

Abstract
The pathogenesis of inflammatory bowel disease (IBD) is still unclear, but includes both inflammatory and autoimmune reactions. Current methodological approaches could better elucidate the cytokine pathways and the genetics involved in the etiopathogenesis of this disease. Interferons (IFNs) are cytokines that play a key role in autoimmune/inflammatory disorders because of their pro- and anti-inflammatory properties as well as their immunoregulatory functions. An increased expression of IFN-regulated genes, widely known as an IFN signature, has been reported in blood and tissue from patients with autoimmune disorders. In this review, we present the function as well as the clinical and therapeutic potential of the IFN signature. Current data demonstrate that the IFN signature can be used as a biomarker that defines disease activity in autoimmune diseases, although this has not been thoroughly studied in IBD. Consequently, further investigation of the IFN signature in IBD would be essential for a better understanding of its actions.
Keywords: Inflammatory bowel disease, interferon, interferon signature
Inflammatory bowel disease (IBD)
IBD, including Crohn’s disease (CD) and ulcerative colitis (UC), is a complicated, chronic, relapsing and heterogeneous disease induced by environmental, genomic, microbial, and immunological factors [1]. CD is a debilitating and incurable chronic idiopathic inflammatory disease of the intestine and is showing an increasing incidence in developing countries [2]. CD is characterized by ulceration and inflammation of the intestinal mucosa, which may affect the whole gastrointestinal tract, but mainly the distal small intestine. Typical characteristics of CD include discontinuous, transmural inflammation of the bowel wall, and the presence of granulomas, fistulas, strictures, and lymphoid aggregates [3]. UC is a chronic, idiopathic inflammatory disease that affects the colon. It is characterized by relapsing and remitting mucosal inflammation, starting in the rectum and extending to proximal segments of the colon. UC is limited to the colon and only affects its innermost lining, while CD can develop anywhere in the gastrointestinal tract in all the layers of the bowel walls [4].
IBD therapy aims to induce and maintain clinical and endoscopic remission. Current treatments include aminosalicylate drugs and antibiotics for mild-to-moderate IBD, conventional anti-inflammatory agents (corticosteroids), immunosuppressants (thiopurines and methotrexate) and biologic therapy for moderate-to-severe disease. Recent data, however, suggest that the early initiation of more aggressive treatment, such as biologic therapy, can modify disease progression and may lead to less damage. Immunomodulatory drugs are often used in combination with biologics in IBD patients with severe disease for the maintenance of remission [5]. Apart from these well-studied treatments, systemic administration of type I interferon ([IFN], e.g., IFN-α, IFN-β) in IBD patients has been evaluated for the suppression of disease burden, with controversial results. Immunoregulatory therapy with IFN type I can inhibit production of tumor necrosis factor (TNF) and IFN-γ, antagonize the IFN-γ signaling pathway, and increase production of the anti-inflammatory cytokine interleukin (IL)-10 [6,7]. IFN-β also has an immunoregulatory action by enhancing regulatory T-lymphocyte and natural killer-cell activity [8,9].
IBD and immunity
IBD is characterized as an immune-mediated disease, although it still remains unknown whether this autoimmunity has a direct pathogenic effect in CD or in UC, despite the presence of autoantibodies that react to bacterial antigens [10].
Innate immunity
The role of innate immunity in the pathogenesis of IBD has been described since the discovery of mutations in the NOD2/CARD15 gene. The NOD2/CARD15 gene is an intracellular bacterial sensor, with reduced expression in the presence of the 3020insC mutation, leading to a vulnerability to bacterial infections in IBD [11]. NOD2/CARD15 mutations result in reduced α-defensin expression. On the other hand, β-defensin levels are increased as a result of enteric inflammation; this is not the case for CD regarding HBD-2 and HBD-3 [12,13]. The combination of NOD2 mutations and the decreased defensin expression may lead to defective resistance against intestinal microorganisms and might result in inflammation of the mucosa [14].
Toll-like receptors (TLRs) also have an important role in innate immunity, through their ability to detect both normal and pathological microbes and to regulate the host’s antimicrobial response [15]. Abnormal TLR expression or function has been implicated in the development or persistence of intestinal inflammation. TLR4 has been found to be upregulated in CD epithelial cells, while TLR3 is downregulated [16]. TLR4 and TLR9 polymorphisms have also been reported in IBD, but their functional significance is poorly understood [17,18].
Because of the involvement of both NOD2 and TLRs in the recognition of and response to bacteria, they may have a reciprocal interaction that could be dysfunctional in IBD. Macrophages homozygous for NOD2 mutations in CD patients show defective production of IL-1 and IL-8 when stimulated with muramyl dipeptide (MDP) or TNF-α [19]. In addition, peripheral blood mononuclear cells from double-mutant patients failed to interact with MDP and TLR ligands, leading to increased TNF-α and IL-1β production [20]. In this way, the generalized inability of the innate immune response to control via pattern recognition receptors may contribute to IBD, particularly to CD [14].
Adaptive immunity
IBD is characterized by increased production of systemic and mucosal antibodies and an altered amount of immunoglobulin classes and subclasses due to the chronic inflammation of the intestine [21-23]. Additionally, IgG1 antibodies against epithelial cells in the colon are highly produced in UC, but not in CD, possibly demonstrating an autoimmune pathway in this case [24]. Lately, adaptive immunity in IBD has been focused on the subsets of T-helper (Th) cells (a lymphocyte subtype) and their soluble regulators, which are important elements of immune responses [25]. They are involved in the activation of other cells of the immune system and determine the specificity of antibodies secreted by B-cells [26]. Following proliferation, a Th cell is differentiated into Th1, Th2, or Th17 [27]. Th1 cells mediate the production of proinflammatory cytokines, such as IFN-γ, TNF-α, and IL-12, whereas Th2 cells lead to the production of anti-inflammatory cytokines, including IL-4, IL-5, IL-6, and IL-10, which constitute the humoral immune response. There is an interaction between Th1 and Th2 lymphocytes. Th1 cytokines lead to the production of Th1 cells and inhibit Th2 cells. Conversely, Th2 cytokines lead to the production of Th2 cells and inhibit Th1 cells. In healthy individuals, there is an equilibrium between the quantities of Th1 and Th2 cells. The development of immune-mediated disorders such as CD is closely linked to a type 1 immune response causing chronic inflammation of the intestine. CD is characterized by the accumulation of Th1 cells and UC by the accumulation of Th2 cells [28]. The most recent class of Th cells to be revealed is Th17 cells, differentiated in the presence of IL-6, IL-1β, IL-21, and IL-23 [29], and may secrete IL-17A, IL-17F, IL -21, IL-22, IL-26, and chemokine CCL20 [30]. The inflamed mucosa of CD patients is characterized by a multi-complex communication network between cytokines, responsible for the changes that occur in immune level [31]. An example of this complicated, unstable process has been described in CD, with the transition from the Th1 response to a mixed phenotype of Th1/Th17 [32].
A great number of types of cytokine dysregulation have been described, in which proinflammatory and immunoregulatory molecules are included [25]. In CD patients, intestinal CD4+ T cells produce increased numbers of IFN-γ, while the transcription factor of Th1 cells, T-bet, is also overexpressed [33]. Mucosal macrophages also produce remarkable levels of IL-12 and IL-18 [34,35]. Regarding UC, different immunological abnormalities have been observed, where natural killer T (NKT) cells produce large amounts of IL-13, and T cells of mucosa produce more IL-15, proliferate less and die more than healthy cells [36].
Genetics
Genome-wide association studies (GWAS) have been crucial in the recognition of over 230 disease loci linked to IBD [37]. The most strongly and consistently implicated loci are associated with intracellular bacteria killing, innate (CARD15/NOD2, IRGM, IL-23R, LRRK2, and ATG16L1) and adaptive immune responses (IL-23), and the Th17 cell pathway (IL-23R, IL-12B, STAT3, JAK2, and TYK2) [38]. Dendritic cells (DCs) followed by CD4 T, natural killer (NK), and NKT cells showed the highest enrichment of these susceptibility gene sets when tested in a panel of immune cell subsets, indicating a major role for these cells in CD pathogenesis [39].
Most of the loci are common for IBD, although some of them are unique for either CD or UC. NOD-like receptors (NOD2), autophagy genes (ATG16L1, IRGM) and intelectins (ITLN1) present high specificity for CD. Considering UC, gene loci involved in regulatory pathways, such as IL-10 and ARPC2, and in intestinal epithelial cell function, such as ECM1 and E3 ubiquitin ligase, seem to be disease-specific. Furthermore, the association of the human leukocyte antigen/major histocompatibility complex region appears to be stronger with UC rather than CD, a genetic feature of IBD shared with several autoimmune diseases [40-42].
GWAS and the meta-analysis of loci have been crucial in the specification of over 230 genetic susceptibility loci linked to IBD [37]; some of these genes are involved in immunity and in barrier function [39]. Various single-nucleotide polymorphisms (SNPs) have been found in genes related to pathogenic cytokine pathways, for example the Th17/IL-23 pathway, IL-10 pathway, and type I IFN signaling [43]. The relevant pathways could not be precisely determined only from the genetic data, as most of the signaling mediators are shared between different cytokine signaling cascades.
Many of the IBD-associated genes are involved in the type I IFN signaling pathway. The rs2284553 SNP is associated with the IFNGR2 gene and with the IFNAR1. SNPs in JAK2, TYK2, STAT1 and STAT3 genes affect the JAK/STAT signaling pathway, deregulating the action of several cytokines, such as IL-22, IL-10, and type I and type III IFN [39]. Moreover, MDA5 or IRF4 gene polymorphisms can modulate the production of type I and type III IFN [39]. Although studies in mice show that minor alterations in type I IFN may contribute to the imbalance of the immune response in the lamina propria, the exact role of type I IFN in IBD pathology remains unclear [44].
IFNs
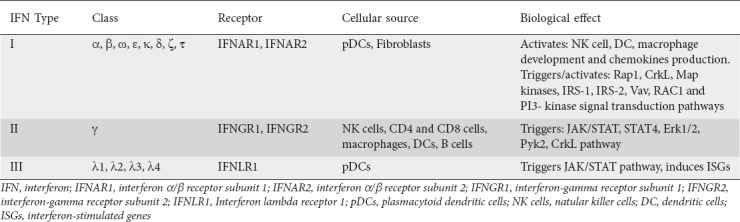
IFNs are cytokines that have antiviral activity. They are classified into 3 categories: type I IFNs (IFN-α, IFN-β and other less explored members), type II IFNs (IFN-γ), and type III IFNs (IFN-λ1, IFN-λ2, IFN-λ3 and IFN-λ4 in humans) [45,46].
The type I multigene family consists of structurally related IFNs, such as IFN-α, IFN-β, IFN-ω, IFN-ε, IFN-κ, IFN-δ, IFN-ζ, and IFN-τ. [45]. The best-studied type I IFN is the IFN-α family, which is encoded by 13 homologous genes [47]. These cytokines are produced after the stimulation of pattern-recognition receptors, as part of the innate immune antiviral response [48]. Data indicate that the initial production of IFN-β and IFN-α4 is dependent upon IRF3 phosphorylation and NF-κB activation [49]. The primary wave of type I IFN then induces IRF7 phosphorylation, leading to a positive feedback loop to increase the release of type I IFN [50].
Type II IFN (also known as IFN-γ), is predominantly produced by NK and NKT cells, as well as by CD4 and CD8 T-lymphocytes. Along with its antiviral activity, IFN-γ also acts against intracellular bacterial infections and tumor progression [51]. There is also evidence to show that type I IFN, IL-12, IL-15 and IL-18 are able to induce production of IFN-γ by NK cells [52].
Type III IFNs are more similar in structure to the IL-10 family than to the other IFN subgroups, but functionally they are similar to type I and type II IFN, as they also contribute to antiviral responses and induce the activation of many common genes [53].
All type I IFN members bind to the same heterodimer, expressed on most cells: the type I IFN receptor (IFNAR). IFNAR is composed of two subunits, IFNAR1 and IFNAR2 [47]. These receptors are endocytosed and activate their related tyrosine kinases, tyrosine kinase 2 (Tyk2) and Janus kinase 1 (JAK1), when bound to IFN type I [54]. The typical signaling cascade results in the phosphorylation of STAT1 and STAT2, which form a complex with IRF9, known as the IFN-stimulated gene factor 3 (ISGF3), leading to the expression of IFN-stimulated genes [54]. Apart from ISGF3, type I IFNs can induce phosphorylation and dimerization of STAT3, STAT4, STAT5 and STAT6 and activate Rap1, CrkL, Map kinases, IRS-1, IRS-2, Vav, RAC1 and PI3-kinase signal transduction pathways [54-59]. IFN-β has been shown to act via the IFNAR1, in an IFNAR2-independent way, through a non-canonical pathway [60].
The 2 other categories of the IFN family, type II and type III, display little similarity to type I IFNs and signal through their own cognate receptors. All IFNs act through various signal transduction pathways, with Janus kinases (JAK)/signal transducer and activator of transcription (STAT) pathway being the best-described [61]. Although type II IFN shares a similar classification with type I IFN, it activates different signal receptors that have other effects than those of type I IFN. IFN-γ signals through the IFN-γ receptor (IFNGR), composed of IFNGR1 and IFNGR2 subunits. In the classical signaling pathway, binding of IFN-γ to IFNGR activates JAK1 and JAK2 and leads to homodimerization and phosphorylation of STAT1 at tyrosine 701 [62]. However, like type I IFN, alternative signaling pathways have also been suggested for IFN-γ, such as STAT4, Erk1/2, Pyk2 and CrkL [63].
Type III IFNs bind to 2 receptors unique to this IFN type that have limited expression in epithelial cells: the low-affinity receptor subunit (IL-10R2) and the high-affinity receptor subunit (IFN-λ receptor 1, IFNLR1) [64]. These receptors trigger the same signaling JAK/STAT pathway as type I IFN receptors and induce a high number of ISGs [65]. All the types of IFNs are summarized in Table 1.
Table 1.
The types of interferons
The role of IFNs has been widely studied in autoimmune diseases; however, there are not many data concerning IFNs and IBD. IFNs have multiple direct and indirect effects on adaptive immunity. IFN-α/β activates NK cells and DCs, with increased expression of major histocompatibility complex class I (MHCI) and other molecules, such as CD40, CD80 and CD86. Type I IFN affects DC targeting in peripheral lymphoid organs by inducing the production of several chemokines, including CXCL8, CXCL9, CXCL10, CXCL11 and their receptors. It boosts the production of IL-12, IL-15, IL-18 and IL-23 by DCs and the heightened expression of B-cell activating factor (BAFF) and a proliferation-inducing ligand (APRIL). Furthermore, type I IFN stimulates macrophage development and inducible nitric oxide (NO) synthase expression in systemic lupus erythematosus (SLE) [66]. IFN-α overproduction has been described in SLE patients; immune complexes containing DNA or RNA in the serum of SLE patients may explain this increased production [67]. Immature plasmacytoid DCs produce increased levels of IFN that can also induce monocyte maturation into DCs [68].
The stimulatory or inhibitory role of type I IFN has been described in various other autoimmune disorders. For example, type I IFN has been suggested as a negative regulator in rheumatoid arthritis (RA), type I insulin-dependent diabetes mellitus, multiple sclerosis (MS), and acute encephalitis. However, IFN-β is used to treat relapsing/remitting forms of MS. Other examples are those of thyroiditis, some forms of autoimmune hemolytic anemia, and Behçet’s disease [67].
Genetic studies in primary Sjögren’s syndrome (pSS) report polymorphisms in the IRF5 gene, whose transcripts may induce the stimulation of IFN-β [69]. A few studies have correlated the increased circulating IFN-α levels with autoimmune/autoimmune-featured diseases such as pSS, diffuse systemic sclerosis and interstitial lung disease [70]. In dermatomyositis, immunohistochemistry methods revealed that IFN-α/β induce the myxovirus resistance protein A in the perifascicular muscle fibers and in capillary cells [71]. IFN-α/β has also been detected in the synovial fluid and tissue of patients with RA. Subsequent treatment approaches using type I IFN have had controversial outcomes in both murine models and humans with RA [72].
It is known that, in healthy individuals, TNF downregulates IFN-α [73], and this leads to an initial theory that the reduction in TNF-mediated IFN-α secretion triggers autoimmunity. However, several studies showed that both IFN-α and TNF are increased in SLE patients and, furthermore, that their levels were indicative of the disease progression [74-78]. It seems that the increased levels of both TNF and IFN-α contribute to the development of autoimmunity [79].
There are studies indicating that type I IFN treatment often has an unpredictable outcome in IBD patients. Some pilot studies have shown that type I IFN may improve the condition of IBD patients [80,81], although some clinical trials found that there is no beneficial outcome of IFN-β1a, IFN-α or IFN-β1 treatment in terms of disease remission [82-84]. According to Mannon et al, UC patients characterized as responders to IFN-β1a therapy presented significant lower levels of mucosal T-cell IL-13 production after treatment. Primary non-responders to IFN-β1a treatment were associated with increased quantities of IL-17 and IL-6, while no other significant decrease in the production of inflammatory cytokines was noticed after clinically and endoscopically effective therapy [85]. We can conclude that, according to the current knowledge concerning type I IFN treatment, no statistical benefit in disease amelioration has been found, although the effect of the treatment seems to be associated with the Th profiles of each patient.
Proinflammatory and anti-inflammatory cytokines, type I IFN included, are significant players in the maintenance of gut microbiota, given their immunoregulatory effects on the growth and renewal of intestinal epithelial cells (IECs) [86-88]. Type I IFN is mainly expressed by lamina propria CD11c+ DCs in the intestine [89]; more specifically, these cells express IFN-α5 and IFN-α9 mRNA, but not IFN-α4 [90]. Moreover, it has been found that CD11c+ DCs express type I IFN-induced genes, including 2’-5’ oligoadenylate synthetase (OAS), OAS-like family members, IRF5, IRF7, CXCL10, RNase L and PKR, as well as IL-15α [91].
IFN signature in IBD
An increased expression of IFN-regulated genes, widely known as an IFN signature, has been reported in blood and tissue from patients with autoimmune disorders. The IFN signature has been studied in multiple autoimmune diseases, such as SLE, subacute cutaneous lupus erythematosus (SCLE), discoid lupus erythematosus (DLE), myositis, pSS, systemic sclerosis (SSc), scleroderma, and RA [92-95].
In SLE, the IFN signature is correlated with disease activity and severity and can be modulated with appropriate therapy [94]. Bauer et al suggest that IFN-induced chemokines can be used as possible biomarkers for predicting SLE [96,97]. The IFN-regulated gene signature is increased in peripheral blood cells and is correlated with disease activity in SCLE and DLE [95]. Furthermore, higher expression levels of IFN-λ1 in serum and tissues induce type I IFN-regulated genes in cutaneous lupus erythematosus [98]. The IFN type I signature also defines a subset of RA patients, with a specific biomolecular phenotype, represented by increased activity of the innate defense system, coagulation and complement cascades, and fatty acid metabolism [99]. Similarly, the IFN signature can identify only a subgroup of pSS patients: those who present higher activity of the disease and a more obvious activation of the immune system, including higher BAFF mRNA expression [100]. However, the IFN signature does not have such a strong association with dermatomyositis, polymyositis, SSc and RA, as with SLE [94,101].
Extended research by Smiljanovic et al studied the impact of a large group of cytokines regulated by IFNs and TNF-α in SLE and RA. Specifically the IFN-α2α signature included the upregulation of the following probe sets: CCL2, CCL8, CD164, CXCL10, CXCL11, FAS, IFI16, IFI27, IFI44, IFI44L, IL-15, IL-15RA, MX1, MX2, OAS1, OAS2, OAS3, OASL, SIGLEC1, SSB, STAT1, STAT2, STAT3 and TNFSF10, and downregulation of the following probe sets: CENTD2, CYP1B1, GPX4, ID2, IER3, IRS2, JUN, KLF13, KLF2, KLF4, PTAFR, TNFAIP2 and TNFRSF10B in SLE. A pronounced IFN-α2α signature was also identified in most RA patients. The IFN-α2α imprint in RA included the upregulation of CD163, CD55, CITED2, IL6ST, FOSL2, MAFF, ATF3 and MT2A and the downregulation of CCNG2, CXCR4, ICAM2, FADD, GPX3, NGRN, PURA, TNFSF8 and TP53. The network that characterizes an IFN-γ signature in SLE contains the following genes: STAT1, STAT3, IFI16, IRF7, CXCL10 and TNFSF10. In contrast, the network that demonstrates an IFN-γ imprint in RA contains genes like CCL3, CCL4, CXCR4 and ATF3 [100].
Appendicitis and appendectomy (AA), when occurring at an early age, have been found to be protective against the development of colitis in adulthood [102]. Cheluvappa et al showed that AA influence the expression of IFN-associated genes using microarray and gene set enrichment analysis in an AA model [103], explaining its beneficial role in trinitrobenzenesulfonic acid (TNBS) colitis. In this study, the expression of 46 distal colonic, IFN activity-associated genes was measured 3 and 28 days after AA. At day 3 after AA, IRF7 and IFI35 were significantly upregulated, while IFNK and IFRD2 were significantly downregulated. At day 28 after AA, IFNZ, IFIT1, IFIT2, IFIT3, IRF9, IFIH1 and IFI44 were found significantly upregulated. However, IRF2BP1, IRF2BP2 and IFI30 were significantly downregulated [104]. The only IFN gene that was upregulated at any time point was IFNZ [105]. The IFNz gene in mice exhibits immunomodulatory and antiviral effects, initiated by its IFN-α/β receptor binding [105]. These data agree with another study that showed imiquimod-induced colonic upregulation of IFN activity-associated genes or their gene sets (IFIT1, IFIT2, IFIT3, IRF7, IFI44 and IFIH1) that provide protection against DSS colitis [106]. A similar study investigated the role of several chemokines in colitis after AA. At day 28 after AA, CCL5 was significantly upregulated, whereas CCL20 was significantly downregulated. Interestingly, CXCL11 was found to be significantly upregulated at days 3 and 28 after AA [107]. The IFN-regulated genes in the IBD model, SLE and RA are presented in Table 2.
Table 2.
Interferon (IFN)-regulated genes in the inflammatory bowel disease (IBD) model, systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA)
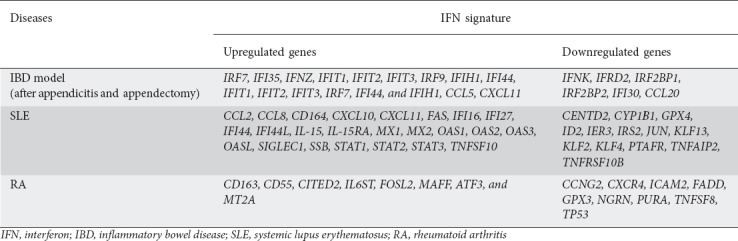
IFIT1, IFIT2, and IFIT3 are induced by IFNs, virus infections, and several molecular patterns, interfere with multiple protein–protein interactions, and present antiproliferative and immunoregulatory properties [108,109]. The IRF family of transcription factors that bind with DNA has the ability to recognize and bind to consensus gene sequences (IFN, stimulated response elements and virus-response elements), helping in IFN production, IFN/feedback-inhibition, management of cell growth, T-/B- lymphocyte activity and IFN-induced gene expression [110,111]. Specifically, IRF7 is a positive IFN regulator and a key mediator of the IFN positive feedback amplification loop, while IRF9 contributes toward a heterotrimeric complex, which induces IRF7 via JAK/STAT signaling, and autocrine IFN-receptor activation [110]. IFI44 binds intracellular GTP and prevents cells from proliferating [112], while IFI30 is an enzyme of lysosomal thiol reductase. It is expressed from antigen-presenting cells (APCs) and is responsible for processing the antigen through the decrease of bisulfide bonds of endocytosed proteins [113]. IFI35 is a leucine zipper protein, expressed in fibroblasts, macrophages and epithelial cells [114]. IFIH1 is an RNA helicase involved in induction of translation, in nuclear/mitochondrial splicing and ribosome assembly [115]. The IFN IFNz in mice exhibits immunoregulatory and antiviral effects, initiated by its IFN-α/β receptor binding [105]. Considering the chemokines involved in the IFN signature, the chemokine CCL5 is a chemoattractant for monocytes, memory Th cells, and eosinophils [116], and is induced by IFN-γ [117]. The chemokine CCL20 is also induced by IFN-γ [117] and is chemoattractant for lymphocytes and neutrophils, which can help lymphoid tissues of mucosa to attract lymphocytes and DCs to epithelial cells [118], while IFN-β and IFN-γ induce the chemokine CXCL11. CXCL11 is a chemoattractant for IL-activated T-cells [119].
There are not many studies concerning the correlation of the IFN-regulated gene expression and the response to anti-TNF therapy. The aforementioned study of Palucka et al suggests the upregulation of IFN-α regulated genes in circulating leukocytes of patients with systemic onset juvenile idiopathic arthritis who receive anti-TNF therapy [73]. A study performed in 33 RA patients revealed that increased IFN response gene activity is associated with a poor clinical response to TNF blockade, most evident 2 months after the initiation of treatment. Interestingly, the combination of five IFN response genes (OAS1, LGALS3BP, Mx2, OAS2 and SERPING1) into one IFN response gene set improved the predictive accuracy compared to OAS1 and LGALS3BP expression separately [120].
Although IBD is not considered as an autoimmune disease, it may trigger autoimmunity caused by the increased antigenic load and mucosal immune activation. It is well established that both innate and adaptive immunity play a major role in IBD development. Several genetic alterations are shared between IBD and autoimmune diseases. Furthermore, chronic intestinal inflammation presented in IBD could trigger autoimmunity. IBD-related inflammation consists of a variety of abnormalities in humoral and cell-mediated immunity, and a generalized enhanced reactivity against intestinal bacteria. The IFN signature in autoimmune diseases represents a useful tool as a biomarker of disease progression and treatment efficiency. Thus, the IFN signature in IBD could serve as an early predictor of disease activity and progression, as well as a supplementary therapeutic target [121,122].
Concluding remarks
Many autoimmune diseases present an IFN signature in cells from peripheral blood and tissues. Current data demonstrate that the IFN signature can be used as a biomarker that defines disease activity. In RA, the IFN signature may be used in the prediction of treatment response to some biological drugs, while inhibition of the IFN signature in other autoimmune disorders, such as SLE, could have a potential role in clinical practice. The IFN signature in IBD has not been thoroughly studied compared to autoimmune diseases. For this reason, further investigation of the IFN signature in IBD would be essential for a better understanding of its actions.
Biography
Medical School, National and Kapodistrian University of Athens, Athens, Greece
Footnotes
Conflict of Interest: None
References
- 1.de Souza HSP, Fiocchi C, Iliopoulos D. The IBD interactome:an integrated view of aetiology, pathogenesis and therapy. Nat Rev Gastroenterol Hepatol. 2017;14:739–749. doi: 10.1038/nrgastro.2017.110. [DOI] [PubMed] [Google Scholar]
- 2.Molodecky NA, Soon IS, Rabi DM, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2012;142:46–54. doi: 10.1053/j.gastro.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 3.Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med. 2009;361:2066–2078. doi: 10.1056/NEJMra0804647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peyrin-Biroulet L, Loftus EV, Jr, Colombel JF, Sandborn WJ. The natural history of adult Crohn's disease in population-based cohorts. Am J Gastroenterol. 2010;105:289–297. doi: 10.1038/ajg.2009.579. [DOI] [PubMed] [Google Scholar]
- 5.Thomas A, Lodhia N. Advanced therapy for inflammatory bowel disease:a guide for the primary care physician. J Am Board Fam Med. 2014;27:411–420. doi: 10.3122/jabfm.2014.03.130224. [DOI] [PubMed] [Google Scholar]
- 6.Panitch HS, Folus JS, Johnson KP. Recombinant IFN-b inhibits IFN-gamma production in MS. Ann Neurol. 1987;22:137. [Google Scholar]
- 7.Brod SA, Marshall GD Jr, Henninger EM, Sriram S, Khan M, Wolinsky JS. Interferon-beta 1b treatment decreases tumor necrosis factor-alpha and increases interleukin-6 production in multiple sclerosis. Neurology. 1996;46:1633–1638. doi: 10.1212/wnl.46.6.1633. [DOI] [PubMed] [Google Scholar]
- 8.Rudick RA, Ransohoff RM, Peppler R, VanderBrug Medendorp S, Lehmann P, Alam J. Interferon beta induces interleukin-10 expression:relevance to multiple sclerosis. Ann Neurol. 1996;40:618–627. doi: 10.1002/ana.410400412. [DOI] [PubMed] [Google Scholar]
- 9.Schnaper HW, Aune TM, Pierce CW. Suppressor T cell activation by human leukocyte interferon. J Immunol. 1983;131:2301–2306. [PubMed] [Google Scholar]
- 10.Targan SR, Karp LC. Defects in mucosal immunity leading to ulcerative colitis. Immunol Rev. 2005;206:296–305. doi: 10.1111/j.0105-2896.2005.00286.x. [DOI] [PubMed] [Google Scholar]
- 11.Hisamatsu T, Suzuki M, Reinecker HC, Nadeau WJ, McCormick BA, Podolsky DK. CARD15/NOD2 functions as an antibacterial factor in human intestinal epithelial cells. Gastroenterology. 2003;124:993–1000. doi: 10.1053/gast.2003.50153. [DOI] [PubMed] [Google Scholar]
- 12.Wehkamp J, Harder J, Weichenthal M, et al. NOD2 (CARD15) mutations in Crohn's disease are associated with diminished mucosal alpha-defensin expression. Gut. 2004;53:1658–1664. doi: 10.1136/gut.2003.032805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wehkamp J, Harder J, Weichenthal M, et al. Inducible and constitutive beta-defensins are differentially expressed in Crohn's disease and ulcerative colitis. Inflamm Bowel Dis. 2003;9:215–223. doi: 10.1097/00054725-200307000-00001. [DOI] [PubMed] [Google Scholar]
- 14.Kugathasan S, Fiocchi C. Progress in basic inflammatory bowel disease research. Semin Pediatr Surg. 2007;16:146–153. doi: 10.1053/j.sempedsurg.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 15.Cook DN, Pisetsky DS, Schwartz DA. Toll-like receptors in the pathogenesis of human disease. Nat Immunol. 2004;5:975–979. doi: 10.1038/ni1116. [DOI] [PubMed] [Google Scholar]
- 16.Cario E, Podolsky DK. Differential alteration in intestinal epithelial cell expression of toll-like receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect Immun. 2000;68:7010–7017. doi: 10.1128/iai.68.12.7010-7017.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Franchimont D, Vermeire S, El Housni H, et al. Deficient host-bacteria interactions in inflammatory bowel disease?The toll-like receptor (TLR)-4 Asp299gly polymorphism is associated with Crohn's disease and ulcerative colitis. Gut. 2004;53:987–992. doi: 10.1136/gut.2003.030205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Török HP, Glas J, Tonenchi L, Bruennler G, Folwaczny M, Folwaczny C. Crohn's disease is associated with a toll-like receptor-9 polymorphism. Gastroenterology. 2004;127:365–366. doi: 10.1053/j.gastro.2004.05.051. [DOI] [PubMed] [Google Scholar]
- 19.Li J, Moran T, Swanson E, et al. Regulation of IL-8 and IL-1beta expression in Crohn's disease associated NOD2/CARD15 mutations. Hum Mol Genet. 2004;13:1715–1725. doi: 10.1093/hmg/ddh182. [DOI] [PubMed] [Google Scholar]
- 20.van Heel DA, Ghosh S, Butler M, et al. Muramyl dipeptide and toll-like receptor sensitivity in NOD2-associated Crohn's disease. Lancet. 2005;365:1794–1796. doi: 10.1016/S0140-6736(05)66582-8. [DOI] [PubMed] [Google Scholar]
- 21.Swidsinski A, Ladhoff A, Pernthaler A, et al. Mucosal flora in inflammatory bowel disease. Gastroenterology. 2002;122:44–54. doi: 10.1053/gast.2002.30294. [DOI] [PubMed] [Google Scholar]
- 22.D'Haens GR, Geboes K, Peeters M, Baert F, Penninckx F, Rutgeerts P. Early lesions of recurrent Crohn's disease caused by infusion of intestinal contents in excluded ileum. Gastroenterology. 1998;114:262–267. doi: 10.1016/s0016-5085(98)70476-7. [DOI] [PubMed] [Google Scholar]
- 23.Campieri M, Gionchetti P. Probiotics in inflammatory bowel disease:new insight to pathogenesis or a possible therapeutic alternative? Gastroenterology. 1999;116:1246–1260. doi: 10.1016/s0016-5085(99)70029-6. [DOI] [PubMed] [Google Scholar]
- 24.Takahashi F, Das KM. Isolation and characterization of a colonic autoantigen specifically recognized by colon tissue-bound immunoglobulin G from idiopathic ulcerative colitis. J Clin Invest. 1985;76:311–318. doi: 10.1172/JCI111963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Podolsky DK, Fiocchi C. Cytokines, chemokines, growth factors, eicosanoids and other bioactive molecules in IBD. W.B. Saunders; 1999. Inflammatory Bowel Disease; pp. 191–207. [Google Scholar]
- 26.Janeway CA, Jr, Travers P, Walport M, et al. B-cell activation by armed helper T cells. 5th edition. New York: Garland Science; 2001. Immunobiology:The Immune System in Health and Disease. [Google Scholar]
- 27.Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM. Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity. 2006;24:677–688. doi: 10.1016/j.immuni.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 28.Romagnani S. Th1/Th2 cells. Inflamm Bowel Dis. 1999;5:285–294. doi: 10.1097/00054725-199911000-00009. [DOI] [PubMed] [Google Scholar]
- 29.Iwakura Y, Ishigame H. The IL-23/IL-17 axis in inflammation. J Clin Invest. 2006;116:1218–1222. doi: 10.1172/JCI28508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McGeachy MJ, Cua DJ. The link between IL-23 and Th17 cell-mediated immune pathologies. Semin Immunol. 2007;19:372–376. doi: 10.1016/j.smim.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 31.Neurath MF. Cytokines in inflammatory bowel disease. Nat Rev Immunol. 2014;14:329–342. doi: 10.1038/nri3661. [DOI] [PubMed] [Google Scholar]
- 32.Zorzi F, Monteleone I, Sarra M, et al. Distinct profiles of effector cytokines mark the different phases of Crohn's disease. PLoS One. 2013;8:e54562. doi: 10.1371/journal.pone.0054562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neurath MF, Weigmann B, Finotto S, et al. The transcription factor T-bet regulates mucosal T cell activation in experimental colitis and Crohn's disease. J Exp Med. 2002;195:1129–1143. doi: 10.1084/jem.20011956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Monteleone G, Biancone L, Marasco R, et al. Interleukin 12 is expressed and actively released by Crohn's disease intestinal lamina propria mononuclear cells. Gastroenterology. 1997;112:1169–1178. doi: 10.1016/s0016-5085(97)70128-8. [DOI] [PubMed] [Google Scholar]
- 35.Pizarro TT, Michie MH, Bentz M, et al. IL-18, a novel immunoregulatory cytokine, is up-regulated in Crohn's disease:expression and localization in intestinal mucosal cells. J Immunol. 1999;162:6829–6835. [PubMed] [Google Scholar]
- 36.Fuss IJ, Heller F, Boirivant M, et al. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. J Clin Invest. 2004;113:1490–1497. doi: 10.1172/JCI19836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Uhlig HH, Muise AM. Clinical genomics in inflammatory bowel disease. Trends Genet. 2017;33:629–641. doi: 10.1016/j.tig.2017.06.008. [DOI] [PubMed] [Google Scholar]
- 38.Franke A, McGovern DP, Barrett JC, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat Genet. 2010;42:1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jostins L, Ripke S, Weersma RK, et al. International IBD Genetics Consortium (IIBDGC) Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gregersen PK, Olsson LM. Recent advances in the genetics of autoimmune disease. Annu Rev Immunol. 2009;27:363–391. doi: 10.1146/annurev.immunol.021908.132653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Van Limbergen J, Wilson DC, Satsangi J. The genetics of Crohn's disease. Annu Rev Genomics Hum Genet. 2009;10:89–116. doi: 10.1146/annurev-genom-082908-150013. [DOI] [PubMed] [Google Scholar]
- 42.Zhernakova A, van Diemen CC, Wijmenga C. Detecting shared pathogenesis from the shared genetics of immune-related diseases. Nat Rev Genet. 2009;10:43–55. doi: 10.1038/nrg2489. [DOI] [PubMed] [Google Scholar]
- 43.Liu TC, Stappenbeck TS. Genetics and pathogenesis of inflammatory bowel disease. Annu Rev Pathol. 2016;11:127–148. doi: 10.1146/annurev-pathol-012615-044152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Giles EM, Sanders TJ, McCarthy NE, et al. Regulation of human intestinal T-cell responses by type 1 interferon-STAT1 signaling is disrupted in inflammatory bowel disease. Mucosal Immunol. 2017;10:184–193. doi: 10.1038/mi.2016.44. [DOI] [PubMed] [Google Scholar]
- 45.Lee AJ, Ashkar AA. The Dual Nature of Type I and Type II Interferons. Front Immunol. 2018;9:2061. doi: 10.3389/fimmu.2018.02061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prokunina-Olsson L, Muchmore B, Tang W, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet. 2013;45:164–171. doi: 10.1038/ng.2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rönnblom L, Eloranta ML. The interferon signature in autoimmune diseases. Curr Opin Rheumatol. 2013;25:248–253. doi: 10.1097/BOR.0b013e32835c7e32. [DOI] [PubMed] [Google Scholar]
- 48.Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Honda K, Takaoka A, Taniguchi T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity. 2006;25:349–360. doi: 10.1016/j.immuni.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 50.Tamura T, Yanai H, Savitsky D, Taniguchi T. The IRF family transcription factors in immunity and oncogenesis. Annu Rev Immunol. 2008;26:535–584. doi: 10.1146/annurev.immunol.26.021607.090400. [DOI] [PubMed] [Google Scholar]
- 51.Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma:an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75:163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 52.Pegram HJ, Andrews DM, Smyth MJ, Darcy PK, Kershaw MH. Activating and inhibitory receptors of natural killer cells. Immunol Cell Biol. 2011;89:216–224. doi: 10.1038/icb.2010.78. [DOI] [PubMed] [Google Scholar]
- 53.Zhou Z, Hamming OJ, Ank N, Paludan SR, Nielsen AL, Hartmann R. Type III interferon (IFN) induces a type I IFN-like response in a restricted subset of cells through signaling pathways involving both the Jak-STAT pathway and the mitogen-activated protein kinases. J Virol. 2007;81:7749–7758. doi: 10.1128/JVI.02438-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005;5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 55.Uddin S, Lekmine F, Sharma N, et al. The Rac1/p38 mitogen-activated protein kinase pathway is required for interferon alpha-dependent transcriptional activation but not serine phosphorylation of Stat proteins. J Biol Chem. 2000;275:27634–27640. doi: 10.1074/jbc.M003170200. [DOI] [PubMed] [Google Scholar]
- 56.Uddin S, Yenush L, Sun XJ, Sweet ME, White MF, Platanias LC. Interferon-alpha engages the insulin receptor substrate-1 to associate with the phosphatidylinositol 3'-kinase. J Biol Chem. 1995;270:15938–15941. doi: 10.1074/jbc.270.27.15938. [DOI] [PubMed] [Google Scholar]
- 57.Uddin S, Majchrzak B, Woodson J, et al. Activation of the p38 mitogen-activated protein kinase by type I interferons. J Biol Chem. 1999;274:30127–30131. doi: 10.1074/jbc.274.42.30127. [DOI] [PubMed] [Google Scholar]
- 58.Platanias LC, Sweet ME. Interferon alpha induces rapid tyrosine phosphorylation of the vav proto-oncogene product in hematopoietic cells. J Biol Chem. 1994;269:3143–3146. [PubMed] [Google Scholar]
- 59.Ahmad S, Alsayed YM, Druker BJ, Platanias LC. The type I interferon receptor mediates tyrosine phosphorylation of the CrkL adaptor protein. J Biol Chem. 1997;272:29991–29994. doi: 10.1074/jbc.272.48.29991. [DOI] [PubMed] [Google Scholar]
- 60.de Weerd NA, Vivian JP, Nguyen TK, et al. Structural basis of a unique interferon-βsignaling axis mediated via the receptor IFNAR1. Nat Immunol. 2013;14:901–907. doi: 10.1038/ni.2667. [DOI] [PubMed] [Google Scholar]
- 61.O'Shea JJ, Plenge R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity. 2012;36:542–550. doi: 10.1016/j.immuni.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shuai K, Schindler C, Prezioso VR, Darnell JE., Jr Activation of transcription by IFN-gamma:tyrosine phosphorylation of a 91-kD DNA binding protein. Science. 1992;258:1808–1812. doi: 10.1126/science.1281555. [DOI] [PubMed] [Google Scholar]
- 63.Gotthardt D, Sexl V. STATs in NK-Cells:The Good, the Bad, and the Ugly. Front Immunol. 2016;7:694. doi: 10.3389/fimmu.2016.00694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sommereyns C, Paul S, Staeheli P, Michiels T. IFN-lambda (IFN-lambda) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog. 2008;4:e1000017. doi: 10.1371/journal.ppat.1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bolen CR, Ding S, Robek MD, Kleinstein SH. Dynamic expression profiling of type I and type III interferon-stimulated hepatocytes reveals a stable hierarchy of gene expression. Hepatology. 2014;59:1262–1272. doi: 10.1002/hep.26657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Meyer O. Interferons and autoimmune disorders. Joint Bone Spine. 2009;76:464–473. doi: 10.1016/j.jbspin.2009.03.012. [DOI] [PubMed] [Google Scholar]
- 67.Baccala R, Kono DH, Theofilopoulos AN. Interferons as pathogenic effectors in autoimmunity. Immunol Rev. 2005;204:9–26. doi: 10.1111/j.0105-2896.2005.00252.x. [DOI] [PubMed] [Google Scholar]
- 68.Blanco P, Palucka AK, Gill M, Pascual V, Banchereau J. Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science. 2001;294:1540–1543. doi: 10.1126/science.1064890. [DOI] [PubMed] [Google Scholar]
- 69.Miceli-Richard C, Comets E, Loiseau P, Puechal X, Hachulla E, Mariette X. Association of an IRF5 gene functional polymorphism with Sjögren's syndrome. Arthritis Rheum. 2007;56:3989–3994. doi: 10.1002/art.23142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kim D, Peck A, Santer D, et al. Induction of interferon-alpha by scleroderma sera containing autoantibodies to topoisomerase I:association of higher interferon-alpha activity with lung fibrosis. Arthritis Rheum. 2008;58:2163–2173. doi: 10.1002/art.23486. [DOI] [PubMed] [Google Scholar]
- 71.Greenberg SA, Pinkus JL, Pinkus GS, et al. Interferon-alpha/beta-mediated innate immune mechanisms in dermatomyositis. Ann Neurol. 2005;57:664–678. doi: 10.1002/ana.20464. [DOI] [PubMed] [Google Scholar]
- 72.Conigliaro P, Perricone C, Benson RA, et al. The type I IFN system in rheumatoid arthritis. Autoimmunity. 2010;43:220–225. doi: 10.3109/08916930903510914. [DOI] [PubMed] [Google Scholar]
- 73.Palucka AK, Blanck JP, Bennett L, Pascual V, Banchereau J. Cross-regulation of TNF and IFN-alpha in autoimmune diseases. Proc Natl Acad Sci U S A. 2005;102:3372–3377. doi: 10.1073/pnas.0408506102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Maury CP, Teppo AM. Tumor necrosis factor in the serum of patients with systemic lupus erythematosus. Arthritis Rheum. 1989;32:146–150. doi: 10.1002/anr.1780320206. [DOI] [PubMed] [Google Scholar]
- 75.Studnicka-Benke A, Steiner G, Petera P, Smolen JS. Tumour necrosis factor alpha and its soluble receptors parallel clinical disease and autoimmune activity in systemic lupus erythematosus. Br J Rheumatol. 1996;35:1067–1074. doi: 10.1093/rheumatology/35.11.1067. [DOI] [PubMed] [Google Scholar]
- 76.Gabay C, Cakir N, Moral F, et al. Circulating levels of tumor necrosis factor soluble receptors in systemic lupus erythematosus are significantly higher than in other rheumatic diseases and correlate with disease activity. J Rheumatol. 1997;24:303–308. [PubMed] [Google Scholar]
- 77.Aringer M, Stummvoll GH, Steiner G, et al. Serum interleukin-15 is elevated in systemic lupus erythematosus. Rheumatology (Oxford) 2001;40:876–881. doi: 10.1093/rheumatology/40.8.876. [DOI] [PubMed] [Google Scholar]
- 78.Mavragani CP, Niewold TB, Moutsopoulos NM, Pillemer SR, Wahl SM, Crow MK. Augmented interferon-alpha pathway activation in patients with Sjögren's syndrome treated with etanercept. Arthritis Rheum. 2007;56:3995–4004. doi: 10.1002/art.23062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Aringer M, Smolen JS. The role of tumor necrosis factor-alpha in systemic lupus erythematosus. Arthritis Res Ther. 2008;10:202. doi: 10.1186/ar2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Madsen SM, Schlichting P, Davidsen B, et al. An open-labeled, randomized study comparing systemic interferon-alpha-2A and prednisolone enemas in the treatment of left-sided ulcerative colitis. Am J Gastroenterol. 2001;96:1807–1815. doi: 10.1111/j.1572-0241.2001.03875.x. [DOI] [PubMed] [Google Scholar]
- 81.Nikolaus S, Rutgeerts P, Fedorak R, et al. Interferon beta-1a in ulcerative colitis:a placebo controlled, randomised, dose escalating study. Gut. 2003;52:1286–1290. doi: 10.1136/gut.52.9.1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pena Rossi C, Hanauer SB, Tomasevic R, Hunter JO, Shafran I, Graffner H. Interferon beta-1a for the maintenance of remission in patients with Crohn's disease:results of a phase II dose-finding study. BMC Gastroenterol. 2009;9:22. doi: 10.1186/1471-230X-9-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tilg H, Vogelsang H, Ludwiczek O, et al. A randomised placebo controlled trial of pegylated interferon alpha in active ulcerative colitis. Gut. 2003;52:1728–1733. doi: 10.1136/gut.52.12.1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pena-Rossi C, Schreiber S, Golubovic G, et al. Clinical trial:a multicentre, randomized, double-blind, placebo-controlled, dose-finding, phase II study of subcutaneous interferon-beta-la in moderately active ulcerative colitis. Aliment Pharmacol Ther. 2008;28:758–767. doi: 10.1111/j.1365-2036.2008.03778.x. [DOI] [PubMed] [Google Scholar]
- 85.Mannon PJ, Hornung RL, Yang Z, et al. Suppression of inflammation in ulcerative colitis by interferon-β-1a is accompanied by inhibition of IL-13 production. Gut. 2011;60:449–455. doi: 10.1136/gut.2010.226860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.McAleer JP, Kolls JK. Maintaining poise:commensal microbiota calibrate interferon responses. Immunity. 2012;37:10–12. doi: 10.1016/j.immuni.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 87.Santaolalla R, Abreu MT. Innate immunity in the small intestine. Curr Opin Gastroenterol. 2012;28:124–129. doi: 10.1097/MOG.0b013e3283506559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Guarner F, Malagelada JR. Gut flora in health and disease. Lancet. 2003;361:512–519. doi: 10.1016/S0140-6736(03)12489-0. [DOI] [PubMed] [Google Scholar]
- 89.Chirdo FG, Millington OR, Beacock-Sharp H, Mowat AM. Immunomodulatory dendritic cells in intestinal lamina propria. Eur J Immunol. 2005;35:1831–1840. doi: 10.1002/eji.200425882. [DOI] [PubMed] [Google Scholar]
- 90.Kole A, He J, Rivollier A, et al. Type I IFNs regulate effector and regulatory T cell accumulation and anti-inflammatory cytokine production during T cell-mediated colitis. J Immunol. 2013;191:2771–2779. doi: 10.4049/jimmunol.1301093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cho H, Kelsall BL. The role of type I interferons in intestinal infection, homeostasis, and inflammation. Immunol Rev. 2014;260:145–167. doi: 10.1111/imr.12195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bennett L, Palucka AK, Arce E, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. 2003;197:711–723. doi: 10.1084/jem.20021553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Baechler EC, Batliwalla FM, Karypis G, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A. 2003;100:2610–2615. doi: 10.1073/pnas.0337679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Higgs BW, Liu Z, White B, et al. Patients with systemic lupus erythematosus, myositis, rheumatoid arthritis and scleroderma share activation of a common type I interferon pathway. Ann Rheum Dis. 2011;70:2029–2036. doi: 10.1136/ard.2011.150326. [DOI] [PubMed] [Google Scholar]
- 95.Braunstein I, Klein R, Okawa J, Werth VP. The interferon-regulated gene signature is elevated in subacute cutaneous lupus erythematosus and discoid lupus erythematosus and correlates with the cutaneous lupus area and severity index score. Br J Dermatol. 2012;166:971–975. doi: 10.1111/j.1365-2133.2012.10825.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bauer JW, Petri M, Batliwalla FM, et al. Interferon-regulated chemokines as biomarkers of systemic lupus erythematosus disease activity:a validation study. Arthritis Rheum. 2009;60:3098–3107. doi: 10.1002/art.24803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bauer JW, Baechler EC, Petri M, et al. Elevated serum levels of interferon-regulated chemokines are biomarkers for active human systemic lupus erythematosus. PLoS Med. 2006;3:e491. doi: 10.1371/journal.pmed.0030491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zahn S, Rehkämper C, Kümmerer BM, et al. Evidence for a pathophysiological role of keratinocyte-derived type III interferon (IFNλ) in cutaneous lupus erythematosus. J Invest Dermatol. 2011;131:133–140. doi: 10.1038/jid.2010.244. [DOI] [PubMed] [Google Scholar]
- 99.van der Pouw Kraan TC, Wijbrandts CA, van Baarsen LG, et al. Rheumatoid arthritis subtypes identified by genomic profiling of peripheral blood cells:assignment of a type I interferon signature in a subpopulation of patients. Ann Rheum Dis. 2007;66:1008–1014. doi: 10.1136/ard.2006.063412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Smiljanovic B, Grün JR, Biesen R, et al. The multifaceted balance of TNF-αand type I/II interferon responses in SLE and RA:how monocytes manage the impact of cytokines. J Mol Med (Berl) 2012;90:1295–1309. doi: 10.1007/s00109-012-0907-y. [DOI] [PubMed] [Google Scholar]
- 101.Reynier F, Petit F, Paye M, et al. Importance of correlation between gene expression levels:application to the type I interferon signature in rheumatoid arthritis. PLoS One. 2011;6:e24828. doi: 10.1371/journal.pone.0024828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Koutroubakis IE, Vlachonikolis IG, Kouroumalis EA. Role of appendicitis and appendectomy in the pathogenesis of ulcerative colitis:a critical review. Inflamm Bowel Dis. 2002;8:277–286. doi: 10.1097/00054725-200207000-00007. [DOI] [PubMed] [Google Scholar]
- 103.Watson Ng WS, Hampartzoumian T, Lloyd AR, Grimm MC. A murine model of appendicitis and the impact of inflammation on appendiceal lymphocyte constituents. Clin Exp Immunol. 2007;150:169–178. doi: 10.1111/j.1365-2249.2007.03463.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cheluvappa R, Eri R, Luo AS, Grimm MC. Modulation of interferon activity-associated soluble molecules by appendicitis and appendectomy limits colitis-identification of novel anti-colitic targets. J Interferon Cytokine Res. 2015;35:108–115. doi: 10.1089/jir.2014.0091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Oritani K, Kanakura Y. IFN-zeta/limitin:a member of type I IFN with mild lympho-myelosuppression. J Cell Mol Med. 2005;9:244–254. doi: 10.1111/j.1582-4934.2005.tb00353.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sainathan SK, Bishnupuri KS, Aden K, et al. Toll-like receptor-7 ligand Imiquimod induces type I interferon and antimicrobial peptides to ameliorate dextran sodium sulfate-induced acute colitis. Inflamm Bowel Dis. 2012;18:955–967. doi: 10.1002/ibd.21867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cheluvappa R, Thomas DG, Selvendran S. The role of specific chemokines in the amelioration of colitis by appendicitis and appendectomy. Biomolecules. 2018:8. doi: 10.3390/biom8030059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Guo J, Peters KL, Sen GC. Induction of the human protein P56 by interferon, double-stranded RNA, or virus infection. Virology. 2000;267:209–219. doi: 10.1006/viro.1999.0135. [DOI] [PubMed] [Google Scholar]
- 109.Fensterl V, Sen GC. The ISG56/IFIT1 gene family. J Interferon Cytokine Res. 2011;31:71–78. doi: 10.1089/jir.2010.0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Honda K, Taniguchi T. IRFs:master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol. 2006;6:644–658. doi: 10.1038/nri1900. [DOI] [PubMed] [Google Scholar]
- 111.Zhao GN, Jiang DS, Li H. Interferon regulatory factors:at the crossroads of immunity, metabolism, and disease. Biochim Biophys Acta. 2015;1852:365–378. doi: 10.1016/j.bbadis.2014.04.030. [DOI] [PubMed] [Google Scholar]
- 112.Hallen LC, Burki Y, Ebeling M, et al. Antiproliferative activity of the human IFN-alpha-inducible protein IFI44. J Interferon Cytokine Res. 2007;27:675–680. doi: 10.1089/jir.2007.0021. [DOI] [PubMed] [Google Scholar]
- 113.West LC, Cresswell P. Expanding roles for GILT in immunity. Curr Opin Immunol. 2013;25:103–108. doi: 10.1016/j.coi.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bange FC, Vogel U, Flohr T, Kiekenbeck M, Denecke B, Böttger EC. IFP 35 is an interferon-induced leucine zipper protein that undergoes interferon-regulated cellular redistribution. J Biol Chem. 1994;269:1091–1098. [PubMed] [Google Scholar]
- 115.Moura R, Araujo J, Guimarães R, Crovella S, Brandão L. Interferon induced with helicase C domain 1 (IFIH1):trends on helicase domain and type 1 diabetes onset. Gene. 2013;516:66–68. doi: 10.1016/j.gene.2012.11.066. [DOI] [PubMed] [Google Scholar]
- 116.Stone KD, Prussin C, Metcalfe DD. IgE, mast cells, basophils, and eosinophils. J Allergy Clin Immunol. 2010;125:S73–S80. doi: 10.1016/j.jaci.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rauch I, Müller M, Decker T. The regulation of inflammation by interferons and their STATs. JAKSTAT. 2013;2:e23820. doi: 10.4161/jkst.23820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Cheluvappa R. Experimental appendicitis and appendectomy modulate the CCL20-CCR6 axis to limit inflammatory colitis pathology. Int J Colorectal Dis. 2014;29:1181–1188. doi: 10.1007/s00384-014-1936-5. [DOI] [PubMed] [Google Scholar]
- 119.Müller M, Carter S, Hofer MJ, Campbell IL. Review:The chemokine receptor CXCR3 and its ligands CXCL9, CXCL10 and CXCL11 in neuroimmunity-a tale of conflict and conundrum. Neuropathol Appl Neurobiol. 2010;36:368–387. doi: 10.1111/j.1365-2990.2010.01089.x. [DOI] [PubMed] [Google Scholar]
- 120.van Baarsen LG, Wijbrandts CA, Rustenburg F, et al. Regulation of IFN response gene activity during infliximab treatment in rheumatoid arthritis is associated with clinical response to treatment. Arthritis Res Ther. 2010;12:R11. doi: 10.1186/ar2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bar Yehuda S, Axlerod R, Toker O, et al. The association of inflammatory bowel diseases with autoimmune disorders:a report from the epi-IIRN. J Crohns Colitis. 2019;13:324–329. doi: 10.1093/ecco-jcc/jjy166. [DOI] [PubMed] [Google Scholar]
- 122.Geremia A, Biancheri P, Allan P, Corazza GR, Di Sabatino A. Innate and adaptive immunity in inflammatory bowel disease. Autoimmun Rev. 2014;13:3–10. doi: 10.1016/j.autrev.2013.06.004. [DOI] [PubMed] [Google Scholar]