


Abstract
The spatiotemporal expression of genes is controlled by enhancer sequences that bind transcription factors. Identifying the target genes of enhancers remains difficult because enhancers regulate gene expression over long genomic distances. To address this, we used an evolutionary approach to build two genome-wide maps of predicted enhancer–gene associations in the human and zebrafish genomes. Evolutionary conserved sequences were linked to their predicted target genes using PEGASUS, a bioinformatics method that relies on evolutionary conservation of synteny. The analysis of these maps revealed that the number of predicted enhancers linked to a gene correlate with its expression breadth. Comparison of both maps identified hundreds of putative vertebrate ancestral regulatory relationships from which we could determine that predicted enhancer–gene distances scale with genome size despite strong positional conservation. The two maps represent a resource for further studies, including the prioritization of sequence variants in whole genome sequence of patients affected by genetic diseases.
INTRODUCTION
Enhancers are short DNA sequences that bind transcription factors and contact promoters in cis to activate or repress the transcription of genes into RNA (1). This control—or regulation—of gene expression by enhancers ensures the fine tuning of mRNA abundance in cells. Disruption of enhancer function has been shown to lead to abnormal gene expression and thus to disease (2–4). In addition, the majority of variants identified in Genome Wide Association Studies (GWAS) are found outside coding sequences (5). Together with the observation that many patients remain undiagnosed after genome sequencing because no plausible coding variant can be incriminated (6), these considerations underscore the importance of identifying enhancers and their target genes to better understand genome function.
Numerous methods have been developed to identify enhancers across entire genomes. Early methods were based on the analysis of the evolutionary conservation of non-coding sequences (7–9). More recently, the rise of next generation sequencing technologies has enabled large-scale epigenomics projects to map regulatory regions in a genome, e.g. enhancer-associated histone modifications (10,11), open chromatin regions (12) or binding of enhancer-associated proteins on the genome (13,14). Of note, these approaches predict enhancers through indirect evidence for regulatory function, and do not associate predicted enhancers to their target genes. Although choosing the nearest gene is often used as default (15), the fraction of enhancers regulating their nearest flanking gene is not known. In fact, it is known that enhancers can regulate genes over long distances, sometimes several hundreds of kilobases (kb) away, sometimes bypassing other genes (16,17). The classical case of the Shh gene in mouse demonstrates this quite directly as mutations affecting its expression in the intron of the lmbr1 gene located approximately 1 Mb away (16).
Linking long distance regulatory regions to the genes they regulate is important to study and understand the function of enhancers. Three main categories of experimental methods have been developed to assign enhancers to target genes in a genome-wide manner. The first uses chromosomal conformation capture techniques to identify physical interaction between two loci in the genome (18–23). The second measures the correlation of transcription activity between non-coding sequences and nearby genes (24), assuming the two are signatures of a coordinated regulatory function. The third applies a similar approach to link regions that show correlated open chromatin status across tissues and cell-types (25,26). These experimental methods are—by definition—specific to cell-types, tissue or groups thereof where the experiment is carried out and have been applied mostly in human and mouse genomes, while most sequenced vertebrate genomes (e.g. fish) have no such predictions available yet. The use of methods based on evolutionary principles could solve these difficulties, because they do not depend on the specific biological contexts required by experimental assays and are more easily applicable to multiple species (27–29).
We previously developed such a method called PEGASUS (Predicting Enhancer Gene Associations Using Synteny), a computational method to predict enhancers and their target genes using signals of evolutionary conserved linkage (or synteny) (30). The rationale underlying PEGASUS postulates that an evolutionary genomic rearrangement would dissociate a cis-acting enhancer from its target gene, and would therefore be deleterious. Negative selection would hence result in the preservation of local synteny between enhancers and their target gene, leading to the establishments of so-called Genomic Regulatory Blocs (GRBs) (31,32). PEGASUS is agnostic to cell-types or tissues, as it provides a view of evolutionary conserved enhancer-target interactions active in at least one tissue during the development and lifetime of an individual. It can be applied to any sequenced genome among vertebrates. It is therefore complementary to experimental assays restricted to a specific tissue or cell-type of a given species, as it is able to reveal interactions that would otherwise be hard to investigate experimentally. It was originally tested on the human X chromosome followed by experimental validations of >1000 predicted interactions using transgenic assays (30).
Here, we applied PEGASUS on the entire human and zebrafish genomes to generate two independent genome-wide maps of predicted enhancer–gene interactions. We exploit these resources to uncover evidence for a direct link in the human genome between the number of predicted enhancers associated with a gene and the number of tissues it is expressed in. By comparing these maps, we outline a set of genes with conserved cis-regulation in vertebrates enriched in brain and development functions. We find that the average distance separating predicted enhancer and their target genes scales with genome size, suggestive of weak selective pressure preserving this distance. Finally, our collections of predicted enhancers-gene associations are a valuable resource for the community, represent testable hypotheses that should facilitate genomic studies (e.g. linking transcription factor ChIP-seq peaks to predicted targets) and accelerate the interpretation non-coding variants in whole genome sequences from patient.
MATERIALS AND METHODS
Defining conserved non-coding elements and their most likely target genes
We used a previously published method to predict enhancers and their most likely target genes (30). This method first predicts enhancers as conserved non-coding elements (or CNEs for short) in multiple genome alignments, and second links a CNE to its most likely target gene(s) as the gene in its vicinity with the most conserved synteny, through the computation of a linkage score measuring this conservation.
We identified CNEs in the human and zebrafish genomes in multiple alignments as described previously (30). Briefly, we first identified seeds of 10 bp with at least nine alignment columns conserved between all species considered. These seeds were then extended on both sides, allowing up to three non-conserved alignments columns. We allowed up to 40% of mismatches in a column to consider it as conserved for zebrafish and up to 12% for human. The more relaxed criteria used for zebrafish accounts for the larger phylogenetic distance between this genome and the most closely related genome in the multiple alignment (last common ancestor ∼300 million years old, Supplementary Figure S1) compared to the human situation (last common ancestor ∼90 million years old with the nearest non-primate genome). For the human genome (GRCH37-hg19 version), we used the UCSC 100-way multiple alignments restricted to 35 Sarcopterygii species with a scaffold N50 of at least 1 Mb (a full list is available in Supplementary Table S3). Alignment blocks had to contain at least six species (including human) with one non-primate species to be considered. For the zebrafish genome (danRer7/Zv9 version), we generated multiple alignments that include six other Neopterygii species (a full list is available in Supplementary Table S4). Multiple alignments were built first by pairwise alignments between zebrafish and other species using LastZ (33), then by using these to build multiple alignments with Multiz (34). Alignment parameters can be found in Text S1. Alignments blocks had to contain at least 3 species (including zebrafish) to be considered. Because of the requirement for evolutionary conservation of CNEs, PEGASUS does not identify species-specific CNEs as candidate enhancers.
We used PEGASUS (30) to identify target genes in both genomes. This method first identifies all protein coding genes (Ensembl 75) (35) in a 1 Mb radius around CNEs. It then computes a linkage score for each gene, reflecting the evolutionary conservation of synteny between a CNE and a particular gene. For each gene around a CNE present in N species, the linkage score is computed as follows (equation 1 from (30)):
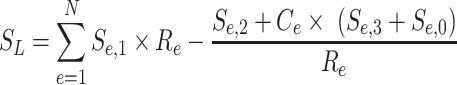 |
where Ce is a corrective factor to take assembly errors from low-coverage sequences into account, Re the rearrangements rate between human or zebrafish and the species e, Se,0, Se,1, Se,2 and Se,3 the respective status of the orthologous gene considered in species e (absent or mis-annotated, present and within the correct radius, present and outside the radius, present and on a different chromosome, respectively). The radius in each species is 1 Mb corrected by the genome size of species e normalized by the human or zebrafish genome size. Re is computed as (Equation (2) in (30)):
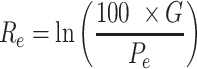 |
where G is the number of gene pairs in the human or zebrafish genome and Pe the number of these pairs that are direct neighbours in species e. The linkage score is then normalized in a [0,1] interval using a sigmoid transformation (Equations (3)–(5) in (30)). For a given CNE, the gene with the highest linkage score is defined as its most likely target gene. If more than one gene have the highest linkage score, they all are defined as most likely targets. Adjacent CNEs targeting identical gene(s), present in the same species, having identical linkage scores and distant by less than 100 bp were merged together. CNEs located at 100 bp or less from an exon were discarded.
Overlap with functional marks and enhancer predictions
We investigated the link between PEGASUS predictions and functional marks and previous in vivo enhancer annotations. We computed the overlap with (a) ChIP-seq peaks of histone modifications (namely H3K27ac, H3K4me1 and H3K4me3) in embryonic stem cells in human (10) and across various developmental stages in zebrafish (36), (b) enhancer predictions from the FANTOM project (24) or from the Vista database (37) for human, and from differentially methylated regions (38) in zebrafish development and (c) ATAC-seq peaks in zebrafish (39) and DNase 1 hypersensitive sites in human (10). For all computations, all overlap of at least 1 bp were considered. Overlap (Figure 2) was computed as the percentage of PEGASUS CNEs that overlap a ChIP-seq peak or annotated enhancer. Recall rates (Figure 3A) were computed as the fraction of enhancers predicted by any given method that overlap PEGASUS CNEs. We also computed recall rates for enhancer regions inferred from Capture Hi-C data in four cell types and compared these recall rates with those computed from FOCS data (see next section, Figure 3B). Recall rates can directly be used to compute the false negative rate as recall = 1 – false negative rate.
Figure 2.

Overlap between PEGASUS CNEs and functional annotations. (A) Percentage of CNEs overlapping histone modification ChIP-seq peaks from embryonic stem cells in human (left) (10) and various developmental stages in zebrafish (right) (36). (B) Percentage of CNEs overlapping enhancer predictions from FANTOM5 (24) or Vista (37) in human (left) and from differentially methylated regions during development in zebrafish (right). ds.DMR stands for developmental stage-specific differentially methylated regions, regions which exhibit tissue-specific differences in methylation levels and overlap distal cis-regulatory regions (38). dsDMRs.All: all sites at all stages; dsDMRs.Dec: sites with decrease in methylation from 6 to 24 hpf embryos, shown to be specific to early development (38). (C) Percentage of CNEs overlapping open chromatin regions, DNase1 peaks in human embryonic stem cells (10) (left) and overlap of ATAC-seq regions with a normalized read count higher than five in zebrafish embryos (39) (right). CNEs were divided into 10 deciles of equal size according to their linkage scores. Class 0 represents CNEs with no associated target gene.
Figure 3.

Overlap between enhancer-target gene prediction methods. (A) Recall rates (1 – false negative rate)for predicted enhancer regions in the human genome. Predicted enhancer regions are the same as in Figure 2. (B) Recall rates for enhancer regions of cHi-C data in adipocytes (20), GM12878 cells (21), embryonic stem cell-derived cardiomyocytes (22) and pancreatic islets (23). Recall rates were computed from PEGASUS enhancers (orange) or FOCS enhancers (25) using the same number of interactions in both sets. Mean recall rates and 2.5–97.5 percentiles (error bar) of 1000 random samplings were plotted for PEGASUS. For each dataset, recall rates for FOCS predictions are significantly different from recall rates for PEGASUS predictions (all P-values < 10−35). (C) Recall rates for enhancer-target gene interactions of cHi-C data in the same cell types as (B). The colour code is identical as (B) and the same number of interactions were used in both sets. Mean recall rates and 2.5–97.5 percentiles (error bar) of 1000 random samplings were plotted for PEGASUS. For each dataset, recall rates for FOCS predictions are significantly different from recall rates for PEGASUS predictions (all P-values < 10−36). (D) Percentage of agreement between in-silico predictions methods (PEGASUS or FOCS) and in-vitro cHi-C interactions, computed as the percentage of one to one overlapping enhancers predicted to target the same gene (see Materials and Methods for more details). We analysed 1755 interactions for pancreatic islets, 1751 interactions for hESC-CMs, 326 interactions for GM12878 cells and 767 interactions for adipocytes.
Comparing target gene predictions with experimental predictions
We compared enhancer–gene interactions predicted by PEGASUS and FOCs (25) with interactions predicted experimentally by Capture Hi-C (cHi-C) in human adipocytes (20), GM12878 cells (21), embryonic stem cell-derived cardiomyocytes (22) and pancreatic islets (23). In the FOCS dataset, we focused on interactions with protein coding genes, merged predictions from the four sources (Fantom, Roadmap, Gro-seq and Encode) and removed redundant interactions to obtain a dataset of 118 021 interactions. To avoid issues with dataset size differences (∼6 million unique (CNE–gene) interactions in PEGASUS), we randomly sampled 1000 times the same number of PEGASUS interactions as for FOCs (Figure 3C). Separately, we also computed the recall rates between PEGASUS and the four cHi-C datasets using the complete set of PEGASUS predictions (Supplementary Figure S6). Recall rates are the fraction of correctly predicted cHi-C enhancer–gene interactions where two conditions are met: candidate enhancers overlap by at least one base and the target gene of this overlapping enhancer is identical with PEGASUS (resp. FOCs). To evaluate more specifically the target prediction recall rate, we determined the percentage of identical predicted target among the subset of overlapping candidate enhancers. Here we considered only candidate enhancers with a one-to-one overlap (one PEGASUS/FOCs candidate enhancer overlapping only one cHi-C candidate enhancer), and both targeting only one gene. Indeed, including CNEs spanning several predicted enhancers or CNEs targeting multiple genes artificially increase the chances of observing overlaps between methods. In this set of overlapping PEGASUS/FOCs enhancers and cHi-C enhancers, the recall was computed as the percentage of overlapping enhancers with the same target gene prediction.
Gene expression data
Gene expression values and calls for the human genome were downloaded from the Bgee database (40) which collects expression calls in 311 human adult tissues. For each gene, we computed the number of human tissues in which a gene is called as expressed. To avoid redundancy, we filtered out terms describing tissues that had daughter terms for the same gene.
Defining orthologous enhancers and target genes between human and zebrafish
We downloaded human–zebrafish and zebrafish–human pairwise chain alignments from UCSC. We defined orthologous CNEs as human and zebrafish CNEs that overlapped by at least 10 bp on either pairwise alignment. We next downloaded human–zebrafish orthologous genes from the Ensembl database (version 75) (35) to identify orthologous enhancers targeting orthologous genes.
Because of the evolutionary distance between human and zebrafish, some orthologous regions are difficult to align and are thus impossible to detect. To circumvent this problem, we used the spotted gar genome (41) to identify additional orthologous CNEs. We downloaded human-spotted gar pairwise chain alignments and used our custom-made zebrafish-spotted gar pairwise chain alignments to respectively map human and zebrafish CNEs onto the spotted gar genome. We considered human and zebrafish CNEs as orthologous if they overlapped by at least 10 bp on the spotted gar genome. No information other than orthology of CNEs on the spotted gar genome was used. We identified orthologous targets by looking at the orthologous genes set used above. Orthologous CNEs identified both directly and via the spotted gar were combined.
Null distribution for CNE–TSS orientation
We computed null distributions to investigate the conservation of CNE and TSS orientation. We first downloaded phastCons conserved elements defined separately in the human and zebrafish genomes (42) from the UCSC database. We filtered these elements to keep only elements that were orthologous between the two genomes (i.e. overlapping on pairwise genome-wise alignments). We then sampled inside a 1 Mb around each pair of human–zebrafish orthologous phastCons elements a pair of human–zebrafish orthologous genes and looked at the TSS orientation relative to the phastCons element. We repeated the sampling 500 times to obtain null distributions of ratios. We compared these distributions to observed ratios by computing Z-scores.
Gene enrichment analysis
We performed anatomical terms enrichment analyses using the TopAnat webtool of the Bgee database (40) and the PantherDB webtool (43). The test set was defined as human–zebrafish orthologous genes with conserved CNEs defined above. The control set was defined in both species as all genes targeted by at least one CNE.
Distance to transcription start sites
CNE–TSS distances were computed only for CNEs with one predicted target gene. We downloaded transcription start sites (or TSS) locations from the Ensembl database (version 75) (35). For each gene, we considered only the transcript giving the longest protein. We computed for each enhancer–gene the distance to the TSS as the shortest distance from enhancer boundary to the target's TSS.
CNE activity breadth prediction
We computed the activity breadth of each CNE by computing the number of tissues where a particular CNE overlaps a histone modification ChIP-seq peak. We focused on H3K27ac and H3K4me1, using ChIP-seq data from the ENCODE project (10).
Topologically associating domains
We downloaded topologically associating domains (or TADs) coordinates for two cell types, human embryonic stem cells (hESCs) and IMR90 fibroblasts (44). We converted these coordinates from hg18 to hg19 using the liftOver utility available at the UCSC genome browser (45). For CNEs targeting only one gene, we computed for each cell type whether both an enhancer and its target gene were located within the same TAD. As control, we shuffled TADs by performing random permutations of TAD genomic localisations using the ‘shuffle’ program from the bedtool package (46), keeping the same distribution of intervals sizes.
In vivo validation
Vector and cloning
The predicted Irx1b CNE (chr19_2681: chr19:28 704 114–28 704 349, danRer7 version of the zebrafish genome) was amplified from zebrafish genomic DNA using the following primers: CNE-Irx1b-Forward: 5′-TGAATGCTCATCCGGAACATCCACTGCTGCTCCCAAAG-3′; CNE-Irx1b-Reverse: 5′-GACCTGCAGACTGGCAGTTCCTCGCCAGAGCTCAG-3′ and cloned into pZED plasmid (47) upstream of the minimal GATA2 promoter/GFP reporter.
Zebrafish egg injections for transgenesis
The Tol2 transposon/transposase method of transgenesis (48) was used with minor modifications. Two nanoliters containing 20 ng/μl of transposase mRNA and 30 ng/μl of phenol/chloroform purified pZED construct were injected in one-cell stage embryos.
In situ hybridization
In situ hybridization were performed as described (49), using an Irx1b probe corresponding to exon 2.
Zebrafish egg injections for mutagenesis
Three RNAs targeting three ultra-conserved sequences in the CNE were designed as follows: CNE-Irx1b-guide1: TCCGTCACGCTGAGATAATC; CNE-Irx1b-guide2: TCAAACACTTTGGGGAACAA; CNE-Irx1b-guide3: TGACCTCTCACCTCGGGCTA. Similarly, three RNAs targeting three ultra-conserved sequences in a random genomic region were designed as follows: Control-guide1: TTGCTTCTGCGCTGAAATAA; Control-guide2: ATGGACTAAAAATTTCACTT; Control-guide3: GAATGTTGATTGTAATTACA. They were purchased from Integrated DNA Technologies as ‘crRNA’, hybridized with their ‘tracrRNA’, forming the guide RNA (gRNA) and incubated with a Cas9 protein (gift from J.-P. Concordet). Three nanoliters containing a mix of the three resulting ribonucleoproteins (Cas9/gRNA) targeting either the control or the predicted Irx1b enhancer were injected at 15 μM each.
Thirty four embryos showed a signal for decreased gene activity over 37 embryos tested.
RESULTS
Enhancers–target genes maps in the human and zebrafish genomes
We predicted enhancers in the human and zebrafish genomes as conserved non-coding elements (CNEs) and applied the PEGASUS method (30) to predict their most likely target genes. PEGASUS assigns to a CNE the gene(s) within a pre-defined radius (set arbitrarily to 1 Mb in both human and zebrafish as a compromise between the number of predicted interactions and their quality, see Text S3 and Supplementary Figure S7 for more details) with the most conserved synteny (linkage between a gene and its CNE), which we quantify using an evolutionary linkage score (Figure 1A). For the human genome, we first analysed the UCSC 100-way multiple genome alignment restricted to 35 non-teleost fish vertebrates with good genome reconstruction quality (methods) to identify 1 376 482 human CNEs. We applied PEGASUS on these elements and assigned over 95% of these CNEs (1 311 643) to 18 339 human genes (out of 20 342 protein coding genes in the human genome, Figure 1D). Human CNEs cover 2.5% of the genome. Out of the ∼1.3 million CNEs identified in the human genome, 394 179 (30%) have only one target gene. For zebrafish, we generated a multiple alignment of seven teleost fish genomes (methods), leading to the identification of 111 281 CNEs, 50% of which (55 515) could be linked to 17 363 genes (out of 26 427 protein coding genes in the zebrafish genome). 9919 (or 17.9%) of zebrafish CNEs have a single target gene. These CNEs cover 0.5% of the zebrafish genome (Supplementary Figure S2). The lower sensitivity in identifying zebrafish CNEs can be explained by phylogenetic sampling differences between the two groups of genome sequences included in the multiple alignments (see Discussion and Text S1). The majority of CNEs are close to their target genes: the median CNE–TSS distance is 353 kb in human and 289 kb in zebrafish (Figure 1B). More details on CNEs, targets and linkage scores can be found in Supplementary Figures S3–S5.
Figure 1.

Application of the PEGASUS method and on the complete human and zebrafish genomes. (A) Schematic summary of the PEGASUS method. 1) CNEs (conserved non-coding elements, in red) are identified by cross-species conservation and all genes in a 1 Mb radius are selected as candidate targets. 2) For each gene, the method will look in every species where the CNE was defined if the gene is present in the genome and in the radius (scaled by relative genome size, green ticks), present but outside the radius (hash) or absent from the genome (red crosses). Genes are free to move around within this radius. 3) This information is used to compute a linkage score between a CNE and each gene within a 1 Mb radius. 4) The gene(s) with the highest linkage score is(are) considered to be the most probable target(s). (B) Distribution of CNE–target gene TSS distances. (C) proportion of intronic, flanking and jumping CNEs. (D) Map of CNE–gene interactions in the human genome. For the sake of visibility, only the 174 465 CNE–gene interactions with a PEGASUS score comprised between 0.9 and 1.0 are shown as red arcs. Black blocs alongside chromosomes are protein-coding genes. Grey rectangles are sequences replaced by ‘Ns’ in the hg19 assembly. An expanded region centered on the FAM71C gene is shown. Green rectangles are protein-coding genes, arcs connect a CNE to the TSS of the predicted target gene and are coloured according to their corresponding linkage score.
The zebrafish enhancer–gene map presented here is the first genome wide resource of its kind. Of note, the human and zebrafish analyses were performed using distinct sets of genomes, enabling rigorous comparisons between phylogenetically independent datasets. We also point out that in both maps, one gene can be associated to more than one CNE (99% of genes in human, 82% of genes in zebrafish), while one CNE can be associated to more than one protein coding gene (70% of CNEs in human, 82% in zebrafish).
PEGASUS can predict enhancer–gene interactions that skip over neighbouring genes, also called ‘bystander’ genes (31). We found a large fraction of these ‘jumping’ interactions in the human and zebrafish genomes, 27% and 49% respectively (Figure 1C). Moreover, 34% of these ‘jumping’ CNEs in human and 37% in zebrafish are located in an intron of a gene that is not their target gene.
PEGASUS is an in-silico method entirely based on evolutionary signals to identify the target genes of CNEs. We evaluated how our predictions coincide with in-vivo inferences of regulatory regions (histone modifications (10,36) experimental enhancer predictions (24,37,38), or open chromatin regions (10,39)). Overlap with inferred regulatory regions is positively associated with linkage score in both species, which support the regulatory role of PEGASUS CNEs (Figure 2). Interestingly, in both human and zebrafish, CNEs show a stronger link with histone modifications associated with enhancer activity (H3K27ac and H3K4me1) than with modifications thought to be enriched in promoter regions (H3K4me3) Figure 2A), consistent with their distal regulatory role predicted by PEGASUS. A positive association between linkage score and experimentally predicted enhancers predictions (Figure 2B) and open chromatin regions (Figure 2C) further supports this regulatory role. Overall, the PEGASUS recall rate against several sources of candidate enhancers ranges from 0.21 to 0.90 (Figure 3A). Experimentally verified VISTA sequences (37) show the highest recall rate while the remaining resources (histone marks, transcribed regions, open chromatin regions) show lower recall rates but also possess an unknown rate of functional enhancers.
We further computed the recall rate between PEGASUS in-silico target gene assignments and in-vitro inferences obtained by capture Hi-C (20–23), as well as how these rates compare to FOCS, another recent method designed to predict enhancer–gene interactions using functional data (25). Of note, these in-vitro inferences are currently available only for the human genome. We randomly sampled the same number of PEGASUS interactions as available for FOCS, and show that on average the recall rate is similar between PEGASUS and FOCS (0.005–0.038; Figure 3B). If the complete set of PEGASUS interactions is used however, the recall rate reaches 0.24–0.45 in the four capture Hi-C datasets (Supplementary Figure S6). To gain a finer insight into the ability of PEGASUS to recapitulate in-vitro predictions, we computed the percentage of identically predicted target genes between PEGASUS and capture Hi-C datasets, but restricted them to common (overlapping) candidate enhancers. We show that 28–42% of capture Hi-C targets are predicted by PEGASUS. These results show that PEGASUS, which works in a cell-type or tissue agnostic way, predicts target genes of putative enhancers in a manner consistent with experimental and tissue specific methods such as capture Hi-C.
Finally, we show that enhancer–gene associations predicted by PEGASUS are consistent with the 3D organisation of the human genome, because they are located inside topologically associating domains (TADs (44)) more often than expected by chance. For CNEs linked to a single gene, 57% and 66% of predicted interactions indeed reside within a TAD in hESCs and IMR90 cells respectively, compared to an average of 32% and 41% respectively when we shuffle TAD intervals (proportion test P-values < 10−10 for both cell types, Figure 4; see Text S2 for more details).
Figure 4.

PEGASUS predicted interactions and TADs. (A) Percentage of CNE-gene associations located in the same TAD as a function of PEAGSUS linkage score. (B) Percentage of CNE-gene associations located in the same TAD as a function of CNE–TSS distance. TADs locations are available for hESCs and IMR90 cells (44). Full lines correspond to observed TAD locations, dashed lines to shuffled TAD locations.
Genes with more enhancers are expressed in more tissues
Genes cover a broad range of tissue specificities, from broadly expressed genes required for generic cellular functions and expressed in most tissues to tightly regulated developmental genes sometimes expressed in just a few cells in a short window of time. It naturally follows that the number of enhancers regulating a gene might directly influence the breadth of a its expression pattern. Although this has never been demonstrated in vertebrates, indirect evidence exists in non-vertebrate organisms with compact genomes, like Drosophila or Caenorhabditis elegans, where genes expressed in a greater number of tissues and spatial domains are flanked by more non-coding DNA than other genes (50). We used candidate enhancer–gene interactions predicted by PEGASUS in the human genome to investigate this question, using expression data from the Bgee database (40). We first distinguished target genes with a transcription start site (TSS) overlapping a CpG island (referred to as CpG genes) and other genes (referred to as non-CpG genes) because CpG genes are usually broadly expressed while other genes are more tissue-specific (51–53). Results show that genes targeted by more CNEs tend to be expressed in more tissues in both CpG and non-CpG genes, (Figure 5A; all quartile distributions significantly different, Wilcoxon rank sum tests between successive quartile distributions P-values < 10−5).
Figure 5.

Regulation complexity is positively linked with expression breadth. (A) Genes targeted by more CNEs are expressed in more tissues: distribution of the number of tissues a gene is expressed in four quantiles of genes based on their number of. Distributions were plotted separately for CpG-genes and non-CpG genes. Expression calls were obtained from the Bgee database (40). The data represents 17 951 genes. (B) CNEs closer to their target genes are active in more tissues: distribution of the number of tissues with a histone modification ChIP-seq peak overlapping PEGASUS CNEs in four quartiles of CNEs based on their distances to their target TSS. Distributions were plotted separately for two histone modifications associated with enhancer activity. ChIP-seq peaks were recovered from ENCODE (10). The data represents 374 421 CNEs.
Finally, we asked if the genomic distance between a candidate enhancer and its target gene is linked to the enhancer's tissue specificity. For each candidate enhancer, we computed the number of tissues where it is active (overlaps ChIP-seq peaks of H3K27ac and H3K4me1 histone modifications (10)). We observe that candidate enhancers closer to their targets are active in a higher number of tissues while more distant enhancers tend to show a more restricted tissue specificity (Figure 5B, all but one Wilcoxon rank sum tests between successive quartile distributions show P-values < 10−5, H3K27ac third versus fourth quartile P-value = 0.17)
Function of regulatory interactions conserved in vertebrates
We defined orthologous CNE-gene associations between the human and zebrafish genomes to study features associated with this conservation of regulatory linkage. Such conserved linkage between enhancers and target genes is consistent with a common origin in the ancestor of Euteleostomi (bony vertebrates), the last common ancestor of human and zebrafish. We identified ∼2000 CNEs conserved between human and zebrafish (1986 in human, 1949 in zebrafish) associated by PEGASUS to ∼600 human-zebrafish orthologous genes (567 human genes, 607 zebrafish genes, see Materials and Methods). Functional enrichment analyses show that these ancestral regulatory associations are highly enriched in neuronal functions and development (Supplementary Tables S1 and S2). Thirty percent of these predicted associations are annotated as ‘jumping’ over one or more bystander genes in both species. This includes DMRTA2, a transcription factor involved in female germ cell development (54) and in brain development (55), or TSHZ1, a member of the teashirt gene family involved in olfactory bulb development (56). The strong enrichment in core developmental functions observed with orthologous PEGASUS predictions (Supplementary Table S2) is consistent with earlier observations, as enhancers identified through sequence conservation are often found to be active during development, especially in the nervous system (7,8,57,58).
We validated a predicted ancestral association using a CRISPR–Cas9 mediated knock-out approach. We focused on one CNE of the zebrafish genome (chr19_2681), predicted to be associated with a single gene named irx1b. This gene plays multiple roles during pattern formation of vertebrate embryos (59,60), and we expect its expression pattern to be tightly regulated by a complex array of enhancers. The CNE has evidence for a functional activity during development: it overlaps H3K27ac and H3K4me1 marks as well as ATAC-seq peaks (Figure 6A) and is conserved in all vertebrates. The human orthologous CNE is associated by PEGASUS to IRX1 and IRX2 and also shows evidence for a functional role in this species (H3K4me1 and H3K27ac (10)) as well as sequence conservation. The deletion of the CNE greatly decreases the expression of the endogenous gene in several structures of the zebrafish embryo (Figure 6B, C) establishing it as a bona fide developmental enhancer. Interestingly, the CNE targeted by the deletion is closer to another gene, irx4b, without being associated to it by PEGASUS (Figure 6D), and the expression of this gene is unaffected by the absence of the CNE (Figure 6E). This further illustrates that choosing the nearest gene as a target of a putative enhancer can lead to false predictions and that PEGASUS can distinguish the correct gene target among closely spaced genes.
Figure 6.

In vivo inactivation of a predicted ancestral enhancer for irx1b affects its expression. (A) Evidence for the regulatory potential of the chr19_2681 CNE. The figure shows the normalized read counts for a ChIP-seq analysis of histone modifications (H3K4me1 & H3K27ac) in four developmental stages (dashed & dotted lines) (36) and for an ATAC-seq analysis in 24 hpf embryos (full lines) (39) in a 60 kb region around chr19_2681 (red rectangle). (B) 24 h old F0 zebrafish embryos injected with a Tol2 transposon containing the predicted irx1b CNE positioned 5′ of the gata2 minimal promoter driving green fluorescent protein (GFP) expression. (C) In situ hybridization for irx1b mRNA performed on 24 h old wild type embryos (WT) or embryos injected with a mix of three CRISPR/Cas9 ribonucleoprotein complexes targeted at the predicted irx1b enhancer. The CNE activity profile overlaps with the expression profile of irx1b, which comprises the acousticovestibular ganglia, the caudal diencephalon, the tectum, the hindbrain, the spinal cord and the anterior part of the otic vesicle but not the mid-hindbrain boundary. irx1b expression level is greatly decreased in all these structures when the CRISPR/Cas9 complex is targeted to the CNE compared to the control, establishing it as a bona fideirx1b enhancer. (D) The chr19_2681 CNE, predicted to target irx1b is located closer to irx4b (37 kb) than to irx1b (88 kb). (E) In contrast to (B), irx4b’s expression profile which includes the anterior part of the otic vesicle and a few cells in the hindbrain is not affected by the CRISPR/Cas9 complex showing that this CNE is specific to irx1b and does not regulate irx4b.
Enhancers-gene distances scale with genome size
The ‘action range’ of enhancers is known to encompass a wide span, from within the target gene itself to more than 1 Mb away (16,17). Importantly, it has been shown that enhancers can change localization within a TAD without affecting downstream gene regulation (61). Together with results showing high rates of enhancer turnover between species (62,63), these specific examples suggest little selective constraints exist on maintaining enhancers in a specific position relative to their target genes. We tested this hypothesis genome wide using the ∼2000 predicted gene-enhancer associations conserved between human and zebrafish (two genomes with different sizes, 3.1 and 1.5 Gb respectively). We estimated the relative neutral evolution of genomic distances using the sizes of orthologous introns, which are thought to be under negligible size constraint. Results show that distances between orthologous CNEs and their orthologous target genes scale with intron size (median CNE–TSS distance ratio = 2.23, median intron length ratio = 2.39, Figure 7A), consistent with an absence of functional constraint on CNE–gene distances.
Figure 7.

Distances between CNEs and target genes scale with genome size. (A) Pairwise comparison of CNE-TSS distances and intron lengths between human and zebrafish. All comparisons were made using the set of human-zebrafish orthologous genes with conserved CNEs. For enhancer–TSS distances, we compared the CNE–TSS distances (DH and DZ for human and zebrafish) for each conserved pair of gene and CNE. For intron lengths, we compared the total intron length (sum of a gene's intron lengths, IH and IZ for human and zebrafish) for each orthologous gene pair. Comparisons were computed as log2(human/zebrafish) ratios. (B) Deep conservation of CNE-TSS relative orientation between human and zebrafish. For the 3570 conserved interactions we analysed, we determined if CNEs were on the 5′ side of the TSS in both species (top left panel), both on the 3′ side of the TSS in both species (bottom left panel), or in different orientations (top and bottom right panels). Numbers represent corresponding percentages of conserved interactions in each category. Z-scores were computed by comparing observed ratios to expected ratios. We computed expected ratios by considering human–zebrafish orthologous non-exonic phastCons elements (42) and the relative orientation of human–zebrafish orthologous genes located in a 1 Mb radius around these elements.
Perhaps surprisingly, despite this absence of selective constraint on interaction distances, we note that the positions of CNEs relative to their target gene TSS (i.e. whether a CNE lies on the 5′ or the 3′ side of the TSS) seems highly conserved. We found that >91% of orthologous CNEs are located on the same side of their TSS in the human and zebrafish species (30.6% on the 5′ side and 60.9% on the 3′ side, Figure 7B). To establish if this is due to an evolutionary conserved topological constraint instead to the average rate of human–zebrafish genomic conservation in these regions, we computed a null distribution by sampling human–zebrafish orthologous conserved sequences and arbitrarily assigning them to a gene within 1 Mb of their position, thus mimicking PEGASUS results but without any influence of its linkage score. We find that PEGASUS interactions with conserved orientations in human and zebrafish are observed more than expected (P-values < 10−23 and 0.05, Figure 7B). Second, CNEs located on the 5′ side of their target TSS are observed less than expected (P-value < 10−13). Finally, CNEs located on the 3′ side of their target TSS are observed more often than expected (positive Z-score, P-value < 10−23, Figure 7B), indicating our observations are unlikely to be due to chance alone.
DISCUSSION
We applied the PEGASUS method to identify ∼1 300 000 human and ∼55 000 zebrafish predicted enhancers (conserved non-coding elements) targeting the majority of the genes in their respective genomes. We find strong evidence for a regulatory role of these interactions, consistent with previous studies that concentrated on highly conserved sequences in vertebrates and involved in genomic regulatory blocks (GRBs) (64). We further show that regulatory interactions ancestral to vertebrates concentrate on core functions necessary to build an organism, that the number of predicted enhancers associated to a gene positively correlates with its breadth of expression and that the distance between predicted enhancers and their target gene evolves neutrally. Our catalogue of enhancer–gene associations contributes to the study of gene regulation by enhancers in vertebrates, can be easily used in a variety of studies and can improve our understanding of gene functions in particular biological contexts.
The first PEGASUS published set of associations was restricted to the human × chromosome (30). Here, we significantly improve our knowledge of enhancers in vertebrates by applying PEGASUS to the entire human genome, and in the zebrafish genome where no set of enhancer–gene association exists to-date. Moreover, this catalogue can be used to guide and improve the interpretation of epigenomics data such as histone modifications or open chromatin regions or of sequence variants found to be associated to a particular disease in large-scale sequencing projects.
Effects of phylogenetic sampling
We identified a contrasted number of CNEs between human and zebrafish (∼1 300 000 and ∼55 000, respectively). This difference can be explained by differences in phylogenetic sampling, i.e. the number of species and their phylogenetic relationships used for predicting enhancers and linking them to their target gene. Zebrafish was compared to only six other genomes, with zebrafish being an outgroup to all but the spotted gar (Supplementary Figure S1). In contrast, human was compared to 35 other genomes (Supplementary Figure S1). We tested the influence of this phylogenetic sampling by comparing the human genome to six other genomes that mirror the phylogenetic relationships in the zebrafish study (human being an outgroup to all but one species and equivalent phylogenetic distances as in the fish phylogeny, Supplementary Table S3). In this reduced set, we identify approximately 253 620 CNEs, of which 193 085 (∼82%) target 13 398 genes, a sharp reduction compared to the set identified with a full phylogenetic sampling. The relatively small number of CNEs identified in the zebrafish genome can therefore be explained by the lower number of fish species that can be used for comparative analyses. The addition of more fish species will improve predicted enhancers identifications in the near future.
The challenges of predicting long-distance regulatory interactions
PEGASUS genome-wide in-silico enhancer-target gene predictions allow us to directly compare PEGASUS with genome-wide in vitro assays: we found that PEGASUS predictions and in vitro predictions agree up to 42% of the time. Most functional assays currently employed to predict long-range regulatory interactions in the human genome rely on specific cell lines or tissues (20–23). This might limit expectations to observe overlaps in their predictions, especially given that many enhancers are tissue-specific (11,65,66). It is interesting to observe that recall rates for capture Hi-C data computed with a computational method that relies on the analysis of epigenetics data from hundreds of cell types (25) is equivalent to rates computed with PEGASUS. The sole rationale underlying PEGASUS predictions is that the interactions are functional, therefore under sufficient evolutionary conservation to be identified by comparisons with other genomes. This feature allows PEGASUS to be applied in genomes for which limited epigenetics data is available. Moreover, PEGASUS is able to predict enhancer–gene regulatory interactions that ‘jump’ over one or more bystander genes, which reflects the biology of gene expression regulation more accurately than ‘nearest gene’ approaches. Indeed, experimental methods often find a large fraction of candidate enhancers targeting a distal (non-nearest) gene. This is the case for example for 60% of the FANTOM5 enhancer-promoter interactions (24) or 48–58% of capture Hi-C datasets used in this work (21,23). This suggests that a ‘nearest gene’ strategy, which is still often used to define target genes when studying predicted regulatory regions from epigenomics data (e.g. GREAT (15)) is likely to miss a large fraction of relevant interactions.
PEGASUS identifies >40% of enhancer–gene interactions observed in experimental assays carried out in human cell lines (Figure 3C). A much higher overlap may not be expected because the reliance of PEGASUS on evolutionary constraints tend to enrich for interactions active during development (7,13), and these are typically harder to identify in differentiated cell lines. In addition, given the rapid evolutionary turnover of enhancer regions during evolution (62,63,67), it is likely that a fraction of cell-type specific enhancers have had little time to leave detectable footprints of selection in a genome. For the same reasons, PEGASUS will fail to capture species-specific or recently evolved regulatory interactions.
No evidence for natural selection acting on enhancer–gene distances
Enhancer regulation is mediated through the 3D organisation of the genome. Enhancer-gene interactions occur mostly within TADs (68), large units of chromosomal interactions largely conserved between cell types and species (44,69), via DNA looping (70). Consistent with observations that the distance between an enhancer and its target within a TAD has no effect on its regulatory potential (61), we show that CNE–TSS interaction evolution between human and zebrafish follows the same pattern as intron size evolution. A recent analysis of GRBs in metazoans based on the analysis of clusters of conserved non-coding elements showed that these blocks correlate well with known TADs and their sizes seem to correlate well with genome size (71), providing further evidence that interaction distances between enhancers and target genes are under the same forces that affect genome size in metazoans. Interestingly, our results show that this lack of selective constraint on interaction distances comes with a strong conservation of relative CNE–TSS orientation.
This study provides a unique view of the conservation and evolution of enhancers in vertebrate genomes. Our results based on evolutionary and comparative genomics are complementary to and consistent with genome-wide experimental observations. They support a model where the number of enhancers controlling a gene drives its expression breadth. They also highlight the biological functions with conserved regulation since the vertebrate ancestor. Moreover, the PEGASUS method provides a robust tissue and life stage agnostic target gene prediction method that opens research possibilities in the study of gene regulation in a wide number of species.
DATA AVAILABILITY
PEGASUS predictions for the human genome (hg19), the zebrafish genome (danRer7) as well as interactions predicted to be conserved between both genomes are available here: ftp://ftp.biologie.ens.fr/pub/dyogen/PEGASUS/.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Pierre Vincens for the coordination of computing resources, Morgane Thomas-Chollier and Camille Berthelot for fruitful comments and remarks on earlier versions of this manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
French Government and implemented by Agence Nationale de la Recherche [ANR-14-CE13-0004-02, ANR-10-BINF-01-03 Ancestrome, ANR-10-LABX-54 MEMOLIFE, ANR-10-IDEX-0001-02 PSL* Research University]. Funding for open access charge: Agence Nationale de la Recherche.
Conflict of interest statement. None declared.
REFERENCES
- 1. Shlyueva D., Stampfel G., Stark A.. Transcriptional enhancers: from properties to genome-wide predictions. Nat. Rev. Genet. 2014; 15:272–286. [DOI] [PubMed] [Google Scholar]
- 2. Kleinjan D.A., van Heyningen V.. Long-range control of gene expression: emerging mechanisms and disruption in disease. Am. J. Hum. Genet. 2005; 76:8–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Smemo S., Campos L.C., Moskowitz I.P., Krieger J.E., Pereira A.C., Nobrega M.A.. Regulatory variation in a TBX5 enhancer leads to isolated congenital heart disease. Hum. Mol. Genet. 2012; 21:3255–3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lupiáñez D.G., Kraft K., Heinrich V., Krawitz P., Brancati F., Klopocki E., Horn D., Kayserili H., Opitz J.M., Laxova R. et al.. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell. 2015; 161:1012–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Edwards S.L., Beesley J., French J.D., Dunning A.M.. Beyond GWASs: illuminating the dark road from association to function. Am. J. Hum. Genet. 2013; 93:779–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang F., Lupski J.R.. Non-coding genetic variants in human disease. Hum. Mol. Genet. 2015; 24:R102–R110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Woolfe A., Goodson M., Goode D.K., Snell P., McEwen G.K., Vavouri T., Smith S.F., North P., Callaway H., Kelly K. et al.. Highly conserved non-coding sequences are associated with vertebrate development. PLoS Biol. 2005; 3:e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Visel A., Prabhakar S., Akiyama J.A., Shoukry M., Lewis K.D., Holt A., Plajzer-Frick I., Afzal V., Rubin E.M., Pennacchio L.A.. Ultraconservation identifies a small subset of extremely constrained developmental enhancers. Nat. Genet. 2008; 40:158–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lindblad-Toh K., Garber M., Zuk O., Lin M.F., Parker B.J., Washietl S., Kheradpour P., Ernst J., Jordan G., Mauceli E. et al.. A high-resolution map of human evolutionary constraint using 29 mammals. Nature. 2011; 478:476–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Project Consortium ENCODE. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Consortium Roadmap Epigenomics, Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J. et al.. Integrative analysis of 111 reference human epigenomes. Nature. 2015; 518:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shibata Y., Sheffield N.C., Fedrigo O., Babbitt C.C., Wortham M., Tewari A.K., London D., Song L., Lee B.-K., Iyer V.R. et al.. Extensive evolutionary changes in regulatory element activity during human origins are associated with altered gene expression and positive selection. PLoS Genet. 2012; 8:e1002789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Visel A., Blow M.J., Li Z., Zhang T., Akiyama J.A., Holt A., Plajzer-Frick I., Shoukry M., Wright C., Chen F. et al.. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009; 457:854–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Blow M.J., McCulley D.J., Li Z., Zhang T., Akiyama J.A., Holt A., Plajzer-Frick I., Shoukry M., Wright C., Chen F. et al.. ChIP-Seq identification of weakly conserved heart enhancers. Nat. Genet. 2010; 42:806–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McLean C.Y., Bristor D., Hiller M., Clarke S.L., Schaar B.T., Lowe C.B., Wenger A.M., Bejerano G.. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010; 28:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lettice L.A., Heaney S.J.H., Purdie L.A., Li L., de Beer P., Oostra B.A., Goode D., Elgar G., Hill R.E., de Graaff E.. A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum. Mol. Genet. 2003; 12:1725–1735. [DOI] [PubMed] [Google Scholar]
- 17. Sanyal A., Lajoie B.R., Jain G., Dekker J.. The long-range interaction landscape of gene promoters. Nature. 2012; 489:109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mifsud B., Tavares-Cadete F., Young A.N., Sugar R., Schoenfelder S., Ferreira L., Wingett S.W., Andrews S., Grey W., Ewels P.A. et al.. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat. Genet. 2015; 47:598–606. [DOI] [PubMed] [Google Scholar]
- 19. Jäger R., Migliorini G., Henrion M., Kandaswamy R., Speedy H.E., Heindl A., Whiffin N., Carnicer M.J., Broome L., Dryden N. et al.. Capture Hi-C identifies the chromatin interactome of colorectal cancer risk loci. Nat. Commun. 2015; 6:6178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pan D.Z., Garske K.M., Alvarez M., Bhagat Y.V., Boocock J., Nikkola E., Miao Z., Raulerson C.K., Cantor R.M., Civelek M. et al.. Integration of human adipocyte chromosomal interactions with adipose gene expression prioritizes obesity-related genes from GWAS. Nat. Commun. 2018; 9:1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cairns J., Freire-Pritchett P., Wingett S.W., Várnai C., Dimond A., Plagnol V., Zerbino D., Schoenfelder S., Javierre B.-M., Osborne C. et al.. CHiCAGO: robust detection of DNA looping interactions in capture Hi-C data. Genome Biol. 2016; 17:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Choy M.-K., Javierre B.M., Williams S.G., Baross S.L., Liu Y., Wingett S.W., Akbarov A., Wallace C., Freire-Pritchett P., Rugg-Gunn P.J. et al.. Promoter interactome of human embryonic stem cell-derived cardiomyocytes connects GWAS regions to cardiac gene networks. Nat. Commun. 2018; 9:2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Miguel-Escalada I., Bonàs-Guarch S., Cebola I., Ponsa-Cobas J., Mendieta-Esteban J., Atla G., Javierre B.M., Rolando D.M.Y., Farabella I., Morgan C.C. et al.. Human pancreatic islet three-dimensional chromatin architecture provides insights into the genetics of type 2 diabetes. Nat. Genet. 2019; 51:1137–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Andersson R., Gebhard C., Miguel-Escalada I., Hoof I., Bornholdt J., Boyd M., Chen Y., Zhao X., Schmidl C., Suzuki T. et al.. An atlas of active enhancers across human cell types and tissues. Nature. 2014; 507:455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hait T.A., Amar D., Shamir R., Elkon R.. FOCS: a novel method for analyzing enhancer and gene activity patterns infers an extensive enhancer-promoter map. Genome Biol. 2018; 19:56–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Thurman R.E., Rynes E., Humbert R., Vierstra J., Maurano M.T., Haugen E., Sheffield N.C., Stergachis A.B., Wang H., Vernot B. et al.. The accessible chromatin landscape of the human genome. Nature. 2012; 489:75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ahituv N., Prabhakar S., Poulin F., Rubin E.M., Couronne O.. Mapping cis-regulatory domains in the human genome using multi-species conservation of synteny. Hum. Mol. Genet. 2005; 14:3057–3063. [DOI] [PubMed] [Google Scholar]
- 28. Mongin E., Dewar K., Blanchette M.. Mapping association between long-range cis-regulatory regions and their target genes using synteny. J. Comput. Biol. 2011; 18:1115–1130. [DOI] [PubMed] [Google Scholar]
- 29. He B., Chen C., Teng L., Tan K.. Global view of enhancer-promoter interactome in human cells. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:E2191–E2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Naville M., Ishibashi M., Ferg M., Bengani H., Rinkwitz S., Krecsmarik M., Hawkins T.A., Wilson S.W., Manning E., Chilamakuri C.S.R. et al.. Long-range evolutionary constraints reveal cis-regulatory interactions on the human X chromosome. Nat. Commun. 2015; 6:6904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kikuta H., Laplante M., Navratilova P., Komisarczuk A.Z., Engström P.G., Fredman D., Akalin A., Caccamo M., Sealy I., Howe K. et al.. Genomic regulatory blocks encompass multiple neighboring genes and maintain conserved synteny in vertebrates. Genome Res. 2007; 17:545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Akalin A., Fredman D., Arner E., Dong X., Bryne J.C., Suzuki H., Daub C.O., Hayashizaki Y., Lenhard B.. Transcriptional features of genomic regulatory blocks. Genome Biol. 2009; 10:R38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schwartz S., Kent W.J., Smit A., Zhang Z., Baertsch R., Hardison R.C., Haussler D., Miller W.. Human-mouse alignments with BLASTZ. Genome Res. 2003; 13:103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Blanchette M., Kent W.J., Riemer C., Elnitski L., Smit A.F.A., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D. et al.. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 2004; 14:708–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Flicek P., Amode M.R., Barrell D., Beal K., Billis K., Brent S., Carvalho-Silva D., Clapham P., Coates G., Fitzgerald S. et al.. Ensembl 2014. Nucleic Acids Res. 2014; 42:D749–D755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bogdanović O., Fernández-Miñán A., Tena J.J., la Calle-Mustienes E., Hidalgo C., van Kruysbergen I., van Heeringen S.J., Gómez-Skarmeta J.L.. Dynamics of enhancer chromatin signatures mark the transition from pluripotency to cell specification during embryogenesis. Genome Res. 2012; 22:2043–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Visel A., Minovitsky S., Dubchak I., Pennacchio L.A.. VISTA Enhancer Browser–a database of tissue-specific human enhancers. Nucleic Acids Res. 2007; 35:D88–D92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lee H.J., Lowdon R.F., Maricque B., Zhang B., Stevens M., Li D., Johnson S.L., Wang T.. Developmental enhancers revealed by extensive DNA methylome maps of zebrafish early embryos. Nat Commun. 2015; 6:6315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gehrke A.R., Schneider I., la Calle-Mustienes E., Tena J.J., Gomez-Marin C., Chandran M., Nakamura T., Braasch I., Postlethwait J.H., Gómez-Skarmeta J.L. et al.. Deep conservation of wrist and digit enhancers in fish. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:803–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bastian F., Parmentier G., Roux J., Moretti S., Laudet V., Robinson-Rechavi M.. Bgee: Integrating and Comparing Heterogeneous Transcriptome Data Among Species. Data Integration in the Life Sciences, Lecture Notes in Computer Science. 2008; 5109:Berlin, Heidelberg: Springer; 124–131. [Google Scholar]
- 41. Braasch I., Gehrke A.R., Smith J.J., Kawasaki K., Manousaki T., Pasquier J., Amores A., Desvignes T., Batzel P., Catchen J. et al.. The spotted gar genome illuminates vertebrate evolution and facilitates human-teleost comparisons. Nat. Genet. 2016; 48:427–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Siepel A., Bejerano G., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S. et al.. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005; 15:1034–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mi H., Muruganujan A., Thomas P.D.. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013; 41:D377–D386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J.S., Ren B.. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012; 485:376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D.. The human genome browser at UCSC. Genome Res. 2002; 12:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bessa J., Tena J.J., la Calle-Mustienes E., Fernández-Miñán A., Naranjo S., Fernández A., Montoliu L., Akalin A., Lenhard B., Casares F. et al.. Zebrafish enhancer detection (ZED) vector: a new tool to facilitate transgenesis and the functional analysis of cis-regulatory regions in zebrafish. Dev. Dyn. 2009; 238:2409–2417. [DOI] [PubMed] [Google Scholar]
- 48. Kawakami K., Takeda H., Kawakami N., Kobayashi M., Matsuda N., Mishina M.. A transposon-mediated gene trap approach identifies developmentally regulated genes in zebrafish. Dev. Cell. 2004; 7:133–144. [DOI] [PubMed] [Google Scholar]
- 49. Hauptmann G., Gerster T.. Two-color whole-mount in situ hybridization to vertebrate and Drosophila embryos. Trends Genet. 1994; 10:266. [DOI] [PubMed] [Google Scholar]
- 50. Nelson C.E., Hersh B.M., Carroll S.B.. The regulatory content of intergenic DNA shapes genome architecture. Genome Biol. 2004; 5:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Saxonov S., Berg P., Brutlag D.L.. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:1412–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ramsköld D., Wang E.T., Burge C.B., Sandberg R.. An abundance of ubiquitously expressed genes revealed by tissue transcriptome sequence data. PLoS Comput. Biol. 2009; 5:e1000598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhu J., He F., Hu S., Yu J.. On the nature of human housekeeping genes. Trends Genet. 2008; 24:481–484. [DOI] [PubMed] [Google Scholar]
- 54. Poulain M., Frydman N., Tourpin S., Muczynski V., Mucsynski V., Souquet B., Benachi A., Habert R., Rouiller-Fabre V., Livera G.. Involvement of doublesex and mab-3-related transcription factors in human female germ cell development demonstrated by xenograft and interference RNA strategies. Mol. Hum. Reprod. 2014; 20:960–971. [DOI] [PubMed] [Google Scholar]
- 55. Urquhart J.E., Beaman G., Byers H., Roberts N.A., Chervinsky E., O'Sullivan J., Pilz D., Fry A., Williams S.G., Bhaskar S.S. et al.. DMRTA2 (DMRT5) is mutated in a novel cortical brain malformation. Clin. Genet. 2016; 89:724–727. [DOI] [PubMed] [Google Scholar]
- 56. Ragancokova D., Rocca E., Oonk A.M.M., Schulz H., Rohde E., Bednarsch J., Feenstra I., Pennings R.J.E., Wende H., Garratt A.N.. TSHZ1-dependent gene regulation is essential for olfactory bulb development and olfaction. J. Clin. Invest. 2014; 124:1214–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Plessy C., Dickmeis T., Chalmel F., Strähle U.. Enhancer sequence conservation between vertebrates is favoured in developmental regulator genes. Trends Genet. 2005; 21:207–210. [DOI] [PubMed] [Google Scholar]
- 58. Visel A., Taher L., Girgis H., May D., Golonzhka O., Hoch R.V., McKinsey G.L., Pattabiraman K., Silberberg S.N., Blow M.J. et al.. A high-resolution enhancer atlas of the developing telencephalon. Cell. 2013; 152:895–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bosse A., Zülch A., Becker M.B., Torres M., Gómez-Skarmeta J.L., Modolell J., Gruss P.. Identification of the vertebrate Iroquois homeobox gene family with overlapping expression during early development of the nervous system. Mech. Dev. 1997; 69:169–181. [DOI] [PubMed] [Google Scholar]
- 60. Lecaudey V., Anselme I., Dildrop R., Rüther U., Schneider-Maunoury S.. Expression of the zebrafish Iroquois genes during early nervous system formation and patterning. J. Comp. Neurol. 2005; 492:289–302. [DOI] [PubMed] [Google Scholar]
- 61. Symmons O., Pan L., Remeseiro S., Aktas T., Klein F., Huber W., Spitz F.. The Shh Topological Domain Facilitates the Action of Remote Enhancers by Reducing the Effects of Genomic Distances. Dev. Cell. 2016; 39:529–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Schmidt D., Wilson M.D., Ballester B., Schwalie P.C., Brown G.D., Marshall A., Kutter C., Watt S., Martinez-Jimenez C.P., Mackay S. et al.. Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science. 2010; 328:1036–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Villar D., Berthelot C., Aldridge S., Rayner T.F., Lukk M., Pignatelli M., Park T.J., Deaville R., Erichsen J.T., Jasinska A.J. et al.. Enhancer evolution across 20 mammalian species. Cell. 2015; 160:554–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Polychronopoulos D., King J.W.D., Nash A.J., Tan G., Lenhard B.. Conserved non-coding elements: developmental gene regulation meets genome organization. Nucleic Acids Res. 2017; 45:12611–12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Javierre B.M., Burren O.S., Wilder S.P., Kreuzhuber R., Hill S.M., Sewitz S., Cairns J., Wingett S.W., Várnai C., Thiecke M.J. et al.. Lineage-specific genome architecture links enhancers and non-coding disease variants to target gene promoters. Cell. 2016; 167:1369–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Heinz S., Romanoski C.E., Benner C., Glass C.K.. The selection and function of cell type-specific enhancers. Nat. Rev. Mol. Cell Biol. 2015; 16:144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Liao B.-Y., Weng M.-P.. Natural selection drives rapid evolution of mouse embryonic heart enhancers. BMC Syst Biol. 2012; 6:S1.(Suppl 2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Symmons O., Uslu V.V., Tsujimura T., Ruf S., Nassari S., Schwarzer W., Ettwiller L., Spitz F.. Functional and topological characteristics of mammalian regulatory domains. Genome Res. 2014; 24:390–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Cournac A., Koszul R., Mozziconacci J.. The 3D folding of metazoan genomes correlates with the association of similar repetitive elements. Nucleic Acids Res. 2016; 44:245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Deng W., Lee J., Wang H., Miller J., Reik A., Gregory P.D., Dean A., Blobel G.A.. Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor. Cell. 2012; 149:1233–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Harmston N., Ing-Simmons E., Tan G., Perry M., Merkenschlager M., Lenhard B.. Topologically associating domains are ancient features that coincide with Metazoan clusters of extreme noncoding conservation. Nat. Commun. 2017; 8:441. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
PEGASUS predictions for the human genome (hg19), the zebrafish genome (danRer7) as well as interactions predicted to be conserved between both genomes are available here: ftp://ftp.biologie.ens.fr/pub/dyogen/PEGASUS/.