

Abstract
Alagille syndrome (ALGS) is an autosomal dominant multisystem disorder with cholestasis as a defining clinical feature. We sought to characterize hepatic outcomes in a molecularly defined cohort of children with ALGS‐related cholestasis. Two hundred and ninety‐three participants with ALGS with native liver were enrolled. Participants entered the study at different ages and data were collected retrospectively prior to enrollment, and prospectively during the study course. Genetic analysis in 206 revealed JAGGED1 mutations in 91% and NOTCH2 mutations in 4%. Growth was impaired with mean height and weight z‐scores of <−1.0 at all ages. Regression analysis revealed that every 10 mg/dL increase in total bilirubin was associated with a decrease in height z‐score by 0.10 (P = 0.03) and weight z‐score by 0.15 (P = 0.007). Total bilirubin was higher for younger participants (P = 0.03) with a median of 6.9 mg/dL for those less than 1 year old compared with a median of 1.3 mg/dL for participants 13 years or older. The median gamma glutamyl transferase also dropped from 612 to 268 in the same age groups. After adjusting for age, there was substantial within‐individual variation of alanine aminotransferase. By 20 years of age, 40% of participants had developed definite portal hypertension. Estimated liver transplant–free survival at the age of 18.5 years was 24%. Conclusions: This is the largest multicenter natural history study of cholestasis in ALGS, demonstrating a previously underappreciated burden of liver disease with early profound cholestasis, a second wave of portal hypertension later in childhood, and less than 25% of patients reaching young adulthood with their native liver. These findings will promote optimization of ALGS management and development of clinically relevant endpoints for future therapeutic trials.

Abbreviations
- ALGS
Alagille syndrome
- ALT
alanine aminotransferase
- ChiLDReN
Childhood Liver Disease Research Network
- CSS
clinician scratch scale
- DXA
dual energy X‐ray absorptiometry
- GGT
gamma‐glutamyl transferase
- JAG1
JAGGED1
- TB
total bilirubin
Alagille syndrome (ALGS) is an autosomal dominant, multisystem disorder with highly variable clinical features.1, 2, 3, 4 Mutations in JAGGED1 (JAG1) are identified in more than 90% of clinically defined patients with ALGS, and NOTCH2 mutations have been reported in a small minority.5, 6, 7, 8 Cholestasis is a key clinical defining feature, and ALGS is, in fact, the most common cause of inherited cholestasis in children.9 Current knowledge of the characteristics and outcomes of cholestasis in ALGS is based on retrospective single‐center studies from more than 15 years ago.4, 10, 11 These studies suffer from a lack of consistent clinical and molecular definitions of ALGS and may be reporting on patients with other causes of cholestasis or on siblings with milder ALGS phenotypes. In addition, these older studies include a spectrum of individuals ranging from severe cholestasis to minimal hepatic manifestations. Based on these heterogeneous and loosely classified data, liver involvement in ALGS has been regarded as having a relatively good prognosis, with only approximately 20%‐30% of patients requiring liver transplantation.10, 11 There is a paucity of data regarding the frequency of hepatic complications in ALGS, especially in the current era.
The goal of this study was to characterize the burden and natural history of liver disease in children with ALGS‐related cholestasis within a large, ongoing, multicentered study of childhood cholestatic liver diseases. We sought to determine the frequency of hepatic complications, examine transplant‐free survival, and explore the determinants of growth impairment in ALGS. It is imperative to better understand the natural history of cholestasis in ALGS in order to optimize management strategies, such as the timing of liver transplantation, to provide improved prognostic information for patients and their families, and to design clinical trials to evaluate novel therapies for this disorder.
Methods
Study Design and Participants
This is a longitudinal study of hepatic disease progression in a prevalent cohort of patients with ALGS, all of whom had cholestasis. Participants entered the study at different ages, and data were collected retrospectively prior to enrollment, and prospectively during the study course (Fig. 1). Participants included in this study were enrolled in the Longitudinal Study of Genetic Causes of Intrahepatic Cholestasis (LOGIC; NCT00571272) protocol organized within the Childhood Liver Disease Research Network (ChiLDReN), a National Institute of Diabetes and Digestive and Kidney Diseases/National Institutes of Health–funded network of 14 pediatric academic medical centers across North America. This study was approved by institutional review boards at each center, and informed consent was obtained from parents/guardians or participants 18 years or older, and assent from children of appropriate age.
Figure 1.
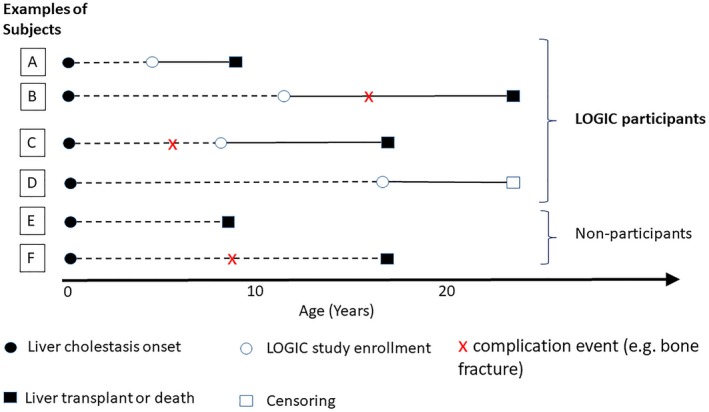
Schematic to represent study design. This schematic illustrates the time of study entry, time to complication event, time to liver transplant or death, and the censoring of six classes of example subjects. The solid black dot represents the onset of cholestasis. The horizontal dotted lines indicate the time period prior to study entry. The white circle represents the time of study enrollment. The red “x” represents the time of complication event (e.g., bone fracture). The black and white squares represent the time of liver transplant/death and censoring, respectively. Although example participants A, B, C, and D were enrolled and observed in the LOGIC study at various time points after birth, the subjects in groups E and F experienced liver transplant/death before the opportunity to be enrolled, and therefore were not observed in the study. In addition, not all events were observed in A, B, C, and D due to right censoring and left censoring. For example, the event time of complication was censored by liver transplant/death for participant A. Participant B experienced the complication event before liver transplant/death. Subject C experienced the complication event before being enrolled, and the time of the complication event was retrospectively collected. For subject D, the complication did not happen before he or she was censored (dropped out of the LOGIC study or the data were cut off to retrieve the analysis data set). Abbreviation: LOGIC, Longitudinal Study of Genetic Causes of Intrahepatic Cholestasis.
LOGIC is a longitudinal observational study of the natural history of several genetic causes of intrahepatic cholestasis, including ALGS. Defined data elements are collected systematically and prospectively at yearly intervals for up to 20 years, or until death, liver transplant, or dropout (Fig. 1). Pertinent data are recorded about past history at enrollment, and interval medical history is collected annually, including surgery and major events or complications such as ascites, bone fractures, and gastrointestinal bleeding. Growth parameters and standard laboratory evaluations are recorded at enrollment and annually at follow‐up visits. This data analysis focuses on natural history prior to liver transplantation.
All participants with ALGS in LOGIC were less than 25 years of age at enrollment, had confirmed ALGS by set diagnostic clinical criteria (Supporting Table S1), and had evidence of cholestasis. Only probands were included (no affected siblings). For the purposes of this study, LOGIC cholestasis enrollment criteria were defined by the presence of one or more of the following: fasting total serum bile acid greater than 3 times the upper limit of normal for age, direct/conjugated bilirubin greater than 2 mg/dL, fat‐soluble vitamin deficiency otherwise unexplainable, gamma‐glutamyl transferase (GGT) greater than 3 times the upper limit of normal for age, and/or intractable pruritus explainable only by liver disease. The onset of definite clinically evident portal hypertension in this cohort was assessed using the following criteria, which have recently been published by the ChiLDReN12: (1) ascites and the use of diuretics, or (2) documented esophageal or gastric varices on endoscopy, or (3) presence of both splenomegaly (spleen palpable > 2 cm below costal margin) and platelets < 150,000/mm3. Pruritus was documented using the clinician scratch scale (CSS), which is a physician‐observed assessment tool devised by Whitington.13 The CSS scores the physical manifestations of pruritus as follows: 0 = none, 1 = rubbing or mild scratching when undistracted, 2 = active scratching without evident skin abrasions, 3 = abrasions evident, and 4 = cutaneous mutilation, hemorrhage, and scarring evident.
Participants were confirmed as ALGS by standard clinical criteria and/or the presence of a disease‐causing mutation in JAG1 or NOTCH2. The clinical criteria were at least three of the following: bile duct paucity on liver biopsy, heart murmur, or cardiac anomaly; posterior embryotoxon or other anterior chamber defect; butterfly vertebrae; and/or characteristic facial features and renal anomalies. The details of the inclusion criteria are described in Supporting Table 1 At the time of data analysis, the total number of patients with ALGS in LOGIC was 377. Participants with a liver transplant prior to enrollment were excluded (n = 74) from this analysis of pretransplant natural history, and an additional 10 participants were excluded for having a non‐ALGS gene mutation (ABCB4, ABCB11, or HNF1beta), resulting in a sample of 293 participants with ALGS.
Table 1.
Characteristics at Baseline of Participants With ALGS
| Characteristic | Group or Statistic | |
|---|---|---|
| Age (years) | n | 293 |
| Median (Q1, Q3) | 3.0 (1.0‐8.0) | |
| Range (min, max) | (0, 25) | |
| Gender | Male | 175 (60%) |
| Mutational analysis | JAG1 | 187 (64%) |
| NOTCH2 | 9 (3%) | |
| Neither JAG1 or NOTCH2 | 10 (3%) | |
| Not tested | 87 (30%) | |
| Race | White | 181 (62%) |
| Black or African American | 33 (11%) | |
| Asian | 13 (4%) | |
| Other | 39 (13%) | |
| Missing or refused | 27 (9%) | |
| Ethnicity | Hispanic | 65 (22%) |
| Non‐Hispanic | 219 (75%) | |
| Missing or refused | 9 (3%) | |
| Cardiac features | Severe* heart defect | 89 (30%) |
| Mild† heart defect | 170 (58%) | |
| No cardiac involvement | 34 (12%) | |
| Renal features | Any renal anomaly | 63 (22%) |
| Height z‐score | n | 266 |
| Mean (SD) | −1.9 (1.4) | |
| Weight z‐score | n | 269 |
| Mean (SD) | −1.9 (1.5) | |
| BMI z‐score‡ | n | 171 |
| Mean (SD) | −0.4 (1.2) |
Severe heart defect = any intracardiac or valvar defect, coarctation of the aorta.
Mild heart defect = peripheral pulmonary stenosis, murmur with normal echocardiogram.
Not available for participants younger than 2 years old.
Abbreviation: BMI, body mass index.
Statistical Analysis
Summary statistics were calculated for baseline participant characteristics. Graphs of laboratory values collected longitudinally over the course of the study were plotted by participant age. A statistical challenge for analyzing this data set is left‐truncated survival data, due to the prevalent cohort study design. For studying rare diseases, a prevalent cohort design allows faster enrollment and is more feasible than an incident cohort design. However, because the LOGIC study enrolls patients with ALGS at any age, a subset of our target population (a representative population of all patients with ALGS‐related cholestasis) was not captured due to the occurrence of liver transplant or death before being encountered by this study. We therefore ascertained a select cohort that was potentially healthier than our actual target population. In all survival analyses, age was used as the time variable, and appropriate statistical methods were used to adjust for left truncation so that all estimates derived referred to our target population, namely, all patients with ALGS‐related cholestasis rather than the observed prevalent cohort. We calculated the transplant‐free survival and cumulative incidence functions for transplant‐free survival (transplant or death before transplant, whichever happened first), accounting for left truncation. To calculate cumulative incidence rates over age for ascites, bone fracture, and variceal bleeding, the methods of Wang14 and Peng and Fine15 for nonparametric estimation with left‐truncated semicompeting risk data were used, treating liver transplant and death as competing risk events. When the method of Peng and Fine was applied, a bootstrapping method was used to calculate 95% confidence intervals (CIs) for the cumulative incidence rates for these complications. To study the impact of cholestasis, the presence of a cardiac defect or renal anomaly on growth, we conducted a growth curve analysis using a linear mixed‐effects model to repeated measures of height z‐score with total bilirubin as time‐dependent covariate and cardiac defect/renal anomaly as a time‐independent covariate. For this analysis, we used time from study entry as the time variable. To study laboratory markers and symptoms of cholestasis among survivors over time, we calculated the medians and interquartile range and examined their changes over age visually. In addition, we used generalized estimation equations to test whether median or mean levels changed significantly over age for each of the laboratory markers among study participants, assuming a linear relationship between median level and age.
Results
Between December 2007 and September 2018, 293 participants with ALGS meeting the enrollment criteria (mean age of 5 years; range: 2 weeks to 25 years) were enrolled and prospectively followed. The median duration of follow‐up was 2.7 years (range 0‐10 years). The duration of follow‐up was similar across different ages in the study cohort. Baseline characteristics of the 293 participants with ALGS are included in Table 1 All participants had cholestasis, as specified by the inclusion criteria. Eighty‐eight percent of the cohort had a cardiac or cardiovascular anomaly and 22% had a renal anomaly, in keeping with other series of patients with ALGS.10, 16 Additional details regarding the ALGS disease characteristics are presented in Table 2.
Table 2.
Features of ALGS at Enrollment
| Disease Features | n | Count (%) |
|---|---|---|
| Cardiac | ||
| Peripheral pulmonary stenosis | 263 | 194 (74%) |
| Pulmonary valve stenosis | 247 | 34 (13%) |
| Tetralogy of fallot | 251 | 16 (6%) |
| Ventricular septal defect | 249 | 33 (13%) |
| Atrial septal defect | 246 | 32 (13%) |
| Other cardiac anomaly | 276 | 92 (33%) |
| Facies | ||
| Deep‐set eyes | 265 | 209 (79%) |
| Broad forehead | 273 | 240 (88%) |
| Pointed chin | 269 | 238 (88%) |
| Eyes | ||
| Posterior embryotoxon | 263 | 114 (43%) |
| Axenfeld’s anomaly | 244 | 3 (1%) |
| Other eye condition | 260 | 15 (6%) |
| Skeletal | ||
| Butterfly vertebrae | 268 | 104 (39%) |
| Other skeletal condition | 269 | 11 (4%) |
| Renal | ||
| Dysplastic kidney | 265 | 10 (4%) |
| Single kidney | 264 | 3 (1%) |
| Renal tubular acidosis | 261 | 24 (9%) |
| Other renal structural anomaly | 264 | 34 (13%) |
Genetics
Mutational analysis was available in 206 of 293 participants, revealing mutations in JAG1 in 91% and in NOTCH2 in 4% of those tested. No mutation in JAG1 or NOTCH2 was identified in 5% of participants, even though they met strict diagnostic clinical criteria, as described previously. The types of mutation identified in this cohort (Table 3) reflected the published data, with a preponderance of protein‐truncating mutations.17, 18 An analysis of genotype–phenotype relationships was unrevealing for JAG1. There was no impact of JAG1 mutation type on transplant‐free survival (P = 0.11) in this cohort, nor on severity of cardiac defect (P = 0.34). No analysis was performed on the NOTCH2 genotypes and phenotype due to the small numbers of patients with these mutations.
Table 3.
Classification of JAG1 and NOTCH2 Mutations Identified in the Study Cohort (n = 196)
| Classification | JAG1 (n = 187) | NOTCH2 (n = 9) |
|---|---|---|
| Insertion/deletion | 70 (37%) | 2 (22%) |
| Missense | 35 (19%) | 5 (56%) |
| Nonsense | 48 (26%) | 1 (11%) |
| Splice site | 18 (10%) | |
| Whole/partial gene deletion | 4 (2%) | |
| Other | 11 (6%) | |
| Missing | 1 (1%) | 1 (11%) |
Growth
Growth was impaired in participants with ALGS at enrollment (Table 1). Mean height and weight z‐scores were lower than −1 at all ages in the study population (Supporting Table S2).
A growth curve analysis, adjusted for the presence of a cardiac defect and age at enrollment, demonstrated the impact of total bilirubin (TB) on growth. This model shows that every 10 mg/dL increase in TB was associated with a decrease in height z‐score by 0.10 (P = 0.03) and a decrease in weight z‐score by 0.15 (P = 0.007). The same model was used to assess the impact of any cardiac defect on growth in ALGS. After adjusting for TB and age at enrollment, the presence of a severe cardiac defect was not associated with either height z‐score (P = 0.15) nor weight z‐score (P = 0.32).
Hearing Assessment
Audiology testing was performed in 110 patients with ALGS. Of these, 62 (56%) passed air conduction testing in both ears at all frequencies, 42 (38%) failed at least one frequency in at least one ear, and 6 (5%) were inconclusive due to missing tests. Those who failed air conduction then went on to have tympanometry testing. Eleven of 42 (26%) passed both ears and 28 of 42 (67%) failed one or both ears. In 2 of 42 tympanometry was not performed, and in 1 of 42 the data were not interpretable due to ear tubes.
Natural History and Outcomes of Liver Involvement
In this ALGS cohort, 10 of 293 (3%) had undergone Kasai portoenterostomy in infancy. Twenty‐nine (10%) participants had a partial biliary diversion procedure either before enrollment or during follow up.
Serial biochemistry and hematologic parameters were recorded for all participants during follow‐up (Fig. 2). TB was higher for younger participants (P = 0.03), with a median of 6.9 mg/dL for those less than 1 year old, and was lower with a median of 1.3 mg/dL for participants 13 years or older surviving with their native liver. This was mirrored by a fall in median total cholesterol (P < 0.001) (Fig. 2). The median level of GGT was also higher in the younger participants (P = 0.002), with a median of 612 for those younger than 1 year old, and a median of 268 for participants 13 years or older surviving with their native liver. The change in median alanine aminotransferase (ALT) and aspartate aminotransferase (AST) with age reached statistical significance (Fig. 1); however, these changes represented a small drop in clinical terms, as the median ALT was 154 in those younger than 1 year old and 131 in participants aged 13 or older, and the median AST fell from 176 to 116 in the same age groups. After adjusting for age, the biological variation of ALT within an individual was noted to be quite large. The SD of the variation of the log10 base–transformed ALT was approximately 0.18, which means that ALT can vary from 56% lower to 129% higher for 95% of the time in a given individual.
Figure 2.
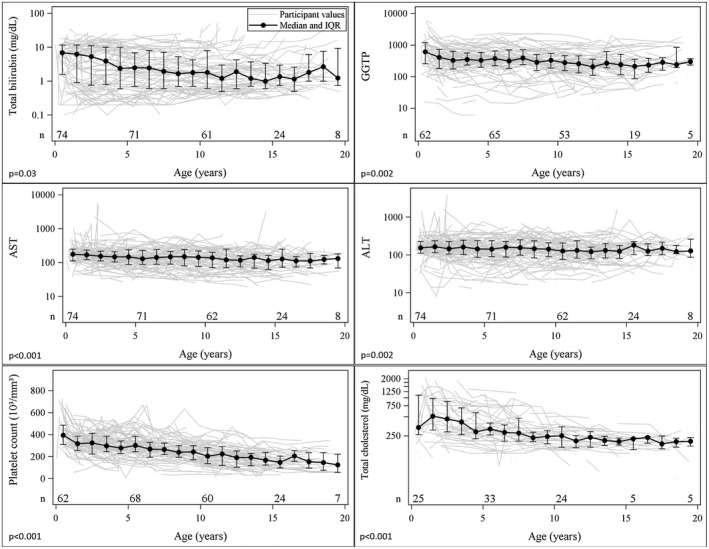
Laboratory characteristics among survivors by age. Individual participant laboratory values are shown over time with the median and interquartile range shown in blue and P values. The P values are from tests for trends in laboratory values by age in generalized estimating equation models accounting for correlation among repeated measures within participants. Serum bilirubin, cholesterol, and GGT are higher earlier in life but improve in many children. The platelet count progressively falls over childhood. Due to sparse data after age 20 years, only laboratory values measured at age 20 or younger are presented. Abbreviations: GGTP, gamma‐glutamyl transpeptidase; and IQR, interquartile range.
Platelet count was higher for younger participants (P < 0.001) (Fig. 2), with a median of 395,000/mL for those younger than 1 year old, and it was lower for participants aged 13 years or older surviving with their native liver, with a median of 177,000/mL. The international normalized ratio varied very little with age in this population, and the median value was 1.0 across all ages.
The improvement in biochemical markers of cholestasis among survivors with native livers over time was accompanied by a reduction in the prevalence of pruritus and xanthomas by age in this cohort (Fig. 3). Pruritus was recorded on an annual basis using the CSS.13> The highest percentage of participants with ALGS with pruritus occurred in those 2 years of age and tended to be lower for older participants. The prevalence of xanthomas among survivors in the cohort also declined with age, with no participants having extensive xanthomas after the age of 12 years. The frequency of xanthomas was highest among participants aged 2 years.
Figure 3.
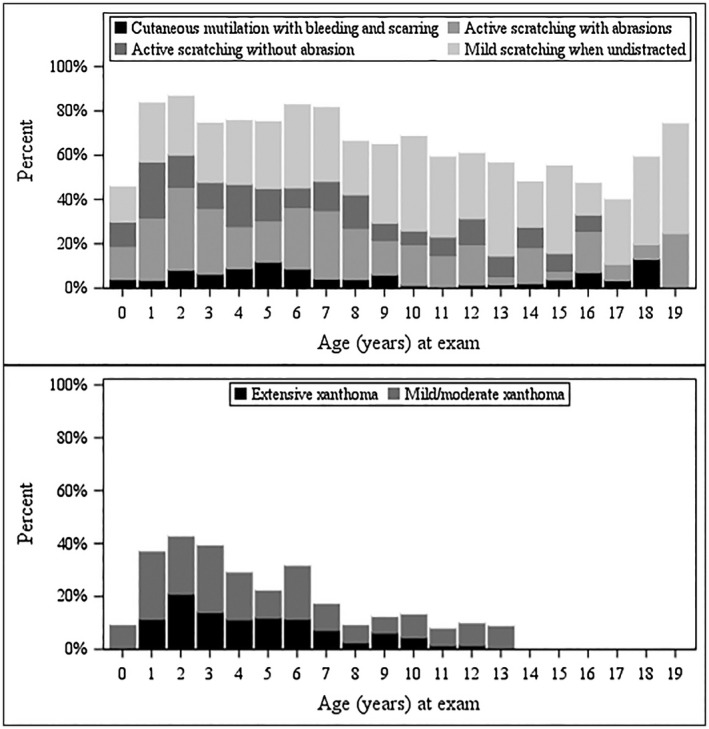
Prevalence of pruritus and xanthoma among survivors by age. Pruritus, as measured by CSS, and the burden of xanthomas improve over time in ALGS survivors with native livers. CSS scores the physical manifestations of pruritus as follows: 0 = none, 1 = rubbing or mild scratching when undistracted, 2 = active scratching without evident skin abrasions, 3 = abrasion evident, 4 = cutaneous mutilation, hemorrhage, and scarring evident.
The risk of hepatic complications was carefully assessed in the cohort (Fig. 4), accounting for left truncation in the study sample. Among patients with ALGS‐related cholestasis, the estimated cumulative incidence of ascites at the age of 20 years was 36% (95% CI: 16%, 57%). Thirty‐seven participants reported to have developed their first episode of ascites prior to study enrollment, and another 36 did so during follow‐up. One or more episodes of gastrointestinal bleeding were reported to have occurred in 23 patients prior to enrollment and 17 during the study period, which corresponded to a 16% (95% CI: 4%, 28%) cumulative incidence of first gastrointestinal bleeding in participants with ALGS‐related cholestasis. Splenomegaly was reported in 62 patients prior to study enrollment and another 49 during follow‐up. Thrombocytopenia (platelet count < 150,000/mL) was present in 29 patients prior to enrollment and 45 developed it during follow‐up for a cumulative incidence of 33% (95% CI: 15%, 50%). The estimated cumulative incidence of splenomegaly was 50% (95% CI: 39%, 59%) by 20 years of age. By 20 years of age, 40% (95% CI: 20%, 60%) of patients developed definite clinically evident portal hypertension.
Figure 4.
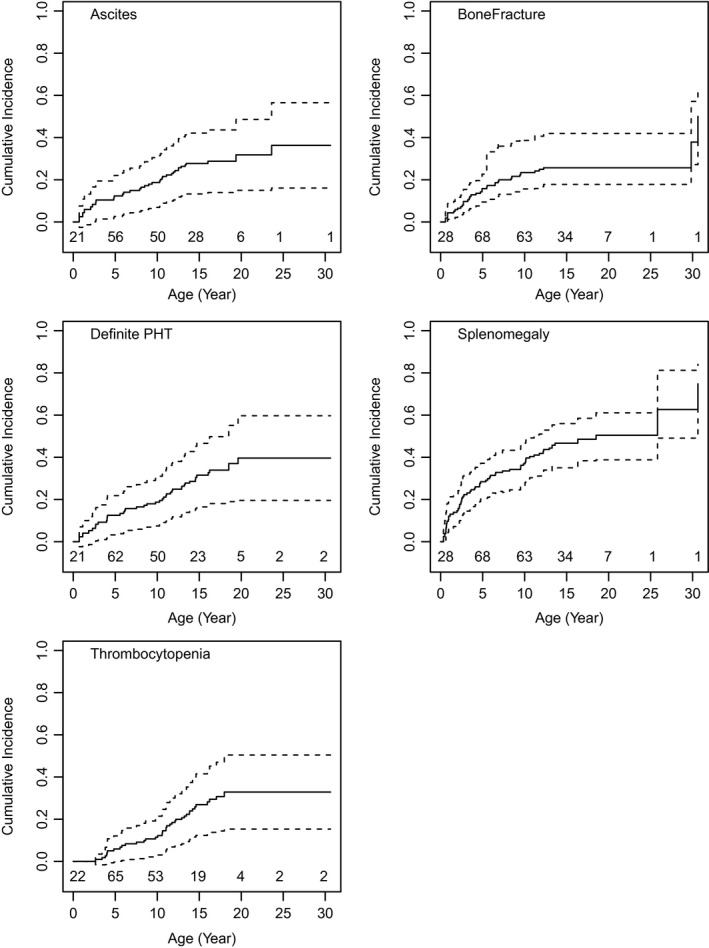
Cumulative incidence of hepatic complications and portal hypertension. The cumulative incidence of ascites, splenomegaly, and thrombocytopenia increase with age, resulting in an increase in the onset of definite clinically evident portal hypertension as defined by (1) ascites treated by diuretics, (2) presence of esophageal or gastric varices, or (3) both splenomegaly (spleen palpable > 2 cm below costal margin) and fewer than 150,000/mm3 platelets. Abbreviation: PHT, portal hypertension.
Finally, 44 of the participants with ALGS reported to have experienced a bone fracture prior to study enrollment and another 27 thereafter; the estimated cumulative incidence of fracture by age of 20 years was 26% (95% CI: 17%, 55%). Interestingly, most fractures occurred in younger children, with 67 of the 71 reported events occurring before the age of 13 years.
Survival
Figure 5 represents a survival curve of this cohort with ALGS‐related cholestasis. Fifty‐eight participants had liver transplant and 11 died with native liver during the study follow‐up. Estimated transplant‐free survival at the age of 18.5 years was 24% (95% CI: 16%, 36%). There was no center effect impacting the rate of transplant‐free survival (P = 0.38).
Figure 5.
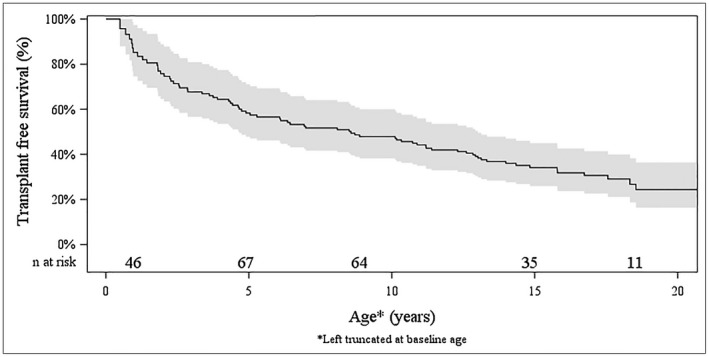
Transplant‐free survival in ALGS. The method for calculating survival accounted for left truncation and estimated the transplant‐free survival at the age of 18.5 years as 24% (95% CI: 16%, 36%). There was no liver or transplant/death observed at age 20 or older in this cohort. The estimated survival curve was truncated at the age of 20 years.
Among the 11 participants who died with native liver during the study, only three deaths were directly attributable to liver disease. The remainder were due to cardiac involvement, pulmonary hemorrhage, and “other” (Supporting Table S3).
Discussion
This large, multicenter, natural history study of patients with ALGS, who presented with cholestasis, demonstrates a previously underappreciated burden of liver disease. Prior studies have included participants with ALGS with heterogeneous clinical features10, 11, 19; our study characterizes a cohort of rigorously defined children with ALGS, all of whom have cholestasis, and therefore represents a focused assessment of the outcomes of liver disease in ALGS. In this cohort, survival to early adulthood with native liver occurred in only 24% of children at 18.5 years. As expected, clinical manifestations of cholestasis (pruritus and xanthomas) were prevalent in early childhood and improved in transplant‐free survivors after the age of 5 years. Given the prevalent nature of our study population, this change could be due to spontaneous improvement and/or survival selection. Spontaneous improvement in cholestasis has been described previously in early childhood in ALGS and led to the supposition that only 20%‐30% of individuals with ALGS require liver transplantation.10, 20 However, here we identify a “second wave” of hepatic disease in later childhood in ALGS with the development of portal hypertension, despite improvement of cholestasis. This portal hypertensive phase is associated with clinically impactful complications (such as ascites) and the need for liver transplantation later in childhood.
Laboratory parameters reflected this clinical pattern of liver disease evolution in ALGS. Serum total and conjugated bilirubin and cholesterol were higher earlier in life but improved in many children who did not undergo liver transplant. In contrast, the platelet count progressively fell in survivors through childhood. Of note, although serum transaminases did not change over time in a clinically meaningful way in the entire cohort, we did identify tremendous intrapatient ALT variation in patients with ALGS‐related cholestasis. Further studies are needed with more frequently collected laboratory data to better capture and characterize this variation in serum transaminases in individual patients with ALGS. This will be crucial to the design of clinical trials for new therapies targeting ALGS.
Growth failure has been widely reported in ALGS,21, 22 and multiple proposed mechanisms have included growth hormone insensitivity and suboptimal nutrition.23, 24 In this cohort, the clinical determinants of growth impairment in ALGS were not easily identified. Total bilirubin had a modest negative effect on height and weight z‐scores, and the potential effect of a significant cardiac defect was not statistically significant. This suggests that the growth impairments in ALGS may be intrinsic and related to the underlying genetic defects, rather than to secondary manifestations of clinical disease in the liver or heart. These data are somewhat surprising, especially when it has been shown previously that children with ALGS who undergo liver transplantation demonstrate a larger degree of catch‐up growth compared with matched biliary atresia counterparts, suggesting that correction of cholestasis ameliorates growth impairments even more so in ALGS than biliary atresia.25 It is possible that the effects of cholestasis on growth could not be fully explored in this cohort, as all participants were ascertained on the basis of cholestasis. These data also further underscore the need for an ALGS‐specific growth chart, so we can understand the growth patterns and better interpret the impact of liver transplantation or new medications.
Bone fractures were common in this cohort, as has been noted in ALGS previously.26 Most of these occurred early, before the age of 13 years. It is difficult to attribute this clustering of fractures to the more profound cholestasis that is seen in the first few years of life in ALGS, as by the age of 13 years the median TB had fallen considerably in this cohort. Although chronic cholestatic liver disease may predispose one to poor bone health related to malabsorption and fat‐soluble vitamin deficiencies, it has also been proposed that the direct effects of decreased Notch signaling in bone may be contributing. This is supported by a recent study from ChiLDReN, in which 49 patients with ALGS and 99 children with other inherited chronic liver diseases underwent dual energy X‐ray absorptiometry (DXA) scans.27 In ALGS, DXA measures were found to be low, but improved after adjustment for weight and height. Of note, DXA z‐scores in the ALGS population correlated negatively with measures of cholestasis, including TB and serum bile acid levels. These data support multifactorial influences on bone density in ALGS, with possible contribution of impaired Notch signaling.
Although chronic otitis media4, 20 and hearing loss20 have been noted in ALGS previously, here we present the largest set of audiology testing in this population. It is clear that hearing loss is common in ALGS and that many of these patients have some evidence of middle ear pathology on tympanometry. Continued prospective follow‐up of this cohort will be needed to determine whether these findings persist or resolve over time and the true prevalence of sensorineural hearing loss in ALGS. Further characterization of hearing loss in ALGS is necessary to explore the etiology; recurrent otitis media related to underlying craniofacial anomalies have been cited as one possible cause as well as defects of the middle ear bones.28 These data suggest that tympanometry, at a minimum, should be included as routine surveillance in ALGS.
There are several limitations to this study. Patients were enrolled at different ages, and data before study enrollment were retrospectively collected and therefore likely less reliable. This also led to a wide variation in the duration of follow‐up. Mutational analysis was not available for the entire cohort, although the strict clinical diagnostic criteria were carefully applied at each center to compensate for these missing genetic data. Finally, it could be argued that there was an ascertainment bias, as all participants were enrolled at tertiary‐level liver centers and therefore the population was enriched with more severe ALGS liver disease. Despite this, the primary objective of the study to characterize liver disease in participants with ALGS‐related cholestasis was fulfilled. The intent was not to determine outcomes in all patients with ALGS but to study a carefully selected cohort with ALGS‐related cholestasis.
In conclusion, this study represents a comprehensive assessment of liver disease in a carefully defined population of children with ALGS in the current era. Specifically, we highlight a previously underrecognized and high burden of progressive liver disease in cholestatic ALGS manifested first by pruritus, xanthomas, and fractures during toddlerhood, with a second wave of portal hypertension and associated complications later in adolescence. Together, this burden of liver disease results in a high rate of liver transplantation in those presenting with cholestasis, such that less than 25% of children with ALGS‐related liver disease will reach the age of 19 years with their native liver. These data highlight the need for age‐related therapies that not only address ALGS‐related cholestasis, but also those that target fibrogenesis and the development of portal hypertension.
Supporting information
Supported by the National Institutes of Health, National Center for Advancing Translational Sciences, Clinical and Translational Sciences Award (Grant/Award Nos. UL1 RR025014, UL1 TR000423, UL1 TR000454, UL1 TR001108, UL1 TR001425, UL1 TR001857, UL1 TR001872, UL1 TR001878, UL1 TR002535, and UL1TR00130); the National Institute of Diabetes, Digestive and Kidney Diseases (Grant/Award Nos. DK 62436 [Ann & Robert H. Lurie Children’s Hospital], DK 62445, DK 62453 [University of Colorado, Denver], DK 62456 [The University of Michigan], DK 62466 [Children’s Hospital of Pittsburgh], DK 62470 [Children’s Healthcare of Atlanta], DK 62481 [The Children’s Hospital of Philadelphia], DK 62497 [Cincinnati Children’s Hospital Medical, DK 62500 [UCSF Children’s Hospital], DK 84536 [Riley Hospital for Children], DK 84538 [Children’s Hospital Los Angeles], DK 84575 [Seattle Children’s Hospital], DK103135 [The Hospital for Sick Children], DK103140 [University of Utah], and DK103149 [Texas Children’s Hospital]).
Potential conflict of interest: Dr. Thompson consults for, advises, received grants from, and owns stock in Generation Bio and Qing Bile Therapeutics. He consults for, advises, and received grants from Albireo and Mirum. He consults for and advises Alnylam, Horizon, and Sana. Dr. Rosenthal consults for and received grants from Gilead, AbbVie, and Retrophin. He consults for Albireo, Mirum, and Audentes. He received grants from BMS and Merck. Dr. Heubi consults for and is on the speakers’ bureau for Retrophin. He consults for and received grants from Mirum. He advises Alnylam. He received grants from Friesland Campina. He has equity interest in Asklepion. Dr. Karpen consults for Albireo, Intercept, and Mirum. Dr. Loomes consults for Mirum and Albireo. Dr. Mack consults for Albireo. Dr. Kamath consults and received grants from Mirum. She consults for Albireo and DCI. Dr. Leung consults for Merck. He received grants from AbbVie and Gilead. Dr. Molleston received grants from Shire, Mirum, Gilead, and AbbVie. Dr. Murray received grants from Gilead. Dr. Sokol consults for Retrophin, Shire, Mirum, and Albireo. Dr. Romero reports research grants from Merck and Gilead. Dr. Spinner consults for Retrophin. All other authors have nothing to report.
References
- 1. Alagille D. Alagille syndrome today. Clin Invest Med 1996;19:325‐330. [PubMed] [Google Scholar]
- 2. Krantz ID, Piccoli DA, Spinner NB. Alagille syndrome. J Med Genet 1997;34:152‐157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kamath BM. Consequences of JAG1 mutations. J Med Genet 2003;40:891‐895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Quiros‐Tejeira RE, Ament ME, Heyman MB, Martin MG, Rosenthal P, Hall TR, et al. Variable morbidity in alagille syndrome: a review of 43 cases. J Pediatr Gastroenterol Nutr 1999;29:431‐437. [DOI] [PubMed] [Google Scholar]
- 5. Li L, Krantz ID, Deng Y, Genin A, Banta AB, Collins CC, et al. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet 1997;16:243‐251. [DOI] [PubMed] [Google Scholar]
- 6. Leonard LD, Chao G, Baker A, Loomes K, Spinner NB. Clinical utility gene card for: Alagille syndrome (ALGS). Eur J Hum Genet 2014;22:435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bauer RC, Laney AO, Smith R, Gerfen J, Morrissette JJ, Woyciechowski S, et al. Jagged1 (JAG1) mutations in patients with tetralogy of Fallot or pulmonic stenosis. Hum Mutat 2010;31:594‐601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kamath BM, Bauer RC, Loomes KM, Chao G, Gerfen J, Hutchinson A, et al. NOTCH2 mutations in Alagille syndrome. J Med Genet 2012;49:138‐144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Balistreri WF, Bezerra JA. Whatever happened to “neonatal hepatitis”? Clin Liver Dis 2006;10:27‐53. [DOI] [PubMed] [Google Scholar]
- 10. Emerick KM, Rand EB, Goldmuntz E, Krantz ID, Spinner NB, Piccoli DA. Features of Alagille syndrome in 92 patients: frequency and relation to prognosis. Hepatology 1999;29:822‐829. [DOI] [PubMed] [Google Scholar]
- 11. Lykavieris P. Outcome of liver disease in children with Alagille syndrome: a study of 163 patients. Gut 2001;49:431‐435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bass LM, Shneider BL, Henn L, Goodrich NP, Magee JC. Clinically evident portal hypertension. J Pediatr Gastroenterol Nutr 2019;68:763‐767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Whitington PF, Whitington GL. Partial external diversion of bile for the treatment of intractable pruritus associated with intrahepatic cholestasis. Gastroenterology 1988;95:130‐136. [DOI] [PubMed] [Google Scholar]
- 14. Wang M‐C. Nonparametric estimation from cross‐sectional survival data. J Am Stat Assoc 1991;86:130‐143. [Google Scholar]
- 15. Peng L, Fine JP. Nonparametric estimation with left‐truncated semicompeting risks data. Biometrika 2006;93:367‐383. [Google Scholar]
- 16. Subramaniam P, Knisely A, Portmann B, Qureshi SA, Aclimandos WA, Karani JB, et al. Diagnosis of Alagille syndrome‐25 years of experience at King's College Hospital. J Pediatr Gastroenterol Nutr 2011;52:84‐89. [DOI] [PubMed] [Google Scholar]
- 17. Spinner NB, Colliton RP, Crosnier C, Krantz ID, Hadchouel M, Meunier‐Rotival M. Jagged1 mutations in alagille syndrome. Hum Mutat 2001;17:18‐33. [DOI] [PubMed] [Google Scholar]
- 18. Colliton RP, Bason L, Lu FM, Piccoli DA, Krantz ID, Spinner NB. Mutation analysis of Jagged1 (JAG1) in Alagille syndrome patients. Hum Mutat 2001;17:151‐152. [DOI] [PubMed] [Google Scholar]
- 19. Hoffenberg EJ, Narkewicz MR, Sondheimer JM, Smith DJ, Silverman A, Sokol RJ. Outcome of syndromic paucity of interlobular bile ducts (Alagille syndrome) with onset of cholestasis in infancy. J Pediatr 1995;127:220‐224. [DOI] [PubMed] [Google Scholar]
- 20. Crosnier C, Lykavieris P, Meunier‐Rotival M, Hadchouel M. Alagille syndrome: the widening spectrum of arteriohepatic dysplasia. Clin Liver Dis 2000;4:765‐778. [DOI] [PubMed] [Google Scholar]
- 21. Quiros‐Tejeira RE, Ament ME, Heyman MB, Martin MG, Rosenthal P, Gornbein JA, et al. Does liver transplantation affect growth pattern in Alagille syndrome? Liver Transpl 2000;6:582‐587. [DOI] [PubMed] [Google Scholar]
- 22. Wasserman D, Zemel BS, Mulberg AE, John HA, Emerick KM, Barden EM, et al. Growth, nutritional status, body composition, and energy expenditure in prepubertal children with Alagille syndrome. J Pediatr 1999;134:172‐177. [DOI] [PubMed] [Google Scholar]
- 23. Bucuvalas JC, Horn JA, Carlsson L, Balistreri WF, Chernausek SD. Growth hormone insensitivity associated with elevated circulating growth hormone‐binding protein in children with Alagille syndrome and short stature. J Clin Endocrinol Metab 1993;76:1477‐1482. [DOI] [PubMed] [Google Scholar]
- 24. Rovner AJ, Schall JI, Jawad AF, Piccoli DA, Stallings VA, Mulberg AE, et al. Rethinking growth failure in Alagille syndrome: the role of dietary intake and steatorrhea. J Pediatr Gastroenterol Nutr 2002;35:495‐502. [DOI] [PubMed] [Google Scholar]
- 25. Kamath BM, Yin W, Miller H, Anand R, Rand EB, Alonso E, et al. Outcomes of liver transplantation for patients with Alagille syndrome: the studies of pediatric liver transplantation experience. Liver Transpl 2012;18:940‐948. [DOI] [PubMed] [Google Scholar]
- 26. Bales CB, Kamath BM, Munoz PS, Nguyen A, Piccoli DA, Spinner NB, et al. Pathologic lower extremity fractures in children with Alagille syndrome. J Pediatr Gastroenterol Nutr 2010;51:66‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Loomes KM, Spino C, Goodrich NP, Hangartner TN, Marker AE, Heubi JE, et al. Bone density in children with chronic liver disease correlates with growth and cholestasis. Hepatology 2019;69:245‐257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Teng CS, Yen HY, Barske L, Smith B, Llamas J, Segil N, et al. Requirement for Jagged1‐Notch2 signaling in patterning the bones of the mouse and human middle ear. Sci Rep 2017;7:2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials