


Abstract
Diplonemids are highly abundant heterotrophic marine protists. Previous studies showed that their strikingly bloated mitochondrial genome is unique because of systematic gene fragmentation and manifold RNA editing. Here we report a comparative study of mitochondrial genome architecture, gene structure and RNA editing of six recently isolated, phylogenetically diverse diplonemid species. Mitochondrial gene fragmentation and modes of RNA editing, which include cytidine-to-uridine (C-to-U) and adenosine-to-inosine (A-to-I) substitutions and 3′ uridine additions (U-appendage), are conserved across diplonemids. Yet as we show here, all these features have been pushed to their extremes in the Hemistasiidae lineage. For example, Namystynia karyoxenos has its genes fragmented into more than twice as many modules than other diplonemids, with modules as short as four nucleotides. Furthermore, we detected in this group multiple A-appendage and guanosine-to-adenosine (G-to-A) substitution editing events not observed before in diplonemids and found very rarely elsewhere. With >1,000 sites, C-to-U and A-to-I editing in Namystynia is nearly 10 times more frequent than in other diplonemids. The editing density of 12% in coding regions makes Namystynia’s the most extensively edited transcriptome described so far. Diplonemid mitochondrial genome architecture, gene structure and post-transcriptional processes display such high complexity that they challenge all other currently known systems.
INTRODUCTION
Diplonemids are heterotrophic marine flagellates belonging to the phylum Euglenozoa, which also includes the well-studied parasitic kinetoplastids and free-living euglenids (1,2) (Figure 1A). Diplonemids have been largely overlooked due to technical limitations, because the SSU rRNA V4 region, typically amplified in the metabarcoding approach, has expanded beyond typical lengths in diplonemids. In a recent survey, which targeted the more conserved V9 region, they have been detected in virtually every sample of seawater (3) and are currently ranked among the most diverse and abundant eukaryotic groups in the world’s oceans (4–7). The ecological role of diplonemids in marine environments has only recently begun being appreciated (6,8,9).
Figure 1.
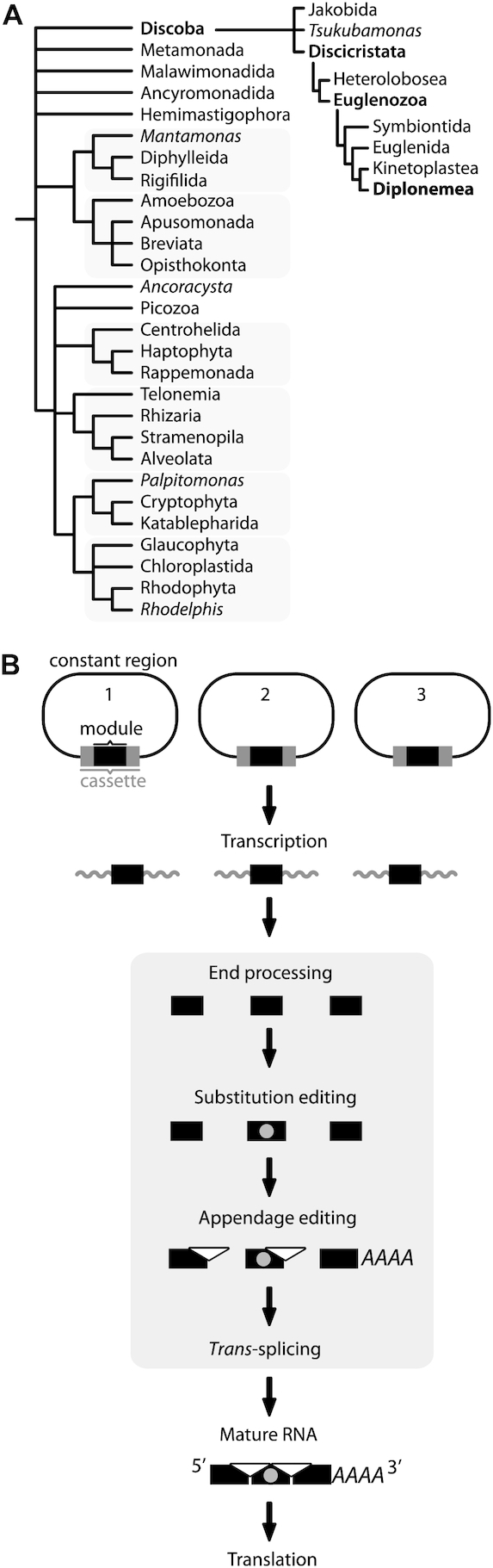
Phylogenetic position of diplonemids and their mitochondrial DNA structure and gene expression. (A) Recent classification of eukaryotes (based on (1)) highlighting the position of diplonemids. (B) The various steps of mitochondrial gene expression in diplonemids. The model gene consists of three pieces, also referred to as modules, each encoded in a unique region (cassette) on a different chromosome. Modules, together with surrounding regions, are transcribed separately from a promoter located in the constant region (25). Primary transcripts are end-processed, removing 5′ and 3′ non-coding regions from the primary transcripts. Certain module transcripts undergo substitution RNA editing and/or appendage RNA editing (nucleotide additions at the module's 3′ end). The module transcript that will constitute the transcript’s 3′ end is poly-adenylated (mRNAs and mtLSU rRNA) or poly-uridylated (mtSSU rRNA) (16,19). Finally, modules are joined together (trans-spliced) yielding mature RNA (mRNA or rRNA). Note that all post-transcriptional processes (gray background) occur in parallel in the diplonemid mitochondrion (19); thus, the arrows do not imply strict sequentiality.
We know close to nothing about the lifestyle of diplonemids (10). Their varied morphology and the recent finding of various bacterial endosymbionts in their cells (11–13) indicate that they have a versatile modus vivendi, likely enabling them to occupy widely different niches within the oceanic ecosystem. According to the 18S rRNA V9 region-based phylogenies, diplonemids fall in four major lineages: (i) ‘classical’ diplonemids (Diplonemidae) including both benthic and planktonic species of the genera Diplonema, Rhynchopus, Lacrimia, Flectonema and Sulcionema; (ii) hemistasiids (Hemistasiidae), a small planktonic clade composed of the genera Hemistasia, Artemidia and Namystynia; (iii) an extremely diverse clade of deep-sea pelagic diplonemids (or DSPD I, recently named Eupelagonemidae); and (iv) a second but relatively small clade of deep-sea pelagic diplonemids (DSPD II) (2,11,12,13,14).
The most conspicuous features of diplonemids are their unique and complex mitochondrial genome architecture and gene expression (Figure 1B), studied in depth in the type species Diplonema papillatum (reviewed in (15–17)). For example, the amount of mitochondrial DNA (mtDNA), estimated at 250 Mbp, is so far the highest recorded for an organelle (18). Further, its mtDNA is composed of >80 covalently closed non-catenated 6 and 7 kbp-long circular chromosomes. Except for a short unique region called the ‘cassette’, chromosomes consist mostly of repetitive sequence termed ‘constant region’, which is essentially identical across chromosomes of a given size class (Figure 1B). It is the unique cassette that typically encloses a single gene fragment, also called module, the size of which ranges from 40 to 540 bp. Chromosomes including cassettes are transcribed separately, then the non-coding portions are removed leaving behind module-only transcripts that are subsequently joined to their cognate neighboring modules derived from other circles (16,19). In this way, mature transcripts (mRNAs and rRNAs) are assembled via massive trans-splicing, the mechanism of which remains unknown.
Trans-splicing in diplonemid mitochondria differs from that observed in organelles and in the nucleus of nematodes and other eukaryotes including diplonemids (20–22), because the former process is apparently catalyzed by neither spliceosomes nor Group I or Group II splicing machineries (16,19,23–26). With the exception of the mitochondrial small subunit ribosomal RNA (mtSSU rRNA), all D. papillatum genes undergo this assembly process, making the extent of trans-splicing unprecedented.
Moreover, in addition to gene fragmentation compensated by trans-splicing, D. papillatum mitochondrial transcripts are subject to extensive RNA editing of two fundamentally different types: post-transcriptional uridine addition (U-appendage) at 3′ ends of certain modules (unique to diplonemids), and deaminations of adenosines to inosine (A-to-I) and cytidines to uridines (C-to-U) at numerous positions within coding regions (16,27).
Studies of three other diplonemid species (D. ambulator, Flectonema neradi and Rhynchopus euleeides) showed little deviation from the features observed in the type species (27,28), except that across these taxa, the size of mitochondrial chromosomes ranges from 2 to 12 kbp (27,28). However, it was reported recently that gene fragmentation, as well as U-additions and A and C substitutions in transcripts are much more frequent in Hemistasia phaeocysticola, the single hemistasiid species examined until now (27,29). While D. papillatum has at most 11 modules per gene (16), the genes of H. phaeocysticola are fragmented twice as much (29).
Does the single examined hemistasiid species represent an exceptional case, or can fragmentation and RNA editing of mitochondrial transcripts reach even higher levels of complexity? To address this question, we examined species that have recently become available in culture, with their morphology, ultrastructure and life cycles described (11–13). Here, we present the most extensive comparative study of diplonemid mtDNA performed thus far. We show that the degree of mitochondrial RNA editing and gene fragmentation can reach unprecedented complexity, highlighting several questions about the role and evolution of these remarkable features.
MATERIALS AND METHODS
Strains, culture conditions and nucleic acids extraction
The six diplonemid species used in this study (Supplementary Table S1) were recently isolated from marine water collected in aquaria, lagoons and sandy beaches of Japan (11–13). The species were axenically cultivated in a medium containing 3.6% sea salts (Sigma-Aldrich, S9883), supplemented with 1% (v/v) heat-inactivated horse serum (Sigma-Aldrich, H0146) and 0.025 g/l LB broth powder (Sigma, L3522). The medium was filter-sterilized using a 0.22-μm filter.
Total DNA from exponentially growing cultures was isolated using MasterPure Complete DNA and RNA Purification Kit (Lucigen, MC85200) specially designed for the isolation of DNA from marine organisms. RNA was extracted from whole cells using TriReagent (MRC, TR118) to prevent the loss of small RNAs corresponding to processing and trans-splicing intermediates. Residual DNA was removed by DNase treatment followed by extraction with a homemade Trizol substitute (30).
Reverse transcription, RT-PCR, 5′ and 3′ RACE, and poly-A tail site mapping
Reverse transcription was performed with First Strand cDNA Synthesis Kit for subsequent PCR (Roche) or with SuperScript IV Reverse Transcriptase (Thermo). Complementary DNA was amplified with Q5 High-Fidelity DNA Polymerase (New England Biolabs). PCR products were purified using the QIAquick Gel Extraction kit (Qiagen), Wizard SV Gel and PCR Clean-Up system (Promega) or Monarch DNA Gel Extraction Kit (New England BioLabs). Mapping of 3′ polyadenylation sites was performed using an oligo-dT primer and a gene-specific primer. To determine the 5′ and 3′ ends of modules (5′ and 3′ RACE), the RNA adapter-oligonucleotide dp124 and the 5′ RACE Adapter (from FirstChoice RLM-RACE Kit, Invitrogen) was ligated to the RNA using T4 RNA ligase I (New England Biolabs) and EcRtcB RNA ligase (New England Biolabs), respectively. Detailed protocols are available at https://www.protocols.io/researchers/matus-valach. RT-PCR was performed using specific primers, and amplicons were sequenced at the IRIC Genomics Core Facility (Montreal, Canada) or at Eurofins Genomics (Ebersberg, Germany). Primer and adaptor sequences are listed in Supplementary Table S2.
Genome and transcriptome sequencing and assembly
Both library preparation and sequencing of genomes and transcriptomes were outsourced to the Genome Quebec Innovation Centre (Montreal, Canada). Single DNA-Seq and RNA-Seq library per species were produced due to limited material availability. Illumina genomic paired-end libraries were constructed from total DNA and sequenced in a single Illumina MiSeq lane. DNA reads were assembled using SPAdes v3.11.1 (31) and alternatively, the Tadpole assembler (part of the BBTools suite; https://jgi.doe.gov/data-and-tools/bbtools/).
To avoid the huge variety of trans-splicing and RNA editing intermediates present in diplonemid mitochondria (16,19), which complicate analysis and interpretation, we opted for the enrichment of mature mitochondrial transcripts, i.e. the polyadenylated (poly-A) RNA fraction, which was isolated from total RNA to construct strand-specific RNA-Seq libraries with an average insert size of ∼200 nt. Libraries were sequenced on an Illumina HiSeq platform. For de novo assembly, Trinity v2.2.0 software was used with default parameters (32). Read counts and lengths for both DNA and RNA sequencing are listed in Supplementary Table S1. The raw sequencing data are available at NCBI (https://www.ncbi.nlm.nih.gov/) as BioProject PRJNA525750.
Two of the examined species (D. japonicum and N. karyoxenos) contain endosymbiotic bacteria (12,13). For the purpose of this study focusing on mitochondrial sequences, it was not necessary to estimate relative abundance of bacterial sequences in the datasets. However, we did perform RNA-Seq read mapping to DNA contigs and found only negligible differences between mapping rates to sequences of endosymbiont-bearing and -lacking species, which suggested that possessing an endosymbiont did not introduce any significant bias to our strategy.
Identification of transcripts and annotation of genomic modules
Candidate contigs originating from the mitochondrial transcriptome were identified by BLASTx searches (33) using protein sequences of previously identified mature mitochondrial mRNAs from D. papillatum, D. ambulator, H. phaeocysticola, F. neradi and R. euleeides as queries, and the transcriptome assemblies of the new diplonemids as queried databases. Genomic modules for each species were annotated based on BLASTn searches using predicted transcript sequences as queries. To validate module assignments, modules were aligned with the respective transcript using the built-in aligner of the Geneious 10.1.3 software (34) and visually inspected. To infer protein-coding ORFs, the nucleotide sequences were conceptually translated using NCBI’s genetic code Table 4 (TGA = Trp). Identified modules are cataloged in Supplementary Table S3.
Completion of mitochondrial transcripts
RNA-Seq reads were mapped to reference sequences with Bowtie2 (35). Especially in the case of nad genes, terminal modules were missing. They were recovered by mapping RNA-Seq reads onto partial transcript contigs, and subsequently by extending the sequences via RT-PCR up to the polyA tail using oligo-dT and gene-specific primers (see above). To screen the contigs across the investigated species for the highly divergent nad genes (the previously designated y genes (36), we employed HMMER 3.1b2, a most sensitive method based on profile hidden Markov models (37).
Chromosome classification
The module-containing contigs were first extended by read mapping using the mapper software implemented in the Geneious 10.1.3 package (34) and then compared with each other by BLASTn. Cassettes were identified by the same criteria as described previously (27). Briefly, cassettes are unique sequences surrounding modules, and are flanked by the constant regions of a chromosome, which we define as sequences with >90% identity over >100 bp adjacent to cassettes (Figure 1B). Contigs bearing the same cassette-flanking regions were assigned to the same chromosome class. For multiple sequence alignments, we used MAFFT v7.388 (38). Chromosome classes were ordered from the highest to the lowest member count and named A, B etc. To calculate mean read coverage of cassettes or modules, DNA-Seq reads were first mapped onto mitochondrial contigs by the Geneious 10.1.3 software. Aligning reads were merged with BBMerge (rem k = 62 extend2 = 50 ecct) and deduplicated with Dedupe (k = 31 ac = f) from BBTools. The resulting reads were mapped with Bowtie2 onto the reference sequence, and mean coverage was calculated using the Pileup tool from BBTools. For N. karyoxenos mitochondrial chromosome sequences, which are highly polymorphic, DNA-Seq reads were mapped with the Geneious 10.1.3 software without subsequent Bowtie2 mapping. The final list of identified chromosomes has been compiled in Supplementary Table S4.
In silico identification of RNA editing and DNA polymorphic sites
RNA editing clusters and longer insertions were identified by comparing the genomic contigs and mature transcript sequences by BLASTn. To distinguish between RNA editing sites and genomic polymorphisms, RNA-Seq and DNA-Seq reads were mapped onto mitochondrial transcripts and genomic contigs, respectively, with the built-in aligner of the Geneious 10.1.3 software. DNA polymorphic sites were identified as those exhibiting two (or sometimes more) nucleotides in the mapped reads, while in the case of RNA editing sites, the consensus nucleotide in genomic reads differed from that in RNA-Seq reads. A genomic position with >10% reads displaying difference from the reference was considered a polymorphic site. For RNA editing, a position was annotated as an editing site if at least 50% reads carried a base change. Note that a vast majority of sites was edited to >90%. RNA editing and DNA polymorphic sites of each transcript are detailed in Supplementary Table S5.
Phylogenomic analysis
We used all 15 assigned mitochondrially encoded protein sequences (Atp6, Cob, Cox1/2/3 and Nad1/2/3/4/4L/5/6/7/8/9) from 11 diplonemids and the corresponding homologs from other discobans, namely Trypanosoma brucei, Bodo saltans and Perkinsela sp. (Kinetoplastida); Euglena gracilis (Euglenida), Acrasis kona, Naegleria gruberi and Stachyamoeba lipophora (Heterolobosea); Tsukubamonas globosa (Tsukubamonadida); and Andalucia godoyi, Reclinomonas americana (ATCC 50394) and Ophirina amphinema (Jakobida). Sequences were downloaded from the NCBI GenBank Protein database except those of B. saltans. We retrieved the latter through tblastn searches (using T. brucei mitochondrial proteins as queries) from transcripts that we assembled by Trinity v2.2.0 using RNA-Seq data deposited in the NBCI Bioproject PRJEB3146. Multiple sequence alignments (MSAs) of proteins were generated with MAFFT v7.38 (38) using the E-INS-i algorithm and default parameters. Protein alignments were stripped of hyper-variable sites (20% gap threshold) with trimAl v1.4.22 (39). Subsequently, protein sequences were concatenated for each species, with the final MSA containing 22 taxa and 5,063 positions. Phylogenetic inferences were performed by a Bayesian approach using posterior probabilities as support values (PhyloBayes v4.1 (40), MrBayes v3.2.6 (41)) and by maximum likelihood with bootstrapping (IQ-TREE v1.6.10 (42) and RAxML v8.2.11 (43)). Bayesian methods were executed in two independent chains and the first 25% cycles were discarded as burn-in. For Phylobayes, we chose the substitution model CAT-GTR and site rate variation modeled as a Dirichlet process (ratecat option); the chains were stopped after they converged (i.e. maxdiff below 0.1 at ∼750 cycles corresponding to ∼25,000 generations). For MrBayes, we chose the GTR model with six discrete categories of gamma rate variation and 200,000 MCMC generations. For ML computations, we chose the substitution matrix LG for amino acid frequencies, which was determined as the best model by Model Finder (44). For IQ-TREE, we used default parameters with the option to calculate 1,000 ultrafast bootstrap replicates. For RAxML, additional parameters were: 50 categories for rate heterogeneity (CAT option), the algorithm ‘rapid bootstrap analysis’ and 100 distinct alternative runs on distinct starting trees for bootstrap support values. To evaluate the reliability of the inferred tree, we further analyzed the gene- and site-concordance factors (gCF and sCF, respectively) for each branch, as implemented in IQ-TREE v1.7 (45), with default parameters and the option to merge models across loci.
RESULTS
Gene repertoire
We examined mitochondrion-encoded genes from four Diplonemidae species (Diplonema japonicum strain YPF1604, Rhynchopus humris YPF1608, Lacrimia lanifica YPF1601 and Sulcionema specki YPF1618) and two Hemistasiidae species (Artemidia motanka YPF1610 and Namystynia karyoxenos YPF1621). In all six species, we identified the same set of genes described earlier in four Diplonemidae species (27), namely genes encoding ATP synthase subunit 6 (atp6), cytochrome b (cob), three cytochrome c oxidase subunits (cox1, cox2 and cox3), 10 NADH dehydrogenase subunits (nad1, nad2 [previously y3], nad3 [y1], nad4, nad4L [y6], nad5, nad6 [y5], nad7, nad8 and nad9 [y2] (36), as well as small and large subunit mitoribosomal RNAs (rns and rnl) (Figure 2; Supplementary Tables S3 and S4). In the two hemistasiids, we failed to detect nad6 [y5], but this was presumably due to the gene’s high divergence and not to its genuine absence. Moreover, Lacrimia, Sulcionema and Namystynia also encoded y4, a gene first discovered in D. papillatum; in R. humris, we found a candidate corresponding to module 2 from D. papillatum (y4-m2), but not m1. With homologs of the latter gene at hand, we revisited data from a previous study (27), which enabled us to detect the two y4 modules in R. euleeides, unrecognized previously because of extensive overlaps with cox3-m1 and nad5-m11 in this species. In Lacrimia, we found an additional gene, named y7 (single module), which potentially codes for a protein of 67 amino acid residues. No tRNAs were found; as in other euglenozoan species, they are apparently not mitochondrially encoded, but reside on nuclear DNA and are imported to the mitochondria.
Figure 2.

Mitochondrial gene fragmentation and RNA editing sites. Modular structure of mitochondrial genes and modules undergoing RNA editing and trans-splicing in the six species studied here. For symbols, see inset.
The inferred mitochondrion-encoded proteins of all species analyzed here and those studied previously (27) displayed an exceptionally low level of sequence conservation, which made detection of most genes challenging. Cox1 was the most conserved protein across the diplonemids (32.7% identity across 11 species), while Nad3 with a mere 2.5% sequence identity was on the other end of the spectrum (Supplementary Figure S1).
Module numbers and sizes
The four Diplonemidae species investigated here build their 17–19 identified mitochondrial genes from essentially the same number of modules as does the type species D. papillatum. The sole exception is Sulcionemarns, which is encoded by two modules instead of one in all other ‘classical’ diplonemids. In the two Hemistasiidae species, the total number of modules is doubled (Table 1 and Figure 2), while module sizes are halved (Figure 3). In fact, a given module observed in Diplonemidae is typically split into two to four modules in Hemistasiidae, since gene breakpoints are typically conserved across diplonemids (for sequences that can be confidently aligned, occasional shifts are less than 6 bp).
Table 1.
Mitochondrial chromosomes in studied diplonemids
| Chromosomes classes | Unclassified chromosomes | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Group | Species | No. of classes (types) | No. of mono-module chromosomes (types) | No. of multi-module chromosomes (types) | No. of empty chromosomes (types) | No. of mono-module chromosomes | No. of multi-module chromosomes | No. of chromosomes with one module | No. of chromosomes with multiple modules (No. of modules) | Total chromosome count (No. of modules) |
| Diplonemidae | D. japonicum | 5 (A – E) | 4 (C, D) | 20 (A, B, E) | – | 3 | 3 | 7 | 23 (73a) | 30 (80a) |
| R. humris | 5 (A – E) | 59 (A – E) | 5 (A, B, D) | 2 (A) | 6 | 1 | 65 | 6 (16) | 73 (81) | |
| L. lanifica | 8 (A – H) | 71 (A – H) | 4 (A, B, D) | 7 (B, C, E) | 4 | – | 75 | 4 (8) | 86 (83) | |
| S. specki | 4 (A – D) | 38 (A) | 9 (A – D) | 16 (A) | 1 | 1 | 39 | 10 (44b) | 65 (83b) | |
| Hemistasiidae | A. motanka | 17 (A – Q) | 107 (A – K, M – Q) | 15 (A, B, E – G, J, L, N, P) | n.d. | 22 | 2 | 128 | 18 (37c) | 146 (165c) |
| N. karyoxenos | 1d (X) | 137 (X) | 5 (X) | n.d. | 20 | 1 | 157 | 6 (13) | 163 (170) | |
n.d., not determined.
a atp6-m1 on two different chromosomes (counted once).
b nad2-m2 on two different chromosomes (counted once).
c nad3-m5 on three, and nad4-m3 and nad4-m5 on two different chromosomes (counted once).
dChromosomes assigned to a class based on different criteria than in other species.
Figure 3.

Average gene module size in diplonemids. The average module sizes of mitochondrial genes from all diplonemids for which data are available. Modules in Hemistasiidae species (right) are about half the size compared to those from the Diplonemidae clade (left). Four diplonemid species and H. phaeocysticola (light hues) were studied previously. Asterisk for H. phaeocysticola, the structure of only four genes is known (cob, cox1, cox2 and nad7).
About 4% of modules in hemistasiids are shorter than 20 bp—even as short as 3 bp—and referred to in the following as mini-modules (Figure 4A and Supplementary Figure S2). It should be noted that mini-modules cannot be unambiguously distinguished from appendage RNA editing (see also below) by inspection of DNA–RNA sequence differences alone. Still, two lines of evidence support mini-modules. First, we confirmed by 3′ RACE and subsequent sequencing of Artemidia cox3 the existence of an mRNA trans-splicing intermediate containing the putative cox3-m9 mini-module. The 3′ RACE RT-PCR sampled two amplicon populations: a minor one with 5 Us appended to the 3′ terminus of the upstream module 8 and a major one with the triplet CAG, corresponding to the mini-module 9, joined to the aforementioned U-tract (Figure 4B). The triplet thus appeared to have been added as a whole, i.e. trans-spliced, rather than as a succession of unrecognized editing events. However, to completely rule out the latter alternative, a more extensive sampling of 3′-end RNA processing and editing intermediates by RNA-Seq would be necessary. Second, in the available RNA-Seq data, we detected RNA processing and trans-splicing intermediates, which contained in addition to the diminutive module its flanking sequence, thus indicating where in the genome it resided. This way, we could trace back the genomic source of four and eight such gene pieces in RNA-Seq reads of Artemidia and Namystynia, respectively. (Note, however, that for six additional short segments of 2–6 nt in five transcripts of Namystynia this was not possible and the corresponding regions have been marked as unresolved [Figure 2F].) For the putative cox3-m9 mini-module, we could detect two RNA-Seq reads containing the CAG triplet joined upstream to the cognate cox3-m8 (with the appended U-tract) and downstream to cox1-m21 (Figure 4C). This indicated that cox3-m9 and cox1-m21 of Artemidia were actually juxtaposed on the same chromosome U01. The observation of the intermediates containing mini-modules with their flanking sequences led us to hypothesize a possible assembly scenario for mini-modules, where a larger precursor acts as a mini-module carrier (Figure 4D).
Figure 4.

Mini-modules and new RNA editing types. (A) Example of the putative 3 bp-long mini-module cox3-m9 from Artemidia. The upper map shows the 3′ RACE approach and the location of the mini-module in the transcript. (B) Sequence chromatogram of a 3′ RACE amplicon including cox3-m9. Note the mixed chromatogram peaks downstream of the T-tract indicating a mixture of RT-PCR products with or without the CAG triplet, causing a 3-nt phase shift. (C) The cox3-m9 mini-module is encoded adjacent to cox1-m21 in the chromosome U01. The mini-module-encoding locus was inferred from the shown RNA-Seq reads that cover cox3-m8 with its appended U-tract, followed by the CAG triplet flanked by the cox1-m21 sequence. The non-coding region of the chromosome is set in italics. (D) Hypothetical scenario of the RNA processing pathway of the adjacent cox3-m9 and cox1-m21 modules and trans-splicing to their cognate partners. In this model, the larger precursor acts as a mini-module ‘carrier’. The yellow box indicates the RNA intermediate identified in panel (C). The intermediates in black frames illustrate the expected, cognate modules up- and downstream of cox3-m9 and cox1-m21.
Classes of mitochondrial chromosomes
Knowing the module sequences from transcriptome data allowed us to identify the corresponding regions in genomic contigs, while the repetitive sequences adjacent to the unique module-flanking regions allowed us to delimit cassettes (see Figure 1). Further, cassette-flanking repetitive sequences were presumed to be part of the constant regions of chromosomes, according to the classification scheme of mitochondrial chromosomes in D. papillatum (16), and thus allowed categorization of chromosomes into multiple classes (Table 1; see ‘Materials and Methods’ section for details). In this way, 4 classes were established in Sulcionema, 5 in D. japonicum and R. humris, 8 in Lacrimia and 17 in Artemidia.
For Namystynia chromosomes, in contrast to the other species, we could not employ the criterium of recurring cassette-flanking sequences (representing the constant regions), because sequences around modules frequently consisted of unequally spaced tandem and dispersed repeated homooligomeric motifs that could not be unambiguously aligned. However, numerous chromosomes shared motif 1 (5′-GGGCCAAAAA-3′) upstream and motif 2 (5′-TTTTGGGCC-3′) downstream of the cassettes. Consequently, all chromosomes bearing these motifs were classified as class X, which is much more diverse than the classes from the other diplonemids. Finally, in every species, a handful of chromosomes did not fit into a defined class and therefore were grouped into the category ‘unclassified’. Total counts of classified and unclassified chromosomes for each species are summarized in Table 1 and Supplementary Table S4.
Since the sequence repeats prevented the assembly of whole chromosomes from the available short reads (except for two cases in Sulcionema; see below), chromosome sizes remain unknown. Nevertheless, the assembled genomic contigs indicate that the overall chromosome architecture conformed to that previously observed in other diplonemids (24,27,28), namely cassette sizes varied from ∼0.2 to ∼2 kbp with median length ∼330 (±50) bp. Two types of deviations were observed. First, the median size of D. japonicum cassettes was ∼1 kbp; as we detail below, this was due to the unusually high number of modules per cassette. Second, Sulcionema and Artemidia chromosomes contained cassettes with sizes well above the 2 kbp mark (from 3.2 to 12.3 kbp). Based on the complete assembly of three Sulcionema class D chromosomes (Supplementary Table S4), we calculated that these long cassettes covered 83–90% of the circles, the complete opposite of the situation in other analyzed diplonemid chromosomes, where a cassette represents only 5–10% of the chromosome length (24,27,28).
Module content and arrangement
Diplonema papillatum has 81 distinct mitochondrial chromosomes, 76 of which carry a single cassette that in turn contains a single module (mono-module/mono-cassette organization). The remaining chromosomes contain one cassette each that encloses two modules (three instances; multi-module/mono-cassette organization) or cassettes without any identified module (two instances). In the other diplonemids examined previously and here, additional arrangements coexist, notably three or more (up to 11) modules per cassette and also two cassettes per chromosome (multi-module/multi-cassette organization) (27). In these latter instances, modules are either separated, overlapping or nested.
The six species analyzed here differed considerably in their total number of distinct chromosomes, ranging from 30 in D. japonicum to 163 in Namystynia (Table 1 and Supplementary Table S4). These differences were not only due to a different number of modules in a given species, but also to the fact that some chromosomes encoded multiple modules. For example, D. japonicum contained 23 multi-module chromosomes, the highest number in this category among all species analyzed (27), but only 7 mono-module chromosomes (Table 1). In contrast, among the 86 chromosomes of Lacrimia, only four contained multiple modules. The highest number of modules detected in a single chromosome was 11 in Sulcionema; this species also contained by far the largest number of apparently module-less cassettes (all 16 from its A class chromosomes; Supplementary Table S4).
About 60% of multi-module chromosomes of the diplonemids examined here contained partially overlapping or nested modules (Supplementary Table S6), an arrangement also noted before in other classical diplonemids (27). Overlaps were only conserved among closely related species (nad5-m10 + nad5-m3, nad6-m2 + nad4L-m2 and nad9-m2 + nad6-m3 in D. ambulator and D. japonicum; nad9-m3 + nad9-m4 in R. euleeides and R. humris), and most overlapping modules were encoded on the same strand. No embedded modules were detected in Sulcionema (similarly to D. papillatum), whereas in Namystynia, all overlapping modules were completely nested (Figure 5 and Supplementary Table S6).
Figure 5.
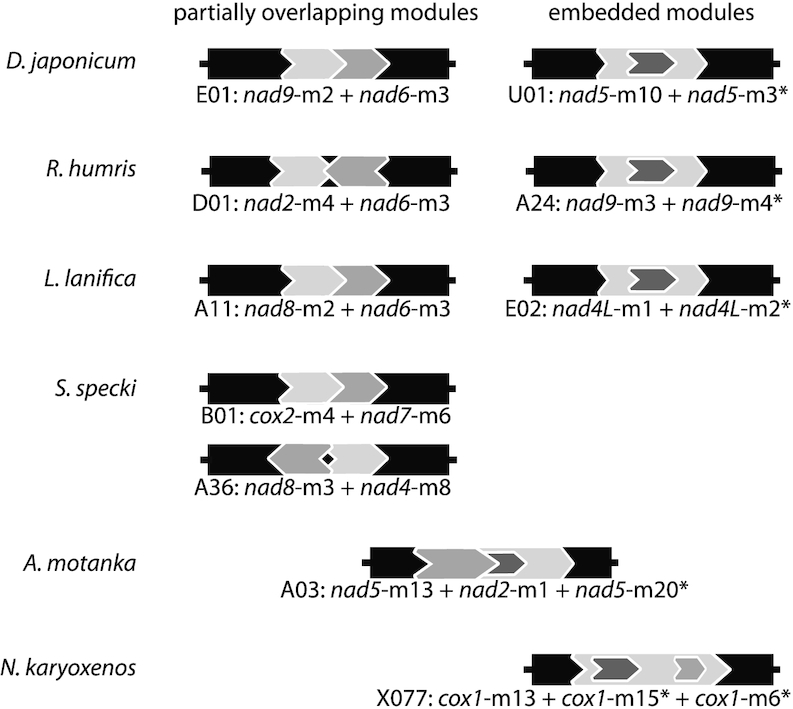
Overlapping gene module arrangements. Scheme of representative cassettes with overlapping modules detected in the six diplonemids studied here. Note that S. specki does not contain embedded modules and that N. karyoxenos lacks partially overlapping modules. Constant regions of chromosomes (indicated by chromosome IDs E01, U01, etc.; see also Supplementary Table S4) are depicted as black rectangles. Modules are represented by dark- and light-gray filled arrows. The arrow tip indicates the direction of module transcription. >>, modules encoded on the same strand; <>, 5′-ends of modules encoded on opposite strands overlap; ><, 3′-ends of modules encoded on opposite strands overlap.
Conserved types of mitochondrial RNA editing and their distribution
In all species studied here, we identified mitochondrial transcripts that underwent multiple events of C-to-U and A-to-I substitution editing and U-appendage editing (Figures 2 and 6; Supplementary Table S5). These types of editing had also been described previously in four Diplonemidae species and H. phaeocysticola (16,27). In the type species, the presence of inosines in transcripts has been demonstrated experimentally indicating that post-transcriptional A-to-G DNA-RNA differences arose by deamination, a process that most certainly applies to C-to-U changes as well (16). Substitution editing sites occurred in clusters in similar gene regions across species, although individual sites did not necessarily coincide. Earlier reported substitution editing clusters (e.g. nad2-m4, nad3-m2, nad4-m1, nad6-m1, nad7-m3 and m5, nad9-m1 and rns) were present in the species studied here as well, although with some exceptions, such as the complete absence of substitution editing sites in nad3, nad7 and nad9 of Lacrimia (Figure 2). We also identified new sites of C-to-U and A-to-I substitutions and U appendage. These included one and two new C-to-U editing sites in Lacrimia cob-m5 and cox3-m1, respectively. Further, Sulcionema possessed a novel A-to-I substitution site in cox3-m3, and in R. humris, we discovered two new editing sites at the junction of m1 and m2 of cox3 (Supplementary Table S5).
Figure 6.

Phylogenetic distribution of mitochondrial RNA editing in diplonemids. Names of species analyzed in this study are set in bold. (A) RNA editing types for each gene are compared. Phylogenetic relationships shown on the left are inferred from nuclear 18S rRNA and taken from (5). The tree on the right is based on concatenated mitochondrial proteins (this study); dashed lines indicate the uncertainty in positioning the Eupelagonemidae and DSPD II clades. (B) Cumulative counts of RNA editing events per type for each species.
All the above editing types were much more frequent in the hemistasiids and affected every single transcript (Figures 2 and 6; Supplementary Table S5). In addition to the editing clusters documented previously (16,27), we identified several novel instances, mostly located at the ends of modules (which complicated the recognition of the corresponding modules). Although more numerous, substitution editing sites in Artemidia (493 A-to-I and 620 C-to-U sites in >100 editing clusters) were amassed in half the number of clusters compared to Namystynia (458 A-to-I and 588 C-to-U sites in >210 editing clusters; Supplementary Table S5). Interestingly, certain editing sites in Namystynia coincided with one of the >300 genomic polymorphisms (mostly single-nucleotide polymorphisms, SNPs) dispersed across modules (Supplementary Table S5 and Supplementary Figure 3A).
Novel types of RNA editing
By inspecting DNA–RNA differences, we detected two new types of editing not documented before in diplonemids. First, the two hemistasiids carried G-to-A substitutions (Figure 7A; Supplementary Figure S3A,B), notably 14 unambiguous sites in six different transcripts of Namystynia and one such site in nad5 of Artemidia (Figures 2 and 6; Supplementary Table S5); a second site may exist in Artemidia nad1; however, the corresponding A in RNA could have also originated by A-appendage (see below).
Figure 7.

Hyper-edited region with novel types of RNA editing. (A) The junction of the nad1 modules 7 and 8 from N. karyoxenos combines frequent RNA editing and the newly observed G-to-A substitution and A+U appendage events. Note also numerous genome-encoded single-nucleotide polymorphisms. (B) Alignment of a minor population of RNA-Seq reads to the 3′ end of nad5-m12. The majority of >300 RNA-Seq mate 1 reads extending to or spanning the nad5-m12/m13 junction (not shown) map exactly to the sequence of the transcript containing nad5-m12, the RNA editing-appended tract and nad5-m13. A minor population shown here represents nad5-m12 modules (4.6%) containing a 3′ terminal A-tract, which we interpreted as RNA editing intermediates. The apparent G-appendage in N. karyoxenos might thus originate from an A-appendage followed by A-to-I deamination.
The second new type of editing was detected in Sulcionema and Namystynia. The nad4 transcript of the former contained between m4 and m5 not only a non-encoded U but also an additional A (Figure 2), which we confirmed by 3′ RACE (Supplementary Figure S3C). Such A-appendage editing appeared far more frequent in Namystynia, in which we spotted one to four such sites in eight different transcripts, summing up to a total of 17 editing positions and 41 post-transcriptionally added As (Figure 2 and Supplementary Table S5). The A-appendage site of Sulcionema nad4 also occurred in the same transcript of Namystynia (m13/m14 junction). Curiously, at all sites except the one in nad4L, A-appendage in Namystynia coincided with U-appendage, occasionally in an interspersed fashion, as for example between nad4-m1 and m2 (5′-UUAAUUUUUUUUU-3′).
In Namystynia, we also observed six cases of apparent G-additions between modules, and again intermixed with other post-transcriptionally added nucleotides, for example 5′-GAUUU-3′ between nad5-m12 and m13 (Supplementary Table S5). The Gs could have arisen in two ways, by (i) genuine G-appendage editing or (ii) A-appendage followed by A-to-I deamination. While editing by rare G-insertions had been observed in an amoebozoan and a heterolobosean (46,47), we considered the second alternative more likely because it would not imply an additional machinery required to specifically add G residues. Further, RNA-Seq read mapping to the junction of nad5-m12 and nad5-m13, where such a G-addition was noted, revealed a minor population of RNA-Seq reads that displayed 3′ terminal A-tracts, which we interpret as not-yet edited intermediates (Figure 7B). It will be interesting to validate this hypothesis by biochemical assays once Namystynia becomes more amenable to experimental work.
Phylogenetic relationships among diplonemids
In molecular phylogenies based on 18S rRNA (11,12,14) (Figure 6), the genera Diplonema, Rhynchopus, Lacrimia, Flectonema and Sulcionema formed Diplonemidae, which are also referred to as ‘classical’ diplonemids, to the exclusion of Hemistasiidae, Eupelagonemidae (formerly DSPD I (48)), and the lineage currently described by its acronym ‘DSPD II’. To build a more robust phylogeny, we used here the concatenated protein sequences inferred from 15 different mitochondrial transcripts, identified in this and previous studies. The resulting tree (Figure 6, right tree; Supplementary Figure S4A) resolved the relationships between diplonemids with high confidence but differs in topology from the nuclear 18S rRNA-based trees including the same species (Figure 6, left tree). First, in the mitochondrial phylogeny, Lacrimia grouped together with Diplonema, while it is placed at the base of the Diplonema + Rhynchopus clade in the 18S rRNA trees. Second, Namystynia, and not Artemidia, was the sister taxon of Hemistasia. Finally, the most significant deviation was the position of Sulcionema, as it branched together with Diplonemidae in the nuclear trees, but with Hemistasiidae in the mitochondrial tree.
To examine possible reasons for this incongruence, we calculated gene- and site-concordance factors for each branch in the tree (45) (Supplementary Figure S4B). Compared to the concatenated dataset, single-locus phylogenies showed comparably low support for the positions of Lacrimia and D. papillatum; however, the majority of informative sites in the concatenated dataset supported the tree topology. The conflicting positions within Hemistasiidae were mainly due to the limited data currently available for H. phaeocysticola (i.e. four proteins instead of 15). More importantly, most single-protein phylogenies placed Sulcionema prior to the divergence of hemistasiids, i.e. the topology shown here (Supplementary Figure S4A), but the site-wise support for this topology was almost identical with that of the two other mutually exclusive topologies, in which Sulcionema formed a sister group to either all diplonemids or the Diplonemidae clade. Further taxon sampling of basal diplonemids should allow to resolve the described inconsistencies.
DISCUSSION
In the past, most studies were performed on the type species D. papillatum which, incidentally, has recently become genetically tractable (49). Together with three other classical diplonemids that had formerly been examined at the molecular level (11–14), these species represent a tiny and ecologically restricted fraction of the highly diverse group (4,14). Here, we have considerably expanded the knowledge about the diplonemid mitochondrial genomes by examining six recently isolated species from both the Diplonemidae and Hemistasiidae clades, thus covering a substantial part of diplonemid diversity (11–13).
Diplonemids are record holders in mitochondrial genome content and organization
It is worth noting that these protists, which were neglected until recently, carry the largest amount of mtDNA documented in an organelle, which in D. papillatum even exceeds that of nuclear DNA (18). In comparison to human mtDNA, which typically constitutes only about 1% of total cellular DNA and consists of a single, circular-mapping 16.5 kbp molecule encoding complete protein-coding genes (13 in human versus 16 in diplonemids), mtDNA in diplonemids is unprecedented in its magnitude, while its gene expression mode adds a supplementary layer of complexity. Most of the diplonemid mtDNA is non-coding, with the extensive constant regions apparently carrying only the origin of replication and transcription initiation signals. The baroque organization of the mitochondrial genome and transcriptome in diplonemids is partially met by the well-studied case of sister trypanosomatids, which tells us that sustaining this extravagancy must require an enormously complex cellular machinery. Moreover, observing such unusual features in the free-living diplonemids challenges the common view that extreme oddities are synonymous with a parasitic lifestyle.
The enigmatic y4 gene
All diplonemids analyzed in this and previous studies (27) encode the same set of mitochondrial genes (Figure 6). The only gene with patchy distribution, encountered in half of the species, is y4 encoding a hypothetical protein, which is poorly conserved at the sequence level and for which no homolog was found outside diplonemids. The Y4 protein of D. papillatum was recently detected by mass spectrometry in a respirasome supercomplex and, therefore, might represent a novel diplonemid-specific subunit of one of the respiratory chain complexes (36). Alternatively, Y4 might specify a highly derived mitochondrion-encoded mitoribosomal protein. For example, the kinetoplastid RPS12 (encoding the mitoribosomal uS12m) and MURF5 are renowned for their extreme sequence divergence, and the latter has been uncovered as RPS3 (uS3m) only by structure determination of the Trypanosoma brucei mitoribosome (50). No homologs encoding uS12m and uS3m were detected in the D. papillatum nuclear genome (our unpublished observations), which suggests that Y4 may be an extremely divergent mitoribosomal protein. Structure determination of the Diplonema respirasome and mitoribosome will be the ultimate test of these hypotheses.
Mitochondrial gene fragmentation at new heights in hemistasiids
An earlier study of four genes indicated high fragmentation in Hemistasia mtDNA (29). Our more systematic investigation of complete mitochondrial transcriptomes from two other hemistasiids generalized this finding, uncovering putative mini-modules as short as 3 bp. The vast majority of mini-modules is embedded in another module, which indicates that increasing gene fragmentation facilitates double use of coding sequence for distinct genes. Importantly, reuse can be even multiple: in Namystynia, cox1-m15 and cox1-m6 are both embedded in cox1-m13, while in Artemidia, nad2-m1 and nad5-m20 extensively overlap with nad5-m13, with a 9-bp region contributing to all three gene pieces (Figure 5 and Supplementary Table S6).
Ultra-short (1–30 nt) coding sequences are found in nuclear and mitochondrial genomes of numerous organisms (51–53). These micro-exons are joined to their neighbors—by the spliceosome or the Group I or Group II splicing machineries—via cis-splicing, thus relying on a physical connection between exons for proper joining. The hemistasiid case suggests that mini-module joining might proceed through an intermediate where a larger precursor acts as a ‘carrier’ of the mini-module (Figure 4C and D), ensuring the correct trans-splicing of a sequence that alone is presumably too short to ensure specificity. The actual mechanism of module transcript match-making still remains an intriguing puzzle.
RNA editing at an unprecedented level
Among diplonemids, the largest number of mitochondrial editing sites was counted in Namystynia, notably over 1,000 A-to-I and C-to-U substitutions, 14 G-to-A changes, and 94 U+A-tracts that sum up to >600 nt added to modules (Supplementary Table S5). As in previously studied diplonemids (16), RNA editing had an overall restorative effect on coding sequences, allowing production of functional proteins from a priori defective gene pieces.
The myxomycete Physarum polycephalum, several dinoflagellates, and the lycophytes Isoetes engelmannii and Selaginella uncinata, are renowned for extensive organellar editing with 1,333 sites in P. polycephalum mitochondria, 1,782 in I. engelmannii mitochondria and 3,415 in S. uncinata plastids (47,54–56). When comparing the number of edits per number of residues in a given transcriptome, the editing is most pervasive in Isoetes (6.7%), several dinoflagellates (5.4–6.5%), followed by Selaginella (4.3%) and Physarum (3.5%). Diplonemidae rank lower (1.9–2.5%; Supplementary Table S7), yet Hemistasiidae surpass all previous records. With an editing density of 12.2%, Namystynia has the most extensively edited transcriptome documented so far (Supplementary Table S7).
Physarum polycephalum is also one of the few species known to employ more than one mode of mitochondrial RNA editing: co-transcriptional nucleotide insertions and occasional deletions (57), and post-transcriptional C-to-U substitutions (47,58), while diplonemids feature substitution (C-to-U, A-to-I and G-to-A) as well as U- and A-appendage editing.
New types of RNA editing
In hemistasiids, we detected two types of editing novel for diplonemid mitochondria, which involve G-to-A substitutions and A-appendage to internal modules. G-to-A editing has been only rarely reported in mitochondria, e.g. in dinoflagellates such as Hematodinium (59), while such events are extremely uncommon in the nucleus (60,61). Attesting to the importance of this type of editing in hemistasiids, the G-to-A substitution site in nad5-m6 of Artemidia is also conserved in Namystynia. The editing event contributes to the replacement of a Ser by an Asp codon that corresponds to the function-critical residue at position 179 in mammalian Nad5, an amino acid involved in the proton relay of complex I (62).
The molecular mechanism of G-to-A editing remains a matter of speculation; while C and U interconversion can proceed by transamination (U-to-C) and deamination (C-to-U)—since the two bases differ only in the absence or presence of an amino group—G and A differ in two groups, and no single chemical reaction is known to interconvert these two bases.
More concrete notions exist about A-appendage editing, which is a crucial step in the maturation of the dinoflagellate cox3 transcript (63,64). The reaction is presumably catalyzed by the poly(A) polymerase that otherwise adds poly-A tails to mitochondrial transcripts. The corresponding enzymes have been characterized in mammals and trypanosomes (65). In the latter, the 3′ tails are actually a mix of A+U residues, generated in two steps. Prior to editing, which in trypanosomes involves U-insertions and U-deletions (66–68), a 20–25 residue-long 3′ A-tail is added. Once editing is completed, this tail is elongated to a 200–300 nt-long A+U heteropolymer, earmarking the transcript for translation and allowing its association with the mitochondrial ribosome (69). Both short and long tails are synthesized by the kinetoplast poly(A) polymerase 1 (70), which forms a complex with two pentatricopeptide proteins called kinetoplast polyadenylation/uridylation factors (KPAFs) 1 and 2 (71). RNA-editing terminal uridylyl transferase 1, which forms a complex with 3′ exonuclease (72), is involved in the formation of long mRNA 3′ tails (73,74), and possibly also in uridylation of rRNAs and gRNAs.
We expect a similar protein complex to operate in diplonemid mitochondria. However, in diplonemids, the addition of As—frequently together with Us—takes place in two fundamentally different contexts: not only at the 3′ end of terminal modules, thus generating mRNA tails, but also of internal modules, as we documented here for Namystynia (Figures 2 and 7A; Supplementary Table S5). It will be interesting to examine in diplonemids whether a single protein complex is responsible for generating both A-tails and A-appendages or whether distinct specialized complexes have evolved for the two purposes.
Mitochondrial genes of Sulcionema—primitively simple or reduced upon divergence?
The mitochondrial system of Sulcionema appears in several aspects less complex than that of the other diplonemids. Furthermore, this species has the shortest branch in both mitochondrial (Figure 6A and Supplementary Figure 4A) and nuclear phylogenies (11). Some of these features might have been reduced upon divergence, while others might be primitive.
Of particular interest is the absence of the post-transcriptionally added Us between modules m4 and m5 of Sulcionema cox1 (Supplementary Figure S5). Besides being the first editing site identified in a diplonemid (23), this U-appendage had apparently an important evolutionary impact on the Cox1 protein structure. In all species except diplonemids, the protein region corresponding to junction m4/m5 (loop 1) is positively charged and, in the folded protein, interacts with a downstream loop 2 composed of small and hydrophobic residues. The inverse situation applies to all diplonemids that feature cox1 U-appendage editing. Here, the U-tract added at the m4/m5 junction and its environs specify a hydrophobic patch, whereas the downstream loop contains a polar Arg residue (28). Interestingly, the Sulcionema Cox1 protein has exactly the same hydropathy pattern in loops 1 and 2 as the other diplonemids, only the Us at the m4/m5 junction are genome-encoded as part of m4. More extensive taxon sampling will be necessary to untangle the order of the two evolutionary events, loop 1/loop 2-polarity switching and U-appendage editing.
Other less complex features in Sulcionema include the lack of nested modules (Supplementary Table S6) and the low RNA editing frequency (Supplementary Table S7) compared to the other diplonemids. Furthermore, the range of copy numbers across its module-bearing chromosomes is only ∼13 (∼30 when including its 16 module-less chromosomes), but 50–150 in the other Diplonemidae, and even ∼600 in the hemistasiid Artemidia (up to ∼300 of its B class chromosomes alone) (Supplementary Table S4) (27). It was noted that unequal copy numbers of chromosomes in multipartite genomes may cause chromosome loss during random mtDNA segregation to daughter cells (75). One solution to this problem is to over-amplify mtDNA, which in D. papillatum represents >50% of total cellular DNA (18,75). It would thus be interesting to see whether the more even chromosome copy number distribution in Sulcionema correlates with a lower mtDNA to nuclear DNA ratio.
Gene fragmentation and complexity in the most diverse diplonemids
Given the very high estimate of diplonemid species in the ocean (5,7), it can be safely predicted that species with even more complex mitochondrial genomes and transcriptomes will eventually be discovered, especially among hemistasiids and eupelagonemids. For example, when reanalyzing the published genome sequences from 10 single-cell eupelagonemids (4), we detected in the data from ‘cell 13’ three potential mitochondrial modules encoding highly conserved regions of cox1 and nad7. One candidate module corresponds exactly to the hemistasiid nad7-m2, the second to the upstream half of the hemistasiid nad7-m3, and the third is a homolog of cox1-m11 from Hemistasia (Supplementary Figure S5). This indicates that the mitochondrial genomes of Eupelagonemidae species are similar to those of the Hemistasiidae clade with respect to module sizes and RNA editing (Figure 2 and Table 1; Supplementary Table S5).
CONCLUSIONS AND OUTLOOK
Since diplonemids are a highly successful group as to their geographic distribution and habitat diversity, their extravagantly complex mitochondrial system has apparently little if any impact on their fitness. We see in it an excellent example of constructive neutral evolution (76–79), which postulates that a stepwise increase in the complexity of a given cellular machinery can occur with no associated selective consequence. Regardless of whether the flexible gene module structure allows sequence reuse for unrelated genes and/or de novo generation of ‘improved’ gene pieces, the variety of splicing and editing events makes the mitochondrial genome of diplonemids a laboratory for new inventions. Indeed, in a diplonemid cell, it takes ‘the whole village’ to decode the handful of mitochondrial genes. Our next challenge is to identify the individual players involved in decoding and to establish how the complex gene expression is coordinated.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Jan Votýpka (Charles University, Prague) and Akinori Yabuki (JAMSTEC, Yokohama) for help with the isolation of strains.
Author contributions: M.V., G.B., J.L.: conceptualization; K.Z., M.V.: methodology; K.Z.: software; K.Z., M.V., B.K.: data curation; K.Z., M.V.: formal analysis; B.K., K.Z., M.V.: investigation; K.Z., M.V., B.K.: visualization; B.K., K.Z., D.F., M.V.: writing—original draft; M.V., G.B., J.L.: writing—review and editing; G.P., G.B.: resources; J.L., G.B., M.V., D.F.: supervision; J.L.: project administration; J.L., G.B., B.K.: funding acquisition.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
ERC CZ grant [LL1601 to J.L.]; Czech Ministry of Education (ERD Funds) [OPVVV16_019/ 0000759 to J.L.]; Gordon and Betty Moore Foundation [GBMF-4983.01 to J.L., G.B.]; Natural Sciences and Engineering Research Council of Canada [RGPIN-2014-05286, RGPIN-2019-04024 to G.B.]; Grant Agency of the University of South Bohemia [094/2018/P to B.K.]. Funding for open access charge: Gordon and Betty Moore Foundation.
Conflict of interest statement. None declared.
REFERENCES
- 1. Burki F., Roger A.J., Brown M.W., Simpson A.G.B.. The new tree of eukaryotes. Trends Ecol. Evol. 2019; 35:43–55. [DOI] [PubMed] [Google Scholar]
- 2. Adl S.M., Bass D., Lane C.E., Lukeš J., Schoch C.L., Smirnov A., Agatha S., Berney C., Brown M.W., Burki F. et al.. Revisions to the classification, nomenclature, and diversity of eukaryotes. J. Eukaryot. Microbiol. 2019; 66:4–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. de Vargas C., Audic S., Henry N., Decelle J., Mahe F., Logares R., Lara E., Berney C., Le Bescot N., Probert I. et al.. Eukaryotic plankton diversity in the sunlit ocean. Science. 2015; 348:1261605–1261605. [DOI] [PubMed] [Google Scholar]
- 4. Gawryluk R.M.R., del Campo J., Okamoto N., Strassert J.F.H., Lukeš J., Richards T.A., Worden A.Z., Santoro A.E., Keeling P.J.. Morphological identification and single-cell genomics of marine diplonemids. Curr. Biol. 2016; 26:3053–3059. [DOI] [PubMed] [Google Scholar]
- 5. Flegontova O., Flegontov P., Malviya S., Audic S., Wincker P., de Vargas C., Bowler C., Lukeš J., Horák A.. Extreme diversity of diplonemid eukaryotes in the ocean. Curr. Biol. 2016; 26:3060–3065. [DOI] [PubMed] [Google Scholar]
- 6. David V., Archibald J.M.. Evolution: Plumbing the depths of diplonemid diversity. Curr. Biol. 2016; 26:R1290–R1292. [DOI] [PubMed] [Google Scholar]
- 7. Flegontova O., Flegontov P., Malviya S., Poulain J., de Vargas C., Bowler C., Lukeš J., Horák A.. Neobodonids are dominant kinetoplastids in the global ocean. Environ. Microbiol. 2018; 20:878–889. [DOI] [PubMed] [Google Scholar]
- 8. Sibbald S.J., Archibald J.M.. More protist genomes needed. Nat. Ecol. Evol. 2017; 1:0145. [DOI] [PubMed] [Google Scholar]
- 9. Keeling P.J., Campo J. del. Marine protists are not just big bacteria. Curr. Biol. 2017; 27:R541–R549. [DOI] [PubMed] [Google Scholar]
- 10. Lukeš J., Flegontova O., Horák A.. Diplonemids. Curr. Biol. 2015; 25:R702–R704. [DOI] [PubMed] [Google Scholar]
- 11. Tashyreva D., Prokopchuk G., Yabuki A., Kaur B., Faktorová D., Votýpka J., Kusaka C., Fujikura K., Shiratori T., Ishida K.-I. et al.. Phylogeny and morphology of new diplonemids from Japan. Protist. 2018; 169:158–179. [DOI] [PubMed] [Google Scholar]
- 12. Tashyreva D., Prokopchuk G., Votýpka J., Yabuki A., Horák A., Lukeš J.. Life cycle, ultrastructure, and phylogeny of new diplonemids and their endosymbioticbacteria. mBio. 2018; 9:e02447-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Prokopchuk G., Tashyreva D., Yabuki A., Horák A., Masařová P., Lukeš J.. Morphological, ultrastructural, motility and evolutionary characterization of two new hemistasiidae species. Protist. 2019; 170:259–282. [DOI] [PubMed] [Google Scholar]
- 14. Okamoto N., Gawryluk R.M.R., Campo J., Strassert J.F.H., Lukeš J., Richards T.A., Worden A.Z., Santoro A.E., Keeling P.J.. A revised taxonomy of diplonemids including the Eupelagonemidae n. fam. and a type species, Eupelagonema oceanica n. gen. & sp. J. Eukaryot. Microbiol. 2019; 66:519–524. [DOI] [PubMed] [Google Scholar]
- 15. Valach M., Moreira S., Faktorová D., Lukeš J., Burger G.. Post-transcriptional mending of gene sequences: Looking under the hood of mitochondrial gene expression in diplonemids. RNA Biol. 2016; 13:1204–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Moreira S., Valach M., Aoulad-Aissa M., Otto C., Burger G.. Novel modes of RNA editing in mitochondria. Nucleic Acids Res. 2016; 44:4907–4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Faktorová D., Valach M., Kaur B., Burger G., Lukeš J.. Cruz-Reyes J, Gray M. Mitochondrial RNA editing and processing in diplonemid protists. RNA Metabolism in Mitochondria. Nucleic Acids and Molecular Biology. 2018; 34:Cham: Springer; 145–176. [Google Scholar]
- 18. Lukeš J., Wheeler R., Jirsová D., David V., Archibald J.M.. Massive mitochondrial DNA content in diplonemid and kinetoplastid protists. IUBMB Life. 2018; 70:1267–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kiethega G.N., Yan Y., Turcotte M., Burger G.. RNA-level unscrambling of fragmented genes in Diplonema mitochondria. RNA Biol. 2013; 10:301–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sturm N.R., Maslov D.A., Grisard E.C., Campbell D.A.. Diplonema spp. possess spliced leader RNA genes similar to the kinetoplastida. J. Eukaryot. Microbiol. 2001; 48:325–331. [DOI] [PubMed] [Google Scholar]
- 21. Lasda E.L., Blumenthal T.. Trans-splicing. Wiley Interdiscip. Rev. RNA. 2011; 2:417–434. [DOI] [PubMed] [Google Scholar]
- 22. Glanz S., Kück U.. Trans-splicing of organelle introns–a detour to continuous RNAs. BioEssays. 2009; 31:921–934. [DOI] [PubMed] [Google Scholar]
- 23. Marande W., Lukeš J., Burger G.. Unique mitochondrial genome structure in diplonemids, the sister group of kinetoplastids. Eukaryot. Cell. 2005; 4:1137–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Marande W., Burger G.. Mitochondrial DNA as a genomic jigsaw puzzle. Science. 2007; 318:415. [DOI] [PubMed] [Google Scholar]
- 25. Vlcek C., Marande W., Teijeiro S., Lukeš J., Burger G.. Systematically fragmented genes in a multipartite mitochondrial genome. Nucleic Acids Res. 2011; 39:979–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Valach M., Moreira S., Kiethega G.N., Burger G.. Trans-splicing and RNA editing of LSU rRNA in Diplonema mitochondria. Nucleic Acids Res. 2014; 42:2660–2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Valach M., Moreira S., Hoffmann S., Stadler P.F., Burger G.. Keeping it complicated: mitochondrial genome plasticity across diplonemids. Sci. Rep. 2017; 7:14166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kiethega G.N., Turcotte M., Burger G.. Evolutionarily conserved cox1 trans-splicing without cis-motifs. Mol. Biol. Evol. 2011; 28:2425–2428. [DOI] [PubMed] [Google Scholar]
- 29. Yabuki A., Tanifuji G., Kusaka C., Takishita K., Fujikura K.. Hyper-eccentric structural genes in the mitochondrial genome of the algal parasite Hemistasia phaeocysticola. Genome Biol. Evol. 2016; 8:2870–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rodríguez-Ezpeleta N., Teijeiro S., Forget L., Burger G., Lang B.F.. Parkinson J. Construction of cDNA libraries: Focus on protists and fungi. Expressed Sequence Tags (ESTs). Methods in Molecular Biology (Methods and Protocols). 2009; 533:Totowa, NJ: Humana Press; 33–47. [DOI] [PubMed] [Google Scholar]
- 31. Bankevich A., Nurk S., Antipov D., Gurevich A.A., Dvorkin M., Kulikov A.S., Lesin V.M., Nikolenko S.I., Pham S., Prjibelski A.D. et al.. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012; 19:455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Haas B.J., Papanicolaou A., Yassour M., Grabherr M., Blood P.D., Bowden J., Couger M.B., Eccles D., Li B., Lieber M. et al.. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013; 8:1494–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Altschul S. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997; 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kearse M., Moir R., Wilson A., Stones-Havas S., Cheung M., Sturrock S., Buxton S., Cooper A., Markowitz S., Duran C. et al.. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012; 28:1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Valach M., Léveillé-Kunst A., Gray M.W., Burger G.. Respiratory chain complex I of unparalleled divergence in diplonemids. J. Biol. Chem. 2018; 293:16043–16056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Eddy S.R. A new generation of homology search tools based on probabilistic inference. Genome Inform. 2009; 23:205–211. [PubMed] [Google Scholar]
- 38. Katoh K., Standley D.M.. MAFFT Multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013; 30:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Capella-Gutiérrez S., Silla-Martínez J.M., Gabaldón T.. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009; 25:1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lartillot N., Lepage T., Blanquart S.. PhyloBayes 3: a Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics. 2009; 25:2286–2288. [DOI] [PubMed] [Google Scholar]
- 41. Ronquist F., Teslenko M., van der Mark P., Ayres D.L., Darling A., Höhna S., Larget B., Liu L., Suchard M.A., Huelsenbeck J.P.. MrBayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012; 61:539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nguyen L.-T., Schmidt H.A., von Haeseler A., Minh B.Q.. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015; 32:268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stamakis A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014; 30:1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kalyaanamoorthy S., Minh B.Q., Wong T.K.F., von Haeseler A., Jermiin L.S.. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods. 2017; 14:587–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Minh B.Q., Hahn M., Lanfear R.. New methods to calculate concordance factors for phylogenomic datasets. 2018; 05 December 2018, preprint: not peer reviewed 10.1101/487801. [DOI] [PMC free article] [PubMed]
- 46. Yang J., Harding T., Kamikawa R., Simpson A.G.B., Roger A.J.. Mitochondrial genome evolution and a novel RNA editing system in deep-branching heteroloboseids. Genome Biol. Evol. 2017; 9:1161–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bundschuh R., Altmüller J., Becker C., Nürnberg P., Gott J.M.. Complete characterization of the edited transcriptome of the mitochondrion of Physarum polycephalum using deep sequencing of RNA. Nucleic Acids Res. 2011; 39:6044–6055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lara E., Moreira D., Vereshchaka A., López-García P.. Pan-oceanic distribution of new highly diverse clades of deep-sea diplonemids. Environ. Microbiol. 2009; 11:47–55. [DOI] [PubMed] [Google Scholar]
- 49. Kaur B., Valach M., Peña-Diaz P., Moreira S., Keeling P.J., Burger G., Lukeš J., Faktorová D.. Transformation of Diplonema papillatum, the type species of the highly diverse and abundant marine microeukaryotes Diplonemida (Euglenozoa). Environ. Microbiol. 2018; 20:1030–1040. [DOI] [PubMed] [Google Scholar]
- 50. Ramrath D.J.F.F., Niemann M., Leibundgut M., Bieri P., Prange C., Horn E.K., Leitner A., Boehringer D., Schneider A., Ban N.. Evolutionary shift toward protein-based architecture in trypanosomal mitochondrial ribosomes. Science. 2018; 362:eaau7735. [DOI] [PubMed] [Google Scholar]
- 51. Weyn-Vanhentenryck S.M., Mele A., Yan Q., Sun S., Farny N., Zhang Z., Xue C., Herre M., Silver P.A., Zhang M.Q. et al.. HITS-CLIP and integrative modeling define the Rbfox splicing-regulatory network linked to brain development and autism. Cell Rep. 2014; 6:1139–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Osigus H.-J., Eitel M., Schierwater B.. Deep RNA sequencing reveals the smallest known mitochondrial micro exon in animals: The placozoan cox1 single base pair exon. PLoS One. 2017; 12:e0177959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ustianenko D., Weyn-Vanhentenryck S.M., Zhang C.. Microexons: discovery, regulation, and function. Wiley Interdiscip. Rev. RNA. 2017; 8:e1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Grewe F., Herres S., Viehöver P., Polsakiewicz M., Weisshaar B., Knoop V.. A unique transcriptome: 1782 positions of RNA editing alter 1406 codon identities in mitochondrial mRNAs of the lycophyte Isoetes engelmannii. Nucleic Acids Res. 2011; 39:2890–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Oldenkott B., Yamaguchi K., Tsuji-Tsukinoki S., Knie N., Knoop V.. Chloroplast RNA editing going extreme: more than 3400 events of C-to-U editing in the chloroplast transcriptome of the lycophyte Selaginella uncinata. RNA. 2014; 20:1499–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Klinger C.M., Paoli L., Newby R.J., Wang M.Y.W., Carroll H.D., Leblond J.D., Howe C.J., Dacks J.B., Bowler C., Cahoon A.B. et al.. Plastid transcript editing across dinoflagellate lineages shows lineage-specific application but conserved trends. Genome Biol. Evol. 2018; 10:1019–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gott J.M. Discovery of new genes and deletion editing in Physarum mitochondria enabled by a novel algorithm for finding edited mRNAs. Nucleic Acids Res. 2005; 33:5063–5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schallenberg-Rüdinger M., Lenz H., Polsakiewicz M., Gott J.M., Knoop V.. A survey of PPR proteins identifies DYW domains like those of land plant RNA editing factors in diverse eukaryotes. RNA Biol. 2013; 10:1549–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jackson C.J., Gornik S.G., Waller R.F.. The mitochondrial genome and transcriptome of the basal dinoflagellate Hematodinium sp.: Character evolution within the highly derived mitochondrial genomes of dinoflagellates. Genome Biol. Evol. 2012; 4:59–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang I.X., Grunseich C., Chung Y.G., Kwak H., Ramrattan G., Zhu Z., Cheung V.G.. RNA–DNA sequence differences in Saccharomyces cerevisiae. Genome Res. 2016; 26:1544–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Daneck P., Nellaker C., McIntyre R.E., Buendia-Buendia J.E., Bumpstead S., Ponting C.P., Flint J., Durbin R., Keane T.M., Adams D.J.. High levels of RNA-editing site conservation amongst 15 laboratory mouse strains. Genome Biol. 2012; 13:R26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Agip A.-N.A., Blaza J.N., Bridges H.R., Viscomi C., Rawson S., Muench S.P., Hirst J.. Cryo-EM structures of complex I from mouse heart mitochondria in two biochemically defined states. Nat. Struct. Mol. Biol. 2018; 25:548–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jackson C.J., Norman J.E., Schnare M.N., Gray M.W., Keeling P.J., Waller R.F.. Broad genomic and transcriptional analysis reveals a highly derived genome in dinoflagellate mitochondria. BMC Biol. 2007; 5:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Burger G., Jackson C.J., Waller R.F.. Bullerwell C. Unusual mitochondrial genomes and genes. Organelle Genetics. 2012; Berlin, Heidelberg: Springer; 41–77. [Google Scholar]
- 65. Chang J.H., Tong L.. Mitochondrial poly(A) polymerase and polyadenylation. Biochim. Biophys. Acta. 2012; 1819:992–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zimmer S.L., Simpson R.M., Read L.K.. High throughput sequencing revolution reveals conserved fundamentals of U-indel editing. Wiley Interdiscip. Rev. RNA. 2018; 9:e1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Cruz-Reyes J., Mooers B.H.M., Doharey P.K., Meehan J., Gulati S.. Dynamic RNA holo-editosomes with subcomplex variants: Insights into the control of trypanosome editing. Wiley Interdiscip. Rev. RNA. 2018; 9:e1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Alfonzo J., Thiemann O., Simpson L.. The mechanism of U insertion/deletion RNA editing in kinetoplastid mitochondria. Nucleic Acids Res. 1997; 25:3751–3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Aphasizhev R., Aphasizheva I.. Uridine insertion/deletion editing intrypanosomes: a playground for RNA-guided information transfer. Wiley Interdiscip. Rev. RNA. 2011; 2:669–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Etheridge R.D., Aphasizheva I., Gershon P.D., Aphasizhev R.. 3′ adenylation determines mRNA abundance and monitors completion of RNA editing in T. brucei mitochondria. EMBO J. 2008; 27:1596–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Aphasizheva I., Maslov D., Wang X., Huang L., Aphasizhev R.. Pentatricopeptide repeat proteins stimulate mRNA adenylation/uridylation to activate mitochondrial translation in trypanosomes. Mol. Cell. 2011; 42:106–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zhang L., Sement F.M., Suematsu T., Yu T., Monti S., Huang L., Aphasizhev R., Aphasizheva I.. PPR polyadenylation factor defines mitochondrial mRNA identity and stability in trypanosomes. EMBO J. 2017; 36:2435–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ryan C.M., Read L.K.. UTP-dependent turnover of Trypanosoma brucei mitochondrial mRNA requires UTP polymerization and involves the RET1 TUTase. RNA. 2005; 11:763–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Aphasizheva I., Aphasizhev R.. RET1-catalyzed uridylylation shapes the mitochondrial transcriptome in Trypanosoma brucei. Mol. Cell. Biol. 2010; 30:1555–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Burger G., Valach M.. Perfection of eccentricity: Mitochondrial genomes of diplonemids. IUBMB Life. 2018; 70:1197–1206. [DOI] [PubMed] [Google Scholar]
- 76. Gray M.W., Lukes J., Archibald J.M., Keeling P.J., Doolittle W.F.. Irremediable complexity?. Science. 2010; 330:920–921. [DOI] [PubMed] [Google Scholar]
- 77. Flegontov P., Gray M.W., Burger G., Lukeš J.. Gene fragmentation: a key to mitochondrial genome evolution in Euglenozoa?. Curr. Genet. 2011; 57:225–232. [DOI] [PubMed] [Google Scholar]
- 78. Lukeš J., Archibald J.M., Keeling P.J., Doolittle W.F., Gray M.W.. How a neutral evolutionary ratchet can build cellular complexity. IUBMB Life. 2011; 63:528–537. [DOI] [PubMed] [Google Scholar]
- 79. Stoltzfus A. Constructive neutral evolution: Exploring evolutionary theory's curious disconnect. Biol. Direct. 2012; 7:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.