


Abstract
Tight and coordinate regulation of virulence determinants is essential for bacterial biology and involves dynamic shaping of transcriptional regulatory networks during evolution. The horizontally transferred two-partner secretion system ExlB–ExlA is instrumental in the virulence of different Pseudomonas species, ranging from soil- and plant-dwelling biocontrol agents to the major human pathogen Pseudomonas aeruginosa. Here, we identify a Cro/CI-like repressor, named ErfA, which together with Vfr, a CRP-like activator, controls exlBA expression in P. aeruginosa. The characterization of ErfA regulon across P. aeruginosa subfamilies revealed a second conserved target, the ergAB operon, with functions unrelated to virulence. To gain insights into this functional dichotomy, we defined the pan-regulon of ErfA in several Pseudomonas species and found ergAB as the sole conserved target of ErfA. The analysis of 446 exlBA promoter sequences from all exlBA+ genomes revealed a wide variety of regulatory sequences, as ErfA- and Vfr-binding sites were found to have evolved specifically in P. aeruginosa and nearly each species carries different regulatory sequences for this operon. We propose that the emergence of different regulatory cis-elements in the promoters of horizontally transferred genes is an example of plasticity of regulatory networks evolving to provide an adapted response in each individual niche.
INTRODUCTION
Horizontal gene transfer (HGT) is a major mechanism for the evolution of Prokaryotes facilitating their ability to adapt to specific environmental niches. The majority of genes expressed by bacteria are regulated at the transcriptional level through the action of transcription factors (TFs). In many instances, the genes encoding TFs and their target regulated genes are genetically linked; consequently, they are acquired as a single unit during HGT (1). However, the environment of the recipient, whether they are distinct species or even different strains of the same species, may be sufficiently different from that of the donor to make it necessary to rewire the regulation of the acquired structural genes and integrate them into existing regulatory networks. Mutations in the regulatory sequences near the promoters of horizontally acquired genes readily alter the specificity of recognition by the TFs and represent a simple way of placing them under the control of new regulatory elements that respond to input signals of the specific environment.
A new taxonomic group of the human pathogen P. aeruginosa has recently emerged, characterized by major features in their virulence factor repertoire; namely the absence of several important toxins, including the Type III Secretion System (T3SS) effectors and the associated secretion and regulatory machinery (2). Instead, they express ExlB–ExlA, a Two-Partner Secretion (TPS) system secreting a potent cytotoxin (3). ExlB (PSPA7_4641) is the cognate outer membrane transporter of the 172 kDa pore-forming cytotoxin, the Exolysin ExlA (PSPA7_4642) (4). In strains harboring the exlBA operon, apparent genetic scars at the T3SS-encoding locus can be identified, suggesting an unfavorable functional incompatibility between the two secretion systems or their respective exported toxins resulting in the evolutionary selection of a single secretion system (5). Whole-genome-based population studies demonstrated that the exlBA operon is present in two distinct phylogenetic groups, one sharing an average nucleotide identity (ANI) of ∼98% with the T3SS+ major group, and another representing clonal outliers with an ANI of ∼93% (2,5–8). The current cohort of strains with the exlBA operon and lacking the T3SS-encoding genes comprises isolates found in the environment or recovered from both acute and chronic human infections (5,6,9,10).
The presence of the exlBA operon in specific P. aeruginosa phylogenetic groups, as well as in some other Pseudomonas species, implies its acquisition by HGT and therefore its expression might be controlled by TFs of the recipients, found at other locations on the chromosome. We recently investigated the exlBA regulation in the human urinary tract isolate P. aeruginosa IHMA879472 (IHMA87 (11)). We showed that the operon is under direct control of the global regulator Vfr, a member of the cyclic AMP receptor (CRP) family, which together with the co-activator cAMP stimulates exlBA expression (12). The consensus recognition sites for the CRP proteins in different bacterial species, including the P. aeruginosa Vfr (13), are well conserved and can be identified immediately upstream of the exlBA core promoter. This sequence is required for the expression of exlBA and was shown to specifically bind Vfr (12). Therefore, after the acquisition of the exlBA operon by HGT, it became part of the global cAMP/Vfr regulatory network that controls the expression of a number of virulence factors and biofilm determinants in P. aeruginosa. Here, we further probed the regulatory mechanisms controlling exlBA expression by attempting to identify additional regulators, assess their distribution and function in several P. aeruginosa groups and compare these to other Pseudomonas species.
MATERIALS AND METHODS
Bacterial strains
The bacterial strains used in this study are listed in Supplementary Table S5. P. aeruginosa and E. coli strains were grown in Lysogeny Broth (LB) at 37°C under agitation. P. chlororaphis, P. protegens and P. putida were cultivated at 28°C. P. aeruginosa strains were selected on LB plates supplemented with 25 μg/ml irgasan. Antibiotics for P. aeruginosa were added when needed at the following concentrations: 75 μg/ml gentamicin and 75 μg/ml tetracycline. For P. chlororaphis, 25 μg/ml rifampicin and 25 μg/ml gentamicin were used.
Genome sequencing and assembly
The genome of P. aeruginosa IHMA87 was sequenced using Illumina HiSeq (11) and completed with PacBio (Base Clear, Leiden, Netherlands) technology. Reads from both platforms were assembled using the hybrid assembler Unicycler version 0.4.0 (14) in normal mode to obtain two circular contigs with an average read depth of 136.5X. Genome annotation was carried out using Prokka version 1.12 (15) and annotation was manually curated to include or correct known gene names. The average nucleotide identity (ANI) between the chromosomes of PA7 and IHMA87 was calculated as the OrthoANIu value (16), while the synteny between the two genomes was identified and visualized using Mauve version snapshot_2015-02-13 by aligning them using the progressive Mauve algorithm with default parameters (17).
Transposon mutagenesis
A transposon mutant library was constructed in P. aeruginosa IHMA87 exlBA::lacZ using the Himar-1 mariner transposon on pBTK24 plasmid, which carries an outward-directed Ptac promoter, making it able to either disrupt or overexpress adjacent genes. The library was generated by triparental mating the P. aeruginosa strain with the E. coli donor strain carrying pBTK24 and a pRK2013-containing helper strain. After overnight culture on LB agar plates with appropriate antibiotics, P. aeruginosa and E. coli were resuspended in LB at OD600 = 1. After incubation of P. aeruginosa at 42°C without agitation for 2 h, the three strains were then combined at a 1:2:2 recipient-to-donor/helper ratio, concentrated 30× and spotted for a total of 16 50-μl puddles on LB agar plates. After 4 h of incubation at 37°C (allowing one bacterial doubling, as checked by CFU counting), the puddles were scraped off, pooled and stored at −80°C. The mutant library size was estimated at 100 000 mutants by CFU counting, allowing a complete coverage of the genome (transposon insertions every 65 bp on average). Blue colonies were isolated after plating on LB agar plate containing 25 μg/ml irgasan, 75 μg/ml gentamicin and 40 μg/ml 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-Gal) and overnight incubation at 37°C followed by 48 h incubation at 4°C. Among 236 mutants selected on plates and re-tested in an ONPG-based β-galactosidase activity assay, 36 displayed at least a 2-fold increase of the reporter activity in comparison to the parental strain. Transposon insertion regions in these candidate mutants were amplified by semi-random PCR and sequenced, leading to the identification of 13 genes potentially involved in exlBA regulation (Supplementary Table S1). All primers are listed in Supplementary Table S6.
Cell culture and cytotoxicity assay
A549 epithelial cells (ATCC CCL-185) were grown in Roswell Park Memorial Institute medium (RPMI) 1× supplemented with 10% fetal bovine serum (FBS) at 37°C, 5% CO2. Cells were seeded in 96-well plates (50 000 cells/well, 200 μl/well). One hour before infection, the medium was replaced with non-supplemented RPMI medium. Ten minutes before infection, it was supplemented with 0.25 μM Syto24 (Thermofisher) and 0.5 μg/ml Propidium Iodide (PI). In parallel, bacteria were diluted to OD600 = 0.1 from 16 h-grown liquid cultures and further incubated for 3 h. For the infection, bacterial cultures at OD600 = 1.0 were diluted and added to monolayers to fit the multiplicity of infection (MOI) of 10. Live-cell microscopy and image analysis were done using an Incucyte S3 Live-cell Imaging System (Essen Bioscience). Total cell population was automatically counted at T = 0 in each well by monitoring the Syto24 labeling and further used for the normalization of the PI incorporation data. The number of cells with incorporated PI was counted every 45 min. Areas under the curves were estimated from PI incorporation curves by linear trapezoidal integration.
Routine cytotoxicity assays were performed by measuring only the PI fluorescence (excitation 544 nm/emission 590 nm) every 10 min with Fluoroskan Ascent FL2.5 (Thermo Corporation), during 8 h at 37°C. Cell shrinkage assays were performed as described previously (18). Briefly, A549 cells were stained with Dii dye (Life Technologies) for 1 h then washed and placed in RPMI containing 10% FBS before infection. Dii staining detection was done by image acquisition every hour on an Incucyte S3 Live-cell Imaging System. Percent Red Object Confluence in RED channel was measured to quantify cell surface and normalized to t0 to obtain cell shrinkage values.
Galleria mellonella infection assay
Larvae of G. mellonella were obtained from Sud-Est Appâts (Queige, France) and kept in a dark container at room temperature. White larvae of 2.5- to 3-cm size were selected and kept until infection the next day. Pseudomonas strains were diluted to OD600 = 0.1 from overnight cultures and grown in LB under agitation until OD600 = 1 was reached. Bacteria were then pelleted and resuspended in sterile PBS and diluted to ∼ 5.10−1 bacteria/μl or ∼ 6.103 bacterial/μl for P. aeruginosa and P. chlororaphis, respectively. An insulin pen (HumaPen Luxura, Lilly Nederland) was used to inject 10 μl of bacterial suspension to the last proleg of the larvae. Animals injected with sterile PBS served as a control for physical trauma. CFUs for each dilution were systematically counted from the insulin pen to ensure proper bacterial loads. Forty larvae were injected per condition. Infection development was followed for 24 h at 37 or 30°C for P. aeruginosa or P. chlororaphis, respectively, and the animals were considered dead when they failed to react to touch. Strains were independently randomized and counting was blinded and done by another person, ensuring no bias in spotting and counting of CFU, as well as in counting dead larvae. Statistical significance was assessed using a Log-rank test.
RNA isolation
Total RNA was isolated from liquid cultures at OD600 = 1 using the hot phenol–chloroform extraction method. Briefly, after cell lysis in hot Lysis-phenol solution (40 mM sodium acetate, 1% SDS, 2 mM EDTA in acid phenol solution), RNA was isolated by sequential phenol–chloroform extractions and ethanol precipitation. Residual DNA was removed by treatment with DNAse following manufacturer’s instructions (Invitrogen). Quantification of RNA was done using a QuBit 3.0 Fluorimeter. RNA sample quality was then assessed on an Agilent Bioanalyzer, yielding RINs of 9 or higher.
Construction of libraries for RNA-seq, sequencing and data analysis
After RNA isolation and DNAse treatment, ribosomal RNAs were depleted using the Ribo-Zero rRNA Removal Kit (Illumina) following manufacturer’s instructions. The cDNA libraries were constructed from 50 ng of depleted RNA using the NEBNext Ultra II Directional RNA library prep kit following manufacturer’s instructions (NEB). Libraries were size-selected to 200–700 bp fragments using SPRIselect beads, and quality was assessed on the Agilent Bioanalyzer using High Sensitivity DNA chips. Sequencing was performed at the Biopolymers Facility at Harvard Medical School on an Illumina NextSeq500. Approximately 18 million single-end 50 bp reads per sample were generated on average. More than 90% of reads were uniquely aligned for each sample to the IHMA87 or PAO1 genomes using Bowtie2 (19). Read counts per feature were then obtained with htseq-count (20). Differential gene expression between mutant and wild-type strains was assessed using DESeq2 (21). RNA-seq was performed in biological duplicates for each strain.
Chromatin immuno-precipitation
A VSV-G tag-encoding sequence was inserted in the erfA gene on the chromosomes of IHMA87 and PAO1 by two-step allelic exchange with the pEXG2-erfA-VSVG and pEXG2-PA0225-VSVG plasmids, respectively. The correct production of C-terminal VSV-G-tagged ErfA proteins in the corresponding IHMA87 ErfA-VSVG and PAO1 ErfA-VSVG strains was assessed by immunoblotting, and the correct activity of the tagged protein was verified by measuring the exlA::lacZ transcriptional fusion activity of a strain producing ErfA-VSV-G (Supplementary Figure S3). Each strain was grown in 30 ml LB medium from overnight cultures diluted to OD600 = 0.1 at 37°C under agitation. At OD600 = 1, 600 μl of 37% formaldehyde solution was added and samples were incubated for 10 min at 22°C on a rotating wheel. Crosslinking was stopped by the addition of 3 ml of sterile 1.5 M glycine and further incubation for 10 min at 22°C on a rotating wheel. Cells were collected by centrifugation and washed two times with sterile PBS before resuspension in 600 μl of Lysis Buffer (50 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, pH 7.4, containing EDTA-free Roche protease inhibitor cocktail). Cell lysis and DNA fragmentation to 100–800 bp were done by sonication in a Qsonica bath sonicator for 8 min at 70% of amplitude. Lysates were cleared by centrifugation. Anti-VSV-G antibodies (Sigma) were bound on magnetic Protein A beads according to manufacturer’s instructions (Dynabeads Protein A, Invitrogen) and further washed in Lysis Buffer. Cleared lysates were first incubated with bare beads only for 30 min at 4°C on a rotating wheel to prevent excess of non-specific binding to the beads in the subsequent steps. After removal of the bare beads, the lysates were incubated overnight at 4°C on a rotating wheel with antibodies bound to the beads. Beads were then washed three times using the provided Washing Buffer (Invitrogen) and transferred into a new tube after the last wash. Supernatants were removed and 70 μl of Elution Buffer (50 mM Tris-HCl, 10 mM EDTA, 1% SDS, pH 7.5) added to the beads and incubated for 10 min at 70°C under vigorous agitation (600 rpm). About 1 μl of 100 mg/ml RNase A was added to the supernatants followed by incubation for 30 min at 65°C, after which 5 μl of 20 mg/ml Proteinase K were added for another 1 h incubation at 50°C. The crosslinks were reversed by incubating the samples for 12 h at 65°C. Sample volumes were then adjusted to 100 μl with nuclease-free water before addition of 3 μl of 3 M sodium acetate (pH 5). DNA was purified on Monarch DNA Clean Up columns (NEB). ChIP was done in biological duplicates for each strain with wild-type strains serving as controls. Before constructing the libraries, each sample was tested for proper ErfA expression by western immunoblotting and DNA fragmentation by purification of DNA after lysis and agarose gel electrophoresis.
ChIP-seq library construction, sequencing and data analysis
After ChIP, DNA was quantified on a QuBit 3.0 Fluorimeter and 3.5 ng of DNA was used to construct libraries using the NEBNext Ultra II DNA library prep kit (NEB). The quality of DNA libraries was assessed on an Agilent Bioanalyzer. Sequencing was performed at the Biopolymers Facility at Harvard Medical School on an Illumina NextSeq500, generating an average of 18 million single-end 50 bp reads per sample with >90% of reads from each sample uniquely aligning to the IHMA87 or PAO1 genomes using Bowtie2 (19). Peak calling was done using MACS2 (22) for each duplicate against the two corresponding negative control samples. Peaks found in both replicates were selected using the Intersect tool from BEDTools (23) with a minimum overlap of 50% on each compared peaks. The closest gene and distance from it were identified for each peak using the ClosestBed tool from BEDTools (23). MEME-ChIP (24) was used to find motifs, using the 100 bp DNA regions centered on the summits of all detected peaks.
Electrophoretic mobility shift assay
Target DNA regions were amplified by PCR using Cy5-labeled primers and purified on DNA Clean up columns (NEB). The resulting 80-bp DNA probes were incubated at 0.5 nM for 5 min at 37°C in EMSA Buffer (10 mM Tris-HCl, 50 mM KCl, 10 mM MgCl2, 10% glycerol, 0.1 mg/ml BSA, pH 8) containing 25 ng/μl poly(dI-dC). For competition assays, 100 nM unlabeled DNA probes (200-fold excess) were incubated with the labeled probes. ErfA protein was added at the indicated concentrations in a final reaction volume of 20 μl and incubated for an additional 15 min at 37°C. Samples were then loaded on a native 5% Tris-borate (TB) polyacrylamide gel and run at 100 V and 4°C in cold 0.5× TB Buffer. Fluorescence imaging was performed using a Chemidoc MP.
RT-qPCR
After total RNA isolation and DNase treatment, cDNA synthesis was carried out using 2 μg of RNA with the SuperScript IV first-strand synthesis system (Invitrogen) in the presence or absence of reverse transcriptase to assess the absence of genomic DNA. The CFX96 real-time system (Bio-Rad) was used to PCR amplify the cDNA, and the quantification was based on use of SYBR green fluorescent molecules. About 2 μl of cDNA were incubated with 10 μl of Luna Universal qPCR 2X Master Mix (NEB) and forward and reverse specific primers at final concentrations of 250 nM in a total volume of 20 μl. The real-time PCR was done according to manufacturer’s instructions. To generate standard curves for each pair of primers, serial dilutions of the cDNA were used. The experiments were performed with three biological replicates for each strain, and the relative expression of mRNAs was analyzed with the CFX Manager software (Bio-Rad) using the Pfaffl method relative to rpoD reference Cq values. Statistical analyses were performed by T-test. The sequences of primers are listed in Supplementary Table S6.
DAP-seq
To construct DNA libraries, genomic DNA (gDNA) was extracted from overnight cultures of P. aeruginosa IHMA87 and PAO1, P. chlororaphis PA23, P. putida KT2440 and P. protegens CHA0 using the GenElute Bacterial Genomic DNA kit (Sigma). Purified gDNAs were fragmented to 100–500 bp by sonication in a Qsonica bath sonicator for 6 min at 70% of amplitude. DNA end-repair was performed on 5 μg fragmented DNA using the NEBNext End-Repair Module (NEB). The dA-tailing was then performed using the NEBNext dA-tailing Module (NEB). Truncated Y adaptors were annealed by mixing adaptors A and B (Supplementary Table S6) in sterile water to a final concentrations of 30 μM each and incubating at 96°C for 2 min before allowing the samples to cool down to room temperature. Ligation of Y adaptors to the dA-tailed libraries was performed using T4 DNA Ligase (NEB) following manufacturer’s instructions. DNA was purified on Monarch Clean Up columns between each of these steps. The quality of the libraries was checked on High Sensitivity DNA chips on an Agilent Bioanalyzer and by PCR tests with primers specific to adaptors A and B before and after adaptors ligation.
DAP-seq experiments were conducted in triplicates, and negative controls were performed by not adding any protein to the beads. About 20 μl of Dynabeads His-Tag Isolation and Pulldown magnetic beads (Invitrogen) were washed three times in 500 μl of Binding Buffer (sterile PBS containing 0.01% Tween20). About 500 ng of His-tagged ErfA protein was diluted in 500 μl of Binding Buffer and incubated for 20 min with the washed beads on a rotating wheel at room temperature. After binding, the bead–protein complexes were washed six times in 500 μl of Binding Buffer and then resuspended in 80 μl of Binding Buffer containing 50 ng of adaptor-ligated gDNA libraries and further incubated on a rotating wheel at room temperature for 1 h. The bead–protein–DNA complexes were washed six times in 500 μl of Binding Buffer to eliminate unbound DNA before transferring into a new tube following the final wash. The beads were then resuspended in 25 μl of sterile 10 mM Tris-HCl pH 8.5, and incubated for 10 min at 98°C for elution. After incubation, the samples were placed on ice for 5 min, beads were magnetically removed and the released DNA was used for PCR amplification as previously described (25), using a different indexed pair of primers in each sample to allow pooling for sequencing. PCR products were purified using SPRIselect beads at a 1:1 ratio. Quality of each library was assessed using High Sensitivity DNA chips on an Agilent Bioanalyzer.
DAP-seq sequencing and data analysis
Sequencing was performed by the high-throughput sequencing core facility of I2BC (Centre de Recherche de Gif – http://www.i2bc.paris-saclay.fr) using an Illumina NextSeq500 instrument. Approximately 5 million single-end 75 bp reads per sample were generated on average with >90% of reads uniquely aligning to the IHMA87, PAO1, PA23, CHA0 or KT2440 genomes using Bowtie2 (19). Peak calling was done using MACS2 (22) for each triplicate against the three corresponding negative control samples. Peaks found in the three replicates were selected using the Intersect tool from BEDTools (23) with a minimum overlap of 50% on each compared peaks. The closest gene and distance from it were identified for each peak using the ClosestBed tool from BEDTools (23).
exlBA promoter analysis
The ExlA sequence from P. aeruginosa PA7 was searched against the 4846 Pseudomonas genomes from the Pseudomonas database version 18.1 (http://pseudomonas.com, (26)) using a best bi-directional blast hits approach. A total of 446 Pseudomonas strains were found to possess an exlA homolog in their genomes, with BLASTp coverage >95% and sequence identity >30%. In these strains, the presence of an exlB-5′ adjacent gene was confirmed and the 200 bp sequence upstream of exlB start codon was retrieved using Python scripts. A multiple alignment was done between the 446 obtained sequences using Clustal Omega (27) with 5 guide-tree and HMM iterations. The corresponding phylogenetic tree was visualized and annotated with iTOL (28). The presence of erfA, ergAB and vfr homologs in the corresponding strains was assessed using P. aeruginosa PA7 protein sequences as a query, with coverage >95% and sequence identity >30%. The presence of putative transcription factor binding sites in the 200-bp sequences upstream from the exlB start codon was determined using RSAT matrix-scan (29) with default parameters using either Vfr core consensus binding motif (5′-TGNGANNNAGNTCACAT-3′) or ErfA binding motif from ChIP-seq results. Core promoter predictions were performed using BPROM (30). Conservation rates of PexlBA, exlB and exlA nucleotide sequences were obtained using pairwise alignments on all 446 corresponding sequences with Clustal Omega.
See Supplementary Data for Materials and Methods section on genetic manipulations, β-galactosidase activity assays, proteins purifications and western blot analysis.
RESULTS
exlBA promoter sequence shows differences in several Pseudomonas species
A number of Pseudomonas species carry the exlBA operon; this provides an opportunity to examine the evolution of regulatory pathways following acquisition of genes by HGT. Toward this goal, we analyzed the 5′ regulatory sequences at the exlBA promoters in four representative species carrying exlBA. A search of these sequences using Regulatory Sequence Analysis Tools (RSAT) ((31); http://embnet.ccg.unam.mx/rsa-tools/) identified the Vfr-binding site only in the exlBA-carrying lineage of P. aeruginosa and not in other Pseudomonas (Figure 1), although orthologues encoding Vfr (CRP) are found in all species. Binding sites for various TFs have been identified in all sequences; however, none is shared by any two species. Therefore it appears that, in these four species, the exlBA operon might be regulated differently, reflecting a potential evolution of different regulatory sequences in each of them.
Figure 1.
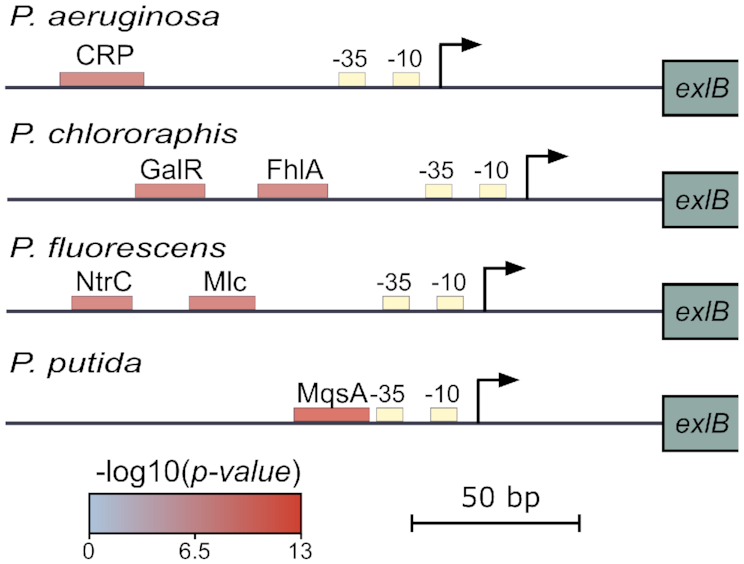
exlBA promoters display diverse predicted cis-regulatory elements across Pseudomonas species. Schematic representation of predicted cis-regulatory elements in the exlBA promoter regions of P. aeruginosa PA7, P. chlororaphis PA23, P. fluorescens Pt14 and P. putida KT2440. Core promoter sequences indicated as ‘-10’ and ‘-35’ boxes were identified by BPROM (30). Predicted binding sites for TFs (CRP, GalR, FhlA, NtrC, Mlc and MqsA) are indicated as boxes colored according to the P-value for each predicted site using RSAT matrix-scan (29).
Screen for regulators of exlBA identifies a novel repressor
To further characterize exlBA regulation, we built and screened a comprehensive transposon mutant library in P. aeruginosa IHMA87 carrying a lacZ gene incorporated into the exlA gene, providing a transcriptional readout for mutants with altered expression of the reporter (see ‘Materials and Methods’ section for details). The location of transposon insertions in 13 candidate genes that resulted in an increased expression of the lacZ reporter was determined (Supplementary Table S1). The insertion with the highest effect on expression was in the gene IHMA87_00215 (PSPA7_0311), encoding an uncharacterized transcriptional regulator containing a Cro/CI-type DNA binding domain, which we named ErfA, for Exolysin Regulatory Factor A. We deleted this gene from the chromosome of P. aeruginosa IHMA87 and confirmed that the loss of erfA gene product leads to a significant increase in the transcription of the exlBA operon and that the various phenotypic changes expected from the rise in ExlA production are correspondingly enhanced. In the ΔerfA strain, we observed an approximately 40-fold increase in β-galactosidase activity of the exlA::lacZ transcriptional fusion, which was restored to the wild-type levels by trans-complementation (Figure 2A). The increase in transcription of exlA in the erfA mutant was accompanied by an increase of both ExlA synthesis and secretion (Figure 2B and C), and consequently of bacterial cytotoxicity during infection of epithelial cells (Figure 2D and E). To assess the impact of de-repression of Exolysin expression in vivo, we compared the infectivity of strains in a wax moth Galleria mellonella model of infection. Here again, the erfA mutant displayed faster killing kinetics of the larvae than the wild-type and complemented strains (Figure 2F) indicating that ErfA negatively regulates ExlA-dependent virulence of IHMA87 in vivo. Finally, to test whether ErfA plays the same inhibitory role in the second group of P. aeruginosa strains phylogenetically closer to the T3SS+ groups (5,8), we deleted the erfA gene in PA70, a strain isolated from expectoration of a non-CF bronchiectasis patient, and in AL-198, a CF isolate. Both mutants exhibited higher level of exlB and exlA expression, and even the completely innocuous PA70 strain became cytotoxic when ErfA was absent (Figure 2G–I), showing the conserved function of ErfA on exlBA repression across two subgroups of P. aeruginosa strains.
Figure 2.

ErfA regulates ExlA-dependent virulence through inhibition of exlBA expression in P. aeruginosa. (A) β-Galactosidase activities of IHMA87 strains harboring exlBA::lacZ. The strains were grown in LB, and activities of the transcriptional fusion were measured at OD600 = 1. The enzyme activities are represented as mean ± standard deviation (SD) from three independent experiments. (B andC) Immunodetection of ExlA in bacterial cell extracts (B) and supernatants (C). Whole bacteria and culture supernatants were sampled and analyzed by SDS-PAGE for ExlA, ErfA, EF-Tu and FliC content using appropriate antibodies. (D) Kinetics of bacterial cytotoxicity on epithelial cells. A549 epithelial cells grown in 96-well plates were infected with indicated strains at a multiplicity of infection (MOI) of 10 in the presence of propidium iodide (PI). PI incorporation, which reflects membrane permeabilization, was monitored every 45 min by automated live-cell microscopy and normalized to the total number of cells measured at the start of the experiment by Syto24 staining. Data are represented as mean ± s.e.m. from three independent experiments. (E) The areas under each curve presented in (D) were calculated from the values obtained after 380 min of infection. ns: not significant. (F) Survival curves of Galleria mellonella larvae infected with an average of 5 bacteria per larva. Forty larvae were infected per strain. Significance testing was performed using log-rank test (P < 0.05). (G) RT-qPCR analysis of exlA expression in PA70 and AL-198 strains and erfA isogenic mutants. All strains were grown in LB medium to OD600 = 1. Expressions were normalized to the abundance of rpoD mRNA. Error bars indicate the SD. The P-value was determined using one-tailed T-test and is indicated by * (P < 0.05) or ** (P < 0.01). (H andI) Kinetics of PI incorporation (A.U.: Arbitrary Unit) into A549 epithelial cells infected at a MOI of 10 with PA70 (H) and AL-198 (I) strains and isogenic erfA mutants.
The binding repertoire of ErfA in P. aeruginosa
An ortholog of erfA can be identified in the genomes of P. aeruginosa strains lacking exlBA, including the reference strain PAO1 (PA0225; www.pseudomonas.com (26)). We therefore assumed that ErfA might have other regulatory targets in these strains. In order to apply genome-wide approaches toward the identification of genes under ErfA control, we sequenced and assembled the genome of IHMA87, which represents the third closed genome of the PA7-like clade after PA7 and CR1 (6,32). In addition to a 6.53 Mb chromosome, IHMA87 harbors a 185 kb mega plasmid (Supplementary Figure S1). With an average nucleotide identity of 98.95% between them, the genomes of PA7 and IHMA87 are also generally syntenic (Supplementary Figure S2).
To examine the global role of ErfA in IHMA87 (exlBA+) and PAO1 (T3SS+), we combined RNA-seq and ChIP-seq approaches. A comparison of the transcriptomes of wild-type and erfA mutants identified 8 and 2 genes that were differentially expressed (log2(fold change)>2) in IHMA87 and PAO1, respectively (Figure 3A and B; Supplementary Table S2). ChIP-seq performed with a functional VSV-G-tagged ErfA (Supplementary Figure S3) led to the identification of 2 and 1 regions exhibiting significant enrichment in IHMA87 and PAO1, respectively (Figure 3C–E and Supplementary Table S2). The analysis of all detected peaks allowed us to identify a conserved 17-bp palindromic motif (5′-ATGACACntnGTGTCAT-3′) as a likely ErfA DNA-binding site (EBS) (Figure 3F). In IHMA87, the second highest peak observed by ChIP-seq was centered on the exlBA promoter, showing that ErfA directly exerts its negative control on the operon. In addition to exlBA, one additional region was enriched in ChIP-seq in both strains, corresponding to the intergenic region between erfA and a two-gene operon transcribed in the opposite direction. These two genes (PA0224/IHMA87_00214 and PA0223/IHMA87_00213) were also the most upregulated genes in the erfA mutants in RNA-seq, as confirmed by RTqPCR (Figure 3G,H), and we propose to name them ergA and ergB for ErfA regulated gene A and B. Mutagenesis experiments showed that ErfA binding on this EBS only slightly diminished erfA expression (Supplementary Figure S4), excluding any auto-regulatory mechanism, as suggested by the location of ErfA binding. Overall, exlBA and ergAB were the only two ErfA targets found in both RNA-seq and ChIP-seq experiments. In addition, genes encoding proteins of the T3SS, including PopB and PopD translocators and exotoxin ExoT, were found slightly downregulated in PAO1 erfA mutant. The downregulation was confirmed for popB by RTqPCR and analysis of a PpopN-lacZ transcriptional fusion, although it has no impact on cytotoxicity toward epithelial cells (Supplementary Figure S5). This slight regulatory effect seems to be indirect and would need further investigation. Therefore, we conclude that ErfA directly regulates two operons: exlBA and ergAB.
Figure 3.

ErfA directly regulates a second operon in addition to exlBA. (A andB) Volcano plots displaying the RNA-seq results of the genes differentially expressed in respective ΔerfA mutants versus IHMA87 (A) and PAO1 (B) wild-type strains. Genes with q-value < 0.05 are depicted in red (IHMA87) or orange (PAO1). (C–E) Enrichment of normalized mapped reads after ChIP-seq on the whole IHMA87 chromosome (C) and IHMA87 plasmid (D), and the PAO1 genome (E). (F) Enriched DNA motif obtained with MEME-ChIP on relevant ChIP-seq peaks corresponding to near-summit regions. (G and H) RT-qPCR analysis of ergA, ergB, exlA and exlB mRNA levels in wild-type, erfA mutant and complemented strains in IHMA87 (G) and PAO1 (H). Experiments were performed in triplicates, with RNA extracted from bacteria at OD600 = 1 in LB and normalized to the rpoD transcript. Error bars indicate the SD. (I) Enrichment of normalized mapped reads after ChIP-seq and location of ErfA binding site on the 7.5 kb region encompassing exlBA. Black arrows indicate transcription start sites. The position of the summit of the ChIP-seq peak is denoted as a bold bright red letter. Bases changed in the PexlEBS2 mutation of the binding site are shown. (J andK) Electrophoretic mobility shift assay of ErfA on exlBA promoter (PexlBA). Recombinant ErfA-His10 protein (0–200 nM) was incubated with 0.5 nM Cy5-labeled PexlBA 80-mer probe (J) or the mutated Cy5-PexlEBS2 probe (K) for 15 min before electrophoresis. For competition assays, excess of unlabeled PexlBA or PexlEBS2 probes (100 nM) are denoted ‘+’ for the corresponding probe. (L) β-Galactosidase activities of the wild-type (WT) and ΔerfA strains harbouring exlBA::lacZ transcriptional fusion. The strain IHMA87exlBA::lacZ PexlBA-EBS2 carries the PexlEBS2 mutation indicated in I on ErfA binding site. Experiments were performed in triplicates, on bacteria in LB at OD600 = 1. Error bars indicate the SD. (M–P) These panels display the same experiments described in I,J,K,L, but focused on PergAB.
The EBS identified by ChIP-seq was centered at +25 nucleotides downstream of the transcription start site of exlBA (12) (Figure 3I). The direct interaction between ErfA and exlBA promoter (PexlBA) as well as specificity to the EBS were confirmed in vitro by EMSA, using either wild-type or mutated probes (Figure 3J,K). The same mutation that abolished ErfA binding in vitro, when introduced in the promoter of the IHMA87 exlBA::lacZ strain, led to increased β-galactosidase activity similar to that measured in the erfA mutant (Figure 3L). This confirmed that the inhibitory effect of ErfA on exlBA expression is the consequence of its direct and specific binding to a palindromic DNA motif found on exlBA promoter. In addition, ErfA binding to this regulatory site was found to counteract the positive effect of the cAMP-responsive Vfr activator on exlBA transcription (Supplementary Figure S6). The configuration of the Vfr- and ErfA-binding sites (VBS and EBS) would allow independent bindings, and thus independent signal integration of both the activator and the repressor, which is reminiscent of what is seen with CRP and LacI regulation of the lac operon (33). The EBS situated upstream of ergAB overlaps the ‘-10’ box of the putative promoter and is conserved in IHMA87 and PAO1 strains (Figure 3M). EMSA revealed a much higher affinity of ErfA for the ergAB promoter than for the exlBA promoter (Figure 3N), fitting well with the higher effect seen on transcription, and the mutation of the site also prevented the binding of the protein both in vitro and in vivo (Figure 3O,P), confirming the direct control of these genes by ErfA.
ErfA regulates two functionally unrelated operons
The product of the ergA gene is predicted to be a class II aldolase while ergB encodes a putative dihydrodipicolinate synthase. However, two independent studies tested ErgB activity as a dihydrodipicolinate synthase without any conclusive results (34,35), leaving both gene products with unknown functions. We investigated whether ergA and ergB play a role in regulation or function of ExlBA by analyzing the transcription of exlBA (Figure 4A), the cytotoxicity on epithelial cells and the virulence in the Galleria model of infection (Figure 4B,C) of ergAB mutant. Collectively, these experiments indicated that ErgA and ErgB have no effect on ExlBA function, at least in the conditions tested herein. Therefore, we conclude that ErfA directly regulates two operons with unrelated functions.
Figure 4.

ErgA and ErgB are functionally unrelated to ExlBA. (A) β-Galactosidase activities of IHMA87 wild-type (WT) and ergAB mutant (ΔergAB) carrying exlBA::lacZ. Activities were measured after growth in LB at OD600 = 1 and are represented as mean (in Miller Units) ± SD from three independent experiments. Statistical significance was assessed using two-tailed T-test. (B) Kinetics of bacterial cytotoxicity on A549 epithelial cells. Cells were infected at a MOI of 10 with the indicated strains in presence of propidium iodide (PI). PI incorporation is represented as mean ± s.e.m. from three independent experiments. (C) Survival curves of Galleria mellonella larvae infected with an average of 5 bacteria per larva. Forty larvae were used for each strain. Statistical testing was performed using log-rank test (P < 0.05).
ErfA targets uniquely ergAB in other Pseudomonas species
Orthologues of the exlBA operon have been identified in several soil and plant dwelling Pseudomonas such as P. fluorescens, P. protegens, P. putida and P. chlororaphis ((36,37), see later this work). In order to investigate the role of ErfA in these species, we determined the ErfA pan-regulon using an in vitro genome-wide binding assay called DAP-seq (25), utilizing a purified recombinant ErfAIHMA87 protein from P. aeruginosa IHMA87. We first confirmed that DAP-seq yielded the same profiles of DNA binding as ChIP-seq in both PAO1 (Figure 5A and Supplementary Table S3) and IHMA87 (Figure 5B and C; Supplementary Table S3). Indeed, only exlBA and ergAB promoters were found significantly enriched. Notably, the difference in fold enrichment between ergAB and exlBA correlated well with in vitro affinity for the two sites observed by EMSA and the in vivo impact on levels of transcripts measured by RTqPCR, making DAP-seq a convenient and reliable tool to assess ErfA binding. As ErfA proteins from the selected Pseudomonas species share between 72 and 76% amino acid identity, we also purified the recombinant ErfAPA23 from P. chlororaphis PA23, which is 75% identical to ErfAIHMA87, and carried out DAP-seq with both proteins on P. aeruginosa IHMA87 and P. chlororaphis PA23 chromosomes to further validate our approach. Both proteins revealed the same binding patterns in the genomes of both of these organisms (Figure 5D–G and Supplementary Table S3), i.e. two promoter regions (exlBA and ergAB) in the P. aeruginosa genome and exclusively the ergAB promoter in P. chlororaphis PA23, confirming that differences in their protein sequences did not impact binding specificities. Furthermore, those data showed that the ErfA regulon is restricted to ergAB in P. chlororaphis PA23. ErfAIHMA87 was then used to determine EBSs on the genomes of P. chlororaphis PA23, P. protegens CHA0 and P. putida KT2440 (Figure 5G–I and Supplementary Table S3). This analysis further revealed the promoter of ergAB as the only target in the three species. Interestingly, while the ergAB operon is adjacent to erfA in P. aeruginosa, P. chlororaphis and P. protegens, it is 1.2 Mb apart in the chromosome of P. putida. This conserved regulation of ErfA stresses the indelible evolutionary conserved link between erfA and ergAB, and not exlBA (Figure 5J and K). In agreement with the DAP-seq data, we confirmed in vivo that, in P. chlororaphis, ErfA controls the expression of ergAB but not of exlBA (Figure 5L). Accordingly, the deletion of erfA affect neither cytotoxicity of P. chlororaphis on epithelial cells (Figure 5M) nor its virulence during G. mellonella larvae infection (Figure 5N). Altogether, ErfA represses ExlBA-dependent virulence specifically in P. aeruginosa.
Figure 5.

exlBA is regulated by ErfA only in P. aeruginosa. (A–C) Enrichment of normalized mapped reads after DAP-seq with purified ErfA from IHMA87 (ErfAIHMA87) on genomes of PAO1 (A), IHMA87 chromosome (B) and plasmid (C). (D–F) Enrichment of normalized mapped reads after DAP-seq with purified ErfA from P. chlororaphis PA23 (ErfAPA23) on whole IHMA87 chromosome (D), and plasmid (E), and PA23 genome (F). (G–I) Enrichment of normalized mapped reads after DAP-seq performed with ErfAIHMA87 on genomic DNA of P. chlororaphis PA23 (G), P. protegens CHA0 (H) and P. putida KT2440 (I). (J andK) Alignment of enrichments of normalized mapped reads for all DAP-seq experiments on the 3 and 7.5 kb region encompassing erfA-ergAB (J) and exlBA (K), respectively. Colors correspond to the ones from the genome-wide read density maps with either ErfAIHMA87 or ErfAPA23. (L) RT-qPCR analysis of ergA and exlA mRNA levels in wild-type and isogenic erfA mutant in P. chlororaphis PA23. Experiments were performed in triplicates, with RNA extracted from bacteria at OD600 = 1 in LB and normalized to rpoD mRNA. Error bars indicate the SD. (M) Kinetics of PI incorporation in A549 epithelial cells infected at a MOI of 10 with P. chlororaphis PA23 strains at 30°C. (N) Survival curves of Galleria mellonella larvae infected with P. chlororaphis PA23 strains. Larvae were infected with an average of ∼6.104 bacteria and incubated at 30°C. Significance testing was performed using log-rank test (P < 0.05).
exlBA promoter shows a wide diversity of regulatory sequences across Pseudomonas species
Interrogation of public databases showed that the exlBA operon is present in nearly 10% of all sequenced Pseudomonas genomes (Supplementary Table S4). In light of the differences observed in exlBA regulation between the experimentally tested species (P. aeruginosa versus P. chlororaphis, P. putida and P. protegens), we performed a phylogenetic analysis and compared exlBA promoter sequences (PexlBA) in all 446 exlBA+ strains (Figure 6A). First, the analysis of their genomes revealed a strong co-occurrence of the three genes erfA, ergA and ergB, corroborating the functional relationship between them. In addition, all erfA+ bacteria carry a conserved EBS at the ergAB promoter. On the contrary, EBS on exlBA promoter was found in only 6.9% of the scanned genomes, all corresponding to P. aeruginosa strains. Consequently, ergAB is the unique member of ErfA ‘core’ regulon, and exlBA is part of its ‘accessory’ regulon that would be P. aeruginosa specific. A similar imbalance was found with Vfr that is conserved in nearly all strains, as only P. aeruginosa strains, with a few exceptions, harbor a conserved VBS in the PexlBA region, further supporting the idea of an evolutionary acquired adapted regulatory network. Indeed, the Vfr protein is conserved in different lineages of P. aeruginosa; it was shown to be important in an acute murine lung infection model (38) and regulates a number of virulence-related factors including the T3SS (39,40), elastase, and exotoxin A (41). Additionally, ErfA might respond to other signals through its C-terminal cupin domain, probably linked to ErgA and ErgB functions, which are beneficial to exlBA expression specifically in P. aeruginosa. It is likely that environmental species respond to signals that are different from those encountered by a human pathogen such as P. aeruginosa, and that they control exlBA expression using unique and specific molecular mechanisms that are best suited for their particular niches. Also, the synteny of the exlBA locus is not conserved in different species, indicating that the genetic environment is variable and might influence the operon expression (Figure 6B). For instance, in P. chlororaphis, a plant-dwelling bacterium proposed as biocontrol agent, exlBA is found between genes encoding a two-component regulatory system and an operon involved in secondary messenger regulation. In P. aeruginosa, these two operons (PA3040 to PA3045) are contiguous (Figure 6C) while exlBA is found in another unrelated location, along with a P. aeruginosa-specific sequence upstream of exlBA containing the promoter encompassing both ErfA- and Vfr-binding sites (Figure 6D). Furthermore, we found that PexlBA is much less conserved than exlB and exlA coding sequences, demonstrating the strain-specific evolution of promoter sequence after acquisition of exlBA (Figure 6E). It is established that DNA-binding regulatory proteins can readily acquire new targets, and we propose that the emergence of binding sites for Vfr and ErfA in the exlBA promoter region of P. aeruginosa wired the operon to the activator and the repressor regulons, balancing the operon transcription in response to signals specific to P. aeruginosa lifestyle.
Figure 6.

exlBA promoters evolved divergent sequences across Pseudomonas species. (A) Phylogenetic tree of PexlBA sequences in 446 Pseudomonas harboring exlBA-like genes. The background color for the strain name is given for the top five species. Presence of the genes erfA, ergAB and vfr are denoted by solid circles at each species name, empty circles indicate the absence of the gene. The P-values, computed with RSAT matrix-scan (29), supporting the presence of ErfA- and Vfr-binding sites on PergAB and PexlBA, are displayed as heat maps on the external rings of the tree, the light gray color indicating the absence of tested binding sites. (B) Synteny of the exlBA genetic environment in P. aeruginosa IHMA87, P. chlororaphis PA23, P. protegens CHA0, P. putida KT2440 and P. fluorescens Pt14. (C) Schematic comparison of the P. chlororaphis exlBA location to that in P. aeruginosa. (D) Schematic comparison of exlBA locus between T3SS+ (PAO1) and exlBA+ (IHMA87) with a zoom on the 296 bp intergenic region encompassing Vfr-binding site, boxes ‘-10’ and ‘-35’, transcription start site (as determined in (12)), and ErfA-binding site. (E) Conservation rates of PexlBA, exlB and exlA nucleotide sequences in 446 Pseudomonas strains. n = 198 470 pairwise alignments for each sequence. Statistical analysis was performed using one-way ANOVA for exlB and exlA sequences against PexlBA. ****: P < 0.0001.
DISCUSSION
We described a novel twist to HGT, where the expression of newly acquired genes is re-programed by adapting to the control of existing signaling pathways and TFs through concomitant evolution or capture of the appropriate regulatory sequences. This phenomenon arises from the fact that different organisms might need to acquire the same function, but do not need it in the same conditions. This is well exemplified by the difference in conservation rates between exlBA coding sequence and exlBA promoter. Indeed, while ExlBA proteins are well conserved, suggesting no major change in function between the different species, a much wider diversity of promoter sequences is found, including completely unrelated ones. In that regard, once a new trait is acquired, the evolutionary pressure might become stronger on its regulatory sequences so that it quickly falls under adequate expression control. The regulatory sequences found immediately upstream of the newly acquired structural genes may be modified by a series of mutations, leading to appearance of new binding sites for the corresponding TFs present in the recipient organism. This hypothesis is supported by the observed vast heterogeneity of exlBA promoter sequences suggesting a diversity of molecular mechanisms exerting the regulation of similar operons across Pseudomonas found in different environments. A broader examination of the conservation, or lack thereof, in regulatory sequences linked to genes acquired by HGT could reveal how common this adaptive mechanism shapes the outcome of molecular evolution giving an organism the ability to survive and thrive in a particular environment.
DATA AVAILABILITY
The genome of IHMA879472 is available at NCBI under the accession numbers CP041354 and CP041355 for the chromosome and plasmid sequences, respectively. The RNA-seq, ChIP-seq and DAP-seq data are available under the GEO accession numbers GSE137485, GSE137484 and GSE137648, respectively.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Peter Panchev for his help with the G. mellonella experiments. We thank Eric Faudry and Stéphanie Bouillot for their help with cell culture and cytotoxicity assays. J.T. thanks François Parcy, Xuelei Lai and Arnaud Stigliani for their technical advices on DAP-seq. Pseudomonas aeruginosa strain IHMA879472 was kindly provided by International Health Management Association (IHMA; USA).
Notes
Present address: Erwin Sentausa, Evotec ID (Lyon) SAS, Marcy l’Étoile, France.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Agence Nationale de la Recherche [ANR-15-CE11-0018-01]; Laboratory of Excellence GRAL, financed within the University Grenoble Alpes graduate school (Ecoles Universitaires de Recherche) CBH-EUR-GS [ANR-17-EURE-0003]; Fondation pour la Recherche Médicale [Team FRM 2017, DEQ20170336705]; French Ministry of Education and Research (to J.T.); CNRS, INSERM, CEA, and Grenoble Alpes University. Funding for open access charge: Fondation pour la Recherche Médicale [DEQ20170336705].
Conflict of interest statement. None declared.
REFERENCES
- 1. Price M.N., Dehal P.S., Arkin A.P.. Horizontal gene transfer and the evolution of transcriptional regulation in Escherichia coli. Genome Biol. 2008; 9:R4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Freschi L., Vincent A.T., Jeukens J., Emond-Rheault J.G., Kukavica-Ibrulj I., Dupont M.J., Charette S.J., Boyle B., Levesque R.C.. The Pseudomonas aeruginosa Pan-Genome provides new insights on its population structure, horizontal gene transfer, and pathogenicity. Genome Biol. Evol. 2019; 11:109–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Huber P., Basso P., Reboud E., Attree I.. Pseudomonas aeruginosa renews its virulence factors. Environ. Microbiol. Rep. 2016; 8:564–571. [DOI] [PubMed] [Google Scholar]
- 4. Elsen S., Huber P., Bouillot S., Coute Y., Fournier P., Dubois Y., Timsit J.F., Maurin M., Attree I.. A type III secretion negative clinical strain of Pseudomonas aeruginosa employs a two-partner secreted exolysin to induce hemorrhagic pneumonia. Cell Host Microbe. 2014; 15:164–176. [DOI] [PubMed] [Google Scholar]
- 5. Reboud E., Elsen S., Bouillot S., Golovkine G., Basso P., Jeannot K., Attree I., Huber P.. Phenotype and toxicity of the recently discovered exlA-positive Pseudomonas aeruginosa strains collected worldwide. Environ. Microbiol. 2016; 18:3425–3439. [DOI] [PubMed] [Google Scholar]
- 6. Roy P.H., Tetu S.G., Larouche A., Elbourne L., Tremblay S., Ren Q., Dodson R., Harkins D., Shay R., Watkins K. et al.. Complete genome sequence of the multiresistant taxonomic outlier Pseudomonas aeruginosa PA7. PLoS One. 2010; 5:e8842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Freschi L., Bertelli C., Jeukens J., Moore M.P., Kukavica-Ibrulj I., Emond-Rheault J.G., Hamel J., Fothergill J.L., Tucker N.P., McClean S. et al.. Genomic characterisation of an international Pseudomonas aeruginosa reference panel indicates that the two major groups draw upon distinct mobile gene pools. FEMS Microbiol. Lett. 2018; 365:doi:10.1093/femsle/fny120. [DOI] [PubMed] [Google Scholar]
- 8. Ozer E.A., Nnah E., Didelot X., Whitaker R.J., Hauser A.R.. The population structure of pseudomonas aeruginosa is characterized by genetic isolation of exoU+ and exoS+ Lineages. Genome Biol. Evol. 2019; 11:1780–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Boukerb A.M., Marti R., Cournoyer B.. Genome Sequences of Three Strains of the Pseudomonas aeruginosa PA7 Clade. Genome Announc. 2015; 3:e01366-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dingemans J., Ye L., Hildebrand F., Tontodonati F., Craggs M., Bilocq F., De Vos D., Crabbe A., Van Houdt R., Malfroot A. et al.. The deletion of TonB-dependent receptor genes is part of the genome reduction process that occurs during adaptation of Pseudomonas aeruginosa to the cystic fibrosis lung. Pathog. Dis. 2014; 71:26–38. [DOI] [PubMed] [Google Scholar]
- 11. Kos V.N., Deraspe M., McLaughlin R.E., Whiteaker J.D., Roy P.H., Alm R.A., Corbeil J., Gardner H.. The resistome of Pseudomonas aeruginosa in relationship to phenotypic susceptibility. Antimicrob. Agents Chemother. 2015; 59:427–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Berry A., Han K., Trouillon J., Robert-Genthon M., Ragno M., Lory S., Attree I., Elsen S.. cAMP and Vfr control exolysin expression and cytotoxicity of pseudomonas aeruginosa taxonomic outliers. J. Bacteriol. 2018; 200:e00135-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kanack K.J., Runyen-Janecky L.J., Ferrell E.P., Suh S.J., West S.E.. Characterization of DNA-binding specificity and analysis of binding sites of the Pseudomonas aeruginosa global regulator, Vfr, a homologue of the Escherichia coli cAMP receptor protein. Microbiology. 2006; 152:3485–3496. [DOI] [PubMed] [Google Scholar]
- 14. Wick R.R., Judd L.M., Gorrie C.L., Holt K.E.. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017; 13:e1005595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014; 30:2068–2069.24642063 [Google Scholar]
- 16. Yoon S.H., Ha S.M., Lim J., Kwon S., Chun J.. A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek. 2017; 110:1281–1286. [DOI] [PubMed] [Google Scholar]
- 17. Darling A.C., Mau B., Blattner F.R., Perna N.T.. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004; 14:1394–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ngo T.D., Ple S., Thomas A., Barette C., Fortune A., Bouzidi Y., Fauvarque M.O., Pereira de Freitas R., Francisco Hilario F., Attree I. et al.. Chimeric Protein-Protein interface inhibitors allow efficient inhibition of Type III secretion machinery and pseudomonas aeruginosa virulence. ACS Infect. Dis. 2019; 5:1843–1854. [DOI] [PubMed] [Google Scholar]
- 19. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Anders S., Pyl P.T., Huber W.. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015; 31:166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W. et al.. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008; 9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Machanick P., Bailey T.L.. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics. 2011; 27:1696–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bartlett A., O’Malley R.C., Huang S.C., Galli M., Nery J.R., Gallavotti A., Ecker J.R.. Mapping genome-wide transcription-factor binding sites using DAP-seq. Nat. Protoc. 2017; 12:1659–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Winsor G.L., Griffiths E.J., Lo R., Dhillon B.K., Shay J.A., Brinkman F.S.. Enhanced annotations and features for comparing thousands of Pseudomonas genomes in the Pseudomonas genome database. Nucleic Acids Res. 2016; 44:D646–D653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sievers F., Wilm A., Dineen D., Gibson T.J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Soding J. et al.. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011; 7:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Letunic I., Bork P.. Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics. 2007; 23:127–128. [DOI] [PubMed] [Google Scholar]
- 29. Nguyen N.T.T., Contreras-Moreira B., Castro-Mondragon J.A., Santana-Garcia W., Ossio R., Robles-Espinoza C.D., Bahin M., Collombet S., Vincens P., Thieffry D. et al.. RSAT 2018: regulatory sequence analysis tools 20th anniversary. Nucleic Acids Res. 2018; 46:W209–W214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Solovyev V., Salamov A.. Li RW. Metagenomics and its Applications in Agriculture, Biomedicine and Environmental Studies. 2011; NY: Nova Science Publishers; 61–78. [Google Scholar]
- 31. Medina-Rivera A., Defrance M., Sand O., Herrmann C., Castro-Mondragon J.A., Delerce J., Jaeger S., Blanchet C., Vincens P., Caron C. et al.. RSAT 2015: Regulatory sequence analysis tools. Nucleic Acids Res. 2015; 43:W50–W56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sood U., Hira P., Kumar R., Bajaj A., Rao D.L.N., Lal R., Shakarad M.. Comparative genomic analyses reveal Core-Genome-Wide genes under positive selection and major regulatory hubs in outlier strains of pseudomonas aeruginosa. Front. Microbiol. 2019; 10:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Browning D.F., Butala M., Busby S.J.W.. Bacterial Transcription Factors: Regulation by Pick “N” Mix. J. Mol. Biol. 2019; 431:4067–4077. [DOI] [PubMed] [Google Scholar]
- 34. Schnell R., Oehlmann W., Sandalova T., Braun Y., Huck C., Maringer M., Singh M., Schneider G.. Tetrahydrodipicolinate N-succinyltransferase and dihydrodipicolinate synthase from Pseudomonas aeruginosa: structure analysis and gene deletion. PLoS One. 2012; 7:e31133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Impey R.E., Panjikar S., Hall C.J., Bock L.J., Sutton J.M., Perugini M.A., Soares da Costa T.P.. Identification of two dihydrodipicolinate synthase isoforms from Pseudomonas aeruginosa that differ in allosteric regulation. FEBS J. 2019; doi:10.1111/febs.15014. [DOI] [PubMed] [Google Scholar]
- 36. Basso P., Wallet P., Elsen S., Soleilhac E., Henry T., Faudry E., Attree I.. Multiple Pseudomonas species secrete exolysin-like toxins and provoke Caspase-1-dependent macrophage death. Environ. Microbiol. 2017; 19:4045–4064. [DOI] [PubMed] [Google Scholar]
- 37. Job V., Bouillot S., Gueguen E., Robert-Genthon M., Panchev P., Elsen S., Attree I.. Pseudomonas two-partner secretion toxin Exolysin contributes to insect killing. 2019; bioRxiv doi:17 October 2019, preprint: not peer reviewed 10.1101/807867. [DOI]
- 38. Smith R.S., Wolfgang M.C., Lory S.. An adenylate cyclase-controlled signaling network regulates Pseudomonas aeruginosa virulence in a mouse model of acute pneumonia. Infect. Immun. 2004; 72:1677–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wolfgang M.C., Lee V.T., Gilmore M.E., Lory S.. Coordinate regulation of bacterial virulence genes by a novel adenylate cyclase-dependent signaling pathway. Dev. Cell. 2003; 4:253–263. [DOI] [PubMed] [Google Scholar]
- 40. Marsden A.E., Intile P.J., Schulmeyer K.H., Simmons-Patterson E.R., Urbanowski M.L., Wolfgang M.C., Yahr T.L.. Vfr directly activates exsA transcription to regulate expression of the pseudomonas aeruginosa Type III secretion system. J. Bacteriol. 2016; 198:1442–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fuchs E.L., Brutinel E.D., Jones A.K., Fulcher N.B., Urbanowski M.L., Yahr T.L., Wolfgang M.C.. The Pseudomonas aeruginosa Vfr regulator controls global virulence factor expression through cyclic AMP-dependent and -independent mechanisms. J. Bacteriol. 2010; 192:3553–3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The genome of IHMA879472 is available at NCBI under the accession numbers CP041354 and CP041355 for the chromosome and plasmid sequences, respectively. The RNA-seq, ChIP-seq and DAP-seq data are available under the GEO accession numbers GSE137485, GSE137484 and GSE137648, respectively.