

Abstract
Background
Recent evidence has found that lncRNA small nucleolar RNA host gene 16 (SNHG16) was associated with cell carcinogenesis in NSCLC. Here, we further investigated the precise functions and mechanisms of SNHG16 in NSCLC progression.
Methods
The expression of SNHG16, microRNA (miR)‐520a‐3p and EPH Receptor A2 (EphA2) was measured using quantitative real‐time polymerase chain reaction and western blot, respectively. Cell proliferation was determined using 3‐(4, 5)‐dimethylthiahiazo (−z‐y1)‐3, 5‐di‐phenytetrazoliumromide (MTT) assay. The migrated and invaded cells were measured by Transwell assay. Flow cytometry was used to detect apoptotic cells. The interaction between miR‐520a‐3p and SNHG16 or EphA2 was confirmed using a dual‐luciferase reporter assay.
Results
We found that SNHG16 was upregulated in NSCLC tissues and cell lines, knockdown of SNHG16 inhibited cell proliferation, migration, invasion and induced apoptosis in vitro as well as suppressed tumor growth in vivo. MiR‐520a‐3p directly bound to SNHG16 and miR‐520a‐3p, and SNHG16 acted as a ceRNA in regulating EphA2 through competitively binding to miR‐520a‐3p. Additionally, rescue assay exhibited the anticancer activity mediated by SNHG16 knockdown on NSCLC could be reversed by miR‐520a‐3p inhibition or EphA2 overexpression.
Conclusion
SNHG16 promoted NSCLC development by regulating the miR‐520a‐3p/EphA2 axis, suggesting novel insights for the pathogenesis of NSCLC and new potential therapeutic targets for the treatment of NSCLC.
Key points
Knockdown of SNHG16 inhibited NSCLC cell proliferation, migration, invasion and induced apoptosis in vitro as well as suppressed tumor growth in vivo.
SNHG16 directly interacted with miR‐520a‐3p.
EphA2 was a target of miR‐520a‐3p.
SNHG16 could regulate the expression of EphA2 by binding to miR‐520a‐3p.
SNHG16 promoted NSCLC development by regulating the miR‐520a‐3p/EphA2 axis.
Keywords: Development, EphA2, miR‐520a‐3p, NSCLC, SNHG16
Introduction
Non‐small cell lung cancer (NSCLC) is one of the main subtypes of lung cancer, accounting for around 80%–85% of all lung cancers, and has a low five‐year survival rate of approximately 15%.1, 2 Although there has been an improvement in the diagnosis and multimodal therapy, the overall survival rate of NSCLC is still unsatisfactory due to metastasis and recurrence.3 Thus, there is a great demand for us to better understand the pathological mechanisms of NSCLC in order to develop novel effective therapeutic approaches for the improvement of outcome.
Long non‐coding RNAs (lncRNAs) are a class of nonprotein coding RNA molecules with the length over 200 nucleotides and have important effects on the regulation of gene expression via chromatin modification, transcription (or post‐transcriptional) and translational regulation4, 5 Increasing evidence has identified that lncRNAs are crucial contributors in regulating malignant physiological or pathological cellular processes, such as tumorigenesis, angiogenesis, drug resistance, and metastasis.6, 7 In addition, numerous studies have gradually revealed that deregulated lncRNAs are associated with the development of many cancers, including NSCLC. For example, Nie et al. revealed that lncRNA UCA1 acted as an oncogene to promote cell proliferation by regulating miR‐193a‐3p/ERBB4 in NSCLC.8 LncRNA HIT exerted oncogenic functions to promote NSCLC cell growth by interacting with E2F1.9 Thus, lncRNAs may be promising candidates for developing effective therapeutics of NSCLC. Among these lncRNAs, lncRNA small nucleolar RNA host gene 16 (SNHG16) recently was discovered to be upregulated in NSCLC and was associated with cell carcinogenesis in NSCLC.10 However, the precise mechanisms underlying the tumorigenesis role of SNHG16 in NSCLC remain largely unclear.
MicroRNAs (miRNAs) are one type of small non‐coding RNAs which can modulate gene expression at post‐transcriptional level in various cancers.11 In NSCLC, many miRNAs have been revealed to be involved in the cancer carcinogenesis by functioning as tumor suppressors or oncogenes, and miRNAs are potential diagnostic biomarkers and therapeutic targets in NSCLC.12, 13, 14 Recently, the important roles of the lncRNA‐miRNA‐mRNA regulatory network and a protein‐protein interaction network have also been identified in the development of NSCLC.15, 16 However, there are few studies to date which concentrate on the interaction network of SNHG16 in NSCLC.
This research aimed to explore the potential biological functions of SNHG16 in NSCLC cell carcinogenesis, investigate the interaction network of SNHG16, as well as how they affect NSCLC development. This study may provide novel therapeutic targets for the treatment of NSCLC.
Methods
Patients and specimens
Tumor specimens and adjacent nontumor tissues from 30 NSCLC patients who received surgical resection were obtained from Dalian University Affiliated Xinhua Hospital and were stored at −80°C for further analysis. All patients were diagnosed by histopathological examination and did not receive any preoperative treatment. This study was permitted by the Ethics Committee of Dalian University Affiliated Xinhua Hospital and all patients had signed their informed consents for inclusion in the study.
Cell culture and transfection
Human bronchial epithelial (16HBE) cell line and NSCLC cell lines (A549, NCI‐H292, NCI‐H460, NCI‐H1703) were obtained from Shanghai Academy of Life Science (Shanghai, China) and maintained in the Dulbecco's modifed Eagle's medium (DMEM; Gibco, Carlsbad, CA, USA) harboring with 10% fetal bovine serum and 1% penicillin/streptomycin (Gibco) with 5% CO2 at 37°C.
The miR‐520a‐3p mimic (miR‐520a‐3p), miR‐520a‐3p inhibitor (anti‐miR‐520a‐3p) and their corresponding negative control (miR‐NC or anti‐miR‐NC) were obtained from RIBOBIO (Guangzhou, China). The short hairpin RNA (shRNA) targeting SNHG16 (sh‐SNHG16), shRNA scramble control (sh‐NC), the small interfering RNA (siRNA) sequences targeting SNHG16 (si‐SNHG16), siRNA negative control (si‐NC), pcDNA, pcDNA SNHG16 overexpression vector (SNHG16), pcDNA EPH Receptor A2 (EphA2) overexpression vector (EphA2) were synthesized by Invitrogen (Carlsbad, CA, USA). All oligonucleotides or vectors were transfected into A549 and NCI‐H1703 cells using Lipofectamine 2000 (Invitrogen) following the standard protocol.
Quantitative real‐time polymerase chain reaction (qRT‐PCR)
TRIzol reagent (Invitrogen) was utilized to extract total RNA according to the standard procedure. For the detection of SNHG16 and EphA2, total RNA was reversely transcribed into complementary DNA (cDNA) using a high capacity cDNA Reverse Transcription Kit (Qiagen, Valencia, CA, USA), and quantitative PCR was then carried out with SYBR Premix Ex Taq (Qiagen). For miR‐520a‐3p detection, cDNA was synthesized by using miScript Reverse Transcription reagent (Qiagen) and amplified using SYBR Select Master Mix (Qiagen). Fold changes were analyzed by 2‐△△Ct method and normalized by glyceraldehyde 3‐phosphate dehydrogenase (GADPH) or U6 small nuclear B noncoding RNA (U6). The specific primer sequences were listed as follows: SNHG16: F, 5′‐GCAGAATGCCATGGTTTCCC‐3′, R, 5′‐GGACAGCTGGCAAGAGACTT‐3′; EphA2: 5′‐CTGGTCTGCAAGGTGTCTGA‐3′, R 5′‐TTGGACAACTCCCAGTAGGG‐3′; miR‐520a‐3p: F, 5′‐ACACTCCAGCTGGGAAAGTGCTTCCC‐3′, R, 5′‐CTCAACTGGTGTCGT GGA‐3′. GADPH: F 5′‐GATATTGTTGCCATCAATGAC‐3′, R 5′‐TTGATTTTGGAGGGATCTCG‐3′; U6: F, 5′‐CTCGCTTCGGCAGCACA‐3′ and R, 5′‐ACGCTTCACGAATTTGCGT‐3′.
Cell proliferation assay
Transfected cells were grown in the 96‐well plates (5000 cells/well) and incubated overnight. Each well was interacted with 10 μL 3‐(4,5‐Dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) solution (Sigma, St. Louis, MO, USA) for another four hours. After removing the supernatants, 150 μL DMSO was added to each well (Sigma). Finally, the absorbance was detected at 490 nm to plot cell proliferation curves.
Cell migration and invasion
For migrated cells detection, cells suspended in DMEM without serum were planted in the upper chambers and 500 μL DMEM mixed with 10% FBS was added to the lower chambers. After incubation for 24 hours at 37°C, migrated cells were then fixed with polyoxymethylene and stained with 0.1% crystal violet. Finally, cells in five randomly selected fields were counted using a microscope. For invaded cells detection, a matrigel invasion chamber (BD Biosciences, San Jose, CA, USA) was utilized and other measurements were similar to the steps of cell migration detection.
Cell apoptosis assay
Cell apoptosis was detected using Annexin V‐FITC/PI apoptosis detection kit (BD Biosciences) according to the standard protocol. Transfected cells were resuspended with binding buffer, followed by staining with 10 μL fluorescein isothiocyanate (FITC) annexin V and propidium iodide (PI). Finally, flow cytometry was used to detect apoptotic cells.
Dual‐luciferase reporter assay
The WT or MUT SNHG16/EphA2 3′UTR containing a miR‐520a‐3p binding sequence was amplified and cloned into the pmirGLO Vector (Promega, Shanghai, China). Then the construct vectors were transfected into A549 and NCI‐H1703 cells with miR‐520a‐3p or miR‐NC for 48 hours. Subsequently, a dual‐luciferase reporter assay kit (Promega) was used to detect the relative luciferase activity.
Western blot
Western blot assay was carried out as previously described.17 Several primary antibodies against EphA2 (1:500, ab5386, Abcam, Cambridge, MA, USA), β‐actin (1:1000, 4967, Cell Signaling Technology, Danvers, MA, USA) as well as HRP‐conjugated secondary antibody (1:1000; ab9482; Abcam) were used for the immunoblot assays. The protein signals were visualized by an ECL method.
Xenograft experiments in vivo
Athymic BALB/c mice (males, four weeks old) were used to establish xenograft models. A549 cells, stably infected with lentivirus containing sh‐SNHG16 or sh‐NC were subcutaneously inoculated into the flanks of the nude mice. The tumor size in the nude mice were detected and calculated every five days. After inoculation for 27 days, the tumor masses were excised and weighed. The study was permitted by the Animal Research Committee of Dalian University Affiliated Xinhua Hospital and undertaken following the guidelines of the National Animal Care and Ethics Institution.
Statistical analysis
Statistical data from three independent experiments were presented as the mean ± standard deviation (SD) and analyzed using GraphPad Prism 7 software (GraphPad Inc., San Diego, CA, USA). Significant differences were analyzed using one‐way analysis of variance (ANOVA) or Student's t‐test. The correlation analysis was analyzed using Spearman's correlation test. A value of P < 0.05 was considered statistically significant.
Results
SNHG16 is upregulated in NSCLC tissues and cell lines
SNHG16 expression was investigated to determine whether SNHG16 expression had changed in NSCLC tissues and cell lines. In comparison with nontumor tissues and 16HBE cell line, the results indicated that SNHG16 was significantly elevated in NSCLC tissues and cell lines (Fig 1a,b). Additionally, A549 and NCI‐H1703 cells were selected for the following experiments due to the evident upregulation.
Figure 1.
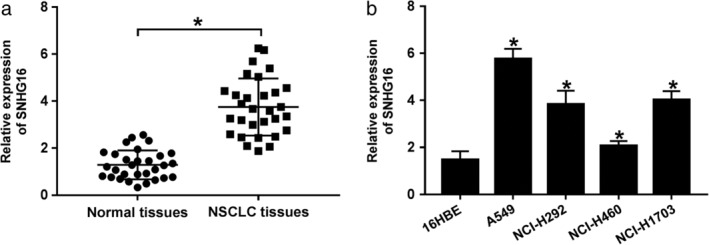
SNHG16 was upregulated in NSCLC tissues and cell lines. (a, b) The expression of SNHG16 was detected using qRT‐PCR in normal tissues, NSCLC tissues, NSCLC cell lines (A549, NCI‐H292, NCI‐H460, and NCI‐H1703), and human bronchial epithelial (16HBE) cell line. *P < 0.05.
SNHG16 silence inhibits proliferation, migration and invasion but induces apoptosis of NSCLC cells
To explore the potential biological functions of SNHG16 in NSCLC cells, SNHG16 was silenced using small interfering RNA (siRNA) sequences targeting SNHG16 and an obvious reduction of SNHG16 level in A549 and NCI‐H1703 cells was observed (Fig 2a). MTT assay showed SNHG16 silence inhibited cell proliferation in NSCLC (Fig 2b,c). Transwell assay subsequently indicated that knockdown of SNHG16 markedly suppressed migration and invasion of A549 and NCI‐H1703 cells (Fig 2d,e). Furthermore, we also found apoptotic cells were significantly increased by SNHG16 deletion in NSCLC (Fig 2f). Therefore, we demonstrated that SNHG16 silence restrained cell carcinogenesis in NSCLC.
Figure 2.
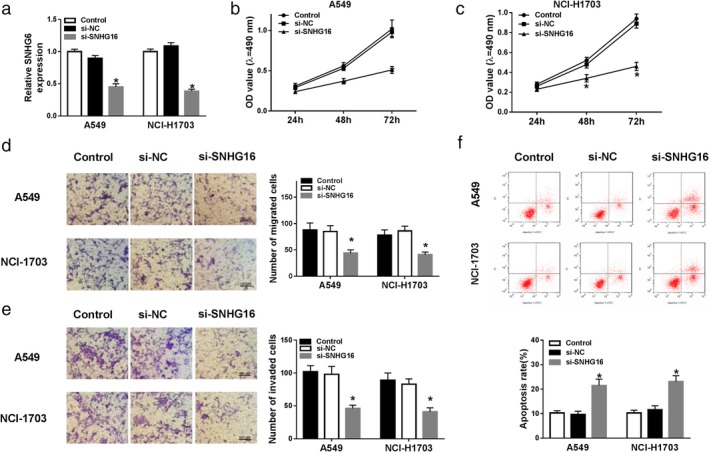
SNHG16 silence inhibited the proliferation, migration and invasion but induced apoptosis of NSCLC cells. SNHG16 was silenced using small interfering RNA sequences targeting SNHG16 in A549 and NCI‐H1703 cells. After transfection, (a) the expression of SNHG16 was measured by qRT‐PCR; (b, c) MTT assay was used to detect cell proliferation; (d, e) migrated and invaded cells were examined using transwell assay; (f) apoptotic cells were detected by flow cytometry. *P < 0.05.
SNHG16 silence exerts antitumor effects in NSCLC cells by directly sponging miR‐520a‐3p
To investigate the underlying mechanism of SNHG16 in the development of NSCLC, we used starBase v2.0 program to predict the potential miRNA targets of SNHG16 and miR‐520a‐3p was identified to have a binding site with SNHG16 (Fig 3a). After that, the dual‐luciferase reporter assay was conducted and miR‐520a‐3p overexpression reduced the luciferase activities of the WT‐SNHG16 reporter vector compared to the control group, while there was no obvious change in MUT‐SNHG16 reporter after overexpression of miR‐520a‐3p in A549 and NCI‐H1703 cells (Fig 3b,c), indicating SNHG16 directly bound to miR‐520a‐3p. Subsequently, the expression of miR‐520a‐3p was detected and we found miR‐520a‐3p was decreased in NSCLC tissues and cell lines (Fig 3d,e); in addition, a negative correlation between miR‐520a‐3p and SNHG16 was demonstrated in NSCLC tissues (Fig 3f), further proving SNHG16 was a sponge of miR‐520a‐3p. Moreover, qRT‐PCR analysis showed the expression of miR‐520a‐3p was promoted by SNHG16 knockdown, while this promotion could be attenuated by miR‐520a‐3p inhibition (Fig 3g). Taken together, SNHG16 directly interacted with miR‐520a‐3p and negatively regulated miR‐520a‐3p expression.
Figure 3.
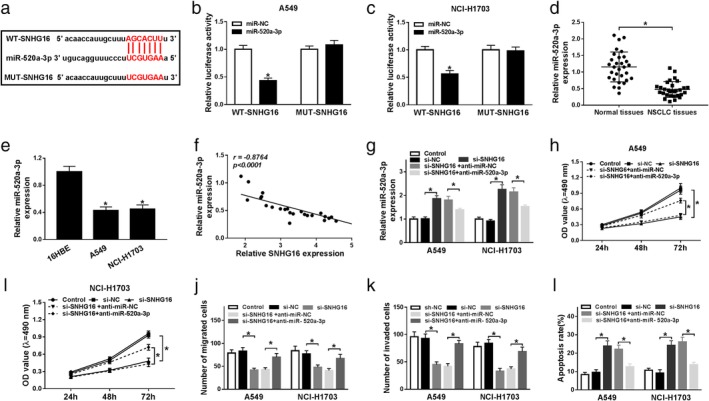
SNHG16 silence exerted antitumor effects in NSCLC cells by directly sponging miR‐520a‐3p. (a) A graphical representation of the miR‐520a‐3p binding site within SNHG16 is presented. (b, c) The interaction between miR‐520a‐3p and SNHG16 was confirmed using a dual‐luciferase reporter assay in A549 and NCI‐H1703 cells. (d, e) The expression of miR‐520a‐3p was detected in normal tissues and NSCLC tissues (d), as well as NSCLC cell lines (A549 and NCI‐H1703) and human bronchial epithelial (16HBE) cell line (e) using qRT‐PCR. (f) The correlation between miR‐520a‐3p and SNHG16 expression was assessed in NSCLC tissues by Spearman's correlation test. A549 and NCI‐H1703 cells were transfected with si‐NC, si‐SNHG16, si‐SNHG16 + anti‐miR‐NC, or si‐SNHG16 + anti‐miR‐520a‐3p. After transfection, (g) the level of miR‐520a‐3p was measured in A549 and NCI‐H1703 cells by qRT‐PCR; (h, i) MTT assay was used to detect cell proliferation; (j, k) cell migration and invasion were determined by transwell assay; (l) apoptotic cells were examined by flow cytometry. *P < 0.05.
Based on the relationship between SNHG16 and miR‐520a‐3p, we further analyzed whether miR‐520a‐3p participated in SNHG16 deletion mediated regulation on cell tumorigenesis in NSCLC. Thus, rescue assay was conducted and the results suggested that miR‐520a‐3p inhibition could reverse SNHG16 silence induced inhibition on the proliferation (Fig 3h,i), migration and invasion (Fig 3j,k), promotion on the apoptosis (Fig 3l) of A549 and NCI‐H1703 cells. These data demonstrated that SNHG16 silence inhibited NSCLC cells carcinogenesis by directly interacting with miR‐520a‐3p.
EphA2 is a target of miR‐520a‐3p
We further investigated the molecular mechanisms by which the SNHG16/miR‐520a‐3p axis regulated NSCLC cell carcinogenesis, and the potential target genes of miR‐520a‐3p were identified using starBase2.0 software and EphA2 was found to contain the putative binding site of miR‐520a‐3p (Fig 4a). Subsequently, the dual‐luciferase reporter assay results suggested that miR‐520a‐3p mimic reduced the luciferase activities of the EphA2‐WT reporter vector, but not mutant reporter vector in A549 and NCI‐H1703 cells (Fig 4b,c), indicating the direct interaction between miR‐520a‐3p and EphA2. Synchronously, we detected the level of EphA2 and found the mRNA and protein levels of EphA2 were significantly elevated in NSCLC tissues and cell lines (Fig 4d–g). In addition, co‐expression analysis exhibited that SNHG16 and EphA2 expression was positively correlated (Fig 4h), moreover, western blot results indicated the protein expression of EphA2 was inhibited by miR‐520a‐3p restoration, but this inhibition could be rescued by following SNHG16 overexpression (Fig 4i,j). Thus, these data suggested EphA2 was a target of miR‐520a‐3p and regulated by SNHG16 through miR‐520a‐3p.
Figure 4.
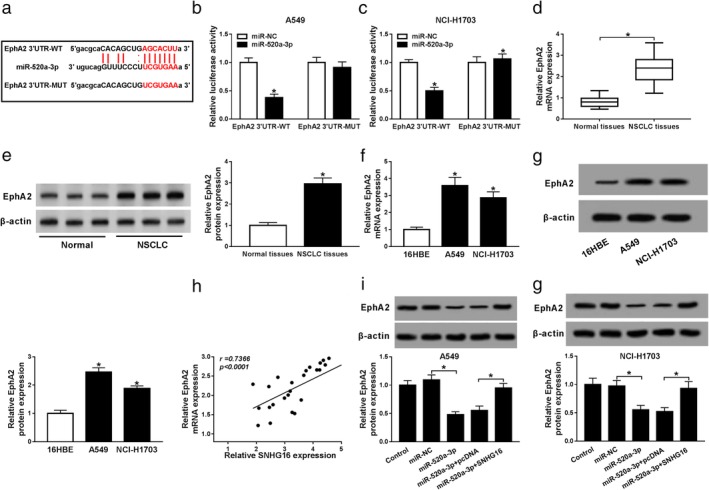
EphA2 is a target of miR‐520a‐3p. (a) The binding site between miR‐520a‐3p and EphA2 is shown. (b, c) The interaction between EphA2 and miR‐520a‐3p was analyzed using a dual‐luciferase reporter assay in A549 and NCI‐H1703 cells. (d–g) The expression of EphA2 was measured in normal tissues and NSCLC tissues (d, e), as well as NSCLC cell lines (A549 and NCI‐H1703) and human bronchial epithelial (16HBE) cell line (f, g) using qRT‐PCR and western blot, respectively. (h) The correlation between SNHG16 and EphA2 expression in NSCLC tissues was assessed by Spearman's correlation test. (i, j) The level of EphA2 was detected in A549 and NCI‐H1703 cells transfected with miR‐NC, miR‐520a‐3p, miR‐520a‐3p + pcDNA, or miR‐520a‐3p + SNHG16 using western blot. *P < 0.05.
SNHG16 silence inhibits cell carcinogenesis in NSCLC by regulating EphA2 expression
Based on the above results, we determined that SNHG16 functioned as a ceRNA in regulating EphA2 through competitively binding to miR‐520a‐3p. Therefore, we wanted to find out whether EphA2 was implicated in SNHG16 deletion mediated antitumor effects. First, A549 and NCI‐H1703 cells were transfected with pcDNA or EphA2, and we found the level of EphA2 was obviously elevated in A549 and NCI‐H1703 cells after transfection (Fig 5a,b). Next, rescue experiments were performed and results confirmed that overexpressed EphA2 could partially overturn SNHG16 deletion‐induced repression on proliferation (Fig 5c,d), migration and invasion (Fig 5e,f), enhancement on apoptosis (Fig 5g) of A549 and NCI‐H1703 cells. Therefore, SNHG16 silence inhibited cell carcinogenesis in NSCLC by regulating EphA2 expression.
Figure 5.
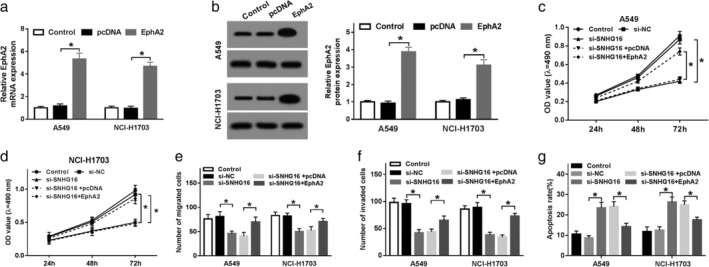
SNHG16 silence inhibited cell carcinogenesis in NSCLC by regulating EphA2 expression. (a, b) The mRNA (a) and protein (b) expression levels of EphA2 were detected in A549 and NCI‐H1703 cells transfected with pcDNA or EphA2 using qRT‐PCR and western blot, respectively. A549 and NCI‐H1703 cells were transfected with si‐NC, si‐SNHG16, si‐SNHG16 + pcDNA, or si‐SNHG16 + EphA2. After transfection, (c, d) MTT assay was used to detect cell proliferation; (e, f) cell migration and invasion were determined by transwell assay; (g) cell apoptosis was analyzed by flow cytometry. *P < 0.05.
Knockdown of SNHG16 inhibits NSCLC tumor growth in vivo
To further uncover the carcinogenesis roles of SNHG16 in vivo, xenograft models were established using A549 cells infected with lentivirus containing sh‐SNHG16 or sh‐NC. As presented in Fig 6a,b, we found knockdown of SNHG16 inhibited NSCLC tumor growth, reflected by the suppression of tumor volume and weight in sh‐SNHG16 group. In addition, molecular analysis showed SNHG16 silence remarkably reduced the expression of SNHG16 and EphA2, but increased miR‐520a‐3p level in the sh‐SNHG16 group compared with sh‐NC group (Fig 6c–e). Altogether, these results indicated that knockdown of SNHG16 might inhibit NSCLC tumor growth in vivo by regulating miR‐520a‐3p and EphA2 expression.
Figure 6.
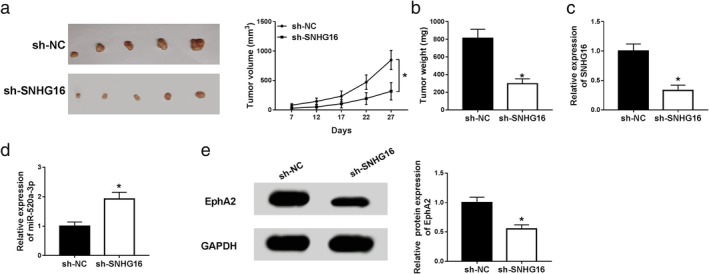
Knockdown of SNHG16 inhibited NSCLC tumor growth in vivo. Xenograft models were established using A549 cells infected with lentivirus containing sh‐SNHG16 or sh‐NC. (a) Tumor volume was calculated every five days. (b) Tumor masses were weighed in each group. (c, d) The levels of SNHG16, miR‐520a‐3p and EphA2 were measured in two groups by qRT‐PCR or western blot. *P < 0.05.
Discussion
To date, thousands of lncRNAs have been widely established in mammalian genomes. LncRNAs play critical roles in a wide range of biological processes and whose deregulation contribute to cell carcinogenesis of human cancers.18, 19 For example, lncRNA UICLM functioned as an oncogene to induce colorectal cancer liver metastasis through regulating miR‐215/ZEB2 axis.20 lncRNA NEAT1 contributed to cell proliferation and epithelial‐mesenchymal transition (EMT) led to breast cancer progression.21 In addition, there is also increasing evidence that lncRNA SNHG16 participates in the regulation of tumorigenesis in many types of cancers. For example, in the study by Cao et al. SNHG16 contributed to bladder cancer cell proliferation by silencing p21 expression and was associated with poor prognosis.22 SNHG16 exerted oncogenic function in glioma by regulating miR‐4518/PRMT5 regulatory axis to promote cell viability and inhibit cell apoptosis in the report by Lu et al.23 Additionally, Xu et al. found SNHG16 performed antitumor effects by inhibiting cell proliferation and chemoresistance in hepatocellular carcinoma.24 Thus, it is necessary for us to detect the roles of SNHG16 in NSCLC. In this study, SNHG16 was found to be upregulated in NSCLC tissues and cells. Functional experiments showed knockdown of SNHG16 inhibited cell proliferation, migration and invasion but induced cell apoptosis in NSCLC. In addition, in vivo experiments also indicated SNHG16 deletion suppressed tumor growth. Therefore, we conclude that SNHG16 is a cell carcinogenesis regulator in NSCLC.
LncRNAs contain multiple miRNA‐binding sites and can serve as sponges of miRNA to regulate miRNAs activity as well as the expression of their downstream target genes.25, 26 Here, we further investigated the miRNA targets of SNHG16 and miR‐520a‐3p was confirmed to be a target of SNHG16. Previous studies have indicated miR‐520a‐3p was decreased and functioned as a tumor suppressor to suppress cell growth and tumorigenesis in diverse human cancers, such as colorectal cancer, breast cancer, gastric cancer as well as NSCLC.27, 28, 29, 30 In this study, we also found miR‐520a‐3p was downregulated in NSCLC tissues and cell lines, and in addition a negative correlation between miR‐520a‐3p and SNHG16 was observed. Subsequently, rescue assay was conducted and results showed SNHG16 silence exerted antitumor effects in NSCLC cells by directly sponging miR‐520a‐3p. Thus, a SNHG16/miR‐520a‐3p regulatory axis in the development of NSCLC was identified.
It is well recognized that transmembrane protein receptors are implicated in the prognosis of various tumors and are more and more critical in cancer treatment.31 EphA2 is one of the protein‐tyrosine kinases that is elevated in almost 90% of NSCLC cell membranes but not in normal lung tissues and cells.32 Overexpressed EphA2 has been found to be related to the poor clinical outcome and mediate cell proliferation and motility in NSCLC.33 In addition, EphA2‐targeted therapy has also been identified to be an effective therapeutic strategy for NSCLC treatment.34, 35 In this study, EphA2 was predicted and proved to be a target of miR‐520a‐3p. Subsequently, miR‐520a‐3p/EphA2 regulatory axis was confirmed using a dual‐luciferase reporter assay and co‐expression analysis. EphA2 was discovered to be overexpressed in NSCLC tissues and cell lines, and overexpressed EphA2 was positively correlated with SNHG16. Thus, we further investigated the relationship among SNHG16, miR‐520a‐3p and EphA2, and the results showed SNHG16 could regulate EphA2 expression by binding to miR‐520a‐3p. Furthermore, rescue assay indicated SNHG16 silence inhibited cell carcinogenesis in NSCLC by regulating EphA2 expression.
In conclusion, this study demonstrated SNHG16 was upregulated in NSCLC tissues and cell lines, and SNHG16 deletion inhibited cell carcinogenesis in NSCLC. Additionally, a SNHG16/miR‐520a‐3p/EphA2 regulatory axis was identified in NSCLC, and SNHG16 silence exerted antitumor effects on NSCLC cells by regulating miR‐520a‐3p/EphA2 axis, indicating new potential therapeutic targets for NSCLC treatment.
Disclosure
The authors declare that they have no conflicts of interest.
References
- 1. Goldstraw P, Ball D, Jett JR et al Non‐small‐cell lung cancer. Lancet 2011; 378: 1727–40. [DOI] [PubMed] [Google Scholar]
- 2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017; 67: 7–30. [DOI] [PubMed] [Google Scholar]
- 3. Rosell R, Bivona TG, Karachaliou N. Genetics and biomarkers in personalisation of lung cancer treatment. Lancet 2013; 382: 720–31. [DOI] [PubMed] [Google Scholar]
- 4. Ren K, Li Y, Lu H et al Long noncoding RNA HOTAIR controls cell cycle by functioning as a competing endogenous RNA in esophageal squamous cell carcinoma. Transl Oncol 2016; 9: 489–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bhan A, Soleimani M, Mandal SS. Long noncoding RNA and cancer: A new paradigm. Cancer Res 2017; 77: 3965–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Peng WX, Koirala P, Mo YY. LncRNA‐mediated regulation of cell signaling in cancer. Oncogene 2017; 36: 5661–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kumar MM, Goyal R. LncRNA as a therapeutic target for angiogenesis. Curr Top Med Chem 2017; 17: 1750–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nie W, Ge HJ, Yang XQ et al LncRNA‐UCA1 exerts oncogenic functions in non‐small cell lung cancer by targeting miR‐193a‐3p. Cancer Lett 2016; 371: 99–106. [DOI] [PubMed] [Google Scholar]
- 9. Yu L, Fang F, Lu S, Li X, Yang Y, Wang Z. lncRNA‐HIT promotes cell proliferation of non‐small cell lung cancer by association with E2F1. Cancer Gene Ther 2017; 24: 221–6. [DOI] [PubMed] [Google Scholar]
- 10. Han W, Du X, Liu M, Wang J, Sun L, Li Y. Increased expression of long non‐coding RNA SNHG16 correlates with tumor progression and poor prognosis in non‐small cell lung cancer. Int J Biol Macromol 2019; 121: 270–8. [DOI] [PubMed] [Google Scholar]
- 11. Jiang C, Hu X, Alattar M, Zhao H. miRNA expression profiles associated with diagnosis and prognosis in lung cancer. Expert Rev Anticancer Ther 2014; 14: 453–61. [DOI] [PubMed] [Google Scholar]
- 12. Florczuk M, Szpechcinski A, Chorostowska‐Wynimko J. miRNAs as biomarkers and therapeutic targets in non‐small cell lung cancer: Current perspectives. Target Oncol 2017; 12: 179–200. [DOI] [PubMed] [Google Scholar]
- 13. Zhou Q, Huang SX, Zhang F et al MicroRNAs: A novel potential biomarker for diagnosis and therapy in patients with non‐small cell lung cancer. Cell Prolif 2017; 50: e12394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yin Z, Xu M, Li P. miRNA‐221 acts as an oncogenic role by directly targeting TIMP2 in non‐small‐cell lung carcinoma. Gene 2017; 620: 46–53. [DOI] [PubMed] [Google Scholar]
- 15. Wang X, Yin H, Zhang L et al The construction and analysis of the aberrant lncRNA‐miRNA‐mRNA network in non‐small cell lung cancer. J Thorac Dis 2019; 11: 1772–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li DY, Chen WJ, Luo L et al Prospective lncRNA‐miRNA‐mRNA regulatory network of long non‐coding RNA LINC00968 in non‐small cell lung cancer A549 cells: A miRNA microarray and bioinformatics investigation. Int J Mol Med 2017; 40: 1895–906. [DOI] [PubMed] [Google Scholar]
- 17. Chen G, Shi Y, Zhang Y, Sun J. CircRNA_100782 regulates pancreatic carcinoma proliferation through the IL6‐STAT3 pathway. Onco Targets Ther 2017; 10: 5783–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Khandelwal A, Bacolla A, Vasquez KM, Jain A. Long non‐coding RNA: A new paradigm for lung cancer. Mol Carcinog 2015; 54: 1235–51. [DOI] [PubMed] [Google Scholar]
- 19. Hu Y, Ma Z, He Y, Liu W, Su Y, Tang Z. LncRNA‐SNHG1 contributes to gastric cancer cell proliferation by regulating DNMT1. Biochem Biophys Res Commun 2017; 491: 926–31. [DOI] [PubMed] [Google Scholar]
- 20. Chen DL, Lu YX, Zhang JX. Long non‐coding RNA UICLM promotes colorectal cancer liver metastasis by acting as a ceRNA for microRNA‐215 to regulate ZEB2 expression. Theranostics 2017; 7: 4836–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang M, Wu WB, Wang ZW, Wang XH. lncRNA NEAT1 is closely related with progression of breast cancer via promoting proliferation and EMT. Eur Rev Med Pharmacol Sci 2017; 21: 1020–6. [PubMed] [Google Scholar]
- 22. Cao X, Xu J, Yue D. LncRNA‐SNHG16 predicts poor prognosis and promotes tumor proliferation through epigenetically silencing p21 in bladder cancer. Cancer Gene Ther 2018; 25: 10–7. [DOI] [PubMed] [Google Scholar]
- 23. Lu YF, Cai XL, Li ZZ et al LncRNA SNHG16 functions as an oncogene by sponging MiR‐4518 and up‐regulating PRMT5 expression in glioma. Cell Physiol Biochem 2018; 45: 1975–85. [DOI] [PubMed] [Google Scholar]
- 24. Xu F, Zha G, Wu Y, Cai W, Ao J. Overexpressing lncRNA SNHG16 inhibited HCC proliferation and chemoresistance by functionally sponging hsa‐miR‐93. Onco Targets Ther 2018; 11: 8855–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Militello G, Weirick T, John D, Doring C, Dimmeler S, Uchida S. Screening and validation of lncRNAs and circRNAs as miRNA sponges. Brief Bioinform 2017; 18: 780–8. [DOI] [PubMed] [Google Scholar]
- 26. Jalali S, Bhartiya D, Lalwani MK, Sivasubbu S, Scaria V. Systematic transcriptome wide analysis of lncRNA‐miRNA interactions. PLOS One 2013; 8: e53823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li J, Wei J, Mei Z et al Suppressing role of miR‐520a‐3p in breast cancer through CCND1 and CD44. Am J Transl Res 2017; 9: 146–54. [PMC free article] [PubMed] [Google Scholar]
- 28. Lv X, Li CY, Han P, Xu XY. MicroRNA‐520a‐3p inhibits cell growth and metastasis of non‐small cell lung cancer through PI3K/AKT/mTOR signaling pathway. Eur Rev Med Pharmacol Sci 2018; 22: 2321–7. [DOI] [PubMed] [Google Scholar]
- 29. Su H, Ren F, Jiang H, Chen Y, Fan X. Upregulation of microRNA‐520a‐3p inhibits the proliferation, migration and invasion via spindle and kinetochore associated 2 in gastric cancer. Oncol Lett 2019; 18: 3323–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang R, Liu R, Liu C et al A novel role for MiR‐520a‐3p in regulating EGFR expression in colorectal cancer. Cell Physiol Biochem 2017; 42: 1559–74. [DOI] [PubMed] [Google Scholar]
- 31. Herbst RS. Review of epidermal growth factor receptor biology. Int J Radiat Oncol Biol Phys 2004; 59: 21–6. [DOI] [PubMed] [Google Scholar]
- 32. Brannan JM, Sen B, Saigal B et al EphA2 in the early pathogenesis and progression of non‐small cell lung cancer. Cancer Prev Res (Phila) 2009; 2: 1039–49. [DOI] [PubMed] [Google Scholar]
- 33. Song W, Ma Y, Wang J, Brantley‐Sieders D, Chen J. JNK signaling mediates EPHA2‐dependent tumor cell proliferation, motility, and cancer stem cell‐like properties in non‐small cell lung cancer. Cancer Res 2014; 74: 2444–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lee HY, Mohammed KA, Kaye F et al EphA2 targeted intratumoral therapy for non‐small cell lung cancer using albumin mesospheres. Am J Transl Res 2017; 9: 3293–303. [PMC free article] [PubMed] [Google Scholar]
- 35. Li N, Liu S, Sun M et al Chimeric antigen receptor‐modified T cells redirected to EphA2 for the immunotherapy of non‐small cell lung cancer. Transl Oncol 2018; 11: 11–7. [DOI] [PMC free article] [PubMed] [Google Scholar]