


Abstract
Allele-specific protospacer adjacent motif (asPAM)-positioning SNPs and CRISPRs are valuable resources for gene therapy of dominant disorders. However, one technical hurdle is to identify the haplotype comprising the disease-causing allele and the distal asPAM SNPs. Here, we describe a novel CRISPR-based method (CRISPR-hapC) for haplotyping. Based on the generation (with a pair of CRISPRs) of extrachromosomal circular DNA in cells, the CRISPR-hapC can map haplotypes from a few hundred bases to over 200 Mb. To streamline and demonstrate the applicability of the CRISPR-hapC and asPAM CRISPR for allele-specific gene editing, we reanalyzed the 1000 human pan-genome and generated a high frequency asPAM SNP and CRISPR database (www.crispratlas.com/knockout) for four CRISPR systems (SaCas9, SpCas9, xCas9 and Cas12a). Using the huntingtin (HTT) CAG expansion and transthyretin (TTR) exon 2 mutation as examples, we showed that the asPAM CRISPRs can specifically discriminate active and dead PAMs for all 23 loci tested. Combination of the CRISPR-hapC and asPAM CRISPRs further demonstrated the capability for achieving highly accurate and haplotype-specific deletion of the HTT CAG expansion allele and TTR exon 2 mutation in human cells. Taken together, our study provides a new approach and an important resource for genome research and allele-specific (haplotype-specific) gene therapy.
INTRODUCTION
Dominant gain-of-function mutations accounts for ∼36% of all currently known human genetic diseases according to the OMIM database. We focus on repeated sequence expansions in neurodegenerative diseases and the autosomal dominant mutation V30M/V112I in TTR, a gene expressed in the liver and causing familial amyloid polyneuropathy (1). The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR-associated protein 9 (Cas9) gene editing technology have paved the way for efficient personalized gene therapy over the past few years (2–4), but gene therapy of dominant disorders is challenging due to the dominant negative effect of the disease causing allele.
The most promising gene-therapy approaches to dominant disorders are via targeted blocking or replacement of the dominant disease-causing allele by allele-specific gene knockout or homology-directed repair (HDR). A number of successful examples of inactivation or correction of dominant mutations by CRISPR/Cas9 have been described such as pathogenic variants of the Dynamin 2 (DNM2) gene, which cause centronuclear myopathies (CNMs) (5); a single-nucleotide mutation (P23H) in rhodopsin (RHO), a mutation causing Retinitis Pigmentosa (RP) (6); a single-nucleotide mutation in the keratin 14 gene (KRT14), which leads to generalized severe epidermolysis bullosa simplex (7); pathogenic variants of DFNA36, causing dominant progressive hearing loss (8); and the Swedish mutation in the amyloid precursor protein (APP) gene, which causes a familiar form of Alzheimer’s disease (AD) (9). However, since CRISPR/Cas9 can tolerate 1–3 mismatches between the gRNA spacer and the target site and most dominantly genetic disorders are caused by point mutations (10), the spacer-mediated allele-specific knockout approach will most likely alter the remaining healthy allele (11). To overcome this disadvantage, a few studies have recently demonstrated that highly specific eradication of the dominant mutation can be accomplished by protospacer adjacent motif (PAM)-mediated allele-specific CRISPR gene editing (12–14). Compared to the spacer-mediated allele-specific knockout approach, the PAM-mediated approach is much more specific, as the preservation and presence of the canonical PAM sequences is critical for the DNA editing activity of CRISPR-Cas9 (15). However, for dominant disorders caused by repeat expansions, targeting the repeat regions (e.g. CAG expansion in the huntingtin gene, HTT) is not feasible.
The presence of PAM-positioning single-nucleotide polymorphisms (SNP) that give rise to opposing PAM functioning (either active or dead PAM) has been demonstrated to be highly valuable for stringent allele-specific gene editing (13). However, one requirement for this approach is to identity the haplotype that comprises the disease-causing mutation and the active PAM allele. It is even more challenging if the allele-specific PAM (asPAM) allele is located at a distant position from the disease-causing mutation. Although long-read next-generation sequencing (NGS) methods provided by e.g. Oxford Nanopore, Bionano Genomics and PacbBio can be used for haplotyping, alternative methods that are affordable and can be adapted by any research group are needed. Previously, we had observed that chromosomal DNA that is deleted by a pair of CRISPRs could form extrachromosomal circular DNA (eccDNA) in cells (16). At the junction of the eccDNA, the two regions (ends) that are originally far apart from each other are now in proximity. We thus have developed an in vivo CRISPR-based method (CRISPR-hapC) for haplotype phasing.
To further combine the CRISPR-hapC and PAM-mediated allele-specific, or haplotype-specific, CRISPR gene editing for a large spectrum of dominant mutations, we developed a database of SNPs for asPAM CRISPR gene editing. This asPAM CRISPR database contains high-frequency (allele frequency > 30%) PAM-presenting SNPs, which give rise to either active or dead PAMs for the four most broadly used CRISPR systems. We applied the asPAM CRISPR and CRISPR-hapC systems to achieve haplotype-specific editing of two human genes (HTT and TTR) in human cells, demonstrating the utility of the asPAM-hapC system and complementary methods in developing and streamlining highly specific and efficient editing of dominant disorders.
MATERIALS AND METHODS
All sequences for CRISPR gRNA spacers, C-Check oligonucleotides, PCR primers, PCR amplification conditions and genotyping/haplotyping of the TTR mutant clones can be found in Supplementary Tables S1–S5.
SaCas9, SpCas9, xCas9 and Cas12a asPAM SNPs
To generate the asPAM SNP and CRISPR database, first, all SNPs with a heterozygous frequency >30% were retrieved from the 1000 human genome database. Next, only those SNPs giving rise to opposing PAM activities were included. The reference and alternative alleles of each SNP must be located in the conserved PAM motif (letters in bold) -NNGRRN for SaCas9, spacer-NGG for SpCas9, spacer-NGK for xCas9 and TTTV-spacer for Cas12a and give rise to either active or dead PAM, respectively. For all the corresponding asPAM CRISPR gRNA spacers, we have aligned them against the human reference genome with bowtie (version 1.1.2) and calculated the number of genomic loci with up to three mismatches. Finally, all asPAM SNPs and CRISPRs were incorporated in the CRISPR database. This database has been made publicly available, and detail of information and instructions on how to use the database can be found on our website (www.crispratlas.com/knockout).
CRISPR asPAM database and HTT and TTR asPAM SNP output
The asPAM-SNP and asPAM CRISPR databases were integrated into our CRISPR Atlas website (www.crispratlas.com), allowing the search of SNPs and CRISPRs for any gene of interest (GOI). To use the database, simply select ‘asPAM CRISPR editing’ from the webtools then output the asPAM-SNPs information of the GOI following the four guided steps in the opened webpage: (i) select GRCh37/h19 as the reference genome; (ii) select the CRISPR and PAM; (iii) select ‘chromosome interval’ as search method; (iv) input the start and end positions of a genome region including the GOI locus. In our test cases, the TTR locus is chr18: 29136875–29202208 and HTT locus is chr4: 3042475–3250766. Alternatively, an Excel sheet containing all the asPAM SNPs and CRISPRs can be requested from the corresponding authors.
In this study, a genome region including a GOI locus comprises GOI DNA sequences and its context excluding the first upstream and downstream genes. Taking the TTR locus as an example, with the first upstream gene DSG-AS1 and downstream gene B4GALT6, we obtain end and start positions as chr18: 29136874 and chr18: 29202209, respectively. Thus, the chromosome interval range used for TTR locus input is chr18: 29136874 + 1 to chr18: 29202209 – 1, which actually is chr18: 29136875–29202208.
Oligonucleotides and plasmids
All DNA oligonucleotides in this study are ordered from either BGI-Qingdao, China (Chromosome1, TTR) or Sigma (HTT). Sequences of these oligonucleotides can be found in the Supplementary Tables. The CRISPR plasmids used in this research are: LentiCRISPRv2 (Addgene plasmid #52961), pUC19 (Addgene plasmid #50005), C-Check vector (Addgene plasmid #66817), pX601-AAV-CMV:NLS-SaCas9-NLS-3xHA-bGHpA;U6:BsaI-sgRNA (Addgene plasmid #61591).
CRISPR gRNA design and vector construction
In this study, as an example, we tested the asPAM CRISPRs and universal gRNAs using the SaCas9 system, for which the spacer length of gRNA is 21 nt. Other CRISPRs used for eccDNA generation and CRISPR-hapC were based on the SpCas9 system as described earlier (16). However, it should be noted that we have also tested the SaCas9 system for eccDNA generation. This also works efficiently.
All the CRISPR gRNA spacers were designed utilizing the online webtools: CRISPOR (http://crispor.tefor.net) and the asPAM CRISPR database. Synthesized gRNA oligonucleotides were annealed and introduced into AAV-SaCas9 or LentiCRISPRv2 (SpCas9) vectors by Golden Gate Assembly (GGA) as described previously (17). Briefly, 1 μl sense strand (SS) and anti-sense strand (AS) gRNA oligonucleotides (100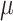 M) were mixed in 2 μl 10 × NEB Buffer 2.1, with a supplement of ddH2O to a final volume of 20 μl. The annealing program was 95°C for 5 min and ramped down to 25°C at a rate of −5°C/min. Then 1 μl annealed gRNA was added into a GGA reaction system that contains 1
M) were mixed in 2 μl 10 × NEB Buffer 2.1, with a supplement of ddH2O to a final volume of 20 μl. The annealing program was 95°C for 5 min and ramped down to 25°C at a rate of −5°C/min. Then 1 μl annealed gRNA was added into a GGA reaction system that contains 1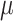 l T4 ligase (NEB), 2
l T4 ligase (NEB), 2 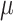 l 10× T4 ligase buffer, 1
l 10× T4 ligase buffer, 1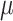 l digest enzyme (Esp3I for lentiCRISPRv2 and Eco31I for AAV-saCas9, ThermoFisher Scientific) and ddH2O in a total volume of 20
l digest enzyme (Esp3I for lentiCRISPRv2 and Eco31I for AAV-saCas9, ThermoFisher Scientific) and ddH2O in a total volume of 20  l. The GGA program was performed in a thermocycler as: 10 cycles of 37°C for 5 min and 22°C for 10 min; 1 cycle of 37°C for 30 min; 1 cycle of 75°C for 15 min. Then 1
l. The GGA program was performed in a thermocycler as: 10 cycles of 37°C for 5 min and 22°C for 10 min; 1 cycle of 37°C for 30 min; 1 cycle of 75°C for 15 min. Then 1  l of the GGA product was used for transformation of competent cells, and colony PCR screening was conducted to select positive colonies carrying the gRNA spacer. All CRISPR plasmids were further validated by Sanger sequencing.
l of the GGA product was used for transformation of competent cells, and colony PCR screening was conducted to select positive colonies carrying the gRNA spacer. All CRISPR plasmids were further validated by Sanger sequencing.
For C-Check (CC) vector construction in this study, oligonucleotides including target sequences (protospacer) and active PAM or dead PAM sequences were synthesized and inserted into CC vectors as described previously (18). All the CC oligonucleotides can be found in Supplementary Tables. The construction steps for CC vectors were the same as that for gRNA ligation, which was also conducted by GGA. CC vectors containing active PAM target sites were named as CC-aPAM and those containing dead PAM sequences were denoted CC-dPAM.
Cell culture and transfection
Cell lines used in this study include human embryonic kidney 239T (HEK293T, ATCC® CRL-3216), liver hepatocellular carcinoma cell line (HepG2, ATCC® HB-8065), human osteorsarcoma cell line (U2OS, ATCC® HTB-96), Hela cells, human ovarian adenocarcinoma cell line (Skov3, ATCC® HTB-77), human lung carcinoma epithelial cell line (A549, ATCC® CCL-185), human fibroblasts (BJ fibroblasts), human bone osteorsarcoma cell line (Saos-2, ATCC® HTB-85), human myelogenous leukemia cell line (K562, ATCC® CCL-243) and human breast cancer cell line (MCF-7, ATCC® HTB-22). All the cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (LONZA) supplemented with 10% fetal bovine serum (FBS) (Gibco), 1% GlutaMAX (Gibco) and penicillin/streptomycin (100 units penicillin and 0.1 mg streptomycin/ml) in a 37°C incubator with 5% CO2 atmosphere and maximum humidity. Cells were re-seeded every 2–3 days when the confluence reached up to 80–90%.
Transfection was conducted with Lipofectamine 2000 transfection reagent (Invitrogen) or X-tremeGene 9 (Roche) in 24-well plates according to the manufacturer’s protocol. Briefly, 60 000 cells/per well were seeded in 24-well plates and the medium was changed when the cells reached 50–70% confluency before transfection (typically 24 h after seeding). For each transfection, 500 ng total plasmid DNA and 1.5 μl Lipofectamine 2000 was diluted separately in Opti-MEM (Gibco) to a total volume of 25 μl. The diluted DNA was added to the diluted Lipofectamine and mixed gently. After 15-min incubation at room temperature, the transfection mixture was homogeneously added to the adherent cells in a dropwise manner. We changed medium 24 h after transfection and harvested cells 48 h later for subsequent assays. For all the co-transfection experiments in the research, including C-Check efficiency test, eccDNA generation and allele-specific knockout, the plasmids co-transfection ratio was 1:1.
Flow cytometry (FCM) analysis
Cells in 24-well plates, dissociated with 100 μl 0.5% trypsin–EDTA, were suspended in 100 μl 5% FBS–PBS and transferred to a 96 deep-well plate on ice. Cells were spun down at 2000 rpm for 5 min and the supernatant was removed. Then the cell pellets were re-suspended in 600 μl PBS and immediately subjected to FCM analysis. FCM was performed using a BD LSRFortessa (supported by the FACS CORE facility, Department of Biomedicine, Aarhus University and FACS CORE, BGI-Qingdao) with at least 30 000 events collected for each sample in triplicate.
TTR mutation cell model establishment
In order to model the TTR mutation genotype that occurs in TTR-FAP patients, we established TTR mutated HepG2 cell line using CRISPR/spCas9. A gRNA targeting exon 2 (near the location of V30M mutation site) of TTR gene was transfected into HepG2 cells. Transfected cells were cultured in selection medium with 1 μg/ml puromycin in 10-cm dishes for 2 weeks, and cell colonies were manually picked and genotyped by PCR and Sanger sequencing. A cell colony (#21) with heterozygous mutation in exon 2 was selected as a model cell line for asPAM CRISPR editing of TTR.
Genomic DNA extraction, PCR and TA cloning
Genomic DNA was extracted using the TIANamp Genomic DNA Kit (for the TTR gene editing, TIANGEN, China) or by cell lysis (for the HTT gene) in accordance with the manufacturer’s instructions. The PCR were conducted using high-fidelity platinum Pfx polymerase in the presence of 2 × enhancer solution (#11708013, Thermo Fisher Scientific). All the primers for SNP validation and eccDNA detection can be found in Supplementary Tables. ECC PCR products were sub-cloned into the pMD-19T vector utilizing TOPO TA Cloning Kit (TaKaRa) according to the manufacturer’s instructions and analyzed by Sanger sequencing.
Generation of eccDNA by CRISPR
A detail protocol for the generation of eccDNA by CRISPR and haplotyping by eccDNA was provided in the Supplementary Methods.
Data analysis
The webtool asPAM CRISPR editing (http://www.crispratlas.com/knockout) was used for GOI SNP-PAM output. Flowjo software was used to gate and output FCM data. Prism 7 was used to analyze FCM data and plot histograms. Sanger sequencing results were deciphered by Snapgene Viewer, and the webtool ICE analysis (https://ice.synthego.com/#/) was used for genotype percentage analysis with Sanger data.
Statistics
Unless stated elsewhere, all experiments were performed in triplicate. The Student’s paired T test was used for statistical analysis, with a P value <0.05 considered as statistically significant.
RESULTS
CRISPR-hapC: a novel method for haplotyping
One technical hurdle when applying CRISPR for allele-specific PAM (asPAM) editing is to identify which of the distal asPAM SNPs is linked to the mutant allele. This kind of linkage analysis, or haplotyping, is even more challenging when mutations are caused by simple repeat expansions (e.g. trinucleotide repeat expansions in the HTT, SCA2 or DMPK genes). Previously, we discovered that the chromosomal DNA deleted by a pair of CRISPR could form extrachromosomal circular DNA (eccDNA) in cells. In our method, called CRISPR-C (16), the eccDNA appears very rapidly (within 48 h after transfection with CRISPR plasmids) and can be retained in some cells for up to 2 weeks. Taking advantage of the eccDNA junction that joins two distal regions in proximity, we proposed that the CRISPR-C technology could be used as an alternative method for haplotyping—a method hereafter called CRISPR-hapC (Figure 1A).
Figure 1.

Haplotyping by CRISPR-mediated DNA circularization (CRISPR-hapC). (A) Schematic illustration of the CRISPR-hapC method and the haplotyping of six single-nucleotide polymorphisms (SNPs) in HEK293 cells. The six SNPs, indicated with red dash lines, are located across the human chromosome 1. The distance between SNP1 and the remaining five SNPs ranges from 660 bp to 221 Mb. Six CRISPR vectors (Chr1-ECC-Cr) were generated, indicated with the blue dashed line. The cleavage of Chromosome 1 with a pair of the CRISPRs will create two double stranded DNA breaks (DSBs). Repair of the DSBs by DNA circularization will lead to the generation of extrachromosomal circular DNAs (eccDNA), or ring chromosome as indicated in the figure. Asterisks indicate SNP1 and SNP6. Bottom: Sanger sequencing results of the eccDNA comprising SNP1 and SNP6 and the resolving of haplotypes. (B) Haplotype of the six SNPs in chromosome 1 of HEK293 cells by CRISPR-hapC.
As a proof-of-concept of the CRISPR-hapC method, we selected six heterozygous SNPs (Supplementary Figure S1A) on Chromosome 1 in HEK293 cells based the HEK293 genome database (http://hek293genome.org/v2/index.php) (19). The heterozygosity of these six SNPs was further confirmed by Sanger sequencing of our HEK293 cells (Supplementary Figure S1B and Figure 1A). The CRISPR-hapC method contains four key steps. Step 1: We followed the guideline of our CRISPR-C method (16), and generated six eccDNA CRISPRs (Chr1-ECC-Cr1 to Chr1-ECC-Cr6) adjacent to the SNPs (SNP1 to SNP6) (Figure 1A and Supplementary Table S2). The Chr1-ECC-Cr1 was upstream of SNP1 and the Chr1-ECC-Cr2 to Chr1-ECC-Cr6 were downstream of SNP2 to SNP6, respectively. This allows us to generate eccDNAs, ranging from 660 bp to 211 Mb, which joins the SNP1 and each of the others SNPs at the junction (Figure 1A). Step 2: To generate eccDNA by CRISPR-C, a pair of CRISPRs (Chr1-ECC-Cr1 and each of the other five CRISPRs) was delivered to HEK293 cells by transfection. Step 3: 48 h after transfection, transfected cells were lysed and eccDNAs were amplified by inversed PCRs with primers flanking the SNPs and the junction (Supplementary Figure S2A). Step 4: the PCR products were subsequently purified, cloned into a TA cloning vector and analyzed by Sanger sequencing (Figure 1A and Supplementary Figure S2B). By analyzing a total of 27 clones (Supplementary Figure S2C), we proved that the CRISPR-hapC methods can be used for haplotype phasing for over 200 Mb (Figure 1B). A step-by-step protocol for haplotyping with CRISPR-hapC is provided in the Supplementary Materials.
The asPAM CRISPR database: Genome-wide distribution of high frequency, allele-specific PAM (asPAM)-positioning SNPs
One attractive application of the CRISPR-hapC method is to enable the haplotype-specific inactivation of dominant disease-causing mutations. We first analyzed the global distribution of canonical PAM motifs for SaCas9 (N1N2G3R4R5N6, N = A/T/C/G, R = A/G), SpCas9 (N1G2G3), xCas9 (an SpCas9 with expanded PAM, N1G2K3, K = G/T) and Cas12a (T-4T-3T-2V-1, V = A/C/G) harboring high frequency SNPs across the human genome (Supplementary Figure S3). For the SaCas9, as minor editing activity is also observed for the sixth nucleotide other than Thymidine (N6) (20), we only selected those SNPs presented at the highly conserved GRR motif to specifically discriminate the two alleles. Another important criterion was that the reference and alternative sequences of each SNP must give rise to an active PAM and a dead PAM, respectively, and vice versa (Figure 2A). For simple classification, we defined all these SNPs and corresponding CRISPR target sites as the allele-specific PAM (asPAM) SNPs and asPAM CRISPRs. Reanalyzing the 1000 human genome database, which comprises 2504 genomes (http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502) (21), we identified all asPAM SNPs and CRISPs for the four most broadly used CRISPR-Cas systems (SaCas9, SpCas9, xCas9 and Cas12a). For example, we identified 645 110 high-frequency (allele frequency > 30%) SaCas9 asPAM SNPs, and most (75%) of the asPAM SNPs were also found in the human SNP database (dbSNP) (Figure 2B). The majority (54.5%) of these asPAM SNPs are in repetitive regions (Figure 2C). For those asPAM SNPs located in gene bodies (including introns) or within 5 kb flanking regions of genes, there is an average of 22 asPAM SNPs/gene, an average of 9.5 kb per SNP, and a coverage of 89% of all known genes (Figure 2D), highlighting that the asPAM CRISPR-based gene editing can be applied to most genes. To facilitate and simplify the application of asPAM CRISPR, we have generated a publicly available database (http://www.crispratlas.com/knockout) allowing the conventional search of asPAM CRISPRs by gene name, gene ID, SNP ID or chromosome regions. The most efficient spacer length of SaCas9 is 21–22 nt (22). For the asPAM CRISPR editing database, we provided a 21-nt spacer length as default, as well as the flanking sequences. Regarding the off-target evaluation of the asPAM CRISPRs, previous studies have found that most CRISPR-Cas9 could not tolerate >2 mismatches (23,24). We aligned each asPAM CRISPR to the human genome with bowtie 2.0 and provided a statistical summary of the genomic loci with up to three mismatches. To select high fidelity asPAM CRISPRs, we recommend combining in silico off-target prediction software (such as Cas-OFFinder (25), CCTop (26) and CRISPor (27)) and genome-wide experimental evaluations (such as GUIDE-seq (28) and CIRCLE-seq (29)) to further analyze the off-target effects of any specific asPAM gRNA of interest.
Figure 2.

Generation of a genome-wide high-frequency, allele-specific PAM-positioning (asPAM) SNP and CRISPR database. (A) Illustration of the SaCas9 CRISPR with active or dead PAMs. The reference and alternative allele of an asPAM SNP must fall in the GRR motif of a NNGRRT PAM and give rise to apposing PAM activities: active PAM (NNGRRT) and dead PAMs: NNHRRT, NNGYRT or NNGRYT. (B) Distribution of all SaCas9 asPAM SNP and CRISPRs across the human genome categorized by SNP frequency and chromosome. (C) Distribution of all SaCas9 asPAM SNP and CRISPRs in human repetitive sequences. Pie plot presents the distribution between asPAM SNPs located at unique and repetitive genome regions. (D) Distribution of distance between two adjacent SaCas9 asPAM SNPs within the gene body (including introns) + 5 kb flanking region.
The asPAM CRISPRs are highly specific for active PAM
Over 40 dominant diseases, most of which are primarily neurodegenerative disorders such as myotonic dystrophy, amyotrophic lateral sclerosis and polyglutamine disorders, are caused by simple repeat expansions across the human genome (30). One advantage of choosing PAM-positioning rather than spacer-positioning SNPs is the specificity of distinguishing the two alleles by the CRISPR-Cas. To validate the specificity of asPAM CRISPRs, we selected the HTT gene to address the specificity of our asPAM CRISPRs. The HTT gene encodes a protein called huntingtin and plays an essential role in nerve cells. Trinucleotide repeat expansions of HTT leads to the development of the devastating neurodegenerative Huntington’s Disease (HD) with no cure today (31–33). One promising therapy for HD is the genetic ablation of the dominant HTT mutation allele (34,35). There are nine SaCas9 asPAM SNPs in the human HTT gene (Figure 3A and Supplementary Figure S4). For all HTT SaCas9 asPAM SNPs, one important feature is that the reference and alternative nucleotide give rise to an active PAM and a dead PAM, respectively, or vice versa.
Figure 3.

Evaluation of asPAM CRISPR specificity by C-Check assay. (A) Schematic illustration of the asPAM SNP and CRISPR for the human HTT locus. Sequences of the PAM sites are presented. aPAM: active PAM; dPAM: dead PAM. (B) Measurement of C-Check cleavage and repair efficiency (% EGFP+/AsRED+ cells, n = 3) by FACS. Transfects are presented in the figure legend. CC-aPAM: C-Check vector carrying the CRISPR target site (protospacer) and an active PAM. CC-dPAM: C-Check vector carrying the CRISPR target site (protospacer) and a dead PAM. asPAM-Cr: all-in-one CRISPR vector expressing SaCas9 and the corresponding HTT targeting gRNA (spacer). PAM sequences are indicated in (A). Asterisks: P < 0.05 in comparison of controls and dPAM. (C) Schematic illustration of the asPAM SNP and CRISPR for the human TTR locus and flanking regions. Sequences of the PAM sites are presented. aPAM: active PAM; dPAM: dead PAM. (D) Measurement of C-Check cleavage and repair efficiency (% EGFP+/AsRED+ cells, n = 3) by FACS. PAM sequences are indicated in (C). Asterisks: P < 0.05 in comparison of controls and dPAM.
Next, we applied the dual-fluorescence surrogate reporter system (C-Check) (36) to investigate whether the asPAM CRISPR can specifically distinguish active PAM and dead PAM sites. For each asPAM CRISPR, we generated two C-Check vectors carrying the corresponding active PAM or dead PAM target site, respectively (Supplementary Figure S5), and performed C-Check analysis. Consistent with previous observations (20,37), much higher editing activity was achieved when the sixth canonical nucleotide of the SaCas9 PAM is a Thymine (NNGRRT). Guanine (NNGRRG) leads to median activity, Cytosine (NNGRRC) to low, and Adenine (NNGRRA) to no detectable activity (Figure 3B). Most importantly, our results showed that CRISPR gene editing activity was only observed in the active PAM C-Check vector for all nine HTT asPAM CRISPRs, suggesting that the HTT asPAM CRISPRs are highly specific (Figure 3B).
We sought to select another locus to further validate the specificity of SaCas9 asPAM CRISPRs. Many dominant disorders are caused by gain-of-function point mutations. One example is the transthyretin familial amyloid polyneuropathy (FAP). FAP is a life-threatening disease that is mainly caused by point mutations in the Transthyretin (TTR) gene. The disease is inherited in an autosomal dominant manner and over 100 disease-causing point mutations have been reported worldwide (38). The misfolded TTR protein leads to deposition of amyloid fibrils extracellularly and typically cause nerve-length dependent polyneuropathy (1). Over 90% of the secretory TTR protein is produced in the liver and, to date, liver transplantation is the only treatment of FAP (39) that makes gene therapy an attractive approach considering the great shortage of transplantable livers. We selected 14 asPAM SNPs within and flanking the human TTR gene (hg19, chr18: 29136875–29202208) and generated 14 TTR asPAM CRISPRs. 14 C-Check vectors were generated for the active PAM and dead PAM loci, respectively (Figure 3C and Supplementary Figure S6). Consistent with what we have observed above for the HTT AsPAM loci, the highest targeting activity was achieved for TTR asPAM CRISPRs with NNGRRT PAMs (Figure 3D; TTR T7, T8 and T11 C-Check efficiency over 70%). Significantly high targeting activities were also observed for some TTR asPAM CRISPRs with noncanonical PAMs: NNGRRA (SNP12, 65%; SNP13, 33%), NNGRRG (SNP6, 42%) or NNGRRC (SNP9, 20%; SNP10, 21%; SNP1, 10%) (Figure 3D and Supplementary Figure S7). Notably, all TTR asPAM CRISPRs only target loci with active PAM, which taken together proves that the asPAM CRISPR is highly specific.
Haplotype-specific deletion of HTT achieved by CRISPR-hapC and asPAM CRISPRs
Allele-specific inactivation of the HTT mutation have been shown to be an attractive approach for curing Huntington’s disease (12,34). As a proof-of-concept of combining the CRISPR-hapC method and asPAM CRISPR for haplotype-specific gene editing, we screened 10 different human cell lines for heterozygous HTT CAG-expansion (Figure 4A). Three cell lines (Hela, HepG2 and MCF-7) contain two different CAG-expansion alleles that can be distinguished by gel electrophoresis. We next sequenced all nine HTT SaCas9 asPAM SNPs in Hela, HepG2 and Skov-3 (Supplementary Figure S8). Both Hela (6 out of 9) and Skov-3 (5 out of 9) cells are heterozygous in most of these asPAM SNPs (Figure 4B). The Hela cells were selected for haplotyping between the HTT CAG allele and the heterozygous HTT asPAM SNPs (2, 3, 5, 7, 8 and 9) by CRISPR-hapC, for identifying asPAM SNPs of which the active PAM is linked to the long CAG expansion allele (Figure 4C).
Figure 4.

Haplotype-specific HTT editing achieved with CRISPR-hapC and asPAM CRISPRs. (A) Genotyping of CAG expansion in exon 1 of the HTT gene in 10 cell lines. (B) Genotyping of the nine HTT asPAM SNPs in three cell lines by sequencing. (C) Schematic illustration of two haplotypes comprising the HTT CAG expansion allele and the asPAM allele. Here, the active PAM (aPAM) is linked to the long CAG allele. (D) Schematic illustration of HTT haplotyping by the CRISPR-hapC approach. Uni-Cr1, a CRISPR that cleaves both alleles. ECC-Cr2 and ECC-Cr3, CRISPR target sites adjacent to the asPAM SNP and used for generating extrachromosomal circular (ECC) DNA. (E) ECC-PCR-based haplotyping of the HTT CAG expansion allele with HTT asPAM SNP2, 3, 4, 7, 8 and 9. (F) Illustration of HTT asPAM CRISPR editing and genotyping with the eccDNA method. The p0, p3 and p9 indicate genotyping primers of ECC-DNA encompassing the ECC-DNA junction. (G) and (H) ECC-DNA PCR genotyping for haplotype-specific deletion of the long CAG expansion allele (29 repeats). Transfections were conducted with a pair of CRISPRs: one universal CRISPR that cleave both alleles and one asPAM-specific CRISPR that only cleave the active PAM allele linked to the long CAG expansion (n = 3). Controls are CRISPR pairs that delete both haplotypes. (I) Summary of single cell colonies edited with a pair of HTT-Uni-Cr and HTT-asPAM-Cr as shown in the table. Note that genotyping was conducted with PCR primers flanking the targeted HTT deletion region. All edited clones have the long CAG allele deleted. Numbers in parentheses are the total number of clones screened for each group.
To achieve this, we first designed two sets of CRISPRs: the asPAM eccDNA CRISPRs (ECC-Cr) and a universal CRISPR (Uni-Cr). The ECC-Crs and Uni-Cr are a pair of CRISPRs flanking the genomic region comprising the mutant and asPAM SNPs (Figure 4D). Six HTT ECC-Crs, denoted HTT-ECC-Cr2, 3, 5, 7, 8 and 9, were generated (Supplementary Table S2). Three HTT-Uni-Crs (HTT-Uni-Cr1, 2 and 3) adjacent to the locus of potential CAG expansion were generated with gene editing activity validated in Hela cells (Supplementary Figure S9). We next transfected Hela cells with the HTT-Uni-Cr1 and one HTT-ECC-Cr. The eccDNA was captured by inverse PCRs for all six pairs of CRISPRs. Linkage analysis between the HTT CAG allele and HTT asPAM SNP was then carried out by sub-cloning the eccDNA into a sequencing vector and analysis by Sanger sequencing. The CRISPR-hapC results showed that the active PAM alleles of HTT asPAM SNP3 and SNP9 are linked to the long allele of CAG expansion in Hela cells (Figure 4E).
We next investigated whether the asPAM CRISPR can be used to achieve haplotype-specific deletion. As depicted in Figure 4F, the asPAM CRISPR based haplotype-specific gene editing should be accomplished by a pair of CRISPRs: a universal CRISPR (Uni-Cr) and an asPAM CRISPR (asPAM-Cr). The asPAM-Cr targets the active PAM SNP that is linked to the mutated allele. Since it is improbable to achieve 100% targeted deletion from a bulk of transfected cells, traditional genotyping of bulk cells by flanking PCR is not feasible for addressing the issue of asPAM CRISPR specificity. Alternatively, a highly labor-intensive approach would have to be employed by genotyping hundreds of single cell colonies with PCR and/or sequencing. To overcome this technical hurdle, we developed an eccDNA-based genotyping method, reasoning that, if the asPAM-Cr is specific, the eccDNA generated by the Uni-Cr and asPAM-Cr should only carry the haplotype comprising the mutant allele (Figure 4F).
Based on the results of haplotyping the HTT CAG expansion and HTT asPAM SNPs in Hela cells, six different combinations of CRISPR pairs (one HTT-Uni-Cr and one HTT-asPAM-Cr) were transfected into Hela cells. Genotyping eccDNAs from transfected cells clearly showed that the long CAG expansion allele, which is linked to the active PAM allele, was present in all eccDNAs (Figure 4G and H). Very weak signal from the short CAG allele was also seen for HTT-asPAM-Cr3, indicating that this asPAM CRISPR might have unspecifically targeted the other allele (Figure 4G). We also analyzed 52 puromycin-resistant single cell colonies by PCR genotyping of the CAG expansion allele. Consistent with the eccDNA-genotyping results, the HTT asPAM CRISPR specifically deleted the haplotype comprising the long CAG expansion allele and the active PAM allele in all the seven clones with deletion (11.5% total efficiency) (Figure 4I).
Highly stringent and haplotype-specific deletion of TTR achieved by CRISPR-hapC and asPAM CRISPRs
To demonstrate the applicability of the CRISPR-hapC and AsPAM CRISPRs for achieving haplotype-specific gene editing in another gene, we analyzed 14 TTR SaCas9 asPAM SNPs in five human cell lines (Hela, LO2, PLC/PRF5, HEK293T and HepG2) (Figure 5A and Supplementary Figure S10) and revealed that the immortalized human liver carcinoma cells (HepG2) are heterozygous in 7 of these 14 SNPs (TTR asPAM SNP2, 3, 4, 10, 11, 12 and 14). We then haplotyped the TTR asPAM SNP3, 11 and 14 with CRISPR-hapC. Using the same procedure as above, we generated three TTR ECC CRISPRs (TTR-ECC-Cr3, ECC-Cr11 and ECC-Cr14) (Figure 5B). The HepG2 cells were then transfected with TTR-ECC-Cr3 and TTR-ECC-Cr11, or TTR-ECC-Cr3 and TTR-ECC-Cr14. TTR eccDNAs that joins SNP3 and SNP11, or SNP3 and SNP14, were generated from the transfected cells (Figure 5C). Sequencing of the TTR eccDNAs showed that all three TTR asPAM SNPs (3,11,14) have the active PAM in one haplotype, and the dead PAM in the other (Figure 5C).
Figure 5.

Haplotype-specific editing with asPAM CRISPR. (A) Genotyping of the 14 TTR asPAM SNPs in five cell lines by Sanger sequencing. (B) Schematic illustration TTR haplotyping of SNP3, SNP11 and SNP14 by the CRISPR-hapC approach. (C) The eccDNA genotyping and linkage results of TTR asPAM SNP3, 11 and 14 by CRISPR-hapC. Letters and symbols in red represent dead PAMs. (D) Schematic illustration of haplotype 1-specific deletion of the TTR allele (+ 1bp insertion in exon 2) by asPAM CRISPR editing. (E) Amplification of TTR ECC-DNAs from cells transfected with paired CRISPRs or only one CRISPR as control. (F) Sanger sequencing of the TTR ECC-DNA from the paired CRISPR (D) treated cells. Data were analyzed by ICE.
To demonstrate the haplotype-specific TTR deletion by asPAM CRISPR in HepG2 cells, we first introduced artificial mutations to TTR exon 2 in HepG2 cells by CRISPR (Supplementary Figure S11A) and performed haplotype analysis between the TTR exon2 mutations and TTR asPAM SNP11 and SNP14 with CRISPR-hapC (Supplementary Table S5). One clone, HepG2–21 (clone #21), which carries +1A and 4bp deletion mutations and, most importantly, one haplotype comprising the active PAM alleles for SNP3, SNP11 and SNP14 and the TTR +1A mutation, was chosen for asPAM-based CRISPR editing (Figure 5D and Supplementary Figure S11B). To enable the genotyping of the target deletion of the TTR +1A allele, a TTR universal CRISPR (TTR-Uni-Cr) targeting the TTR intron 1 and proximal to the artificial mutation was generated (Figure 5D). The HepG2–21 cells were transfected with the TTR-Uni-Cr and TTR-asPAM-Cr11, or TTR-Uni-Cr and TTR-asPAM-Cr14. Inverse PCR amplified the eccDNA junctions from the transfected cells with the pairs of CRISPRs but not the single CRISPR control (Figure 5E). Sanger sequencing of the TTR eccDNA and ICE analysis (40) revealed that only the targeted haplotype comprising TTR exon 2 +1A mutation allele linked to the active PAM was present in the eccDNA (Figure 5F). We also genotyped the transfected cells by flanking PCRs. Consistent with the eccDNA genotyping results, the asPAM CRISPR-mediated TTR deletion only occurs in the haplotype comprising the active PAM allele (Supplementary Figure S12). Taken together, we have proven that a combination of the CRISPR-hapC and the asPAM CRISPR can be used to achieve highly stringent haplotype-specific deletions in human cells.
DISCUSSION
Targeted inactivation and replacement of dominant disease-causing mutations are the leading gene therapy options for dominant disorders. For most dominant disorders caused by repeat expansions such as HTT, there is currently no cure. In this study, we described the allele-specific PAM (asPAM) CRISPR gene editing approach, and demonstrated the successful applications of the asPAM CRISPR system for haplotype-specific deletion of the huntingtin CAG expansion allele and TTR mutation allele in human cell models. To overcome several technical hurdles in haplotype-specific CRISPR gene editing, we (i) developed the CRISPR-hapC method for haplotype phasing between the disease-causing mutation and the distal asPAM SNPs; (ii) generated the asPAM CRISPR database allowing user-friendly selection of the high-frequency asPAM SNPs and CRISPR for any gene; (iii) incorporated an eccDNA-based genotyping method for evaluating the CRISPR gene editing outcome; (iv) demonstrated that the combination of CRISPR-hapC and asPAM CRISPRs can be used to achieve highly specific and haplotype-specific gene editing of HTT and TTR in human cells.
One important technology innovation in this study is the convenient, flexible and affordable CRISPR-C based method (CRISPR-hapC) for haplotyping. As demonstrated in our study, haplotyping of two alleles can be achieved by CRISPR-hapC from a few hundred bp to over 200 Mb. The CRISPR-hapC provides a valuable alternative to the existing methods for haplotyping, such as long-read nanopore sequencing (41) and single molecule real-time (SMRT) sequencing (42). Compared to the single molecule next-generation sequencing (NGS) approaches, the CRISPR-hapC method has low throughput but can serve as a valuable complimentary approach for targeted haplotyping and specific applications, such as haplotype-specific CRISPR gene editing as demonstrated in this study. The adaptation of the CRISPR-hapC method by the scientific community will further broaden the haplotype phasing and CRISPR-based applications.
It should be noted that the copy number of genes or chromosome polyploidy would have an effect on the application and interpretation of the CRISPR-hapC results. Currently, the CRISPR-hapC method is based on the presence of only two haplotypes in cells. In our study, we applied the CRISPR-hapC methods to map the haplotypes of 6 SNPs in chromosome 1 (diploid) in HEK293 cells and the HTT gene (two copies) in Hela cells. For the TTR genes, it seems there are three copies in the HepG2#21 cells (Supplementary Figure S11B). However, two copies of the TTR gene carry the same haplotype, thus enabling the application of CRISPR-hapC for TTR asPAM CRISPR editing. Another important note is that, due to the occurrence of chromosomal crossover during meiosis, de novo mutations or dynamic mutations of repeat expansions such as Fragile X syndrome, the parental haplotype comprising the disease-causing allele and the active PAM allele could be altered. Thus, CRISPR-hapC for haplotype-specific asPAM CRISPR gene editing is a truly personalized approach.
The potential off-target effects caused by CRISPR have been a main issue when the technology is translated into clinical application, as inactivation of tumor-suppressor genes or activation of proto-oncogenes could lead to detrimental consequences (43). Homology-directed repair (HDR) and allele-specific inactivation (ASI) are the two most broadly used approaches for CRISPR editing of dominant mutations. Relying on the endogenous HR-based DNA repair machinery, the HDR approach can restore the normal function of the mutated gene but is hampered by its relatively low efficiency (∼100-fold lower as compared to NHEJ-mediated gene knockout) (44,45). Many improvements have been made, such as addition of NHEJ-repressor or HDR-enhancing molecules (46–49), modification of HDR vector and delivery mode (50–52), and engineering of HDR-proficient Cas9 recombinant proteins (53–55). For dominant disorders, correction or disruption of the mutated allele without altering the remaining wild-type copy is essential. The concept of allele-specific editing has thus been introduced for discriminating the mutation and wild-type alleles by CRISPR/Cas9 (56,57). Most ASI attempts were based on SNPs positioning at the CRISPR gRNA spacer (58). We and other groups have shown that CRISPR/Cas9 can tolerate up to three mismatches at the spacer (18,59), while the CRISPR gene editing activity is highly dependent on the presence of canonical PAM. As now proven in 21 asPAM CRISPR loci, the asPAM CRISPR can clearly distinguish active and dead PAMs and, mostly importantly, only introduces DNA cleavage to the active PAM locus and achieves highly stringent haplotype-specific deletion of the dominant mutated allele. Similar to other CRISPR-based gene editing applications, once the asPAM CRISPRs are going to be used for therapeutic purposes, its specificity should also be carefully addressed with genome-wide off-target detection methods such as GUIDE-seq and CIRCLE-seq (28,29).
We here demonstrate with two diseases models, HTT CAG expansion and TTR exon 2 mutation, that highly stringent haplotype-specific editing can be achieved with the CRISPR-hapC and asPAM CRISPR in human model cell lines. As proof-of-concept, in this study we have chosen a universal gRNA, which is adjacent to the disease-causing locus, to demonstrate the haplotype-specific editing by asPAM CRISPRs. As the universal gRNA will target both alleles, an improved strategy of using pair asPAM CRISPRs would further reduce the potential off-target effect. To facilitate the successful application of asPAM CRISPR gene therapy of genetic disorders and to meet the unmet need of gene therapy in dominant disorders, more in vivo studies will be conducted in the future using viral delivery in animal models. In conclusion, we have generated and demonstrated an alternative CRISPR-based haplotype-specific therapy of dominant disorders.
Supplementary Material
ACKNOWLEDGEMENTS
We also thank Dr Fred Dubee for proofreading and providing comments for the improvement of the manuscript.
Author contributions: Yonglun Luo and Lin Lin conceive the idea and oversaw the study. Jiaying Yu, Xi Xiang, Jinrong Huang and Xue Liang contribute equally to the study of experiment and database generation. All other co-authors contributed to the performance of experiments. Jiaying Yu, Xi Xiang, Lars Bolund, Yonglun Luo and Lin Lin drafted the manuscript, and all authors read and revised the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Lundbeck Foundation [R219–2016-1375 to L.L.]; Sanming Project of Medicine in Shenzhen [SZSM201612074 (in part)]; Aarhus University Strategic Grant (AU-iCRISPR); DFF Sapere Aude Starting [8048-00072A to L.L.]. Funding for open access charge: Lundbeck Foundation [R219–2016-1375].
Conflict of interest statement. None declared.
REFERENCES
- 1. Plante-Bordeneuve V., Said G.. Familial amyloid polyneuropathy. Lancet Neurol. 2011; 10:1086–1097. [DOI] [PubMed] [Google Scholar]
- 2. High K., Gregory P.D., Gersbach C.. CRISPR technology for gene therapy. Nat. Med. 2014; 20:476–477. [DOI] [PubMed] [Google Scholar]
- 3. Gori J.L., Hsu P.D., Maeder M.L., Shen S., Welstead G.G., Bumcrot D.. Delivery and specificity of CRISPR-Cas9 genome editing technologies for human gene therapy. Hum. Gene Ther. 2015; 26:443–451. [DOI] [PubMed] [Google Scholar]
- 4. Mollanoori H., Teimourian S.. Therapeutic applications of CRISPR/Cas9 system in gene therapy. Biotechnol. Lett. 2018; 40:907–914. [DOI] [PubMed] [Google Scholar]
- 5. Rabai A., Reisser L., Reina-San-Martin B., Mamchaoui K., Cowling B.S., Nicot A.S., Laporte J.. Allele-Specific CRISPR/Cas9 Correction of a Heterozygous DNM2 Mutation Rescues Centronuclear Myopathy Cell Phenotypes. Mol. Ther. Nucleic Acids. 2019; 16:246–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li P., Kleinstiver B.P., Leon M.Y., Prew M.S., Navarro-Gomez D., Greenwald S.H., Pierce E.A., Joung J.K., Liu Q.. Allele-Specific CRISPR-Cas9 genome editing of the Single-Base P23H mutation for rhodopsin-associated dominant retinitis pigmentosa. CRISPR J. 2018; 1:55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kocher T., Peking P., Klausegger A., Murauer E.M., Hofbauer J.P., Wally V., Lettner T., Hainzl A.M., Bauer J.W. et al.. Cut and Paste: Efficient Homology-Directed Repair of a Dominant Negative KRT14 mutation via CRISPR/Cas9 Nickases. Mol. Ther. 2017; 25:2585–2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gyorgy B., Nist-Lund C., Pan B., Asai Y., Karavitaki K.D., Kleinstiver B.P., Garcia S.P., Zaborowski M.P., Solanes P., Spataro S. et al.. Allele-specific gene editing prevents deafness in a model of dominant progressive hearing loss. Nat. Med. 2019; 25:1123–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gyorgy B., Lööv C., Zaborowski M.P., Takeda S., Kleinstiver B.P., Commins C., Kastanenka K., Mu D., Volak A., Giedraitis V. et al.. CRISPR/Cas9 mediated disruption of the swedish APP allele as a therapeutic approach for Early-Onset Alzheimer's disease. Mol. Ther. Nucleic Acids. 2018; 11:429–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A. et al.. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013; 339:819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gorter de Vries A.R., Couwenberg L.G.F., van den Broek M., de la Torre Cortés P., Ter Horst J., Pronk J.T., Daran J.G.. Allele-specific genome editing using CRISPR-Cas9 is associated with loss of heterozygosity in diploid yeast. Nucleic Acids Res. 2019; 47:1362–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Monteys A.M., Ebanks S.A., Keiser M.S., Davidson B.L.. CRISPR/Cas9 Editing of the mutant huntingtin allele in vitro and in vivo. Mol. Ther. 2017; 25:12–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li Y., Mendiratta S., Ehrhardt K., Kashyap N., White M.A., Bleris L.. Exploiting the CRISPR/Cas9 PAM constraint for single-Nucleotide resolution interventions. PLoS One. 2016; 11:e0144970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Courtney D.G., Moore J.E., Atkinson S.D., Maurizi E., Allen E.H., Pedrioli D.M., McLean W.H., Nesbit M.A., Moore C.B.. CRISPR/Cas9 DNA cleavage at SNP-derived PAM enables both in vitro and in vivo KRT12 mutation-specific targeting. Gene Ther. 2016; 23:108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jiang F., Doudna J.A.. CRISPR-Cas9 Structures and Mechanisms. Annu. Rev. Biophys. 2017; 46:505–529. [DOI] [PubMed] [Google Scholar]
- 16. Moller H.D., Lin L., Xiang X., Petersen T.S., Huang J., Yang L., Kjeldsen E., Jensen U.B., Zhang X., Liu X., Xu X. et al.. CRISPR-C: circularization of genes and chromosome by CRISPR in human cells. Nucleic Acids Res. 2018; 46:e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xiang X., Luo L., Nodzyński M., Li C., Han P., Dou H., Petersen T.S., Liang X., Pan X., Qu K. et al.. LION: a simple and rapid method to achieve CRISPR gene editing. Cell. Mol. Life Sci. 2019; 76:2633–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhou Y., Liu Y., Hussmann D., Brøgger P., Al-Saaidi R.A., Tan S., Lin L., Petersen T.S., Zhou G.Q., Bross P. et al.. Enhanced genome editing in mammalian cells with a modified dual-fluorescent surrogate system. Cell. Mol. Life Sci. 2016; 73:2543–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lin Y.C., Boone M., Meuris L., Lemmens I., Van Roy N., Soete A., Reumers J., Moisse M., Plaisance S., Drmanac R. et al.. Genome dynamics of the human embryonic kidney 293 lineage in response to cell biology manipulations. Nat. Commun. 2014; 5:4767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xie H., Tang L., He X., Liu X., Zhou C., Liu J., Ge X., Li J., Liu C., Zhao J. et al.. SaCas9 requires 5′-NNGRRT-3′ PAM for sufficient cleavage and possesses higher cleavage activity than SpCas9 or FnCpf1 in human cells. Biotechnol. J. 2018; 13:e1700561. [DOI] [PubMed] [Google Scholar]
- 21. Genomes Project, C. Abecasis G.R., Altshuler D., Auton A., Brooks L.D., Durbin R.M., Gibbs R.A., Hurles M.E., McVean G.A.. A map of human genome variation from population-scale sequencing. Nature. 2010; 467:1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Friedland A.E., Baral R., Singhal P., Loveluck K., Shen S., Sanchez M., Marco E., Gotta G.M., Maeder M.L., Kennedy E.M. et al.. Characterization of Staphylococcus aureus Cas9: a smaller Cas9 for all-in-one adeno-associated virus delivery and paired nickase applications. Genome Biol. 2015; 16:257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Anderson E.M., Haupt A., Schiel J.A., Chou E., Machado H.B., Strezoska Ž., Lenger S., McClelland S., Birmingham A., Vermeulen A. et al.. Systematic analysis of CRISPR-Cas9 mismatch tolerance reveals low levels of off-target activity. J. Biotechnol. 2015; 211:56–65. [DOI] [PubMed] [Google Scholar]
- 24. Zheng T., Hou Y., Zhang P., Zhang Z., Xu Y., Zhang L., Niu L., Yang Y., Liang D., Yi F. et al.. Profiling single-guide RNA specificity reveals a mismatch sensitive core sequence. Sci. Rep. 2017; 7:40638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bae S., Park J., Kim J.S.. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014; 30:1473–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stemmer M., Thumberger T., Del Sol Keyer M., Wittbrodt J., Mateo J.L.. CCTop: An intuitive, flexible and reliable CRISPR/Cas9 Target Prediction Tool. PLoS One. 2015; 10:e0124633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Haeussler M., Schönig K., Eckert H., Eschstruth A., Mianné J., Renaud J.B., Schneider-Maunoury S., Shkumatava A., Teboul L., Kent J. et al.. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016; 17:148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tsai S.Q., Zheng Z., Nguyen N.T., Liebers M., Topkar V.V., Thapar V., Wyvekens N., Khayter C., Iafrate A.J., Le L.P. et al.. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015; 33:187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tsai S.Q., Nguyen N.T., Malagon-Lopez J., Topkar V.V., Aryee M.J., Joung J.K.. CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat. Methods. 2017; 14:607–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Paulson H. Repeat expansion diseases. Handb. Clin. Neurol. 2018; 147:105–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Anderson K.E., Eberly S., Marder K.S., Oakes D., Kayson E., Young A., Shoulson I. for the PHAROS Investigators . The choice not to undergo genetic testing for Huntington disease: Results from the PHAROS study. Clin. Genet. 2019; 96:28–34. [DOI] [PubMed] [Google Scholar]
- 32. Kumar A., Kumar S.S., Kumar V., Kumar D., Agarwal S., Rana M.K.. Huntington's disease: an update of therapeutic strategies. Gene. 2015; 556:91–97. [DOI] [PubMed] [Google Scholar]
- 33. Pierzynowska K., Gaffke L., Hać A., Mantej J., Niedziałek N., Brokowska J., Węgrzyn G.. Correction of huntington's Disease Phenotype by Genistein-Induced autophagy in the cellular model. Neuromol. Med. 2018; 20:112–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shin J.W., Kim K.H., Chao M.J., Atwal R.S., Gillis T., MacDonald M.E., Gusella J.F., Lee J.M.. Permanent inactivation of Huntington's disease mutation by personalized allele-specific CRISPR/Cas9. Hum. Mol. Genet. 2016; 25:4566–4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Miller J.R.C., Pfister E.L., Liu W., Andre R., Träger U., Kennington L.A., Lo K., Dijkstra S., Macdonald D., Ostroff G. et al.. Allele-Selective suppression of mutant huntingtin in primary human blood cells. Sci. Rep. 2017; 7:46740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lin L., Luo Y.. Functional evaluation of CRISPR activity by the dual-fluorescent surrogate system: C-Check. Methods Mol. Biol. 2019; 1961:67–77. [DOI] [PubMed] [Google Scholar]
- 37. Ran F.A., Cong L., Yan W.X., Scott D.A., Gootenberg J.S., Kriz A.J., Zetsche B., Shalem O., Wu X., Makarova K.S. et al.. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015; 520:186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mazzeo A., Russo M., Di Bella G., Minutoli F., Stancanelli C., Gentile L., Baldari S., Carerj S., Toscano A., Vita G.. Transthyretin-Related familial amyloid polyneuropathy (TTR-FAP): A Single-Center experience in sicily, an italian endemic area. J. Neuromuscul. Dis. 2015; 2:S39–S48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kavousanaki M., Tzagournissakis M., Zaganas I., Stylianou K.G., Patrianakos A.P., Tsilimbaris M.K., Mantaka A., Samonakis D.N.. Liver transplantation for familial amyloid polyneuropathy (Val30Met): Long-Term Follow-up prospective study in a nontransplant center. Transplant. Proc. 2019; 51:429–432. [DOI] [PubMed] [Google Scholar]
- 40. Hsiau T., Conant D., Rossi N., Maures T., Waite K., Yang J., Joshi S., Kelso R., Holden K., Enzmann B.L. et al.. Inference of CRISPR edits from sanger trace data. 2019; bioRxiv doi:10 August 2019, preprint: not peer reviewed 10.1101/251082. [DOI] [PubMed]
- 41. Gigante S., Gouil Q., Lucattini A., Keniry A., Beck T., Tinning M., Gordon L., Woodruff C., Speed T.P., Blewitt M.E. et al.. Using long-read sequencing to detect imprinted DNA methylation. Nucleic Acids Res. 2019; 47:e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ardui S., Ameur A., Vermeesch J.R., Hestand M.S.. Single molecule real-time (SMRT) sequencing comes of age: applications and utilities for medical diagnostics. Nucleic Acids Res. 2018; 46:2159–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kosicki M., Tomberg K., Bradley A.. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018; 36:765–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liu M., Rehman S., Tang X., Gu K., Fan Q., Chen D., Ma W.. Methodologies for improving HDR Efficiencsy. Front. Genet. 2018; 9:691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Aird E.J., Lovendahl K.N., St Martin A., Harris R.S., Gordon W.R.. Increasing Cas9-mediated homology-directed repair efficiency through covalent tethering of DNA repair template. Commun. Biol. 2018; 1:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Aksoy Y.A., Nguyen D.T., Chow S., Chung R.S., Guillemin G.J., Cole N.J., Hesselson D.. Chemical reprogramming enhances homology-directed genome editing in zebrafish embryos. Commun. Biol. 2019; 2:198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Riesenberg S., Maricic T.. Targeting repair pathways with small molecules increases precise genome editing in pluripotent stem cells. Nat. Commun. 2018; 9:2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Song J., Yang D., Xu J., Zhu T., Chen Y.E., Zhang J.. RS-1 enhances CRISPR/Cas9- and TALEN-mediated knock-in efficiency. Nat. Commun. 2016; 7:10548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Robert F., Barbeau M., Éthier S., Dostie J., Pelletier J.. Pharmacological inhibition of DNA-PK stimulates Cas9-mediated genome editing. Genome Med. 2015; 7:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhang J.P., Li X.L., Li G.H., Chen W., Arakaki C., Botimer G.D., Baylink D., Zhang L., Wen W., Fu Y.W. et al.. Efficient precise knockin with a double cut HDR donor after CRISPR/Cas9-mediated double-stranded DNA cleavage. Genome Biol. 2017; 18:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gutierrez-Triana J.A., Tavhelidse T., Thumberger T., Thomas I., Wittbrodt B., Kellner T., Anlas K., Tsingos E., Wittbrodt J.. Efficient single-copy HDR by 5′ modified long dsDNA donors. Elife. 2018; 7:e39468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lee K., Mackley V.A., Rao A., Chong A.T., Dewitt M.A., Corn J.E., Murthy N.. Synthetically modified guide RNA and donor DNA are a versatile platform for CRISPR-Cas9 engineering. Elife. 2017; 6:e25312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jayavaradhan R., Pillis D.M., Goodman M., Zhang F., Zhang Y., Andreassen P.R., Malik P.. CRISPR-Cas9 fusion to dominant-negative 53BP1 enhances HDR and inhibits NHEJ specifically at Cas9 target sites. Nat. Commun. 2019; 10:2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lin L., Petersen T.S., Jensen K.T., Bolund L., Kühn R., Luo Y.. Fusion of SpCas9 to E. coli Rec A protein enhances CRISPR-Cas9 mediated gene knockout in mammalian cells. J. Biotechnol. 2017; 247:42–49. [DOI] [PubMed] [Google Scholar]
- 55. Charpentier M., Khedher A.H.Y., Menoret S., Brion A., Lamribet K., Dardillac E., Boix C., Perrouault L., Tesson L., Geny S.. CtIP fusion to Cas9 enhances transgene integration by homology-dependent repair. Nat. Commun. 2018; 9:1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Christie K.A., Courtney D.G., DeDionisio L.A., Shern C.C., De Majumdar S., Mairs L.C., Andrew Nesbit M., Tara Moore C.B.. Towards personalised allele-specific CRISPR gene editing to treat autosomal dominant disorders. Sci. Rep. 2017; 7:16174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Smith C., Abalde-Atristain L., He C., Brodsky B.R., Braunstein E.M., Chaudhari P., Jang Y.Y., Cheng L., Ye Z.. Efficient and allele-specific genome editing of disease loci in human iPSCs. Mol. Ther. 2015; 23:570–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Keough K.C., Lyalina S., Olvera M.P., Whalen S., Conklin B.R., Pollard K.S.. AlleleAnalyzer: a tool for personalized and allele-specific sgRNA design. Genome Biol. 2019; 20:167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E.. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012; 337:816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.