

Abstract
Activated pancreatic stellate cells (PSCs) have been widely accepted as a key precursor of excessive pancreatic fibrosis, which is a crucial hallmark of chronic pancreatitis (CP) and its formidable associated disease, pancreatic cancer (PC). Hence, anti-fibrotic therapy has been identified as a novel therapeutic strategy for treating CP and PC by targeting PSCs. Most of the anti-fibrotic agents have been limited to phase I/II clinical trials involving vitamin analogs, which are abundant in medicinal plants and have proved to be promising for clinical application. The use of phytomedicines, as new anti-fibrotic agents, has been applied to a variety of complementary and alternative approaches. The aim of this review was to present a focused update on the selective new potential anti-fibrotic agents, including curcumin, resveratrol, rhein, emodin, green tea catechin derivatives, metformin, eruberin A, and ellagic acid, in combating PSC in CP and PC models. It aimed to describe the mechanism(s) of the phytochemicals used, either alone or in combination, and the associated molecular targets. Most of them were tested in PC models with similar mechanism of actions, and curcumin was tested intensively. Future research may explore the issues of bioavailability, drug design, and nano-formulation, in order to achieve successful clinical outcomes with promising activity and tolerability.
Key words: Pancreatic stellate cells, Anti-fibrotic, Chronic pancreatitis, Pancreatic cancer, Phytochemicals, Curcumin, Resverastrol, Rhein, Emodin, Green tea catechin
Graphical abstract
Quiescent PSCs are converted to their activated form by the actions of lymphocyte, macrophage and/or via autocrine stimulation. Our selective phytochemicals treat chronic pancreatitis and pancreatic cancer by targeting PSCs via the suppression of the same signaling pathways, including ERK1/2, P38 MAPK, SHH signaling and PI3K/Akt.

1. Introduction
Pancreatic stellate cells (PSCs), which are star-shaped fibroblasts, were only identified and characterized 20 years ago, despite research on stellate cells having begun in the eighteenth century1,2. PSCs are responsible for the synthesis and degradation of extracellular matrix (ECM) proteins, such as tissue inhibitors, matrix metalloproteinases (TIMPs), and metalloproteinases (MMPs). Thus, PSCs can regulate the pancreatic tissue functions and maintain the normal architecture of the pancreas by balancing fibrogenesis and the matrix degradation process3. They comprise about 4% of the local cells in the pancreas and are found in the periacinar and interlobular spaces4. Furthermore, they play a pivotal role in the development of a desmoplastic reaction (a reaction associated with tumors that is characterized by the growth of dense fibrous or connective tissues around a tumor), which is the hallmark of chronic pancreatitis (CP) and pancreatic cancer (PC)4.
Quiescent PSCs are activated by pancreatic injury or inflammation to become myofibroblast-like cells, expressing alpha-smooth muscle actin (α-SMA) and various ECM proteins, growth factors, and cytokines through structural and functional changes2. Pancreatic damage and inflammation could expose PSCs to a variety of soluble factors, which act as regulators of PSCs activation, as evidenced by several in vitro studies. These factors are interleukin-1 (IL-1), IL-6, tumor necrosis factor-alpha (TNF-α), platelet-derived growth factor (PDGF), transforming growth factor (TGF-β1), activin A, and ethanol and its metabolites, which cause oxidative stress and pressure, as well as extensive changes in the composition and components of ECM5, 6, 7, 8, 9, 10, 11, 12, 13. Continuous PSC activation occurs if the inflammation and injury are perpetuated; for example, if the inflammation and injury are limited, PSCs might proceed to apoptosis or deactivate, so that fibrosis will not develop. Repeated and persistent pancreatic injury and inflammation are key to the initiation of fibrogenesis2. Furthermore, cigarette smoke components (cigarette smoke extract (CSE) and/or nicotine (NNK)) in clinically relevant concentrations can activate PSCs, as evidenced by increased migration, proliferation, and collagen production in the presence or absence of ethanol. Ultimately, these findings suggest that the development of alcoholic pancreatic fibrosis could be caused by the combined effects of alcohol and cigarette smoke components via PSCs' mediation14. The comparative roles of PSCs in PC progression are clear. PSCs possess adequate capacity to interact with cancer cells and other stromal cells in order to multiply the stromata and promote the cancer progression. Activated PSCs play important roles in PC, including producing ECM proteins and regulating the formation of desmoplastic reaction, as well as promoting cancer cell proliferation, migration, invasion, angiogenesis, and chemoresistance15. In addition, PSCs stimulate angiogenesis, which is important for tumor growth and metastasis, disruption of the antitumor immune system, and indirect induction of immune cell dysfunction15, causing conventional chemotherapy resistance and severe treatment failure. Furthermore, chemoresistance in PC cells is caused by various molecular mechanisms, including epigenetics, post-translational modifications, altered key signaling pathways, epithelial–mesenchymal transition (EMT), and the involvement of cancer stem cells and the cellular and non-cellular components of the tumor microenvironment16,17. Survival rates in PC are only minimally increased, due to the poor responsiveness of pancreatic tumors to chemotherapy and radiation therapy, affected and regulated by the molecular targets (e.g., mucin 1 (MUC1))18,19. Furthermore, PSCs’ role in radiotherapy resistance, by activating the integrin-focal adhesion kinase (FAK) signaling in PC cells, has been confirmed20.
Targeting stroma cells, particularly PSCs, in conjunction with chemotherapy has become the focus of current PC treatment and the two interventions may reveal potent anticancer activities when they are used concurrently. The current chemotherapeutic regimes for CP and PC remain inadequate, and the more recent treatment protocols under trial are costly and toxic15. Therefore, it is urgently necessary to search for, and identify, an alternative, more economical treatment with lower toxicity to replace the current treatment. Phytochemicals have received increasing interest in the past decade, largely owing to their effectiveness for disease prevention and treatment21. The use of natural products, particularly plant-based remedies, is favored for combating fibrotic-induced diseases, compared to other complementary and alternative approaches; for example, Japanese herbal medicine (Saiko-keishi-to), polyphenol compounds (curcumin), antioxidants (vitamins A and E), protease inhibitor (camostat mesylate), and lovastatin (Monascus or Aspergillus-fermented rice and Dioscorea) are plant-derived pharmacological agents that have been shown to prevent and/or improve CP and PC3,22, 23, 24. In recent years, plant-derived products have undergone clinical trials to evaluate their efficacy as anti-fibrotic agents in the treatment of metastatic PC. Specifically, analogs of vitamins A and D have been claimed as potential anti-fibrotic agents against PSCs; for example, paricalcitol, a synthetic vitamin D analog, is undergoing Phase I and II clinical trials, in combination with different conventional chemotherapeutic drugs, for treating metastatic PC15. Other potential PC treatments in clinical settings involve reprogramming PSCs using vitamin A metabolites, such as all-trans retinoic acid or selective retinoic acid receptor beta (RAR-β) agonist15. In addition, combinations of pirfenidone and N-acetylcysteine, or the use of pirfenidone alone, with regard to PC warrant more extensive studies in human subjects25. The evidence has shown that plant-derived products have significant potential to act as anti-fibrotic agents in treating CP and PC and deserve intensive investigation using in vitro and in vivo models.
This paper presents a detailed review of the anti-fibrotic activity of selective potential phytochemicals, which are new and effective in treating CP and PC, by focusing on the PSCs evidenced in in vitro and in vivo models. Furthermore, it discusses the mechanism(s) that underlie the anti-fibrotic activity, the key molecules involved, and the concentrations used in the CP and PC models.
2. Pancreatic stellate cells
PSCs are the pluripotent cells, located between the pancreatic lobules and the surrounding area of acinar, that maintain the connective tissue architecture26. PSCs have two phenotypes: quiescent and activated. In a normal human pancreas, PSCs comprise approximately 4%–7% of the parenchymal cells and contain cytoplasmic lipid droplets containing vitamin A in its quiescent form27. Under normal physiological conditions, PSCs maintain their quiescence by expressing nestin, vimentin, glial fibrillary acidic protein (GFAP), and desmin. Furthermore, retinoids, sometimes in the form of retinyl palmitate, can be found in the cytosolic droplets of quiescent PSCs. These retinoids can be used as markers to differentiate them from the normal fibroblasts26.
The activation of PSCs can be induced by pathologic conditions, such as CP and PC; hence, activated PSCs are responsible for the excessive fibrotic state in pancreatic pathology28. The inactive PSCs are identified by the abundant vitamin A stored in the cytoplasm, while an injured pancreas lacks cytoplasmic vitamin A-storing lipid droplets. Activated PSCs have been identified using a variety of phenotypic parameters. They were found to be localized interlobularly in fibrotic areas adjacent to the carcinoma cells. Activated PSCs display a loss of fat droplets and a high mitotic index, with intense reticular endoplasmic reticulum (ER) and high motility and contraction. Receptors, such as platelet-derived growth factor receptor (PDGF-R), transforming growth factor beta receptor (TGFβ-R), and intercellular adhesion molecules (ICAM-1), are expressed by the activated PSCs, along with other receptors. Additionally, there is an enhanced expression of ECM proteins (collagens I, III and XI, fibronectin, and periostin (a cell adhesion protein) that stimulates cancer cell growth), which forms the fibrous tissue, and an enhanced release of neurotrophic factors/transmitter, growth factors, and cytokines29. Furthermore, activated PSCs also exhibit differential expression of multiple genes, including a 32.25-fold up-regulation of MMP-3 and a 2.25-fold down-regulation of the basement membrane component, collagen type IV-α1, which may contribute to restructuring of the ECM in the activated form29.
The ECM is composed of collagen, fibronectin, and multiple soluble factors secreted by PSCs, which provide the structural support and promote differentiation, remodeling, and carcinogenesis27. Ben-Harosh et al.30 reported that palmitate fatty acids significantly limited PSCs' activation and fibrosis by halting their proliferation and migration capacity via the suppression of ER. By contrast, PSCs’ activation and differentiation were augmented following the treatment of oleate fatty acids and caerulein-induced stress with increased levels of ER stress markers (X-box binding protein 1 (Xbp1) and CCAAT-enhancer-binding protein homologous protein (CHOP)). Fibrous proteins, such as collagens, laminin, and fibronectin, and non-collagenous proteins, such as glycoproteins, proteoglycans, and glycosaminoglycans, together make-up the ECM constituting the stromal component. This abundant stromal reaction usually surrounds the island of cancer cells and accounts for 50%–80% of tumor volume31.
The cytokines and growth factors secreted by PSCs promote the angiogenesis and the proliferation, migration, and invasion of the epithelial cancer cells that lead to metastasis32, 33, 34. Soluble factors, especially IL-6, have been shown to be involved in transitioning non-invasive into invasive pancreatic ductal adenocarcinoma (PDAC)35,36. Apparently, PDAC is among the stroma-rich and fibrotic malignancies, leading to the conclusion that the ECM process plays a key role in the development of fibrosis and PDAC progression. It is characterized by the formation of dense fibrotic stromata (desmoplasia), formed by the activated PSCs26. Furthermore, the continuous interaction between PSCs and the ECM can lead to further PSC activation due to the resulting stiffness of the matrix formation. Such a process is fundamental to pathological fibrosis in both CP and PC37.
3. Chronic pancreatitis
CP is a fibro-inflammatory disease that causes the pancreatic parenchyma to be progressively replaced by fibrous connective tissue, potentially leading to exocrine and endocrine pancreatic inadequacy38. Continuing pancreatic damage is caused by oxidative stress or recurrent episodes of inflammation, leading to irreversible functional and morphological changes in the pancreas, which may or not be clinically evident, and resulting in the development of CP39,40. The activation of digestive enzymes in pancreatitis occurs before they are released into the small intestine and cause glandular damage, leading to CP by inducing a progressive, destructive inflammatory process that ends in the destruction of the pancreas41. The clinical presentation of CP includes abdominal pain, steatorrhea, diabetes, weight loss, and obstructive jaundice, all of which are very similar to PDAC40.
Nearly 70% of CP cases are caused by alcohol abuse, and the remaining cases are attributed to genetic disorders, pancreatic duct obstruction, recurrent acute pancreatitis (AP), autoimmune pancreatitis, or unknown mechanisms42. Laboratory studies have highlighted a proliferation of reactive oxygen species (ROS) as the trigger and potentiator of inflammation, since they activate the signaling cascades that convert the damaged acinar cells into a production site for chemokines and cytokines. ROS have several physiological roles, including signal transduction, but an excess of ROS relative to the antioxidant capacity (electrophilic stress) is potentially harmful. Depending on the concentration, ROS facilitate mild to moderate levels of carcinogenesis and cancer progression, while the excessive ROS damage to cancer cells is dramatic and leads to cell death. Increased ROS are observed in CP, which increases the PC incidents via PSCs’ activation43. Furthermore, the exocytosis blockade seems to be caused by the disruption of the methionine trans-sulphuration pathway that produces the essential methyl and thiol moieties. This condition occurs in both clinically acute and acute-on-chronic pancreatitis44. When this physiological process is disrupted, it may result in the development of pathological fibrosis, with significant adverse effects on the anatomy and physiology of affected tissues. Abnormal deposition of fibrous tissue is a characteristic histological feature of two major diseases of the pancreas: CP and PC45. Fibrosis is a sign that interstitial PSCs have been activated in CP, due to the increase in lipid peroxidation products and the release of mast cell degranulation products:44 hence, it is evident that PSCs are associated with CP, and that targeting PSCs maybe a promising treatment option for CP.
4. Pancreatic cancer
By 2030, it is expected that PC will be the second leading cause of deaths from types of cancer46. Deaths result from PDAC, the major and most aggressive type of PC, and rank fourth among the cancer-related deaths in USA. PC is more prevalent among the elderly (occurring mainly between the ages of seventy and eighty) than in younger people, and less than 20% of patients have localized and potentially curable tumors46,47. PC is very difficult to diagnose and often remains undetected until the disease has reached an advanced stage48. The etiology of PC is poorly understood, but several factors are known to increase the risk. The preventable risk factors include cigarette smoking, obesity, and a high intake of animal fat, while non-preventable risk factors include CP, an inherited genetic predisposition, and cystic fibrosis.
Activated PSCs were found to produce the ECM proteins that comprised the pancreatic stroma. PC cells are closely interacting PSCs that cause an increase in ECM and fibrosis, in turn stimulating cancer cell proliferation and inhibiting cancer cell apoptosis34. PC cells recruit PSCs to their immediate vicinity and promote a fibrogenic response in the PSCs. Inflammatory markers, including IL-6, increase in patients with PDAC49,50. Tumor-associated macrophages are the main source of IL-6 in PC tissue, but IL-6 secreted by the activated PSCs has been reported to regulate PDAC phenotypes.35 CP is a risk factor for PC, but most cases of PC develop in patients without clinical symptoms of CP; however, there is evidence of inflammation in tissue samples51. Kristen rat sarcoma 2 viral oncogene homolog (KRAS) mutations and activations in PC cells have been seen, in mice models, to promote inflammatory signaling and precancerous lesion development. Subsequently, the inflammatory stimulus activates PSCs in the periacinar area, leading to the recruitment of immune cells (monocytes, T cells, neutrophils, macrophages, and mast cells)52. Despite significant advances in understanding tumor biology and developing novel therapies, survival rates remain discouraging. Targeted therapies based on advances in precision medicine, such as immunotherapy, engineered T-cells, tumor vaccines, myeloid-based immunotherapy, stromal modulating immunotherapy, and gene therapy, are nowadays available for use in dedicated healthcare centers, while others are still under preclinical investigation53. In view of the dominant role played by PSCs in the initiation and progression of PC, it is crucial to identify and develop an anti-fibrotic agent to fight PSCs in order to inhibit devastating PC.
5. The roles of pancreatic stellate cells in chronic pancreatitis and pancreatic cancer
One major similar property shared by CP and PC is that both possess a large proportion of stromata. Pancreatic stromata play an important role in hereditary PC, with most cases resulting from the progression of hereditary pancreatitis to CP. PSCs play a key role in supporting and promoting various aspects of PC, such as proliferation, migration, invasion, colony formation, and angiogenesis, in addition to other promoting factors34,54. Liu et al.55 isolated, identified, and cultured human PSCs and discovered that activated PSCs are present in PC tissues. Their results further showed that PSCs can promote the invasive ability of PC cells and reduce the apoptosis rate induced by gemcitabine.
The EMT is necessary for many physiological developmental steps; however, it contributes to tumorigenesis (the broad spectrum of trans-differentiation in tumors) and to metastatic spread56. During tumorigenesis, PSCs transform into active myofibroblast-like phenotypes, which are involved in several processes. They create a suitable microenvironment for facilitating most cases of cancer progression and invasion. PSCs secrete MMPs, including MMP2, MMP9, and MMP13, as well as TIMP1 and TIMP2, suggesting that PSCs contribute to maintaining the balance in the ECM in a healthy organ. However, they disrupt this balance upon their activation in pancreatitis and PC28. The epidermal growth factor receptor (EGFR) pathway is involved in pancreatic fibrosis; the overexpression of heparin-binding epidermal growth factor-like HB-EGF in the pancreatic islet has been claimed as one of mechanisms contributing to the massive fibrotic state in cancer and CP. PSCs express EGFR (which is activated by HB-EGF), leading in an autocrine manner to an increase in PSCs’ activation and migration, thereby modulating the stromata to support PC growth28. Furthermore, Komar et al.57 demonstrated that the JAK/STAT pathway plays a prominent role in PSC proliferation and activation, secreted as an abundance of several immunomodulatory factors, including IL-6 and monocyte chemoattractant protein-1 (MCP-1). Inhibition of this pathway led to reduced caerulein-induced CP in vivo.
6. Application of phytochemicals as new anti-fibrotic agents in treating chronic pancreatitis and pancreatitis cancer
In recent decades, plant-based/herbal constituents and natural products have been widely used as complementary and alternative medicines to increase longevity and treat diseases58. The phytochemicals extracted from the medicinal plants or herbs play important roles in preventing or treating CP and PC via different mechanistic pathways. For this review, we have selected several phytochemicals that have shown potential anti-fibrotic activity against PSCs in recent years. They are curcumin, resveratrol, rhein, emodin, green tea catechin derivatives, ellagic acid, embelin, eruberin A, and metformin, and their respective chemical structures are shown in Fig. 1. The anti-fibrotic activity of these phytochemicals, as well as their mechanistic actions, are elaborated in the following sections.
Figure 1.

Chemical structures of selective phytochemicals that possess potent anti-fibrotic activity. The phytochemicals selected for this review include curcumin, rhein, green tea catechin (EGCG), resveratrol, emodin, ellagic acid, embelin, eruberin A and metformin.
6.1. Curcumin
Curcumin belongs to the Zingiberaceae family and is a turmeric polyphenol derived from the rhizomes of Curcuma longa, which is cultivated in most parts of Southeast Asia59. Curcumin is a lipophilic agent and stable in the acidic pH environment of the stomach. Curcumin is well-known for its medicinal value; in particular, its antioxidant and anti-inflammatory properties. It has been reported that curcumin is responsible for the suppression of cell proliferation, invasion, and angiogenesis60. Furthermore, it also induces apoptosis via deactivation of nuclear factor-kappa B (NF-κB) and its regulated gene products61. Additionally, it can also suppress several inflammatory cytokines, such as TNF-α, interleukins (IL-1, IL-6, IL-8 and IL-1β), and cyclooxygenase-2 (COX-2)62. Curcumin has direct effects on pancreatic beta cells, which could contribute to the hypoglycemic effects of this compound and decreased beta cell volume, suggesting that this is another novel attribute of curcumin. Additionally, it increases the islet content of glutathione (GSH, a product of the modulatory subunit of gamma-glutamate cysteine ligase (γ-GCL)) and basal insulin secretion and protects them from oxidative stress63.
Thus far, a few in vitro and in vivo studies have been performed to evaluate the anti-fibrotic activity of curcumin against PSCs in CP and/or PC models, and the mechanisms of their actions are depicted in Fig. 2. An in vitro study using obese mice livers showed that curcumin decreased inflammation in adipose liver steatosis through the phosphorylation of the signal transducer and activation of transcription 3 (STAT3) signaling, as well as reduction of the cytokine signaling 3 (SOCS3) suppressor and sterol regulatory element-binding protein-1c (SREBP-1c). These findings indicated the ability of curcumin to mediate anti-inflammatory effects for the treatment of liver steatosis62. In activated PSCs, curcumin decreased the pancreatic beta cell volume, which could be associated with hypoglycemic effects64. In addition, curcumin inhibited PDGF-induced PSCs proliferation, and it reduced α-SMA gene expression, IL-1β and TNF-α-induced MCP-1 production, type I collagen production, and activator protein-1 (AP-1) activation in the activated PSCs65. The activation of AMP-activated protein kinase (AMPK) by curcumin is important for the inhibition of differentiation in adipocytes and cancer cells. This results in the attenuation of peroxisome proliferator-activated receptor gamma in 3T3-L1 adipocytes, and decreased COX-2 expression66. Schwer et al.67 provided the first evidence that curcumin can inactivate PSCs by inhibiting their proliferation. This action is mediated by a decrease in extracellular signal-regulated protein kinase 1 and 2 (ERK1/2) activation in parallel with heme oxygenase-1 (HO-1) up-regulation, increasing the level of cellular carbon monoxide and thereby activating P38 mitogen-activated protein kinases (MAPK), leading to a reduction of PSC proliferation. Moreover, curcumin and three phenolic compounds were shown to significantly suppress the mRNA and protein levels of several fibrotic mediators in primary PSCs activated by TGF-β, including α-SMA, type I collagen, and fibronectin, and the underlying mechanism was associated with the down-regulation of the NF-κB signaling pathway. These findings suggested that curcumin may serve as an anti-fibrotic agent for treating pancreatic fibrosis and PSC-related pathologies, including PDAC58. A newly synthesized curcumin analog (L49H37) was used as an intervention to target the stromal component of PC and compared to traditional curcumin. It was found that L49H37 was more potent for inducing PSCs’ apoptosis at a concentration 10 times lower than curcumin. The results68 showed that the anti-proliferative effect of L49H37 (2.5 μmol/L) significantly inhibited PSC proliferation compared to curcumin (25 μmol/L), as evidenced by the observation of changes in the cell cycle regulatory protein levels of the potent cyclin-dependent kinase inhibitor (CKI), P21WAF1/Clip1.
Figure 2.

Proposed mechanisms involved in the anti-fibrotic activity of curcumin by inhibiting the activation of PSCs to acquire myofibroblast-like phenotypes. Curcumin attenuates the production of TNF-α-induced MCP-1. Besides, it can also significantly reduce the activation of MAPKs signaling, such as c-Jun N-terminal kinase (JNK), P38 MAPK and ERK, which are pivotal in stimulating the production of inflammatory cytokines and mediators. Additionally, curcumin further down-regulates NF-κB signaling pathway by reducing its subunit P65. Apart from these, curcumin can notably reduce the gene expression of α-SMA, IL-1β, Col I and Col III as well as diminish PSCs activation by downregulating the mRNA expression levels of several fibrogenic mediators, including Acta 2, Col-α1 and FN1, under the stimulating effects of TGF-β.
The efficacy of curcumin for clinical application has been tested in several clinical phase trials. A well-performed clinical trial revealed that combination therapy with gemcitabine-based chemotherapy and oral curcumin administration (8 g) proved to be feasible and safe for PC patients69. In addition, a phase II clinical trial on 25 patients with advanced PC confirmed the safety and efficacy of curcumin, despite its low bioavailability. Both clinical studies have completed phase II trials70. A randomized placebo-controlled pilot study, involving 20 tropical pancreatitis patients, showed that an oral combination of curcumin (500 mg) and piperine (5 mg) was effective in relieving pain and beneficially modulating the markers of oxidative stress, including malonyldialdehyde (MDA) and GSH71. In advanced PC, a dose of 8 g of curcumin per day was administered for 2 months. This study discovered that curcumin was well-tolerated and signs of biological activity were found in most patients72. Another clinical trial, involving 21 patients, showed stabilized disease progression in advanced PC after administration of an 8 g dose of curcumin per day. One patient maintained disease stabilization for 18 months and a second patient experienced a significant increase in serum cytokine levels, accompanied by brief, but marked, tumor regression (73%)68.
6.2. Rhein
Rhein is a natural anthraquinone derivative, extracted from the rhizomes of several traditional medicinal plants; for example, Rheum palmatum, also known as “da huang”, is commonly used as purgative73. Rhein has been reported to exert various pharmacological effects, such as antimicrobial74, anti-inflammatory75, anti-angiogenic76,77, and anticancer78,79 effects. In fact, the anticancer activity of rhein has been tested in both in vitro and in vivo models; for instance, rhein inhibited PC cell growth by arresting the expression of hypoxia-inducible factor-1 alpha (HIF-1α) via the decrease in phosphorylation of phosphorylated protein kinases B (p-AKT) and ERK1/2 (p-ERK1/2) in vitro80.
The anti-fibrotic activity of rhein has been reported in both CP and PC models, as evidenced in several studies, and the mechanisms of their actions are described in Fig. 3. Tsang et al.81 demonstrated that treatment with rhein (50 mg/kg/day) in a mouse model of cerulein-induced CP significantly attenuated fibrogenesis by decreasing the immunoreactivity of fibrotic activators, including α-SMA and TGF-β, in pancreatic tissues, followed by the reduction of fibronectin (FN1) and type 1 collagen deposition in the exocrine. In addition, rhein was found to suppress various fibrotic and tumorigenic markers, such as α-SMA, fibronectin, type I collagen, N-cadherin, and MMPs, in cultured PSCs and tested mammalian cells by modulating both the sonic hedgehog (SHH) and serine–threonine kinase signaling pathways81,82. Like other phenolic compounds, the underlying mechanisms of the anti-fibrotic and anti-tumorigenic effects of rhein are associated with the downregulation of the NF-κB and STAT3 signaling pathways58,83. The subunit of NF-κB (P65), which is involved in an inflammatory response, was significantly reduced by 20 μmol/L of rhein in PSCs58.
Figure 3.

Proposed mechanisms involved in the anti-fibrotic activity of rhein. Rhein suppresses the activity of PSCs by targeting several signaling pathways and regulating fibrotic and tumorigenic markers. It can inhibit PSCs proliferation and migrations by decreasing the STAT3 pathway-induced signaling, which plays an important role in malignant transformation and tumor progression. Furthermore, rhein suppresses NF-κB signaling pathway by reducing its subunit P65. In additional, rhein can inactivate PSCs by attenuating various fibrotic and tumorigenic markers, such as α-SMA, fibronectin, type I collagen, N-cadherin and MMPs by modulating both SHH and AKT signaling pathways. With these, rhein plays a pivotal role in the process of pancreatic fibrosis, and PSCs cell proliferation and migration.
6.3. Green tea catechin derivatives
Green tea is also known as Camellia sinensi. In Asian countries, the leaves are common, popular, and widely consumed by the people for their health benefits or medicinal effects, such as antioxidant, anti-inflammatory, anti-proliferative, anti-atherosclerotic, and anti-cancer effects84,85. Green tea extracts contain both polyphenolic and non-phenolic components. The phenolic compound in green tea mainly consists of phenolic catechins, including (−)-epigallocatechin gallate (EGCG), (−)-epigallocatechin (ECG), (−)-epicatechin gallate (ECG), (−)-gallocatechin (GC), and (+)-catechin (C)86. EGCG is one of the most important catechins, due to its high content and antioxidant activity.
EGCG has been shown to inhibit the PDGF-induced proliferation and migration of PSCs87. EGCG was proved to inhibit the PDGF-induced tyrosine phosphorylation of the PDGF β-receptor, downstream activation of ERK, and phosphoinositide 3-kinase (PI3K)/AKT pathways. Furthermore, pre-treatment with EGCG inhibited the ethanol-induced activation of PSCs in vitro. Ethanol significantly increased the production of α-SMA protein (type-I procollagen), activated TGF-β1, and induced phosphorylation of P38 MAPK. Interestingly, treatment with EGCG significantly decreased the production of all these factors and thoroughly abolished P38 MAPK phosphorylation. In addition, ethanol-stimulated transformation of PSCs from normal quiescent phenotypes into myofibroblast-like phenotypes was inhibited by EGCG88. In addition, Shen et al.89 demonstrated that EGCG can reduce liver fibrosis by attenuating ROS-induced hepatocyte cell death, and down-regulate the gene expression of pro-fibrotic markers, such as collagen I, fibronectin, and α-SMA in stellate cells.
6.4. Resveratrol
Resveratrol, or 3,5,4′-trihydroxystilbene, is a polyphenolic stilbene compound that can be found in grapes, raspberries, blueberries, cocoa, and peanuts90. It is synthesized in response to injury or attacks from pathogens of the plants91. A large number of in vitro and in vivo studies have demonstrated the protective effects of resveratrol with regard to several pathological diseases, such as cardiovascular diseases92, diabetes93, neurological disorders94,95, and different types of cancer96, 97, 98. The uses of resveratrol in treating PC were evidenced by several in vitro studies. It was shown to induce PC apoptosis by promoting caspase-3 activation, while remaining nontoxic to the normal pancreatic cells99. Furthermore, resveratrol increased the chemosensitivity of PC cells by targeting nutrient-deprived autophagy factor-1 (NAF-1) via ROS/nuclear factor erythroid 2 (NRF2) signaling100. Zhou et al.101 also reported that resveratrol could enhance the sensitivity of PC cells (MiaPaCa-2 and Panc-1) and decrease the markers of cancer stem cells via suppression of SREBP1 in both in vitro and in vivo models. Intriguingly, Yang et al.102 discovered that, in addition to acting as a tumor suppressant via BAX up-regulation, resveratrol can act as a tumor activator by up-regulating vascular endothelial growth factor B (VEGF-B) in Capan-2 cells.
The activation of PSCs is an important process in the development of pancreatic fibrogenesis, leading to CP and PC. Resveratrol was found to impede the activation, invasion, migration, and glycolysis of PSCs induced by ROS by down-regulating the expression of microRNA 21 (miR-21) and increasing the phosphatise and tensin homolog (PTEN) protein levels103. In the same study, the results further demonstrated that resveratrol inhibited the invasion and migration of PC by suppressing ROS/miR-21 mediated activation and glycolysis in PSCs. In addition, resveratrol was shown to suppress the PSCs’ viability via the reduction of several major fibrogenic mediators, such as α-SMA, type I collagen, and fibronectin, which are associated with down-regulation of the NF-κB signaling pathway58.
6.5. Emodin
Emodin (1,3,8-trihydroxy-6-methylanthraquinone), an important component of Aloe vera, can be extracted from members of the Polygonaceae plant family, such as Palmatum Rheum. Diverse studies have found that emodin exerts various pharmacological effects, including anti-inflammatory, anti-angiogenic, and anti-dyslipidemic actions, in addition to having anticancer potential in vitro and in vivo58,104, 105, 106, 107. Emodin has shown potent anticancer activity in PC, as evidenced in several in vitro and in vivo studies, by targeting cell proliferation and inducing apoptosis via various mechanisms58,108. Furthermore, the combination of emodin and existing chemotherapeutic drugs for PC further enhanced the chemosensitivity of PC cells105.
In the literature, only one study investigated the anti-fibrotic activity of emodin against PSCs. Treatment with emodin (4 μmol/L) significantly decreased the primary PSCs’ cell viability by down-regulating the mRNA and protein expression of several fibrotic mediators, including α-SMA, type I collagen, and fibronectin58. The authors suggested that the concentration used in in vitro experiments should not be higher than 5 μmol/L.
6.6. Ellagic acid
Ellagic acid is a plant-derived non-flavonoid polyphenol that is mainly found in fruits and nuts, such as berries, grapes, pomegranates, and walnuts109. In recent decades, the health benefits of ellagic acid have been reported and shown to be effective, not only for the treatment of chronic metabolic diseases, such as dyslipidemia, non-alcoholic fatty liver diseases, insulin resistance, and type-2 diabetes110,111, but also as anticancer and antitumor treatments112,113.
Several studies, in either CP and/or PC models, have evaluated the anti-fibrotic activity of ellagic acid, and its underlying mechanisms are depicted in Fig. 4. Treatment with ellagic acid in an experimental CP model, using Wistar Bonn/Kodori rats, significantly abolished the development of pancreatic fibrosis. The mRNA expression of α-SMA and TGF-β1 were markedly reduced. In addition, the infiltration of macrophages or monocytes and ROS production in isolated PSCs were significantly decreased in the rats after ellagic acid treatment114, and ellagic acid inhibited the PSC proliferation and migration induced by PDGF-BB. Although the PDGF-β receptor protein levels did not change, ellagic acid inhibited the tyrosine phosphorylation of the receptors, such as ERK and AKT. Thus, ellagic acid diminished the activation of the downstream signaling pathway of RAF proto-oncogene serine/threonine-protein kinase (c-Raf)/MAPK/ERK and PI3K/AKT, which are important for PSC cell proliferation and migration115. Masamune et al.115 further reported that ellagic acid inactivated PSCs by attenuating the protein levels of α-SMA and ECM procollagen types I and III. Moreover, ellagic acid treatment inhibited IL-1β- and TNF-α-induced MCP-1 in PSCs correlated with AP-1, but not NF-κB. Ellagic acid was also found to prevent the transformation of PSCs from quiescent into myofibroblast-like phenotypes.
Figure 4.
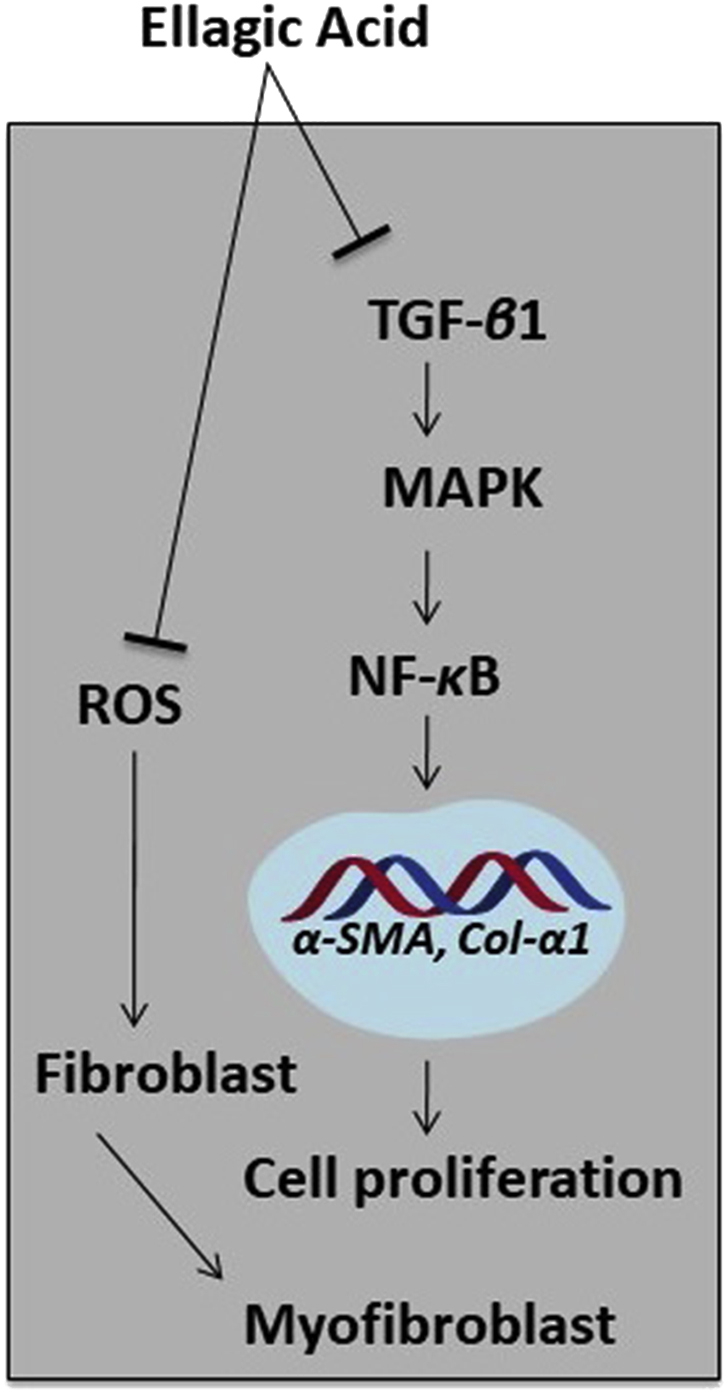
Proposed mechanisms involved in the anti-fibrotic activity of ellagic acid. Ellagic acid can reduce the myeloperoxidase activity and collagen content. Moreover, it attenuates the expression of TGF-β1 as well as modulates its downstream signaling pathway. The amount of α-SMA and macrophages monocytes (ED)-positive cells is decreased after treating with ellagic acid, and it inhibits ROS production that is stimulated by TNF-β1 and PDGF. Other than these, ellagic acid can downregulate α-SMA and collagen genes (α1(I) procollagen and α1(III) procollagen). In PC cells, ellagic acid can decrease the NF-κB transcriptional activity and stimulate apoptosis and reduce cell proliferation.
6.7. Embelin
Embelin is a naturally occurring benzoquinone that can be extracted from the fruits (berries) of the Embelia ribes Burm. plant (Myrsinaceae). It has been used for various traditional medicinal remedies in India. Embelin had been shown to exert different pharmacological effects, including as an anticancer agent via inhibition of cell migration, invasion, and induction of apoptosis in colon, lung, and lung cancer cells116, 117, 118.
To date, the anti-fibrotic activity of embelin has been indicated in a PC model113, with the results showing that embelin inhibited both PC cells and PSCs in a dose-dependent manner. Interestingly, embelin in combination with ellagic acid at low concentrations (0.5–3 μmol/L) synergistically increased apoptosis and reduced cell proliferation compared to the individual treatments. A similar observation was also made in relation to a subcutaneous xenograft mouse model of PC, in which embelin alone, or in combination with ellagic acid, significantly reduced a tumor's size and cellularity. The mechanism underlying the action of embelin is via STAT3 dephosphorylation and the reduced expression of its downstream target, survivin, in PC cells113. In PC cells (MIA PaCa-2 and HPAF-II), embelin was demonstrated to stimulate apoptosis and inhibit cancer cell proliferation dose-dependently. Embelin reversed the anti-apoptotic effect of the X-linked apoptosis protein (XIAP) by preventing the interaction of XIAP with caspases, and it was also found to downregulate the expression of XIAP, survivin, the inhibitor of apoptosis 1 and 2 (IAP1/2), tumor necrosis factor receptor-associated factor 1 (TRAF1), cellular FLICE (a FADD-like IL-1β-converting enzyme), inhibitory protein (cFILP), B-cell lymphoma 2 (BCL-2), and B-cell lymphoma-x (BCL-X) by suppressing the activation of NF-κB113,119. In a xenograft mouse model of PC, embelin was reported to reduce tumor growth and the results also showed that embelin metabolized rapidly after oral administration in the xenograft mouse model113. Furthermore, embelin was found to inhibit cancer cell proliferation by down-regulating the SHH signaling pathway, with implications, not only for promoting cell proliferation, but also cell invasion, metastasis, and tumor growth. It was assumed that targeting SHH might inactivate PSCs, since the expression of SHH was reported to promote tumor growth via PSC-induced desmoplasia formation, as well as to affect the mobility and differentiation of PSCs120,121. The mechanisms of the actions underlying the anti-fibrotic activity of embelin are shown in Fig. 5.
Figure 5.
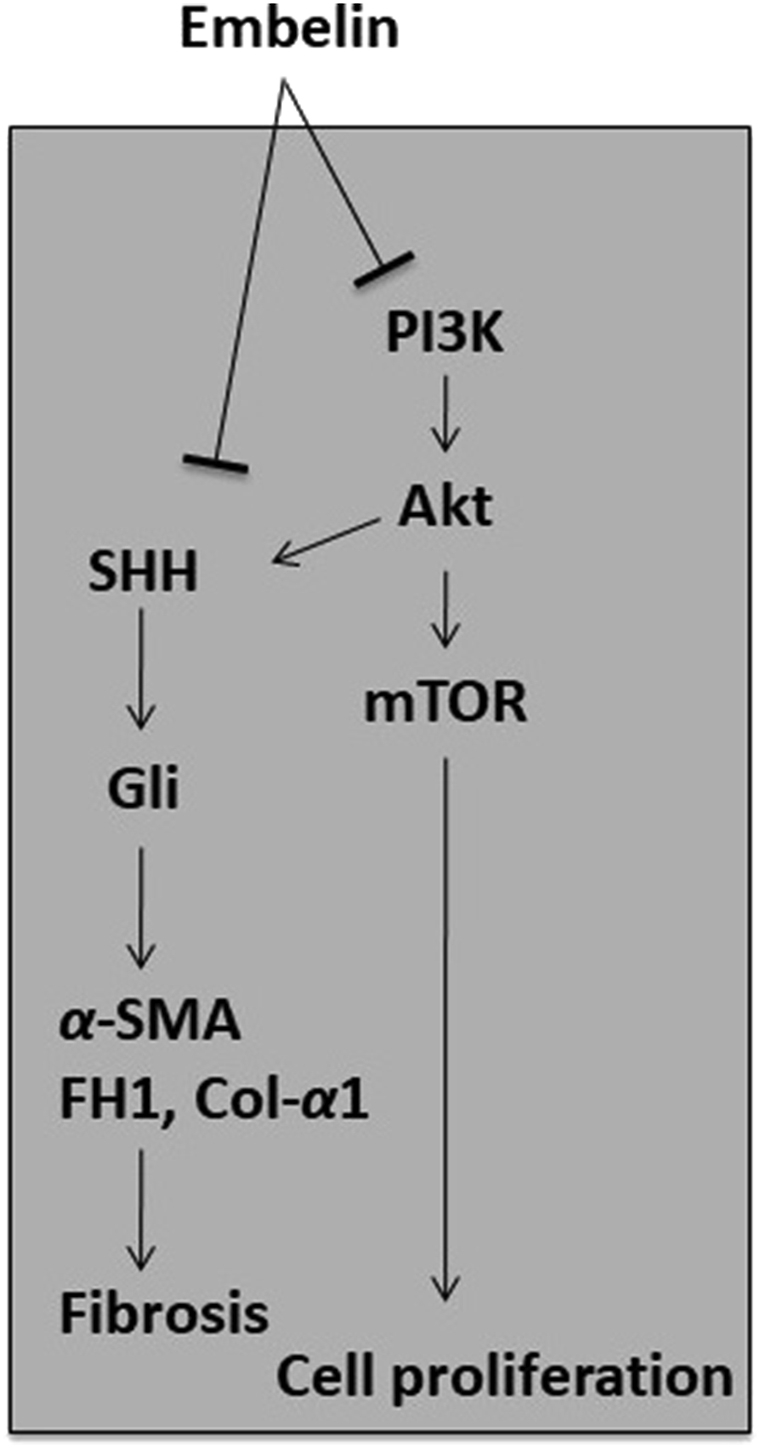
Proposed mechanisms involved in the anti-fibrotic activity of embelin. The anti-fibrotic activity of embelin is thus far reported in PC model, where it can inhibit PSC survival in a dose-dependent manner. Embelin down-regulated the SHH signaling pathway and consequently, the expression of fibrogenic mediators, such as α-SMA, fibronectin, type I collagen is decreased. This suggests that embelin able to alleviate the development of fibrosis.
6.8. Eruberin A
Eruberin A is a pure compound extracted from the fern plant, Pronephrium penangianum. It is an organic flavanol glycoside and has been reported to exert antioxidant effects and reduce proinflammatory cytokine production and diabetic-associated oxidants, such as hydrogen peroxide122,123. Eruberin A has also been demonstrated to have potent cytotoxic effects on L929 fibroblasts and HeLa cells124. Thus far, only one study has investigated the anti-fibrotic activity of eruberin A against primary PSCs; namely, LTC-14. The growth rate of LTC-14 cells decreased after treatment with eruberin A in a dose-dependent manner and significantly suppressed the gene expression of the major fibrotic filaments and ECM mediators, including smooth muscle α actin (Acta 2), collagen type I-alpha 1 (Col I-α1), and fibronectin 1 (FN1) at 20 μg/mL: a concentration that did not cause cytotoxic effects125. Similar results were observed in eruberin A-treated PANC-1 cells. Eruberin A inhibited NF-κB activation and the SHH signaling components, and suppressed the activation of the PI3K/AKT pathway linked to inflammatory and fibrogenesis downstream cascades125. A 20 μg/mL dose of eruberin A inhibited TGF-β-induced AKT phosphorylation and attenuated α-SMA at both protein and cytoplasmic levels in LTC-14 cells and PANC-1 cells125. The effects of eruberin A on PSCs and PC were less reported, so more research and investigations are needed to gain further understanding.
6.9. Metformin
Metformin is one of the guanidine derivatives that are rich in Galega officinalis (a well-known European goat rue) and, in 1918, were shown to lower blood glucose levels126. Metformin is an oral anti-diabetic medicine for type-2 diabetic patients and helps to control the amount of glucose produced by the liver. It has also been reported to exert anticancer effects on different cancer types, including breast, ovarian, pancreatic, and colon cancer, by modulating the inflammatory responses and cancer stem cells127, 128, 129, 130.
Metformin has been shown to inhibit desmoplastic reaction and enhance the chemosensitivity of PDAC towards gemcitabine by activating AMPK. Furthermore, in vitro and in vivo studies have demonstrated that metformin activated AMPK, in both human PSCs and nude mice with subcutaneous pancreatic cancer, by up-regulating p-AMPK expression, significantly decreasing TGF-β1, α-SMA, and collagen in tumor microenvironments, and inhibiting PSC proliferation131,132. In addition, metformin enhanced the sensitivity of PSCs and PC cells in response to gemcitabine treatment. Combined treatment with metformin and gemcitabine significantly reduced the expression of SHH by inhibiting the production of VEGF and tumor neovascularisation, thus improving the chemosensitivity of PC cells to gemcitabine131,133. However, the resistance of PSCs to metformin can be increased by a higher glucose intake, and it was suggested that the tumor microenvironment plays a pivotal role in determining the effect of metformin134. The anti-fibrotic activity of these phytochemicals, as well as their mechanistic actions, are summarized in Table 1.
Table 1.
The mechanisms of actions of selective phytochemicals in inactivating PSCs in chronic pancreatitis and/or pancreatic cancer models.
| Phytochemical | Disease | Test model | Dosage | Metabolic responses/mechanisms | Ref. |
|---|---|---|---|---|---|
| Curcumin | CP | In vitro, TGF-β stimulated cultured primary PSCs (LTC-14) | 20 μmol/L | 1. Down-regulated NF-κB signaling by reducing subunit P65; | 58 |
| 2. Inhibited the production of fibrogenic mediators (Acta 2, Col α1 and FN1) induced by TGF-β. | |||||
| PC | In vitro, cultured PSCs | 5–25 μmol/L | 1. Inhibited PDGF-induced proliferation; | 65 | |
| 2. Reduced gene expression of α-SMA, IL-1β, Col I and Col III; | |||||
| 3. Inhibited production of TNF-α-induced MCP-1. | |||||
| PC | In vitro, cultured human PSCs | 1–25 μmol/L | 1. Inhibited cell proliferation and induced cell apoptosis; | 69 | |
| 2. Increased phosphorylation of ERK1/2 at lower concentrations (1 and 10 μmol/L). | |||||
| Rhein | CP | In vivo, cerulein-induced CP mouse model | 50 mg/kg/day | 1. Attenuated fibrogenesis by decreasing immunoreactivity of fibrotic activators (α-SMA and TGF-β) and reduction of fibronectin (FN1 and type 1 collagen deposition). | 82 |
| CP | In vitro, TGF-β stimulated cultured LTC-14 cells | 20 μmol/L | 1. Reduced P65 (subunit of NF-κB); | 58 | |
| 2. Inhibited the production of fibrogenic mediators (Acta 2, Col α1and Fnl); | |||||
| 3. Down-regulated NF-κB signaling pathway. | |||||
| PC | In vitro, human pancreatic cancer cells BxPC-3, PANC-1, Patu8988T and AsPC-1 | 60 μmol/L | Suppressed constitutive STAT3 tyrosine phosphorylation and induces apoptosis in pancreatic cancer cells. | 84 | |
| In vivo, xenograft BALB/c female mice model | 60 mg/kg | 1. Inhibited tumor growth; | |||
| 2. Reduced expression of p-STAT-3 and p-EGFR; | |||||
| 3. Downregulated STAT-3 signaling pathway. | |||||
| PC | In vitro, cultured PSCs and testing mammalian cells | 20 μmol/L | 1. Suppressed various fibrotic and tumorigenic markers (α-SMA, fibronectin, type I collagen, N-cadherin, and MMPs); | 83 | |
| 2. Modulating both sonic hedgehog (SHH) and serine–threonine kinase signaling pathways. | |||||
| Green tea [(−)-epigallo-catechin3-gallate (EGCG)] | CP | In vitro, cultured PSCs | 25 μmol/L | 1. Inhibited ethanol-induced morphological changes of PSCs from normal quiescent-phenotype to myofibroblast-like; | 89 |
| 2. Decreased production of α-SMA; suppressed type-I procollagen production; | |||||
| 3. Activated TGF-β1 secretion; | |||||
| 4. Abolished ethanol-induced increases in P38 MAP kinase phosphorylation. | |||||
| PC | In vitro, rat PSCs | 1–25 μmol/L | 1. Inhibited PDGF-induced proliferation and migration; | 88 | |
| 2. Inhibited cell cycle progression beyond G1 phase; | |||||
| 3. Inhibited PDGF-induced phosphorylation ERK and Akt. | |||||
| *Liver fibrosis | In vitro, cultured stellate cells | 10 μmol/L | 1. Significantly reduced metallopeptidase inhibitor (TIMP-1), that is an inhibitor of enzymes in mouse fibrosis; | 90 | |
| 2. Downregulated the gene expression of pro-fibrotic markers such as collagen I, fibronectin, and α-SMA. | |||||
| Resveratrol | PC | In vitro, cultured MiaPaC-2 and PANC-1 cell lines | 50 μmol/L | 1. Increased sensitivity of PCs to gemcitabine; inhibited lipid synthesis; 2. Rescued the stemness induced by gemcitabine via suppressing SREBP1. |
102 |
| In vivo, genetically engineered mouse model, KPC mouse model | 50 mg/kg/day | ||||
| PC | In vitro, cultured human PSCs from patient | 1–200 μmol/L | 1. Inhibited H2O2-promoted PSCs activation, migration, and invasion; | 104 | |
| 2. Reduced ROS-induced miR-21 expression and increased PTEN expression. | |||||
| Emodin | CP | In vivo, TGF-β stimulated cultured LTC-14 cells | 4 μmol/L | 1. Reduced P65 (subunit of NF-κB); | 58 |
| 2. Inhibited the production of fibrogenic mediators (Acta2, Col α1, and Fnl); | |||||
| 3. Downregulated NF-κB signaling pathway. | |||||
| Ellagic acid | CP | In vivo, male wistar Bonn/Kobori rats | 100 mg/kg/day | 1. Attenuated myeloperoxidase activity; | 115 |
| 2. Decreased in collagen content, reduced TGF-β1 expression, and reduced the amount of α-SMA and macrophages monocytes (ED)-positive cells; | |||||
| 3. Inhibited TNF-β1 and platelet derived growth factor (PDGF)-induced reactive oxygen species (ROS). | |||||
| PC | In vitro, rat PSCs | 25 μg/mL | 1. Down-regulated α-SMA, collagen (α1(I) procollagen and α1(III) procollagen); | 116 | |
| 2. Inhibited transformation of quiescent freshly isolated PSCs into myofibroblast. | |||||
| PC | In vitro, MIA PaCa-2; HPAF-II cells | 10–30 μmol/L | 1. Decreased NF-κB transcriptional activity; | 114 | |
| 2. Stimulated apoptosis and inhibited proliferation in pancreatic cancer cells. | |||||
| In vivo, nude mice xenograft model of pancreatic cancer | 150 mg/kg in diet | Reduced tumor growth in mouse. | |||
| Embelin | PC | In vitro, AsPC-1, PANC-1, MIA PaCa-2 and Hs766T cell lines | 1–15 μmol/L | Inhibited cell growth; suppressed SHH signaling pathway. | 127 |
| In vivo, Balb/c nude mice xenograft model of pancreatic cancer | 40 mg/kg | 1. Inhibited tumor cell proliferation and induced apoptosis; 2. Inhibited angiogenesis. |
|||
| (AsPC-1) | |||||
| PC | In vitro, MIA PaCa-2; HPAF-II cells | 10–30 μmol/L | Stimulated apoptosis and inhibited proliferation in pancreatic cancer cells. | ||
| In vivo, nude mice xenograft model of pancreatic cancer | 450 mg/kg in diet | Reduced tumor growth in mouse. | 114 | ||
| Metformin | PC | In vivo, genetically engineered mouse model, KPC mice | 200 mg/kg/day | 1. Suppressed the growth and the progression of the tumor in PDAC; | 134 |
| 2. Reduced production of α-SMA and ECM; | |||||
| 3. Anti-PSCs effect via the reduction of SHH expression, thus decreasing VEGF, tumor neovascularization and desmoplastic reaction. | |||||
| PC | In vitro, human pancreatic cancer cell AsPC-1, BxPC3, CFPAC-1, Panc-1 and SW1990 | 5 mmol/L | 1. Significantly reduced mRNA expression of CTGF, TGF-β1, and PDGF-A; | 132 | |
| 2. Suppressed secretion of TGF-β1 through activation of AMPK signaling pathway; | |||||
| 3. Inhibited invasion and migration ability of PSC. | |||||
| In vivo, Balb/c nude mice orthotopic pancreatic cancer model | 100 mg/kg | 1. Reduced the α-SMA-positive cell and collagen in the tumor microenvironment; | |||
| 2. Enhanced chemo sensitivity of gemcitabine by inhibited ECM deposition. | |||||
| Eruberin A | PC | In vitro, rat LTC-14 cell line; human PDAC PANC-1 cell line | 20 μg/mL | 1. Anti-fibrotic effect; suppressed TGF-β-induced fibrogenic mediator; | 135 |
| 2. Downregulated the activation of NF-κB and SHH signaling components; | |||||
| 3. Suppressed the activation of PI3K/AKT signaling pathway. |
7. Conclusions
Phytochemicals extracted from plant-based foods have shown multifaceted bioactivity in maintaining human health and disease prevention. In this present review, we have provided insight into several selective phytochemicals that have recently been evaluated for their anti-fibrotic activity, as well as the underlying mechanisms and key molecules involved, which may serve as potent and novel anti-fibrotic agents in targeting PSCs for their mediated CP and PC diseases. PSC activation can be stimulated by cell damage or an inflamed pancreas, inducing the PCSs’ proliferation and secretion of cytokines and ECM proteins, supporting fibrosis formation, and stimulating cellular microenvironments that are favorable for the pathologic development of CP and PC. With regard to the reviewed phytochemicals, most studies examining their potential anti-fibrotic activity were carried out using PC models, and rhein and ellagic acid treatments were only tested in CP models. Additionally, curcumin has been tested intensively, compared to other selective phytochemicals. Intriguingly, it was noted that most of the selective phytochemicals exerted similar mechanisms of action, which underpinned their anti-fibrotic action against PSCs in both CP and PC models. Specifically, the selective phytochemicals were shown to deactivate the PSCs by decreasing their proliferation, via the regulation of ERK1/2, P38 MAPK, and the SHH and PI3K/AKT signaling pathways, to suppress PSC migration and fibrogenesis. Comparatively, rhein and ellagic acid treatment deactivated PSCs by reducing the expression of their fibrogenic markers (α-SMA), and their soluble factors, such as ECM (fibronectin and collagens) and TGF-β, which are associated with pancreatic fibrosis. Involvement of the above-mentioned mechanisms supported the potential of these selective phytochemicals to act as novel anti-fibrotic agents for combating CP and PC diseases by targeting PSCs.
Given their potential to serve as new anti-fibrotic agents, based on preclinical studies, it is important to consider a few issues relating to drug delivery and low bioavailability before using them in clinical settings. One of the treatment strategies for improving their therapeutic efficacy is to synthesize their analogs by modifying their chemical structures. Taking curcumin as an example, the curcumin analog L49H37 has been proved to exert potent anti-proliferative activity on PSCs at a concentration that is much lower than curcumin itself. To the best of our knowledge, all the synthetic analogs of the other selective phytochemicals, except eruberin A, have been synthesized and tested, mainly in cancer and other disease models135, 136, 137, 138, 139, 140. It is, therefore, crucial to test their treatment efficacy against PSCs in both CP and PC models. Furthermore, curcumin was reportedly subjected to a clinical trial and the advanced PC patients showed good tolerance and attested to its treatment efficacy. However, the issue of low bioavailability may impede its therapeutic efficacy. In recent years, the use of nanoparticle systems, including polymers, liposomes, and some special moieties, have emerged in relation to drug delivery to patients141. With the exception of eruberin A, these selective phytochemicals have been encapsulated into nanoparticles and their therapeutic efficacy against various diseases, mainly cancers, has been tested142. Of these tests, treatment of PC with curcumin, metformin, and ellagic acid showed promising outcomes143, 144, 145. Given the economic and non-toxic properties of the selective phytochemicals, as well as their promising treatment outcomes with regard to PSCs in CP and PC models, further investigation of potential plants or their derived compounds should be encouraged, in order to isolate, identify, and evaluate their benefits, with the aim of discovering more potential anti-fibrotic agents to treat patients with CP-associated PC.
Acknowledgments
This work was supported by two research university grants, from University of Malaya (grant No. GPF002C-2018, Malaysia) and MAHSA University (grant No. RP165-05/19, Malaysia).
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
Contributor Information
Maw Shin Sim, Email: garethsim@um.edu.my.
Yuan Seng Wu, Email: sywu@mahsa.edu.my.
Author contributions
Puvanesswaray Ramakrishnan, Wei Mee Loh, Srinivasa Reddy Bonam, and Yuan Seng Wu contributed to the writing. Yuan Seng Wu conceived and designed the manuscript while, Yuan Seng Wu, Maw Shin Sim, Srinivasa Reddy Bonam, and Subash C.B. Gopinath revised the manuscript. Ismail M. Fareez, Rhanye Mac Guard, and Maw Shin Sim provided vital guidance and insight for the writing.
Conflict of interests
The authors have no conflicts of interest to declare.
References
- 1.Bynigeri R.R., Jakkampudi A., Jangala R., Subramanyam C., Sasikala M., Rao G.V. Pancreatic stellate cell: Pandora's box for pancreatic disease biology. World J Gastroenterol. 2017;23:382–405. doi: 10.3748/wjg.v23.i3.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Masamune A., Watanabe T., Kikuta K., Shimosegawa T. Roles of pancreatic stellate cells in pancreatic inflammation and fibrosis. Clin Gastroenterol Hepatol. 2009;7:S48–S54. doi: 10.1016/j.cgh.2009.07.038. [DOI] [PubMed] [Google Scholar]
- 3.Talukdar R., Tandon R.K. Pancreatic stellate cells: new target in the treatment of chronic pancreatitis. J Gastroenterol Hepatol. 2008;23:34–41. doi: 10.1111/j.1440-1746.2007.05206.x. [DOI] [PubMed] [Google Scholar]
- 4.Dunér S., Lopatko Lindman J., Ansari D., Gundewar C., Andersson R. Pancreatic cancer: the role of pancreatic stellate cells in tumor progression. Pancreatology. 2011;10:673–681. doi: 10.1159/000320711. [DOI] [PubMed] [Google Scholar]
- 5.Luttenberger T., Schmid-Kotsas A., Menke A., Siech M., Beger H., Adler G. Platelet-derived growth factors stimulate proliferation and extracellular matrix synthesis of pancreatic stellate cells: implications in pathogenesis of pancreas fibrosis. Lab Invest. 2000;80:47–55. doi: 10.1038/labinvest.3780007. [DOI] [PubMed] [Google Scholar]
- 6.Schneider E., Schmid-Kotsas A., Zhao J., Weidenbach H., Schmid R.M., Menke A. Identification of mediators stimulating proliferation and matrix synthesis of rat pancreatic stellate cells. Am J Physiol Cell Physiol. 2001;281:C532–C543. doi: 10.1152/ajpcell.2001.281.2.C532. [DOI] [PubMed] [Google Scholar]
- 7.Mews P., Phillips P., Fahmy R., Korsten M., Pirola R., Wilson J. Pancreatic stellate cells respond to inflammatory cytokines: potential role in chronic pancreatitis. Gut. 2002;50:535–541. doi: 10.1136/gut.50.4.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ohnishi N., Miyata T., Ohnishi H., Yasuda H., Tamada K., Ueda N. Activin A is an autocrine activator of rat pancreatic stellate cells: potential therapeutic role of follistatin for pancreatic fibrosis. Gut. 2003;52:1487–1493. doi: 10.1136/gut.52.10.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Apte M.V., Haber P.S., Darby S.J., Rodgers S.C., McCaughan G.W., Korsten M.A. Pancreatic stellate cells are activated by proinflammatory cytokines: implications for pancreatic fibrogenesis. Gut. 1999;44:534–541. doi: 10.1136/gut.44.4.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Apte M.V., Phillips P.A., Fahmy R.G., Darby S.J., Rodgers S.C., McCaughan G.W. Does alcohol directly stimulate pancreatic fibrogenesis? Studies with rat pancreatic stellate cells. Gastroenterology. 2000;118:780–794. doi: 10.1016/s0016-5085(00)70148-x. [DOI] [PubMed] [Google Scholar]
- 11.Masamune A., Kikuta K., Satoh M., Satoh A., Shimosegawa T. Alcohol activates activator protein-1 and mitogen-activated protein kinases in rat pancreatic stellate cells. J Pharmacol Exp Ther. 2002;302:36–42. doi: 10.1124/jpet.302.1.36. [DOI] [PubMed] [Google Scholar]
- 12.Watanabe S., Nagashio Y., Asaumi H., Nomiyama Y., Taguchi M., Tashiro M. Pressure activates rat pancreatic stellate cells. Am J Physiol Gastrointest Liver Physiol. 2004;287:G1175–G1181. doi: 10.1152/ajpgi.00339.2004. [DOI] [PubMed] [Google Scholar]
- 13.Masamune A., Kikuta K., Satoh M., Kume K., Shimosegawa T. Differential roles of signaling pathways for proliferation and migration of rat pancreatic stellate cells. Tohoku J Exp Med. 2003;199:69–84. doi: 10.1620/tjem.199.69. [DOI] [PubMed] [Google Scholar]
- 14.Lee A.T., Xu Z., Pothula S.P., Patel M.B., Pirola R.C., Wilson J.S. Alcohol and cigarette smoke components activate human pancreatic stellate cells: implications for the progression of chronic pancreatitis. Alcohol Clin Exp Res. 2015;39:2123–2133. doi: 10.1111/acer.12882. [DOI] [PubMed] [Google Scholar]
- 15.Kanat O., Ertas H. Shattering the castle walls: anti-stromal therapy for pancreatic cancer. World J Gastrointest Oncol. 2018;10:202–210. doi: 10.4251/wjgo.v10.i8.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Swayden M., Iovanna J., Soubeyran P. Pancreatic cancer chemo-resistance is driven by tumor phenotype rather than tumor genotype. Heliyon. 2018;4 doi: 10.1016/j.heliyon.2018.e01055. e01055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang C., Liu B., Xu X., Zhuang B., Li H., Yin J. Toward targeted therapy in chemotherapy-resistant pancreatic cancer with a smart triptolide nanomedicine. Oncotarget. 2016;7:8360–8372. doi: 10.18632/oncotarget.7073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gunda V., Souchek J., Abrego J., Shukla S.K., Goode G.D., Vernucci E. MUC1-mediated metabolic alterations regulate response to radiotherapy in pancreatic cancer. Clin Cancer Res. 2017;23:5881–5891. doi: 10.1158/1078-0432.CCR-17-1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gunda V., Souchek J., Abrego J., Goode G., Vernucci E., Shukla S.K. Abstract 459: Targeting MUC1 mediated nucleotide metabolism sensitizes pancreatic tumors to radiation therapy. Proceedings: American association for cancer research annual meeting 2017, Washington, DC. Cancer Res. 2017 AM2017–A2459. [Google Scholar]
- 20.Mantoni T.S., Lunardi S., Al-Assar O., Masamune A., Brunner T.B. Pancreatic stellate cells radioprotect pancreatic cancer cells through beta1-integrin signaling. Cancer Res. 2011;71:3453–3458. doi: 10.1158/0008-5472.CAN-10-1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bonam S.R., Wu Y.S., Tunki L., Chellian R., Halmuthur M.S.K., Muller S. What has come out from phytomedicines and herbal edibles for the treatment of cancer? ChemMedChem. 2018;13:1854–1872. doi: 10.1002/cmdc.201800343. [DOI] [PubMed] [Google Scholar]
- 22.Azimi H., Khakshur A.A., Abdollahi M., Rahimi R. Potential New pharmacological agents derived from medicinal plants for the treatment of pancreatic cancer. Pancreas. 2015;44:11–15. doi: 10.1097/MPA.0000000000000175. [DOI] [PubMed] [Google Scholar]
- 23.Vuong Q.V., Hirun S., Phillips P.A., Chuen T.L., Bowyer M.C., Goldsmith C.D. Fruit-derived phenolic compounds and pancreatic cancer: perspectives from Australian native fruits. J Ethnopharmacol. 2014;152:227–242. doi: 10.1016/j.jep.2013.12.023. [DOI] [PubMed] [Google Scholar]
- 24.Yang T., Liu J., Luo F., Lin Q., Rosol T.J., Deng X. Anticancer properties of Monascus metabolites. Anti Cancer Drugs. 2014;25:735–744. doi: 10.1097/CAD.0000000000000102. [DOI] [PubMed] [Google Scholar]
- 25.Suklabaidya S., Das B., Ali S.A., Jain S., Swaminathan S., Mohanty A.K. Characterization and use of HapT1-derived homologous tumors as a preclinical model to evaluate therapeutic efficacy of drugs against pancreatic tumor desmoplasia. Oncotarget. 2016;7:41825. doi: 10.18632/oncotarget.9729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xue R., Jia K., Wang J., Yang L., Wang Y., Gao L. A rising star in pancreatic diseases: pancreatic stellate cells. Front Physiol. 2018;9:754. doi: 10.3389/fphys.2018.00754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lafaro K.J., Melstrom L.G. The paradoxical web of pancreatic cancer tumor microenvironment. Am J Pathol. 2019;189:44–57. doi: 10.1016/j.ajpath.2018.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Allam A., Thomsen A.R., Gothwal M., Saha D., Maurer J., Brunner T.B. Pancreatic stellate cells in pancreatic cancer: in focus. Pancreatology. 2017;17:514–522. doi: 10.1016/j.pan.2017.05.390. [DOI] [PubMed] [Google Scholar]
- 29.Haqq J., Howells L.M., Garcea G., Metcalfe M.S., Steward W.P., Dennison A.R. Pancreatic stellate cells and pancreas cancer: current perspectives and future strategies. Eur J Cancer. 2014;50:2570–2582. doi: 10.1016/j.ejca.2014.06.021. [DOI] [PubMed] [Google Scholar]
- 30.Ben-Harosh Y., Anosov M., Salem H., Yatchenko Y., Birk R. Pancreatic stellate cell activation is regulated by fatty acids and ER stress. Exp Cell Res. 2017;359:76–85. doi: 10.1016/j.yexcr.2017.08.007. [DOI] [PubMed] [Google Scholar]
- 31.Pothula S.P., Xu Z., Goldstein D., Pirola R.C., Wilson J.S., Apte M.V. Key role of pancreatic stellate cells in pancreatic cancer. Cancer Lett. 2016;381:194–200. doi: 10.1016/j.canlet.2015.10.035. [DOI] [PubMed] [Google Scholar]
- 32.Tang D., Wang D., Yuan Z., Xue X., Zhang Y., An Y. Persistent activation of pancreatic stellate cells creates a microenvironment favorable for the malignant behavior of pancreatic ductal adenocarcinoma. Int J Cancer. 2013;132:993–1003. doi: 10.1002/ijc.27715. [DOI] [PubMed] [Google Scholar]
- 33.Madro A., Celinski K., Slomka M. The role of pancreatic stellate cells and cytokines in the development of chronic pancreatitis. Med Sci Monit. 2004;10:RA166–RA170. [PubMed] [Google Scholar]
- 34.Wu Y.S., Looi C.Y., Subramaniam K.S., Masamune A., Chung I. Soluble factors from stellate cells induce pancreatic cancer cell proliferation via Nrf2-activated metabolic reprogramming and ROS detoxification. Oncotarget. 2016;7:36719. doi: 10.18632/oncotarget.9165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu Y.S., Chung I., Wong W.F., Masamune A., Sim M.S., Looi C.Y. Paracrine IL-6 signaling mediates the effects of pancreatic stellate cells on epithelial-mesenchymal transition via Stat3/Nrf2 pathway in pancreatic cancer cells. Biochim Biophys Acta Gen Subj. 2017;1861:296–306. doi: 10.1016/j.bbagen.2016.10.006. [DOI] [PubMed] [Google Scholar]
- 36.Schnittert J., Bansal R., Prakash J. Targeting pancreatic stellate cells in cancer. Trends Cancer. 2019;5:128–142. doi: 10.1016/j.trecan.2019.01.001. [DOI] [PubMed] [Google Scholar]
- 37.Pang T.C.Y., Wilson J.S., Apte M.V. Pancreatic stellate cells: what's new? Curr Opin Gastroenterol. 2017;33:366–373. doi: 10.1097/MOG.0000000000000378. [DOI] [PubMed] [Google Scholar]
- 38.Kuhlmann L., Olesen S.S., Olesen A.E., Arendt-Nielsen L., Drewes A.M. Mechanism-based pain management in chronic pancreatitis—is it time for a paradigm shift? Expert Rev Clin Pharmacol. 2019;12:249–258. doi: 10.1080/17512433.2019.1571409. [DOI] [PubMed] [Google Scholar]
- 39.Chand S.K., Pendharkar S.A., Bharmal S.H., Bartlett A.S., Pandol S.J., Petrov M.S. Frequency and risk factors for liver disease following pancreatitis: a population-based cohort study. Dig Liver Dis. 2019;51:551–558. doi: 10.1016/j.dld.2018.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Narkhede R.A., Desai G.S., Prasad P.P., Wagle P.K. Diagnosis and management of pancreatic adenocarcinoma in the background of chronic pancreatitis: core issues. Dig Dis. 2019;37:315–324. doi: 10.1159/000496507. [DOI] [PubMed] [Google Scholar]
- 41.Rawla P., Sunkara T., Gaduputi V. Epidemiology of pancreatic cancer: global trends, etiology and risk factors. World J Oncol. 2019;10:10–27. doi: 10.14740/wjon1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shimizu K. Mechanisms of pancreatic fibrosis and applications to the treatment of chronic pancreatitis. J Gastroenterol. 2008;43:823–832. doi: 10.1007/s00535-008-2249-7. [DOI] [PubMed] [Google Scholar]
- 43.Zhang L., Li J., Zong L., Chen X., Chen K., Jiang Z. Reactive oxygen species and targeted therapy for pancreatic cancer. Oxid Med Cell Longev. 2016;2016 doi: 10.1155/2016/1616781. 1616781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Braganza J.M., Lee S.H., McCloy R.F., McMahon M.J. Chronic pancreatitis. Lancet. 2011;377:1184–1197. doi: 10.1016/S0140-6736(10)61852-1. [DOI] [PubMed] [Google Scholar]
- 45.Apte M., Pirola R., Wilson J. The fibrosis of chronic pancreatitis: new insights into the role of pancreatic stellate cells. Antioxidants Redox Signal. 2011;15:2711–2722. doi: 10.1089/ars.2011.4079. [DOI] [PubMed] [Google Scholar]
- 46.Sanabria Mateos R., Conlon K.C. Pancreatic cancer. Surgery. 2016;34:282–291. [Google Scholar]
- 47.Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362:1605–1617. doi: 10.1056/NEJMra0901557. [DOI] [PubMed] [Google Scholar]
- 48.Maisonneuve P. Epidemiology and burden of pancreatic cancer. Presse Med. 2019;48:e113–e123. doi: 10.1016/j.lpm.2019.02.030. [DOI] [PubMed] [Google Scholar]
- 49.Wörmann S.M., Diakopoulos K.N., Lesina M., Algül H. The immune network in pancreatic cancer development and progression. Oncogene. 2013;33:2956. doi: 10.1038/onc.2013.257. [DOI] [PubMed] [Google Scholar]
- 50.Kim H.W., Lee J.C., Paik K.H., Kang J., Kim J., Hwang J.H. Serum interleukin-6 is associated with pancreatic ductal adenocarcinoma progression pattern. Medicine (Baltim) 2017;96 doi: 10.1097/MD.0000000000005926. e5926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Whitcomb D., Greer J. Germ-line mutations, pancreatic inflammation, and pancreatic cancer. Clin Gastroenterol Hepatol. 2009;7:S29–S34. doi: 10.1016/j.cgh.2009.07.032. [DOI] [PubMed] [Google Scholar]
- 52.Muniraj T., Jamidar P.A., Aslanian H.R. Pancreatic cancer: a comprehensive review and update. Dis Mon. 2013;59:368–402. doi: 10.1016/j.disamonth.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 53.Salvia R., Casciani F., Sereni E., Bassi C. Pancreatic cancer—what’s next? Presse Med. 2019;48:e187–e197. doi: 10.1016/j.lpm.2019.02.031. [DOI] [PubMed] [Google Scholar]
- 54.Xie D., Xie K. Pancreatic cancer stromal biology and therapy. Genes Dis. 2015;2:133–143. doi: 10.1016/j.gendis.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu S.L., Cao S.G., Li Y., Sun B., Chen D., Wang D.S. Pancreatic stellate cells facilitate pancreatic cancer cell viability and invasion. Oncol Lett. 2019;17:2057–2062. doi: 10.3892/ol.2018.9816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Petersen O.W., Nielsen H.L., Gudjonsson T., Villadsen R., Rank F., Niebuhr E. Epithelial to mesenchymal transition in human breast cancer can provide a nonmalignant stroma. Am J Pathol. 2003;162:391–402. doi: 10.1016/S0002-9440(10)63834-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Komar H.M., Serpa G., Kerscher C., Schwoegl E., Mace T.A., Jin M. Inhibition of Jak/STAT signaling reduces the activation of pancreatic stellate cells in vitro and limits caerulein-induced chronic pancreatitis in vivo. Sci Rep. 2017;7:1787. doi: 10.1038/s41598-017-01973-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lin Z., Zheng L.C., Zhang H.J., Tsang S.W., Bian Z.X. Anti-fibrotic effects of phenolic compounds on pancreatic stellate cells. BMC Complement Altern Med. 2015;15:259. doi: 10.1186/s12906-015-0789-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kocaadam B., Sanlier N. Curcumin, an active component of turmeric (Curcuma longa), and its effects on health. Crit Rev Food Sci Nutr. 2017;57:2889–2895. doi: 10.1080/10408398.2015.1077195. [DOI] [PubMed] [Google Scholar]
- 60.Yang C.L., Liu Y.Y., Ma Y.G., Xue Y.X., Liu D.G., Ren Y. Curcumin blocks small cell lung cancer cells migration, invasion, angiogenesis, cell cycle and neoplasia through Janus kinase-STAT3 signalling pathway. PLoS One. 2012;7 doi: 10.1371/journal.pone.0037960. e37960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bharti A.C., Donato N., Singh S., Aggarwal B.B. Curcumin (diferuloylmethane) down-regulates the constitutive activation of nuclear factor-kappa B and IkappaBalpha kinase in human multiple myeloma cells, leading to suppression of proliferation and induction of apoptosis. Blood. 2003;101:1053–1062. doi: 10.1182/blood-2002-05-1320. [DOI] [PubMed] [Google Scholar]
- 62.Shehzad A., Rehman G., Lee Y.S. Curcumin in inflammatory diseases. Biofactors. 2013;39:69–77. doi: 10.1002/biof.1066. [DOI] [PubMed] [Google Scholar]
- 63.Aggarwal B.B. Targeting inflammation-induced obesity and metabolic diseases by curcumin and other nutraceuticals. Annu Rev Nutr. 2010;30:173–199. doi: 10.1146/annurev.nutr.012809.104755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang D.W., Fu M., Gao S.H., Liu J.L. Curcumin and diabetes: a systematic review. Evid Based Complement Alternat Med. 2013;2013:636053. doi: 10.1155/2013/636053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Masamune A., Suzuki N., Kikuta K., Satoh M., Satoh K., Shimosegawa T. Curcumin blocks activation of pancreatic stellate cells. J Cell Biochem. 2006;97:1080–1093. doi: 10.1002/jcb.20698. [DOI] [PubMed] [Google Scholar]
- 66.Shehzad A., Ha T., Subhan F., Lee Y.S. New mechanisms and the anti-inflammatory role of curcumin in obesity and obesity-related metabolic diseases. Eur J Nutr. 2011;50:151–161. doi: 10.1007/s00394-011-0188-1. [DOI] [PubMed] [Google Scholar]
- 67.Schwer C.I., Guerrero A.M., Humar M., Roesslein M., Goebel U., Stoll P. Heme oxygenase-1 inhibits the proliferation of pancreatic stellate cells by repression of the extracellular signal-regulated kinase1/2 pathway. J Pharmacol Exp Ther. 2008;327:863–871. doi: 10.1124/jpet.108.136549. [DOI] [PubMed] [Google Scholar]
- 68.Gundewar C., Ansari D., Tang L., Wang Y., Liang G., Rosendahl A.H. Antiproliferative effects of curcumin analog L49H37 in pancreatic stellate cells: a comparative study. Ann Gastroenterol. 2015;28:391–398. [PMC free article] [PubMed] [Google Scholar]
- 69.Kanai M., Yoshimura K., Asada M., Imaizumi A., Suzuki C., Matsumoto S. A phase I/II study of gemcitabine-based chemotherapy plus curcumin for patients with gemcitabine-resistant pancreatic cancer. Cancer Chemother Pharmacol. 2011;68:157–164. doi: 10.1007/s00280-010-1470-2. [DOI] [PubMed] [Google Scholar]
- 70.Shehzad A., Qureshi M., Anwar M.N., Lee Y.S. Multifunctional curcumin mediate multitherapeutic effects. J Food Sci. 2017;82:2006–2015. doi: 10.1111/1750-3841.13793. [DOI] [PubMed] [Google Scholar]
- 71.Durgaprasad S., Pai C.G., Vasanthkumar Alvres JF., Namitha S. A pilot study of the antioxidant effect of curcumin in tropical pancreatitis. Indian J Med Res. 2005;122:315–318. [PubMed] [Google Scholar]
- 72.Goel A., Kunnumakkara A.B., Aggarwal B.B. Curcumin as “Curecumin”: from kitchen to clinic. Biochem Pharmacol. 2008;75:787–809. doi: 10.1016/j.bcp.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 73.Sun H., Luo G., Chen D., Xiang Z. A comprehensive and system review for the pharmacological mechanism of action of rhein, an active anthraquinone ingredient. Front Pharmacol. 2016;7:247. doi: 10.3389/fphar.2016.00247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang J., Zhao H., Kong W., Jin C., Zhao Y., Qu Y. Microcalorimetric assay on the antimicrobial property of five hydroxyanthraquinone derivatives in rhubarb (Rheum palmatum L.) to Bifidobacterium adolescentis. Phytomedicine. 2010;17:684–689. doi: 10.1016/j.phymed.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 75.Cong X.D., Ding M.J., Dai D.Z., Wu Y., Zhang Y., Dai Y. ER stress, p66shc, and p-Akt/Akt mediate adjuvant-induced inflammation, which is blunted by argirein, a supermolecule and rhein in rats. Inflammation. 2012;35:1031–1040. doi: 10.1007/s10753-011-9407-4. [DOI] [PubMed] [Google Scholar]
- 76.Fernand V.E., Losso J.N., Truax R.E., Villar E.E., Bwambok D.K., Fakayode S.O. Rhein inhibits angiogenesis and the viability of hormone-dependent and -independent cancer cells under normoxic or hypoxic conditions in vitro. Chem Biol Interact. 2011;192:220–232. doi: 10.1016/j.cbi.2011.03.013. [DOI] [PubMed] [Google Scholar]
- 77.He Z.H., Zhou R., He M.F., Lau C.B., Yue G.G., Ge W. Anti-angiogenic effect and mechanism of rhein from Rhizoma Rhei. Phytomedicine. 2011;18:470–478. doi: 10.1016/j.phymed.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 78.Chang C.Y., Chan H.L., Lin H.Y., Way T.D., Kao M.C., Song M.Z. Rhein induces apoptosis in human breast cancer cells. Evid Based Complement Alternat Med. 2012;2012 doi: 10.1155/2012/952504. 952504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yang X., Sun G., Yang C., Wang B. Novel rhein analogues as potential anticancer agents. ChemMedChem. 2011;6:2294–2301. doi: 10.1002/cmdc.201100384. [DOI] [PubMed] [Google Scholar]
- 80.Hu L., Cui R., Liu H., Wang F. Emodin and rhein decrease levels of hypoxia-inducible factor-1alpha in human pancreatic cancer cells and attenuate cancer cachexia in athymic mice carrying these cells. Oncotarget. 2017;8:88008–88020. doi: 10.18632/oncotarget.21330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tsang S.W., Zhang H., Lin C., Xiao H., Wong M., Shang H. Rhein, a natural anthraquinone derivative, attenuates the activation of pancreatic stellate cells and ameliorates pancreatic fibrosis in mice with experimental chronic pancreatitis. PLoS One. 2013;8 doi: 10.1371/journal.pone.0082201. e82201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tsang S.W., Bian Z.X. Anti-fibrotic and anti-tumorigenic effects of rhein, a natural anthraquinone derivative, in mammalian stellate and carcinoma cells. Phytother Res. 2015;29:407–414. doi: 10.1002/ptr.5266. [DOI] [PubMed] [Google Scholar]
- 83.Yang L., Lin S., Kang Y., Xiang Y., Xu L., Li J. Rhein sensitizes human pancreatic cancer cells to EGFR inhibitors by inhibiting STAT3 pathway. J Exp Clin Cancer Res. 2019;38:31. doi: 10.1186/s13046-018-1015-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chacko S.M., Thambi P.T., Kuttan R., Nishigaki I. Beneficial effects of green tea: a literature review. Chin Med. 2010;5:13. doi: 10.1186/1749-8546-5-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hosseini A., Ghorbani A. Cancer therapy with phytochemicals: evidence from clinical studies. Avicenna J Phytomed. 2015;5:84–97. [PMC free article] [PubMed] [Google Scholar]
- 86.Lee L.S., Kim S.H., Kim Y.B., Kim Y.C. Quantitative analysis of major constituents in green tea with different plucking periods and their antioxidant activity. Molecules. 2014;19:9173–9186. doi: 10.3390/molecules19079173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Masamune A., Kikuta K., Satoh M., Suzuki N., Shimosegawa T. Green tea polyphenol epigallocatechin-3-gallate blocks PDGF-induced proliferation and migration of rat pancreatic stellate cells. World J Gastroenterol. 2005;11:3368–3374. doi: 10.3748/wjg.v11.i22.3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Asaumi H., Watanabe S., Taguchi M., Tashiro M., Nagashio Y., Nomiyama Y. Green tea polyphenol (–)-epigallocatechin-3-gallate inhibits ethanol-induced activation of pancreatic stellate cells. Eur J Clin Investig. 2006;36:113–122. doi: 10.1111/j.1365-2362.2006.01599.x. [DOI] [PubMed] [Google Scholar]
- 89.Shen K., Feng X., Su R., Xie H., Zhou L., Zheng S. Epigallocatechin 3-gallate ameliorates bile duct ligation induced liver injury in mice by modulation of mitochondrial oxidative stress and inflammation. PLoS One. 2015;10 doi: 10.1371/journal.pone.0126278. e0126278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Aggarwal B.B., Bhardwaj A., Aggarwal R.S., Seeram N.P., Shishodia S., Takada Y. Role of resveratrol in prevention and therapy of cancer: preclinical and clinical studies. Anticancer Res. 2004;24:2783–2840. [PubMed] [Google Scholar]
- 91.Hasan M., Bae H. An overview of stress-induced resveratrol synthesis in grapes: perspectives for resveratrol-enriched grape products. Molecules. 2017;22 doi: 10.3390/molecules22020294. E294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cho S., Namkoong K., Shin M., Park J., Yang E., Ihm J. Cardiovascular protective effects and clinical applications of resveratrol. J Med Food. 2017;20:323–334. doi: 10.1089/jmf.2016.3856. [DOI] [PubMed] [Google Scholar]
- 93.Bhatt J.K., Thomas S., Nanjan M.J. Resveratrol supplementation improves glycemic control in type 2 diabetes mellitus. Nutr Res. 2012;32:537–541. doi: 10.1016/j.nutres.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 94.Moussa C., Hebron M., Huang X., Ahn J., Rissman R.A., Aisen P.S. Resveratrol regulates neuro-inflammation and induces adaptive immunity in Alzheimer's disease. J Neuroinflammation. 2017;14:1. doi: 10.1186/s12974-016-0779-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Turner R.S., Thomas R.G., Craft S., van Dyck C.H., Mintzer J., Reynolds B.A. A randomized, double-blind, placebo-controlled trial of resveratrol for Alzheimer disease. Neurology. 2015;85:1383–1391. doi: 10.1212/WNL.0000000000002035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Howells L.M., Berry D.P., Elliott P.J., Jacobson E.W., Hoffmann E., Hegarty B. Phase I randomized, double-blind pilot study of micronized resveratrol (SRT501) in patients with hepatic metastases—safety, pharmacokinetics, and pharmacodynamics. Cancer Prev Res. 2011;4:1419–1425. doi: 10.1158/1940-6207.CAPR-11-0148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kulkarni S.S., Canto C. The molecular targets of resveratrol. Biochim Biophys Acta. 2015;1852:1114–1123. doi: 10.1016/j.bbadis.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 98.Patel K.R., Brown V.A., Jones D.J., Britton R.G., Hemingway D., Miller A.S. Clinical pharmacology of resveratrol and its metabolites in colorectal cancer patients. Cancer Res. 2010;70:7392–7399. doi: 10.1158/0008-5472.CAN-10-2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhou J.H., Cheng H.Y., Yu Z.Q., He D.W., Pan Z., Yang D.T. Resveratrol induces apoptosis in pancreatic cancer cells. Chin Med J. 2011;124:1695–1699. [PubMed] [Google Scholar]
- 100.Cheng L., Yan B., Chen K., Jiang Z., Zhou C., Cao J. Resveratrol-induced downregulation of NAF-1 enhances the sensitivity of pancreatic cancer cells to gemcitabine via the ROS/Nrf2 signaling pathways. Oxid Med Cell Longev. 2018;2018 doi: 10.1155/2018/9482018. 9482018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhou C., Qian W., Ma J., Cheng L., Jiang Z., Yan B. Resveratrol enhances the chemotherapeutic response and reverses the stemness induced by gemcitabine in pancreatic cancer cells via targeting SREBP1. Cell Prolif. 2019;52 doi: 10.1111/cpr.12514. e12514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yang L., Yang L., Tian W., Li J., Liu J., Zhu M. Resveratrol plays dual roles in pancreatic cancer cells. J Cancer Res Clin Oncol. 2014;140:749–755. doi: 10.1007/s00432-014-1624-4. [DOI] [PubMed] [Google Scholar]
- 103.Yan B., Cheng L., Jiang Z., Chen K., Zhou C., Sun L. Resveratrol inhibits ROS-promoted activation and glycolysis of pancreatic stellate cells via suppression of miR-21. Oxid Med Cell Longev. 2018;2018 doi: 10.1155/2018/1346958. 1346958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gaman L., Dragos D., Vlad A., Robu G.C., Radoi M.P., Stroica L. Phytoceuticals in acute pancreatitis: targeting the balance between apoptosis and necrosis. Evid Based Complement Alternat Med. 2018;2018 doi: 10.1155/2018/5264592. 5264592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Guo H.C., Bu H.Q., Luo J., Wei W.T., Liu D.L., Chen H. Emodin potentiates the antitumor effects of gemcitabine in PANC-1 pancreatic cancer xenograft model in vivo via inhibition of inhibitors of apoptosis. Int J Oncol. 2012;40:1849–1857. doi: 10.3892/ijo.2012.1389. [DOI] [PubMed] [Google Scholar]
- 106.Han J.W., Shim D.W., Shin W.Y., Heo K.H., Kwak S.B., Sim E.J. Anti-inflammatory effect of emodin via attenuation of NLRP3 inflammasome activation. Int J Mol Sci. 2015;16:8102–8109. doi: 10.3390/ijms16048102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tzeng T.F., Lu H.J., Liou S.S., Chang C.J., Liu I.M. Emodin, a naturally occurring anthraquinone derivative, ameliorates dyslipidemia by activating AMP-activated protein kinase in high-fat-diet-fed rats. Evid Based Complement Alternat Med. 2012;2012 doi: 10.1155/2012/781812. 781812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Liu A., Chen H., Tong H., Ye S., Qiu M., Wang Z. Emodin potentiates the antitumor effects of gemcitabine in pancreatic cancer cells via inhibition of nuclear factor-kappaB. Mol Med Rep. 2011;4:221–227. doi: 10.3892/mmr.2011.414. [DOI] [PubMed] [Google Scholar]
- 109.Saldanha E., Joseph N., Ravi R., Kumar A., Shetty V., Fayad R. Polyphenols in the prevention of acute pancreatitis: preclinical observations. In: Watson R.R., Preedy V.R., Zibadi S., editors. Polyphenols in human health and disease. Academic Press; San Diago, CA: 2014. pp. 427–433. [Google Scholar]
- 110.Kang I., Buckner T., Shay N.F., Gu L., Chung S. Improvements in metabolic health with consumption of ellagic acid and subsequent conversion into urolithins: evidence and mechanisms. Adv Nutr. 2016;7:961–972. doi: 10.3945/an.116.012575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Polce S.A., Burke C., Franca L.M., Kramer B., de Andrade Paes A.M., Carrillo-Sepulveda M.A. Ellagic acid alleviates hepatic oxidative stress and insulin resistance in diabetic female rats. Nutrients. 2018;10 doi: 10.3390/nu10050531. E531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ceci C., Tentori L., Atzori M.G., Lacal P.M., Bonanno E., Scimeca M. Ellagic acid inhibits bladder cancer invasiveness and in vivo tumor growth. Nutrients. 2016;8 doi: 10.3390/nu8110744. E744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Edderkaoui M., Lugea A., Hui H., Eibl G., Lu Q.Y., Moro A. Ellagic acid and embelin affect key cellular components of pancreatic adenocarcinoma, cancer, and stellate cells. Nutr Cancer. 2013;65:1232–1244. doi: 10.1080/01635581.2013.832779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Suzuki N., Masamune A., Kikuta K., Watanabe T., Satoh K., Shimosegawa T. Ellagic acid inhibits pancreatic fibrosis in male Wistar Bonn/Kobori rats. Dig Dis Sci. 2009;54:802–810. doi: 10.1007/s10620-008-0423-7. [DOI] [PubMed] [Google Scholar]
- 115.Masamune A., Satoh M., Kikuta K., Suzuki N., Satoh K., Shimosegawa T. Ellagic acid blocks activation of pancreatic stellate cells. Biochem Pharmacol. 2005;70:869–878. doi: 10.1016/j.bcp.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 116.Huang Y., Lu J., Gao X., Li J., Zhao W., Sun M. PEG-derivatized embelin as a dual functional carrier for the delivery of paclitaxel. Bioconjug Chem. 2012;23:1443–1451. doi: 10.1021/bc3000468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kim S.W., Kim S.M., Bae H., Nam D., Lee J.H., Lee S.G. Embelin inhibits growth and induces apoptosis through the suppression of Akt/mTOR/S6K1 signaling cascades. The Prostate. 2013;73:296–305. doi: 10.1002/pros.22574. [DOI] [PubMed] [Google Scholar]
- 118.Xu M., Cui J., Fu H., Proksch P., Lin W., Li M. Embelin derivatives and their anticancer activity through microtubule disassembly. Planta Med. 2005;71:944–948. doi: 10.1055/s-2005-871250. [DOI] [PubMed] [Google Scholar]
- 119.Ahn K.S., Sethi G., Aggarwal B.B. Embelin, an inhibitor of X chromosome-linked inhibitor-of-apoptosis protein, blocks nuclear factor-kappaB (NF-kappaB) signaling pathway leading to suppression of NF-kappaB-regulated antiapoptotic and metastatic gene products. Mol Pharmacol. 2007;71:209–219. doi: 10.1124/mol.106.028787. [DOI] [PubMed] [Google Scholar]
- 120.Bailey J.M., Swanson B.J., Hamada T., Eggers J.P., Singh P.K., Caffery T. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin Cancer Res. 2008;14:5995–6004. doi: 10.1158/1078-0432.CCR-08-0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Huang M., Tang S.N., Upadhyay G., Marsh J.L., Jackman C.P., Shankar S. Embelin suppresses growth of human pancreatic cancer xenografts, and pancreatic cancer cells isolated from KrasG12D mice by inhibiting Akt and Sonic hedgehog pathways. PLoS One. 2014;9 doi: 10.1371/journal.pone.0092161. e92161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chen J., Chen X., Lei Y., Wei H., Xiong C., Liu Y. Vascular protective potential of the total flavanol glycosides from Abacopteris penangiana via modulating nuclear transcription factor-kappaB signaling pathway and oxidative stress. J Ethnopharmacol. 2011;136:217–223. doi: 10.1016/j.jep.2011.04.052. [DOI] [PubMed] [Google Scholar]
- 123.Zhao Z., Ruan J., Jin J., Zou J., Zhou D., Fang W. Flavan-4-ol glycosides from the rhizomes of. Abacopteris penangiana. J Nat Prod. 2006;69:265–268. doi: 10.1021/np050191p. [DOI] [PubMed] [Google Scholar]
- 124.Fang J.B., Chen J.C., Duan H.Q. Constituents from Abacopteris penangiana and their cytotoxicity activity. Chin Tradit Herb Drugs. 2010;10:1601–1604. [Google Scholar]
- 125.Tsang S.W., Zhang H.J., Chen Y.G., Auyeung K.K., Bian Z.X., Eruberin A. A natural flavanol glycoside, exerts anti-fibrotic action on pancreatic stellate cells. Cell Physiol Biochem. 2015;36:2433–2446. doi: 10.1159/000430204. [DOI] [PubMed] [Google Scholar]
- 126.Bailey C.J. Metformin: historical overview. Diabetologia. 2017;60:1566–1576. doi: 10.1007/s00125-017-4318-z. [DOI] [PubMed] [Google Scholar]
- 127.Algire C., Amrein L., Zakikhani M., Panasci L., Pollak M. Metformin blocks the stimulative effect of a high-energy diet on colon carcinoma growth in vivo and is associated with reduced expression of fatty acid synthase. Endocr Relat Cancer. 2010;17:351–360. doi: 10.1677/ERC-09-0252. [DOI] [PubMed] [Google Scholar]
- 128.Gou S., Cui P., Li X., Shi P., Liu T., Wang C. Low concentrations of metformin selectively inhibit CD133+ cell proliferation in pancreatic cancer and have anticancer action. PLoS One. 2013;8 doi: 10.1371/journal.pone.0063969. e63969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Chae Y.K., Arya A., Malecek M.K., Shin D.S., Carneiro B., Chandra S. Repurposing metformin for cancer treatment: current clinical studies. Oncotarget. 2016;7:40767–40780. doi: 10.18632/oncotarget.8194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Song C.W., Lee H., Dings R.P., Williams B., Powers J., Santos T.D. Metformin kills and radiosensitizes cancer cells and preferentially kills cancer stem cells. Sci Rep. 2012;2:362. doi: 10.1038/srep00362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Duan W., Chen K., Jiang Z., Chen X., Sun L., Li J. Desmoplasia suppression by metformin-mediated AMPK activation inhibits pancreatic cancer progression. Cancer Lett. 2017;385:225–233. doi: 10.1016/j.canlet.2016.10.019. [DOI] [PubMed] [Google Scholar]
- 132.Wu C., Qiu S., Zhu X., Lin H., Li L. OCT1-mediated metformin uptake regulates pancreatic stellate cell activity. Cell Physiol Biochem. 2018;47:1711–1720. doi: 10.1159/000491003. [DOI] [PubMed] [Google Scholar]
- 133.Qian W., Li J., Chen K., Jiang Z., Cheng L., Zhou C. Metformin suppresses tumor angiogenesis and enhances the chemosensitivity of gemcitabine in a genetically engineered mouse model of pancreatic cancer. Life Sci. 2018;208:253–261. doi: 10.1016/j.lfs.2018.07.046. [DOI] [PubMed] [Google Scholar]
- 134.Zechner D., Burtin F., Albert A.C., Zhang X., Kumstel S., Schonrogge M. Intratumoral heterogeneity of the therapeutical response to gemcitabine and metformin. Oncotarget. 2016;7:56395–56407. doi: 10.18632/oncotarget.10892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gu L., Zhang H., Liu T., Draganov A., Yi S., Wang B. Inhibition of MDM2 by a rhein-derived compound AQ-101 suppresses cancer development in SCID mice. Mol Cancer Ther. 2018;17:497–507. doi: 10.1158/1535-7163.MCT-17-0566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kalyanaraman B., Cheng G., Hardy M., Ouari O., Sikora A., Zielonka J. Modified metformin as a more potent anticancer drug: mitochondrial inhibition, redox signaling, antiproliferative effects and future EPR studies. Cell Biochem Biophys. 2017;75:311–317. doi: 10.1007/s12013-017-0796-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chen K.C., Juang S.H., Lien J.C. Identification of antiproliferative emodin analogues as inhibitors of epidermal growth factor receptor in cancer. Int J Mol Med. 2019;43:1281–1288. doi: 10.3892/ijmm.2019.4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Viault G., Babu K.S., Gautier F., Barille-Nion S., Juin P., Tasseau O. Hemisynthesis of selected embelin analogs and investigation of their proapoptotic activity against cancer cells. Med Chem. 2013;9:1028–1034. doi: 10.2174/1573406411309080003. [DOI] [PubMed] [Google Scholar]
- 139.Chatterjee A., Ronghe A., Padhye S.B., Spade D.A., Bhat N.K., Bhat H.K. Antioxidant activities of novel resveratrol analogs in breast cancer. J Biochem Mol Toxicol. 2018;32 doi: 10.1002/jbt.21925. e21925. [DOI] [PubMed] [Google Scholar]
- 140.Stadlbauer S., Steinborn C., Klemd A., Hattori F., Ohmori K., Suzuki K. Impact of green tea catechin ECG and its synthesized fluorinated analogue on prostate cancer cells and stimulated immunocompetent cells. Planta Med. 2018;84:813–819. doi: 10.1055/s-0044-102099. [DOI] [PubMed] [Google Scholar]
- 141.Poilil Surendran S., George Thomas R., Moon M.J., Jeong Y.Y. Nanoparticles for the treatment of liver fibrosis. Int J Nanomed. 2017;12:6997–7006. doi: 10.2147/IJN.S145951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sharma N., Bhandari S., Deshmukh R., Yadav A.K., Mishra N. Development and characterization of embelin-loaded nanolipid carriers for brain targeting. Artif Cells Nanomed Biotechnol. 2017;45:409–413. doi: 10.3109/21691401.2016.1160407. [DOI] [PubMed] [Google Scholar]
- 143.Snima K.S., Jayakumar R., Lakshmanan V.K. In vitro and in vivo biological evaluation of O-carboxymethyl chitosan encapsulated metformin nanoparticles for pancreatic cancer therapy. Pharm Res. 2014;31:3361–3370. doi: 10.1007/s11095-014-1425-0. [DOI] [PubMed] [Google Scholar]
- 144.Wei Y., Wang Y., Xia D., Guo S., Wang F., Zhang X. Thermosensitive liposomal codelivery of HSA-paclitaxel and HSA-ellagic acid complexes for enhanced drug perfusion and efficacy against pancreatic cancer. ACS Appl Mater Interfaces. 2017;9:25138–25151. doi: 10.1021/acsami.7b07132. [DOI] [PubMed] [Google Scholar]
- 145.Yallapu M.M., Ebeling M.C., Khan S., Sundram V., Chauhan N., Gupta B.K. Novel curcumin-loaded magnetic nanoparticles for pancreatic cancer treatment. Mol Cancer Ther. 2013;12:1471–1480. doi: 10.1158/1535-7163.MCT-12-1227. [DOI] [PMC free article] [PubMed] [Google Scholar]