

Abstract
The natural history, prognostication and optimal treatment of Richter transformation developed from chronic lymphocytic leukemia (CLL) are not well defined. We report the clinical characteristics and outcomes of a large series of biopsy-confirmed Richter transformation (diffuse large B-cell lymphoma or high grade B-cell lymphoma, n=204) cases diagnosed from 1993 to 2018. After a median follow up of 67.0 months, the median overall survival (OS) was 12.0 months. Patients who received no prior treatment for CLL had significantly better OS (median 46.3 vs. 7.8 months; P<0.001). Patients with elevated lactate dehydrogenase (median 6.2 vs. 39.9 months; P<0.0001) or TP53 disruption (median 8.3 vs. 12.8 months; P=0.046) had worse OS than those without. Immunoglobulin heavy chain variable region gene mutation, cell of origin, Myc/Bcl-2 double expression and MYC/BCL2/BCL6 double-/triple-hit status were not associated with OS. In multivariable Cox regression, elevated lactate dehydrogenase [Hazard ratio (HR) 2.3, 95% Confidence Interval (CI): 1.3-4.1; P=0.01], prior CLL treatment (HR 2.0, 95%CI: 1.2-3.5; P=0.01), and older age (HR 1.03, 95%CI: 1.01-1.05; P=0.01) were associated with worse OS. Twenty-four (12%) patients underwent stem cell transplant (20 autologous and 4 allogeneic), and had a median post-transplant survival of 55.4 months. In conclusion, the overall outcome of Richter transformation is poor. Richter transformation developed in patients with untreated CLL has significantly better survival. Stem cell transplant may benefit select patients.
Introduction
Richter transformation (RT) refers to the transformation of chronic lymphocytic leukemia (CLL) to an aggressive lymphoma. It was first described by Dr. Maurice Richter in 1928 with a rapidly fatal case of “reticular cell sarcoma of lymph nodes” arising in the background of “lymphatic leukemia”.1 RT presents with diffuse large B-cell lymphoma (DLBCL) in over 90% of the cases, and classical Hodgkin lymphoma in 5% or less. The incidence of DLBCL type of RT is approximately 0.5-1% per year in newly diagnosed CLL patients,2 and the overall prevalence of RT is about 2-10% in CLL patients according to multiple published studies.3–5 The reported risk factors associated with RT include: advanced stage, large lymph nodes (> 3 cm), unmutated immunoglobulin heavy chain variable region gene (IGHV), del(17p), TP53 mutation, NOTCH1 mutation, and stereotyped B-cell receptor (BCR).2,6–11
Clinically, RT often presents aggressively with rapidly enlarging lymphadenopathy, prominent constitutional symptoms (fevers, night sweats, and unintentional weight loss), elevated LDH, and frequent extranodal tissue involvement.3 Treatment of RT has been challenging. The standard R-CHOP regimen used for treatment of de novo DLBCL has limited efficacy in DLBCL-type RT.12 Higher intensity chemotherapy does not improve outcomes.13–18 Stem cell transplant (SCT) has been studied in RT and appears to be associated with relative long-term survival in select cases.19–23 Overall, RT has a poor prognosis, with a median survival of only 1-2 years.3,5
The landscape of CLL management has changed dramatically with the emergence of several novel targeted agents, such as Bruton’s tyrosine kinase (BTK) inhibitors ibrutinib and acalabrutinib, phosphoinositide 3-kinase δ (PI3Kδ) inhibitors idelalisib and duvelisib, and the B-cell lymphoma 2 (BCL-2) inhibitor venetoclax. It is unclear whether these novel agents affect the risk, prognosis and management of RT. In de novo DLBCL, the prognostic roles of cell of origin (COO), Myc and Bcl-2 double expression, and MYC, BCL2 and/or BCL6 gene rearrangements have been well recognized.24–29 However, the potential impact of these molecular markers on the outcome of DLBCL-type RT has not been well studied.
In this study, we report the clinical characteristics, treatment pattern, and outcomes of a large series of RT patients (n=204) from a single center over more than two decades including the era of novel agents (from 2012 to the present). The potential prognostic impact of prior CLL treatment as well as CLL- and RT-related molecular markers were also explored.
Methods
Patients
This study was approved by the Mayo Clinic Institutional Review Board. All patients were identified from the Mayo Clinic CLL database which includes consecutive CLL patients evaluated in the Division of Hematology at Mayo Clinic, Rochester, MN, USA.2,30,31 CLL patients who developed biopsy-proven RT between April 1993 and April 2018 were identified from the database. For this study, the focus was RT to DLBCL (including high grade B-cell lymphoma, such as double-/triple-hit lymphoma which is now known as high-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements); transformations to Hodgkin lymphoma or other histology were excluded. Clinical, pathological, and molecular characteristics [IGHV mutation, CLL fluorescence in situ hybridization (FISH)] and all treatment information during the CLL phase were abstracted from the database. Clinical, pathological and molecular characteristics [CLL FISH, TP53 somatic mutation, COO by Hans algorithm, Myc/Bcl-2 expression by immunohistochemistry (IHC), MYC/BCL2/BCL6 rearrangement by FISH, CLL and RT clonal relationship by immunoglobulin gene rearrangement], treatment course, clinical response to treatment as determined by treating physician, and survival information after RT was abstracted by chart review. On IHC, the cut-off value for positivity was 40% for Myc, and 50% for Bcl-2. Our institution started to routinely test for Myc expression by IHC and MYC rearrangement by FISH in all DLBCL cases in 2012; therefore, we have missing information on IHC as well as FISH results in patients diagnosed with RT prior to this.
Statistical analysis
The date of RT diagnosis was defined as the date of the biopsy which led to the pathological diagnosis of RT. The time to transformation was defined as the time from CLL diagnosis to RT diagnosis. Overall survival (OS) was defined as the time from RT diagnosis to death from any cause. Time-to-event data were analyzed using the Kaplan-Meier method. Cox proportional hazards models were used to analyze associations between OS and various factors. P<0.05 was considered statistically signifi cant. All statistical analyses were carried out in SAS 9.4 (SAS Institute, Cary, NC, USA).
Results
Clinical characteristics in the chronic lymphocytic leukemia phase
A total of 204 patients with CLL who developed RT were identified. Baseline characteristics at CLL diagnosis are shown in Online Supplementary Table S1. The median age at CLL diagnosis was 62 years (range 22-85), and 148 (72.5%) were male. Seventy-one (71.0%) of 100 patients tested had unmutated IGHV. CLL FISH detected del(17p) in 33 (25.4%), del(11q) in 18 (13.8%), and trisomy 12 in 19 (14.6%) of 130 patients. Forty-seven (66.2%) of 71 patients were high or very high risk by CLL-IPI score (≥4).
Clinical characteristics at Richter transofrmation diagnosis
Median time to transformation was 4.7 years (range 0-34.5) (Table 1). Prior to RT, 69 (33.8%) patients received no treatment for CLL, 108 (52.9%) received chemoimmunotherapy (CIT) only, and 27 (13.2%) received at least one novel agent (idelalisib, ibrutinib, or venetoclax) for CLL; 19 patients received CIT previously and developed RT on novel agents (17 ibrutinib, 1 idelalisib, 1 venetoclax), 6 patients received novel agents previously and developed RT on subsequent treatment (1 rituximab-bendamustine, 3 rituximab-corticosteroid, and 2 rituximab alone), and 2 other patients developed RT on frontline ibrutinib. Median lines of CLL therapy prior to RT was 2 (range 0-13).
Table 1.
Clinical characteristics at Richter transformation diagnosis in 204 patients.
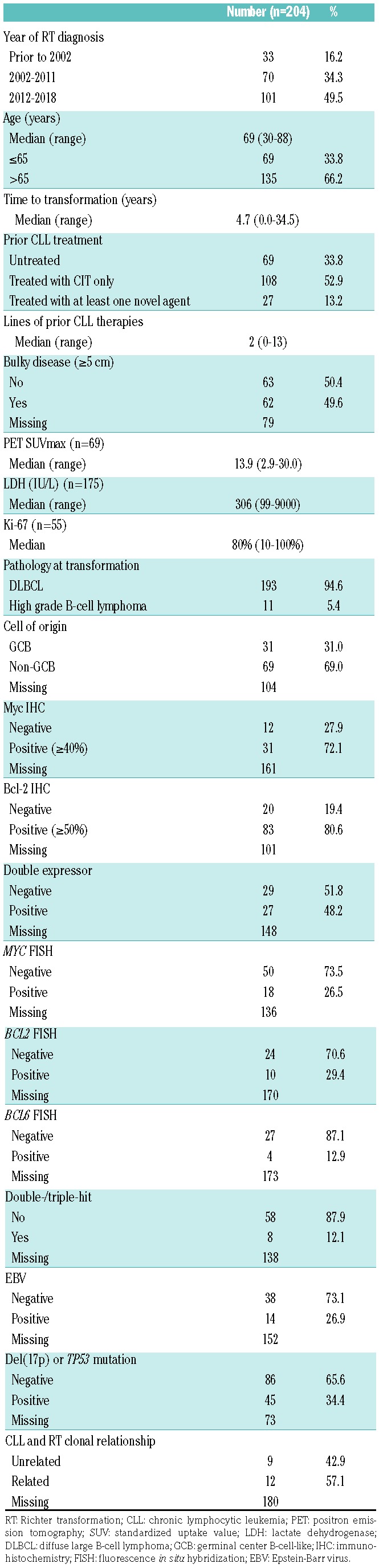
Thirty-three patients were diagnosed with RT prior to 2002 (when rituximab was not routinely available), 70 patients were diagnosed between 2002 and 2011, and 101 patients were diagnosed in 2012 or later (when ibrutinib had become available). The median age at RT diagnosis was 69 years (range 30-88). Sixty-two (49.6%) of 125 patients had bulky disease (≥ 5 cm). COO by Hans algorithm was germinal center B-cell-like (GCB) and non-GCB in 31 of 100 (31.0%) and 69 of 100 (69.0%) patients, respectively. Myc and Bcl-2 were positive by IHC in 31 of 43 (72.1%) and 83 of 103 (80.6%) cases, respectively; 27 of 56 (48.2%) were double-expressors. MYC, BCL2, and BCL6 rearrangement was positive by FISH in 18 of 68 (26.5%), 10 of 34 (29.4%), and 4 of 31 (12.9%) cases, respectively; 8 of 66 (12.1%) were double-/triple-hit. Forty-five (34.4%) of 131 patients had del(17p) or TP53 mutation, i.e. TP53 disruption. CLL and RT were clonally unrelated in 9 (42.9%) of 21 patients.
Richter transformation treatment and outcome
Pattern of first-line treatment for RT is shown in Table 2. The most commonly used first-line treatment was an R-CHOP-like regimen (n=114, 65.5%). Twelve (6.9%) patients received platinum or high-dose cytarabine containing chemotherapy; 21 (12.1%) patients received other chemotherapy (6 with DA-EPOCH-R-like regimen, 15 with others including ProMACE-CytaBOM, R-CEPP, infusional CDE, R-CVP, R-bendamustine, R-gemcitabine/prednisone, high dose methotrexate-based regimen). Nineteen (10.9%) patients received novel agents: ibrutinib (n=4), venetoclax (n=1), ibrutinib plus venetoclax (n=3), pembrolizumab (n=7), pembrolizumab plus ibrutinib (n=1), CD19 monoclonal antibody (n=1), everolimus (n=1), everolimus plus panobinostat (n=1). Eight (4.6%) patients received palliative therapy defined as rituximab, corticosteroids, radiation therapy, alone or in combination.
Table 2.
First-line treatment approaches for Richter transformation (RT) in patients with chronic lymphocytic leukemia (CLL).

Clinical response (assessed by treating physician) to first-line treatment was complete response (CR) in 54 (36.0%), partial response (PR) in 37 (24.0%), stable disease (SD) in 18 (12.0%), and progressive disease (PD) in 42 (28.0%) of 150 patients. The median follow up after RT was 67.0 months, and there were a total of 150 deaths. The median OS after RT diagnosis was 12.0 months (Figure 1A).
Figure 1.

Overall survival (OS) after Richter transformation (RT) diagnosis of the entire cohort and by prior chronic lymphocytic leukemia (CLL) treatment status. (A) OS for all patients. (B) OS by previous CLL treatment status. (C) OS by temporal relationship between CLL and RT in patients with previously untreated CLL. (D) OS by lines of therapy in patients with previously treated CLL. (E) OS by prior CLL treatment category.
Survival by clinical and molecular factors and treatment is summarized in Table 3. The median OS was significantly better in patients who received no CLL treatment than those who received any CLL treatment, with a median OS of 46.3 versus 7.8 months (P<0.001) (Figure 1B). Among the 69 patients who received no CLL treatment, 31 had concurrent CLL and RT (i.e. RT diagnosis within 3 months of CLL diagnosis), with a median OS of 66.9 months; in the other 38 patients with sequential CLL and RT (median time to transformation 55.5 months), the median OS was 29.4 months (P=0.25) (Figure 1C). Among the 135 patients who had received treatment for CLL, patients with only one line of CLL treatment (n=31) had a trend of better OS compared to those with two or more lines of CLL treatment (n=104), with a median OS of 15.3 versus 5.8 months (P=0.09) (Figure 1D). Patients who received CIT only and those who received at least one novel agent for CLL had a median OS of 7.1 and 12.0 months, respectively (P=0.28) (Figure 1E).
Table 3.
Median overall survival (OS) after Richter transformation (RT) diagnosis by clinical characteristics at RT diagnosis or first-line treatment approach for RT.
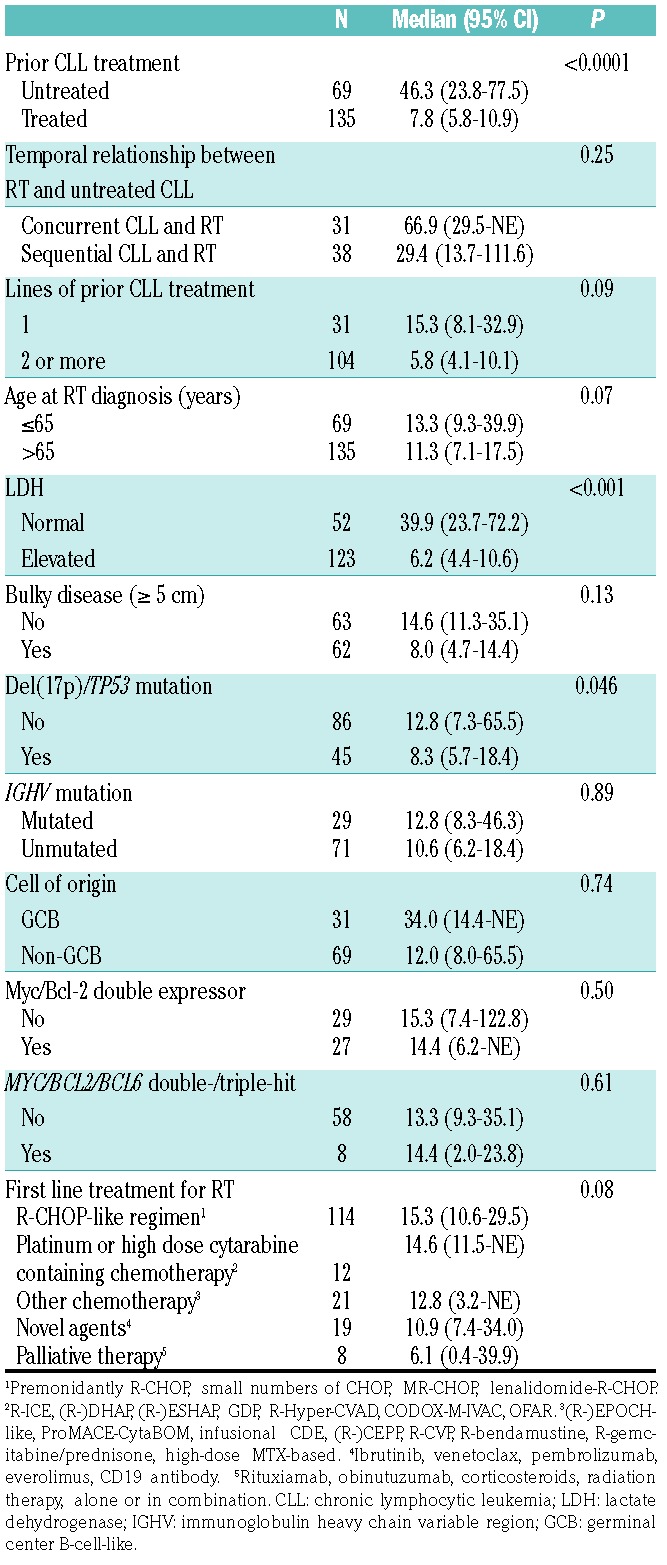
There was no significant difference in median OS between younger (age≤65) and older (age>65) RT patients, with a median OS of 13.3 versus 11.3 months (P=0.07) (Figure 2A). Patients with elevated LDH had worse OS compared to those with normal LDH, with a median OS of 6.2 versus 39.9 months (P<0.0001) (Figure 2B). Bulky disease (nodal size ≥5 cm) was not associated with a worse OS (median OS of 8.0 vs. 14.6 months; P=0.13) (Online Supplementary Figure S1). Patients with TP53 disruption had a worse OS than those without (median OS 8.3 vs. 12.8 months; P=0.046) (Figure 2C). Other molecular characteristics, including IGHV mutation (Online Supplementary Figure S2), DLBCL COO (Online Supplementary Figure S3), Myc/Bcl-2 double expression (Figure 2D), MYC/BCL2/BCL6 double-/triple-hit status (Online Supplementary Figure S4), and CLL and RT clonal relationship (Online Supplementary Figure S5) did not impact RT survival.
Figure 2.

Overall survival (OS) after Richter transformation (RT) diagnosis by clinical and molecular factors. (A) OS by age at RT diagnosis. (B) OS by lactate dehy-drogenase (LDH) at RT diagnosis. (C) OS by TP53 disruption status. (D) OS by Myc and Bcl-2 double expression status.
In a multivariable Cox regression model, we examined the association of age (continuous), LDH (normal vs. elevated), prior CLL treatment (untreated vs. treated) and TP53 disruption with RT survival; we found that elevated LDH (HR 2.3, 95%CI: 1.3-4.1; P=0.01), prior CLL treatment (HR 2.0, 95%CI: 1.2-3.5; P=0.01), and to a lesser extent older age (HR 1.03, 95%CI: 1.01-1.05; P=0.01), but not TP53 disruption (HR 1.3, 95%CI:0.8-2.1; P=0.31), were associated with worse OS.
Patients treated with an R-CHOP-like regimen (n=114) had a median OS of 15.3 months (Online Supplementary Figure S6). Patients treated with platinum or high-dose cytarabine containing chemotherapy (n=12) had a median OS of 14.6 months (P=0.82 vs. R-CHOP-like). Patients treated with other chemotherapy (n=21) had a median OS of 12.8 months (P=0.66 vs. R-CHOP-like). The 19 patients who received novel agents as first-line RT treatment had a median OS of 10.9 months (P=0.12 vs. R-CHOP-like). The median OS for patients receiving palliative therapy (n=8) was 6.1 months (P=0.01 vs. R-CHOP-like).
After achieving PR or better, 24 (11.8%) patients underwent SCT: 20 autologous and 4 allogenic. Details of the clinical characteristics of the 24 patients who underwent SCT are shown in Online Supplementary Table S2. The median age at RT diagnosis was 62 years (range 41-73). Ten patients did not receive any prior CLL treatment. Nineteen patients achieved a PR or better with 1-2 lines of treatment before proceeding to SCT. The median time from RT diagnosis to SCT was 6.8 months (range 3.3-42.3). None of the four allogeneic SCT patients died although the post-SCT follow up was still relatively short for three of them (Figure 3A). Thirteen of the 20 autologous SCT patients had a post-SCT survival greater than two years (Figure 3A). Overall, the 24 SCT patients had a median post-SCT survival of 55.4 months (Figure 3B).
Figure 3.

Survival outcomes of the 24 Richter transformation (RT) patients who underwent stem cell transplantation (SCT). (A) Swimmers plot showing time from RT diagnosis to SCT (blue) and post-SCT survival (red; numbers indicate post-SCT survival in months). (B) Post-SCT survival for all patients who underwent SCT. CI: Confidence Interval.
Discussion
To the best of our knowledge, this is the largest series of CLL patients with biopsy-proven DLBCL-type RT with a long follow up. Our results showed that patients with DLBCL-type RT overall had a poor prognosis with a median OS of only 12 months. Patients with RT who received no prior CLL treatment had a significantly better OS, with a median OS of approximately four years.
Our singular finding is that patients who received no prior CLL therapy had a favorable outcome. Further proof of this finding was provided in a prior phase II trial of ofa-tumumab in combination with CHOP for newly diagnosed RT, where patients who received no prior CLL treatment had significantly better OS (median unreached at 24 months vs. approx. 6 months).32 A recent Danish National CLL Registry study of RT (over 8 years with multiple histologies) also showed similar results, with a median OS of 6.16 year in patients with untreated CLL versus 1.49 years in patients with treated CLL.11 The observed OS differences in these studies may be due to different biology in untreated patients. Further to this finding, RT patients who received only one line of CLL therapy had a trend of better OS compared to those who received two or more lines of prior CLL therapies in our study, and two other studies also demonstrated that fewer lines of prior CLL therapy was associated with better RT survival,18,19 supporting the hypothesis that less therapy of CLL may be associated with less chemoresistance of RT. Of note, patients who were diagnosed with CLL and RT within three months (defined as concurrent RT) had a particularly favorable outcome, with a median OS of approximately six years. We suspect that the concurrent RT cases were more likely clonally unrelated (to the CLL) and resemble de novo DLBCL. This aspect warrants future studies. Based on our data, patients with concurrent RT may benefit from the typical therapy for de novo DLBCL.
TP53 disruption (i.e. del(17p) and/or TP53 mutation) was associated with a worse prognosis of RT in the uni-variate analysis, consistent with a number of prior studies.8,12,18,32–34 Of note, TP53 disruption was not an independent prognostic factor of OS in the multivariable analysis in our study. Rossi et al. showed that TP53 disruption was an independent prognostic factor of RT survival.8 Four other studies did not test for TP53 somatic mutation and reported inconsistent results regarding the independent prognostic role of del(17p).8,12,33,34 Given only 27 (13%) of our patients underwent the TP53 somatic mutation test, we likely underestimated the proportion of RT patients who had TP53 disruption, and thus might have underestimated the negative impact of this molecular abnormality on RT outcomes in our cohort.
The prognostic roles of COO, double expressor and double-hit status in de novo DLBCL have been well established.24–29 In this study, we found that these molecular markers were not prognostic for RT survival. Indeed, Eyre et al. showed that COO and Myc expression status did not influence RT survival in the O-CHOP trial,32 and Fidai et al. showed that MYC and/or BCL2 genetic alterations did not impact RT outcome in a retrospective study.33 RT and de novo DLBCL are likely different diseases given the known distinct genomic abnormalities,5,35 and the impacts of COO and double expressor/double-hit status may therefore be different. We should note that molecular characterizations of COO and double expressor/double-hit status were incomplete in our dataset, and interpretation of these results should be made with caution.
In terms of other relevant prognostic factors, older age and elevated LDH were associated with worse OS in RT, consistent with the MD Anderson data.19 Clonal relationship between CLL and RT was reported to be a critical prognostic factor, with a much better outcome in clonally unrelated RT.8,36 Due to the difficulty of obtaining paired CLL and RT samples (at RT diagnosis) and a lack of universal assessment in our routine clinical practice, we only have a limited number of cases (<5%) in which the CLL and RT clonal relationship was reported. This difficulty is consistent with clinical experiences shared among several different academic centers. While we did not see a statistically significant association of clonal relationship with RT survival, this should not be taken as evidence that goes against prior studies. CLL IGHV mutation status was not associated with RT survival in our study. Prior studies were inconsistent regarding the prognostic role of CLL IGHV mutation in RT survival, with positive association reported in two studies (one in univariate analysis only, the other with only 16 RT patients)11,34 but not others.8,18 It remains unclear whether CLL IGHV mutation status is associated with RT outcome.
In our cohort, RT patients who were exposed to at least one novel agent (predominantly ibrutinib) for CLL treatment had a median OS of 10.9 months, similar to those whose CLL were treated with CIT only, and compares favorably to prior data.37–39 A number of studies have shown that RT that developed on novel agents had poor outcome, with a median OS of approximately 2-3.5 months if developed on ibrutinib37–39 and approximately 12 months if developed on venetoclax.40 In these studies, RT was primarily treated with R-CHOP- or R-EPOCH-like regimens. For our RT patients previously exposed to novel agents for CLL, approximately two-thirds were treated with novel agents (e.g. pembrolizumab, ibrutinib and venetoclax, etc.) and only one-third were treated with R-CHOP-like or other chemotherapy at first line. The efficacy of novel agents in treating RT has been reported by a number of studies.41–46 For example, pembrolizumab demonstrated encouraging efficacy in patients with RT, particularly those with prior exposure of ibrutinib.43 Nivolumab in combination with ibrutinib and pembrolizumab in combination with umbralisib and ublituximab also demonstrated encouraging activity in treating RT.45–47 In addition to the above studies, BTK inhibitors (NCT03899337), PI3K inhibitors (NCT03884998) and/or venetoclax (NCT03054896) based combination regimens (with or without chemotherapy) and other agents such as immunomodulatory drug lenalidomide (NCT03113695, NCT02005289), engineered anti-CD19 monoclonal antibody MOR208 (NCT02005289), and CD3/CD19 bispecific antibody blinatumumab (NCT03121534, NCT03931642) are actively being tested in clinical trials for RT. Chimeric antigen receptor (CAR) T cells are also showing promise in improving the outcome of RT48 and are further tested in clinical trials (NCT03484702). Taken together, novel agents (e.g. pembrolizumab/nivolumab, ibrutinib and venetoclax) would seem to be very reasonable choices in patients who develop RT while receiving one of the targeted agents (e.g. ibrutinib, venetoclax, idelalisib) for CLL. However, it is important to note that despite promising results from multiple studies,41–48 further improvements to increase the efficacy and select optimal patients for different novel agents are clearly needed.
Patients who underwent SCT (n=24, 11.8%) in our cohort overall had a favorable outcome, with a median post-SCT survival of 55.4 months. The role of SCT in RT management has been explored previously.19–22 In the MD Anderson cohort, 20 (13.5%) of 148 RT patients were able to proceed to SCT (3 autologous, 17 allogeneic), and seven patients who underwent allogeneic SCT for consolidation had a favorable outcome with a 3-year OS of 75%.19 A retrospective study by the European Group for Blood and Marrow Transplantation (EBMT) showed that post-remission SCT may benefit a subset of RT patients, with a 3-year OS of 59% for autologous SCT and 36% for allogeneic SCT.20 Two recent single institution studies also reported somewhat favorable outcome of allogenic SCT in RT patients, with a 2-year OS of 44% in one study and a 4-year OS of 50% in the other.21,22 Collectively, these data suggest that select RT patients can benefit from SCT. One should be aware of the potential selection bias when interpreting the data, e.g. patients need to be relatively young and in good condition to proceed to SCT. For example, 10 of the 24 patients in our SCT cohort received no prior CLL treatment; patients were relatively younger, and most patients achieved a favorable response with 1-2 lines of RT therapy and then went on to SCT. We used substantially more autologous (n=20) than allogeneic (n=4) SCT. While allogeneic SCT target both the CLL and RT clones, an autologous SCT primarily eradicates the RT clone and spares the undesired high non-relapse mortality associated with allogeneic SCT.
Our study shows that RT has poor clinical outcomes in general. However, as a heterogeneous disease, its outcome is influenced significantly by prior CLL therapy status. One reason for the poor survival observed in RT after prior CLL therapy may be the known potential of CLL clones to undergo clonal evolution under the pressure of therapy.49,50 We propose a newer approach to manage RT based on their prior CLL therapy status. In treatment-naïve patients and patients with clonally unrelated DLBCL, immunochemotherapy, in particular R-CHOP-like regimens, is the preferred approach in treating these RT. In patients exposed to targeted CLL therapies (including kinase inhibitors and BCL-2 inhibitor) or prior chemoimmunotherapies, the management of RT would likely need to incorporate novel agents, immunotherapy, and/or cellular therapy in clinical trials. SCT consolidation should still be considered in RT patients who achieve a good response to therapy. We fully expect that RT biology will continue to evolve with the changing landscape of CLL as management with novel agents are robustly moving to the front line. In support of this, recent data indicated that 70-80% of RT that developed on novel agents had TP53 disruption and/or complex karyotypes, both of which were prognostic of poor outcomes in RT.39 It is hoped that our study can provide more valuable information on current management of RT and also point to areas of interest for future clinical trials and biological studies.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/105/3/765
Funding
This study was supported by grants from the National Institute of Health Lymphoma SPORE CA 97274 and K23 CA160345, K12 CA090628, Richard M. Schulze Family Foundation for Awards in Cancer Research, the Fraternal Order of Eagles Cancer Research Fund, and the Predolin Foundation. SAP and SSK are recipients of the K12 CA090628 grant from the National Cancer Institute (Paul Calabresi Career Development Award for Clinical Oncology).
References
- 1.Richter MN. Generalized Reticular Cell Sarcoma of Lymph Nodes Associated with Lymphatic Leukemia. Am J Pathol. 1928; 4(4):285–292. [PMC free article] [PubMed] [Google Scholar]
- 2.Parikh SA, Rabe KG, Call TG, et al. Diffuse large B-cell lymphoma (Richter syndrome) in patients with chronic lymphocytic leukaemia (CLL): a cohort study of newly diagnosed patients. Br J Haematol. 2013; 162(6):774–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parikh SA, Kay NE, Shanafelt TD. How we treat Richter syndrome. Blood. 2014; 123(11):1647–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rossi D, Gaidano G. Richter syndrome: pathogenesis and management. Semin Oncol. 2016;43(2):311–319. [DOI] [PubMed] [Google Scholar]
- 5.Rossi D, Spina V, Gaidano G. Biology and treatment of Richter syndrome. Blood. 2018;131(25):2761–2772. [DOI] [PubMed] [Google Scholar]
- 6.Rossi D, Cerri M, Capello D, et al. Biological and clinical risk factors of chronic lymphocytic leukaemia transformation to Richter syndrome. Br J Haematol. 2008; 142(2):202–215. [DOI] [PubMed] [Google Scholar]
- 7.Rossi D, Spina V, Cerri M, et al. Stereotyped B-cell receptor is an independent risk factor of chronic lymphocytic leukemia transformation to Richter syndrome. Clin Cancer Res. 2009;15(13):4415–4422. [DOI] [PubMed] [Google Scholar]
- 8.Rossi D, Spina V, Deambrogi C, et al. The genetics of Richter syndrome reveals disease heterogeneity and predicts survival after transformation. Blood. 2011;117(12): 3391–3401. [DOI] [PubMed] [Google Scholar]
- 9.Rossi D, Rasi S, Spina V, et al. Different impact of NOTCH1 and SF3B1 mutations on the risk of chronic lymphocytic leukemia transformation to Richter syndrome. Br J Haematol. 2012;158(3):426–429. [DOI] [PubMed] [Google Scholar]
- 10.Rossi D, Rasi S, Fabbri G, et al. Mutations of NOTCH1 are an independent predictor of survival in chronic lymphocytic leukemia. Blood. 2012;119(2):521–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ben-Dali Y, Hleuhel MH, Andersen MA, et al. Risk Factors Associated with Richter’s Transformation in Patients with Chronic Lymphocytic Leukemia. Blood. 2018; 132(Suppl 1):1697. [Google Scholar]
- 12.Langerbeins P, Busch R, Anheier N, et al. Poor efficacy and tolerability of R-CHOP in relapsed/refractory chronic lymphocytic leukemia and Richter transformation. Am J Hematol. 2014;89(12):E239–243. [DOI] [PubMed] [Google Scholar]
- 13.Dabaja BS, O’Brien SM, Kantarjian HM, et al. Fractionated cyclophosphamide, vin-cristine, liposomal daunorubicin (daunoXome), and dexamethasone (hyperCVXD) regimen in Richter’s syndrome. Leuk Lymphoma. 2001;42(3):329–337. [DOI] [PubMed] [Google Scholar]
- 14.Tsimberidou AM, Kantarjian HM, Cortes J, et al. Fractionated cyclophosphamide, vin-cristine, liposomal daunorubicin, and dex-amethasone plus rituximab and granulo-cyte-macrophage-colony stimulating factor (GM-CSF) alternating with methotrexate and cytarabine plus rituximab and GM-CSF in patients with Richter syndrome or flu-darabine-refractory chronic lymphocytic leukemia. Cancer. 2003;97(7):1711–1720. [DOI] [PubMed] [Google Scholar]
- 15.Tsimberidou AM, Wierda WG, Plunkett W, et al. Phase I-II study of oxaliplatin, fludara-bine, cytarabine, and rituximab combination therapy in patients with Richter’s syndrome or fludarabine-refractory chronic lymphocytic leukemia. J Clin Oncol. 2008;26(2):196–203. [DOI] [PubMed] [Google Scholar]
- 16.Tsimberidou AM, Wierda WG, Wen S, et al. Phase I-II clinical trial of oxaliplatin, flu-darabine, cytarabine, and rituximab therapy in aggressive relapsed/refractory chronic lymphocytic leukemia or Richter syndrome. Clin Lymphoma Myeloma Leuk. 2013;13(5):568–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Durot E, Michallet A-S, Leprêtre S, Le Q-H, Leblond V, Delmer A. Platinum and high-dose cytarabine-based regimens are efficient in ultra high/high-risk chronic lym-phocytic leukemia and Richter’s syndrome: results of a French retrospective multicenter study. European Journal of Haematology. 2015;95(2):160–167. [DOI] [PubMed] [Google Scholar]
- 18.Rogers KA, Huang Y, Ruppert AS, et al. A single-institution retrospective cohort study of first-line R-EPOCH chemoim-munotherapy for Richter syndrome demonstrating complex chronic lympho-cytic leukaemia karyotype as an adverse prognostic factor. Br J Haematol. 2018;180(2):259–266. [DOI] [PubMed] [Google Scholar]
- 19.Tsimberidou AM, O’Brien S, Khouri I, et al. Clinical outcomes and prognostic factors in patients with Richter’s syndrome treated with chemotherapy or chemoimmunother-apy with or without stem-cell transplantation. J Clin Oncol. 2006;24(15):2343–2351. [DOI] [PubMed] [Google Scholar]
- 20.Cwynarski K, van Biezen A, de Wreede L, et al. Autologous and allogeneic stem-cell transplantation for transformed chronic lymphocytic leukemia (Richter’s syndrome): a retrospective analysis from the chronic lymphocytic leukemia subcommittee of the chronic leukemia working party and lymphoma working party of the European Group for Blood and Marrow Transplantation. J Clin Oncol. 2012; 30(18):2211–2217. [DOI] [PubMed] [Google Scholar]
- 21.Kharfan-Dabaja MA, Kumar A, Stingo FE, et al. Allogeneic hematopoietic cell transplantation for Richter syndrome: a single-center experience. Clin Lymphoma Myeloma Leuk. 2018;18(1):e35–e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bounaix L, Nguyen S, Blaise D, et al. Allogeneic Hematopoietic Stem-Cell Transplantation for Patients with Richter’s Syndrome:the SFGM-TC Experience. Blood. 2018;132(Suppl 1):3457. [Google Scholar]
- 23.El-Asmar J, Kharfan-Dabaja MA. Hematopoietic cell transplantation for Richter syndrome. Biol Blood Marrow Transplant. 2016;22(11):1938–1944. [DOI] [PubMed] [Google Scholar]
- 24.Alizadeh AA, Eisen MB, Davis RE, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403(6769):503–511. [DOI] [PubMed] [Google Scholar]
- 25.Rosenwald A, Wright G, Chan WC, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med. 2002; 346(25):1937–1947. [DOI] [PubMed] [Google Scholar]
- 26.Hans CP, Weisenburger DD, Greiner TC, et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood. 2004;103(1):275–282. [DOI] [PubMed] [Google Scholar]
- 27.Scott DW, Wright GW, Williams PM, et al. Determining cell-of-origin subtypes of diffuse large B-cell lymphoma using gene expression in formalin-fixed paraffin-embedded tissue. Blood. 2014;123(8):1214–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Riedell PA, Smith SM. Double hit and double expressors in lymphoma: Definition and treatment. Cancer. 2018;124(24):4622–4632. [DOI] [PubMed] [Google Scholar]
- 29.Friedberg JW. How I treat double-hit lymphoma. Blood. 2017;130(5):590–596. [DOI] [PubMed] [Google Scholar]
- 30.Shanafelt TD, Jenkins G, Call TG, et al. Validation of a new prognostic index for patients with chronic lymphocytic leukemia. Cancer. 2009;115(2):363–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nabhan C, Chaffee KG, Slager SL, et al. Analysis of racial variations in disease characteristics, treatment patterns, and outcomes of patients with chronic lymphocytic leukemia. Am J Hematol. 2016;91(7):677–680. [DOI] [PubMed] [Google Scholar]
- 32.Eyre TA, Clifford R, Bloor A, et al. NCRI phase II study of CHOP in combination with ofatumumab in induction and maintenance in newly diagnosed Richter syndrome. Br J Haematol. 2016;175(1):43–54. [DOI] [PubMed] [Google Scholar]
- 33.Fidai S, Sukhanova M, Chiu BC-H, et al. <em>TP53</em> Aberrations By FISH in CLL and Complex Karyotype at Transformation Predict for Worse Outcome in Diffuse Large B-Cell Lymphoma - Richter Transformation: A Single Institution Series of 75 DLBCL-RT Cases. Blood. 2018;132(Suppl 1):2984. [Google Scholar]
- 34.Fan L, Wang L, Zhang R, et al. Richter transformation in 16 of 149 Chinese patients with chronic lymphocytic leukemia. Leuk Lymphoma. 2012;53(9):1749–1756. [DOI] [PubMed] [Google Scholar]
- 35.Fabbri G, Khiabanian H, Holmes AB, et al. Genetic lesions associated with chronic lymphocytic leukemia transformation to Richter syndrome. J Exp Med. 2013; 210(11):2273–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abrisqueta P, Delgado J, Alcoceba M, et al. Clinical outcome and prognostic factors of patients with Richter’s syndrome: retrospective multicenter study of the Spanish Chronic Lymphocytic Leukemia (CLL) Study Group (GELLC). Blood. 2017; 130(Suppl 1):2995. [Google Scholar]
- 37.Maddocks KJ, Ruppert AS, Lozanski G, et al. Etiology of Ibrutinib therapy discontinuation and outcomes in patients with chronic lymphocytic leukemia. JAMA Oncol. 2015;1(1):80–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jain P, Keating M, Wierda W, et al. Outcomes of patients with chronic lymphocytic leukemia after discontinuing ibrutinib. Blood. 2015;125(13):2062–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davids MS, Huang Y, Rogers KA, et al. Richter’s syndrome (RS) in patients with chronic lymphocytic leukemia (CLL) on novel agent therapy. J Clin Oncol. 2017; 35(15_suppl):7505. [Google Scholar]
- 40.Anderson MA, Tam C, Lew TE, et al. Clinicopathological features and outcomes of progression of CLL on the BCL2 inhibitor venetoclax. Blood. 2017; 129(25):3362–3370. [DOI] [PubMed] [Google Scholar]
- 41.Tsang M, Shanafelt TD, Call TG, et al. The efficacy of ibrutinib in the treatment of Richter syndrome. Blood. 2015; 125(10):1676–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hillmen P, Schuh A, Eyre TA, et al. Acalabrutinib monotherapy in patients with Richter transformation from the Phase 1/2 ACE-CL-001 clinical study. Blood. 2016;128(22):60.27222478 [Google Scholar]
- 43.Ding W, LaPlant BR, Call TG, et al. Pembrolizumab in patients with CLL and Richter transformation or with relapsed CLL. Blood. 2017;129(26):3419–3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Davids MS, Roberts AW, Seymour JF, et al. Phase I First-in-human Study of Venetoclax in patients with relapsed or refractory non-Hodgkin Lymphoma. J Clin Oncol. 2017; 35(8):826–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jain N, Ferrajoli A, Basu S, et al. A Phase II Trial of Nivolumab combined with Ibrutinib for patients with Richter transformation. Blood. 2018;132(Suppl 1):296. [Google Scholar]
- 46.Mato AR, Svoboda J, Luning Prak ET, et al. Phase I/II Study of Umbralisib (TGR-1202) in Combination with Ublituximab (TG-1101) and Pembrolizumab in patients with relapsed/refractory CLL and Richter’s transformation. Blood. 2018;132(Suppl 1):297. [Google Scholar]
- 47.Younes A, Brody J, Carpio C, et al. Safety and activity of ibrutinib in combination with nivolumab in patients with relapsed non-Hodgkin lymphoma or chronic lym-phocytic leukaemia: a phase 1/2a study. Lancet Haematol. 2019;6(2):e67–e78. [DOI] [PubMed] [Google Scholar]
- 48.Turtle CJ, Hay KA, Hanafi LA, et al. durable molecular Remissions in chronic lympho-cytic leukemia treated with CD19-specific chimeric antigen receptor-modified T cells afterfailure of Ibrutinib. J Clin Oncol. 2017; 35(26):3010–3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barrio S, Shanafelt TD, Ojha J, et al. Genomic characterization of high-count MBL cases indicates that early detection of driver mutations and subclonal expansion are predictors of adverse clinical outcome. Leukemia. 2017;31(1):170–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Popp HD, Flach J, Brendel S, et al. Accumulation of DNA damage and alteration of the DNA damage response in monoclonal B-cell lymphocytosis and chronic lymphocytic leukemia. Leuk Lymphoma. 2018;1–10. [DOI] [PubMed] [Google Scholar]