

Abstract
Nonalcoholic steatohepatitis (NASH) is recognized by hepatic lipid accumulation, inflammation, and fibrosis. No studies have evaluated the prolonged‐release pirfenidone (PR‐PFD) properties on NASH features. The aim of this study is to evaluate how PR‐PFD performs on metabolic functions, and provide insight on a mouse model of human NASH. Male C57BL/6J mice were fed with either normo diet or high‐fat/carbohydrate diet for 16 weeks and a subgroup also fed with PR‐PFD (300 mg/kg/day). An insulin tolerance test was performed at the end of treatment. Histological analysis, determination of serum hormones, adipocytokines measurement, and evaluation of proteins by western blot was performed. Molecular docking, in silico site‐directed mutagenesis, and in vitro experiments using HepG2 cultured cells were performed to validate PR‐PFD binding to peroxisome proliferator–activated receptor alpha (PPAR‐α), activation of PPAR‐α promoter, and sirtuin 1 (SIRT1) protein expression. Compared with the high‐fat group, the PR‐PFD‐treated mice displayed less weight gain, cholesterol, very low density lipoprotein and triglycerides, and showed a significant reduction of hepatic macrosteatosis, inflammation, hepatocyte ballooning, fibrosis, epididymal fat, and total adiposity. PR‐PFD restored levels of insulin, glucagon, adiponectin, and resistin along with improved insulin resistance. Noteworthy, SIRT1–liver kinase B1–phospho‐5′ adenosine monophosphate–activated protein kinase signaling and the PPAR‐α/carnitine O‐palmitoyltransferase 1/acyl‐CoA oxidase 1 pathway were clearly induced in high fat + PR‐PFD mice. In HepG2 cells incubated with palmitate, PR‐PFD induced activation and nuclear translocation of both PPARα and SIRT1, which correlated with increased SIRT1 phosphorylated in serine 47, suggesting a positive feedback loop between the two proteins. These results were confirmed with both synthetic PPAR‐α and SIRT1 activators and inhibitors. Finally, we found that PR‐PFD is a true agonist/ligand for PPAR‐α. Conclusions: PR‐PFD provided an anti‐steatogenic effect and protection for inflammation and fibrosis.
Pirfenidone is an agonist/ligand for PPAR‐α and improves inflammation, fibrosis, insulin sensitivity, and fat metabolism in a mouse model of human NASH through the SIRT1‐LKB1‐pAMPK noncanonical pathway.
Abbreviations
- ACOX1
acyl‐CoA oxidase 1
- ALA333
alanine 333
- AMPK
adenosine monophosphate–activated protein kinase
- AUC
area under the curve
- CPT1A
carnitine O‐palmitoyltransferase 1
- EMSA
electrophoretic mobility‐shift assay
- EX527
Selisistat, a selective SIRT1 inhibitor
- FA
fatty acid
- FFB
Fenofibrate
- H&E
hematoxylin and eosin
- HF
high fat
- HFP
high fat + Pirfenidone
- IL
interleukin
- ITT
insulin tolerance test
- LKB1
liver kinase B1
- MET
methionine
- mRNA
messenger RNA
- NAD+
nicotinamide adenine dinucleotide
- NAFLD
nonalcoholic fatty liver disease
- NAM
nicotinamide
- NASH
nonalcoholic steatohepatitis
- ND
normo diet
- pAMPK
phospho‐5′ adenosine monophosphate–activated protein kinase
- PFD
pirfenidone
- PLBD
pocket of the ligand binding domain
- PPAR
peroxisome proliferator–activated receptor
- PPRE
PPAR‐α response elements
- PR‐PFD
prolonged‐release pirfenidone
- SDP
switch‐diet + PDF
- Ser47
serine 47
- SIRT
sirtuin
- TYR334
tyrosine 334
Nonalcoholic fatty liver disease (NAFLD) is currently one of the most common chronic liver diseases.1 Nonalcoholic steatohepatitis (NASH) pathogenesis suggests multiple potential therapeutic targets,2 as dietary changes and lifestyle modifications are the first‐line therapy for patients with NASH.3 However, the constantly growing number of pharmacological approaches being developed reveals the concern of scientists around the world.4 We have focused this study on the peroxisome proliferator–activated receptor (PPAR) modulators and their upstream regulators.
It has been well‐documented by both pharmacological and genetic studies that sirtuin 1 (SIRT1), a class III nicotinamide adenine dinucleotide (NAD+)‐dependent histone/protein deacetylase, is a crucial regulator of liver fat metabolism that prevents and improves NAFLD features and many other obesity‐related diseases.5 SIRT1 works as a main energy sensor and regulates homeostatic transcriptional responses.6 SIRT1 deacetylates liver kinase B1 (LKB1), which in turn affects the adenosine monophosphate–activated protein kinase (AMPK) phosphorylation on threonine (Thr) 172, an event that is required for its activation7 and further action on cellular energy state, mitochondrial function, and lipid metabolism.8 Human SIRT1 is phosphorylated by c‐Jun N‐terminal kinase 1 on serine 47 (Ser47), and this SIRT1 phosphorylation increases its nuclear localization and enzymatic activity.9 PPARs are nuclear receptors that function as transcriptional factors and are also able to regulate expression levels of SIRT1. It is known that Sirt1 promoter has several PPAR‐αresponse elements (PPREs), which mediate increased SIRT1 protein. In addition, Sirt1 can enhance the activity of PPAR‐α through its co‐activators, suggesting a positive feedback loop.10, 11 PPAR‐α controls the expression of genes related to lipid metabolism in the liver, including genes involved in mitochondrial b‐oxidation, fatty acid (FA) uptake and binding, and lipoprotein transport.12 Thus, PPAR‐α activation provides a number of metabolic benefits triggering the up‐regulation of key lipid‐burning enzymes in mitochondria and peroxisomes as carnitine O‐palmitoyltransferase 1 (CPT1) and acyl‐CoA oxidase 1 (ACOX1).13
Pirfenidone (PFD) is an antifibrogenic molecule used for the treatment of idiopathic pulmonary fibrosis and liver fibrosis.14 PFD has been reported to markedly attenuate liver fibrosis in NASH‐like animal models, using western diet–fed melanocortin 4 receptor–deficient mice (MC4R‐KO) mice15 and decreased steatohepatitis by polarizing M2 macrophages in C57BL/6J mice.16 The aim of this study was to investigate the prolonged‐release PFD (PR‐PFD) effect in NASH metabolic alterations by evaluating the prevention or amelioration of insulin resistance, hepatic lipid accumulation and histological changes, as well as molecular mechanisms in an obesity‐related NASH model. Furthermore, PFD was found to be an agonistic ligand for PPAR‐α by using in silico analysis and corroborated by in vitro cell culture transfections with the PPAR‐α promoter.17
Materials and Methods
Chemicals
PR‐PFD was kindly donated by Cell Pharma S.A. de C.V. (Cuernavaca, Mexico). This new formulation of pirfenidone is delivered over a 10‐hour period. All other chemicals used were of reagent grade.
Cell Culture
HepG2 cells were transiently transfected with the pα(H‐H)‐pGL3 plasmid, which contains the complete PPAR‐α promoter sequence, or with pα(X‐H)‐pGL3 containing a PPAR‐α 5′‐end truncated promoter sequence.18 Forty‐eight hours after transfection, cells were serum‐starved for 8 hours with 500 µM PFD. Additionally, HepG2 cells were incubated with palmitate as indicated in Parra‐Vargas et al.19 with 2 mM nicotinamide (NAM) for 16 hours and treated afterward with 500 μM PFD for 30 minutes, 2 hours, 4 hours, and 8 hours to monitor the PFD effect on SIRT1 protein expression. NAM exerts a potent end‐product inhibition on SIRT1 activity in a noncompetitive fashion with NAD+. In addition, cells were incubated for 8 hours with 10 µM SIRT1720 and 40 µM EX527 (Selisistat, a selective SIRT1 inhibitor), used as specific SIRT1 activator and inhibitor, respectively. Incubation was performed after 8 hours of fetal bovine serum starving.
Dual Luciferase Activity Assay and Electrophoretic Mobility‐Shift Assay
After transfection for 24 hours, a dual‐luciferase assay kit (Promega, Madison, WI) was used to detect firefly luciferase activity by the Veritas Microplate Luminometer (Santa Clara, CA). Renilla luciferase activity was used to normalize the firefly luciferase signal. The results were expressed as the normalized firefly luciferase activity of pα(H‐H)‐pGL3 and pα(X‐H)‐pGL3 transfected cells relative to that of pGL3‐Basic transfected cells, with an arbitrary unit set as one. The assay was performed at least 3 times to confirm the results. Electrophoretic mobility‐shift assay (EMSA) was performed according to previously published work.18
Molecular Docking and Structure Viewing
SwissDock web server based on the EADock DSS engine was used for molecular docking. University of California at San Francisco CHIMERA software was used to visualize the molecular structure and predicted docking results. Crystal structure of human PPAR‐α‐ligand binding domain was obtained from the Protein Data Bank (PDB) with accession code 1I7G. Fenofibrate (FFB) and PFD 3D structures were obtained from the ZINC database of commercially available compounds (accession number 584092 and 1958, respectively). Site direct mutations in PPAR‐α aminoacids binding to PFD were performed using the SWISS‐MODEL server.
Animals and Diet
Four‐week‐old male C57BL/6J mice weighing 20 g to 25 g were fed standard diet and water before the experiment. Mice received care according to Official Mexican Norm NOM‐062‐ZOO‐1999, Guidelines of the University of Guadalajara Animal Facility, and the criteria outlined in the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health. Mice were housed in a 12‐hour light/dark cycle and fed ad libitum with free access to beverage. After 1‐week acclimatization, mice were randomly allocated into five groups and fed for 8 weeks either with standard diet (Envigo T.2018S.15) and water (normo diet [ND], n = 5) or high‐fat diet (Envigo TD.06414) plus high‐carbohydrate beverage (2.31% fructose, 1.89% sucrose) (HF). After 8 weeks, the HF animals were divided in two subgroups (n = 5): The HF group continued with the HF diet until 16 weeks, to induce NASH without treatment, whereas another five cognate animals (HF + PFD [HFP] group) received approximately 300 mg/kg body weight/day PR‐PFD from week 8 to week 16. Additionally, another two HF‐fed groups underwent diet switch from week 8 to week 16, feeding them with regular diet and pure water as a control for standard recommended NASH management (switch diet). A set of mice (switch‐diet + PDF [SDP]) received, in addition to the switch diet, PR‐PDF supplementation (~300 mg/kg body weight/day) in food. Animals were sacrificed under anesthesia after 4 hours of fasting. Liver and epididymal adipose tissue were immediately collected and weighed. For histological examination, liver tissue of the three main lobes and part of the epididymal fat pad were fixed with 4% paraformaldehyde. A liver section was cryopreserved in tissue‐freezing medium to detect the presence of fat by Oil Red O staining.
Energy Intake, Weight, Fasting Glucose, and Insulin Sensitivity
Food intake was measured systematically 3 times per week at 9:00‐10:00 am, and energy ingestion was calculated from food and beverage consumed. Ingestion‐related data were analyzed in both phases: before and during PR‐PFD treatment. Weight and 4‐hour fasting glucose in mice were recorded weekly during the study. Glucose levels were measured with a blood glucose meter in tail vein blood. At the end of the treatment, 48 hours before sacrifice, mice were short‐fasted for 4 hours for analysis with an insulin tolerance test (ITT) after intraperitoneal injection of human‐recombinant short‐acting insulin at a standardized dose of 0.025 U/mice. Glucose levels were measured at 0, 30, 60, and 90 minutes after insulin injection. Once the protocol was finished, all animals were given free access to food and water. Finally, the area under the curve (AUC) was calculated.
Serum Biomarkers, Liver, and Fat Histological Analysis
Leptin, insulin, glucagon, adiponectin, and resistin were measured in serum by multiplex detection immunoassay. Two independent pathologists performed histology analysis in Masson's and hematoxylin and eosin (H&E) slides. Hepatocyte ballooning, inflammation nodules, microsteatosis, and macrosteatosis were quantified. In addition, periportal, centrilobulilar fibrosis and bridging score were scrutinized. Oil Red O staining was performed in frozen liver sections using Image‐ProPlus 6.0 in 10 microscopic fields/tissue section. Total adiposity was calculated as the mean adipocyte area in square micrometers in 10 microscopic fields at ×40.
Immunoblotting and Immunofluorescence
Liver or cell homogenates (50 μg of total protein) were separated by 8% sodium dodecyl sulfate polyacrylamide gel electrophoresis gel, proteins were transferred to polyvinylidene fluoride membranes and immunoblotted with specific antibodies (phosphorylated AMPK [pAMPK] Thr172, 1:500; SIRT1, LKB1, AMPK‐α1/2, PPAR‐α, CPT1A, ACOX1, beta‐tubulin, and beta‐actin) overnight at 4°C, washed and then revealed using anti‐Mouse‐ immunoglobulin G(IgG)‐peroxidase (POD)/anti‐Rabbit‐IgG‐POD. Band intensities were quantified with the ChemiDoc XRS+ and Image Lab 5.0 software. Beta‐actin or tubulin were used as loading control. Immunofluorescence for SIRT1, PPAR‐α, and luciferase in HepG2 cells was performed. Nuclei were stained with 4′,6‐diamidino‐2‐phenylindole. Images were analyzed using Image‐ProPlus 6.0. Marker‐positive staining was determined by taking representative images.
Statistical Analysis
Data are presented as the mean ± SD or SEM. Shapiro‐Wilk test was used to establish normality of variables. Statistical significance was determined for parametric data with one‐way analysis of variance and Tukey's or Bonferroni post hoc test, and Kruskal‐Wallis and Mann‐Whitney U test for nonparametric data (Graph Prism 5.0). P value less than 0.05 was considered to be statistically significant.
Results
PFD Reduces Animal and Fat Weight and Improves Insulin Resistance in HF Mice
As shown in Fig. 1A, HF‐diet groups showed significant increase in weight from the fourth week onward. As seen in switch diet + SDP groups, a reduced body weight was achieved as early as 1 week after the diet switch, reaching ND weight by the end of the study. At the moment of sacrifice, animals in the HF group had the highest weight (49.2 ± 4.7 g), followed by HFP animals (42.2 ± 6.5 g) showing a significant difference (P < 0.05) (Fig. 1B). The control diet group and switch diet groups showed no statistical difference in animal weight. Liver weight/body weight ratio was calculated and no difference was found between groups (Supporting Fig. S1F). Epididymal fat pad in Fig. 1C also showed a statistical difference between the HF and HFP groups (P < 0.05) and no difference between normo‐diet group and switch diet groups.
Figure 1.
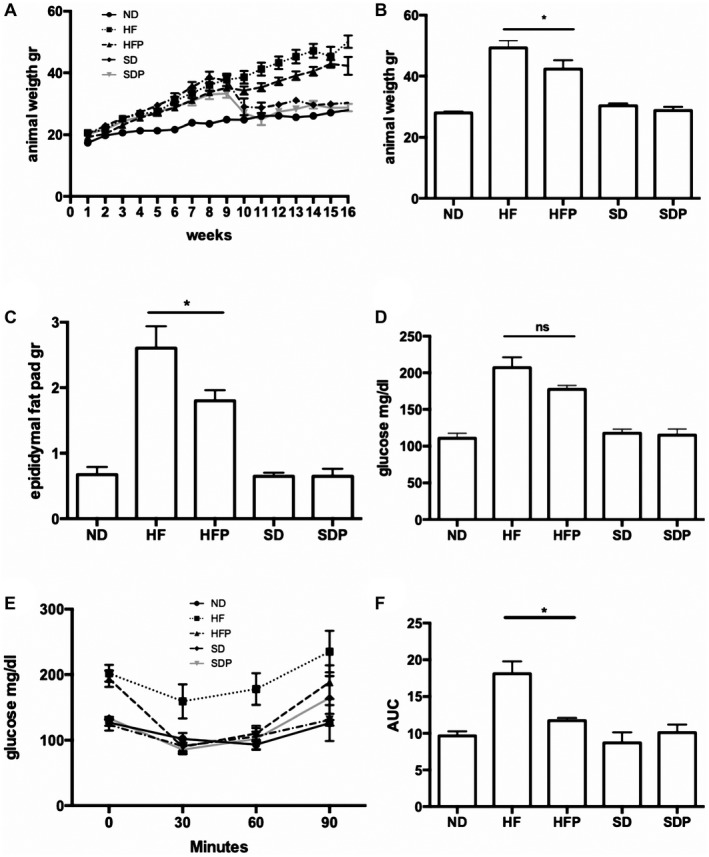
Animal weight and glucose determination. (A) Weekly weight gain. Switch diet and SDP groups changed diet at week 8, with dramatically diminishing weight. (B) Weight at sacrifice. (C) Epididymal adipose fat pad weight. The HFP group showed a statistical diminution in animal and fat weight compared with the HF animals (P < 0.01). (D) Fasting glucose at week 16. HFP animals presented a slight decrease in serum glucose compared with the HF group. (E) ITT. The HFP group showed insulin sensitivity comparable to the ND group. (F) The AUC for ITT is improved in HFP animals (P < 0.01). Data are expressed as the media of the group ± SEM. Abbreviations: ns, not significant; SD, switch diet.
PR‐PFD‐HF mice displayed an increase in insulin sensitivity measured by ITT (Fig. 1E,F) as shown by decreased AUC (P < 0.05). The lowest AUC value was observed in ND mice, while the highest values were detected in HF mice.
PFD Does Not Modify Daily Energy Intake
Supporting Table S1 indicates that daily energy intake was comparable across all groups during the study, as calories from dietary intake (food plus beverage) were used to calculate the total daily energy intake. Analysis of daily energy intake was registered before and during treatment. All animal groups gradually increased in body weight during the experimental phase. After being fed an HF diet for 16 weeks, a significant difference in weight gain compared with ND mice was noted. The HF and HFP groups showed higher energy intake values (13.5 ± 1.3 and 12.7 ± 1.5, respectively). Furthermore, during the treatment phase, values in the HF and HFP groups remained unaltered. Similar values to HF diets in daily energy intake in the switch diet and SDP groups were noted (15.1 ± 6.8 and 14.5 ± 5.4, respectively), even when the diet was switched. After 8 weeks of PR‐PFD intervention, food‐intake values of each group remained essentially unaltered between experimental phases. The daily energy intake from fat was lower in the ND control group due to the fat energy percentage of the experimental standard diet.
PFD Attenuates Liver Injury in HF Mice
Histological analysis of the HF group showed substantial inflammatory changes, predominantly in the periportal area with neutrophils and mononuclear cells. A reduction in inflammation foci was achieved after PR‐PFD treatment from 14.8 ± 4.5 to 8.0 ± 7.1 (P < 0.001). Livers from HF animals had an increased number in fat droplets (P < 0.01, Fig. 2A), and microsteatosis and macrosteatosis had a specific distribution pattern: The first was localized in acinar zones 2 and 3 (central/perivenular), and the second was circumscribed to acinar zone 1 (periportal zone). In comparison, HFP mice had the same distribution pattern, although macrosteatosis significantly decreased (P < 0.0001). Microsteatosis showed a tendency to decrease in HFP mice, but this was not significant. There was also evidence of moderate hepatocyte‐ballooning degeneration in such area, which decreased with PR‐PFD. Periportal and pericentral fibrosis were higher in the HF group (P < 0.01 and P < 0.05), while histological changes that were consistent with liver histological improvement were observed in HFP animals (Fig. 2B,C). Fibrotic bridges were counted in each microscopic field, and a statistical difference was achieved (P < 0.05) (Fig. 2A).
Figure 2.
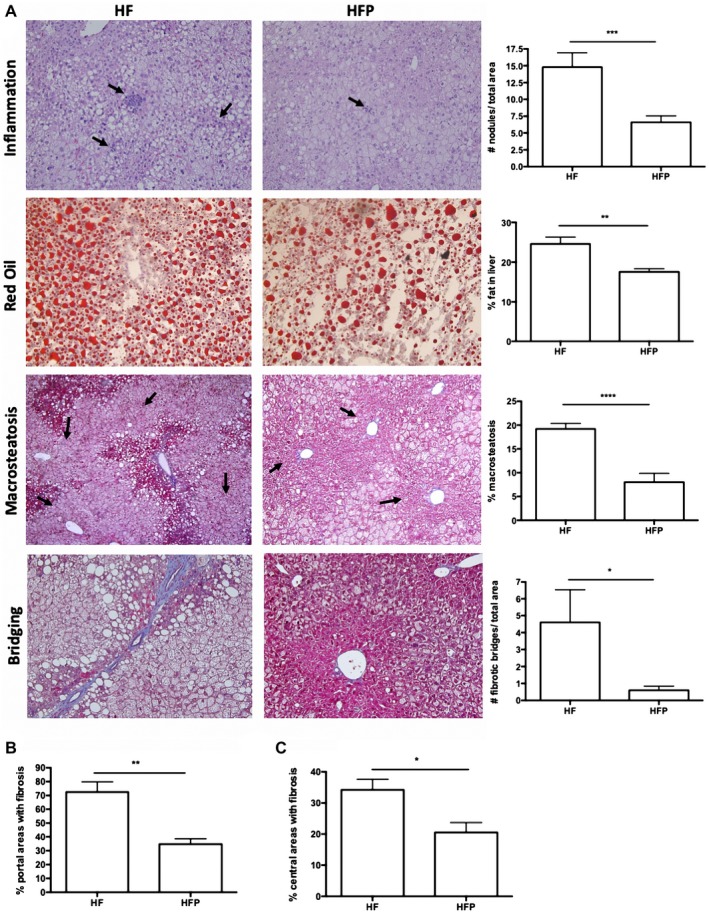
Histological analysis of liver in NASH obesity–induced animals treated with PR‐PFD. (A) HF animals had a 1‐fold increase in inflammation nodules (P < 0.001). Arrows indicate inflammation nodules. Oil Red O staining in fresh samples showed a decrease in fat staining in HFP animals (P < 0.01). Percentage of macrosteatosis was dramatically diminished in liver samples of animals treated with PR‐PFD (P < 0.0001). Representative pictures are displayed at ×10. Total bridges were counted by an independent pathologist. Representative pictures are shown at ×20. Periportal (B) and centrilobulillar (C) areas were evaluated for fibrosis (P < 0.01 and P < 0.05, respectively); ×40. Data are expressed as the median of the group ± SEM.
PFD Reduces Biochemical Markers of Liver Damage and Improves Lipid and Adipokine Serum Levels
Systemic markers of hepatic and lipid function, IL‐17 and adipokines were measured to correlate with the histological results. As indicated in Supporting Fig. S1A, statistically lower aspartate aminotransferase values were found in comparison to HF animals (P > 0.0001), although did not reach similar levels to control animals (233.3 ± 20.2; P > 0.05). Alanine aminotransferase values were 91.2 ± 5.2 and 42.0 ± 8.6 (P > 0.0001) for the HF and HFP groups, respectively (Supporting Fig. S1B). Higher serum triglycerides (P > 0.0001), cholesterol (P > 0.0001), and very low density lipoprotein (P > 0.0001) levels (Supporting Fig. S1C‐E) were found in the HF group compared with the HFP or control groups (P > 0.05). This amelioration after PR‐PFD treatment in these lipid serum markers correlates with histological determination of total adiposity (Fig. 4B). Regarding adipocytokines, resistin (Fig. 3A) was significantly reduced in the HFP group compared with the HF group (169,481 ± 36,275 and 298,199 ± 46,268 pg/mL, P > 0.01, respectively). On the other hand, adiponectin (3,455 ± 298 vs. 1,750 ± 213 pg/mL, P > 0.001) increased in the HFP animals (Fig. 3B). Insulin and glucagon remained unaltered (Fig. 3D,E). Noteworthy, IL‐17 quantities decreased dramatically in the PR‐PFD‐treated mice (Fig. 3D; P > 0.0001).
Figure 4.
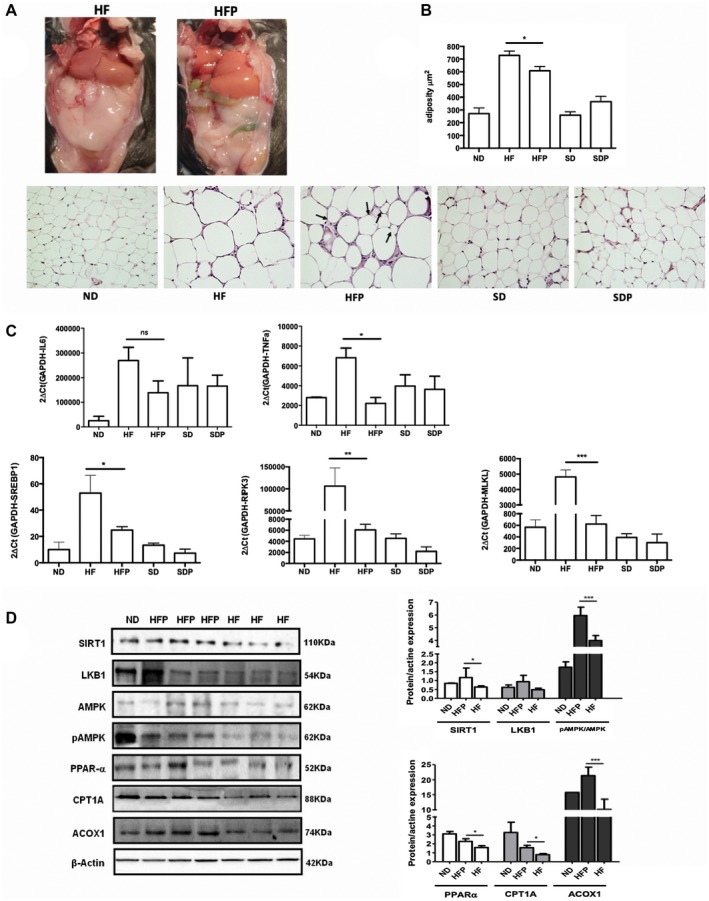
PR‐PFD modifies the adiposity and gene expression, and induces PPAR‐α signaling and SIRT1. (A) Fat pads in mice were diminished in the HPF groups. (B) Representative pictures of adipose tissue stained with H&E are displayed at ×20. Fewer adipocytes in the ND, switch diet, SDP, and HFP animals compared with the HF mice. Morphometric analysis showed a statistically significant diminution in adipocyte area in HFP (P < 0.05). (C) IL6‐mRNA levels showed a tendency to decrease. Tumor necrosis factor α, SREBP1, RIPK3, and MLKL gene expression are diminished in mice treated with PR‐PFD (P < 0.05, P < 0.01, P < 0.01 and P < 0.001, respectively). (D) Representative western blots (three of six liver tissues of animals) and analysis of SIRT1, LKB1, AMPK, pAMPK, PPAR‐α, CPT1A, ACOX1, and beta‐actin in the mouse model. Abbreviations: MLKL, mixed lineage kinase domain‐like; ns, not significant; RIPK3, receptor‐interacting serine/threonine‐protein kinase 3; SD, switch diet.
Figure 3.
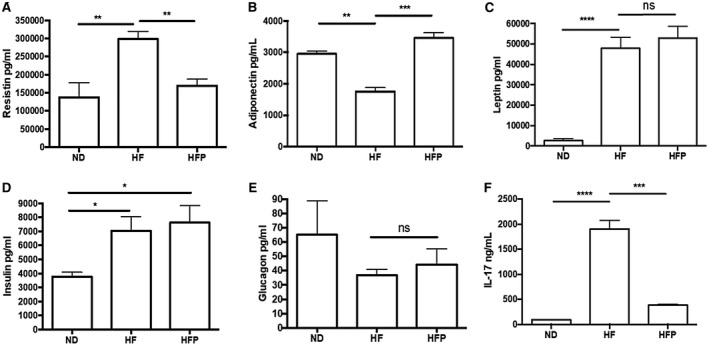
Biochemical markers and adipokines serum levels. (A) Resistin levels demonstrated a 5‐fold decrease in the HFP animals (P < 0.01). (B) Adiponectin (P < 0.001) increases in the HFP group (C) Leptin showed no statistical difference between high‐fat groups. (D) insulin was increased in HF and HFD animals compared with the ND group (P < 0.01). (E) Glucagon showed no statistical difference between groups. (F) IL‐17 levels decreased (P < 0.0001) in the HFP animals. Data are expressed as the median of the group ± SEM. Abbreviation: ns, not significant.
PFD Reduces Adiposity and Gene Expression of Inflammatory and Necroptosis Molecules
Epididymal fat tissue was removed from each mouse, and weighed (Fig. 1C) and stained slides with H&E were analyzed to measure the adipocyte area. The ND and switch diet groups had smaller fat cells, whereas HF animals had larger adipocytes (Fig. 4B). Remarkably, PR‐PFD significantly reduced the total adiposity area (729.0 ± 75.4 vs. 608.4 ± 73.6 μm2; P < 0.05). Liver levels of messenger RNA (mRNA) of two main inflammatory molecules like IL‐6 reduced 1.9‐fold times and tumor necrosis factor α (TNF‐α) diminished 3.1‐fold times in HF animals treated with PR‐PFD. Noteworthy, markers for lipogenesis (sterol regulatory element‐binding transcription factor 1 [SREBP1], P < 0.05) and necroptosis (receptor‐interacting serine/threonine‐protein kinase 3 and mixed lineage kinase domain‐like, P < 0.01 and P < 0.001, respectively) were clearly decreased in HFP mice (Fig. 4C).
PR‐PFD Induces PPAR‐α Signaling and SIRT1 Both In Vivo and In Vitro
Molecules implicated in lipolysis pathway were examined by western blot (Fig. 4D). A clear increase in SIRT1, LKB1, and pAMPK was noted in livers from PFD‐treated animals (P < 0.05). Moreover, axis PPAR‐α/CPT1A/ACOX1 resulted in overexpression after PFD treatment (P < 0.05, P < 0.001). The lipotoxicity model in HepG2 cells indicates that PFD incubation diminished intracellular lipid accumulation (P < 0.001, Fig. 5A). Also, in vitro induction of SIRT1 expression by PFD was corroborated using SIRT1 activator and inhibitors. Figure 5B indicates that incubation with PFD increases SIRT1 expression in a time‐dependent fashion beginning at 4 hours, whereas 2‐mM NAM, which exerts a potent end‐product inhibition on SIRT1 activity in a noncompetitive fashion with NAD+, prevents SIRT1 overexpression. Simultaneous use of PFD plus specific SIRT1 activator and inhibitor demonstrated that PFD increases SIRT1 levels similarly to the SIRT1720 activator. PFD also restores SIRT1 expression even in the presence of inhibitor EX527 (Fig. 5C). Phosphorylated SIRT1 in Ser47 was increased by SIRT1720 and PFD and decreased by EX527, an effect that was overridden by PFD itself (Fig. 5C). Noteworthy, immunofluorescence images showed that PFD increases nuclear translocation of both PPAR‐α and SIRT1 in HepG2 cells, peaking at 8 hours (Fig. 5D).
Figure 5.
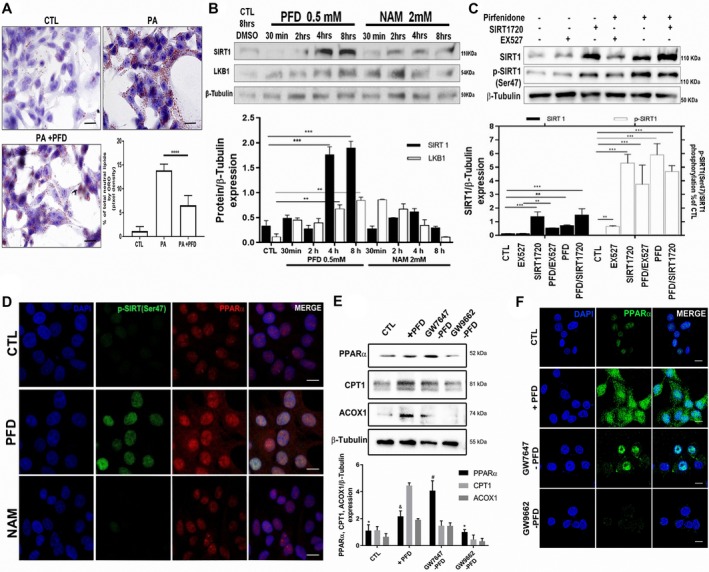
PR‐PFD induces PPAR‐α signaling and SIRT1 both in vivo and in vitro. (A) Representative microphotographs and analysis of Oil Red O staining of HepG2 cells incubated with 1‐mM palmitate with or without PFD. Scale bars = 20 μm. (B) In HepG2 cells, SIRT1 and LKB1 are overexpressed after 4 and 8 hours of PFD incubation, whereas NAM kept the expression down‐regulated. (C) PFD increases SIRT1 expression to similar levels than those obtained with SIRT1720 activator, whereas EX527 as a specific SIRT1 inhibitor decreased SIRT1; diminished SIRT1 phosphorylation in Ser47 is also shown. (D) Positive co‐localization in nuclei of phosphorylated SIRT1(Ser47) and PPAR‐α is observed in cells incubated with PFD. (E) Effect of PPAR‐α agonist (GW7647) and antagonist (GW9662) on HepG2 cell protein expression. (F) Representative confocal images. Scale bars = 10 µm. Data are expressed as the median of the group ± SEM. Abbreviations: CTL, control; PA, palmitate.
Figure 5E depicts the effect of PPAR‐α agonist (GW7647) and antagonist (GW9662) on HepG2 cells' protein expression, in which a predominance of the three protein targets is clear in the PFD and GW7647 cases. GW9662 decreased their expression. Figure 5F shows the representative confocal images corroborating and extending our data.
Molecular Docking Analysis Shows PFD as an Agonistic Ligand for PPAR‐α
Figure 6A shows that PFD contains a carbonyl group attached to the phenyl‐pyridine group. Representative PDB structure of PPAR‐α shows four main domains. In the 3D reconstruction of the ligand binding domain (LBD), we see that it is composed of 13 α‐helixes and four β‐sheets. The pocket of the LBD (PLBD) is located between alpha‐helix H2′, H3, H5, H6, H7, H11, and β‐sheets (Fig. 6A,B).20
Figure 6.
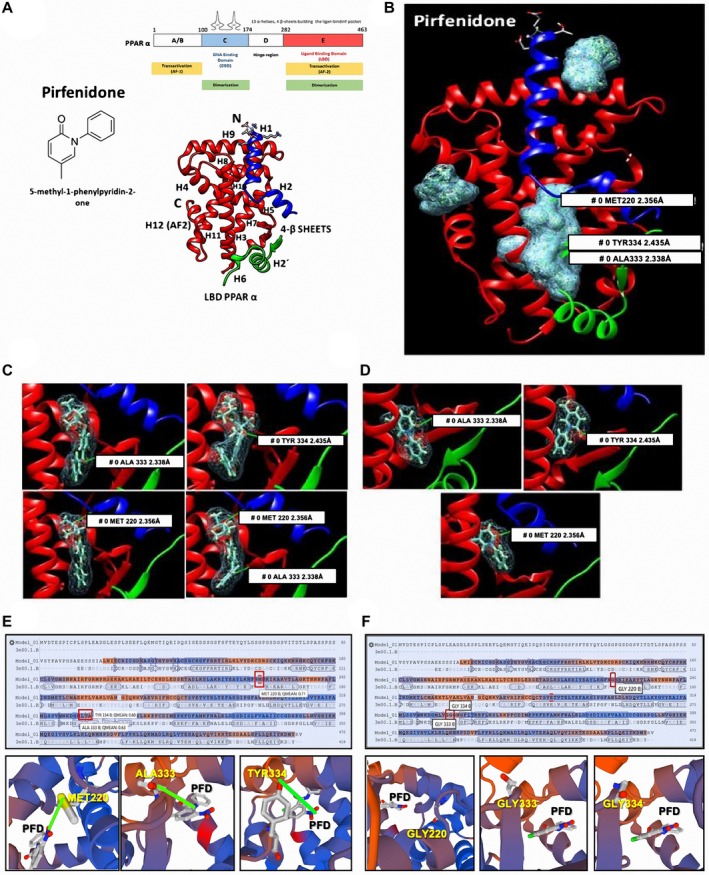
Molecular docking analysis of PFD interaction with PPAR‐α. (A) Chemical structure of PFD and schematic representation PPAR‐α, and 3D structure of the PLBD. (B) SwissDock 3D reconstruction of all possible molecular interactions of PFD with PPAR‐α PLBD. Covalent bonds established between the carbonyl group of FFB (C) and PFD (D) with amino acids MET220, ALA333, and TYR334 of PPAR‐α. Sequence and 3D visualizations of PFD interactions in MET220, ALA333, and TYR334 of PPAR‐α PLBD (E) and annulled interactions when these amino acids are mutated to glycine (F).
A total of 256 possible interactions were obtained for PFD, 17 of which were in the amino domain, 28 in the carboxyl domain, and 211 in the PLBD. A total of 250 possible interactions were obtained for FFB (natural synthetic ligand of PPAR‐α), 8 of which were in the amino domain, 40 in the carboxyl domain, and 202 in the PLBD. Structurally and electrochemically, PFD can interact in the PLBD activating the protein. In addition, software analysis indicates the Gibbs energy of each of the possible protein‐ligand interactions. In the case of the FFB, ΔG values were between −3.7 and −8.4 in the PLBD, and the PFD Gibbs free energy values oscillated between −5.7 and −7.1, clearly showing that PFD has a similar thermodynamic affinity to FFB. The 3D image viewer CHIMERA, which transforms the algorithms generated in SwissDock and produces 3D images, showed all possible links between the protein and the ligand test. Figure 6D shows the electron density of both ligands covalently bound in the PLBD. Three amino acids, methionine 220 (MET220), alanine 333 (ALA333) and tyrosine 334 (TYR334), were identified. Previous analyzes using similar tools showed that fibrates bind primarily to MET220 and to TYR334.21, 22 Figure 6C shows that FFB binds at the same time to two amino acids of PPAR‐α PLBD through its carbonyl and methyl propanoate groups, whereas PFD binds through its carbonyl group to one of the three amino acids mentioned previously (Fig. 6D). These results show that PFD binds to the same amino acids as the synthetic ligand. Then, to corroborate the PFD‐specificity union to PPAR‐α PLBD, the amino acids MET220, ALA333, and TYR334 were mutated to glycine in all mentioned positions using the SWISS‐MODEL server.23 As indicated in Fig. 6F, PFD union to PPAR‐α PLBD is abolished.
PFD Induces the Expression of PPAR‐α, CPT1, and ACOX1 In Vitro
To support our findings and validate that PFD is a ligand for PPAR‐α, we performed a functional assay using DNA constructs.18 We used pα(H‐H)‐pGL3, which keeps the entire PPAR‐α promoter region intact, and pα(X‐H)‐pGL3 pGL3, which has truncated 458 base pair at the 5′‐end. An empty plasmid was used as control (pGL3‐Basic; Fig. 7A). A greater expression of luciferase in HepG2 cells transfected with the vector containing the PPAR‐α complete promoter and treated with PR‐PFD was found (Fig. 7A). The percentage of luminescence obtained after pGL3‐Basic transfection was set as the unit. Figure 7B shows an EMSA using an excised Hind III/Xho I fragment, which contains an hepatocyte nuclear factor 4 and a core AGGTCA consensus PPRE sequence. Shifted DNA proteins bands were visualized with silver staining. It is clear that cell nuclear extracts from PFD‐treated HepG2 cells contained more PPRE‐binding proteins. Furthermore, preincubation of nuclear cell extracts with anti‐PPAR‐α antibody prevented the presence of shifted bands, indicating the true nature of PPAR‐α DNA‐binding proteins.
Figure 7.
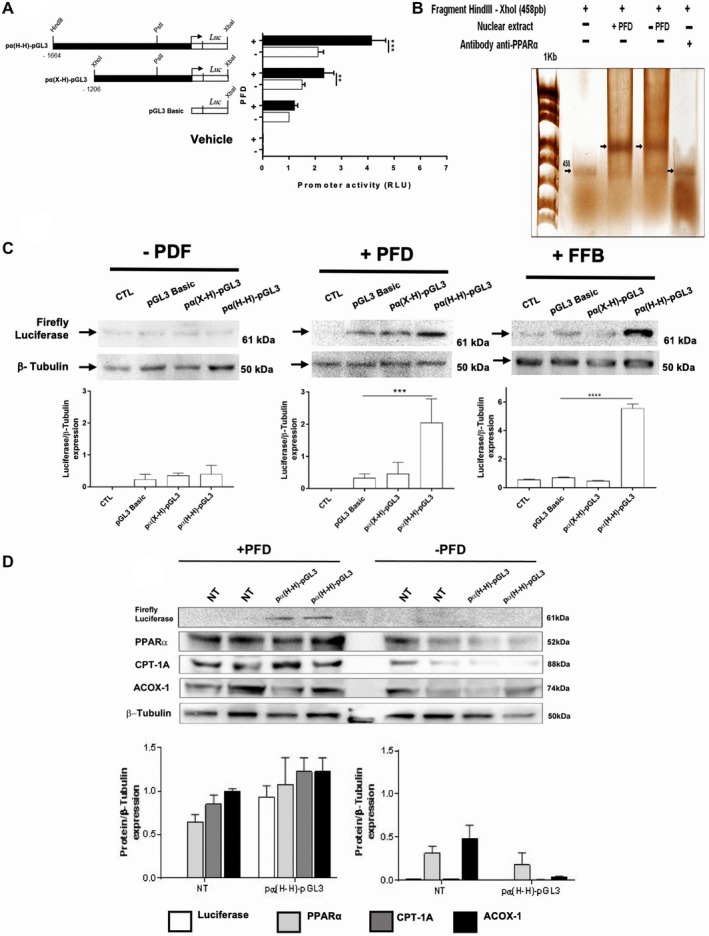
PFD induces activation of PPAR‐α promoter and expression in vitro of PPAR‐α, CPT1, and ACOX1. (A) Schematic representation of human PPAR‐α promoter constructs transfected in HepG2 cells and subsequently treated with PFD or 0.1% DMSO (−PDF). Luciferase expression was quantified using a luminometer. (B) Silver‐stained EMSA. (C) Western blots for quantification of luciferase expression. FFB was used as positive control. (D) PPAR‐α, CPT1, and ACOX1 protein expression in HepG2 cells. Values represent the mean ± SD. ****P < 0.0001 and ***P < 0.001. Abbreviations: CTL, control; NT, Non Treated cells.
Figure 7C shows a western blot analysis of cells transfected with plasmids pα(H‐H)‐pGL3 and incubated with PFD, showing increased expression of luciferase compared to those transfected with pGL3‐Basic. Figure 7D shows that 500‐µM PFD incubation for 24 hours induces expression of PPAR‐α, CPT1, and ACOX1 in HepG2 transfected cells. In control cells, expression of the three proteins remained at baseline. This finding indicates that PFD increases the expression of PPAR‐α and the signaling pathways of beta oxidation.
Discussion
Regular PFD has been demonstrated to reduce liver injury in different models of fibrosis and in rodent models of lipotoxicity and NASH.14, 24, 25 Here, we demonstrated using an in silico analysis/molecular docking approach that PR‐PFD acts as an agonist ligand for PPAR‐α, and we provided evidence of activation of the axis SIRT1‐LKB1‐AMPK phosphorylation and increased PPRA‐α‐CPT1A and ACOX by PR‐PFD. These cascade events resulted in a dramatic reduction of intrahepatic macrosteatosis. To be fair, however, a previous study16 did not find beneficial effects in molecules implicated in lipogenesis and/or lipolysis processes, either in the presence or absence of PFD. There are a number of reasonable explanations regarding this point: First, in the mentioned study, Chen et al. used a rapid‐release PFD that has a normal metabolism reaching serum peak at approximately 2 hours. In our study, we used a formulation of prolonged‐release PFD that has an extended half‐life (approximately 10 hours). Second, Komiya et al. measured these metabolic sensors only at the mRNA level.15 It has been described previously that most molecules involved in hepatic lipolytic processes can be regulated at different gene‐expression levels, not only the transcriptional one.26 Therefore, we examined the hepatic tissue key enzymes and upstream effectors in lipid metabolism at the protein level, using western blot and immunofluorescence. Third, another explanation for this discrepancy is that Komiya et al. used MC4R‐KO mice, rodents with no membrane‐bound receptor of the melanocortin receptor family associated with autosomal dominant obesity.15 Such a mice model would strictly resemble a genetic morbid disease that causes obesity since the neonatal stages.27 They did not observe differences by regular PFD treatment in mRNA levels of molecules implicated in lipid metabolism, probably due to the fact that genetic predisposition to obesity is present since birth in these animals and could be masking differences in fat metabolism.
Another important point to consider is the fasting period during the measurements. Most reports in literature agree that 1‐hour fasting (as used in Komiya et al.) is not enough time to observe biochemical differences. Therefore, we used a 4‐hour fasting period.28 Recently and noteworthy, Chen et al. demonstrated similar results to our findings in terms of improvement in hepatic insulin resistance and steatohepatitis by regular PFD in HF‐fed C57BL/6J mice.16
Adiponectin and leptin have well‐determined actions in terms of NAFLD pathophysiology. Adiponectin deficiency is associated with a pro‐inflammatory condition, as it is observed in obesity and other metabolic disorders. In our NASH animals treated with PR‐PFD, adiponectin increases significantly, reaching normal control levels. Adiponectin normalization then could be contributing to improve insulin sensitivity.29 On the other hand, increased leptin levels reflect the amount of fat stored in adipose tissue, and above the normal levels, act as a pro‐inflammatory stimulus.30 In our animals, leptin did not show diminution, suggesting that its role in prevention of lipid accumulation in nonadipose tissues is still available. The relationship between resistin and NASH is inconclusive, as some studies have claimed that serum resistin levels were associated with neither the presence of NASH nor its severity; others have declared that serum resistin is related to inflammation and fibrosis in NASH.31, 32 Antiresistin antibody therapy improves insulin sensitivity in a mice model of diet‐induced obesity.33 Then, resistin diminution can also be responsible, at least in part, for the reduction in insulin resistance observed in our PR‐PFD animals, in which is resistin diminished.
The portal triad is the main supplier of nutrients and oxygen to the liver. Different zones exist according to the proximity to the portal vein, but also take into account glucose and FA metabolism.34 The periportal area (also zone 1) consists of hepatocytes involved in gluconeogenesis and beta oxidation of FAs. However, the hepatocytes closest to the central vein (pericentral or zone 3) carry out glycolysis and lipogenesis at a higher rate. Regarding NASH, steatosis and oxidative damage to cells are chiefly localized to pericentral regions.35 In this context, our results are favorable, as macrosteatosis is diminished in zone 1, where hepatocytes have the opportunity to regenerate and achieve beta oxidation of FAs.36
PPAR‐α is a nutritional sensor, which allows adaptation of the rates of FA catabolism, lipogenesis, and ketone body synthesis, in response to feeding and starvation.37 As a transcriptional factor, PPAR‐α targets genes involved in FA metabolism (peroxisomal and mitochondrial beta oxidation) and FA transport in tissues such as the muscle, heart, and liver.38 PPAR‐α activation, in combination with PPAR‐β/delta agonism, improves steatosis, inflammation, and fibrosis in preclinical models of NAFLD.39 In our study, animals treated with PR‐PFD augmented PPAR‐αα expression, thus increasing FA oxidation. CPT1A is implicated in mitochondrial beta oxidation. The observed increase in PR‐PFD animals augments free FA oxidation. ACOX1 is the first enzyme of the FA peroxisome beta‐oxidation pathway, which catalyzes the desaturation of acyl‐CoAs to 2‐trans‐enoyl‐CoAs contributing to FA metabolism. ACOX1 was also increased after PR‐PDF treatment in our mice. Taking these data together, increased expression of PPAR‐α, CPT1A, and ACOX led to the previously described benefits like reduced epididymal fat accumulation, body weight, and macrosteatosis.
Important to state is the fact that most patients with NASH display insulin resistance independent of body weight.40 In fact, peripheral insulin resistance is now considered a better predictor of hepatic injury in NAFLD than visceral adiposity or the commonly used fibrosis scoring system.41 Therefore, one of our main goals in this study was to determine whether PR‐PFD was able to induce any change in this parameter. Noteworthy, an enhanced insulin sensitivity was observed by ITT in the group treated with PR‐PFD.
As shown here, PFD is a ligand for PPAR‐α. Our molecular docking analysis, along with site direct mutations in PPAR‐α amino acids binding to PFD, indicates that interaction between PFD with the PLBD structure of PPAR‐α is thermodynamically stable.
Reinforcing these assumptions, HepG2 cells transfected with constructs containing PPAR‐α promoter or its truncated version treated with PFD increases coupled‐luciferase expression. As previously reported by Pineda‐Torra et al., fibrates that interact with PPAR‐α PLBD activated the entire PPAR‐α promoter and induced the highest expression of luciferase, indicating that PPAR‐α has a positive feedback expression.18 These data corroborate that PFD could interact with PLBD and consequently induce PPAR‐α overexpression as well as proteins downstream of the signaling pathway, as CPT1 is implicated in mitochondrial beta oxidation and ACOX1 is involved in peroxisomal beta oxidation42 (Fig. 7D). Moreover, our EMSA corroborates and extends the notion that PPAR‐α does bind to PPRE. Furthermore, it is known that SIRT1 promoter has several PPREs that mediate increased SIRT1 protein. SIRT1 can also enhance the activity of PPAR‐α through its co‐activators, suggesting a positive feedback loop.10, 11 Our data clearly demonstrate a positive PFD effect on SIRT1 phosphorylation on Ser47, which has been associated with augmented nuclear translocation and enzymatic activity (Fig. 5). Thus, a plausible PR‐PFD action would be stimulating either PPAR‐α or SIRT1 and creating a positive feedback among them. On the other hand, our study shows that PFD is a ligand agonist for PPAR‐α and modulates hepatic lipid metabolism in the HF‐induced NASH model.
Supporting information
Financial support: Ciencia Básica CONACYT CB‐2015‐01 awarded to Juan Armendariz Borunda (Grant/Award No. 259096).
Potential conflict of interest: Dr. Armendariz‐Borunda consults for Cell Pharma.
[Correction added 27 January 2020 after original publication. In the Abstract, mice described as PR‐PFD were corrected where appropriate.]
References
- 1. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease—meta‐analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016;64:73‐84. [DOI] [PubMed] [Google Scholar]
- 2. Sumida Y, Yoneda M. Current and future pharmacological therapies for NAFLD/NASH. J Gastroenterol 2018;53:362‐376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Promrat K, Kleiner DE, Niemeier HM, Jackvony E, Kearns M, Wands JR, et al. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology 2010;51:121‐129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Oseini AM, Sanyal AJ. Therapies in non‐alcoholic steatohepatitis (NASH). Liver Int 2017;37:97‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ding RB, Bao J, Deng CX. Emerging roles of SIRT1 in fatty liver diseases. Int J Biol Sci 2017;13:852‐867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC‐1alpha and SIRT1. Nature 2005;434:113‐118. [DOI] [PubMed] [Google Scholar]
- 7. Sanders MJ, Grondin PO, Hegarty BD, Snowden MA, Carling D. Investigating the mechanism for AMP activation of the AMP‐activated protein kinase cascade. Biochem J 2007;403:139‐148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ruderman N, Prentki M. AMP kinase and malonyl‐CoA: targets for therapy of the metabolic syndrome. Nat Rev Drug Discov 2004;3:340‐351. [DOI] [PubMed] [Google Scholar]
- 9. Nasrin N, Kaushik V, Fortier E, Wall D, Pearson K, de Cabo R, et al. JNK1 phosphorylates SIRT1 and promotes its enzymatic activity. PLoS One 2009;4:8414‐8423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hayashida S, Arimoto A, Kuramoto Y, Kozako T, Honda S, Shimeno H, et al. Fasting promotes the expression of SIRT1, an NAD+ ‐dependent protein deacetylase, via activation of PPARalpha in mice. Mol Cell Biochem 2010;339:285‐292. [DOI] [PubMed] [Google Scholar]
- 11. Purushotham A, Schug TT, Xu Q, Surapureddi S, Guo X, Li X. Hepatocyte‐specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab 2009;9:327‐338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Masternak MM, Bartke A. PPARs in calorie restricted and genetically long‐lived mice. PPAR Res 2007;2007:1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rakhshandehroo M, Knoch B, Muller M, Kersten S. Peroxisome proliferator‐activated receptor alpha target genes. PPAR Res 2010;2010:1‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Armendariz‐Borunda J, Islas‐Carbajal MC, Meza‐Garcia E, Rincon AR, Lucano S, Sandoval AS, et al. A pilot study in patients with established advanced liver fibrosis using pirfenidone. Gut 2006;55:1663‐1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Komiya C, Tanaka M, Tsuchiya K, Shimazu N, Mori K, Furuke S, et al. Antifibrotic effect of pirfenidone in a mouse model of human nonalcoholic steatohepatitis. Sci Rep 2017;7:44754‐44766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen G, Ni Y, Nagata N, Xu L, Zhuge F, Nagashimada M, et al. Pirfenidone prevents and reverses hepatic insulin resistance and steatohepatitis by polarizing M2 macrophages. Lab Invest 2019;99:1335‐1348. [DOI] [PubMed] [Google Scholar]
- 17. Grosdidier S, Fernandez‐Recio J. Docking and scoring: applications to drug discovery in the interactomics era. Expert Opin Drug Discov 2009;4:673‐686. [DOI] [PubMed] [Google Scholar]
- 18. Pineda Torra I, Jamshidi Y, Flavell DM, Fruchart JC, Staels B. Characterization of the human PPARalpha promoter: identification of a functional nuclear receptor response element. Mol Endocrinol 2002;16:1013‐1028. [DOI] [PubMed] [Google Scholar]
- 19. Parra‐Vargas M, Sandoval‐Rodriguez A, Rodriguez‐Echevarria R, Dominguez‐Rosales JA, Santos‐Garcia A, Armendariz‐Borunda J. Delphinidin ameliorates hepatic triglyceride accumulation in human HepG2 cells, but not in diet‐induced obese mice. Nutrients 2018;10:1060‐1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cronet P, Petersen JF, Folmer R, Blomberg N, Sjoblom K, Karlsson U, et al. Structure of the PPARalpha and ‐gamma ligand binding domain in complex with AZ 242; ligand selectivity and agonist activation in the PPAR family. Structure 2001;9:699‐706. [DOI] [PubMed] [Google Scholar]
- 21. Capelli D, Cerchia C, Montanari R, Loiodice F, Tortorella P, Laghezza A, et al. Structural basis for PPAR partial or full activation revealed by a novel ligand binding mode. Sci Rep 2016;6:34792‐34804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yamamoto Y, Takei K, Arulmozhiraja S, Sladek V, Matsuo N, Han SI, et al. Molecular association model of PPARalpha and its new specific and efficient ligand, pemafibrate: structural basis for SPPARMalpha. Biochem Biophys Res Commun 2018;499:239‐245. [DOI] [PubMed] [Google Scholar]
- 23. Benkert P, Biasini M, Schwede T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2011;27:343‐350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Garcia L, Hernandez I, Sandoval A, Salazar A, Garcia J, Vera J, et al. Pirfenidone effectively reverses experimental liver fibrosis. J Hepatol 2002;37:797‐805. [DOI] [PubMed] [Google Scholar]
- 25. Kitade H, Chen G, Ni Y, Ota T. Nonalcoholic fatty liver disease and insulin resistance: new insights and potential new treatments. Nutrients 2017;9:387‐400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Saponaro C, Gaggini M, Carli F, Gastaldelli A. The subtle balance between lipolysis and lipogenesis: a critical point in metabolic homeostasis. Nutrients 2015;7:9453‐9474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cordero P, Li J, Nguyen V, Pombo J, Maicas N, Novelli M, et al. Developmental programming of obesity and liver metabolism by maternal perinatal nutrition involves the melanocortin system. Nutrients 2017;9:1041‐1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moebus S, Gores L, Losch C, Jockel KH. Impact of time since last caloric intake on blood glucose levels. Eur J Epidemiol 2011;26:719‐728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K, et al. The fat‐derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med 2001;7:941‐956. [DOI] [PubMed] [Google Scholar]
- 30. Meek TH, Morton GJ. The role of leptin in diabetes: metabolic effects. Diabetologia 2016;59:928‐932. [DOI] [PubMed] [Google Scholar]
- 31. Senates E, Colak Y, Yesil A, Coskunpinar E, Sahin O, Kahraman OT, et al. Circulating resistin is elevated in patients with non‐alcoholic fatty liver disease and is associated with steatosis, portal inflammation, insulin resistance and nonalcoholic steatohepatitis scores. Minerva Med 2012;103:369‐376. [PubMed] [Google Scholar]
- 32. Lee JH, Chan JL, Yiannakouris N, Kontogianni M, Estrada E, Seip R, et al. Circulating resistin levels are not associated with obesity or insulin resistance in humans and are not regulated by fasting or leptin administration: cross‐sectional and interventional studies in normal, insulin‐resistant, and diabetic subjects. J Clin Endocrinol Metab 2003;88:4848‐4856. [DOI] [PubMed] [Google Scholar]
- 33. Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, et al. The hormone resistin links obesity to diabetes. Nature 2001;409:307‐312. [DOI] [PubMed] [Google Scholar]
- 34. Hijmans BS, Grefhorst A, Oosterveer MH, Groen AK. Zonation of glucose and fatty acid metabolism in the liver: mechanism and metabolic consequences. Biochimie 2014;96:121‐129. [DOI] [PubMed] [Google Scholar]
- 35. Yeh MM, Brunt EM. Pathological features of fatty liver disease. Gastroenterology 2014;147:754‐764. [DOI] [PubMed] [Google Scholar]
- 36. Takahashi Y, Fukusato T. Histopathology of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J Gastroenterol 2014;20:15539‐15548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hashimoto T, Cook WS, Qi C, Yeldandi AV, Reddy JK, Rao MS. Defect in peroxisome proliferator‐activated receptor alpha‐inducible fatty acid oxidation determines the severity of hepatic steatosis in response to fasting. J Biol Chem 2000;275:28918‐28928. [DOI] [PubMed] [Google Scholar]
- 38. Lefebvre P, Chinetti G, Fruchart JC, Staels B. Sorting out the roles of PPAR alpha in energy metabolism and vascular homeostasis. J Clin Invest 2006;116:571‐580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pawlak M, Lefebvre P, Staels B. Molecular mechanism of PPARalpha action and its impact on lipid metabolism, inflammation and fibrosis in non‐alcoholic fatty liver disease. J Hepatol 2015;62:720‐333. [DOI] [PubMed] [Google Scholar]
- 40. Chitturi S, Abeygunasekera S, Farrell GC, Holmes‐Walker J, Hui JM, Fung C, et al. NASH and insulin resistance: Insulin hypersecretion and specific association with the insulin resistance syndrome. Hepatology 2002;35:373‐379. [DOI] [PubMed] [Google Scholar]
- 41. Rosso C, Mezzabotta L, Gaggini M, Salomone F, Gambino R, Marengo A, et al. Peripheral insulin resistance predicts liver damage in nondiabetic subjects with nonalcoholic fatty liver disease. Hepatology 2016;63:107‐116. [DOI] [PubMed] [Google Scholar]
- 42. Mandard S, Muller M, Kersten S. Peroxisome proliferator‐activated receptor alpha target genes. Cell Mol Life Sci 2004;61:393‐416. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials