


Abstract
Dysregulated splicing is a common event in cancer even in the absence of mutations in the core splicing machinery. The aberrant long non-coding transcriptome constitutes an uncharacterized level of regulation of post-transcriptional events in cancer. Here, we found that the stress-induced long non-coding RNA (lncRNA), LINC02657 or LASTR (lncRNA associated with SART3 regulation of splicing), is upregulated in hypoxic breast cancer and is essential for the growth of LASTR-positive triple-negative breast tumors. LASTR is upregulated in several types of epithelial cancers due to the activation of the stress-induced JNK/c-JUN pathway. Using a mass-spectrometry based approach, we identified the RNA-splicing factor SART3 as a LASTR-interacting partner. We found that LASTR promotes splicing efficiency by controlling SART3 association with the U4 and U6 small nuclear ribonucleoproteins (snRNP) during spliceosome recycling. Intron retention induced by LASTR depletion downregulates expression of essential genes, ultimately decreasing the fitness of cancer cells.
INTRODUCTION
Hypoxia is a common micro-environmental feature that favors tumor progression and acquired drug resistance (1). Long non-coding RNAs (lncRNAs) have recently emerged as key players in hypoxia-driven cancer progression. lncRNAs represent a large and heterogeneous class of non-coding RNAs broadly defined as RNA molecules longer than 200 nucleotides without protein-coding potential. Most lncRNAs are predominantly localized within the cell nucleus, exhibit scarce evolutionary conservation (2), and low expression levels (3). Although <1% of the identified human lncRNAs has been functionally characterized, these particular lncRNAs participate in a variety of physiological and pathological processes (4–6). Hypoxia-induced lncRNAs play pivotal roles in regulating hypoxic responses at chromatin, transcriptional, and post-transcriptional levels by acting as either direct modulators of the hypoxia-inducible factor (HIF) transcriptional cascade, or as HIF-independent effectors. The aberrant expression of hypoxia-induced lncRNAs is associated with aggressive tumor phenotypes, showing to be promising for future utility as a tumor marker and/or therapeutic target (reviewed in (7)).
Recent studies have demonstrated that synthetic nucleic acids represent a valuable approach for drug development (8–10). Antisense oligonucleotides (ASOs), synthetic single-stranded oligonucleotides that bind to the complementary cellular RNA sequences and block their expression, have been successfully tested in preclinical in vivo models. ASO-mediated depletion of MALAT1 impedes tumor growth and reduces lung metastases in a mouse model of mammary carcinoma (11). ASO-based knockdown of the lncRNA SAMMSON decreases survival of melanoma cells in vitro and renders melanomas to be more sensitive to MAPK-targeting therapeutics in patient-derived xenografts (12). An alternative approach for lncRNA targeting could be based on blocking lncRNA–protein interactions. As an example, the small-molecule inhibitor ellipticine targets HOTAIR–EZH2 interaction (13), thereby blocking epigenetic changes that could drive cancer metastasis (14,15).
Here, we searched for hypoxia-regulated lncRNAs that could serve as putative targets in breast cancer therapy. We found that the stress-induced lncRNA LINC02657, which we named LASTR (lncRNA associated with SART3 regulation of splicing), is upregulated in hypoxic breast cancer and is essential for the growth of triple-negative breast cancer cells. LASTR depletion affects splicing efficiency leading to increased intron retention of essential genes and decreased cancer cell fitness. Altogether, we propose LASTR inhibition as a novel therapeutic approach for triple-negative breast cancer treatment.
MATERIALS AND METHODS
Plasmid constructs and GapmeRs
HA-tagged SART3, PURA, PURB, TRA2A, TRA2B or FIP1L1 were cloned into pcDNA 3.1 (+) plasmid. pGIPZ lentiviral vectors containing shControl, shHIF1α and shHIF2α were purchased from Open Biosystems. shRNAs against SART3 or GFP were obtained from the RNAi Consortium (TRC). Scramble shRNA or shRNA against LASTR were cloned into Tet-pLKO-puro. Tet-pLKO-puro was a gift from Dmitri Wiederschain (Addgene plasmid #21915). Wild-type or mutated LASTR promoter was cloned into the pGL4.20 (Promega) vector upstream of the Firefly luciferase ORF sequence. GapmeRs were purchased from Exiqon (Supplementary Table S1).
Cell culture
All cell lines were purchased from ATCC. MDA-MB-468 and MDA-MB-231 cell lines were cultured in DMEM–Glutamax (Gibco) supplemented with 10% fetal bovine serum (FBS) (Hyclone), 100 μg/ml streptomycin and 100 U/ml penicillin (Gibco). BT-549 cells were cultured in RPMI supplemented with 10% FBS (Hyclone), 100 μg/ml streptomycin and 100 U/ml penicillin (Gibco). MCF10A cells were cultured in MEBM (Lonza) supplemented with BPE, hEGF, insulin, hydrocortisone (Lonza) and 50 μg cholera toxin (Sigma-Aldrich).
Lentiviral infections were performed as described by the RNAi Consortium (TRC). Infected cells were selected using puromycin (InvivoGen). Plasmid transfection was performed using FuGENE HD transfection reagent (Promega); transfection of GapmeRs (10 nM) was performed using Lipofectamine 2000 (Thermo Fisher Scientific). The following drugs were used: SP600125 (Sigma-Aldrich); doxycycline-hyclate (Sigma-Aldrich); doxorubicin (Sigma-Aldrich).
Rapid amplification of cDNA ends
Rapid Amplification of cDNA Ends was performed using the SMARTer 5′/3′ RACE kit from Takara, according to the manufacturer's recommendations. Briefly, first strand cDNA was synthesized by SMARTScribe Reverse Transcriptase using a modified oligo (dT) primer, containing an additional sequence that is used as a primer binding site for downstream 3′ PCR reactions. On the 5′ end, the SMARTer II A Oligonucleotide, containing several non-templated residues, is annealed and serves as an additional template for SMARTScribe RT and as a primer binding site for downstream 5′ PCR reactions. A forward gene specific primer for LASTR was designed for the 3′ end amplification, and a reverse primer was designed for the 5′ end amplification.
Gene expression and splicing analyses
RNA from total cell lysates was isolated using the NucleoSpin RNA kit (Machery Nagel). Nuclear and cytoplasmic extracts were isolated using the Nuclei EZ prep kit (Sigma-Aldrich). RNA extraction was then performed using the Trizol/chloroform protocol. cDNA was reverse-transcribed with the Sensifast cDNA synthesis kit (Bioline). Quantitative real-time PCR was performed with the LightCycler 480 SYBR Green I Master reagent using the LightCycler 480 system (Roche). All primer sequences are reported in Supplementary Table S1.
For RNA-seq analysis, RNA was extracted with the Nucleospin RNA kit (Machery Nagel). RSEM (16) was used to estimate gene abundance. Raw sequencing data were aligned by STAR (17) with hg38 as the reference genome and GENCODE v22 as gene annotation. Differentially expressed genes (baseMean > 50, |log2FoldChange| > 1, Padj < 0.05) were determined by the R/Bioconductor package DESeq2 (18). Biotypes of genes were annotated according to GENCODE v22. The TPM (transcripts per million) values of differentially expressed genes were used to generate a heatmap by the R/Bioconductor package ‘pheatmap’ using the ‘Euclidean’ distance measure and ‘ward.D2’ clustering method.
For the TCGA BRCA analysis, the hierarchical clustering analysis was performed using the hypoxia gene signature (19). RNAseq data for breast cancer samples were downloaded from the TCGA GDC Data Portal. Clustering method ‘ward.D2’ and distance ‘Euclidean’ were used to generate a dendrogram (20). The samples were separated depending on the expression of the hypoxia gene signature. R/Bioconductor package DESeq2 was used to identify differentially expressed genes between hypoxia and normoxia. The significance was determined by Padj < 0.05 and |log2FoldChange| > 1.
Alternative splicing analysis was performed using the R/Bioconductor package ASpli (21). Briefly, raw FASTQ reads were mapped to the human genome using STAR with default parameters. Exon skipping, intron retention, alternative 5′ splicing site, and alternative 3′ splicing site were analyzed. To measure alternative splicing levels, PSI (percent spliced-in or percent of inclusion) and PIR (percent of intron retention) metrics were calculated for each sample. Two types of exon–exon or exon–intron junction spanning reads were counted: (i) reads that support sequence inclusion, (ii) reads that support sequence exclusion. Only reads of which >10% of the length overlapped with junctions were counted. PIR/PSI was defined as the ratio between type 1 and the sum of types 1 and 2. To include low expression genes in the analysis, we did not set a minimum number of reads supporting a junction, but we excluded events with a sum of exclusion reads and inclusion reads lower than 5. For intron retention, we further checked the balance of two exon–intron junctions by applying a binomial test to assess if the ratio of the smaller one of two exon–intron junctions reads and inclusion reads is 50%. Events with NA or 0 variance were removed from the list. After obtaining the list of spliced events, we performed a binomial test to test the null-hypothesis that the proportion of upregulated splicing events in one condition is 50%. The distribution of difference in PIR/PSI of spliced events between conditions are shown by histograms. The mean of PIRs/PSIs was used to represent the splicing level for one condition. Differentially spliced events were determined by t-test (P < 0.05).
For splicing analysis in the TCGA samples, the BRCA RNA-seq bam files were downloaded from the TCGA GDC Data Portal. 25th and 75th percentiles were used to define LASTR high and low expression. Intron retention analysis in the TCGA samples was performed as described above, but we only considered the intron retention events that were detected in at least 10 TCGA samples of both the low and high LASTR expression group.
Dual luciferase reporter assay
Firefly luciferase reporters containing the LASTR promoter constructs and the pRL-TK Renilla luciferase reporter were transfected using FuGENE HD transfection reagent (Promega). The luciferase activity was detected using the Dual Luciferase Reporter Assay kit (Promega) according to the manufacturer's protocol. Luminescence was measured using the VICTOR multilabel plate reader (Perkin Elmer).
Cellular assays
2D colony formation was performed by seeding 1 × 103 cells (MDA-MB-231 and BT-549) or 2 × 103 cells (MDA-MB-468) per well in a six-well plate. Colonies were fixed and stained using 1% crystal violet in 35% methanol and counted with the ColCount colony counter (Oxford Optronix). Soft agar colony formation was performed by seeding 1 × 104 cells in 0.35% agar (Sigma) on a 0.5% agar bottom layer. The number of colonies was quantified using the NIH ImageJ software. Cell growth was analyzed by seeding 5000 cells/well in a 96-well plate. The growth was measured using the Cell Titer Glo kit (Promega) according to the manufacture's protocol.
For the radio-sensitization assay, cells transfected with GapmeRs for 24 h, were exposed to increasing doses of irradiation (2–6 Gy). After irradiation, cells were plated into 10 cm dishes. After 2–3 weeks cells were fixed with 2.5% glutaraldehyde in PBS and stained with 0.4% crystal violet. The colonies containing 50 or more cells were counted with the ColCount colony counter (Oxford Optronix).
In vivo tumor xenografts
Xenograft experiments were approved by the Ethical Committee for Animal Experimentation (117/2016). Cells were injected orthotopically into the mammary gland of immunodeficient NMRI nude mice. 5 × 105 cells were resuspended in 100 μl of a 1:1 mixture of PBS and Matrigel (Corning) and injected on both sides. Tumor volumes were monitored every 2 days. Once the tumor volume reached 100 mm3, expression of shRNA against LASTR was achieved by administering the mice drinking water supplemented with 2 mg/ml doxycycline-hyclate (Sigma) in 10% sucrose (Sigma). Drinking water with doxycycline was refreshed every 48 h and protected from light. Mice bearing tumors excessing 1000 mm3 were sacrificed.
Immunohistochemistry and immunoblotting
All antibodies are reported in Supplementary Table S1. IHC was performed on paraffin-embedded tumors. After fixation in 4% paraformaldehyde (Bioconnect), 7 μm paraffin slides were rehydrated and treated with hydrogen peroxide. Antigen retrieval was induced by heat in Tris–EDTA (pH 9). The sections were incubated with a specific antibody and diaminobenzidine (Dako) was used as a detection method followed by hematoxylin counterstaining. Quantification was done using the ImageJ IHC Profiler plugin (22).
For immunoblotting, cells were lysed and proteins were subjected to SDS-PAGE (Invitrogen) and transferred to nitrocellulose membranes followed by immunoblotting with specific antibodies. The signal was visualized with a chemiluminescence detection kit (Amersham Pharmacia Biotech) using an automated digital developer.
Immunoprecipitation and RNA pull-down
Pull-down of in vitro transcribed LASTR was performed as previously published (23). Briefly, in vitro transcribed biotinylated sense or antisense LASTR transcripts were synthesized using the AmpliScribe T7-Flash Biotin–RNA Transcription Kit, according to the manufacturer's instructions. Cells were lysed in ice-cold RNA Immunoprecipitation (RIP) lysis buffer. Upon centrifugation at 15 000 g for 15 min at 4°C, total protein extract was pre-cleared with streptavidin agarose beads (Thermo Fisher Scientific). 1.5 mg of pre-cleared cell lysates was incubated with 10 pmol of the biotinylated sense or antisense LASTR transcript in RNA structure buffer (10 mM Tris–HCl pH 7.0, 100 mM KCl, 10 mM MgCl2) overnight at 4°C. Streptavidin agarose beads (Thermo Fisher Scientific) were then added for 1 h at 4°C. The beads were washed three times with ice-cold 1× complete NT2 (50 mM Tris–HCl pH 7.4, 150 mM NaCl, 1 mM MgCl2, 20 mM EDTA, 0.05% NP‐40 and 1 mM TPEC, 100 U/ml RNase inhibitor (Roche), protease inhibitor, PhosSTOP (Roche)) buffer, three times with trypsin digestion buffer (20 mM Tris–HCl, pH 8.0 and 2 mM CaCl2), and used for mass spectrometry (MS) analysis.
Pull-down of endogenous LASTR was performed as previously described in (12) with few modifications (for a detailed step-by-step protocol, refer to Barra et al. ‘M(R)apping RNA–protein interactions’, Methods Mol Biol, 2020 (in press)). Briefly, 100 μg of streptavidin magnetic beads were incubated overnight at 4°C with 900 pmol of biotinylated LASTR sense or antisense RNA antisense purification (RAP) probes (Biosearch Technologies, Probe sequences are shown in Supplementary Table S1). 4 × 107 cells were washed twice in PBS and fractionated using the Nuclei EZ prep (Sigma-Aldrich) according to the manufacturer's instructions. Nuclei were lysed in 3.5 ml of pull-down buffer (20 mM Tris–HCl pH 8.0, 200 mM NaCl, 2.5 mM MgCl2, 0.05% Triton X-100 in DEPC water) supplemented with a cocktail of protease inhibitors (Halt Protease and Phosphatase Inhibitor Single-Use Cocktail (100×) – Thermo Fisher), 1 mM dithiothreitol and 60 U/ml of SUPERase In RNase Inhibitor (Invitrogen). Lysates were incubated with the beads coupled to the RAP probes at 4°C for 3 h. Samples were then washed five times in pull-down buffer and then divided in two, to elute either proteins or RNA. RNA was eluted from the beads using Trizol/chlorophorm and precipitated with isopropanol. The purified RNA was treated with DNAse and stored at −80°C. The RAP efficiency was estimated by RT-qPCR using 0.5 μg of RNA for the inputs and equal elution volumes for each pull-down sample.
For RNA immunoprecipitation (RIP), cells were washed with ice-cold PBS and lysed in polysome buffer (20 mM Tris–HCl pH 8, 200 mM NaCl, 2.5 mM MgCl2, 0.5% Triton X-100, 60 U/ml SUPERase In RNase Inhibitor (Invitrogen), 1 mM dithiothreitol (DTT) and 1× Halt Protease and Phosphatase Inhibitor Single-Use Cocktail (Life Technologies)) and incubated on ice for 15 min. The soluble fraction obtained by centrifugation at 3220 g for 15 min at 4°C was precleared with 20 μl of Pierce protein A/G magnetic beads (Thermo Fisher Scientific) for 1 h at 4°C. A fraction of the lysate was used for RNA extraction using Trizol/chloroform. The rest of the sample was analysed by immunoblotting. The lysates were incubated with 5 μg of antibody against SART3 or IgG specific overnight at 4°C, and then with 50 μl of Pierce protein A/G magnetic beads for 3 h at 4°C. Beads were then washed four times with polysome buffer and divided in two parts. One fraction was used to confirm immunoprecipitation by immunoblotting, and the other fraction was used for RNA extraction using Trizol/chloroform and isopropanol precipitation.
Expression of the genes of interest was calculated using the ΔCt method, correcting the Ct values by their dilution factor. The Ct value of the input was subtracted from the Ct values of the RIP/RAP, and this normalized Ct value was used to calculate the yield as % of input by the following equation:
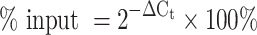 |
Fluorescent in situ hybridization (FISH)
Cells were plated on an eight‐well chamber glass slide (Nalge Nunc International), fixed with 4% paraformaldehyde and permeabilized in 70% ethanol. Hybridization was carried out using a pool of 27 FISH probes designed against LASTR using the Stellaris probe designer software (Biosearch Technologies). The incubation was performed overnight at 37°C in 2× SSC, 10% formamide and 10% dextran. Cells were counterstained with DAPI and images were obtained by using a confocal Nikon C2 microscope (VIB Bioimaging Core).
Chromatin immunoprecipitation
ChIP was performed using the Magna Chip kit (Millipore) following manufacturer's protocol. Briefly, cells were fixed in 1% formaldehyde, lysed and subjected to differential centrifugation to extract nuclei. Nuclei were then lysed and cross-linked chromatin was sonicated to obtain optimal DNA fragment size. Immunoprecipitation was performed by incubating 2 μg of DNA material with 3 μg of antibody against c-JUN and phosphorylated c-JUN (Ser73) or control IgG and protein A/G coupled magnetic beads, overnight at 4°C. Elution, reversal of cross-linking of protein/DNA complexes and DNA extraction was done following the kit's protocol. qPCR analysis was performed using primers specific to the LASTR promoter region (Supplementary Table S1).
RNA native gel electrophoresis
The U4/U6 complex formation was analyzed by performing a native RNA gel electrophoresis after in solution hybridization as described in (24). Briefly, a U6snRNA fluorescent probe (Supplementary Table S1) was designed as described in (24). 0.1 pmol of the U6snRNA probe was incubated with 1 μl of nuclear RNA (3 μg/μl) in the presence of hybridization buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA) for 15 min at 37°C. The reaction was stopped by placing the samples on dry ice. 1 μl of 5× Hi-Density TBE sample buffer was added (Novex, Invitrogen). The samples were then subjected to a non-denaturing 8% TBE gel (Novex, Invitrogen) in TBE running buffer (Novex, Invitrogen) at 250 V on ice. After electrophoresis, the gel was developed using a Typhoon biomolecular imager. Subsequent staining of the gel with Midori Green was used to control the loading.
Mass spectrometry analysis
For the shotgun analysis after RNA immunoprecipitation, washed beads were re-suspended in 150 μl digestion buffer and incubated for 4 h with 1 μg of trypsin (Promega) at 37°C. Beads were removed, another 1 μg of trypsin was added and proteins were further digested overnight at 37°C. Peptides were purified on Omix C18 tips (Agilent), dried and re-dissolved in 20 μl of 0.1% trifluoroacetic acid in water/acetonitrile (98:2, v/v) of which 2 μl was injected for LC–MS/MS analysis on an Ultimate 3000 RSLCnano system (Thermo) in line connected to a Q Exactive mass spectrometer (Thermo). Trapping was performed at 10 μl/min for 4 min in solvent A (0.1% formic acid in water/acetonitrile (98:2, v/v)) on a 100 μm internal diameter (I.D.) × 20 mm trapping column (5 μm beads, C18 Reprosil-HD, Dr Maisch, Germany) and the sample was loaded on a reverse-phase column (made in-house, 75 μm I.D. × 150 mm, 3 μm beads C18 Reprosil-HD, Dr Maisch). Peptides were eluted by a linear increase from 2 to 55% solvent B (0.08% formic acid in water/acetonitrile (2:8, v/v)) over 120 min at a constant flow rate of 300 nl/min. The mass spectrometer was operated in data-dependent mode, automatically switching between MS and MS/MS acquisition. Full-scan MS spectra (400–2000 m/z) were acquired at a resolution of 70 000 in the orbitrap analyzer after accumulation to a target value of 3 000 000. The ten (LASTR) or five (SART3) most intense ions above a threshold value of 17 000 or 13 000 were isolated (window of 2.0 Th) for fragmentation at a normalized collision energy of 25% after filling the trap at a target value of 50 000 for maximum 60 or 80 ms, respectively. MS/MS spectra (200–2000 m/z) were acquired at a resolution of 17 500 in the orbitrap analyzer. The S-lens RF level was set at 50, and we excluded precursor ions with single, unassigned and charge states above five from fragmentation selection. Data analysis was performed with MaxQuant (version 1.5.3.30) using the Andromeda search engine with default search settings including a false discovery rate set at 1% on both the peptide and protein level. Spectra were searched against the human proteins in the SwissProt database, supplemented with the sequences of protein A/G. The mass tolerance for precursor and fragment ions were set to 4.5 and 20 ppm, respectively, during the main search. Enzyme specificity was set as C-terminal to arginine and lysine, also allowing cleavage at proline bonds with a maximum of two missed cleavages. Only proteins with at least one unique or razor peptide were retained leading to the identification of 959 (LASTR) and 252 proteins (SART3). Proteins were quantified by the MaxLFQ algorithm integrated in the MaxQuant software. A minimum ratio count of two unique or razor peptides was required for quantification. Further data analysis was performed with the Perseus software (version 1.5.3.0) after loading the protein groups file from MaxQuant. Proteins only identified by site and reverse database hits were removed, LFQ intensity values were log2 transformed and replicate samples of both conditions were grouped. In the SART3 experiment, protein intensity values in each replicate were normalized based on the expression level of the SART3 bait protein. Proteins with less than three valid values in at least one group were removed and missing values were imputed from a normal distribution around the detection limit. Then, a T-test was performed for pairwise comparison of both conditions. For each protein, the log2 (LASTR sense/LASTR antisense (LASTR) or LASTR GapmeR1/Scramble GapmeR (SART3)) fold change value is indicated on the X-axis, while the statistical significance (−log P value) is indicated on the Y-axis. Proteins outside the curved lines, set by an FDR value of 0.05 and an S0 value of 1 (LASTR) or 1.5 (SART3) in the Perseus software, are considered as putative targets.
For the shotgun analysis on whole cell lysates, frozen cell pellets were dissolved in 1 ml lysis buffer (20 mM HEPES pH 8.0, 8 M urea) and lysis was performed by sonication at high intensity with 10 cycles of 30 s in a water bath at 10°C using a Diagenode Bioruptor Plus. After centrifugation for 15 min at 20 000 × g at room temperature to remove insoluble components, proteins were reduced by addition of 5 mM DTT and incubation for 30 min at 55°C and then alkylated by addition of 10 mM iodoacetamide for 15 min at room temperature in the dark. The protein concentration was measured using a Bradford assay (Bio-Rad) and from each sample 200 μg of protein was used to continue the protocol. Samples were further diluted with 20 mM HEPES pH 8.0 to a final urea concentration of 4 M and proteins were digested with 2 μg of LysC (Wako) (1/100, w/w) for 4 h at 37°C. Samples were again diluted to 2 M of urea and digested with 2 μg of trypsin (Promega) (1/100, w/w) overnight at 37°C. The resulting peptide mixture was acidified by addition of 1% trifluoroacetic acid (TFA) and after 15 min incubation on ice, samples were centrifuged for 15 min at 1780 × g at room temperature to remove insoluble components. Next, peptides were purified on SampliQ SPE C18 cartridges (Agilent). Columns were first washed with 1 ml of 100% acetonitrile (ACN) and pre-equilibrated with 3 ml of solvent A (0.1% TFA in water/ACN (98:2, v/v)) before samples were loaded on the column. After peptide binding, the column was washed again with 2 ml of solvent A and peptides were eluted twice with 75 μl of elution buffer (0.1% TFA in water/ACN (40:60, v/v)). Purified peptides were dried, re-dissolved in solvent A and ∼3 μg of each sample was injected for LC–MS/MS analysis on an Ultimate 3000 RSLCnano system (Thermo) in-line connected to a Q Exactive HF mass spectrometer (Thermo) equipped with a Nanospray Flex Ion source (Thermo). Trapping was performed at 10 μl/min for 4 min in solvent A on a 20 mm trapping column (made in-house, 100 μm internal diameter (I.D.), 5 μm beads, C18 Reprosil-HD, Dr Maisch, Germany) and the sample was loaded on a 200 cm long micro pillar array column (PharmaFluidics) with C18-endcapped functionality mounted in the Ultimate 3000’s column oven at 50°C. For proper ionization, a fused silica PicoTip emitter (10 μm inner diameter) (New Objective) was connected to the μPAC™ outlet union and a grounded connection was provided to this union. Peptides were eluted by a non-linear increase from 1 to 55% MS solvent B (0.1% FA in water/ACN (2:8, v/v)) over 145 min, first at a flow rate of 750 nl/min, then at 300 nl/min, followed by a 15-min wash reaching 99% MS solvent B and re-equilibration with MS solvent A (0.1% FA in water/ACN (2:8, v/v)). The mass spectrometer was operated in data-dependent mode, automatically switching between MS and MS/MS acquisition for the 16 most abundant ion peaks per MS spectrum. Full-scan MS spectra (375–1500 m/z) were acquired at a resolution of 60 000 in the orbitrap analyzer after accumulation to a target value of 3 000 000. The 16 most intense ions above a threshold value of 13 000 were isolated (window of 1.5 Th) for fragmentation at a normalized collision energy of 28% after filling the trap at a target value of 100 000 for maximum 80 ms. MS/MS spectra (200–2000 m/z) were acquired at a resolution of 15 000 in the orbitrap analyzer. The S-lens RF level was set at 50 and we excluded precursor ions with single, unassigned and >7 charge states from fragmentation selection. Data analysis was performed with MaxQuant (version 1.6.2.6) using the Andromeda search engine with default search settings including a false discovery rate set at 1% on both the peptide and protein level. Spectra were searched against the human proteins in the Swiss-Prot database. The mass tolerance for precursor and fragment ions was set to 4.5 and 20 ppm. Enzyme specificity was set as C-terminal to arginine and lysine, also allowing cleavage at proline bonds with a maximum of two missed cleavages. Matching between runs was enabled with a matching time window of 1.5 min and an alignment time window of 20 min. Only proteins with at least one unique or razor peptide were retained leading to the identification of 5528 proteins. Proteins were quantified by the MaxLFQ algorithm integrated in the MaxQuant software. A minimum ratio count of two unique or razor peptides was required for quantification. Further data analysis was performed with the Perseus software (version 1.6.2.1) after loading the protein groups file from MaxQuant. Proteins with less than three valid values in at least one group were removed and missing values were imputed from a normal distribution around the detection limit leading to a list of 2940 quantified proteins used for further data analysis. Differentially expressed proteins were identified by a t-test.
RESULTS
Hypoxia-induced lncRNA LASTR is upregulated in epithelial cancer
Although hypoxia can occur at different stages of breast tumorigenesis (25,26), it is not clear how hypoxia affects cells in the earlier stages of transformation. To identify non-coding transcripts involved in the cellular adaptation to limited oxygen supply in early pre-malignant lesions, we used the non-tumorigenic MCF10A breast epithelial cell line to replicate conditions found within early hyperplastic breast lesions. As MCF10A cells are non-transformed, this setup allows us to study the effects of hypoxia independently from the contribution of cancer-associated genetic and epigenetic changes. To gain a global overview of the transcriptional response to hypoxia, we assessed the expression profiles of MCF10A cells cultured under normoxic or hypoxic conditions. The RNA sequencing (RNAseq) analysis identified 45 lncRNAs that were upregulated by hypoxia in MCF10A cells (Figure 1A). Several of the identified non-coding RNAs, such as MALAT1, NEAT1 andLUCAT1 have been previously reported as hypoxia-induced lncRNAs (27,28).
Figure 1.

The lncRNA LASTR is commonly upregulated in epithelial cancers. (A) Heatmap showing differentially expressed lncRNAs in normoxic and hypoxic (1% O2, 48 h) MCF10A cells as determined by RNA-seq. The scale bar indicates the z-score of transcripts per million (TPM). (B) Overlap between hypoxia-associated lncRNAs in hypoxic TCGA BRCA tumors and hypoxic MCF10A cells. (C) Graphical representation of the zoomed LASTR genomic region along with LASTR CAGE expression data of the LASTR-1 isoform obtained from the FANTOM5 project and the results from a 5′ and 3′ RACE analysis in MCF10A and MDA-MB-231 cells. (D) Boxplot showing LASTR expression in the TCGA BRCA patients stratified by hypoxia status as described in (19). n = number of patients. (E) RT-qPCR analysis of mRNA expression analysis in breast cells cultured under normoxic and hypoxic (1% O2) conditions. Data are presented as mean ± s.e.m.; P-values were determined by two-sided t-tests, n = 3. (F) LASTR expression in the TCGA BRCA patients stratified according to their PAM50 status. P‐values were determined by a two‐sided t‐test. n = number of patients. (G) LASTR expression in the TCGA BRCA patients stratified by tumor stages (T1–T4). n = number of patients. (H) LASTR expression in the TCGA tumor samples. Kidney Renal Clear Cell Carcinoma (KIRC), Head and Neck Squamous Cell Carcinoma (HNSC), Lung Squamous Cell Carcinoma (LUSC), Lung Adenocarcinoma (LUAD) and Cervical Squamous Cell Carcinoma and Endocervical Adenocarcinoma (CESC). P‐values were determined by two‐sided t‐tests. (D, F, G, H) Boxplots show the 25th and 75th percentile of values. The horizontal line indicates the median expression value with whiskers expanding to 1.5 times the interquartile range of the data. P‐values were determined by two‐sided t‐tests.
We next assessed which of these lncRNAs were also overexpressed in hypoxic breast cancers. We stratified The Cancer Genome Atlas (TCGA) breast cancer patients according to their hypoxia gene signature (19) (Supplementary Figure S1A) and identified 742 lncRNAs that were differentially expressed in hypoxic versus normoxic tumors. Expression of 17 out of 45 lncRNAs, upregulated by hypoxia in MCF10A cells was also altered in the hypoxic TCGA breast cancer samples (Figure 1B). For further studies, we decided to focus on the intergenic lncRNA LINC02657, which we named LASTR, as it was described in the atlas of human lncRNAs from the FANTOM5 project (29) (Figure 1C, Supplementary Figure S1B); does not overlap with other genes; and was one the most abundantly expressed lncRNAs in MCF10A cells and breast cancer cell lines (Supplementary Figure S3C). We confirmed that hypoxic TCGA BRCA tumors showed higher levels of LASTR expression (Figure 1D). An RT-qPCR analysis validated that LASTR was upregulated by hypoxia in MCF10A cells along with hypoxia-induced genes, LOX and NFIL3 (30) (Figure 1E). We also observed that hypoxia increased expression of LASTR in the triple-negative MDA-MB-231 and BT-549 cell lines, even though the hypoxic response was less significant in breast cancer cells when compared to non-tumorigenic MCF10A cells (Figure 1E).
The LASTR gene is located on the minus strand of chromosome 10, flanked by CALML3 and ASB13 loci, and has three isoforms (Figure 1C and Figure S1B). Consistently with the FANTOM5 data, RT-qPCR analysis demonstrated that the shortest LASTR-1 isoform was the most abundant (Supplementary Figure S1D). A RACE analysis revealed that the LASTR transcript was 704 nucleotides long, composed of two exons, and polyadenylated (Figure 1C). DeepBase v2.0 analysis (http://biocenter.sysu.edu.cn/deepBase/) of the LASTR-1 evolutional pattern showed that it is a primate specific lncRNA (Supplementary Figure S1D), whereas ORF Finder analysis (www.ncbi.nlm.nih.gov/gorf/gorf.html) revealed that the shortest LASTR transcript is composed of non-translatable regions for at least 46% of its length. Consistent with this observation, PepBank search failed to identify any human peptide matching any ORF in this locus, and PhyloCSF and CPAT software further confirmed that LASTR does not have protein-coding potential (PhyloCSF score < 0.364 and CPAT Fickett TESTCODE statistic P < 0.01).
Analysis of LASTR expression in the TCGA BRCA cohort demonstrated that LASTR was overexpressed in all breast cancer subtypes when compared to normal breast tissue. The highest levels of LASTR upregulation were observed in the most aggressive HER2-positive and triple-negative or basal breast cancers (Figure 1F). LASTR expression did not correlate with stages of breast cancer (Figure 1G), suggesting that LASTR overexpression is a relatively early event during breast cancer progression. Further analysis of the TCGA datasets unveiled that LASTR was also significantly upregulated in several types of epithelial cancers (Figure 1H). Moreover, the survival analyses showed that high levels of LASTR expression were associated with poor survival of patients with renal clear cell carcinoma and lung adenocarcinoma (Supplementary Figure S1F, G). Altogether, these findings suggest that LASTR might be associated with cancer development.
LASTR expression is induced by the stress-activated JNK/c-JUN pathway
Hypoxia-responsive lncRNAs can be categorized as HIF-dependent and HIF-independent (7). HIFs could directly regulate lncRNAs at the transcriptional level through hypoxia response elements (HREs). However, in silico prediction and ENCODE ChIP (chromatin immunoprecipitation) analyses did not reveal HREs in the LASTR promoter. Concordantly, suppression of either HIF1α or HIF2α did not significantly affect induction of LASTR under hypoxic conditions (Supplementary Figure S2A, B), indicating that LASTR upregulation by hypoxia is HIF-independent.
The transcription factor ChIPseq data from ENCODE revealed binding of several transcription factors upstream of the LASTR transcription start site. However, only c-JUN showed consistent binding to the LASTR promoter in three different cancer cell lines, K562, HelA-S3 and A549. The ChIPBase depository (http://deepbase.sysu.edu.cn/chipbase/) also demonstrated the presence of the c-JUN binding site in a close proximity to the LASTR transcription start site in the breast cancer cell line BT-549. Moreover, the c-JUN canonical motif sequence (TGACTCACC) is located ∼90 nucleotides upstream of the LASTR transcription start site (Figure 2A). We also found a higher expression of LASTR in the TCGA BRCA patients with higher levels of c-JUN phosphorylated at Ser73 (Figure 2B). Altogether, these observations suggest that LASTR expression might be regulated by the JNK/c-JUN pathway.
Figure 2.

Stress induces expression of the lncRNA LASTR in a JNK/c-JUN-dependent manner. (A) Graphical representation of the LASTR-1 promoter region along with the ENCODE CHIP-seq data from Hela-S3, HUVECs and K562 cells summarized in a black bar. The red bar shows a canonical c-JUN binding site. (B) Boxplot showing LASTR expression in TCGA BRCA patients stratified by levels of c-JUN phosphorylated at Ser73. Median was used to define high and low levels of phospho-c-JUN. P‐value was determined by a Mann–Whitney test. n = number of patients. (C) Immunoblot analysis of phosphorylated and total c-JUN in MCF10A cells treated with DMSO or SP600125 (20 μM, 18 h). (D) RT-qPCR analysis of LASTR expression in MCF10A cells treated with DMSO or SP600125 (20 μM) for the indicated time periods, n = 3. (E) Immunoblot analysis of phosphorylated and total c-JUN in MCF10A cells treated with DMSO or SP600125 (20 μM, 18 h) under normoxia or hypoxia (1% O2, 16 h). (F) RT-qPCR analysis of LASTR expression in normoxic or hypoxic (1% O2, 16 h) MCF10A cells treated for 18 h with DMSO or SP600125 (20 μM), n = 3. (G) Wild-type LASTR and mutant LASTR promoter activity in MCF10A cells treated with DMSO or SP600125 (20 μM, 18 h) under normoxia or hypoxia (1% O2, 16 h) as determined by a dual-luciferase reporter assay. P-value was determined by a one-way ANOVA test, n = 3. (H) Immunoblot analysis of phosphorylated and total c-JUN in MCF10A cells treated for 1 h with DMSO or doxorubicin (0.3 μM). (I) RT-qPCR analysis of LASTR expression in MCF10A cells treated for 24 h with DMSO or doxorubicin (0.3 μM). n = 3. (J) RT-qPCR analysis of LASTR expression in MCF10A cells treated for 24 h with doxorubicin (0.3 μM), or the combination of doxorubicin (0.3 μM) and SP600125 (20 μM), n = 3 (K) qPCR analysis of LASTR promoter recovered in ChIP assays conducted with the indicated antibodies. Chromatin precipitation was performed using MCF10A cells under normoxia or hypoxia (1% O2, 16 h), n = 3. (D, F, I, J, K) Data are presented as mean ± s.e.m.; P-values were determined by two-sided t-tests.
The phosphorylation of c-JUN at Ser63 and Ser73 by stress-induced c-JUN-NH2-terminal kinase (JNK) prevents ubiquitin-dependent degradation of c-JUN and potentiates its activity (31–33). Therefore, we tested how pharmacological blocking of JNK by the SP600125 inhibitor affected LASTR expression. We found that treatment with SP600125 not only reduced levels of phosphorylated and total c-JUN as expected, but also inhibited LASTR expression (Figure 2C, D). On the other hand, hypoxia led to increased phosphorylation at Ser63 and stability of c-JUN, as well as increased expression of LASTR, whereas JNK inhibition blocked hypoxia-mediated LASTR induction (Figure 2E, F).
We next assessed the activity of a 315 bp DNA fragment of the LASTR promoter, which overlaps with the predicted c-JUN binding site (Figure 2A), by cloning this fragment into the pGL4 luciferase reporter plasmid. We found that hypoxia triggered the LASTR promoter activity, whereas SP600125 treatment suppressed its activity (Figure 2G). The effect of the JNK inhibition on LASTR promoter activity was even more significant under hypoxic conditions (Figure 2G). Furthermore, mutations in the canonical c-JUN binding motif abolished both basal and hypoxia-induced activity of the LASTR promoter (Figure 2G). Taken together, these results indicate that LASTR upregulation by hypoxia is JNK/c-JUN-dependent. Given that DNA damage is also known to activate the JNK/c-JUN signaling pathway (34), we also explored whether the DNA-damaging drug doxorubicin could also induce LASTR expression in a JNK/c-JUN-dependent manner. Consistent with previous reports, we found that doxorubicin treatment increased levels of phosphorylated and total c-JUN (Figure 2H). Moreover, doxorubicin increased LASTR expression in MCF10A cells in a JNK-dependent manner (Figure 2I, J), indicating that doxorubicin treatment activates the JNK/c-JUN pathway and promotes LASTR expression.
To validate c-JUN binding to the LASTR genomic region, we performed a ChIP-qPCR using antibodies against total c-JUN or c-JUN phosphorylated at Ser73. Both c-JUN and phospho-c-JUN showed a stronger binding to the LASTR promoter region in response to hypoxia (Figure 2K). Taken together, these results indicate that LASTR expression is triggered by the stress-activated JNK/c-JUN pathway, suggesting that overexpression of LASTR in human cancers could be caused by upregulation of c-JUN activity, a process commonly occurring in epithelial cancer.
LASTR depletion affects splicing efficiency by regulating the function of the RNA-splicing factor SART3
To assess the cellular function of the lncRNA LASTR, we first examined its subcellular localization by performing cellular fractionation. Consistent with previous reports (35), the lncRNA MALAT1 was strictly retained in the nucleus. On the other hand, we found that LASTR was predominantly localized in the nucleus although it could be also detected in the cytosolic fraction (Figure 3A). This finding was confirmed by performing LASTR-Fluorescent In Situ Hybridization (LASTR-FISH). LASTR showed a diffused nuclear pattern of expression with an enrichment in nucleoli-like structures (Figure 3B).
Figure 3.

LASTR modulates SART3 interactions with U4snRNP and U6snRNP. (A) Subcellular localization of LASTR. LASTR expression was analyzed by RT-qPCR in cellular fractions of MCF10A. Data are presented as mean ± s.e.m.; P-values were determined by two-tailed t-tests, n = 3. (B) LASTR fluorescent in situ hybridization of MCF10A. LASTR-specific probe signals are shown in red. Scale bar, 10 μm. (C) Volcano plot showing LASTR interactors from triplicate pull-downs of total protein extracts from MCF10A cells. Proteins interacting with biotinylated LASTR sense or antisense strands were determined by MS. Putative nuclear interactors of LASTR are shown in red. (D) Validation of LASTR nuclear interactors. Biotinylated sense or antisense LASTR was used for pull-down from MCF10A cells ectopically expressing HA-tagged candidate proteins. The pulldown was followed by immunoblotting using anti-HA antibody. (E) SART3 was immunoprecipitated from MCF10A total protein extracts using anti-SART3 antibody. The presence of SART3 in the immunoprecipitate was detected by immunoblotting, the presence of the indicated non-coding RNAs in the SART3 immunoprecipitate was determined by RT-qPCR analysis. Data are presented as mean ± s.e.m.; P-values were determined by two-tailed t-test, n = 3. (F) LASTR was pulled down from MCF10A cell lysates using biotinylated Stellaris probes. Stellaris probes against antisense LASTR were used as a negative control. SART3 and NONO were detected in the LASTR pull-downs by immunoblotting. (G) Volcano plot of SART3 interactors from triplicate pull-downs of total protein extracts from MCF10A cells treated with Scramble GapmeR or LASTR GapmeR1. U4-associated proteins are shown in red; U6-associated proteins are shown in blue. (H) Presence of U4snRNA and U6snRNA in SART3 immunoprecipitates as detected by RT-qPCR. SART3 immunoprecipitated from total protein extracts of MCF10A cells treated with Scramble GapmeR or LASTR GapmeR1 using anti-SART3 antibody. Data are presented as mean ± s.e.m.; P-values were determined by two-tailed t-tests, n = 3.
As multiple lncRNAs have been shown to act in a complex with proteins (36), we screened for putative protein interactors of LASTR. We incubated in vitro transcribed biotinylated LASTR sense or antisense RNA strands with MCF10A protein extracts. Subsequently, the lncRNA–protein complexes were pulled down using streptavidin beads. Quantitative mass spectrometry (MS) analysis identified several proteins enriched in the LASTR sense fraction (Figure 3C).
We focused on nuclear proteins for further characterization, as LASTR was mostly observed in the nucleus (Figure 3A, B). We incubated in vitro transcribed biotinylated LASTR sense or antisense RNA strands with MCF10A cell lysates overexpressing the different HA-tagged candidates. The lncRNA–protein complexes were pulled down using streptavidin beads followed by immunoblotting with anti-HA antibody. SART3 was the best hit as it showed the strongest enrichment with the LASTR sense strand and no detectable interaction with the antisense strand (Figure 3D).
Previous reports demonstrated that SART3 interacts transiently with the U6 and U4/U6 small nuclear ribonucleoprotein (RNP) particles and promotes the assembly of the U4/U6snRNP complex (snRNPs) (37,38). In fact, we detected a significant enrichment for U6 snRNA when we pulled down endogenous SART3. LASTR was also present in the SART3 pull-downs, whereas we did not detect any enrichment for the more abundant nuclear lncRNAs, MALAT1 and NEAT1 (Figure 3E). Using an RNA affinity purification (RAP), we further validated the binding of LASTR to SART3. Endogenous LASTR was pulled down with biotinylated Stellaris probes specific for either the sense, or antisense transcript of LASTR. Whereas we detected SART3 in the LASTR pull-downs, we did not observe an enrichment of the abundant nuclear protein NONO (Figure 3F). Altogether, our results indicate that LASTR (lncRNA associated with SART3 regulation of splicing) is a nuclear lncRNA that forms a complex with the SART3 protein.
Although we observed a strong binding of SART3 to U6 snRNA in the RNA immunoprecipitation (RIP) experiment (Figure 3E), we could not detect any U6 snRNA interaction with SART3 bound to LASTR, suggesting that LASTR might affect SART3 association with the splicing machinery (Figure 3F). To test this idea, we purified SART3 complexes isolated from MCF10A cells treated with either Scramble, or LASTR specific GapmeR1 and analyzed the SART3 interactome by MS (Figure 3G, Supplementary Figure S2C). The proteins identified in the SART3 immunoprecipitates considerably overlapped (P-value < 2.2e−16 as determined by a Fisher's exact test) with the previously reported SART3 interactome, validating our assay (39). The analysis of proteins differentially interacting with SART3 demonstrated that treatment with the LASTR GapmeR1 increased the interaction of SART3 with the components of U4snRNP, SNRPD1, SNRPD2 and SNRPD3 (Figure 3G). On the other hand, LASTR depletion did not affect SART3 binding to the components of the U6snRNP, such as LSM2–8 proteins, PRPF31, and PRPF4 (40,41). Concordantly with the MS results, LASTR depletion increased association of SART3 with U4snRNA, but did not affect the interaction of SART3 with U6snRNA (Figure 3H). We also found that suppression of LASTR did not affect the formation of the U4/U6 complex (Supplementary Figure S2E). Together, these results suggest that LASTR does not affect the formation of the U4/U6 complex, but could promote SART3 dissociation from the U4/U6snRNP complex during the splicesome cycle.
To examine the impact of LASTR on splicing, we performed transcriptome analysis using a recently developed R package ‘ASpli’ for an integrative splicing analysis. ASpli allows an unbiased evaluation of annotated and novel splicing events and calculates the differences in the percentage of exon inclusion (PSI) and percentage of intron retention (PIR) (Supplementary Figure S3A). We first utilized ASpli to assess the effect of SART3 suppression on splicing (Supplementary Figure S3B). We found that depletion of SART3 in MDA-MB-231 breast cancer cells using two different shRNAs led to increased intron retention and exon skipping (Figure 4A; Supplementary Figure S3C), whereas we did not observe a consistent alteration in alternative 5′ and 3′ splicing (Supplementary Figure S3D, E). These results indicate that SART3 suppression reduces the efficiency of pre-mRNA processing, but does not alter alternative 5′ and 3′ splicing.
Figure 4.

LASTR depletion leads to widespread intron retention. (A) Histogram showing the distributions of differences in percent of intron retention (PIR) and exon skipping (PSI) events in MDA-MB-231 expressing shGFP or shSART3-2. (B) Histogram showing the distributions of differences in percent of intron retention (PIR) and exon skipping (PSI) events in MCF10A cells transfected with Scramble GapmeR or LASTR GapmeRs 1 and 2. (C) RT-qPCR analysis of LASTR expression in MDA-MB-231 cells infected with tetracycline-inducible shRNAs against LASTR, untreated or treated with doxycycline (1 μg/ml, 8 days). Data are presented as mean ± s.e.m.; P-value was determined by a two-sided t-test, n = 3. (D–F) Histograms showing the distribution of differences in percent of intron retention (PIR) and exon skipping (PSI) in MDA-MB-231 cells infected with tetracycline-inducible shRNAs against LASTR, untreated or treated with doxycycline (1 μg/ml, 8 days). (G) Histogram showing the distribution of differences in percent of the indicated splicing events in hypoxic (1% O2, 48 h) MCF10A cells transfected with Scramble GapmeR or LASTR GapmeRs 1 and 2. (A, B, D–G) The number of identified intron retaining and exon skipping events is shown. P-value was determined by Binomial test.
Similar to SART3 suppression, we observed increased intron retention and exon skipping in MCF10A after transfection with LASTR-specific GapmeRs (Figure 4B) and in MDA-MB-231 cells upon induction of three different shRNAs targeting LASTR (Figure 4C–F; Supplementary Figure S4A, B). On the other hand, LASTR suppression did not affect the usage of alternative 5′ and 3′ splice sites (Supplementary Figure S4C, D). We also assessed the role of LASTR in splicing regulation under hypoxic conditions, which is known to affect alternative splicing (42,43). We found that hypoxia altered both splicing efficiency and alternative splicing in MCF10A cells (Supplementary Figure S5). Strikingly, LASTR suppression in hypoxic MCF10A cells treated with LASTR GapmeRs further decreased splicing efficiency and altered splicing patterns (Figure 4G), indicating that stress-mediated LASTR upregulation might counteract decreased splicing efficiency under hypoxic stress. Given that LASTR depletion leads to an increased association of SART3 with U4snRNP and decreased splicing efficiency, we speculate that LASTR might be implicated in the dissociation of SART3 from the U4/U6snRNP complex during spliceosome recycling.
Intron retention mediated by LASTR loss decreases the fitness of cancer cells
The impairment of the splicing machinery could be deleterious for cancer cells (44). Concordantly, suppression of LASTR by inducible shRNAs impaired the ability of triple-negative breast cancer cells to form 2D colonies (Figure 5A; Supplementary Figure S6A, B) and inhibited anchorage-independent growth in MDA-MB-231 cells (Figure 5B). Induction of the expression of the LASTR shRNA also blocked the growth of MDA-MB-231 under hypoxic conditions (Figure 5C). To measure the effect of LASTR suppression on tumor growth, we orthotopically implanted MDA-MB-231/tet-shLASTR-2 cells into the breast pads of immunodeficient mice. When the average volume of the tumors reached 100 mm3, we induced the expression of the shRNA against LASTR by adding doxycycline to the drinking water. Of note, MDA-MB-231 xenografts contained multiple hypoxic regions (Supplementary Figure S6C). Knockdown of LASTR dramatically inhibited MDA-MB-231 xenograft growth despite the incomplete depletion of LASTR (Figure 5D-F). Ki67 immunostaining confirmed decreased proliferation in LASTR-depleted MDA-MB-231 xenografts (Figure 5G). As DNA damage also upregulates LASTR expression (Figure 2I), we also investigated whether LASTR modified the irradiation response of cancer cells. We found that LASTR knockdown led to radio-sensitization of MCF10A-HER2 and MDA-MB-468 cells (Figure 5H). Taken together, these results indicate that LASTR fosters the fitness of cancer cells. (Figure 5D, E and Supplementary Figure S6C)
Figure 5.

LASTR fosters cell fitness under stress conditions. (A) 2D colony formation of the indicated cell lines untreated and treated with doxycycline (1 μg/ml). Representative images of 2D colonies and quantification of the colony number. P-values were determined by two-tailed t-tests, n = 3. (B) Anchorage-independent growth of the indicated cell lines untreated and treated with doxycycline (1 μg/ml). P-values were determined by two-tailed t-tests, n = 3. (C) Cell growth of hypoxic MDA-MB-231/tet-shLASTR-2 cells untreated and treated with doxycycline (1 μg/ml) as measured by the CellTiter-Glo assay. Data are presented as mean ± s.e.m.; P-value was determined by two-way ANOVA, n = 3. (D) Tumor growth of MDA-MB-231/tet-shLASTR-2 xenografts. Cells were implanted into the breast pads of immunodeficient mice. When the average tumor volume reached 100 mm3, expression of the shRNA against LASTR was induced by adding doxycycline to the drinking water (2 mg/ml). Data are presented as mean ± s.e.m.; P-value was determined by two-way ANOVA, n = 9. (E) Tumor weight of MDA-MB-231/tet-shLASTR-2 xenografts, untreated or treated with doxycycline (2 mg/ml) at the end point. Data are presented as mean ± s.e.m.; P-value was determined by a two-tailed t-test. (F) RT-qPCR analysis of LASTR expression in MDA-MB-231/tet-shLASTR-2 xenografts. Data are presented as mean ± s.e.m.; P-value was determined by a two-tailed t-test. (G) Ki67 immunostaining of MDA-MB-231/tet-shLASTR-2 xenografts. Scale bar, 100 μm. Data are presented as mean ± s.e.m.; P-value was determined by a two-tailed t-test. (H) Colony formation assay of MCF10A-HER2 and MDA-MB-468 cells after exposure to increasing doses of ionizing radiation. The cells were pre-treated with the indicated GapmeRs 24 h prior irradiation. Survival is presented as percentage ± s.e.m. of colonies formed relative to untreated cells; P-values were determined by two-tailed t-tests, n = 3.
Dysregulated intron retention, which is a key process underlying tumorigenesis, represented the most salient and consistent feature across samples with decreased LASTR expression. The analysis of the TCGA BRCA cohort also showed that low LASTR expression was associated with increased intron retention (Figure 6A). Therefore, we explored whether decreased cell fitness triggered by LASTR depletion could be caused by aberrant intron retention. We observed an extremely low correlation between PIR changes in the analyzed models, indicating that there was no overlap in significant intron retention events between different model systems (Supplementary Figure S6D). Nonetheless, we noticed that highly expressed genes have a stronger negative correlation with PIR changes when compared to genes with low expression (Supplementary Figure S6E). This indicates that highly expressed genes are more prone to intron retention. Importantly, core fitness genes, which are either essential genes or genes that promote cell proliferation (45), have a significantly higher expression level in breast cancer cells than the STOP genes that negatively regulate cell growth (46) (Supplementary Figure S6F). Consistently to these observations, we found a significant enrichment for core fitness genes among the genes with significantly increased intron retention in cells with either suppressed SART3 or LASTR expression and in the TCGA samples with low LASTR expression (Figure 6B–D). We also observed a similar enrichment for core fitness genes among the genes with significantly increased intron retention in MCF10A cells depleted for LASTR under hypoxic conditions (Figure 6E).
Figure 6.

LASTR loss declines fitness of cancer cells due to decreased expression of essential genes. (A) Histograms showing the distribution of differences in percent of significant intron retention (PIR) in TCGA BRCA tumors with low or high LASTR expression (n = 547). The number of differentially intron retaining genes is shown. P-values were determined by Binomial tests. 25th and 75th percentiles were used to define LASTR high and low expression in the TCGA samples. (B–E) Genes showing increased intron retention in cells with suppressed SART3 or LASTR expression or in the TCGA BRCA samples with low LASTR expression were compared to the lists of core fitness genes (45) or STOP genes (46) using Fisher's exact test. 25th and 75th percentiles were used to define LASTR high and low expression in the TCGA BRCA samples. (F) Gene set enrichment analysis of differentially expressed mRNAs in MDA-MB-231/tet-shLASTR-2 untreated and treated with doxycycline (1 μg/ml, 8 days) among the core fitness genes with significantly increased intron retention. (G) Gene set enrichment analysis of differentially expressed proteins in MDA-MB-231/tet-shLASTR-2 untreated and treated with doxycycline (1 μg/ml, 8 days) among the core fitness genes with significantly increased intron retention. (H, I) RT-qPCR analysis of intron retention/exon expression and mRNA expression levels of the indicated transcripts in MDA-MB-231/tet-shLASTR-2 untreated or treated with doxycycline (1 μg/ml, 8 days). Data are presented as mean ± s.e.m.; P-value was determined by a two-sided t-test, n = 3.
Intron retention leads to downregulation of gene expression through the nonsense-mediated decay pathway (42,47). We suggested that LASTR loss may reduce the fitness of cancer cells by affecting the splicing efficiency and downregulating expression of multiple core fitness genes. In fact, global analyses of mRNA and protein expression upon LASTR depletion in MDA-MB-231 cells demonstrated that the core fitness genes with significantly increased intron retention were downregulated at both mRNA and protein levels in LASTR-suppressed MDA-MB-231 cells (Figure 6F, G). RT-qPCR experiments confirmed increased intron retention and decreased expression of several core fitness genes, such as HAUS5, TONSL, NDOR1, PSMG1 and HNRNPH1, upon LASTR suppression (Figure 6H, I), suggesting that concurrent downregulation of multiple core fitness genes leads to decreased growth of LASTR-depleted cells. Taken together, LASTR fosters fitness of cancer cells by inhibiting intron retention and downregulating expression of essential genes and could serve as a potential target for anti-cancer therapy.
DISCUSSION
Alternative pre-mRNA splicing is an essential cellular process, which enables multi-exonic genes to generate diverse mRNA isoforms. This shapes the transcriptome size and complexity and eventually leads to increased proteome diversity. A growing number of lncRNAs have been reported to regulate splicing by different mechanisms. lncRNAs such as MALAT1 or asFGFR2 affect the splicing output of target genes by modulating chromatin structure. Natural antisense transcripts affect splicing by forming RNA-RNA duplexes with pre-mRNA. By interacting with specific splicing factors, lncRNAs could affect their subcellular localization, posttranslational modification, and activity (reviewed in (48).
Here, we found that LASTR regulates splicing efficiency by binding and regulating SART3, a recycling factor of the splicing machinery. Specifically, LASTR loss regulates the interaction of SART3 with the U4snRNP and U6snRNP. Given that LASTR depletion increases the association of SART3 with the snRNP, it is likely that LASTR might promote dissociation of SART3 from a transient SART3-U4/U6snRNP complex. Studies of the yeast homolog of SART3, Prp24, propose a model in which Prp24 first interacts with U6snRNP and facilitates its association with U4snRNP through partial U4/U6snRNA annealing, but then dissociates from a transient Prp24-U4/U6snRNP complex to allow further spliceosome assembly. Importantly, Prp24 binds more tightly to the annealed U4/U6 complex than the U6 snRNP to stabilize the annealed product, suggesting that additional factors are required to displace Prp24 from U4/U6 di-snRNP (49,50). We could speculate that LASTR may serve as such a factor in human cells.
Depletion of LASTR leads to splicing defects. The impairment of the splicing machinery itself could be beneficial for anticancer therapy as it can have deleterious effects for cancer cells due to general splicing dysregulation. For instance, it has been shown that small natural molecules called pladienolides (SSA and E7107), that specifically target SF3B, a heptameric protein complex of the spliceosomal U2snRNP, can inhibit cancer cell proliferation and induce tumor growth regression in xenograft models due to the induction of defective pre-mRNA splicing (51). Interestingly, the same effect was accomplished using siRNA treatment against SF3B (52).
GapmeR-mediated LASTR depletion induces splicing defects in multiple genes that are essential for cancer cells, thereby specifically affecting cancer cell fitness. Moreover, in an immune competent environment, intron retention may generate neoantigens that could help the clearance of the tumor by the immune system and/or be used for vaccine production (53). Antisense oligonucleotides have already been utilized to induce defective splicing of some essential genes, e.g. in the case of a splicing switch of STAT3α to STAT3β mRNA isoform, which leads to cancer cell death and tumor regression in a xenograft model (54). Antisense oligonucleotide–mediated skipping of exon 6 decreases MDM4 abundance, inhibits melanoma growth, and enhances sensitivity to MAPK-targeting therapeutics (55). These observations further strengthen the idea of using LASTR inhibition as a suitable drug target in breast cancer treatment. Considering that LASTR overexpression occurs not only in breast cancer but also in the majority of epithelial cancers, this points towards more broad applications of LASTR targeted therapy. Moreover, as LASTR suppression induces radio-sensitization of cancer cells, these findings open new prospects for the design of a beneficial combinatorial therapeutic approach in cancer therapy.
DATA AVAILABILITY
All proteomics data have been deposited with the PRIDE database (Project accession: PXD013334). All NGS data have been deposited to NCBI under accession number GSE129344.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Mariana Ascensão-Ferreira, Bernardo P. de Almeida and Nuno L. Barbosa-Morais for insightful comments on the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Research Foundation Flanders (FWO) postdoctoral fellowship (MFB), the KU Leuven Research Fund (C24/17/073), VIB Core Funding.
Conflict of interest statement. None declared.
REFERENCES
- 1. Wilson W.R., Hay M.P.. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer. 2011; 11:393–410. [DOI] [PubMed] [Google Scholar]
- 2. Ulitsky I. Evolution to the rescue: using comparative genomics to understand long non-coding RNAs. Nat. Rev. Genet. 2016; 17:601–614. [DOI] [PubMed] [Google Scholar]
- 3. Derrien T., Johnson R., Bussotti G., Tanzer A., Djebali S., Tilgner H., Guernec G., Martin D., Merkel A., Knowles D.G. et al.. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012; 22:1775–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bergmann J.H., Spector D.L.. Long non-coding RNAs: modulators of nuclear structure and function. Curr. Opin. Cell Biol. 2014; 26:10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Clark M.B., Mattick J.S.. Long noncoding RNAs in cell biology. Semin. Cell Dev. Biol. 2011; 22:366–376. [DOI] [PubMed] [Google Scholar]
- 6. Quinn J.J., Chang H.Y.. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016; 17:47–62. [DOI] [PubMed] [Google Scholar]
- 7. Shih J.W., Kung H.J.. Long non-coding RNA and tumor hypoxia: new players ushered toward an old arena. J. Biomed Sci. 2017; 24:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lavorgna G., Vago R., Sarmini M., Montorsi F., Salonia A., Bellone M.. Long non-coding RNAs as novel therapeutic targets in cancer. Pharmacol. Res. 2016; 110:131–138. [DOI] [PubMed] [Google Scholar]
- 9. Li C.H., Chen Y.C.. Targeting long non-coding RNAs in cancers: progress and prospects. Int. J. Biochem. Cell B. 2013; 45:1895–1910. [DOI] [PubMed] [Google Scholar]
- 10. Matsui M., Corey D.R.. Non-coding RNAs as drug targets. Nat. Rev. Drug. Discov. 2017; 16:167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Arun G., Diermeier S., Akerman M., Chang K.C., Wilkinson J.E., Hearn S., Kim Y., MacLeod A.R., Krainer A.R., Norton L. et al.. Differentiation of mammary tumors and reduction in metastasis upon Malat1 lncRNA loss. Gene Dev. 2016; 30:34–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Leucci E., Vendramin R., Spinazzi M., Laurette P., Fiers M., Wouters J., Radaelli E., Eyckerman S., Leonelli C., Vanderheyden K. et al.. Melanoma addiction to the long non-coding RNA SAMMSON. Nature. 2016; 531:518–522. [DOI] [PubMed] [Google Scholar]
- 13. Fatemi R.P., Salah-Uddin S., Modarresi F., Khoury N., Wahlestedt C., Faghihi M.A.. Screening for small-molecule modulators of long noncoding RNA-protein interactions using AlphaScreen. J. Biomol. Screen. 2015; 20:1132–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gupta R.A., Shah N., Wang K.C., Kim J., Horlings H.M., Wong D.J., Tsai M.C., Hung T., Argani P., Rinn J.L. et al.. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010; 464:1071–U1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kogo R., Shimamura T., Mimori K., Kawahara K., Imoto S., Sudo T., Tanaka F., Shibata K., Suzuki A., Komune S. et al.. Long noncoding RNA HOTAIR regulates polycomb-dependent chromatin modification and is associated with poor prognosis in colorectal cancers. Cancer Res. 2011; 71:6320–6326. [DOI] [PubMed] [Google Scholar]
- 16. Li B., Dewey C.N.. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011; 12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Buffa F.M., Harris A.L., West C.M., Miller C.J.. Large meta-analysis of multiple cancers reveals a common, compact and highly prognostic hypoxia metagene (vol 102, pg 428, 2010). Br. J. Cancer. 2010; 103:1136–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thienpont B., Steinbacher J., Zhao H., D’Anna F., Kuchnio A., Ploumakis A., Ghesquiere B., Van Dyck L., Boeckx B., Schoonjans L. et al.. Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature. 2016; 537:63–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mancini E, Iserte J., Yanovsky M, Chernomoretz A.. ASpli: Analysis of alternative splicing using RNA-Seq. R package version 1.12.0. 2019; doi:10.18129/B9.bioc.ASpli. [Google Scholar]
- 22. Varghese F., Bukhari A.B., Malhotra R., De A.. IHC Profiler: an open source plugin for the quantitative evaluation and automated scoring of immunohistochemistry images of human tissue samples. PLoS One. 2014; 9:e96801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Feng Y., Hu X., Zhang Y., Zhang D., Li C., Zhang L.. Methods for the study of long noncoding RNA in cancer cell signaling. Methods Mol. Biol. 2014; 1165:115–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brow D.A., Vidaver R.M.. An element in human U6 RNA destabilizes the U4/U6 spliceosomal RNA complex. RNA. 1995; 1:122–131. [PMC free article] [PubMed] [Google Scholar]
- 25. Cai Y., Crowther J., Pastor T., Abbasi Asbagh L., Baietti M.F., De Troyer M., Vazquez I., Talebi A., Renzi F., Dehairs J. et al.. Loss of chromosome 8p governs tumor progression and drug response by altering lipid metabolism. Cancer Cell. 2016; 29:751–766. [DOI] [PubMed] [Google Scholar]
- 26. Vaupel P., Schlenger K., Knoop C., Hockel M.. Oxygenation of human tumors: evaluation of tissue oxygen distribution in breast cancers by computerized O2 tension measurements. Cancer Res. 1991; 51:3316–3322. [PubMed] [Google Scholar]
- 27. Brock M., Schuoler C., Leuenberger C., Buhlmann C., Haider T.J., Vogel J., Ulrich S., Gassmann M., Kohler M., Huber L.C.. Analysis of hypoxia-induced noncoding RNAs reveals metastasis-associated lung adenocarcinoma transcript 1 as an important regulator of vascular smooth muscle cell proliferation. Exp. Biol. Med. (Maywood). 2017; 242:487–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Choudhry H., Schodel J., Oikonomopoulos S., Camps C., Grampp S., Harris A.L., Ratcliffe P.J., Ragoussis J., Mole D.R.. Extensive regulation of the non-coding transcriptome by hypoxia: role of HIF in releasing paused RNApol2. EMBO Rep. 2014; 15:70–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hon C.C., Ramilowski J.A., Harshbarger J., Bertin N., Rackham O.J.L., Gough J., Denisenko E., Schmeier S., Poulsen T.M., Severin J. et al.. An atlas of human long non-coding RNAs with accurate 5′ ends. Nature. 2017; 543:199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mense S.M., Sengupta A., Zhou M., Lan C., Bentsman G., Volsky D.J., Zhang L.. Gene expression profiling reveals the profound upregulation of hypoxia-responsive genes in primary human astrocytes. Physiol. Genomics. 2006; 25:435–449. [DOI] [PubMed] [Google Scholar]
- 31. Binetruy B., Smeal T., Karin M.. Ha-Ras augments C-Jun activity and stimulates phosphorylation of its activation domain. Nature. 1991; 351:122–127. [DOI] [PubMed] [Google Scholar]
- 32. Derijard B., Hibi M., Wu I.H., Barrett T., Su B., Deng T.L., Karin M., Davis R.J.. Jnk1 - a protein-kinase stimulated by Uv-Light and Ha-Ras that binds and phosphorylates the C-Jun activation domain. Cell. 1994; 76:1025–1037. [DOI] [PubMed] [Google Scholar]
- 33. Musti A.M., Treier M., Bohmann D.. Reduced ubiquitin-dependent degradation of c-Jun after phosphorylation by MAP kinases. Science. 1997; 275:400–402. [DOI] [PubMed] [Google Scholar]
- 34. Shinoda C., Maruyama M., Fujishita T., Dohkan J., Oda H., Shinoda K., Yamada T., Miyabayashi K., Hayashi R., Kawagishi Y. et al.. Doxorubicin induces expression of multidrug resistance-associated protein 1 in human small cell lung cancer cell lines by the c-jun N-terminal kinase pathway. Int. J. Cancer. 2005; 117:21–31. [DOI] [PubMed] [Google Scholar]
- 35. Miyagawa R., Tano K., Mizuno R., Nakamura Y., Ijiri K., Rakwal R., Shibato J., Masuo Y., Mayeda A., Hirose T. et al.. Identification of cis- and trans-acting factors involved in the localization of MALAT-1 noncoding RNA to nuclear speckles. RNA. 2012; 18:738–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang X.Q.D., Crutchley J.L., Dostie J.. Shaping the genome with non-coding RNAs. Curr. Genomics. 2011; 12:307–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rader S.D., Guthrie C.. A conserved Lsm-interaction motif in Prp24 required for efficient U4/U6 di-snRNP formation. RNA-A Publ. RNA Soc. 2002; 8:1378–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Raghunathan P.L., Guthrie C.. A spliceosomal recycling factor that reanneals U4 and U6 small nuclear ribonucleoprotein particles. Science. 1998; 279:857–860. [DOI] [PubMed] [Google Scholar]
- 39. Sowa M.E., Bennett E.J., Gygi S.P., Harper J.W.. Defining the human deubiquitinating enzyme interaction landscape. Cell. 2009; 138:389–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Salgado-Garrido J., Bragado-Nilsson E., Kandels-Lewis S., Seraphin B.. Sm and Sm-like proteins assemble in two related complexes of deep evolutionary origin. EMBO J. 1999; 18:3451–3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vidal V.P.I., Verdone L., Mayes A.E., Beggs J.D.. Characterization of U6 snRNA-protein interactions. RNA. 1999; 5:1470–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Boutz P.L., Bhutkar A., Sharp P.A.. Detained introns are a novel, widespread class of post-transcriptionally spliced introns. Gene Dev. 2015; 29:63–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Memon D., Dawson K., Smowton C.S., Xing W., Dive C., Miller C.J.. Hypoxia-driven splicing into noncoding isoforms regulates the DNA damage response. NPJ Genom Med. 2016; 1:16020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lee S.C.W., Abdel-Wahab O.. Therapeutic targeting of splicing in cancer. Nat. Med. 2016; 22:976–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hart T., Chandrashekhar M., Aregger M., Steinhart Z., Brown K.R., MacLeod G., Mis M., Zimmermann M., Fradet-Turcotte A., Sun S. et al.. High-resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell. 2015; 163:1515–1526. [DOI] [PubMed] [Google Scholar]
- 46. Solimini N.L., Xu Q.K., Mermel C.H., Liang A.C., Schlabach M.R., Luo J., Burrows A.E., Anselmo A.N., Bredemeyer A.L., Li M.Z. et al.. Recurrent hemizygous deletions in cancers may optimize proliferative potential. Science. 2012; 337:104–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jacob A.G., Smith C.W.J.. Intron retention as a component of regulated gene expression programs. Hum. Genet. 2017; 136:1043–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Romero-Barrios N., Legascue M.F., Benhamed M., Ariel F., Crespi M.. Splicing regulation by long noncoding RNAs. Nucleic Acids Res. 2018; 46:2169–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Didychuk A.L., Montemayor E.J., Brow D.A., Butcher S.E.. Structural requirements for protein-catalyzed annealing of U4 and U6 RNAs during di-snRNP assembly. Nucleic Acids Res. 2016; 44:1398–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jandrositz A., Guthrie C.. Evidence for a Prp24 binding-site in U6 Snrna and in a putative intermediate in the annealing of U6 and U4 Snrnas. Embo Journal. 1995; 14:820–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kotake Y., Sagane K., Owa T., Mimori-Kiyosue Y., Shimizu H., Uesugi M., Ishihama Y., Iwata M., Mizui Y.. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat. Chem. Biol. 2007; 3:570–575. [DOI] [PubMed] [Google Scholar]
- 52. Kaida D., Motoyoshi H., Tashiro E., Nojima T., Hagiwara M., Ishigami K., Watanabe H., Kitahara T., Yoshida T., Nakajima H. et al.. Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA. Nat. Chem. Biol. 2007; 3:576–583. [DOI] [PubMed] [Google Scholar]
- 53. Smart A.C., Margolis C.A., Pimentel H., He M.X., Miao D.N., Adeegbe D., Fugmann T., Wong K.K., Van Ailen E.M.. Intron retention is a source o neoepitopes in cancer. Nat. Biotechnol. 2018; 36:1056–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zammarchi F., de Stanchina E., Bournazou E., Supakorndej T., Martires K., Riedel E., Corben A.D., Bromberg J.F., Cartegni L.. Antitumorigenic potential of STAT3 alternative splicing modulation. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:17779–17784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dewaele M., Tabaglio T., Willekens K., Bezzi M., Teo S.X., Low D.H.P., Koh C.M., Rambow F., Fiers M., Rogiers A. et al.. Antisense oligonucleotide-mediated MDM4 exon 6 skipping impairs tumor growth. J. Clin. Invest. 2016; 126:68–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All proteomics data have been deposited with the PRIDE database (Project accession: PXD013334). All NGS data have been deposited to NCBI under accession number GSE129344.