e46910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wax MB, Tezel G, Kawase K et al Serum autoantibodies to heat shock proteins in glaucoma patients from Japan and the United States. Ophthalmology 2001; 108: 296–302. [DOI] [PubMed] [Google Scholar]
- 53. Cancino‐Diaz ME, Sanchez‐Becerra M, Elizondo‐Olascoaga C et al Amino acid regions 357–368 and 418–427 of Streptococcus pyogenes 60kDa heat shock protein are recognized by antibodies from glaucomatous patient sera. Microb Pathog 2010; 48: 239–244. [DOI] [PubMed] [Google Scholar]
- 54. Beutgen VM, Perumal N, Pfeiffer N et al Autoantibody biomarker discovery in primary open angle glaucoma using serological proteome analysis (SERPA). Front Immunol 2019; 10: 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Stasi K, Nagel D, Yang X et al Complement component 1Q (C1Q) upregulation in retina of murine, primate, and human glaucomatous eyes. Invest Ophthalmol Vis Sci 2006; 47: 1024–1029. [DOI] [PubMed] [Google Scholar]
- 56. Gunawardena H, Betteridge ZE, McHugh NJ. Myositis‐specific autoantibodies: their clinical and pathogenic significance in disease expression. Rheumatology (Oxford) 2009; 48: 607–612. [DOI] [PubMed] [Google Scholar]
- 57. Targoff IN. Update on myositis‐specific and myositis‐associated autoantibodies. Curr Opin Rheumatol 2000; 12: 475–481. [DOI] [PubMed] [Google Scholar]
- 58. Lee YH, Pang SW, Tan KO. PNMA2 mediates heterodimeric interactions and antagonizes chemo‐sensitizing activities mediated by members of PNMA family. Biochem Biophys Res Commun 2016; 473: 224–229. [DOI] [PubMed] [Google Scholar]
- 59. Tripathi BJ, Tripathi RC. Neural crest origin of human trabecular meshwork and its implications for the pathogenesis of glaucoma. Am J Ophthalmol 1989; 107: 583–590. [DOI] [PubMed] [Google Scholar]
- 60. Funke S, Beutgen VM, Bechter L et al An in‐depth view of the porcine trabecular meshwork proteome. Int J Mol Sci 2019; 20: 2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Musunuru K, Darnell RB. Paraneoplastic neurologic disease antigens: RNA‐binding proteins and signaling proteins in neuronal degeneration. Annu Rev Neurosci 2001; 24: 239–262. [DOI] [PubMed] [Google Scholar]
- 62. Lin LH, Xu YW, Huang LS et al Serum proteomic‐based analysis identifying autoantibodies against PRDX2 and PRDX3 as potential diagnostic biomarkers in nasopharyngeal carcinoma. Clin Proteomics 2017; 14: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mustafa MZ, Nguyen VH, Le Naour F et al Autoantibody signatures defined by serological proteome analysis in sera from patients with cholangiocarcinoma. J Transl Med 2016; 14: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. DeMarshall C, Sarkar A, Nagele EP et al Utility of autoantibodies as biomarkers for diagnosis and staging of neurodegenerative diseases. Int Rev Neurobiol 2015; 122: 1–51. [DOI] [PubMed] [Google Scholar]
- 65. Lorenz K, Beck S, Keilani MM et al Course of serum autoantibodies in patients after acute angle‐closure glaucoma attack. Clin Exp Ophthalmol 2017; 45: 280–287. [DOI] [PubMed] [Google Scholar]
- 66. European Glaucoma Society Terminology and guidelines for glaucoma, 4th edition ‐ chapter 2: classification and terminologysupported by the EGS foundation: part 1: foreword; Introduction; Glossary; Chapter 2 classification and terminology. Br J Ophthalmol 2017; 101: 73–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wade NC, Grierson I, O'Reilly S et al Cross‐linked actin networks (CLANs) in bovine trabecular meshwork cells. Exp Eye Res 2009; 89: 648–659. [DOI] [PubMed] [Google Scholar]
- 68. Alhamdani MS, Schroder C, Werner J et al Single‐step procedure for the isolation of proteins at near‐native conditions from mammalian tissue for proteomic analysis on antibody microarrays. J Proteome Res 2010; 9: 963–971. [DOI] [PubMed] [Google Scholar]
- 69. Mohammed H, Taylor C, Brown GD et al Rapid immunoprecipitation mass spectrometry of endogenous proteins (RIME) for analysis of chromatin complexes. Nat Protoc 2016; 11: 316–326. [DOI] [PubMed] [Google Scholar]
- 70. Schmelter C, Funke S, Treml J et al Comparison of two solid‐phase extraction (SPE) methods for the identification and quantification of porcine retinal protein markers by LC‐MS/MS. Int J Mol Sci 2018; 19: 3847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Funke S, Markowitsch S, Schmelter C et al In‐depth proteomic analysis of the porcine retina by use of a four step differential extraction bottom up LC MS platform. Mol Neurobiol 2017; 54: 7262–7275. [DOI] [PubMed] [Google Scholar]
- 72. da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4: 44–57. [DOI] [PubMed] [Google Scholar]
- 73. da Huang W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009; 37: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zhou Y, Zhou B, Pache L et al Metascape provides a biologist‐oriented resource for the analysis of systems‐level datasets. Nat Commun 2019; 10: 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials