


Abstract
RNA aptamers that bind HIV-1 reverse transcriptase (RT) inhibit RT in enzymatic and viral replication assays. Some aptamers inhibit RT from only a few viral clades, while others show broad-spectrum inhibition. Biophysical determinants of recognition specificity are poorly understood. We investigated the interface between HIV-1 RT and a broad–spectrum UCAA-family aptamer. SAR and hydroxyl radical probing identified aptamer structural elements critical for inhibition and established the role of signature UCAA bulge motif in RT-aptamer interaction. HDX footprinting on RT ± aptamer shows strong contacts with both subunits, especially near the C-terminus of p51. Alanine scanning revealed decreased inhibition by the aptamer for mutants P420A, L422A and K424A. 2D proton nuclear magnetic resonance and SAXS data provided constraints on the solution structure of the aptamer and enable computational modeling of the docked complex with RT. Surprisingly, the aptamer enhanced proteolytic cleavage of precursor p66/p66 by HIV-1 protease, suggesting that it stabilizes the productive conformation to allow maturation. These results illuminate features at the RT-aptamer interface that govern recognition specificity by a broad-spectrum antiviral aptamer, and they open new possibilities for accelerating RT maturation and interfering with viral replication.
INTRODUCTION
Aptamers are nucleic acids that can be selected via Systematic Evolution of Ligands by EXponential Enrichment against specific target(s). RNA aptamers selected to bind HIV-1 reverse transcriptase (RT) inhibit the protein's enzymatic activity in biochemical assays and they block HIV replication in cell culture (1–7). The inhibitory function of aptamers against RT enzymatic activity comes from their ability to compete with viral primer/template (p/t) for RT binding (1,8–12), although many of the molecular details of the interactions between RNA aptamers and various RTs are still poorly understood. In addition to binding nucleic acids, RT contacts a network of viral and host proteins during HIV-1 replication. Alteration of these interactions could potentially provide additional mechanisms for aptamer-mediated interference with viral replication, a possibility that we explore here with respect to protease-mediated maturation. Aptamers can bind the mature heterodimer RT, and recent evidence suggests that they also bind to the precursor homodimer in the cytoplasm during viral assembly (7); therefore, we reasoned that they could modulate protein-protein interaction involving RT.
For DNA aptamers, efforts have been made to elucidate the RT-binding interface using different approaches, including crystallization and mass spectrometry footprinting (9,13). In contrast, information on the interface between RT and RNA aptamers is still limited, with most structural studies only focused on RT complexes with pseudoknot aptamers such as T1.1 (9,14–16), which are known to be sensitive to RT amino acid sequence variations (7,17). Several structural families of anti-HIV RNA aptamers have been described based on conserved signature motifs, including family 1 pseudoknots (F1Pk), family 2 pseudoknots (F2Pk), 6/5 asymmetric loop motif ((6/5)AL) and UCAA-bulge motif (UCAA) (1,6,7,17–19). F1Pk aptamers are highly specific for RTs that encode arginine at position 277, as K277 RTs are not susceptible to inhibition by F1Pk pseudoknots such as aptamer T1.1 (7,17). In contrast, UCAA and (6/5)AL aptamers can inhibit RTs from diverse lentiviruses and thus have been considered as broad-spectrum inhibitors (6,7,19).
Aptamers from each structural family likely make distinct molecular contacts, and the broad-spectrum aptamers may recognize conserved regions among phylogenically diverse RTs. Information on RT-aptamer binding interfaces from different aptamer structural families will provide insight for understanding the mechanism of broad-spectrum inhibition and for engineering nucleic acid tools for differential recognition of HIV-1. Here, we have defined critical RNA–protein molecular interactions for a broad-spectrum RNA aptamer from both the aptamer and RT perspectives, focusing on 148.1, a UCAA-family aptamer that emerged from a ‘PolyTarget’ selection against a panel of RTs from different HIV strains, including HIV-1 Group M subtypes A, B, and A/E, HIV-1 Group O, and HIV-2 (19). The UCAA motif definition includes two conserved base pairs (AC/GU) on the 5′ side of the unpaired UCAA within a relatively simple stem-loop structure. The broad-spectrum aptamer 148.1t1 (19) is the smallest UCAA variant (44 nt) tested to date that satisfied the essential requirements of the motif, making it a promising subject for structural studies. Using biochemical and chemical approaches, we identified the 38 nt-binding core of aptamer 148.1t1 (named 148.1-38m) and elucidated the interface of the complex between RT and aptamer 148.1-38m. Alanine scanning mutagenesis of this region revealed decreases in susceptibility for specific mutant RTs toward inhibition by 148.1-38m. 2D NMR and SAXS established structural features of the apoRNA in solution, and computational modeling revealed a 3D structure of the bound complex. Furthermore, we investigated the ability of the aptamer to interfere with RT-PR interactions. Information on RT-aptamer binding interfaces from different aptamer structural families will provide insight for understanding the mechanism of broad-spectrum inhibition.
MATERIALS AND METHODS
Purification of RT and the RT mutants
Plasmids expressing mutant RT were generated using Phusion site-direct mutagenesis on a modified pRT-Dual plasmid (originally containing HXB2 RT) (19). Mutations were confirmed by Sanger sequencing (University of Missouri DNA Core Facility). RT expressing plasmids were heat shock transformed into Escherichia coli BL21(DE3)pLysS competent cells and cells were incubated overnight at 37°C on LB-Agar plates (35 μg/ml streptomycin). Clonal isolates from each plate were recovered and incubated for ∼16 h at 37°C with 225 rpm shaking in 10 ml of 2xYT broth supplemented with 35 µg/ml streptomycin. Cultures were then diluted into 1 L of 2xYT broth with streptomycin. Bacteria were allowed to grow until A600nm reached 0.8. Protein overexpression was induced with 1 mM IPTG. After a 4-h incubation, samples were centrifuged for 15 min at 4000 xg. Supernatants were decanted and cell pellets were frozen at −80°C until purification. For purification, cell pellets were resuspended in lysis buffer (25 mM Tris–HCl pH 8.0, 500 mM NaCl, 1 mM PMSF, 0.15 mg/ml lysozyme) and subjected to four rounds of ultrasonication, with 30 s of sonication on ice, followed by 2 min of rest on ice in each round. Cell lysates were then centrifuged for 15 min at 4°C and 12 000 × g to remove cell debris. Cell extracts were passed through a 0.45 μm filter to remove insoluble material and applied to a Ni-NTA agarose affinity column (Qiagen) for His tag purification. Purification was performed according to the manufacturer's protocol with an added 40 ml of high salt wash (2 M NaCl) to remove any endogenous nucleic acids bound to RTs. Eluted protein samples were pooled and quantified using UV absorbance at 280 nm on a NanoDrop 1000 spectrophotometer (Thermo Fisher) using an extinction coefficient at 280 nm of 2.6012 × 105 M−1 cm−1. Purified proteins were concentrated and exchanged into 2× storage buffer (100 mM HEPES pH 7.5, 100 mM NaCl) using Amicon Ultra Centrifugal Filters (Millipore Sigma). Proteins were stored at −80°C after addition of glycerol to 50% (v/v).
Primer extension assay
DNA-dependent DNA polymerase activities of RTs were performed as previously described (19). In brief, 20 nM of RT (in-house purified HXB2 and its mutant variants) was incubated with 100 nM of aptamer in extension buffer (50 mM Tris pH 7.5, 50 mM NaCl, 5 mM MgCl2) on ice for 10 min, then a mixture of primer/template/dNTP was added (final concentration: 10 nM primer, 20 nM template and 100 μM of each dNTP) (19). DNA-dependent DNA polymerase activities of RTs were assayed using a 31-nt DNA template (5′-CCATAGATAGCATTGGTGCTCGAACAGTGAC-3′) and a complementary, 5′-Cy3-labeled 18-nt DNA primer (5′-Cy3-GTCACTGTTCGAGCACCA-3′). The extension reaction was incubated at 37°C for 10 min. Reaction was stopped by adding 2× polyacrylamide gel electrophoresis (PAGE) loading dye (95% formamide, 50 mM ethylenediaminetetraacetic acid (EDTA), xylene and bromophenol blue). Samples were separated on 10% denaturing (8M urea) PAGE. Gels were scanned on a Typhoon FLA9000 imager (GE Healthcare) and quantified using Multi Gauge software (Fujifilm). P-values were calculated using unpaired t-test computed by GraphPad Prism.
RT polymerase activity assays:
Polymerase activity of RT was monitored via time course primer extension assays essentially as above, except that reactions were stopped at various time points (0, 1, 2, 3, 4, 5, 6, 7, 8, 9 and 10 min) prior to analysis by denaturing gel electrophoresis as above. The fraction of primer converted to full-length product as a function of time was plotted using GraphPad Prism.
K d determinations
Aptamers of interest were 5′-radiolabeled using [32P]ATP and polynucleotide kinase. Labeled aptamer (∼40 000 cpm) was added to binding reactions containing various concentrations of subtype-B RT. Binding buffer contained 140 mM KCl, 1 mM MgCl2 and 50 mM Tris–HCl pH 7.5 (final concentration). Binding reactions were mixed on ice, then moved to room temperature and incubated for 15 min before loading onto pre-wet nitrocellulose membranes under vacuum and washing with 1× binding buffer. Aptamer–RT complexes were retained on the membrane, and radioactivity was determined via scintillation counting. The ‘no RT’ reaction was used as a control to determine protein-independent contribution to signal (typically <3%) and the amount of radioactivity present in the unfiltered sample was set to 100%. The protein-dependent signal was determined by subtracting the value obtained for the no RT control from the value obtained for the sample. Values at each RT concentration were fit to a one-site specific binding graph Y = Bmax*X/(Kd + X) using Prism software version 6.2. In the equation, Bmax is the maximum fraction bound, Kd is the dissociation constant, X is RT concentration and Y is fraction bound.
Hydroxyl radical footprinting
Hydroxyl radical footprinting reactions were performed using methods similar to those previously described for mapping nucleic acid-RT binding interactions (9,12,20,21). 5′-radiolabeled aptamers (∼40 000 cpm) were incubated in 30 mM KCl, 2 mM MgCl2 30 mM Tris–HCl (pH 7.5) in the presence or absence of 500 nM HIV-1 RT and equilibrated for 15 min on ice in a total volume of 20 μl at the bottom of a 1.7 ml centrifuge tube. The hydroxyl radical footprinting solution was generated by placing 1 μl of a freshly prepared Fe(II)-EDTA solution from powders (1 mM (NH4)2Fe(SO4)2·6H2O and 2 mM EDTA final concentration unless otherwise noted), a 1 μl of 2% hydrogen peroxide (freshly diluted from a 30% stock) and 1 μl of 100 mM sodium ascorbate) as three separate drops on the side of the tube. The reaction was initiated by simultaneously combining the three 1 μl drops with the aptamer solution by briefly spinning the tube in a centrifuge. The reactions were quenched by quickly adding 1 μl of 100 mM thiourea, 2.5 μl of 3 M sodium acetate and 1 μl of 20 μg/μl glycogen after 2 min incubation, unless otherwise noted.
Hydrogen-deuterium exchange
Solution-phase amide HDX experiments were carried out with a fully automated system described previously (22) with slight modifications. Briefly, 4 μl of 10 μM full-length RT alone or in the presence of ligand at a molar ratio of 1.1 and 1.5:1 ligand:RT, respectively, was diluted to 20 μl with D2O and incubated at 4°C for 10, 30, 60, 900 or 3600 s. Following exchange, unwanted forward or back exchange was minimized and the protein was denatured with a quench solution (5 M urea, 50 mM TCEP and 1% v/v TFA) at 1:1 ratio to protein. Samples were then passed through an in-house prepared immobilized pepsin column at 50 μl min−1 (0.1% v/v TFA, 15°C) and the resulting peptides were trapped on a C18 trap column (Hypersil Gold, Thermo Fisher). The bound peptides were then gradient-eluted (5-50% CH3CN w/v and 0.3% w/v formic acid) across a 1 × 50 mm C18 HPLC column (Hypersil Gold, Thermo Fisher) for 5 min at 4°C. The eluted peptides were then analyzed directly using a high resolution Orbitrap mass spectrometer (LTQ Orbitrap XL with ETD, Thermo Fisher). Each HDX experiment was performed in triplicate. To identify peptides, MS/MS experiments were performed with a LTQ Orbitrap mass spectrometer over a 70 min gradient. Product ion spectra were acquired in a data-dependent mode and the five most abundant ions were selected for the product ion analysis. The MS/MS *.raw data files were converted to *.mgf files and then submitted to Mascot (Matrix Science, London, UK; http://www.matrixscience.com) for peptide identification. Peptides with a Mascot score of 20 or greater were included in the peptide set used for HDX detection. The MS/MS Mascot search was also performed against a decoy (reverse) sequence and false positives were ruled out. The MS/MS spectra of all the peptide ions from the Mascot search were further manually inspected and only the unique charged ions with the highest Mascot scores were used in estimating the sequence coverage. The intensity weighted average m/z value (centroid) of each peptide isotopic envelope was calculated with the latest version of our in-house developed software, HDX Workbench (23).
2D1H-1H NMR
Purified RNA samples were lyophilized overnight and dissolved in D2O with 10 mM dTris–HCl, pH 7.0, 140 mM KCl, 10 mM NaCl and 1 mM MgCl2 to a final concentration of ∼700 μM. NMR data were collected at 308K on a Bruker Avance III 800 MHz spectrometer equipped with TCI cryoprobe (University of Missouri, Columbia). Two dimensional 1H-1H NOESY data were processed with NMRPipe and NMR Draw (24), and analyzed with NMRViewJ (One Moon Scientific, NJ, USA).
SAXS data collection and analysis
148.1-38m and 148.1-38m delU were prepared by invitro transcription using DNA templates containing a T7 promoter. DNA templates were prepared by phosphoramidite method oligonucleotide synthesis (Integrated DNA Technologies). Transcription products were subjected to denaturing gel electrophoresis overnight. RNA was visualized by UV-shadowing, extracted from the gel through electro-elution (Elutrap, Whatman) and washed in ultra-centrifugal filters (Amicon, Millipore Sigma). Prior to travel to the beamline the RNA samples were lyophilized. On site the samples were dissolved in 10 mM Tris–HCl (pH 7.5) to a concentration of 100 μM. In order to refold the RNA the samples were boiled for 5 min and cooled quickly in ice. Salts were then added to achieve a final concentration 40 mM KCl, 10 mM NaCl and 1 mM MgCl2 and the RNA was incubated for 5 min.
The samples were then applied to a size exclusion column connected inline to the beamline. SAXS data were collected as the samples eluted from the column (Argonne beamline 12-ID-B). ATSAS (25) was used to average the sample data and the buffer scattering was subtracted. DAMMIF was used to generate ab initio models. Pairwise distribution plots were created using GNOM by goodness of fit to the raw SAXS data and the total quality estimate. Ab initio models were visualized using PyMOL.
3D structural prediction of the apoRNA
Using the template-generated 3D structures by Vfold3D as initial states, we ran coarse-grained Molecular Dynamics simulations in IsRNA model with the secondary structures as constraints, including the tertiary contact suggested by the NMR data (A30-H2 to U14-H1′ distance restraint (distance < 5 Å)), to sample a 3D structure ensemble. Coarse-grained Molecular Dynamics simulations were implemented with Langevin dynamics at temperature T = 300 K with the integration time step set to Δt = 1fs. The simulation length was 5 ns and the structure snapshots were recorded every 5ps. The top 10% snapshot structures with the lowest energies in IsRNA model were collected and then clustered according to pairwise root-mean-square deviation (RMSD) with a threshold of 5 Å. The centroid structures of the clusters were selected as candidate 3D structures, resulting a total of 70 modeled structures for aptamer 148.1-38m.
Computational docking of the aptamer–RT complex
The RNA aptamer structures were docked to HIV-1 RT using our own protein–RNA docking program, MDockRP (26). MDockRP is based on the Fast Fourier Transformation algorithm (27), and is implemented with the pairwise shape complementarity function and electrostatic energy function of the protein–protein docking program, ZDOCK (28). The GPU-based MDockRP created a total of 54 000 docking modes for each model aptamer structure on HIV-1 RT.
Protease cleavage assay
The 2.4 pmol of p66/p66 RT was incubated with 45.4 pmole aptamer on ice in binding buffer (50 mM Tris pH 7.5, 50 mM NaCl, 5 mM MgCl2). Note that a modified version of aptamer 148.1 was used in which the pyrimidines (Y) were replaced with 2′F-substituted pyrimidines (148.1-2′FY) to reduce aptamer degradation during the assay. After 10 min, 45.4 pmole HIV PR (Abcam) was added and the reaction was incubated at 37°C for 6 h. RT without aptamer or PR was used as control for background and was set up in the same reaction conditions. The cleavage reaction was stopped by adding 4× Laemmli protein dye (Biorad). Samples were loaded onto 10% sodium dodecyl sulphate (SDS)-PAGE. Cleavage of p66/p66 by PR was detected via immunoblot. Proteins in the SDS-PAGE were transferred to PVDF membrane (transferring buffer contained 50 mM Tris pH 7.5, 380 mM glycine and 20% methanol) using semi-dry electroblotting method. Transferring current was set at 100 mA for 2 h. Membrane was washed three times with phosphate-buffered saline (PBS)-Tween (80 mM Na2HPO4, 1.5 M NaCl, 20 mM KH2PO4, 30 mM KCl and 0.5% Tween 20, pH 7.5) before blocking with milk (5% milk in PBS-Tween). After 45 min, membrane was incubated in primary rabbit anti-RT antibody (This reagent was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: Cat. #6195 and lot #140226) (1:4000 dilution in 3% PBS-Tween milk) overnight at 4°C. After washing three times with PBS-Tween, membrane was incubated in secondary horseradish peroxidase anti-rabbit (1:10 000 dilution in 4% PBS-Tween milk) for 1 h. Membrane was washed three times with PBS-Tween. Development of the membrane started by loading the Immobilon Classico Western HRP substrate (Millipore Sigma) onto the washed membrane. Luminescence was detected using the UVP Biospectrum Imaging System (Analytikjena). Image was captured using Vision Works software. Intensities of p66 and p51 bands corresponding to each reaction sample were quantified using ImageJ, then the p51:p66 ratio was calculated. The p51:p66 ratio corresponding to p66 homodimer alone (no PR and no aptamer) was set as background and was subtracted from the p51:p66 ratio of each sample. The adjusted p51:p66 ratios were then plotted via GraphPad Prism 6.2.
RESULTS
Determination of the binding core of broad-spectrum aptamer 148.1
To investigate the RNA structural features required for binding of aptamer 148.1-38m to RT, mutated and truncated variants were evaluated for their abilities to inhibit purified RT (Figure 1; Supplementary Table S1 and Figure S1). Based on the predicted secondary structure of aptamer 148.1t1, an additional 9 nt segment on the 5′ end of this aptamer was replaced with GGG fragment (to aid transcription efficiency) to create a 38 nt version denoted 148.1-38m. Aptamer 148.1-38m retained the ability to bind and inhibit RT, with a normalized fraction extended product (NFEP) value <0.2 (Figure 1 and Supplementary Figure S1A). RT inhibition was not significantly sensitive to base pairing or deletion of the 5′GGG overhang (Supplementary Figure S1) or to replacing the AAUCU loop with GGA (Figure 1A and B). In contrast, inhibition was lost upon deletion of the two unpaired Us (U6 and U14) or pairing them with As inserted into the opposite strand (NFEP values 0.78 and 0.88, respectively) (Figure 1B). A representative gel for primer extension assay is shown in Supplementary Figure S2. Deleting U6 or replacing it with C abolished inhibition (Figure 1C), and the variant with U6 deletion (148.1-38m delU6) served as a non-binding control in later binding studies. Although deleting U14 did not affect aptamer inhibition, changing U14 to C abolished the inhibition (Figure 1D) potentially by changing the overall predicted structure. When U14 was replaced by C, the signature UCAA bulge is predicted to become an AC/UUCAA asymmetric internal loop in the U14C mutant but is predicted to be maintained in the U14 deletion mutant (Supplementary Figure S3). The signature UCAA bulge was also sensitive to modifications, as single variations at any of the four position (12 variants) and a scrambled variant of the bulge, AACU, also abolished inhibition of RT (NFEP values > 0.6) (Figure 1C and Supplementary Figure S4). The results from modification studies indicate that the unpaired U6 and the UCAA bulge are critical to 148.1-38m's inhibition of RT, while the stem loop was not highly sensitive to the alteration tested here. Although findings of 148.1-38m's UCAA bulge and stem loop are consistent with the previously identified consensus for UCAA aptamers 80.103 and 80.111 (6), aptamer 148.1-38m differs in the role of the first unpaired U from the aptamers’ 5′-end, as deletion of the unpaired nucleotides of aptamer 80.103 did not affect inhibition (6).
Figure 1.

Binding core and critical structural features of 148.1-38m. (A) Modifications on 148.1-38m aptamer. The 2D predicted structure of the aptamer was generated via Mfold (http://unafold.rna.albany.edu/?q=mfold/RNA-Folding-Form). Red, blue and green sticks depict C-G, A-U and non-Watson–Crick base pairings, respectively. (B), (C) and (D) Inhibition of RT by 148.1-38m variants measured via primer extension. For each RT-aptamer pair, the fraction extended product was measured and normalized to that of the ‘No Aptamer’ reaction. The fraction of primers extended to full-length product was the highest (NFEP value = 1) in the absence of aptamer (designated ‘No Aptamer’). In the absence of RT, p/t was not able to extend. Variants that gave NFEP values higher than 0.6 were considered ‘not inhibitory.’ Values presented on the graphs were the results from three independent experiments. The error bars represent the standard deviation of the NFEP values. (B) Base pairing or deletion of both unpaired Us (U6 and U14) abolished aptamer inhibition whereas replacing AAUCU stem loop with minimum GGA sequence did not affect inhibition of the aptamers. (C) The unpaired U6 and the signature UCAA bulge were also critical to 148.1-38m inhibition. The UCGA, UCUA and AACU variants failed to inhibit RT. (D) Variant with deleted U14 was still inhibitory. However replacing U14 by C14 abolished aptamer inhibition. ns (P > 0.05), *** (P < 0.001).
UCAA bulge of 148.1-38m does not directly interact with RT
We next looked at the RNA–protein interface by identifying nucleotides with changes in solvent accessibility of their sugar moieties upon binding to RT. Aptamers 148.1-38m and 148.1-38m delU6 were subjected to hydroxyl radical cleavage in the presence or absence of RT (Figure 2 and Supplementary Figure S5), and cleavage intensities at each position were compared for free and bound aptamer under identical treatment (9,20,29,30). The non-binding control aptamer, 148.1-38m delU6, failed to bind (Supplementary Figure S5A) or inhibit RT (Figure 1C), and its hydroxyl radical cleavage pattern was unchanged by the addition of RT. A quantitative representation of cleavage patterns clearly identified differences in cleavage intensities between bound and unbound aptamer 148.1-38m (Figure 2 and Supplementary Figure S5B). Addition of protein induced a hypersensitive cleavage site at the sugar of nucleotide A30 within the UCAA bulge. Sugars of the neighboring nucleotides A31 and C29 and nucleotides A19 and U22 of the stem loop were also sensitive to cleavage, albeit to a much lesser degree (Figure 2A). These observations indicated that the sugar moieties of the nucleotides in the UCAA bulge become more exposed to solvent when the aptamer is in complex with RT. In contrast, RT protected nucleotides C9 to C13 opposite the UCAA bulge and G25 to U27 adjacent to the UCAA bulge from cleavage. These data suggest that the UCAA bulge might not be buried in the complex but rather remains exposed and shapes the aptamer's 3D structure, helping the two stems of the aptamer adopt a conformation that is appropriate for docking into the RT binding pocket.
Figure 2.

Hydroxyl radical footprinting profiles of 148.1-38m and its delU6 variant in the presence (magenta) or absence (blue) of RT. (A) Cleavage patterns from hydroxyl radical digestions are shown for 148.1-38m (45) and the delU6 variant (18). When in complex with RT, 148.1-38m was subjected to hyper-sensitive cleavage (‘exposed’) at position A30 of the UCAA bulge. Positions A19, U22, A31 and C29, were also sensitive to cleavage but not to the same level as A30. In contrast, C9 to C13 and G25-U27 experienced decrease in level of cleavage (‘protected’) when RT was present. (B) Protected (green) and exposed (orange) positions are mapped onto predicted structure of 148.1-38m.
RT makes distinct interactions with specific and broad-spectrum inhibitors
Aptamer binding surfaces on RT were identified by hydrogen deuterium exchange coupled with mass spectrometry (HDX/MS), using complexes formed between RT and 148.1-38m. As a reference, HDX/MS was also performed using the minimum binding core of F1Pk aptamer 70.05 (70.05core2) (1,17) (Supplementary Figure S6). We predicted that the two complexes would leave different footprints on RT due to the differences in aptamer structure. For the HDX/MS experiment, the p51 subunit of RT was isotopically labeled with 15N so that digested peptides from p51 could be differentiated from those of p66 (22,31).
Differences in HDX/MS for the RT heterodimer in the presence and absence of aptamers 148.1-38m and 70.05core2 were mapped onto the atomic coordinates of the RT crystal structure without substrate (PDB ID: 1DLO) (32) (Figure 3A). Aptamer 148.1-38m protected p66 in the fingers, palm, and C-terminal region of the thumb and the C-terminal of RNase H domain, in addition to protecting p51 in the fingers, palm and thumb subdomains and near the C-terminal domain (Figure 3A and Supplementary Figure S7). Aptamer 70.05core2 protected p66 peptides located in the fingers, N-terminal region of the palm and the thumb subdomain and the N-terminal region of the RNase H domain, in addition to protecting p51 in the finger and connection subdomains (Figure 3A and Supplementary Figure S8). Although there was some similarity in the footprints of the two aptamers, each aptamer formed a distinct set of contacts with the target RT protein, especially within the p51 subunit. These results are consistent with previous observations that both aptamers compete with p/t for binding to RT (1,6,9,12). Protection at peptides covering position 277 was observed in both complexes but were notably were higher in RT-70.05core2 than in RT–148.1 complex (Supplementary Figure S9), consistent with the finding that R277K mutation abolished the inhibitory function of F1Pk aptamers but not UCAA aptamers (6,7,17). In the absence of nucleic acid, RT exists predominantly in a fingers-and-thumb-together (‘closed’) conformation (32–35). Previous HDX studies of interactions between RT and efavirenz, a non-nucleoside RT inhibitor, observed exposed regions in the p66 fingers and thumb domains that were associated with formation of an ‘open’ conformation between the two domains (31). We found no evidence of similarly exposed peptides in either RT–aptamer complex, suggesting that our observation is not linked to conformational changes in the finger and thumb domains.
Figure 3.

HDX/MS analysis of RT-148.1-38m (left) versus RT–70.05core2 (right) complexes. (A) The difference in HDX of RT in the presence and absence of the 148.1-38m and 70.05core2 aptamers are mapped onto the crystal structure of ligand-free RT (PDB ID: 1DLO). Change in HDX is color-scaled. Lime green to dark blue indicates decreasing percent deuterium uptake (protection) compared to apo RT. Yellow to bright red represents increasing percent deuterium uptake (exposure). Peptides in gray are those having no net change. (B) Highly protected peptides on RT in the presence of 148.1-38m. When in complex with RT, peptide 418-425 of the p51 subunit (magenta box- Region 1) and peptide 524–555 of the p66 subunit (brown box- Region 2) are strongly protected, especially the 418–425 peptide (dark blue), relative to the remaining regions of RT.
P420A, L422A and K424A mutants are less susceptible to 148.1-38m inhibition
Two regions of RT were strongly stabilized against HDX upon binding to aptamer 148.1-38m. Region 1 (positions 418-NTPPLKL-425) in the p51 subunit was protected to a remarkably high degree by aptamer 148.1-38m, and also modestly by aptamer 70.05core2 (Figure 3B and Supplementary Figure S7B). Within this region, positions 419, 420, 422 and 425 are highly conserved among phylogenically diverse RTs. The remaining positions (418, 421, 423 and 424) were conserved within group M (HXB2, NL4-3, 93TH and 94CY) and SIVcpz (SIV-US) (Supplementary Figure S10A). Region 2 (positions 534-AWVPAHGIGGNEQVDKLVSAG-555) in the p66 subunit was only protected by aptamer 148.1-38m but not by aptamer 70.05core2, although the magnitude of the protection was less than in Region 1. Almost all amino acid in this region are conserved among RTs from different HIV-strains (36).
To explore the significance of Region 1 in RT–aptamer interactions, each amino acid was individually mutated to alanine in the p51 subunit (Supplementary Figure S10B). The singly substituted p51 subunits were co-expressed with wild-type p66 and purified to yield eight enzymatically active mutant RTs (N418A, T419A, P420A, P421A, L422A, V423A, K424A and L425A) (Supplementary Figure S11). Aptamers 148.1-38m and 70.05core2 were both able to inhibit all eight mutants, at least to some degree; however, mutants P420A, L422A and K424A were noticeably less susceptible than the other RTs to inhibition by aptamer 148.1-38m (NFEP > 0.7, Figure 4A), while all eight mutants remained susceptible to 70.05core2 inhibition (NFEP 0.15 to 0.45). The difference in inhibition by aptamers 148.1-38m and 70.05core2 is especially evident for RT mutants L422A and K424A (P = 0.0016 and 0.0004, respectively) and for N418A (P = 0.02), even though both aptamers inhibit N418A with NFEP < 0.6. These observations underscore the functional significance of differences in molecular contacts between aptamer structural families. Nevertheless, none of these alanine substitutions yielded the degree of resistance obtained with an R277K mutant RT, which extended essentially all of the input p/t to full-length product in the presence of 70.05core2, even while remaining susceptible to inhibition by 148.1-38m (Figure 4A). Polymerase activities of the three mutants (P420A, L422A and K424A) were similar to, or slightly less than, that of WT RT (Figure 4B and Supplementary Figure S12), ruling out the possibility that their reduced sensitivities to inhibition by 148.1-38m could be due to enhanced abilities to bind or extend the DNA substrate. Mutant T419A was also included as a internal control and its polymerase activity was similar to that of WT RT. Overall, the alanine-scanning mutations of Region 1 caused more severe effects on the enzymatic inhibition by 148.1-38m than by 70.05core2, consistent with the significantly higher protection of this fragment in the 148.1–38m complex than in the RT–70.05core2 complex. Amino acids 420, 422 and 424 of p51 appear to be involved in establishing the RT–148.1–38m interface.
Figure 4.
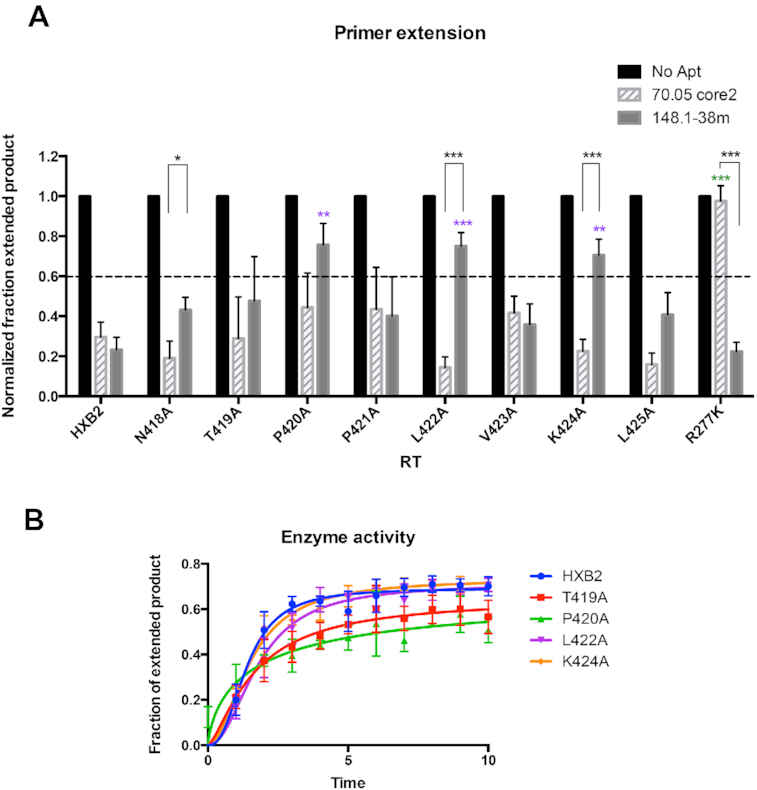
Alanine-scanning mutagenesis of the 418-NTPPLVKL-425 peptide of p51. (A) Values on the y-axis represent the normalized fraction extended product (NFEP). For each RT-aptamer pair, the fraction of primer converted to full-length product was determined by quantifying band intensities using Multigauge software then normalized to that of RT-No Aptamer. In the absence of aptamer (No Aptamer), the fraction of primers extended to full-length product was the highest. This NFEP value is defined as ‘1.’ In the absence of RT, p/t was not able to extend. Reactions in which the NFEP values were higher than 0.6 were considered to be ‘not inhibitory.’ Values presented on the graphs were the results from three independent experiments. The error bars represent the standard deviation of the NFEP values. Inhibition of mutant RTs by 70.05core2 and 148.1-38m via primer extension assay. R277K is fully resistant to 70.05core2 and was used as control for the assay. Among tested mutants, P420A, L422A and K424A had reduced susceptibility to 148.1-38m inhibition. In contrast, 70.05core2 inhibits all mutants except for R277K. Significant difference between inhibition of 148.1-38m against HXB2 and mutant RT is indicated on top of the specific mutant (purple). Significant difference between inhibition of 70.05core2 against HXB2 and mutant RT is indicated on top of the specific mutant (green). In black is the significant difference between inhibition of 148.1-38m and 70.05core2 against the same mutant RT. * (P < 0.05), ** (P < 0.01), *** (P < 0.001). (B) Enzymatic activity of mutant RTs compared to WT RT (HXB2). Activity was measure by primer extension assay and represented as the fraction of extended product over the time of reaction. Values presented on the graphs were the results from three independent experiments. The error bars represent the standard deviation of the fraction of extended product values.
When both aptamers were titrated against four RTs (WT HXB2, T419A, P420A and R277K), aptamer 148.1-38mer inhibited P420A weakly compared to its inhibition of HXB2, T419A and R277K (Figure 5A). Inhibition of P420A was partially rescued at high concentration of aptamer. This weak resistance is consistent with the slight decrease in binding affinity of 148-38m for P420A compared to WT HXB2 (Supplementary Figure S13). In contrast, RT mutant R277K was not susceptible to inhibition by aptamer 70.05core2 at any tested concentration (up to 180 nM), while all other RTs were equivalently inhibited in a concentration-dependent manner by this aptamer (Figure 5B), again highlighting differences in amino acid sequence sensitivities for aptamers from different structural families. Since full-length aptamer 70.05 had been previously demonstrated to be unable to bind R277K (7), the strong resistance phenotype of R277K mutant against 70.05core2 was expected.
Figure 5.
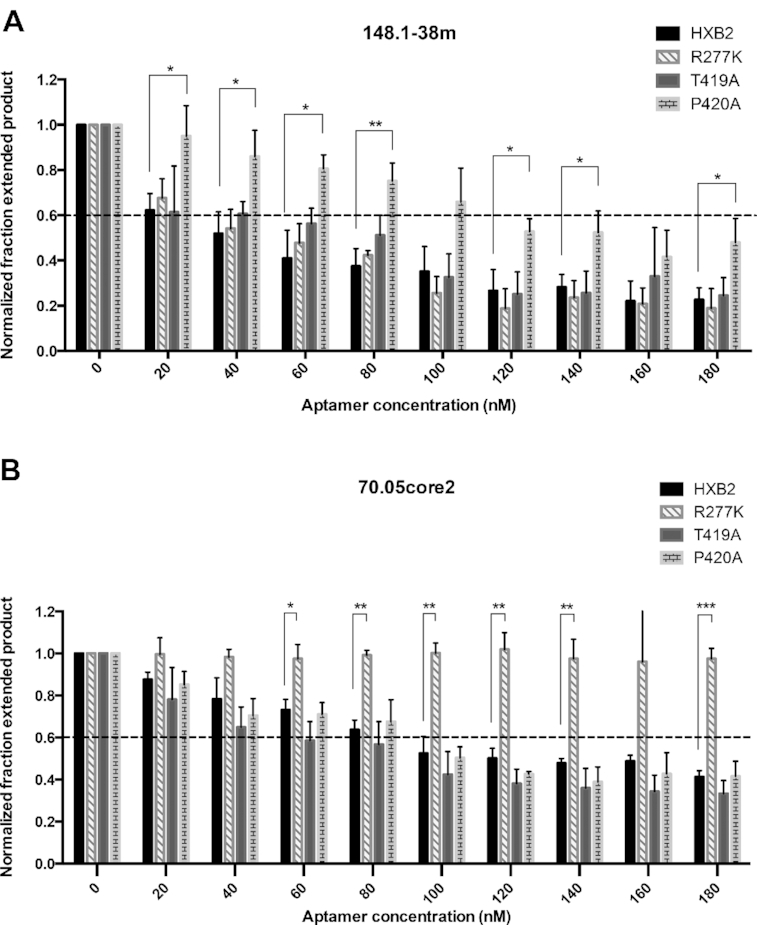
Inhibition profiles of WT and mutant RTs in response to increasing concentration of 148.1-38m (A) and 70.05core2 (B). Values shown represent triplicate measurements (±SD) of three independent experiments. (A) Inhibitory phenotype of 148.1-38m against P420A can be partially rescued with high aptamer concentration (160 and 180 nM). Inhibition profiles of R277K and T419A are similar to WT HXB2. Significant difference between inhibition of 148.1-38m against HXB2 and P420A mutant is represented in the graph. * (P < 0.05), ** (P < 0.01). (B) R277K was used as a control and stayed fully resistant to 70.05core2 under any tested concentration. T419A and P420A behaved similarly to WT HXB2 in the presence of 70.05core2 indicating that change in susceptibility observed with P420A is specific only to 148.1-38m. Significant difference between inhibition of 148.1-38m against HXB2 and P420A mutant is represented in the graph. * (P < 0.05), ** (P < 0.01), *** (P < 0.001).
Structural constraints for aptamer 148.1-38m in solution
2D 1H-1H NOESY NMR spectra were collected for aptamer 148.1-38m and peaks were assigned (Supplemental Figure S14) to establish initial structural constraints for the free (apo) RNA. Base stacking interactions were observed for residues in the lower and upper stems. Residues U6, A19-U22 and C29 gave rise to strong cross-peaks, indicating that these residues are flexible in the structure. Interestingly, cross-peaks of U14 were broad, suggesting that it may be engaged in ordered structure rather than acting as a flexible bulge. An NOE between A30-H2 and U14-H1′ was observed, and the H8 of G25 was shifted unusually upfield (Figure 6A). These data suggest that the UCAA bulge likely makes tertiary contact with U14 and residues nearby. To examine the impact of U6 and U14 on the structure of the aptamer, we collected 2D 1H-1H NOESY data for delU6+delU14. The chemical shifts of residues neighboring U6 and U14 in aptamer 148.1-38m exhibited large changes due to the deletion (Figure 6B), indicating a role for these residues in re-organizing local structure. U16-A24 remained unaffected, demonstrating that these U deletions had minimal impact on the structure of the apical loop region. These observations are consistent with RT inhibitory assays showing that the GGA stem loop mutant did not reduce the RT inhibitory function of the aptamer (Figure 1B). The chemical shifts of UCAA and their neighboring residues were sensitive to the U6 and U14 deletions, even though they were distant from the deleted Us in primary sequence and in the folded secondary structure, consistent with the tertiary contact suggested above.
Figure 6.

NMR study of 148.1-38m and its mutant RNA. (A). Overlay of a representative region of the 2D NOESY NMR spectra collected for 148.1-38m (black) and delU6+delU14 (blue). H2, H6 and H8 connectivities are denoted in vertical dashed lines. The connectivity between A30-H2 and U14-H1′ is shown (horizontal red dashed line). (B) Absolute values of the chemical shift differences (△δ) of each residue in the 148.1-38m and delU6+delU14 are plotted. The chemical shifts of H8 of purines and H6 of pyrimidines are used to represent the chemical shift change of each residue.
To determine whether deletion of U6 and U14 altered the structure of the 148.1-38m, in-line small angle X-ray scattering (SAXS) data were collected. SAXS provides global information about RNA size and shape in solution. Deletion of both U6 and U14 altered the SAXS scattering profile (Figure 7A). Comparison of the pairwise distribution plot shows that 148.1-38m has a slightly longer max dimension (x-axis intercept in Figure 7B). To further understand the structural differences between 148.1-38m and 148.1-38m dU we used DAMMIF to reconstruct ab initio models of each RNA. Comparison of these models shows an alteration of the RNA structure upon deletion of the two Us. Specifically, 148.1-38m has a more compact structure than 148.1-38m dU. Deletion of U6 and U14 disrupts the tertiary contacts between the top stem with the UCAA bulge, leading to a more extended RNA fold not favored by RT binding (Figure 7C).
Figure 7.
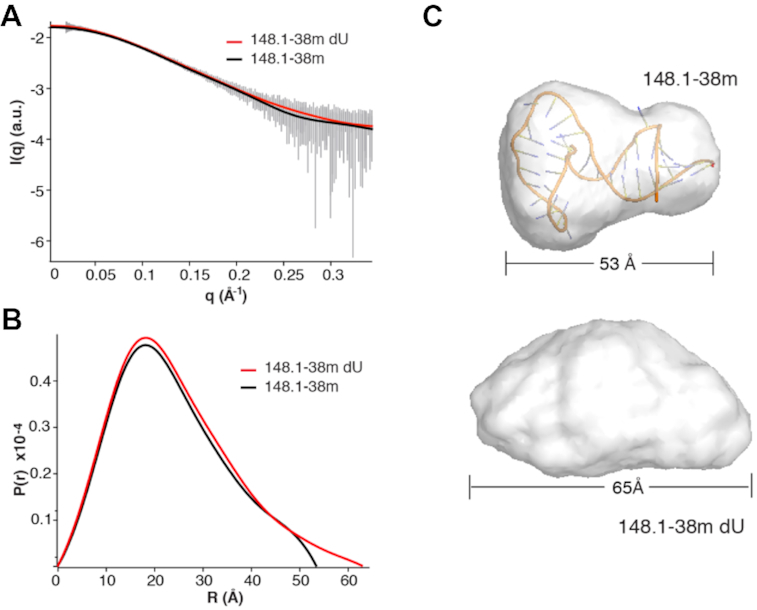
Deletion of uracil alters SAXS scattering profile. (A) Line of best fit of scattering data. 148.1-38m denoted by black line. 148.1-38m dU denoted by red line. Error bars of averaged scattering data in gray. (B) Pair wise distribution plot of 148.1-38m (black line) and 148.1-38m (red line). (C) Ab initio models of 148.1-38m (top, overlaid with docked conformation of RNA from Figure 8) and 148.1-38m dU (bottom).
Docking model of aptamer 148.1-38m with HIV-1 RT
An iterative strategy of experimental constraints and computational modeling was employed to obtain a 3D model of aptamer 148.1-38m in complex with HIV-1 RT. The 3D structure of RNA aptamer 148.1-38m was computationally modeled with the assistance of the observed NMR experimental data. Secondary structures of 148.1-38m derived from NMR data (Supplementary Figure S15A) and predicted from sequence by using the free energy-based approach Vfold2D model (37,38) (Supplementary Figure S15B) essentially agreed with the representation in Figure 1A. Therefore, the secondary structure inferred from NMR data and the initial 3D structures from the templated-based Vfold3D model (38–40) were used as input for the IsRNA coarse-grained molecular dynamics simulation method (41) to generate a set of putative 3D structures (see Supplementary Figure S16 for representative 3D apo RNA conformations). The simulated coarse-grained structures were then used as input to generate refined all-atom 3D structure models using NAMD simulations with energy minimization (42) based on the CHARMM force field (43) for subsequent docking modeling.
The structure of the complex between aptamer 148.1-38m and HIV-1 RT was then predicted through molecular docking. Specifically, 70 modeled all-atom 3D structures of aptamer 148.1-38m obtained from the above computational study were individually docked onto the crystal structure of the HIV-1 RT protein (PDB ID: 1DLO (32)) using our in-house docking program, MDockPR (26). Using a 6-degree orientation sampling interval, 54 000 docking poses of protein–RNA complex structures were generated for each of the 70 modeled aptamer structures, for a total of 3 780 000 potential poses. These docking poses were further filtered with five experimental constraints: First, Region 1 (N418 - L425 in the p51 subunit of HIV-1 RT) should have at least one close contact (≤5Å) with the aptamer, based on the observed HDX protection in this region. Second, U6 of the aptamer should have at least one close contact, based on the observations above that U6 deletion or substitution resulted in loss of the inhibitory function. The filtering resulted in 47 425 candidate poses, which can be categorized into two distinct types of binding modes. One is characterized by a single contact between the RNA aptamer and the protein, where only a cluster of nucleotides that are close to each other in space forms the single binding interface with the protein. The other type of binding mode is characterized by multiple contacts with the protein by more than one nucleotide cluster. Only poses of the multiple-contact mode were retained, because it is more typical of physiological interactions, as evidenced by the supermajority of protein/RNA complexes deposited in the PDB. The remaining 12 400 complex structures were cascaded to two extra filters, which imposed the constraints that the RNA should have at least one close contact (≤5Å) with Region 2 (A534 – G555 of p66) in Figure 3 (based on strong HDX protection in this region), and none of the sugars of the UCAA bulge is in contact with the protein (based on hydroxyl radical probing). At last, the remaining 2807 candidate poses were restricted to having the termini of the aptamer pointing away from the protein to allow for longer stems on either end of the aptamer, based on the observed tolerance for such extensions. The 1255 surviving candidates were ranked by ITScorePR (26,44) and then further consolidated into 301 clusters, defined as groups of structures with an L-RMSD less than 12Å from each other, are represented by a representative. We examined the top 20 best-scored clusters and selected the final complex structure. The resulting final model was locally minimized with the ITScorePR score, and the optimized structure is reported as the predicted complex structure of aptamer 148.1-38m and HIV-1 RT (Figure 8).
Figure 8.
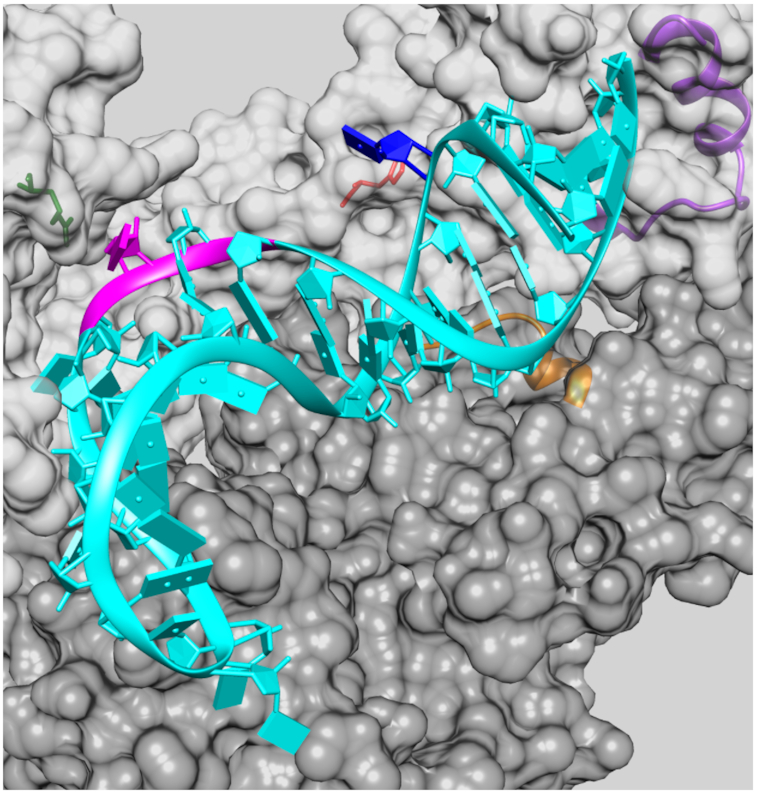
Modeled structure of the aptamer 148.1-38m in complex with the HIV-1 RT protein. The two subunits of protein, i.e. p51 and p66, are presented as dark gray and light gray surface, respectively. Region 1 of p51 is highlighted in golden ribbon representation and Region 2 of p66 in purple ribbon. The side chains of two residues of p66, HIS361 and ARG284, are displayed in stick representation and colored red and green, respectively. The RNA aptamer is depicted in cyan ribbon representation, except for the UCAA segment, which is emphasized as a stretch of a magenta ribbon. The bases of the aptamer are shown as cyan slabs, except for U6 and C29, which are colored blue and magenta, respectively.
As shown in Figure 8, the aptamer contacts both Region 1 and Region 2 of the protein. The overall shape complementarity in the complex structure is moderate, with a buried solvent accessible surface area of ∼1900Å2 upon binding, which is ∼27% less than the 2600Å2 that is buried for a pseudoknot aptamer in complex with RT (15). A number of amino acids are predicted to lie within 5 to 8 Å of the aptamer (Supplementary Table S2) and are thus in position to contact the aptamer directly. These contacts include potential specific interactions between the U6 and the His361 of subunit p66 (<5Å) and between C29 and Arg284 of p66 (<8Å). Fitting the RNA model selected for RT docking against the experimental SAXS data above calculated a χ2 value of 0.2, suggesting that 148.1-38m RNA does not experience a major shape change upon RT binding.
148.1-38m enhanced protease cleavage of p66 homodimer
During the HIV replication cycle, the immature HIV RT is produced as a homodimer with two p66 subunits. Then the p51 subunit is formed by the proteolytic cleavage of one of the p66 subunits (45,46). Although p51 consists of the same amino acids as p66 except for the cleaved region, the domains of two subunits are oriented differently in space. The p66 subunit is responsible for the enzymatic function of RT whereas p51 plays a structural role and aids in binding to nucleic acids. The cleavage of p66/p66 to form p66/p51 is important for the maturation of HIV and is catalyzed by HIV-1 protease (PR) (46,47). Previous studies showed that interruption of this proteolytic cleavage led to virion instability (48). Because the PR cleavage site that gives rise to p51 is buried in the mature heterodimer, it has been suggested that the p66/p66 precursor unfolds or adopts different conformations along its maturation pathway (46,47,49–51).
The strongly protected 418–425 peptide of p51 is located 15 amino acids away from the PR cleavage site of p66. Therefore, in vitro PR cleavage assays were performed to test the impact of aptamer 148.1-38m on RT maturation. When p66/p66 was incubated with PR in the absence of aptamers, cleavage of p66 homodimer was inefficient (Figure 9 and Supplementary Figure S17, lane 2), similar to previous observations (52). A modified version of full-length aptamer 148.1 was used in which the pyrimidines (Y) were replaced with 2′-F-substituted pyrimidines (148.1-2′FY) to reduce aptamer degradation during the assay. The 2′FY modification did not interfere with aptamer-p66/p66 or aptamer-p66/p51 interactions (Supplementary Figure S18). Surprisingly, the presence of 148.1-2′FY significantly enhanced PR cleavage of p66 (Figure 9A, lane 3). Another broad-spectrum aptamer, single-chain DNA RT1t49(-5) (53), may also enhance cleavage slightly, although the stimulation is not statistically significant. Our results suggested that interaction with 148.1 aptamer might drive p66/p66 toward the productive conformation, allowing proteolytic processing of p66/p66 homodimer to p66/p51.
Figure 9.

Effect of aptamers on protease cleavage of p66. Aptamer used in this assay were the RNA aptamer 148.1-38m and DNA aptamer RT1t49(-5). (A) Representative protein immunoblot showing PR-mediated cleavage of p66/p66. The presence of p66 and p51 in each sample was detected by Western blot. Free p66/p66 RT was used as a control for background signal of p51. Additional data can be found in Supplementary Figure 9. (B) Quantitative representation of three individual experiments. Intensities of p66 and p51 bands corresponding to each reaction sample were quantified using ImageJ then the p51:p66 ratio was calculated. The p51:p66 ratio correlated with free p66/p66 was set as background and was subtracted from the p51:p66 ratio of each remaining sample. The adjusted p51:p66 ratio then was plotted via Prism software version 6.2. ns (P > 0.05), * (P < 0.05). Values shown represent triplicate measurements (±SD) of three independent experiments.
DISCUSSION
In vitro inhibition of RT enzymatic activity by nucleic acid aptamers derives from competition with p/t for binding to RT. Thus, details of their interactions with RT are crucial for understanding the molecular basis of inhibition. The UCAA aptamer family was previously identified as broad-spectrum, capable of inhibiting phylogenetically diverse RTs (6,7,19). This work extends those observations, revealing details of the interaction between HIV-1 RT and 148.1-38m, representing the UCAA aptamer family. The complementary structural information obtained in our study from both the RNA and protein perspectives provides critical guidance for understanding the RT–aptamer interface and the structural basis of RT–nucleic acid specificity.
In the absence of nucleic acids, RT is in a ‘closed’ conformation with the fingers and thumb domains together (34,35), but it adopts an ‘open’ conformation with the fingers and thumb domains apart in the presence of dsDNA, DNA/RNA duplex (33,54,55) or non-nucleoside reverse transcriptase inhibitors (NNRTIs) (15,56–60). The rearrangement from closed to open conformation when RT is in complex with an NNRTI was evident in prior HDX/MS analyses (31,61). When RT is in complex with RNA pseudoknot aptamer T1.1, the ‘closed’ conformation is maintained (9,15), with the tips of the finger and thumb domains in contact (15). The fact that no region of increased exposure was evident in the present study when RT binds to either 148.1-38m or 70.05core2 suggests that both of these aptamers bind to the fingers and thumb domains together ‘closed’ conformation of the apo-protein. Aptamer 148.1-38m also appears to remain close to its solution structure upon binding RT. To allow for the possibility of induced-fit binding (RNA changes structure upon RT binding), all of the predicted 3D models for the apo-RNA were included for protein docking. However, the RNA structure in the final complex model is consistent with the SAXS data, suggesting a nearly-rigid fit binding with only minor local rearrangements to expose the sugar of A30 to solvent and enhance its sensitivity to hydroxyl radicals.
Our HDX/MS data show significant differences in the level of protection between footprinting of RT-148.1-38m and that of RT-70.05core2 in peptides comprising Region 1 (418-425 of p51) and Region 2 (534–555 of p66). The fact that footprinting of F1Pk aptamer 70.05core2 was found in p51 is consistent with the prior observation of an extensive interaction between another F1Pk aptamer and p51 in an aptamer-RT co-crystal (15). Amino acids 420, 422 and 424 are especially important in establishing the RT–148.1–38m interaction, since alanine mutation at these positions diminished aptamer inhibition invitro. The partial resistance phenotype was specific to 148.1–38m, and did not impair inhibition by F1Pk aptamer 70.05core2. The amino acid sequence of the 418–425 peptide is highly conserved among naïve and NRTI-treated patients (36). Alignment of this region among phylogenetically diverse RTs also shows a high level of conservation with variations found only at three positions (N418S, V423I and K424R), and even then only for MVP5180 and EHO, the two RTs that are most distant from group M subtype B (HXB2) in the panel (Supplementary Figure S10). The charge and hydrophobicity of the peptide were maintained for each of these natural variations. Interaction with RT at this conserved region might be important for RT recognition of 148.1–38m, allowing the aptamer to inhibit a panel of different RTs and potentially slowing the emergence of viable, aptamer-resistant virus.
Aptamers have been used as tools to study protein–protein interactions (62,63), but never as a tool for exploring interactions between viral proteins. During the viral life cycle, HIV-1 RT interacts with many different viral and host proteins, including HIV-1 integrase (IN), nucleocapsid (NC) and PR. RT-IN interaction during reverse transcription and integration can enhance RT activity by stimulating initiation and elongation of viral DNA synthesis (64–66), while RT interactions with IN can inhibit DNA 3′-end processing by IN (67). RT-NC interaction increases the proportion of long cDNA transcripts by RT (68), stimulates DNA strand transfer reactions and modulates RNase H activity (69,70). Interaction between RT and PR is critical for RT maturation (45,47), proteolytic processing of the RT-IN cleavage site (71), RT activity (72) and PR activity (73). Cleavage of the p66/p66 homodimer by purified PR to yield p66/p51 heterodimer in vitro has been previously reported to be slow and inefficient (52), and this processing may require partial unfolding of the beta strand β1 of the RNaseH domain (46,47,50–52,74). Our data indicate that RNA aptamer 148.1–38m and potentially DNA aptamer RT1t49(-5) both enhance PR cleavage of p66/p66. The ability of anti-RT aptamers to increase PR cleavage of p66/p66 in vitro has not been previously reported. However, tRNA was shown to enhance the proteolytic processing of p66/p66 homodimer to p66/p51 (51). Previous studies reported that non-nucleoside reverse transcriptase inhibitors could induce premature activation of PR via stimulation of Gag-Pol dimerization (75,76). It may be possible to exploit the effect of aptamers on RT–PR interaction for studying and manipulating HIV maturation by PR, a critical process that is still not fully understood. Aptamers have been exploited as inhibitors of reverse transcription. However, their potential to interrupt the network of interactions among viral proteins during integration, maturation, packaging and other steps in viral life cycle could open a new avenue for blocking viruses and for studying biological processes during infection.
Supplementary Material
ACKNOWLEDGEMENTS
We thank all members of the Burke laboratory for helpful discussions and suggestions.
Notes
Present address: Steve Tuske, Amicus Therapeutics, Gene Therapy Center of Excellence, 3675 Market Street, Philadelphia, PA 19104, USA.
Present address: Jie Zheng, Shanghai Institute of Materia Medica, Shanghai, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health (NIH) [R01AI074389 to D.H.B., R01GM109980 to X.Z., R01GM117059 to S.J.C., R01GM063732 to S.J.C., R37AI026790 to EA, U54 GM103368 to EA, R01AI150460 to X.H.]. Funding for open access charge: NIH [R01AI074389].
Conflict of interest statement. None declared.
REFERENCES
- 1. Burke D.H., Scates L., Andrews K., Gold L.. Bent pseudoknots and novel RNA inhibitors of type 1 human immunodeficiency virus (HIV-1) reverse transcriptase. J. Mol. Biol. 1996; 264:650–666. [DOI] [PubMed] [Google Scholar]
- 2. Chaloin L., Lehmann M.J., Sczakiel G., Restle T.. Endogenous expression of a high-affinity pseudoknot RNA aptamer suppresses replication of HIV-1. Nucleic Acids Res. 2002; 30:4001–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fisher T.S., Joshi P., Prasad V.R.. Mutations that confer resistance to Template-Analog inhibitors of human immunodeficiency virus (HIV) type 1 reverse transcriptase lead to severe defects in HIV replication. J. Virol. 2002; 76:4068–4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Held D.M., Kissel J.D., Saran D., Michalowski D., Burke D.H.. Differential susceptibility of HIV-1 reverse transcriptase to inhibition by RNA aptamers in enzymatic reactions monitoring specific steps during genome replication. J. Biol. Chem. 2006; 281:25712–25722. [DOI] [PubMed] [Google Scholar]
- 5. Lange M.J., Sharma T.K., Whatley A.S., Landon L.A., Tempesta M.A., Johnson M.C., Burke D.H.. Robust suppression of HIV replication by intracellularly expressed reverse transcriptase aptamers is independent of ribozyme processing. Mol. Ther. 2012; 20:2304–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Whatley A.S., Ditzler M.A., Lange M.J., Biondi E., Sawyer A.W., Chang J.L., Franken J.D., Burke D.H.. Potent inhibition of HIV-1 reverse transcriptase and replication by nonpseudoknot, “UCAA-motif” RNA aptamers. Mol. Ther. Nucleic Acids. 2013; 2:e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lange M.J., Nguyen P.D.M., Callaway M.K., Johnson M.C., Burke D.H.. RNA-protein interactions govern antiviral specificity and encapsidation of broad spectrum anti-HIV reverse transcriptase aptamers. Nucleic Acids Res. 2017; 45:6087–6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tuerk C., MacDougal S., Gold L.. RNA pseudoknots that inhibit human immunodeficiency virus type 1 reverse transcriptase. Proc. Natl. Acad. Sci. U.S.A. 1992; 89:6988–6992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ditzler M.A., Bose D., Shkriabai N., Marchand B., Sarafianos S.G., Kvaratskhelia M., Burke D.H.. Broad-spectrum aptamer inhibitors of HIV reverse transcriptase closely mimic natural substrates. Nucleic Acids Res. 2011; 39:8237–8247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kensch O., Connolly B.A., Steinhoff H.J., McGregor A., Goody R.S., Restle T.. HIV-1 reverse transcriptase-pseudoknot RNA aptamer interaction has a binding affinity in the low picomolar range coupled with high specificity. J. Biol. Chem. 2000; 275:18271–18278. [DOI] [PubMed] [Google Scholar]
- 11. Kissel J.D., Held D.M., Hardy R.W., Burke D.H.. Single-stranded DNA aptamer RT1t49 inhibits RT polymerase and RNase H functions of HIV type 1, HIV type 2, and SIVCPZ RTs. AIDS Res. Hum. Retroviruses. 2007; 23:699–708. [DOI] [PubMed] [Google Scholar]
- 12. Kissel J.D., Held D.M., Hardy R.W., Burke D.H.. Active site binding and sequence requirements for inhibition of HIV-1 reverse transcriptase by the RT1 family of single-stranded DNA aptamers. Nucleic Acids Res. 2007; 35:5039–5050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Miller M.T., Tuske S., Das K., DeStefano J.J., Arnold E.. Structure of HIV-1 reverse transcriptase bound to a novel 38-mer hairpin template-primer DNA aptamer. Protein Sci. 2016; 25:46–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Green L., Waugh S., Binkley J.P., Hostomska Z., Hostomsky Z., Tuerk C.. Comprehensive chemical modification interference and nucleotide substitution analysis of an RNA pseudoknot inhibitor to HIV-1 reverse transcriptase. J. Mol. Biol. 1995; 247:60–68. [DOI] [PubMed] [Google Scholar]
- 15. Jaeger J., Restle T., Steitz T.A.. The structure of HIV-1 reverse transcriptase complexed with an RNA pseudoknot inhibitor. EMBO J. 1998; 17:4535–4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Aeksiri N., Songtawee N., Gleeson M.P., Hannongbua S., Choowongkomon K.. Insight into HIV-1 reverse transcriptase-aptamer interaction from molecular dynamics simulations. J. Mol. Model. 2014; 20:2380–2388. [DOI] [PubMed] [Google Scholar]
- 17. Held D.M., Kissel J.D., Thacker S.J., Michalowski D., Saran D., Ji J., Hardy R.W., Rossi J.J., Burke D.H.. Cross-clade inhibition of recombinant human immunodeficiency virus type 1 (HIV-1), HIV-2, and simian immunodeficiency virus SIVcpz reverse transcriptases by RNA pseudoknot aptamers. J. Virol. 2007; 81:5375–5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ditzler M.A., Lange M.J., Bose D., Bottoms C.A., Virkler K.F., Sawyer A.W., Whatley A.S., Spollen W., Givan S.A., Burke D.H.. High-throughput sequence analysis reveals structural diversity and improved potency among RNA inhibitors of HIV reverse transcriptase. Nucleic Acids Res. 2013; 41:1873–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Alam K.K., Chang J.L., Lange M.J., Nguyen P.D.M., Sawyer A.W., Burke D.H.. Poly-target selection identifies broad-spectrum RNA aptamers. Mol. Ther. Nucleic Acids. 2018; 13:605–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gotte M., Maier G., Gross H.J., Heumann H.. Localization of the active site of HIV-1 reverse Transcriptase-associated RNase H Domain on a DNA template using site-specific generated hydroxyl radicals. J. Biol. Chem. 1998; 273:10139–10146. [DOI] [PubMed] [Google Scholar]
- 21. Metzger W., Schickor P., Heumann H.. A cinematographic view of Escherichia coli RNA polymerase translocation. EMBO J. 1989; 8:2745–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chalmers M.J., Busby S.A., Pascal B.D., He Y., Hendrickson C.L., Marshall A.G., Griffin P.R.. Probing protein ligand interactions by automated hydrogen/deuterium exchange mass spectrometry. Anal. Chem. 2006; 78:1005–1014. [DOI] [PubMed] [Google Scholar]
- 23. Pascal B.D., Willis S., Lauer J.L., Landgraf R.R., West G.M., Marciano D., Novick S., Goswami D., Chalmers M.J., Griffin P.R.. HDX workbench: software for the analysis of H/D exchange MS data. J. Am. Soc. Mass Spectrom. 2012; 23:1512–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Delaglio F., Grzesiek S., Vuister G.W., Zhu G., Pfeifer J., Bax A.. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995; 6:277–293. [DOI] [PubMed] [Google Scholar]
- 25. Franke D., Petoukhov M.V., Konarev P.V., Panjkovich A., Tuukkanen A., Mertens H.D.T., Kikhney A.G., Hajizadeh N.R., Franklin J.M., Jeffries C.M. et al.. ATSAS 2.8: a comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J. Appl. Crystallogr. 2017; 50:1212–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xu X., Qiu L., Yan C., Ma Z., Grinter S.Z., Zou X.. Performance of MDockPP in CAPRI rounds 28–29 and 31–35 including the prediction of water-mediated interactions. Proteins. 2017; 85:424–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Katchalski-Katzir E., Shariv I., Eisenstein M., Friesem A.A., Aflalo C., Vakser I.A.. Molecular surface recognition: determination of geometric fit between proteins and their ligands by correlation techniques. Proc. Natl. Acad. Sci. U.S.A. 1992; 89:2195–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen R., Li L., Weng Z.. ZDOCK: an initial-stage protein-docking algorithm. Proteins. 2003; 52:80–87. [DOI] [PubMed] [Google Scholar]
- 29. Tullius T.D., Greenbaum J.A.. Mapping nucleic acid structure by hydroxyl radical cleavage. Curr. Opin. Chem. Biol. 2005; 9:127–134. [DOI] [PubMed] [Google Scholar]
- 30. Jain S.S., Tullius T.D.. Footprinting protein–DNA complexes using the hydroxyl radical. Nat. Protoc. 2008; 3:1092–1100. [DOI] [PubMed] [Google Scholar]
- 31. Goswami D., Tuske S., Pascal B.D., Bauman J.D., Patel D., Arnold E., Griffin P.R.. Differential isotopic enrichment to facilitate characterization of asymmetric multimeric proteins using hydrogen/deuterium exchange mass spectrometry. Anal. Chem. 2015; 87:4015–4022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hsiou Y., Ding J., Das K., Clark A.D. Jr, Hughes S.H., Arnold E.. Structure of unliganded HIV-1 reverse transcriptase at 2.7 A resolution: implications of conformational changes for polymerization and inhibition mechanisms. Structure. 1996; 4:853–860. [DOI] [PubMed] [Google Scholar]
- 33. Kensch O., Restle T., Wohrl B.M., Goody R.S., Steinhoff H.J.. Temperature-dependent equilibrium between the open and closed conformation of the p66 subunit of HIV-1 reverse transcriptase revealed by site-directed spin labelling. J. Mol. Biol. 2000; 301:1029–1039. [DOI] [PubMed] [Google Scholar]
- 34. Rodgers D.W., Gamblin S.J., Harris B.A., Ray S., Culp J.S., Hellmig B., Woolf D.J., Debouck C., Harrison S.C.. The structure of unliganded reverse transcriptase from the human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. U.S.A. 1995; 92:1222–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Esnouf R., Ren J., Ross C., Jones Y., Stammers D., Stuart D.. Mechanism of inhibition of HIV-1 reverse transcriptase by non-nucleoside inhibitors. Nat. Struct. Biol. 1995; 2:303–308. [DOI] [PubMed] [Google Scholar]
- 36. Santos A.F., Lengruber R.B., Soares E.A., Jere A., Sprinz E., Martinez A.M., Silveira J., Sion F.S., Pathak V.K., Soares M.A.. Conservation patterns of HIV-1 RT connection and RNase H domains: identification of new mutations in NRTI-treated patients. PLoS One. 2008; 3:e1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cao S., Chen S.J.. Predicting RNA folding thermodynamics with a reduced chain representation model. RNA. 2005; 11:1884–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xu X., Zhao P., Chen S.J.. Vfold: a web server for RNA structure and folding thermodynamics prediction. PLoS One. 2014; 9:e107504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cao S., Chen S.J.. Physics-based de novo prediction of RNA 3D structures. J. Phys. Chem. B. 2011; 115:4216–4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xu X., Chen S.J.. A method to predict the 3D structure of an RNA scaffold. Methods Mol. Biol. 2015; 1316:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang D., Chen S.J.. IsRNA: An iterative simulated reference state approach to modeling correlated interactions in RNA folding. J. Chem. Theor. Comput. 2018; 14:2230–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R.D., Kale L., Schulten K.. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005; 26:1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Brooks B.R., Brooks C.L. 3rd, Mackerell A.D. Jr, Nilsson L., Petrella R.J., Roux B., Won Y., Archontis G., Bartels C., Boresch S. et al.. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009; 30:1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Huang S.Y., Zou X.. A knowledge-based scoring function for protein-RNA interactions derived from a statistical mechanics-based iterative method. Nucleic Acids Res. 2014; 42:e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chattopadhyay D., Evans D.B., Deibel M.R. Jr, Vosters A.F., Eckenrode F.M., Einspahr H.M., Hui J.O., Tomasselli A.G., Zurcher-Neely H.A., Heinrikson R.L. et al.. Purification and characterization of heterodimeric human immunodeficiency virus type 1 (HIV-1) reverse transcriptase produced by in vitro processing of p66 with recombinant HIV-1 protease. J. Biol. Chem. 1992; 267:14227–14232. [PubMed] [Google Scholar]
- 46. Hostomska Z., Matthews D.A., Davies J.F. 2nd, Nodes B.R., Hostomsky Z.. Proteolytic release and crystallization of the RNase H domain of human immunodeficiency virus type 1 reverse transcriptase. J. Biol. Chem. 1991; 266:14697–14702. [PubMed] [Google Scholar]
- 47. Tomasselli A.G., Sarcich J.L., Barrett L.J., Reardon I.M., Howe W.J., Evans D.B., Sharma S.K., Heinrikson R.L.. Human immunodeficiency virus type-1 reverse transcriptase and ribonuclease H as substrates of the viral protease. Protein Sci. 1993; 2:2167–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Abram M.E., Parniak M.A.. Virion instability of human immunodeficiency virus type 1 reverse transcriptase (RT) mutated in the protease cleavage site between RT p51 and the RT RNase H domain. J. Virol. 2005; 79:11952–11961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zheng X., Pedersen L.C., Gabel S.A., Mueller G.A., Cuneo M.J., DeRose E.F., Krahn J.M., London R.E.. Selective unfolding of one Ribonuclease H domain of HIV reverse transcriptase is linked to homodimer formation. Nucleic Acids Res. 2014; 42:5361–5377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. London R.E. Structural maturation of HIV-1 reverse transcriptase-A metamorphic solution to genomic instability. Viruses. 2016; 8:E260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ilina T.V., Slack R.L., Elder J.H., Sarafianos S.G., Parniak M.A., Ishima R.. Effect of tRNA on the maturation of HIV-1 reverse transcriptase. J. Mol. Biol. 2018; 430:1891–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sluis-Cremer N., Arion D., Abram M.E., Parniak M.A.. Proteolytic processing of an HIV-1 pol polyprotein precursor: insights into the mechanism of reverse transcriptase p66/p51 heterodimer formation. Int. J. Biochem. Cell Biol. 2004; 36:1836–1847. [DOI] [PubMed] [Google Scholar]
- 53. Schneider D.J., Feigon J., Hostomsky Z., Gold L.. High-affinity ssDNA inhibitors of the reverse transcriptase of type 1 human immunodeficiency virus. Biochemistry. 1995; 34:9599–9610. [DOI] [PubMed] [Google Scholar]
- 54. Jacobo-Molina A., Ding J., Nanni R.G., Clark A.D. Jr, Lu X., Tantillo C., Williams R.L., Kamer G., Ferris A.L., Clark P. et al.. Crystal structure of human immunodeficiency virus type 1 reverse transcriptase complexed with double-stranded DNA at 3.0 A resolution shows bent DNA. Proc. Natl. Acad. Sci. U.S.A. 1993; 90:6320–6324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chung S., Miller J.T., Lapkouski M., Tian L., Yang W., Le Grice S.F.. Examining the role of the HIV-1 reverse transcriptase p51 subunit in positioning and hydrolysis of RNA/DNA hybrids. J. Biol. Chem. 2013; 288:16177–16184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kohlstaedt L.A., Wang J., Friedman J.M., Rice P.A., Steitz T.A.. Crystal structure at 3.5 A resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science. 1992; 256:1783–1790. [DOI] [PubMed] [Google Scholar]
- 57. Ren J., Esnouf R., Garman E., Somers D., Ross C., Kirby I., Keeling J., Darby G., Jones Y., Stuart D. et al.. High resolution structures of HIV-1 RT from four RT-inhibitor complexes. Nat. Struct. Biol. 1995; 2:293–302. [DOI] [PubMed] [Google Scholar]
- 58. Das K., Martinez S.E., Bauman J.D., Arnold E.. HIV-1 reverse transcriptase complex with DNA and nevirapine reveals non-nucleoside inhibition mechanism. Nat. Struct. Mol. Biol. 2012; 19:253–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sharaf N.G., Ishima R., Gronenborn A.M.. Conformational plasticity of the NNRTI-Binding pocket in HIV-1 Reverse transcriptase: A fluorine nuclear magnetic resonance study. Biochemistry. 2016; 55:3864–3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sarafianos S.G., Marchand B., Das K., Himmel D.M., Parniak M.A., Hughes S.H., Arnold E.. Structure and function of HIV-1 reverse transcriptase: molecular mechanisms of polymerization and inhibition. J. Mol. Biol. 2009; 385:693–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Seckler J.M., Barkley M.D., Wintrode P.L.. Allosteric suppression of HIV-1 reverse transcriptase structural dynamics upon inhibitor binding. Biophys. J. 2011; 100:144–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hartig J.S., Najafi-Shoushtari S.H., Grune I., Yan A., Ellington A.D., Famulok M.. Protein-dependent ribozymes report molecular interactions in real time. Nat. Biotechnol. 2002; 20:717–722. [DOI] [PubMed] [Google Scholar]
- 63. Huang C.C., Cao Z., Chang H.T., Tan W.. Protein-protein interaction studies based on molecular aptamers by affinity capillary electrophoresis. Anal. Chem. 2004; 76:6973–6981. [DOI] [PubMed] [Google Scholar]
- 64. Wu X., Liu H., Xiao H., Conway J.A., Hehl E., Kalpana G.V., Prasad V., Kappes J.C.. Human immunodeficiency virus type 1 integrase protein promotes reverse transcription through specific interactions with the nucleoprotein reverse transcription complex. J. Virol. 1999; 73:2126–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dobard C.W., Briones M.S., Chow S.A.. Molecular mechanisms by which human immunodeficiency virus type 1 integrase stimulates the early steps of reverse transcription. J. Virol. 2007; 81:10037–10046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tekeste S.S., Wilkinson T.A., Weiner E.M., Xu X., Miller J.T., Le Grice S.F., Clubb R.T., Chow S.A.. Interaction between reverse transcriptase and integrase is required for reverse transcription during HIV-1 replication. J. Virol. 2015; 89:12058–12069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hehl E.A., Joshi P., Kalpana G.V., Prasad V.R.. Interaction between human immunodeficiency virus type 1 reverse transcriptase and integrase proteins. J. Virol. 2004; 78:5056–5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Drummond J.E., Mounts P., Gorelick R.J., Casas-Finet J.R., Bosche W.J., Henderson L.E., Waters D.J., Arthur L.O.. Wild-type and mutant HIV type 1 nucleocapsid proteins increase the proportion of long cDNA transcripts by viral reverse transcriptase. AIDS Res. Hum. Retroviruses. 1997; 13:533–543. [DOI] [PubMed] [Google Scholar]
- 69. Cameron C.E., Ghosh M., Le Grice S.F., Benkovic S.J.. Mutations in HIV reverse transcriptase which alter RNase H activity and decrease strand transfer efficiency are suppressed by HIV nucleocapsid protein. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:6700–6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Peliska J.A., Balasubramanian S., Giedroc D.P., Benkovic S.J.. Recombinant HIV-1 nucleocapsid protein accelerates HIV-1 reverse transcriptase catalyzed DNA strand transfer reactions and modulates RNase H activity. Biochemistry. 1994; 33:13817–13823. [DOI] [PubMed] [Google Scholar]
- 71. Nishitsuji H., Yokoyama M., Sato H., Yamauchi S., Takaku H.. Identification of amino acid residues in HIV-1 reverse transcriptase that are critical for the proteolytic processing of Gag-Pol precursors. FEBS Lett. 2011; 585:3372–3377. [DOI] [PubMed] [Google Scholar]
- 72. Bottcher M., Grosse F.. HIV-1 protease inhibits its homologous reverse transcriptase by protein-protein interaction. Nucleic Acids Res. 1997; 25:1709–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Goobar-Larsson L., Luukkonen B.G., Unge T., Schwartz S., Utter G., Strandberg B., Oberg B.. Enhancement of HIV-1 proteinase activity by HIV-1 reverse transcriptase. Virology. 1995; 206:387–394. [DOI] [PubMed] [Google Scholar]
- 74. Wapling J., Moore K.L., Sonza S., Mak J., Tachedjian G.. Mutations that abrogate human immunodeficiency virus type 1 reverse transcriptase dimerization affect maturation of the reverse transcriptase heterodimer. J. Virol. 2005; 79:10247–10257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Figueiredo A., Moore K.L., Mak J., Sluis-Cremer N., de Bethune M.P., Tachedjian G.. Potent nonnucleoside reverse transcriptase inhibitors target HIV-1 Gag-Pol. PLoS Pathog. 2006; 2:e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Jochmans D., Anders M., Keuleers I., Smeulders L., Krausslich H.G., Kraus G., Muller B.. Selective killing of human immunodeficiency virus infected cells by non-nucleoside reverse transcriptase inhibitor-induced activation of HIV protease. Retrovirology. 2010; 7:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.