

Abstract
Degradation of endoplasmic reticulum (ER) by selective autophagy (ER‐phagy) is crucial for ER homeostasis. However, it remains unclear how ER scission is regulated for subsequent autophagosomal sequestration and lysosomal degradation. Here, we show that oligomerization of ER‐phagy receptor FAM134B (also referred to as reticulophagy regulator 1 or RETREG1) through its reticulon‐homology domain is required for membrane fragmentation in vitro and ER‐phagy in vivo. Under ER‐stress conditions, activated CAMK2B phosphorylates the reticulon‐homology domain of FAM134B, which enhances FAM134B oligomerization and activity in membrane fragmentation to accommodate high demand for ER‐phagy. Unexpectedly, FAM134B G216R, a variant derived from a type II hereditary sensory and autonomic neuropathy (HSAN) patient, exhibits gain‐of‐function defects, such as hyperactive self‐association and membrane scission, which results in excessive ER‐phagy and sensory neuron death. Therefore, this study reveals a mechanism of ER membrane fragmentation in ER‐phagy, along with a signaling pathway in regulating ER turnover, and suggests a potential implication of excessive selective autophagy in human diseases.
Keywords: CAMK2B, ER stress, ER‐phagy, FAM134B oligomerization, membrane fragmentation
Subject Categories: Autophagy & Cell Death; Post-translational Modifications, Proteolysis & Proteomics
CAMK2B‐dependent phosphorylation of autophagy receptor FAM134B promotes its oligomerization and membrane‐scission activity, a process deregulated in sensory neuropathy.

Introduction
The endoplasmic reticulum (ER) is the largest intracellular organelle, which constitutes a continuous intracellular network of sheet and tubular membrane structures (Shibata et al, 2006). ER plays essential roles in protein and lipid synthesis, calcium homeostasis, organelle communication and innate immunity (Shibata et al, 2009). The oscillation of ER size and shape in response to varying environmental cues is crucial to cell homeostasis (Walter & Ron, 2011). Elimination of redundant ER is mediated by a selective autophagy pathway, which is coined as ER‐phagy (Bernales et al, 2006, 2007).
Selective autophagy is a cellular quality control pathway through which a variety of autophagy cargoes are specifically engulfed by autophagosomes and delivered to lysosomes for degradation (Stolz et al, 2014; Farre & Subramani, 2016; Zaffagnini & Martens, 2016; Gatica et al, 2018). The specificity of this process is governed by autophagy receptors that simultaneously bind to cargoes and the LC3 family members on the expanding autophagosomal membranes (Khaminets et al, 2016; Gatica et al, 2018). Recent studies have greatly advanced the understanding of ER‐phagy by identifying the key autophagy receptors (Khaminets et al, 2015; Mochida et al, 2015; Fumagalli et al, 2016; Grumati et al, 2017; Smith et al, 2017; An et al, 2019; Chen et al, 2019; Chino et al, 2019).
One essential question that remains unsolved is how the ER membrane is fragmented into “bite size” for subsequent autophagosomal sequestration (Mochida et al, 2015; Nakatogawa & Mochida, 2015). Moreover, little is known about how environmental or intracellular signals are transduced to trigger ER‐phagy in a time‐dependent manner (Rubinsztein, 2015). Lastly, the cause and consequence of excessive ER‐phagy are poorly understood (Rubinsztein, 2015). FAM134B‐mediated ER‐phagy appears to be a good model system to address above questions, because FAM134B induces liposome fragmentation in vitro and mediates ER‐phagy in vivo (Khaminets et al, 2015), and more importantly, the dysfunction of FAM134B causes hereditary sensory and autonomic neuropathy type 2 (HSAN II; Kurth et al, 2009).
In this study, we show that FAM134B oligomerization drives the fragmentation of ER prior to ER‐phagy. ER stress subsequently triggers CAMK2B‐mediated FAM134B phosphorylation, which further enhances FAM134B oligomerization, ER scission, and ER‐phagy. To strengthen our model, we provide evidence that a type II HSAN patient‐derived FAM134B variant, FAM134BG216R, appears to be a gain‐of‐function mutant, as it is hyperactive in oligomerization, ER fragmentation, and ER‐phagy, which results in sensory neuron death.
Results
FAM134B forms oligomers that are required for ER fragmentation and ER‐phagy
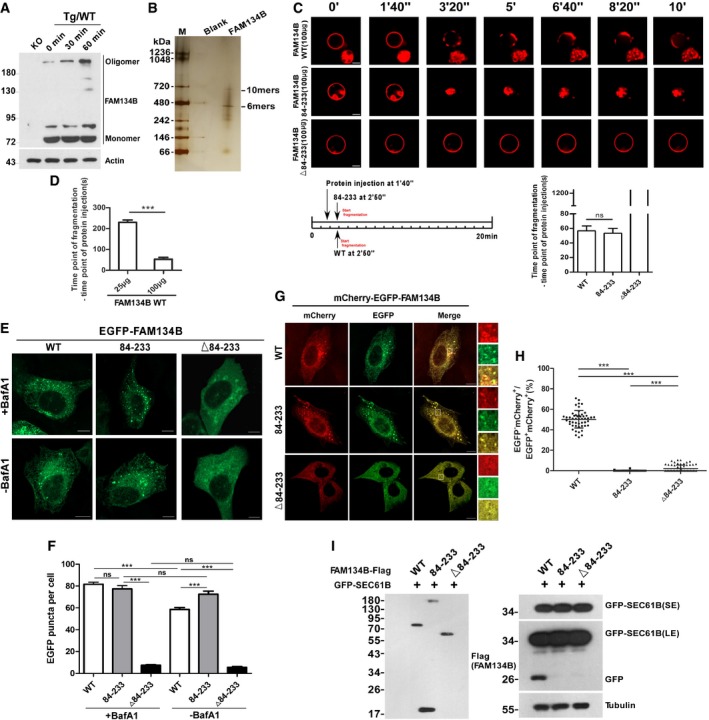
We observed that FAM134B formed oligomers in vivo and that this process was further enhanced by Thapsigargin (Tg)‐induced ER stress (Fig 1A) or starvation (Appendix Fig S1A). These oligomers were partially resistant to denaturing solutions containing SDS and DTT. In contrast, the oligomerization of another reticulon protein, Reticulon 4 (RTN4), was not altered under the same conditions (Appendix Fig S1B). Purified recombinant human FAM134B analysis by native PAGE (Fig 1B) and size‐exclusion chromatography (Appendix Fig S1C) revealed that the molecular weight of FAM134B oligomers was approximately 450–700 kD. Mutational analysis uncovered that the amino acids ranging from 84 to 233, the internal reticulon domain (RTND) of FAM134B, were required and sufficient for its self‐association and oligomerization (Appendix Fig S1D–F). To measure ER fragmentation activity in vitro, we established a liposome fragmentation assay (Appendix Fig S1G), and we observed that purified recombinant FAM134BWT or FAM134B84‐233 (RTND) but not the RTND‐deletion mutant FAM134BΔ84‐233 was able to induce liposome fragmentation in a dose‐dependent manner (Fig 1C and D, and Appendix Fig S1H). In contrast, recombinant proteins for RTN4 WT or its RTND failed to induce liposome fragmentation in vitro at the same conditions (Appendix Fig S1I–L). To measure ER membrane scission in vivo, we chose the U2OS cell line because it expresses low levels of endogenous FAM134B (Khaminets et al, 2015), and we used Bafilomycin A to block lysosomal activity, which resulted in accumulation of ER membrane fragments. We observed that the expression of FAM134BWT or FAM134B84‐233 but not that of FAM134BΔ84‐233 induced the formation of puncta that were positive for both FAM134B and BAP31, an ER marker (Fig 1E and F, and Appendix Fig S1M). Therefore, FAM134B84‐233 appeared to be required and sufficient to induce ER membrane scission in vivo at overexpression conditions. To measure ER‐phagy activity, we applied the mCherry·EGFP tandem tagging strategy and GFP‐cleavage assays (Khaminets et al, 2015; Klionsky et al, 2016). When the ER membrane fragments are digested by autolysosomes, they will either appear as mCherry+EGFP‐ foci under confocal microscope because mCherry is more stable in an acidic environment or they will produce free GFP visualized by Western analysis because of the partial digestion of GFP‐SEC61B, an ER sheet resident protein. We observed that although FAM134B84‐233 could fragmentate ER membrane, it failed to induce ER‐phagy and deliver the fragmentated ER into autolysosomes for degradation (Fig 1G–I) due to the lack of an LC3‐interacting motif (LIR; Appendix Fig S1F). In contrast, the mutant, FAM134BΔ84‐233, which contains LIR but not RTND, was not able to induce ER‐phagy, because of the missing oligomerization and ER‐binding activities (Fig 1G–I). By transmission electron microscopy (TEM) and immunoelectron microscopy analysis, we confirmed that the expression of exogenous FAM134B WT was able to fragmentate ER membrane without inducing apoptotic response (Appendix Fig S1N–P), and these results indicated that the puncta structures positive for exogenous FAM134B appeared not to be ER membrane blebs. Together, RTND mediates FAM134B oligomerization that is required for ER membrane fragmentation and ER‐phagy.
Figure 1. FAM134B forms oligomers that are required for ER membrane scission and ER‐phagy.
-
AEndogenous FAM134B forms oligomers. FAM134B knockout (KO) or wild‐type (WT) 293T cells were treated with 1 μM of Thapsigargin (Tg) for different time, and the cell lysates were prepared and analyzed by Western blot.
-
BAnalysis of the oligomer size of recombinant FAM134B WT using native PAGE.
-
CIn vitro liposome fragmentation assay. After the injection of recombinant proteins (100 μg/100 μl) into the chamber (500 μl), the morphological changes of liposomes were monitored by live imaging for 20 min. The images at different time points as indicated are presented. Scale bars, 10 μm. “ns” means no significance, one‐way ANOVA; error bars indicate SEM (n = 3).
-
DQuantification of the time between protein addition and liposome fragmentation. ***P < 0.001, one‐way ANOVA; error bars indicate SEM (n = 3).
-
E, FMeasurement of the intracellular ER‐scission activity. U2OS cells transiently expressing EGFP‐FAM134B (WT), EGFP‐FAM134B (84–233), or EGFP‐FAM134B (Δ84–233) at same levels, lysosomal degradation of EGFP‐FAM134B was blocked by Bafilomycin A1 (Baf A1) or DMSO. GFP‐positive puncta were quantified for each cell in (F). For each group, at least 30 cells were counted. Scale bars, 10 μm. ***P < 0.001, “ns” means no significance, one‐way ANOVA; error bars indicate SEM.
-
G, HMeasurement of the ER‐phagy activity. U2OS cells transiently expressing mCherry‐EGFP‐FAM134B (WT), mCherry‐EGFP‐FAM134B (84–233), or mCherry‐EGFP‐FAM134B (Δ84–233) at same levels. ER‐phagy is indicated by mCherry‐positive but EGFP‐negative puncta were quantified for each cell in (H). For mCherry‐EGFP‐FAM134B (WT), 50 cells were counted (n = 50); for mCherry‐EGFP‐FAM134B (84–233), n = 56; for mCherry‐EGFP‐FAM134B (Δ84–233), n = 66. Scale bars, 10 μm. The scale bars in the magnification boxes are 2 μm. ***P < 0.001, one‐way ANOVA; error bars indicate SEM.
-
ILysosomal cleavage of GFP was analyzed by Western blot for the cells co‐expressing GFP‐SEC61B and FAM134B WT‐Flag, FAM134B (84–233)‐Flag, or FAM134B (Δ84–233)‐Flag.
Source data are available online for this figure.
Phosphorylation enhances FAM134B oligomerization and its activity in membrane fragmentation and ER‐phagy
To understand how FAM134B oligomerization was increased under autophagy‐stimulating or ER‐stress conditions, we conducted mass spectrometry analysis, which resulted in the identification of serine 149, 151, and 153 (S149, S151, S153) as potential FAM134B phosphorylation sites (Fig 2A, Appendix Fig S2A and B). Notably, all of these three residues reside within RTND. Mimicking de‐phosphorylation by mutating these serines (S) into alanines (A) individually reduced FAM134B self‐interaction (Fig 2B). In contrast, imitating permanent phosphorylation by replacing the serines (S) with aspartate (D) enhanced FAM134B self‐association (Fig 2B). Consistently, simultaneous substitution of all three serines with alanines (FAM134BSA) or aspartates (FAM134BSD) significantly decreased or increased FAM134B self‐binding, respectively (Fig 2C and Appendix Fig S2C). Consistently, purified recombinant protein of FAM134BSD exhibited significantly higher activity in liposome fragmentation than FAM134BWT (Fig 2D and Appendix Fig S2D). Under overexpression conditions, FAM134B phosphorylation significantly increased its activity in membrane fragmentation and ER‐phagy in cultured U2OS cells (Fig 2E; Appendix Fig S2E–H). To further solidify this conclusion, we developed a stringent rescue assay. To do this, we first established FAM134B knockout (KO) cell lines, and we then used the FAM134B KO cells to set up a serial of stable cell lines which individually express different forms of FAM134B proteins in an inducible manner (Appendix Fig S2I and J). By titrating the inducer (Doxycycline, DOX) concentration and the induction time, we were able to express exogenous FAM134B proteins close to the endogenous levels (Appendix Fig S2K). Under these optimized conditions, we further confirmed the important role of FAM134B phosphorylation in membrane fragmentation and ER‐phagy (Fig 2F–I). These results demonstrate that phosphorylation regulates FAM134B's activities in oligomerization, ER scission, and ER‐phagy.
Figure 2. Phosphorylation enhances FAM134B oligomerization and its activity in membrane scission and ER‐phagy.

-
AMass spectrometry analysis uncovered that FAM134B was phosphorylated at serine 151.
-
BComparison of the self‐interaction of FAM134B mono‐phosphorylation mutants using co‐IP.
-
CComparison of the self‐interaction of FAM134 tri‐phosphorylation mutants using co‐IP.
-
DComparison of membrane scission activity using in vitro liposome fragmentation assay. Scale bars, 10 μm. ***P < 0.001, one‐way ANOVA; error bars indicate SEM (n ≧ 3).
-
ELysosomal cleavage of GFP was analyzed by Western blot for the cells co‐expressing GFP‐SEC61B and FAM134B (WT)‐Flag, FAM134B (SA)‐Flag, or FAM134B (SD)‐Flag.
-
F, GMeasurement of the intracellular ER‐scission activity. FAM134B knockout (KO) U2OS cells were engineered to express EGFP‐FAM134B (WT), EGFP‐FAM134B (SA), or EGFP‐FAM134B (SD) at endogenous levels, and lysosomal degradation of EGFP‐FAM134B was blocked by Bafilomycin A1 (Baf A1). GFP‐positive puncta were quantified for each cell in (G). For control, 25 cells were counted (n = 25); for EGFP‐FAM134B (WT), n = 27; for EGFP‐FAM134B (SA), n = 29; for EGFP‐FAM134B (SD), n = 25. Scale bars, 10 μm. ***P < 0.001, one‐way ANOVA; error bars indicate SEM.
-
H, IMeasurement of the ER‐phagy activity. FAM134B knockout (KO) U2OS cells were engineered to express mCherry‐EGFP‐FAM134B (WT), mCherry‐EGFP‐FAM134B (S151A), or mCherry‐EGFP‐FAM134B (S151D) at endogenous levels. Lysosomal mCherry‐positive but GFP‐negative puncta were quantified for each cell in (I). For mCherry‐EGFP‐FAM134B (WT), 25 cells were counted (n = 25); for mCherry‐EGFP‐FAM134B (SA), n = 23; for mCherry‐EGFP‐FAM134B (SD), n = 21. Scale bars, 10 μm. The scale bars in the magnification boxes are 2 μm. ***P < 0.001, one‐way ANOVA; error bars indicate SEM.
Source data are available online for this figure.
CAMK2B phosphorylates FAM134B at serine 151 to promote oligomerization, ER fragmentation, and ER‐phagy
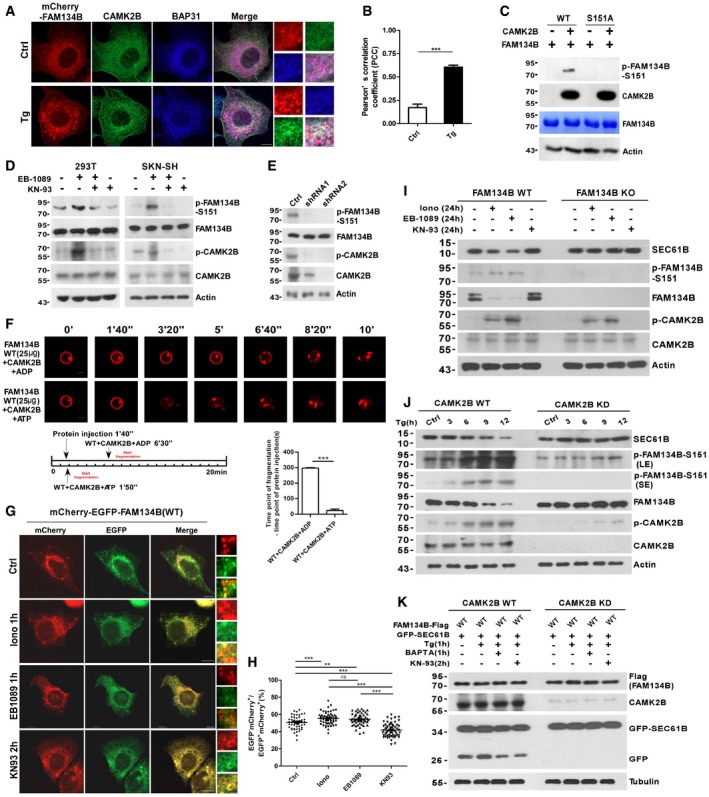
To understand how upstream signals regulate FAM134B phosphorylation, we first selected a panel of candidate kinases based on bioinformatic analysis of the sequence of the phosphorylation sites (S149, S151, S153). These kinases were further screened by in vitro kinase assays and mass spectrometry analyses (Appendix Fig S2L), which resulted in the identification of CAMK2B as the candidate kinase phosphorylating FAM134B at S151 (Appendix Fig S3A). It is reasonable to postulate that ER stress leads to the elevation of cytoplasmic calcium levels, which subsequently activates CAMK2B to trigger ER‐phagy through FAM134B. Indeed, Tg treatment enhanced the interaction between CAMK2B and calmodulin (Appendix Fig S3B), which is the calcium sensor and plays a key role in CAMK2B activation, and Tg treatment also increased the colocalization and the association of CAMK2B with ER membrane structures (Fig 3A and B; Appendix Fig S3C). CAMK2B interacted with FAM134B under physiological conditions (Appendix Fig S3D and E), and CAMK2B phosphorylates FAM134B at S151 in in vitro kinase assays, which were validated by Western blot (Fig 3C) using a specific phosphor‐antibody recognizing phosphorylated S151 of human FAM134B (p‐FAM134B‐S151) and by radioautography (Appendix Fig S3F). In addition, we also showed that mutating S151 to alanine (S151A) totally abolished the phosphorylation signal (Fig 3C and Appendix Fig S3F). Furthermore, the CAMK2B activators Ionomycin and EB1089 enhanced FAM134B phosphorylation at S151 in a time‐ or a dose‐dependent manner in different cell lines (Appendix Fig S3G–N). In contrast, treating cells with the CAMK2B inhibitor KN‐93 or with CAMK2B‐specific shRNA repressed S151 phosphorylation (Fig 3D and E). Indeed, CAMK2B was able to stimulate FAM134B‐mediated liposome fragmentation in vitro (Fig 3F). The CAMK2B activators or inhibitor stimulated or repressed ER scission and ER‐phagy in cultured cells (Fig 3G and H; Appendix Fig S3O–R). More importantly, modulating CAMK2B activity by small molecules was able to dramatically alter ER‐phagy levels in FAM134B or CAMK2B WT cells but not in FAM134B KO or CAMK2B knockdown (KD) cells, which further demonstrated the importance of CAMK2B‐FAM134B signaling axis in ER‐phagy (Fig 3I–K; Appendix Fig S3S and T). Therefore, the CAMK2B‐FAM134B axis relays upstream signals to ER‐phagy machineries to maintain ER homeostasis.
Figure 3. CAMK2B phosphorylates FAM134B at Ser151 to enhance ER fragmentation and ER‐phagy.
-
A, BEndogenous CAMK2B redistribution to ER membrane structures labeled by mCherry‐FAM134B and BAP31 upon Tg treatment. Scale bars, 10 μm. The scale bars in the magnification boxes are 2 μm. The colocalization was analyzed by Pearson's correlation coefficient (PCC) in (B). For Ctrl, 16 cells were counted (n = 16); for Tg treatment, 18 cells were counted (n = 18). ***P < 0.001, one‐way ANOVA; error bars indicate SEM.
-
CIn vitro FAM134B S151 phosphorylation by CAMK2B was detected by Western blot. Recombinant proteins for FAM134BWT and FAM134BS151A purified from E. coli were incubated with purified CAMK2B by IP in kinase buffer. FAM134B phosphorylation was analyzed by a specific antibody recognizing phos‐Serine151 of human FAM134B.
-
DFAM134B S151 phosphorylation in cells treated with CAMK2 activator (100 nM EB1089 for 1 h) or/and inhibitor (10 μM KN93 for 2 h). 293T or SKN‐SH (a cell line derived from neuroblastoma) cells were treated with drugs as indicated, and whole cell lysates were analyzed by phospho‐FAM134B (S151) antibody.
-
EFAM134B S151 phosphorylation in CAMK2B knockdown 293T cells.
-
FIn vitro reconstitution of CAMK2B‐FAM134B‐mediated membrane fragmentation using liposome assay. Purified recombinant FAM134B and CAMK2B were preincubated in kinase buffer with ADP or ATP at 30°C for 10 min, and the resultant protein mixtures were transferred to chamber coated with liposomes. Liposome fragmentation was monitored by live imaging. Scale bars, 10 μm. ***P < 0.001, one‐way ANOVA; error bars indicate SEM (n = 3).
-
G, HMeasurement of the ER‐phagy activity. U2OS cells transiently expressing mCherry‐EGFP‐FAM134B (WT) were treated with compounds as indicated. Lysosomal mCherry‐positive but GFP‐negative puncta were quantified for each cell in (H). For vehicle control, 53 cells were counted (n = 53); for Ionomycin, n = 56; for EB1089, n = 54; for KN‐93, n = 58. Scale bars, 10 μm. The scale bars in the magnification boxes are 2 μm. ***P < 0.001, one‐way ANOVA; error bars indicate SEM.
-
IFAM134B WT or knockout (KO) U2OS cells were treated with CAMK2 activator Ionomycin (Iono), EB1089, or inhibitor KN93 for 24 h, and Western blot was performed to analyze the proteins as indicated.
-
JCAMK2B WT or knockdown (KD) U2OS cells were treated with 1 μM Thapsigargin (Tg) to induce ER stress for different time as indicated. Cells were collected and analyzed for proteins as indicated by Western blot.
-
KGFP‐cleavage assay. CAMK2B WT or knockdown (KD) U2OS cells were co‐transfected with FAM134B‐Flag and GFP‐SEC61B, and 24 h posttransfection, cells were treated with drugs as indicated and analyzed for GFP cleavage by Western blot. Note: The levels of Flag‐FAM134B, CAMK2B, GFP‐SEC61B, and Tubulin were analyzed by Western blot using different membranes.
Source data are available online for this figure.
The patient‐derived variant FAM134BG216R is a gain‐of‐function mutant in oligomerization, membrane scission, and ER‐phagy
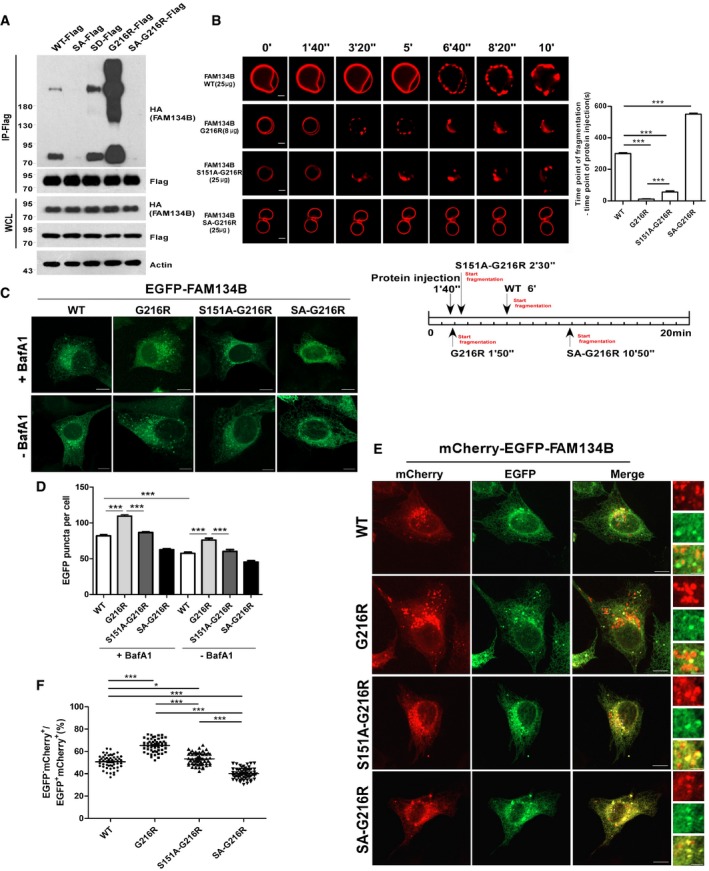
FAM134B mutations cause type II HSAN, which is attributed to cell death of sensory neurons. Most of these type II HSAN patient‐derived FAM134B mutants possess frameshift mutations or premature stop codons, leading to loss‐of‐function defects (Kurth et al, 2009; Davidson et al, 2012; Murphy et al, 2012). However, there is one pathogenic FAM134B variant, FAM134G216R, with uncertain significance (Davidson et al, 2012). Because G216R resides in the RTND of FAM134B, we postulated that this variant may affect FAM134B oligomerization. Surprisingly, FAM134BG216R displayed dramatically enhanced self‐interaction and oligomerization (Fig 4A), which was mitigated by mutating S149, 151, and 153 into alanines either simultaneously (SA‐G216R) (Fig 4A and Appendix Fig S4A) or individually (S149A‐G216R, S151A‐G216R, S153A‐G216R; Appendix Fig S4B). As expected, FAM134BG216R showed significantly higher activity in liposome fragmentation, which was partially neutralized by the S149A, S151A, and S153A mutations (Fig 4B and Appendix Fig S4C–F). Under overexpression conditions, FAM134BG216R was able to induce ER scission and ER‐phagy in a more efficient manner than FAM134BWT and other mutants in cultured cells (Fig 4C–F and Appendix Fig S4G). The puncta structure of FAM134BG216R and other mutants were mostly positive for BAP31 and LC3 (Appendix Fig S4H), excluding the possibility of forming aggregates or ER membrane blebs. Furthermore, we performed a rescue assay by adopting a similar strategy that was described in Appendix Fig S2I–K. By expressing exogenous FAM134B proteins at the endogenous levels (Appendix Fig S2K), we confirmed that FAM134BG216R appeared to be a gain‐of‐function mutant by measurement of ER membrane fragmentation and ER‐phagy (Appendix Fig S4I–L). Together, FAM134BG216R is hyperactive in oligomerization, which results in aberrant ER scission and excessive ER‐phagy.
Figure 4. A HSAN II patient‐derived variant, FAM134BG 216R, is a gain‐of‐function mutant in oligomerization, ER scission, and ER‐phagy.
-
AComparison of the self‐interaction of FAM134B WT with mutants as indicated using co‐IP. SA‐G216R was constructed by simultaneously mutating S149, S151, and S153 to alanine (SA) on the basis of FAM134B‐G216R.
-
BIn vitro liposome fragmentation assay. Scale bars, 10 μm. ***P < 0.001, one‐way ANOVA; error bars indicate SEM (n ≧ 3).
-
C, DMeasurement of the intracellular ER‐scission activity of FAM134B WT, G216R, S151A‐G216R, and SA‐G216R. GFP‐tagged FAM134B proteins were expressed in U2OS cells at same levels. The autolysosomal degradation of ER fragments labeled by GFP‐FAM134B was blocked by Baf A1 or DMSO. GFP‐positive puncta were quantified for each cell in (D). For all of the groups, at least 30 cells were included for quantification; Scale bars, 10 μm. ***P < 0.001, one‐way ANOVA; error bars indicate SEM.
-
E, FMeasurement of the ER‐phagy activity of FAM134BG216R. U2OS cells transiently expressing mCherry‐EGFP‐FAM134B (WT), mCherry‐EGFP‐FAM134B (G216R), mCherry‐EGFP‐FAM134B (S151A‐G216R), or mCherry‐EGFP‐FAM134B (SA‐G216R) at same levels. Lysosomal mCherry‐positive but GFP‐negative puncta were quantified for each cell in (F). For WT, 50 cells were counted (n = 50); for G216R, n = 55; for S151A‐G216R, n = 51; for SA‐G216R, n = 52. Scale bars, 10 μm. The scale bars in the magnification boxes are 2 μm. *P < 0.05, ***P < 0.001, one‐way ANOVA; error bars indicate SEM.
Source data are available online for this figure.
FAM134BG216R induces sensory neuron death in vitro
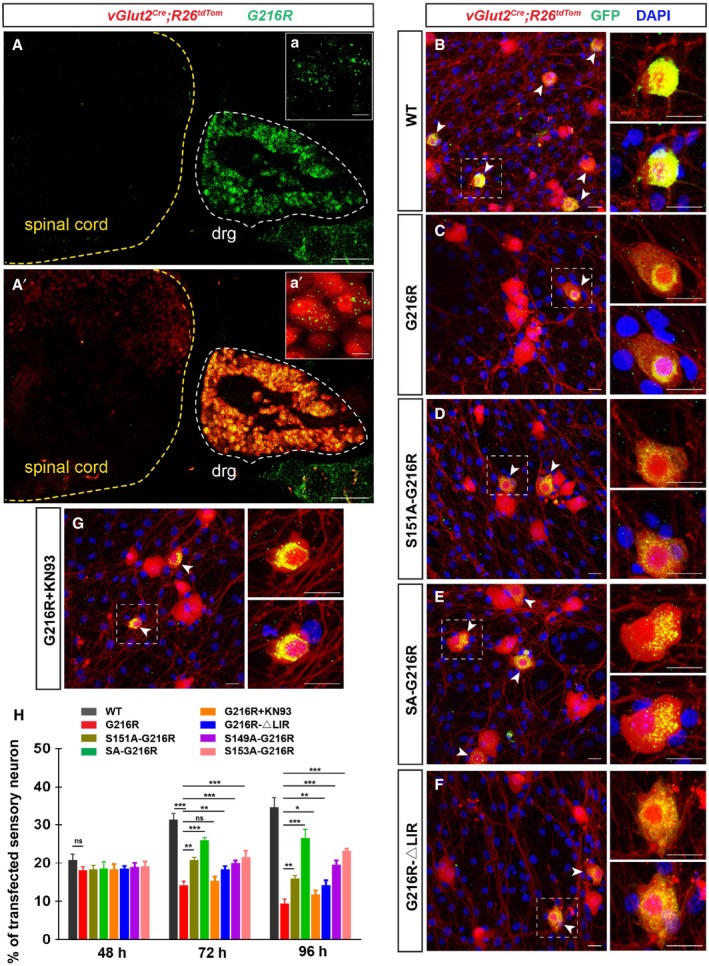
Next, we asked whether FAM134BG216R‐induced excessive ER‐phagy may affect neuronal survival. To test this hypothesis, we first verified the specific high expression of FAM134B in mouse dorsal root ganglion (DRG) in vivo (Fig 5A and A′). We prepared the primary culture of DRG sensory neurons (Wang & Marquardt, 2012) and infected them with lentivirus expressing GFP‐FAM134B WT, G216R, S149A‐G216R, S151A‐G216R, S153A‐G216R, SA‐G216R and G216R‐ΔLIR, respectively. Considering the comparable infection efficiency among various lentivirus and variation in neuron density among different culture experiments, we used the percentages of transfected sensory neuron (GFP+/Tom+) in total sensory neurons (Tom+) to calculate the sensory neuron viability. We observed that mouse DRG neurons infected with lentiviral particles that expressed GFP‐tagged FAM134BG216R proteins underwent progressive cell death, whereas neuronal viability was not altered with FAM134BWT (Fig 5B and C). In addition, the fact that FAM134BS149A‐G216R, FAM134BS151A‐G216R, FAM134B,S153A‐G216R or FAM134BSA‐G216R overexpression in DRG neuron exhibited significant reduced toxicity further indicated that FAM134BG216R‐induced neuronal death was due to its hyperactive oligomerization, and, consequently, excessive ER‐phagy (Fig 5B–E and H; Appendix Fig S5A). Further analysis indicated that FAM134BG216R overexpression resulted in apoptotic neuronal death (Appendix Fig S5B and C), which could be partially mitigated by mutating the LIR motif (Fig 5F and H). We also excluded the possibility that G216R mutation affects FAM134B oligomerization by modulating CAMK2B‐mediated phosphorylation (Appendix Fig S5D–G), although the phosphorylation levels of FAM134B by CAMK2B regulate FAM134BG216R oligomerization. Furthermore, chemically inhibiting CAMK2B activity by KN93 was able to ameliorate the cytotoxicity effect of FAM134BG216R (Fig 5G and H; Appendix Fig S5H and I). Therefore, our results suggest that FAM134BG216R is a gain‐of‐function mutant in causing sensory neuronal cell death by inducing excessive ER scission and ER‐phagy.
Figure 5. FAM134BG 216R induced sensory neuron death.
-
A, A’Specific expression of FAM134B mRNA (green) in sensory neurons (red) of P4d vGlut2 Cre ;R26 lsl‐tdTom mouse. Scale bars, 100 μm. The white dotted line indicates the dorsal root ganglia (DRG) where sensory neurons are genetically labeled with Tomato. Insets of (a) and (a’) are high magnifications of sensory neurons showing the colocalization of FAM134B mRNA and Tomato. Scale bars, 10 μm.
-
B–GCultured sensory neurons from DRGs of E14.5 vGlut2 Cre ;R26 lsl‐tdTom embryos were infected with same titers of recombinant lentivirus expressing GFP‐FAM134B WT (B), G216R (C), S151A‐G216R (D), SA‐G216R (E), G216R‐ΔLIR (F), and G216R supplemented with 5 μM KN93 (G), respectively. Infected sensory neurons (GFP+/Tom+) are indicated with arrowheads. The nuclei of all cells are shown with DAPI staining. Scale bars, 20 μm.
-
HQuantification of the viability of sensory neuron after 48, 72, and 96 h of infection. Sensory neuron viability was calculated as the percentage of transfected sensory neuron (GFP+/Tom+) in total sensory neurons (Tom+). For each time point, four biological replicates were included for statistical analysis. *P < 0.05, **P < 0.01, ***P < 0.001, two‐way ANOVA; error bars indicate SEM. “ns” is the abbreviation for “not significant”. For all of the groups, at least 10 fields of the slides were included.
Discussion
In this study, one factor tuning FAM134B activity in ER membrane fragmentation during ER‐phagy appeared to be CAMK2B, a master regulator of different signal pathways by sensing cytoplasmic calcium concentrations. Therefore, it is conceivable that regional calcium spikes may trigger ER‐phagy through the CAMK2B‐FAM134B signaling axis. In addition, previous studies have shown that CAMK2B regulates the bulk autophagy pathway by phosphorylating Beclin 1 (Li et al, 2017). Therefore, CAMK2B may simultaneously activate the core autophagy machinery and FAM134B to systemically boost ER‐phagy.
We have demonstrated that the concentration and phosphorylation of FAM134B are the two key factors for ER scission and ER‐phagy. Therefore, regional enrichment and activation of FAM134B on ER sheets may determine the timing and magnitude of ER‐phagy. One unsolved puzzle in ER‐phagy is how FAM134B discriminates which part of the ER membrane should be fragmented for autophagic degradation. Presumably, when ER‐phagy is induced, the polarized distribution of FAM134B is mediated by unknown proteins that specify certain microdomains of ER sheets for degradation. Alternatively, the regional enrichment of FAM134B may depend on specific lipids, as previous studies have shown that specific lipid compositions are required for coat protein complex recruitment (Matsuoka et al, 1998; Spang et al, 1998; Crottet et al, 2002).
How ER fragmentation is achieved by other ER‐phagy receptors remains unclear. Yeast Atg40 and mammalian RTN‐3L might adopt a similar strategy, as both molecules also contain RTND and LIR (Mochida et al, 2015; Grumati et al, 2017). However, they differ in subcellular localization, as RTN‐3L localizes to ER tubules and sheet edges in mammalian cells (Hu et al, 2008; Shibata et al, 2010; Grumati et al, 2017) and Atg40 locates to periphery‐ER. In contrast, FAM134B primarily resides on ER sheets (Khaminets et al, 2015; Grumati et al, 2017). Another ER‐phagy receptor, SEC62, also locates on ER sheets (Fumagalli et al, 2016), and interestingly, SEC62 associates with FAM134C (Huttlin et al, 2015), a FAM134 family member that has broader tissue distribution than FAM134B (Kurth et al, 2009). Therefore, SEC62 may cooperate with FAM134 family members to induce ER fragmentation for ER‐phagy. Recently, Atlastins were shown to contribute to ER fragmentation during ER‐phagy (Liang et al, 2018); however, it is unclear how Atlastins seamlessly cooperate with ER‐phagy receptors, which specify certain ER microdomains for autophagic degradation.
In the sensory neuron culture assay, we observed that KN93 treatment reduced the cytotoxicity that was caused by G216R overexpression after 96‐h incubation. Compared with the effect of other mutants such as S149A‐G216R, S151A‐G216R, S153A‐G216R, SA‐G216R, or G216R‐ΔLIR,the mitigation effect of CAMK2B inhibition was weaker. The reason behind this could be that CAMK2B is an essential regulator in neurons, and its inhibition could be toxic to the sensory neurons per se.
In sum, our study not only proposes a model for dynamic regulation of ER membrane scission, but also elucidates a signaling pathway in regulating ER turnover, thereby providing further insights into the spatiotemporal regulation of ER‐phagy. Furthermore, the finding of the link between excessive ER‐phagy and neuronal cell death indicates an essential function of optimal ER‐phagy in maintaining ER equilibrium and neuron homeostasis.
Materials and Methods
Plasmids
Wild‐type HA‐FAM134B was made by cloning cDNA into pcDNA3.1‐HA containing the amino‐terminal HA tag using BamHI and XhoI sites. Wild‐type FAM134B‐Flag was constructed by inserting FAM134B cDNA into pCDNA5/FRT/TO‐3*Flag containing the carboxy‐terminal Flag tag using BamHI. Wild‐type GFP‐FAM134B and GFP‐SEC61B were generated by subcloning FAM134B ORFs into pEGFP‐C1 using XhoI and KpnI. Wild‐type mCherry‐EGFP‐FAM134B was generated by subcloning FAM134B ORFs into pmCherry‐EGFP using KpnI and BamHI. Wild‐type EGFP‐FAM134B was generated by subcloning FAM134B ORFs into FUIPW using AscI and BamHI. HP138‐EGFP‐FAM134B WT and HP138‐mCherry‐EGFP‐FAM134B WT were constructed by seamless cloning. Other FAM134B‐related mutants in this study were generated by site‐directed mutagenesis on the basis of their respective WT constructs.
Antibodies
Anti‐HA‐Tag‐HRP‐direct (M180‐7, MBL, WB:1:5,000), Anti‐Flag‐Tag‐HRP‐direct (M185‐7, MBL, WB:1:5,000), Anti‐GFP (11814460001, Roche, WB:1:4,000), Anti‐GFP (Ab13970, Abcam, IF:1:2,000), Cleaved Caspase‐3 (9661, Cell Signaling, IF:1:1,000), Tuj‐1 (Ab78078, Abcam, IF:1:3,000), Anti‐Beta‐Tubulin (M1305‐2, Huaan Hangzhou, WB:1:5,000), Anti‐Beta‐Actin (M1210‐2, Huaan Hangzhou, WB:1:5,000), Anti‐BAP31 (393810, Santa Cruz, IF:1:500), Anti‐CAMK2 (4436, Cell Signaling, WB:1:1,000), Anti‐pCAMK2 (12716, Cell Signaling, WB:1:1,000), Anti‐Nogo (271878, Santa Cruz, WB:1:5,000), Anti‐Parp1 (ab227244, abcam, WB:1:5,000), Anti‐SEC61B (393633, Santa Cruz, WB:1:1,000), Anti‐LC3 (PM036, MBL, IF:1:200), Alexa Fluor 488 (A11001, Thermo Fisher Scientific, IF:1:1,000), Alexa Fluor 546 (A11003, Thermo Fisher Scientific, IF:1:1,000), Alexa Fluor 594 (A11005, Thermo Fisher Scientific, IF:1:1,000), and Alexa Fluor 405 (A31556, Thermo Fisher Scientific, IF:1:1,000). HRP‐conjugated secondary antibodies were purchased from Invitrogen.
Antibody preparation
FAM134B rabbit polyclonal antibody was raised by DJ Biotech. FAM134B rabbits were immunized against the peptide (C)GYTPQTDTSDDLDRP (amino acids 307‐321; NP_001030023.1) and (C)LPTELKRKKQQLDSAHR (amino acids 352‐368; NP_001030023.1), which were described before (Khaminets et al, 2015). P‐FAM134B (Ser 151) rabbit polyclonal antibody was generated by immunizing four rabbits using the synthesized phosphor‐peptide (C)LWRSL(pS)ESWEV (amino acids 146–156; NP_001030023.1) (WYG Biotech).
Chemicals
Thapsigargin (Tg) was purchased from J&K Scientific. Ionomycin was purchased from Cell Signaling. EB‐1089 was purchased from Santa Cruz, while KN‐93, BAPTA, and staurosporine (STS) were from Selleck. PC, POPS, Biotin‐PE, and Rhodamine‐PE were purchased from Avanti®. Cholesterol, agarose and Bafilomycin A1 (Baf A1), puromycin, and doxycycline hyclate were purchased from Sigma.
Cell culture
U2OS and 293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 2 mM l‐glutamine, and 1% penicillin–streptomycin. The human neuroblastoma cell lines SKN‐SH was a gift by Prof. Xiaofeng Zhu. SKN‐SH cells were grown in DMEM medium supplemented with 10% FBS (heat inactivated at 56°C for 30 min) and 1% penicillin–streptomycin in a 37°C incubator with a humidified 5% CO2 atmosphere. Dorsal root ganglion neurons were isolated from the embryos of a pregnant mouse at E14.5 and were cultured in neurobasal medium (add 5 ml of B‐27 (50×), 2.5 ml of l‐glutamic acid (50 mM), 0.92 g of l‐glutamine and 2.5 ml of penicillin–streptomycin (100×) to 240 ml of neurobasal medium, filter‐sterilize, and store at 4°C; Wang & Marquardt, 2012).
FAM134B knockout U2OS cells
pLKO‐cas9‐FAM134B sgRNA vector was constructed with a target sequence 5′GAGCTCAGCAGCTCGTCGGCG3′. U2OS cells were seeded in a 6‐well plate with 50% confluency 1 day before transfection. Cells were transfected with 3 μg pLKO‐cas9‐FAM134B sgRNA vector by Lipofectamine 3000 (Thermo Fisher Scientific, L3000015). Twenty‐four hours later, regular medium was replaced with medium containing 1 μg/ml puromycin. After 2‐day incubation, cells were diluted and seeded into 15‐cm dishes. Two weeks later, single cell clones were trypsinized and seeded into 96‐well plates. WB was performed to screen single cell clones with anti‐FAM134B antibody. Then, WB verified KO cell clones were sent for sequencing verification.
Stable cell lines construction
FAM134B‐Flag/293FRT stable cell lines were obtained using the Flp‐In™ System from Invitrogen. pOG44 plasmid and pcDNA5/FRT‐FAM134B vectors were co‐transfected into Flp‐In™ 293FRT cells and were selected with 75 μg/ml of Hygromycin B (Sigma, V900372).
Inducible stable cell lines construction
A tet‐on system was used in FAM134B−/− U2OS cell line. Cells were co‐transfected with HP216 vector and HP138‐FAM134B‐related vectors. Cell lines were obtained through 1 μg/ml puromycin selection.
shRNA knockdown
CAMK2B annealed oligonucleotides were cloned into pLV3‐shRNA ‐Puro using BamHI and EcoRI cloning sites. Target sequences are gaccagatgtgatttgttaaa (CAMK2B shRNA1) and gatcattaagaccacggagca (CAMK2B shRNA2), respectively. Scrambled shRNA knockdown 293T or U2OS cells and CAMK2B shRNA knockdown 293T or U2OS cells were obtained by lentivirus infection and were selected with 1 μg/ml of puromycin. Recombinant lentiviruses were produced following the lentiviral packaging protocol.
Immunoprecipitation and Western blot
Cell pellets were homogenized in TAP buffer (20 mM Tris–HCl, pH 7.4, 150 mM NaCl, 0.5% NP‐40, 1 mM NaF, 1 mM Na3VO4, 1 mM EDTA, 10 nM MG132, protease cocktail, phosphatase cocktail) and incubated on ice for 30 min. The cell lysate was cleared by centrifugation at 18,407 g for 10 min. The supernatant was incubated with antibody‐conjugated beads and rotated for 4 h at 4°C. After incubation, the beads were washed 3 times with TAP buffer. Western blot was performed following standard procedures. For each representative figure which was shown, at least three different experiments were performed.
Immunofluorescence
Cells grown on coverslips were transfected with different plasmids, then fixed in 4% paraformaldehyde in PBS for 10 min at room temperature, and permeabilized with 0.4% Triton X‐100 in PBS for 10 min. Following permeabilization, cells were treated with block buffer (5% BSA, 0.1% Triton X‐100 in PBS) for 1 h at room temperature. Cells were incubated with primary antibodies diluted in block buffer overnight at 4°C. Cells were washed three times with PBS, each for 10 min, followed by incubation with Alexa Fluor‐conjugated secondary antibody (Life Technologies) diluted in block buffer for 1 h at room temperature. Before applying the mounting buffer, the slides were washed three times with PBS, each for 10 min. Slides were examined by using a laser scanning confocal microscope (Zeiss LSM 800). For each representative image which was shown, three different experiments were performed to ensure the reproducibility.
Recombinant protein purification
Wild‐type FAM134B was cloned into pGEX6P1 and expressed as glutathione‐S‐transferase (GST) fusion proteins with a TEV protease cleavage site in between. FAM134B mutants were generated by site‐directed mutagenesis based on pGEX6P1‐FAM134B WT. GST fusion proteins were expressed in Escherichia coli BL21 (DE3) at 16°C to achieve maximal soluble expression. Cells were collected by centrifugation and washed three times with cold PBS. The cells were lysed by sonication in lysis buffer (20 mM Tris–HCl, pH 7.5, 500 mM NaCl, 1 mM EDTA, 0.5% Triton‐X100, protease inhibitor cocktail from Roche) and centrifuged at 12,000 g for 15 min. 0.2 ml GST‐Sepharose resin (GE Healthcare) pre‐equilibrated with 20 ml TEV protease cleavage buffer (10 mM Tris–HCl, pH 8.0; 150 mM NaCl; 0.1% NP‐40; 1 mM DTT) was added to the supernatant and rotated at 4°C for 2 h. Next, beads were washed three times with TEV protease cleavage buffer, and then, the recombinant protein was eluted from the resin by incubating at 4°C overnight with 10 μg/ml of TEV protease to cleave off the desired protein from the GST tag, which was still bound to the GST‐Sepharose resin after the overnight cleavage reaction. Purified untagged recombinant proteins were further fractionated using a Mono‐Q column, followed by dialysis against PBS. Proteins were quantified by the Bradford method and analyzed by SDS–PAGE and Coomassie blue staining.
Size‐exclusion chromatography
Size‐exclusion chromatography (SEC) using Superose 6 10/300 GL was performed at 4°Cusing an AKTA PURE according to the Handbooks from GE Healthcare Life Sciences (http://www.gelifesciences.com/handbooks). Briefly, the column was equilibrated with two column volumes of buffer (20 mM Hepes, pH 7.5, 100 mM NaCl, filtration, and ultrasonic defoaming) at a 0.25 ml/min flow rate. 500 μg/500 μl WT FAM134B protein was injected onto the column. Samples were collected and subjected to SDS–PAGE analysis and Coomassie blue staining. The FAM134B oligomer size was deduced from the chromatograms of six standard proteins on Superose 6 10/300 GL.
Native PAGE
To be compatible with other assays, including cryoEM structural analysis of the FAM134B oligomers, after SEC, 100 μl FAM134B protein (10 μg) of the peak was added with 5 μl 2.3% glutaraldehyde and placed at 37°C for 3 min, and the reaction was stopped by addition of 10 μl of 1 M Tris–HCl pH 8.0. Then, a mixture containing 20 μl protein + 2 μl 50% glycerol + 3 μl 0.1% Coomassie blue G‐250 was incubated for 15 min at room temperature and loaded onto a linear 3–12% gradient native PAGE gel (Life Technologies) running in blue cathode/anode buffer for 2 h at 150 V in an ice bath. The gel was stained using the Pillsin silver staining kit.
Mass spectrometry analysis
293FRT cell line stably expressing FAM134B‐Flag was cultured on two 15 cm dishes to 100% confluency in the presence of 1 μg/ml doxycycline. To purify recombinant FAM134B‐Flag, immunoprecipitation was performed as described above using anti‐Flag antibody‐conjugated beads. Approximately, 10 μg purified FAM134B‐Flag was analyzed by SDS–PAGE and visualized by Colloidal blue staining and specific FAM134B bands were subjected to mass spectrometry analysis to identify the post‐translational modifications, including phosphorylation. Mass spectrometry analysis was also applied to confirm the serine 151 phosphorylation using the protein samples derived from in vitro kinase assays.
In vitro kinase assay
In vitro kinase assays were performed as described previously (Rose et al, 2006). Briefly, purified recombinant FAM134B was incubated with CAMK2B purified from 293T in 30 μl of reaction mixture containing 400 μM ATP and/or a trace amount (10 μCi) of [γ‐32P] ATP, 10 μg FAM134B, 250 ng CAMK2B, protease inhibitors, and 1× CAMK2B kinase buffer (10 mM Hepes, pH 7.2, 1 mM EGTA, 5 mM MgCl2, 2 mM CaCl2). After incubation at 30°C for 10 min, the reaction was terminated by the addition of 10 μl of 5× SDS loading buffer and subjected to SDS–PAGE analysis. The phosphorylation of FAM134B at S151 was visualized by three methods, namely, autoradiography, Western blot using the phosphor‐antibody recognizing phosphor‐FAM134B (S151) and mass spectrometry analysis.
Liposome assay
Using 200 μl of 1% agarose to coat one glass slide overnight, meanwhile, the other glass slide was coated by using 7 μg of streptavidin protein (7 μg of streptavidin dissolved in 200 μl of ddH2O) and allowed to dry overnight. Next day, prepare lipid solution (15 μl PC: 30 μl cholesterol: 13.5 μl PE (12.5 μl Biotin‐PE + 1 μl rhodamine‐PE): 1.5 μl POPS = 25: 50: 22.5: 2.5, then add 40 μl of chloroform to a total of 100 μl. Using a glass syringe (HEMILTON) to add the lipid solution to the agarose‐coated glass slide and allow the sample to dry with Argon gas for 3 h. Then, 200 μl 1× PBS was gently added to agarose glass for 10 min to suspend the liposomes, which were subsequently added to the streptavidin‐coated glass slide. After 20‐min incubation, which allowed the liposomes to anchor to glass slides through biotin–streptavidin interaction, recombinant protein was injected into the chamber and the morphological change of the liposomes was observed continuously for 20 min using a laser scanning confocal microscope (Olympus IX2). For each representative image which was shown, three different experiments were performed to ensure the reproducibility.
EGFP‐cleavage assay
U2OS cells were transiently co‐transfected with 1.5 μg EGFP‐SEC61B and 0.5 μg Wild‐type FAM134B‐Flag or FAM134B‐flag mutants. Cell lysates were immunoblotted with antibodies against Flag, GFP, and β‐tubulin.
Endoplasmic reticulum isolation
The ER membrane fractions were isolated following the Endoplasmic Reticulum Isolation Kit (Sigma, 038M4161V). Briefly, detached cells were collected at 600 g for 5 min, wash the cells with 10 volumes of PBS, and centrifuge to obtain the packed cell volume (PCV). PCV was measured and suspended in a volume of 1× Hypotonic Extraction Buffer equivalent to 3 times the PCV for 20 min at 4°C to allow the cells to swell. The cells were collected by centrifugation. The “new” PCV was measured and resuspended in 1× Isotonic Extraction Buffer equivalent to 2 times the “new” PCV. The cells were then treated with 10 strokes of the Dounce homogenizer and then proceeded to the differential centrifugation steps.
Mouse DRG sensory neuron survival assay
Dorsal root ganglia (DRG) from E14.5 vGlut2 Cre ;R26 lsl‐tdTom mouse embryos were isolated and digested with Trypsin (Invitrogen) at 37°C for 20 min. Dissociated sensory neurons were plated on glass coverslips coated with poly‐D‐lysine (1 mg/ml, Chemicon) and laminin (100 μg/ml, Invitrogen), and cultured in a neurobasal‐defined medium supplemented with NGF (1 ng/ml; R&D) for 24 h (Wang & Marquardt, 2012). Following infection with lentivirus (lentiviruses were produced following lentiviral packaging protocol), sensory neurons were fixed for late experiments.
Single‐molecule RNA in situ hybridization
RNAscope ISH FAM134B/Retreg1 probe was designed and manufactured by Advanced Cell Diagnostics (Retreg1, cat no. 545851, Mm‐Retreg1 targeting 209‐1820 of NM_025459.3). Sections from P4d neonatal vGlut2 Cre ;R26 lsl‐tdTom mice were cut at 20 μm and used for in situ hybridization according to the manufacturer's instructions (Wang et al, 2012).
Immunoelectron microscopy
EGFP‐FAM134B cells were fixed with immuno‐EM fixing buffer (4% paraformaldehyde and 0.1% glutaraldehyde in 0.1 M PB) overnight at 4°C. The fixed cells were washed three times with 0.1 M PB, and terminated with 50 mM glycine, then followed by permeabilization for 40 min with 0.1% saponin and 5% fetal bovine serum in 1× PBS. Cells were incubated with anti‐GFP antibody (ab6556, abcam, 1:1,000) overnight at 4°C, followed by nanogold‐labeled Fab'goat anti‐mouse IgG(H + L) antibody (34C918, Nanoprobes, 1:50) overnight at 4°C. Cells were silver enhanced for 2 min and then fixed with 1% aqueous osmium tetraoxide for 40 min. Cells were dyed with 2% uranyl acetate for 40 min and dehydrated through graded alcohols (50–100%) and 100% acetone twice each for 15–20 min. Samples were embedded in EPON 812 resin. Ultrathin (90 nm) sections were obtained by ultrathin slicer machine. Electron microscopy images of the samples were taken using Tecnai G2 Spirit transmission electron microscope (FEI Company).
Transmission electron microscopy
EGFP‐FAM134B cells were fixed with 2.5% glutaraldehyde in 1× PBS overnight at 4°C. Cells were washed three times with 1× PBS, fixed with 1% aqueous osmium tetraoxide for 40 min, dyed with 2% uranyl acetate for 40 min, dehydrated through graded alcohols (50–100%), and 100% acetone twice each for 15–20 min. Samples were embedded in EPON 812 resin. Ultrathin (90 nm) sections were obtained by ultrathin slicer machine and stained with 2% uranyl acetate and 0.3% lead citrate. Electron microscopy images of the samples were taken using Tecnai G2 Spirit transmission electron microscope (FEI Company).
Statistical analysis
All experiments were independently repeated at least three times. Statistically significant differences were determined by Student's t test using Prism 5 (GraphPad). Values were stated as mean ± SEM of at least three independent experiments, unless otherwise noted. P < 0.05 was considered to be statistically significant. NS, not significant.
Author contributions
QS and DN designed the experiments. XJ, XWa, XD, MD, BL, XWe, JZ, RT, LL, QZ, and LW performed the experiments. WL contributed reagents. LF, JZ, LL, and SC performed the mass spectrometry. QS, DN, XJ, and XWa wrote the manuscript. All authors discussed the results and commented on the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Source Data for Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Acknowledgements
We thank Prof. Jiahuai Han from Xiamen University for cDNA. We are grateful to Prof. Xiaofeng Zhu from Sun Yat‐Sen University for SKN‐SH cell line. We thank the center of Cryo‐Electron microscopy Zhejiang University for assistance with transmission electron microscopy. We thank the Imaging Center of Zhejiang University School of Medicine for assistance with confocal microscopy. This study was supported by the National Natural Science Foundation under Grant 31771525 and 91754113 to Q.S., 31770938 and 91854113 to D.N., Ministry of Science and Technology of the People's Republic of China under Grant 2017YFA0503402 (to Q.S.), and the Key Program of Zhejiang Provincial Natural Science Foundation of China (LZ16C050001) to D.N.
The EMBO Journal (2020) 39: e102608
See also: https://doi.org/10.15252/embj.2020104546 (March 2020)
Contributor Information
Lei Fang, Email: njfanglei@nju.edu.cn.
Dante Neculai, Email: dneculai@zju.edu.cn.
Qiming Sun, Email: qmsun@zju.edu.cn.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium database (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (Perez‐Riverol et al, 2019) with the dataset identifier PXD016661.
References
- An H, Ordureau A, Paulo JA, Shoemaker CJ, Denic V, Harper JW (2019) TEX264 is an endoplasmic reticulum‐resident ATG8‐interacting protein critical for ER remodeling during nutrient stress. Mol Cell 74: 891–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernales S, McDonald KL, Walter P (2006) ER‐phagy, Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol 4: e423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernales S, Schuck S, Walter P (2007) ER‐phagy: selective autophagy of the endoplasmic reticulum. Autophagy 3: 285–287 [DOI] [PubMed] [Google Scholar]
- Chen Q, Xiao Y, Chai P, Zheng P, Teng J, Chen J (2019) ATL3 is a tubular ER‐Phagy receptor for GABARAP‐mediated selective autophagy. Curr Biol 29: 846–855 [DOI] [PubMed] [Google Scholar]
- Chino H, Hatta T, Natsume T, Mizushima N (2019) Intrinsically disordered protein TEX264 mediates ER‐phagy. Mol Cell 74: 909–921 [DOI] [PubMed] [Google Scholar]
- Crottet P, Meyer DM, Rohrer J, Spiess M (2002) ARF1.GTP, tyrosine‐based signals, and phosphatidylinositol 4,5‐bisphosphate constitute a minimal machinery to recruit the AP‐1 clathrin adaptor to membranes. Mol Biol Cell 13: 3672–3682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson G, Murphy S, Polke J, Laura M, Salih M, Muntoni F, Blake J, Brandner S, Davies N, Horvath R et al (2012) Frequency of mutations in the genes associated with hereditary sensory and autonomic neuropathy in a UK cohort. J Neurol 259: 1673–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farre JC, Subramani S (2016) Mechanistic insights into selective autophagy pathways: lessons from yeast. Nat Rev Mol Cell Biol 17: 537–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli F, Noack J, Bergmann TJ, Presmanes EC, Pisoni GB, Fasana E, Fregno I, Galli C, Loi M, Solda T et al (2016) Translocon component Sec62 acts in endoplasmic reticulum turnover during stress recovery. Nat Cell Biol 18: 1173–1184 [DOI] [PubMed] [Google Scholar]
- Gatica D, Lahiri V, Klionsky DJ (2018) Cargo recognition and degradation by selective autophagy. Nat Cell Biol 20: 233–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grumati P, Morozzi G, Holper S, Mari M, Harwardt MI, Yan R, Muller S, Reggiori F, Heilemann M, Dikic I (2017) Full length RTN3 regulates turnover of tubular endoplasmic reticulum via selective autophagy. Elife 6: e25555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Shibata Y, Voss C, Shemesh T, Li Z, Coughlin M, Kozlov MM, Rapoport TA, Prinz WA (2008) Membrane proteins of the endoplasmic reticulum induce high‐curvature tubules. Science 319: 1247–1250 [DOI] [PubMed] [Google Scholar]
- Huttlin EL, Ting L, Bruckner RJ, Gebreab F, Gygi MP, Szpyt J, Tam S, Zarraga G, Colby G, Baltier K et al (2015) The BioPlex network: a systematic exploration of the human interactome. Cell 162: 425–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaminets A, Heinrich T, Mari M, Grumati P, Huebner AK, Akutsu M, Liebmann L, Stolz A, Nietzsche S, Koch N et al (2015) Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 522: 354–358 [DOI] [PubMed] [Google Scholar]
- Khaminets A, Behl C, Dikic I (2016) Ubiquitin‐dependent and independent signals in selective autophagy. Trends Cell Biol 26: 6–16 [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K et al (2016) Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 12: 1–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurth I, Pamminger T, Hennings JC, Soehendra D, Huebner AK, Rotthier A, Baets J, Senderek J, Topaloglu H, Farrell SA et al (2009) Mutations in FAM134B, encoding a newly identified Golgi protein, cause severe sensory and autonomic neuropathy. Nat Genet 41: 1179–1181 [DOI] [PubMed] [Google Scholar]
- Li X, Wu XQ, Deng R, Li DD, Tang J, Chen WD, Chen JH, Ji J, Jiao L, Jiang S et al (2017) CaMKII‐mediated Beclin 1 phosphorylation regulates autophagy that promotes degradation of Id and neuroblastoma cell differentiation. Nat Commun 8: 1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang JR, Lingeman E, Ahmed S, Corn JE (2018) Atlastins remodel the endoplasmic reticulum for selective autophagy. J Cell Biol 217: 3354–3367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka K, Orci L, Amherdt M, Bednarek SY, Hamamoto S, Schekman R, Yeung T (1998) COPII‐coated vesicle formation reconstituted with purified coat proteins and chemically defined liposomes. Cell 93: 263–275 [DOI] [PubMed] [Google Scholar]
- Mochida K, Oikawa Y, Kimura Y, Kirisako H, Hirano H, Ohsumi Y, Nakatogawa H (2015) Receptor‐mediated selective autophagy degrades the endoplasmic reticulum and the nucleus‐ATG39 and ATG40. Nature 522: 359–362 [DOI] [PubMed] [Google Scholar]
- Murphy SM, Davidson GL, Brandner S, Houlden H, Reilly MM (2012) Mutation in FAM134B causing severe hereditary sensory neuropathy. J Neurol Neurosurg Psychiatry 83: 119–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatogawa H, Mochida K (2015) Reticulophagy and nucleophagy: new findings and unsolved issues. Autophagy 11: 2377–2378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Riverol Y, Csordas A, Bai J, Bernal‐Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M et al (2019) The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47: D442–D450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose AJ, Kiens B, Richter EA (2006) Ca2+‐calmodulin‐dependent protein kinase expression and signalling in skeletal muscle during exercise. J Physiol 574: 889–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein DC (2015) Cell biology: receptors for selective recycling. Nature 522: 291–292 [DOI] [PubMed] [Google Scholar]
- Shibata Y, Voeltz GK, Rapoport TA (2006) Rough sheets and smooth tubules. Cell 126: 435–439 [DOI] [PubMed] [Google Scholar]
- Shibata Y, Hu J, Kozlov MM, Rapoport TA (2009) Mechanisms shaping the membranes of cellular organelles. Annu Rev Cell Dev Biol 25: 329–354 [DOI] [PubMed] [Google Scholar]
- Shibata Y, Shemesh T, Prinz WA, Palazzo AF, Kozlov MM, Rapoport TA (2010) Mechanisms determining the morphology of the peripheral ER. Cell 143: 774–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MD, Harley ME, Kemp AJ, Wills J, Lee M, Arends M, von Kriegsheim A, Behrends C, Wilkinson S (2017) CCPG1 is a non‐canonical autophagy cargo receptor essential for ER‐Phagy and pancreatic ER proteostasis. Dev Cell 44: 217–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spang A, Matsuoka K, Hamamoto S, Schekman R, Orci L (1998) Coatomer, Arf1p, and nucleotide are required to bud coat protein complex I‐coated vesicles from large synthetic liposomes. Proc Natl Acad Sci USA 95: 11199–11204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolz A, Ernst A, Dikic I (2014) Cargo recognition and trafficking in selective autophagy. Nat Cell Biol 16: 495–501 [DOI] [PubMed] [Google Scholar]
- Walter P, Ron D (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334: 1081–1086 [DOI] [PubMed] [Google Scholar]
- Wang F, Flanagan J, Su N, Wang LC, Bui S, Nielson A, Wu X, Vo HT, Ma XJ, Luo Y (2012) RNAscope: a novel in situ RNA analysis platform for formalin‐fixed, paraffin‐embedded tissues. J Mol Diagn 14: 22–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Marquardt T (2012) Direct live monitoring of heterotypic axon‐axon interactions in vitro . Nat Protoc 7: 351–363 [DOI] [PubMed] [Google Scholar]
- Zaffagnini G, Martens S (2016) Mechanisms of selective autophagy. J Mol Biol 428: 1714–1724 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Source Data for Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium database (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (Perez‐Riverol et al, 2019) with the dataset identifier PXD016661.