

Abstract
The L‐type Ca2+ channel CaV1.2 governs gene expression, cardiac contraction, and neuronal activity. Binding of α‐actinin to the IQ motif of CaV1.2 supports its surface localization and postsynaptic targeting in neurons. We report a bi‐functional mechanism that restricts CaV1.2 activity to its target sites. We solved separate NMR structures of the IQ motif (residues 1,646–1,664) bound to α‐actinin‐1 and to apo‐calmodulin (apoCaM). The CaV1.2 K1647A and Y1649A mutations, which impair α‐actinin‐1 but not apoCaM binding, but not the F1658A and K1662E mutations, which impair apoCaM but not α‐actinin‐1 binding, decreased single‐channel open probability, gating charge movement, and its coupling to channel opening. Thus, α‐actinin recruits CaV1.2 to defined surface regions and simultaneously boosts its open probability so that CaV1.2 is mostly active when appropriately localized.
Keywords: calmodulin, gating charge, IQ motif structure, open probability, surface expression
Subject Categories: Membrane & Intracellular Transport, Neuroscience
The scaffold protein α‐actinin is a positive regulator of CaV1.2 channel open probability and gating.
Introduction
Ca2+ influx through CaV1.2 is critical for the functions of many organs as strikingly illustrated by Timothy syndrome (Splawski et al, 2004). In this disease, a point mutation in CaV1.2 causes, among other symptoms, lethal arrhythmias, autistic‐like behaviors, immune deficiency, and webbing of fingers (Splawski et al, 2004). CaV1.2 is the main L‐type channel in heart (Seisenberger et al, 2000), vascular smooth muscle cells (Ghosh et al, 2017), and brain (Hell et al, 1993; Sinnegger‐Brauns et al, 2004). Ca2+ influx through CaV1.2 triggers cardiac contraction, regulates arterial tone (Ghosh et al, 2017), mediates different forms of synaptic long‐term potentiation (Grover & Teyler, 1990; Patriarchi et al, 2016; Qian et al, 2017), and controls neuronal excitability (Marrion & Tavalin, 1998; Berkefeld et al, 2006). Furthermore, L‐type channels are much more strongly coupled to gene expression than other Ca2+ channels (Dolmetsch et al, 2001; Li et al, 2012; Ma et al, 2014; Cohen et al, 2018). Finally, CaV1.2 forms a physical and functional complex with the β2 adrenergic receptor (Davare et al, 2001) making it a prime target for signaling by norepinephrine (Patriarchi et al, 2016; Qian et al, 2017; Man et al, 2020), which is important for wakefulness, attention, and various forms of learning (Cahill et al, 1994; Berman & Dudai, 2001; Hu et al, 2007; Minzenberg et al, 2008; Carter et al, 2010).
CaV1.2 consists of the pore‐forming α11.2 subunit and auxiliary α2δ and β subunits, which facilitate release from the endoplasmic reticulum and the controlled trafficking of CaV1.2 to the cell surface (Dai et al, 2009; Dolphin, 2012, 2016; Zamponi et al, 2015; Ghosh et al, 2018). However, α2δ and β subunits do not target CaV1.2 to specific sites in the plasma membrane. Rather, CaV1.2 anchoring at defined regions at the cell surface is mediated by α‐actinin, which binds to the IQ motif in the C‐terminus of α11.2 (Hall et al, 2013; Tseng et al, 2017).
A systematic yeast two‐hybrid screen defined three residues in the IQ motif of α11.2, whose mutations to alanine residues affect α‐actinin binding: K1647A, Y1649A, and I1654A. All three mutations reduced surface expression of CaV1.2 by ~35% but current density by 70–80% (Tseng et al, 2017). These results suggest that α‐actinin binding to the IQ motif promotes not only surface localization but also channel activity. Such a multifunctional role would ensure that CaV1.2 is mostly active at its ultimate destinations and much less so when in transit and outside its target areas.
The closely related α11.3 subunit of the L‐type channel CaV1.3 shares nearly 100% sequence identity with α11.2 in its membrane‐proximal 165 residues of its C‐terminus in which the IQ motif is embedded (the eponymous Ile is I1654 of α11.2 and I1609 of α11.3; Appendix Fig S1). Ca2+‐free calmodulin (apoCaM) binds to this IQ motif, and mutation of I1609 in α11.3 impairs both, apoCaM binding and open probability Po of CaV1.3 (Ben Johny et al, 2013; Adams et al, 2014; Ben‐Johny et al, 2014). We solved the NMR structures of the third and fourth EF hands of α‐actinin‐1 (EF3 and EF4, residues 822–892; Fig 1A) and of full‐length apoCaM bound to the α‐helical IQ motif of CaV1.2 (residues 1,646–1,664). This work provided new insight into the structure of CaV1.2 especially as relevant for these two critical binding partners and informed experiments that dissected the exact functions of α‐actinin versus apoCaM binding. Refined analysis of CaV1.2 activity by cell‐attached single‐channel recording revealed that point mutations that affected α‐actinin‐1 but not those that affected apoCaM binding dramatically decreased the channel Po by impairing gating charge movement as well as its coupling to channel opening. We conclude that α‐actinin plays a dual role by anchoring CaV1.2 at specific subcellular domains such as the postsynaptic sites and at the same time boosting its open probability. This mechanism ensures that the activity of CaV1.2 is minimal when in transit during secretory trafficking and outside its intended location at the cell surface, where its Ca2+ conductance could adversely affect cell functions, but maximal at its final destination.
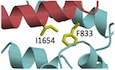
Figure 1. α‐Actinin‐1 forms electrostatic and hydrophobic contacts with CaV1.2.
- Linear model of α‐actinin domains (CH: calponin homology domains; SR: spectrin repeats; EF: EF hands). Bars and numbers on top of the model depict the segments used in this work.
- Ensemble of 10 lowest energy NMR‐derived structures of α‐actinin‐1_EF34 (cyan) bound to IQ peptide (red). Structural statistics are given in Table 1.
- Energy‐minimized average structure of α‐actinin‐1_EF34 (cyan) bound to IQ (red), revealing intermolecular salt bridges between K1647 of IQ and E847/E851 of α‐actinin‐1. The K1647 side‐chain amino nitrogen atom is 2.8 and 2.5 Å away from the side‐chain carbonyl oxygen atoms of E847 and E851, respectively.
- Intermolecular hydrophobic contacts between I1654 of IQ (red) and F833 of α‐actinin‐1 (cyan). The I1654 side‐chain methyl carbon atom is 2.9 Å away from the closest aromatic ring atom of F833. Side‐chain atoms are colored yellow.
- Lack of direct contacts between F1658 of IQ (red) and α‐actinin‐1 (cyan). The aromatic side chain of F1658 is primarily solvent exposed and does not directly contact α‐actinin‐1. The aromatic ring of F1658 is closest to the aromatic ring of Y859 and β‐methylene carbon of Q856 of α‐actinin‐1, which are 5.2 and 4.3 Å apart, respectively.
- FP titrations show binding of WT α‐actinin‐1_EF34 to IQ peptides WT (black) and K1647E (red), of α‐actinin‐1_EF34 mutant E847K/E851K to IQ peptides WT (green) and K1647E (blue), of full‐length α‐actinin‐1 to IQ WT (o), and lack of IQ binding to α‐actinin‐1_CH1‐CH2 (+) and α‐actinin‐1_EF12 (x; see Table 2 for binding parameters and standard errors).
- FP titrations showing binding of wild‐type α‐actinin‐1_EF34 to IQ peptides WT (black) and I1654A (blue) and of ACTN1_EF34 mutant F833 to IQ WT (red; see Table 2 for binding parameters and standard errors).
Results
Binding of α‐actinin‐1 EF3/EF4 to the CaV1.2 IQ motif
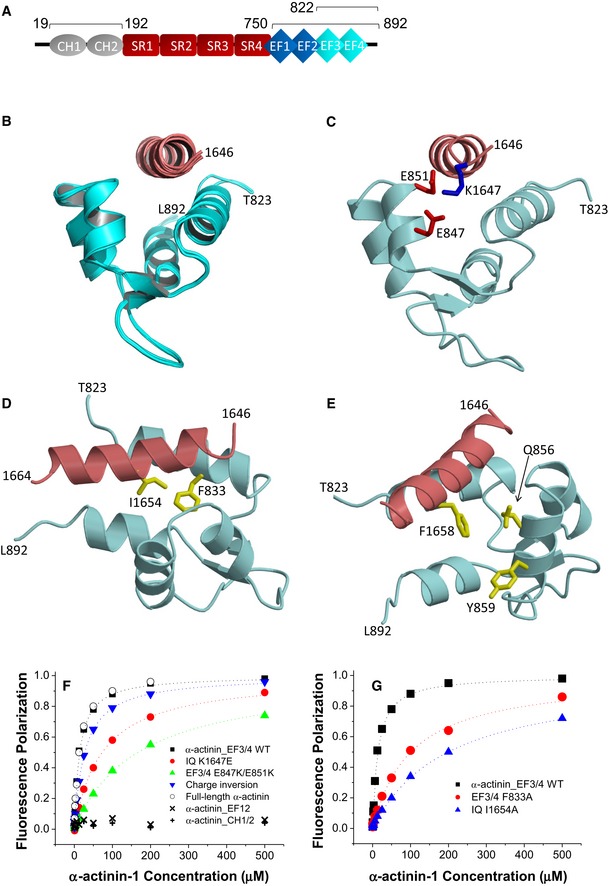
α‐actinin is encoded by four homologous genes with α‐actinin‐1 and α‐actinin‐2 being most prominent in neurons (Wyszynski et al, 1997; Hall et al, 2013; Hell, 2014; Matt et al, 2018). α‐actinins consist of two N‐terminal calponin homology domains (CH1, CH2; residues 19–192), four central spectrin repeats (SR1‐4), which form a rod‐like coiled‐coil structure (Ribeiro Ede et al, 2014), and four EF hands at their C‐termini (residues 750–892; Fig 1A). We first performed NMR spectroscopy with the CH1/CH2 region (residues 19–192). Two‐dimensional NMR spectra of 15N‐labeled CH1/CH2 recorded in the presence and absence of unlabeled IQ peptide appeared to be virtually identical, consistent with a lack of IQ binding to CH1/CH2 under NMR conditions (Fig EV1A). The lack of IQ binding to the CH1/CH2 domain is supported by the absence of detectable binding measured by fluorescence polarization (see below Fig 1F). In stark contrast, the NMR spectrum of 15N‐labeled EF‐hand domain of α‐actinin‐1 (residues 750–892) showed clearly detectable spectral changes upon adding a saturating amount of unlabeled IQ peptide, demonstrating a binding interaction (Fig EV1B). The NMR peaks of α‐actinin‐1 most affected by the binding of IQ (see labeled peaks in Fig EV1B) were assigned to residues in EF3 and EF4 (the C‐lobe of the EF‐hand region; residues 822–892). Consistently, the NMR spectrum of 15N‐labeled α‐actinin‐1 EF3/4 (residues 822–892) exhibited large spectral changes upon the addition of the IQ peptide (Fig EV1C), which are similar to those seen with the construct that contains all four EF hands (Fig EV1B). We conclude that the IQ peptide binds to the α‐actinin‐1 C‐lobe.
Figure EV1. Mapping the CaV1.2 IQ binding site in α‐actinin and apoCaM.
- Overlay of 15N‐1H HSQC spectra of 15N‐labeled α‐actinin‐1 CH1/CH2 domain (residues 19–192) by itself (black peaks) and after addition of saturating, unlabeled IQ peptide (red peaks).
- Overlay of 15N‐1H HSQC spectra of 15N‐labeled α‐actinin‐1 EF‐hand domain (residues 750–892) by itself (black peaks) and after addition of saturating, unlabeled IQ peptide (red peaks).
- Overlay of 15N‐1H HSQC spectra of 15N‐labeled α‐actinin‐1 C‐lobe (α‐actinin‐1 EF34; residues 822–892) by itself (black peaks) and after addition of saturating, unlabeled IQ peptide (red peaks).
- Overlay of 15N‐1H HSQC spectra of 15N‐labeled apoCaM by itself (black peaks) and after addition of saturating, unlabeled IQ peptide (red peaks).
NMR structure of α‐actinin‐1 EF3/4 bound to the IQ motif of CaV1.2
We had previously reported NMR spectral assignments for α‐actinin‐1 EF3/4 (BMRB accession number 25902) (Turner et al, 2016). We used these assignments to obtain NMR‐derived structural restraints for high‐resolution structural analysis of α‐actinin‐1 EF3/4 bound to the CaV1.2 IQ motif (EF3/4‐IQ). Atomic‐level NMR structures were calculated on the basis of distance restraints derived from the analysis of NOESY spectra and long‐range orientational restraints derived from residual dipolar coupling (RDC) data (Fig EV2A). EF34 in the complex was resolved for 69 residues, starting at T823 and ending at L892. The 10 lowest energy NMR structures are overlaid in Fig 1B and structural statistics summarized in Table 1. The overall precision of the ensemble was expressed by an RMSD of 0.3 Å calculated from the coordinates of the main chain atoms. The energy‐minimized average structure of EF3/4‐IQ (Fig 1C, calculated from the ensembles in Fig 1B) contained two EF‐hand motifs (α1: A826–A837; α2: M845–E851; α3: P854–R863; α4: M880–Y887; β1: Y842–T844; β2: A876–D878) bound to an α‐helical IQ motif (CaV1.2 residues 1,646–1,664). The NMR structure of EF3/4‐IQ (Fig 1C) was quite similar (1.8 Å RMSD) to the NMR structure of α‐actinin‐2 bound to the seventh Z‐repeat of titin (Atkinson et al, 2001). It contained important intermolecular contacts that stabilize the EF3/4–IQ interaction (Fig 1C–E). Most striking were a salt bridge between IQ K1647 and EF3/4 E847/E851 (Fig 1C) and hydrophobic contacts involving IQ I1654 and EF3F833 (Fig 1D). The IQ residue F1658 was mostly solvent exposed in the EF3/4‐IQ complex contributing minimally if at all to the EF3/4–IQ interaction (Fig 1E), in contrast to being buried inside apoCaM in the apoCaM/IQ complex (see below).
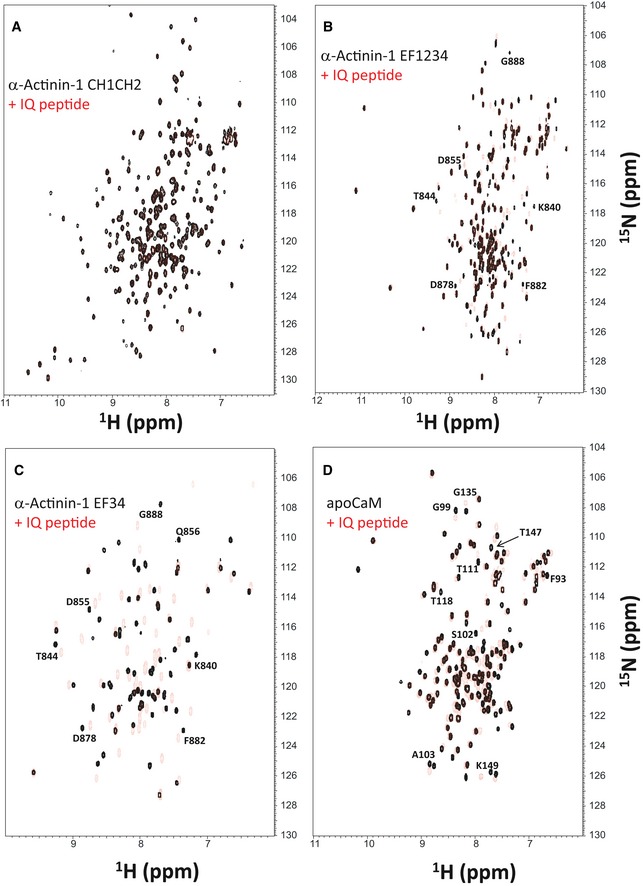
Figure EV2. NMR structural analysis using residual dipolar couplings (RDCs).
-
A, B15N‐1H HSQC‐IPAP spectra (Tjandra & Bax, 1997) of 15N‐labeled α‐actinin‐1_EF34 (A) or apoCaM (B) in the presence of saturating unlabeled IQ peptide. Representative and measured RDC values are marked above‐selected peaks. Samples used in HSQC‐IPAP experiments were prepared by adding 5 mg of 15N‐labeled protein to 0.5 ml of NMR buffer containing 12 mg/ml of filamentous bacteriophage Pf1. HSQC‐IPAP spectra were recorded in the presence of Pf1 (A, B) and absence of Pf1 (not shown). Residual dipolar couplings (RDCs) were measured (in Hz) as the difference in splitting for the 15N‐[1H] doublet components relative to the isotropic 1JNH coupling. RDCs measured for 34 residues (α‐actinin‐1_EF34/IQ) and 36 residues (apoCaM/IQ) served as orientational structural restraints applied during the refinement phase of the structure calculation (Schwieters et al, 2003).
-
C, DPlots showing the correlation between observed versus calculated backbone RDCs predicted from the final NMR‐derived structures for α‐actinin‐1_EF34/IQ and apoCaM/IQ, respectively. The correlation coefficient (r 2) equals 0.99, and Q is 0.095 and 0.089 for α‐actinin‐1_EF34/IQ and apoCaM/IQ, respectively.
Table 1.
NMR structural statistics for α‐actinin‐1 EF34/IQ
| NMR structural restraints | |
| Intermolecular NOEs | 73 |
| Hydrogen bonds | 62 |
| RDC Q‐Factor | 0.095 |
| 1DHNRDC | 34 |
| RDC correlation coefficient (R) | 0.99 |
| Root mean squared deviation from average structure | |
| α‐actinin‐1 backbone atoms | 0.5 Å ± 0.3 for 200 structures |
| α‐actinin‐1 backbone atoms (refined) | 0.3 Å ± 0.2 for 50 structures |
| Haddock scoring | |
| Cluster size | 194 |
| Van der Waals energy | −40.2 ± 6.2 |
| Electrostatic energy | −395.4 ± 37.1 |
| Restraints violation energy | 278.0 ± 6.23 |
| Ramachandran plot | |
| Most favored regions | 96.7% |
| Allowed regions | 2.2% |
| Unfavored regions | 1.1% |
Validation of the α‐actinin‐1 EF3/4–IQ NMR structure by mutagenesis
Mutations in both α‐actinin‐1 EF3/4 and IQ peptide were designed to verify their predicted intermolecular contacts. A synthetic fluorescein‐labeled IQ peptide (residues 1,644–1,666) was titrated with EF3/4 and fluorescence polarization (FP) monitored to determine their K d values. The IQ peptide bound to α‐actinin‐1 EF3/4 with nearly the same affinity as to full‐length α‐actinin‐1 (Fig 1F; Table 2). The IQ peptide did not bind to α‐actinin‐1 EF1/2 (Fig 1F). Accordingly, IQ interacts exclusively with the C‐lobe but not N‐lobe of the EF‐hand domain in α‐actinin‐1.
Table 2.
K d (in μM) for α‐actinin‐1 EF3/EF4 binding to IQ as determined by FP
| α‐actinin‐1 EF3/4 | |||
|---|---|---|---|
| WT | E847K/E851K | F833A | |
| CaV1.2 IQ | |||
| WT | 14 ± 2 | 167 ± 20 | 100 ± 10 |
| K1647E | 73 ± 10 | 27 ± 2 | / |
| K1647A | 67 ± 10 | / | / |
| Y1649A | 42 ± 4 | / | / |
| I1654A | 200 ± 20 | / | / |
| F1658A | 20 ± 2 | / | / |
| K1662E | 20 ± 2 | 150 ± 20 | / |
Shown are mean ± SD (n = 3 for all conditions).
The salt bridges formed between IQ residue K1647 and α‐actinin‐1 residues E847 and E851 (Fig 1C) provided an opportunity for charge inversion experiments. The single‐residue alterations K1647A and K1647E in the IQ peptide increased the K d by ~5‐fold and mutating E847 and E851 in α‐actinin‐1 EF3/4 to lysine (K) by ~12‐fold (Fig 1F; Table 2). Combining the E847K/E851K mutations in α‐actinin‐1 EF3/4 with the K1647E substitution in the IQ peptide mostly but not fully restored the binding affinity to the K1647E IQ peptide. This powerful charge inversion experiment unequivocally identified the importance of those salt bridges for the α‐actinin‐1–IQ interaction. Furthermore, I1654 of the IQ motif was predicted to form hydrophobic interactions with F833 in EF3 (Fig 1D). In fact, the α‐actinin‐1 EF3/4 mutant F833A showed an ~8‐fold decrease in binding affinity, which is consistent with the decrease observed for the I1654A substitution in the IQ peptide (Fig 1G; Table 2). These results confirmed two important intermolecular interactions that had been seen in the NMR structure (Fig 1C): a salt bridge contact between K1647 (CaV1.2) and E847/E851 (α‐actinin‐1) and the linchpin hydrophobic contact between I1654 (CaV1.2) and F833 (α‐actinin‐1).
NMR structure of apoCaM bound to the IQ motif of CaV1.2
ApoCaM is predicted to bind to the IQ motif of CaV1.3 under basal conditions to augment Po (Adams et al, 2014) but no structure has so far been available to aid data interpretation. The 1H‐15N HSQC NMR spectrum of 15N‐labeled apoCaM showed detectable spectral changes upon adding a saturating amount of unlabeled IQ peptide, demonstrating binding (Fig EV1D). The NMR peaks of apoCaM most affected by the binding of IQ (see labeled peaks in Fig EV1D) were assigned to residues in its C‐lobe (CaM EF3/4). Complete NMR spectral assignments for apoCaM bound to the IQ peptide had been reported previously (Lian et al, 2007). We used these assignments to obtain NMR‐derived structural restraints and determine the atomic‐level NMR structure of apoCaM bound to the IQ peptide (called apoCaM‐IQ). NMR‐derived structures were calculated on the basis of NOESY distance restraints and long‐range orientational restraints derived from NMR RDC data (Fig EV2B) (Tjandra & Bax, 1997). Our NMR chemical shift analysis indicated that the IQ peptide contacted residues in the C‐lobe (residues 82–148) but not N‐lobe of apoCaM (residues 1–78; Fig 2). The 10 lowest energy NMR structures are overlaid in Fig 2A and structural statistics summarized in Table 3. The overall precision of the ensemble was expressed by an RMSD of 0.6 Å calculated from the coordinates of the main chain atoms. The energy‐minimized average structure of apoCaM/IQ (Fig 2B, calculated from the ensembles in Fig 2A) contained two EF‐hand motifs (EF3 and EF4) in a semi‐open conformation akin to that observed for apoCaM bound to the IQ motif in voltage‐gated Na+ channels (Chagot & Chazin, 2011; Feldkamp et al, 2011). This semi‐open apoCaM C‐lobe structure was bound to the α‐helical IQ motif (α11.2 residues 1,646–1,665; Fig 2B) with an orientation that was opposite to that observed in the crystal structure of Ca2+‐CaM bound to IQ (Van Petegem et al, 2005).
Figure 2. ApoCaM forms electrostatic and hydrophobic contacts with CaV1.2.
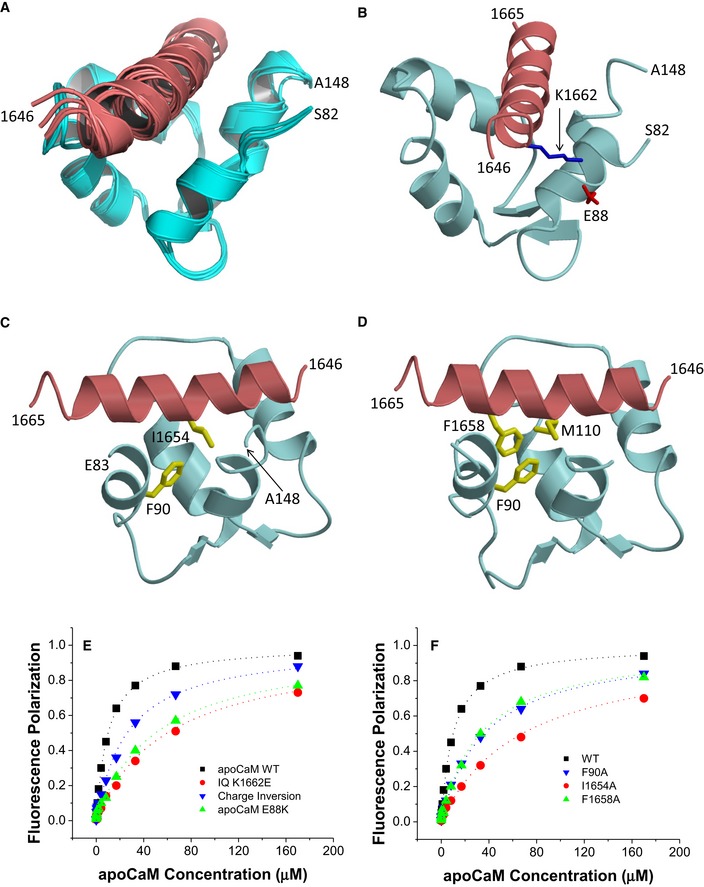
- Ensemble of 10 lowest energy NMR‐derived structures of apoCaM/IQ complex. Structural statistics are given in Table 3.
- Energy‐minimized average structure of apoCaM (cyan) bound to IQ (red), showing an intermolecular salt bridge between IQ K1662 and apoCaM E88. The K1662 side‐chain amino nitrogen atom is 2.7 Å away from the side‐chain carbonyl oxygen of E88.
- Intermolecular hydrophobic contacts between IQ I1654 (red) and apoCaM F90 (cyan). Side‐chain atoms colored yellow. The I1654 side‐chain methyl carbon atom is 2.2 Å away from the closest aromatic ring atom of F90.
- Intermolecular hydrophobic contacts between F1658 of IQ and F90/M110 in apoCaM. The aromatic side‐chain atoms of F1658 are 2.3 and 2.6 Å away from the closest side‐chain atom of F90 and M110, respectively.
- FP titrations showing binding of apoCaM to IQ peptides WT (black) and K1662E (red) and of apoCaM mutant E88K to IQ WT (green) and IQ peptide K1662E (blue, “charge inversion”; see Table 4 for binding parameters and standard errors).
- FP titrations showing binding of apoCaM to IQ peptides WT (black), I1654A (red), and F1658A (green) and of apoCaM mutant F90A to IQ WT (blue; see Table 4 for binding parameters and standard errors).
Table 3.
NMR structural statistics for apoCaM/IQ
| NMR structural restraints | |
| Hydrogen bonds | 62 |
| RDC Q‐Factor | 0.089 |
| 1DHNRDC | 36 |
| RDC Correlation Coefficient (R) | 0.99 |
| Root mean squared deviation from average structure | |
| CaM backbone atoms | 0.6 Å ± 0.3 for 200 structures |
| CaM backbone atoms (refined) | 0.6 Å ± 0.2 for 92 structures |
| Haddock scoring | |
| Cluster size | 200 |
| Van der Waals energy | −56.8 ± 0.6 |
| Electrostatic energy | −463.5 ± 20.7 |
| Restraints violation energy | 99.8 ± 36.2 |
| Ramachandran plot | |
| Most favored regions | 89.8% |
| Allowed regions | 8.0% |
| Unfavored regions | 2.3% |
The NMR‐derived structure of apoCaM bound to α11.2 IQ peptide contained a number of important intermolecular contacts that stabilized this interaction (Fig 2B–D). The most striking intermolecular interactions were as follows: (i) a salt bridge between IQ residue K1662 and apoCaM residue E88 (Fig 2B); (ii) hydrophobic contacts involving the side‐chain atoms of IQ residue I1654 and apoCaM residue F90 (Fig 2C); and (iii) hydrophobic contacts involving the side‐chain atoms of IQ residue F1658 and apoCaM residues F90/M110 (Fig 2D).
Validation of the apoCaM–IQ NMR structure by mutagenesis
Mutations in both apoCaM and IQ peptide were introduced to verify their predicted intermolecular contacts. We titrated fluorescein‐labeled IQ peptide (residues 1,644–1,666) with apoCaM and measured FP. The K d for WT IQ peptide was 10 μM (Fig 2E, Table 4), similar to the K d of 13 μM in earlier work (Evans et al, 2011). To understand why this K d is ~20‐fold higher than the K d of 580 nM deducted previously from isothermal titration calorimetry (ITC) measurements (Findeisen et al, 2013), we performed ITC experiments by adding increments of apoCaM (100 μM) to the IQ peptide (10 μM) under near physiological salt concentration (100 mM KCl, Appendix Fig S2A). No heat signal other than that of dilution was detectable (Appendix Fig S2B). The prior ITC experiments were performed at 5 mM KCl (Findeisen et al, 2013) and may represent a non‐physiological electrostatic attraction between oppositely charged apoCaM and IQ that is suppressed by more physiological salt levels (100 mM KCl). Indeed, apoCaM binds to the IQ peptide with nearly fourfold higher affinity in the absence of salt (Appendix Fig S2C; K d is 2.6 μM at 0 KCl and 10 μM at 100 mM KCl).
Table 4.
K d (in μM; means ± SEM) for apoCaM binding to IQ as determined by FP
| apoCaM | |||
|---|---|---|---|
| WT | E88K | F90A | |
| CaV1.2 IQ | |||
| WT | 10 ± 3 | 50 ± 10 | 33 ± 5 |
| K1647E | 12 ± 3 | 53 ± 1 0 | / |
| K1647A | 11 ± 3 | / | / |
| Y1649A | 16 ± 4 | / | / |
| I1654A | 68 ± 1 0 | / | / |
| F1658A | 33 ± 5 | / | / |
| K1662E | 60 ± 10 | 26 ± 5 | / |
Shown are mean ± SD (n = 3 for all conditions).
According to our FP‐binding assay, the K1662E mutation in the IQ peptide and the E88K mutation in apoCaM each decreased the binding affinity between IQ peptide and apoCaM by more than fivefold (Fig 2E; Table 4). Combining the apoCaM E88K mutation with the IQ peptide alteration K1662E restored the binding affinity to some degree although not completely (Fig 2E). The apoCaM mutant F90A showed a ~3.3‐fold decrease in binding affinity for WT IQ peptide (Fig 2F; Table 4). Consistently, I1654A and F1658A substitutions in the IQ peptide resulted in a comparable reduction in their affinity for apoCaM (Fig 2F). These results confirmed three important intermolecular interactions that had been seen in the NMR structure: (i) a salt bridge between IQ residue K1662 and apoCaM residue E88; (ii) hydrophobic contact between IQ residue I1654 and apoCaM residue F90; and (iii) hydrophobic contact between IQ residue F1658 and apoCaM residues F90 and M110.
α‐Actinin and apoCaM do not form a ternary complex with CaV1.2 IQ
The structures of α‐actinin‐1 (Fig 1) and apoCaM (Fig 2) each bound to the IQ peptide reveal that both binding sites are structurally overlapped and therefore should preclude formation of a ternary complex (α‐actinin‐1/IQ/apoCaM), in contrast to what was suggested previously (Hall et al, 2013). The possibility of ternary complex formation was further ruled out by NMR titrations (Fig EV3). The NMR spectrum of 15N‐labeled α‐actinin‐1 EF‐hand domain (Fig EV3A, black peaks) exhibited spectral changes upon the addition of an equivalent amount of unlabeled IQ peptide (Fig EV3A, red peaks marked by arrows) that are reversed by the addition of fivefold excess of unlabeled apoCaM (Fig EV3A, green peaks marked by arrows). This result indicates that α‐actinin‐1 and apoCaM both compete for binding to the IQ peptide, which implies that α‐actinin‐1 and apoCaM do not simultaneously bind to CaV1.2 IQ and therefore rules out the possibility of a ternary complex. A similar result was observed for the NMR titration with 15N‐labeled apoCaM binding to unlabeled IQ (Fig EV3B). The IQ‐induced spectral changes to apoCaM are reversed by the addition of fivefold excess of unlabeled α‐actinin‐1 (Fig EV3B, green peaks marked by arrows), demonstrating that α‐actinin‐1 and apoCaM do not bind simultaneously to the IQ peptide, which argues against a ternary complex.
Figure EV3. α‐actinin‐1 and apoCaM do not form a ternary complex with IQ.
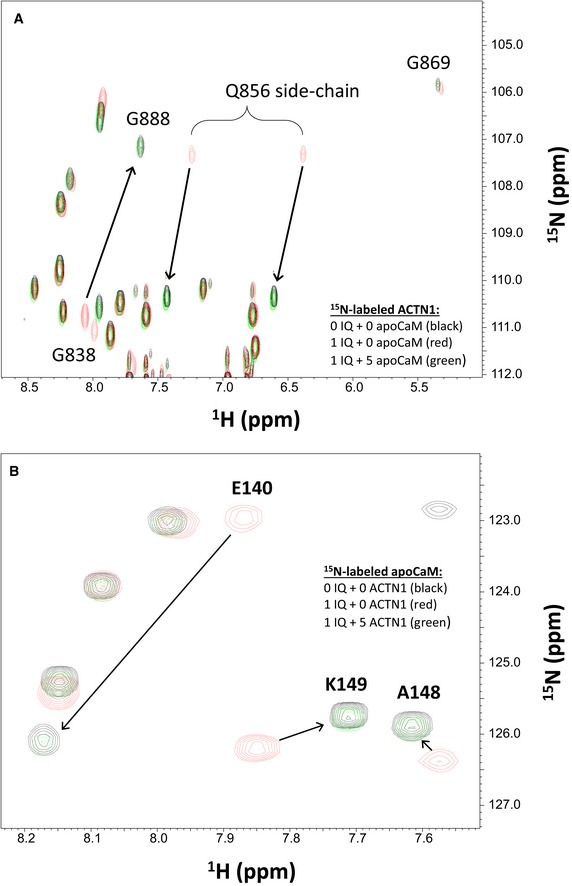
- Overlay of 15N‐1H HSQC spectra of 15N‐labeled α‐actinin‐1 EF‐hand domain (residues 750–892) by itself (black peaks), after adding an equivalent amount of unlabeled IQ peptide (red peaks), and after adding fivefold excess of unlabeled apoCaM combined with the 1 equivalent of unlabeled IQ peptide (green peaks).
- Overlay of 15N‐1H HSQC spectra of 15N‐labeled apoCaM by itself (black peaks), after adding an equivalent amount of unlabeled IQ peptide (red peaks), and after adding fivefold excess of unlabeled α‐actinin‐1 combined with the 1 equivalent of unlabeled IQ peptide (green peaks).
α‐Actinin binding to the CaV1.2 IQ motif augments open probability of individual channels
Point mutations that impaired α‐actinin binding to the IQ motif of α11.2 reduced current density upon reconstitution of CaV1.2 in HEK293 cells by 70–80% but surface expression by only 35–40% as determined by two different methods (Tseng et al, 2017). We performed cell‐attached recordings (Fig 3A) to precisely determine single‐channel parameters of CaV1.2 as before (Davare et al, 2001; Patriarchi et al, 2016; Qian et al, 2017; Bartels et al, 2018). All three mutations in the IQ motif that impaired α‐actinin binding (Table 2) (Tseng et al, 2017), i.e., K1647A, Y1649A, and I1654A, decreased functional availability by 79–93%, ensemble averages by 84‐91%, and single‐channel activity by ~85–92% without affecting unitary current amplitudes (i) (Figs 3B–G and EV4).
Figure 3. CaV1.2 mutations that affect α‐actinin‐1 drastically decrease channel open probability.
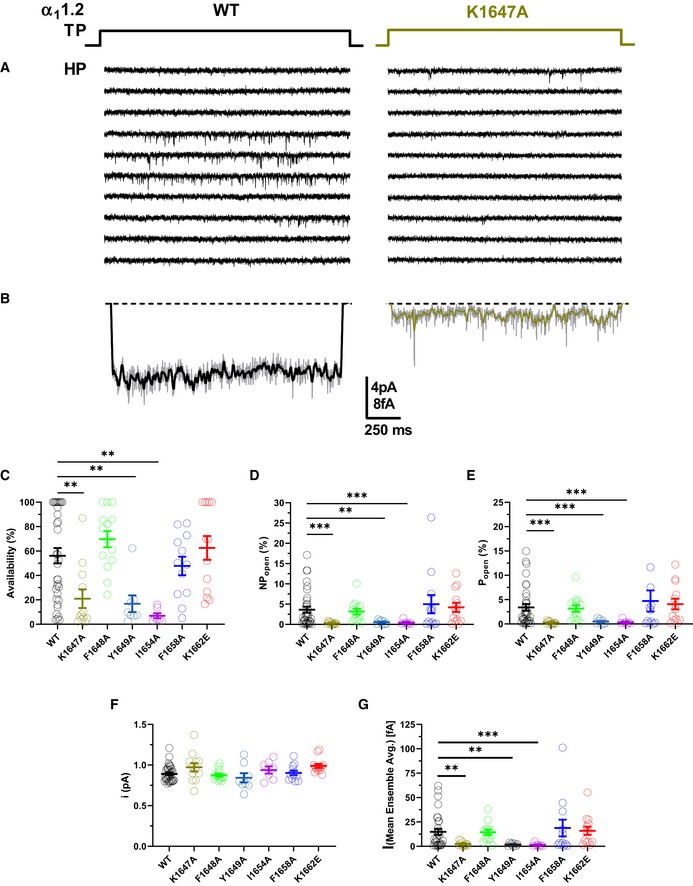
-
ASingle CaV1.2 channel recordings of CaV1.2 WT and K1647A. Holding potential (HP) was −80 mV and test potential (TP) 0 mV. Shown are 10 consecutive sweeps from representative experiments (see Fig EV4 for more sweeps).
-
BMean assemble averages for all experiments with CaV1.2 WT and K1647A, which are based on a total of 2,744 and 868 sweeps, respectively (see Table 5).
-
C–GDot plots and means ± SEM for availability (i.e., likelihood that a sweep had at least one event; C), NPo (D), Po (E), unitary current amplitude i (F), and the mean of the current of a single channel at any point in time calculated from the ensemble averages of each experiment (G) (**P < 0.01, ***P < 0.001 compared to WT; one‐way ANOVA with Bonferroni post hoc test (C) or Welch ANOVA with Tamhane T2 test (D–G); n = 7–35. see Table 5 for more details).
Figure EV4. Representative single‐channel traces for CaV1.2 WT, K1647A, F1648A, Y1649A, I1654A, F1658A, and K1662E.
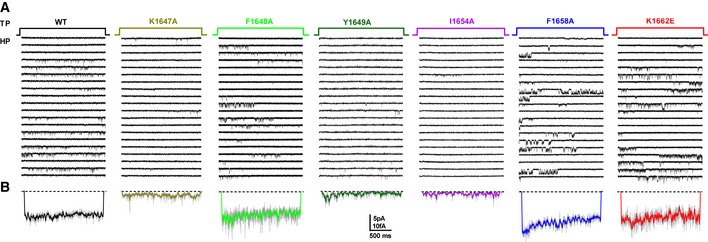
-
A, BHolding potential (HP) was −80 mV and test potential (TP) 0 mV. Shown are 20 consecutive sweeps from representative experiments (A) and mean assemble average currents (B) for all experiments for CaV1.2 WT and each mutant.
The single‐channel activity is the product of the number (N) of channels in a patch, the open probability (Po), and unitary current amplitudes (i) of each individual channel (i.e., N·Po·i). Because i was unaffected (Fig 3F), the mutations must affect NPo. Given that surface expression of all three mutations decreased by only 35–40% versus WT α11.2 under exactly the same conditions (Tseng et al, 2017), the ~90% reduction of NPo for all three mutants to ~10% of WT CaV1.2 suggests that Po of the remaining ~60% channels is only ~1/6 of the Po of WT CaV1.2. These observations suggest a remarkable ~6‐fold decrease in Po upon loss of α‐actinin binding. To corroborate this notion, we determined N for each recording and derived Po for individual channels. Accordingly, Po is reduced by ~90% for individual channels carrying a K1647A, Y1649A, or I1654A mutation (Fig 3E; Table 5a–e). At the same time, the F1658A and K1662E mutations, which diminish apoCaM binding, did not alter NPo (Table 5f,g).
Table 5.
Biophysical properties of CaV1.2 microscopic single‐channel currents for WT versus IQ motif mutants
| Gating parameter | Availability (%) | NPopen (%) | Popen (%) | I (mean ensemble avg.) [fA] | Unitary current (pA) | Sweeps | n |
|---|---|---|---|---|---|---|---|
| WT (a) | 56.2 ± 6.3 | 3.6 ± 0.75 | 3.4 ± 0.7 | 14.8 ± 3.1 | 0.89 ± 0.02 | 2,744 | 32–35 |
| K1647A (b) | 21.0 ± 7.6** | 0.30 ± 0.09*** | 0.27 ± 0.08*** | 2.3 ± 0.6** | 0.97 ± 0.05 | 868 | 12 |
| F1648A (c) | 69.7 ± 6.6 | 3.2 ± 0.7 | 3.2 ± 0.7 | 14.4 ± 2.6 | 0.87 ± 0.02 | 1,221 | 13–14 |
| Y1649A (d) | 16.8 ± 6.8** | 0.53 ± 0.17** | 0.49 ± 0.2*** | 1.9 ± 0.5** | 0.84 ± 0.06 | 556 | 8 |
| I1654A (e) | 7.0 ± 2.1** | 0.44 ± 0.2*** | 0.35 ± 0.2*** | 1.4 ± 0.6*** | 0.94 ± 0.04 | 589 | 7 |
| F1658A (f) | 47.8 ± 7.6 | 5.0 ± 2.2 | 4.7 ± 2.2 | 18.8 ± 8.6 | 0.90 ± 0.03 | 986 | 12 |
| K1662E (g) | 62.5 ± 9.7 | 4.2 ± 1.1 | 4.1 ± 1.1 | 15.9 ± 4.2 | 0.99 ± 0.03 | 1,015 | 13 |
| One‐Way ANOVA | a‐b† | a‐b‡ | a‐b‡ | a‐b‡ | NS | ||
| a‐d† | a‐d‡ | a‐d‡ | a‐d‡ | ||||
| a‐e† | a‐e‡ | a‐e‡ | a‐e‡ |
HEK293 cells were transiently transfected with WT and IQ domain mutant CaV1.2 (α11.2, α2‐δ1, and β2a) for recording of single‐channel activity in 110 mM Ba2+ upon depolarized from a holding potential −80 mV to a test potential (TP) of 0 mV for 2 s at an interpulse rate of 0.14 Hz. Availability is quantified as the fraction of sweeps showing channel activity over the number of total sweeps. Statistical significance was determined by pairwise multiple testing WT (a) against K1647A (b), F1648A (c), Y1649A (d), I1654A (e), F1658A (f), and K1662E (g) by a one‐way ANOVA with Bonferroni† post hoc test or Welch ANOVA with Tamhane‡ T2 test. Given are mean values ± SEM (*P < 0.05,**P < 0.01, ***P < 0.0001).
To further scrutinize the role of α‐actinin in Po of CaV1.2, CaV1.2 was co‐expressed with WT α‐actinin‐1 or its binding‐deficient E847K/E851K mutant (Fig 4A) and availability, NPo, Po, i, and assemble averages determined (Fig 4B–G). Overexpression of WT but not E847K/E851K mutant α‐actinin‐1 increased Po of CaV1.2 WT by more than twofold from 3.4% seen with CaV1.2 alone to 7.9% (Fig 4E; Table 6a–c). Availability, NPo, and assemble averages but not unitary current amplitudes were also increased by WT but not E847K/E851K mutant α‐actinin‐1 (Fig 4B, C, D, F and G; Table 6a–c). Accordingly, in HEK293 cells functional occupancy of CaV1.2 by endogenous α‐actinin appears to be far from complete.
Figure 4. Ectopic α‐actinin‐1 expression increases CaV1.2 open probability.
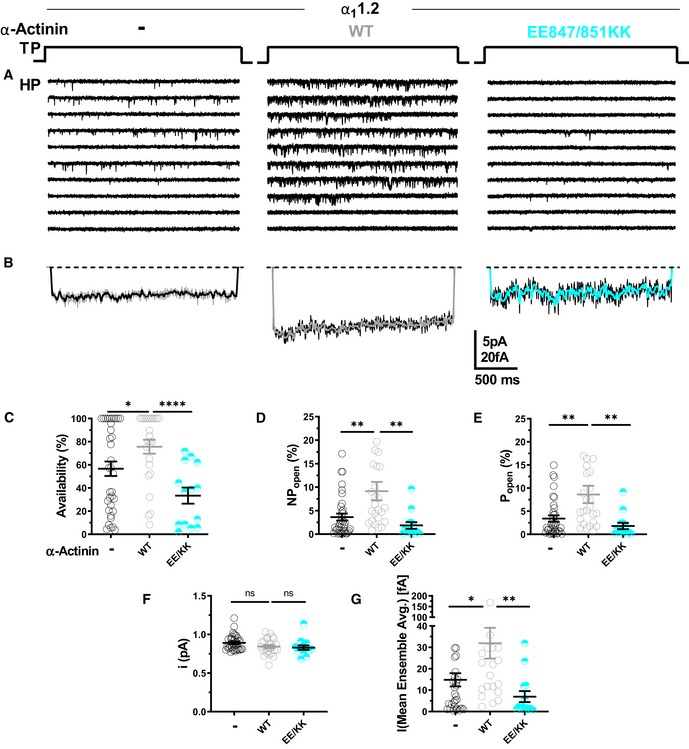
-
ARepresentative single CaV1.2 channel recordings of WT CaV1.2 alone or with WT or E847K/E851K mutant α‐actinin‐1.
-
BMean assemble averages for all experiments for each combination.
-
C–GDot plots and means ± SEM for availability (C), NPo (D), Po (E), unitary current amplitude i (F), and the mean of the current of a single channel calculated from the ensemble averages of each experiment (G) (*P < 0.05, **P < 0.01, ****P < 0.001; unpaired t‐test; n = 13–35; see Table 6 for more details).
Table 6.
Effect of α‐actinin‐1 ectopic expression on biophysical properties of CaV1.2 single‐channel currents
| Gating parameter | Availability (%) | NPopen (%) | Popen (%) | I (Mean ensemble Avg.) [fA] | Unitary current (pA) | Sweeps | n |
|---|---|---|---|---|---|---|---|
| WT (a) | 56.6 ± 6.3 | 3.6 ± 0.75 | 3.4 ± 0.69 | 14.8 ± 3.1 | 0.89 ± 0.02 | 2,744 | 32–35 |
| WT + α‐Actinin (b) | 76.5 ± 6.2* | 9.2 ± 2.0** | 7.9 ± 2.0* | 31.3 ± 7.5* | 0.84 ± 0.02 | 2,388 | 23–25 |
| WT + EE847/851KK (c) | 32.9 ± 7.5**** | 1.9 ± 0.7** | 1.8 ± 0.7* | 7.4 ± 2.8* | 0.83 ± 0.03 | 1,252 | 13 |
| K1647E + α‐Actinin (d) | 43 ± 5.0 | 1.8 ± 0.3 | 1.7 ± 0.3 | 8.5 ± 1.5 | 0.83 ± 0.01 | 2,601 | 30 |
| K1647E + EE847/851KK (e) | 67.9 ± 6.9** | 2.6 ± 0.5 | 2.5 ± 0.5 | 11.3 ± 2.7 | 0.84 ± 0.02 | 1,376 | 16 |
| Unpaired t‐test | a‐b¥ | a‐b¥ | a‐b¥ | a‐b¥ | NS | ||
| b‐c¥ | b‐c¥ | b‐c¥ | b‐c¥ | ||||
| d‐e¥ | d‐e P = 0.7 | d‐e P = 0.7 |
HEK293 cells were transiently transfected with WT CaV1.2 (α11.2, α2‐δ1, and β2a) and, if indicated, with α‐actinin‐1 WT or E847K/E851K. Single‐channel activity was obtained as in Table 5. Statistical significance was determined by pairwise testing of CaV1.2 WT alone (a) versus CaV1.2 WT + α‐actinin WT (b), CaV1.2 WT + α‐actinin‐1 WT versus CaV1.2 + α‐actinin‐1 E847K/E851K (c), and CaV1.2 K1647E + α‐actinin‐1 WT (d) versus CaV1.2 K1647E + α‐actinin‐1 E847K/E851K (e) with an unpaired t‐test¥. Given are mean values ± SEM (*P < 0.05, **P < 0.01, ****P < 0.0001). Values for CaV1.2 WT are provided for comparison from Table 5.
To further test whether α‐actinin needs to bind to the CaV1.2 IQ motif to augment Po, we used a charge inversion experiment by mutating K1647 to the negatively charged glutamyl rather than neutral alanyl residue and then attempted rescue of the expected reduction in Po by pairing expression of K1647E CaV1.2 with charge‐inverted E847K/E851K α‐actinin‐1. Co‐expression of K1647E mutant CaV1.2 with WT α‐actinin‐1 yielded a low availability, NPo, Po, and ensemble average (Fig 5C–E and G; Table 6d), which were well below the values observed upon expression of WT CaV1.2 alone or co‐expression of WT CaV1.2 with E847K/E851K mutant α‐actinin‐1 (Fig 4C–E and G; Table 6a–c). The reduction in availability was statistically significantly, though not fully, rescued when K1647E mutant CaV1.2 was paired with E847K/E851K mutant rather than WT α‐actinin‐1 (Fig 5A; Table 6e). Po also appears to be partially rescued although the P value was 0.07 versus pairing of WT α‐actinin‐1 with K1647E mutant CaV1.2. This rescue of loss of availability and likely Po for K1647E mutant CaV1.2 by E847K/E851K mutant α‐actinin‐1 must be due to either enhanced opening of individual channels, a change in channel surface expression, or both. This rescue is difficult to explain by a mechanism other than that α‐actinin‐1 binding augments channel activity, including binding of endogenous apoCaM. Consistently, impairing apoCaM binding to the CaV1.2 IQ motif by mutating F1658 to Ala or K1662 to Glu had no effect on availability, NPo, and Po, and did not affect ensemble averages (Fig 3C–G; Table 5).
Figure 5. α‐Actinin‐1 E847K/E851K partially rescues open probability for CaV1.2 K1647E.
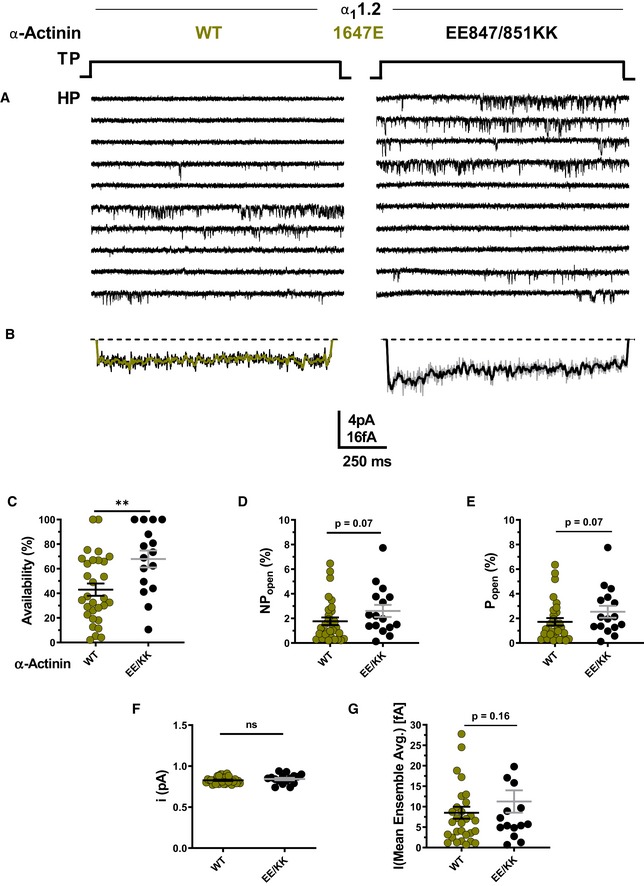
-
ARepresentative single CaV1.2 channel recordings of CaV1.2 K1647E with WT or E847K/E851K α‐actinin‐1.
-
BMean assemble averages for all experiments for both combinations.
-
C–GDot plots and means ± SEM for availability (C), NPo (D), Po (E), unitary current amplitude i (F), and mean of the current of a single channel calculated from the ensemble averages of each experiment (G) (**P < 0.01; unpaired t‐test; n = 16–30; see Table 6 for more details).
One possibility for a reduction in Po is that K1647A, Y1649A, and I1654A shift the voltage dependence of CaV1.2 to more positive potentials such that WT channels open upon depolarization to 0 mV more readily than mutant channels. However, neither the reversal potential nor the voltage dependence of activation was significantly affected by the K1647A and Y1649A mutations and only minimally by the I1654A mutation (Appendix Fig S3; Table 7a–d).
Table 7.
Biophysical properties of CaV1.2 macroscopic whole‐cell patch currents for WT versus IQ motif mutants
| Analysis | I‐V | G‐V | Charge movement | ITail/Qon | ||||
|---|---|---|---|---|---|---|---|---|
| Parameter | V1/2act. (mV) | kact. | Erev. (mV) | V1/2act. (mV) | klow | khigh | Qon (fC/pF) | Slope |
| WT (a) | −18.5 ± 0.5 | −4.2 ± 0.4 (10) | 51.3 ± 1.3 | −6.5.0 ± 0.4 | 7.0 ± 0.3 (16) | −8.5 ± 27.4 | 3.8 ± 0.9 (18) | 3.9 ± 1.5 (16) |
| K1647A (b) | −19.9 ± 0.9 | −5.9 ± 0.6 (11) | 55.2 ± 2.0 | −8.5.0 ± 0.9 | 8.5 ± 0.8 (15) | 9.0 ± 10.5 | 1.3 ± 0.3* (15) | 0.26 ± 1.5 (15) |
| Y1649A (c) | −19.9 ± 0.6 | −4.0 ± 0.5 (10) | 49.2 ± 1.5 | −6.3.0 ± 0.7 | 7.5 ± 0.8 (16) | 28.7 ± 6.9 | 0.98 ± 0.3** (13) | 0.19 ± 1.0 (12) |
| I1654A (d) | −12.6 ± 1.0**** | −5.4 ± 0.7 (5) | 48.2 ± 2.6 | −2.5 ± 0.9** | 7.5 ± 0.9 (13) | 38.5 ± 11.5 | 0.87 ± 0.3** (13) | 1.3 ± 0.6 (12) |
| F1658A (e) | −26.5 ± 1.1** | −4.5 ± 0.9 (6) | 58.6 ± 4.1 | −10.6 ± 1.4** | 5.3 ± 1.6 (11) | 20.2 ± 3.9 | 2.8 ± 0.6 (13) | 3.1 ± 1.4 (12) |
| K1662E (f) | −20.2 ± 0.4 | −4.2 ± 0.3 (12) | 50.1 ± 1.2 | −6.1.0 ± 0.7 | 7.9 ± 0.8 (21) | 49.2 ± 19.4 | 3.5 ± 1.1 (17) | 2.2 ± 1.0 (14) |
| ANOVA/t‐test | a‐d† | a‐d† | a‐b† | |||||
| a‐c† | ||||||||
| a‐e† | NS | NS | a‐e† | NS | NS | a‐d† | ||
HEK293 cells were transiently transfected with WT CaV1.2 (α11.2, α2‐δ1, and β2a). Whole‐cell patch recording was performed in 20 mM Ba2+ (see Fig 7). To obtain current–voltage relationships (I‐V), currents were recorded upon depolarization from a holding potential of −80 mV to increasingly more positive potentials (see Fig 7A for details). The voltage for half maximal activation (V1/2act), activation constant (kact), and reversal potential (Erev) was determined. To obtain conductance–voltage relationships (G‐V), tail currents (ITail) were recorded upon repolarization to −50 mV following depolarization from a holding potential of −80 mV to increasingly more positive potentials (see Fig 7A for details). The activation kinetics for G‐V were best described by double exponential fits, and the rate constants k high and k low were determined accordingly. Total charge movement (Qon) and the ratio of ITail/Qon were determined as in Fig 7. Statistical significance was determined by pairwise multiple testing WT (a) against K1647A (b), Y1649A (c), I1654A (d), F1658A (e), and K1662E (f) by a one‐way ANOVA with a Bonferroni† post hoc test. Given are mean values ± SEM (*P < 0.05, **P < 0.01, ****P < 0.0001).
To test whether this charge inversion rescue for the K1647E mutant promoted surface expression of CaV1.2, we performed surface biotinylation experiments (Fig 6). As expected, co‐expression of WT α‐actinin‐1 with WT CaV1.2 increased α11.2 biotinylation by ~60% versus WT α11.2 alone. Thus, WT CaV1.2 expression at the surface is enhanced by α‐actinin‐1 (Fig 6A). That the increase in surface expression of WT CaV1.2 by WT α‐actinin‐1 overexpression was smaller than the increase in Po is analogous to the smaller effects of the α11.2 K1647A, Y1649A, and I1654A mutations on surface expression compared to charge density (Tseng et al, 2017) and the larger decrease in Po and NPo we report here (Fig 3). Importantly, this increase in CaV1.2 surface expression by α‐actinin‐1 was not seen when WT α11.2 was co‐expressed with E847K/E851K mutant α‐actinin‐1 or K1647E mutant α11.2 with WT α‐actinin‐1 (Fig 6B). However, the charge inversion we performed by combining these mutants failed to increase surface expression of CaV1.2 to a degree that would be detectable (Fig 6B). This result is in contrast to our findings that K1647E α11.2 availability and likely Po were partially rescued by the E847K/E851K α‐actinin‐1 charge reversal (Fig 5). It is conceivable that a small rescue effect did occur but was indiscernible for statistical reasons as 95% confidence intervals (CIs) are larger than a potentially partial rescue of, e.g., 30% of the impairment of ~50% by the K1647E mismatch with WT α‐actinin‐1 (Appendix Table S1; a 30% rescue would translate into only an ~15% increase in surface expression).
Figure 6. Modulation of CaV1.2 surface expression via its interaction with α‐actinin‐1.
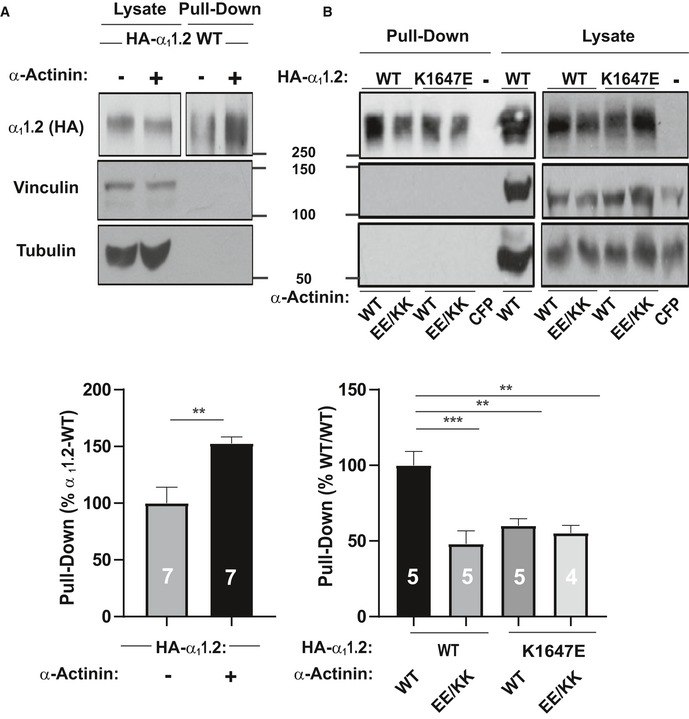
- HEK293 cells were transfected with WT α11.2, α2δ‐1, and β2A ± WT α‐actinin‐1 and cultured for 22–24 h before surface biotinylation. Shown are representative immunoblots of NeutrAvidin pull‐down samples (from lysate containing 600 μg protein) and total lysate samples containing 20 μg protein. α11.2 was detected with an antibody against its HA tag (top), which is present in all constructs used throughout this work. Pull‐down and lysate samples are from the same blot but different exposures because signals from lysate samples were much stronger than from pull‐down samples. Immunoblotting for vinculin (middle) and tubulin (bottom) indicated that comparable amounts of these intracellular control proteins were present in lysate samples. Their absence in pull‐down samples as seen on the same blots showed that these prominent intracellular proteins did not undergo biotinylation as control for membrane integrity during surface biotinylation. Bar graph shows means ± SEM of the pull‐down immunosignals in mutants relative to surface labeling of control α11.2 samples lacking α‐actinin‐1 co‐expression (mean set to 100%; see Materials and Methods; **P < 0.01 two‐tailed t‐test; n = 7).
- HEK293 cells were transfected with WT or K1647E mutant α11.2, α2δ‐1, and β2A, and WT or E847K/E851K mutant α‐actinin‐1, or CFP alone as negative control and cultured for 22–24 h before surface biotinylation. Shown are representative immunoblots of pull‐down and lysate samples as in (A). Bar graph shows means ± SEM of the pull‐down immunosignals in mutants relative to surface labeling in the WT/WT control (mean set to 100%; see Materials and Methods). **P < 0.01, ***P < 0.001; one‐way ANOVA with Tukey post hoc test; n = 4–5).
α‐Actinin binding to the CaV1.2 IQ motif augments gating charge movement as well as its coupling to channel opening
To test the molecular mechanism underlying the α‐actinin‐induced increase in Po in more detail, we analyzed gating charge movement at the Ba2+ current reversal potential (Fig 7) by whole‐cell patch‐clamp recording. This approach measures all of the Ca2+ channels on the surface, thereby ruling out any potential limitation caused by the channel selection inherent in the cell‐attached patch recordings in Figs 3, 4, 5. The K1647A, Y1649A, and I1654A mutations reduced gating currents (Qon) by 66–77% (Fig 7A, B and D; Table 7) suggesting that α‐actinin fosters gating charge movement. This reduction is stronger than the reduction in surface expression (Tseng et al, 2017) but not quite as strong as the 90% reduction in NPo for these mutants (Fig 5). Thus, we investigated whether α‐actinin also augments coupling of gating charge movement to channel opening. We analyzed the relationship of tail currents (Itail) to Qon and determined the slopes of the regression curves as an established parameter for quantification of the coupling of Qon to Itail (Tuluc et al, 2009; Yang et al, 2010). Accordingly, the slopes for K1647A, Y1649A, and I1654A mutant CaV1.2 were strongly reduced compared to WT (Fig 7C; Table 7a–d). Because large gating events were rare for these mutants, we combined all data from those three mutants for further analysis to better cover that range of the curve. This analysis confirmed that the slope of the resulting curve was substantially smaller than for WT CaV1.2 (Fig 7E). Collectively, these data indicate that binding of α‐actinin to the CaV1.2 IQ motif augments not only surface expression but also gating charge movement and its coupling to channel opening. Neither gating charge movements nor their coupling to channel opening were significantly affected by the F1658A or K1662E mutations (Fig 7; Table 7e,f), once more arguing against a role in apoCaM binding to the IQ motif in defining NPo under basal conditions.
Figure 7. CaV1.2 mutations that affect α‐actinin‐1 impair gating charge movement and its coupling to channel opening.
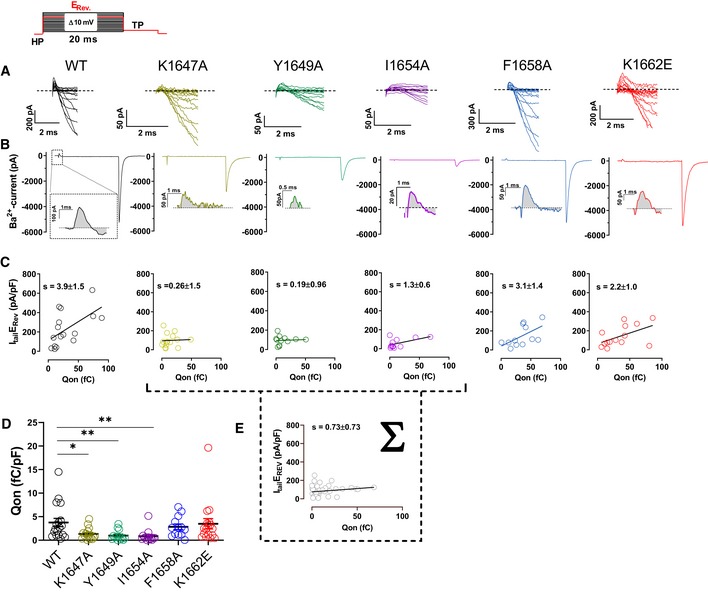
- Representative current traces of the first 2 ms obtained from recordings upon depolarizations from a holding potential of −80 mV to the indicated potentials (the voltage protocol is schematized in the upper left corner).
- Representative current traces upon step depolarizations to the reversal potential (Erev) for 20 ms to obtain movement of the ON‐gating charges (Qon), and subsequent to −50 mV for 10 ms to obtain tail currents (Itail). Insets: magnifications of exemplary Qon for CaV1.2 WT, K1647A, Y1649A, I1654A, F1658A, and K1662E.
- Plots of Itail (in this panel corrected for variations in cell capacitance) versus total detectable charge transfer for Qon. Slopes of regression curves are strongly reduced for CaV1.2 K1647A, Y1649A, and I1654A versus WT (see Table 7 for more details).
- Dot plots and means ± SEM of Qon (in this panel corrected for variations in cell capacitance; *P < 0.05; **P < 0.01; one‐way ANOVA with Bonferroni post hoc test; n = 13–18; see Table 7 for more details).
- The reduction in slope of the regression curve of combined population data for K1647A, Y1649A, and I1654A versus WT indicates reduced coupling of Itail with Qon when α‐actinin binding to the IQ motif is diminished.
Discussion
The NMR structures of α‐actinin‐1 and apoCaM bound to the CaV1.2 IQ motif presented here revealed distinct intermolecular interactions that were verified by mutagenesis experiments involving charge inversion for both interactions. The EF hands in α‐actinin‐1 are not capable of binding to Ca2+ under physiological conditions because their EF‐hand loops lack the proper residues for ligating Ca2+ with high affinity (Backman, 2015). Therefore, the α‐actinin‐1 EF‐hand domain bound to CaV1.2 is in the Ca2+‐free state under basal conditions ([Ca2+]i of ~100 nM) and adopts a semi‐open conformation of Ca2+‐free EF hands like that observed for apoCaM bound to various target proteins (Houdusse et al, 2006; Chagot & Chazin, 2011; Feldkamp et al, 2011). However, the semi‐open EF‐hand conformation of α‐actinin‐1 binds to the IQ peptide with a polarity opposite to that of apoCaM (see black arrows in Fig EV5A and B). The CaV1.2 residue K1647 at the N‐terminal end of the IQ helix forms intermolecular salt bridges with α‐actinin‐1 residues E847 and E851. By stark contrast, the IQ helix is rotated 180 degrees upon binding to apoCaM (see black arrows in Fig EV5). This opposite orientation places CaV1.2 residue K1662 at the C‐terminal end of the IQ helix in close proximity to CaM residues E85 and E88, which are homologous to α‐actinin‐1 residues E847 and E851. Similarly, the IQ helices in voltage‐gated Na+ channels bind to apoCaM and Ca2+‐saturated CaM with opposite polarity (Hovey et al, 2017). Thus, the orientation of the IQ helix bound to the EF‐hand motif may be an important structural determinant of binding specificity and may explain why α‐actinin‐1 and apoCaM have different functional effects.
Figure EV5. IQ peptide binds with opposite polarity to α‐actinin‐1 versus apoCaM.
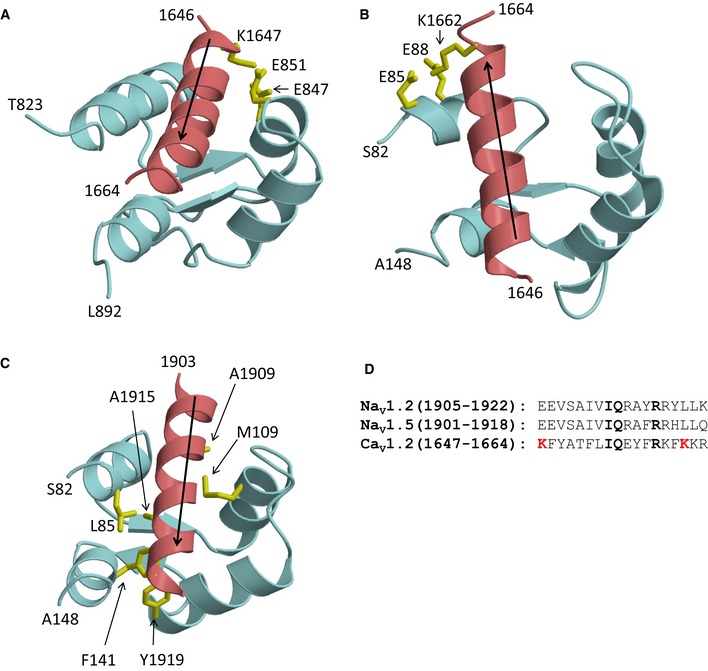
- NMR structure of α‐actinin‐1 (cyan) bound to CaV1.2 IQ motif (red).
- NMR structure of apoCaM (cyan) bound to CaV1.2 IQ motif (red).
- NMR structure apoCaM (cyan) bound to the NaV1.2 IQ motif (red). PDB accession number is 2KXW (Feldkamp et al, 2011). Non‐conserved intermolecular contacts between hydrophobic side‐chain atoms are colored yellow. The β‐methyl side‐chain atoms of NaV1.2 residues A1909 and A1915 are 2.5 Å away from side‐chain methyl atoms of apoCaM residues M109 and L85, respectively. The aromatic side‐chain atoms of NaV1.2 residue Y1919 are 3.3 Å away from aromatic side‐chain atoms of apoCaM residue F141.
- Amino acid sequence alignment of IQ motifs from NaV1.2, NaV1.5, and CaV1.2. Residues that are conserved between NaV and CaV are shown in boldface font. Lysine residues in CaV1.2 that form intermolecular salt bridges in the CaV1.2 IQ peptide complexes with α‐actinin‐1_EF3/4 and apoCaM and are not conserved between the Ca2+ and Na2+ channels are colored red.
Voltage‐gated Na+ channels exhibit Ca2+‐dependent inactivation (CDI) mediated by Ca2+‐CaM (Ben‐Johny et al, 2014; Gabelli et al, 2016), similar to CaV1.2 (Peterson et al, 1999; Zuhlke et al, 1999). However, the structure of apoCaM bound to the CaV1.2 IQ motif (Fig EV5B) is quite different from previous structures of apoCaM bound to NaV1.2 (Fig EV5C) or NaV1.5 (Chagot & Chazin, 2011; Gabelli et al, 2014). The NaV1.2 IQ motif sequence is only 17% identical to that of CaV1.2 (Fig EV5D). NaV1.2 residues A1909, A1915, and Y1919, which are not conserved in CaV1.2, each make critical contacts with apoCaM (Fig EV5C). The side‐chain methyl groups of A1909 and A1915 are each 2.5 Å away from the side‐chain methyl atoms of CaM residues M109 and L85, respectively. The aromatic side chain of Y1919 is in intimate contact with the aromatic side chain of F141 from CaM. Another important difference is that the CaV1.2 IQ helix binds to apoCaM with opposite directionality compared to the NaV1.2 IQ helix (see black arrows in Fig EV5B and C). Hence, the salt bridge that connects CaV1.2 (K1662) to apoCaM (E88) is not conserved in NaV1.2 or NaV1.5 but the large number of non‐conserved intermolecular hydrophobic contacts to NaV1.2 (Fig EV5C) caused by the opposite binding orientation of the IQ helix should outweigh any stabilization from the salt bridge in CaV1.2 and may explain why apoCaM binds to NaV1.2 and NaV1.5 with nanomolar affinity (Hovey et al, 2017) compared to the much weaker micromolar affinity of apoCaM binding to CaV1.2 (Table 4).
The relatively high dissociation constant for apoCaM binding to the α11.2 IQ motif (K d = 10 μM) may be outside the physiological concentration range for apoCaM in neurons. Under basal conditions, the cytosolic apoCaM is kept below 1 μM (Wu & Bers, 2007) by proteins that have a high affinity for apoCaM. For instance, in neurons neurogranin (RC3) (Huang et al, 2000, 2004; Ran et al, 2003; Zhong et al, 2011) and GAP‐43 (P‐57) (Cimler et al, 1985) serve as sinks and reservoirs for apoCaM. In neurons, total concentration of CaM is ~10 μM (Egrie et al, 1977; Zhabotinsky et al, 2006), of neurogranin is 20 μM (Huang et al, 2004; Zhabotinsky et al, 2006), and of GAP‐43 is 18 μM (Cimler et al, 1985; Zhabotinsky et al, 2006), and their K d values for apoCaM are in the range of 1–5 μM (Alexander et al, 1987; Huang et al, 2000; Zhabotinsky et al, 2006), resulting in 0.9 μM free CaM under basal conditions (Zhabotinsky et al, 2006). Therefore, on the basis of the relatively low binding affinity of apoCaM (K d = 10 μM), the fractional binding of apoCaM to α11.2 can be estimated to be < 10% under basal physiological conditions in neurons. Recent work suggests that ectopically expressed WT CaM as well as Ca2+ binding‐deficient CaM1234 is in large excess over endogenous CaM (Iacobucci & Popescu, 2017). As a result, CaM1234 may occupy a much larger fraction of α11.2 than the endogenous WT apoCaM. In this scenario, expression of CaM1234 could impair CDI as seen earlier (Peterson et al, 1999) by mechanisms other than displacement of endogenous apoCaM as invoked earlier (Peterson et al, 1999).
Alterations of I1609 in α11.3 (equivalent to I1654 in α11.2) impair binding of apoCaM to the closely related CaV1.3 channel and Po, which was interpreted to mean that apoCaM binding to this IQ motif augments Po (Adams et al, 2014). Here, we show that the homologous I1654 in the highly conserved IQ motif of CaV1.2 is not only critical for apoCaM but also α‐actinin binding. Accordingly, mutating I1609 could have decreased NPo by impairing α‐actinin rather than apoCaM binding. Indeed, cell‐attached channel recordings showed an ~90% reduction in Po for all three α‐actinin binding‐deficient α11.2 IQ mutants. This effect included K1647, which contacts α‐actinin (Fig 1C) but not apoCaM. Consistently, the K1647A mutation reduced binding of α‐actinin (Table 2) but not apoCaM (Table 4). The same is true for the Y1649A mutation. Although Y1649 does not form a direct interaction with α‐actinin EF3‐EF4, the Y1649A mutation does abrogate α‐actinin binding in the yeast two‐hybrid system (Tseng et al, 2017). We suggest that Y1649 is necessary to stabilize the orientation of L1653 and neighboring I1654, which is required for binding to α‐actinin (Appendix Fig S4A). Thus, our new data indicate that Po is largely determined by α‐actinin and not apoCaM binding. In fact, the F1658A and K1662E mutations, which decreased apoCaM binding (Table 4), did affect neither Po (Fig 3E) nor Qon (Fig 7A, B and D) and minimally if at all α‐actinin binding (Table 2). In support of these results, others found no effect of CaM on CaV1.2 plasma membrane targeting in HEK293 cells (Bourdin et al, 2010). This conclusion is further supported by the charge inversion experiments of K1647E mutant α11.2 and E847K/E851K mutant α‐actinin‐1 in which impaired functional channel availability and likely impaired Po are partially rescued (Fig 5). These findings support the functional relevance of the interaction of the positively charged K1647 in the IQ motif with the negatively charged E847 and E851 of α‐actinin‐1. That the rescue of the reduction in Po of K1647E mutant α11.2 by E847K/E851K mutant α‐actinin‐1 is far from complete can in part be explained by analysis of the E851K rotamers (Appendix Fig S4B–D). Most of the E851K rotamers are predicted to clash with side‐chain and backbone atoms within the α‐actinin region at the channel–α‐actinin interface based on our structure (Fig 1C), which will most likely affect the positioning of the EF‐hand region of α‐actinin relative to α11.2 and thereby the α‐actinin–α11.2 interaction. As a result, binding of E847K/E851K mutant α‐actinin‐1 to α11.2 will be decreased as compared to WT α‐actinin. In fact, binding affinity is also not fully rescued when the K1647E IQ peptide is titrated with E847K/E851K mutant α‐actinin‐1 EF3/4 segment (Table 2). A change in the exact structure of the region surrounding the IQ motif in full‐length α11.2 could be the reason for further impairment of α‐actinin binding furnishing a potential explanation for our finding that functional availability and Po are less effectively rescued by the full charge inversion than the in vitro binding affinity.
Further mechanistic insight is provided by the strong reduction in gating charge movement upon loss of α‐actinin binding (Fig 7). The most striking and very clear finding for Qon is the observation that gating charges for the three α‐actinin binding‐deficient α11.2 mutants are so small that they are often barely if at all detectable and amplitudes are difficult to determine. Accordingly, α‐actinin binding to the IQ motif promotes the outward movement of the S4 segments, which gives rise to the gating charges, presumably by either lowering energy barriers for this motion or by stabilizing the “outward” conformation of S4 segments. In addition, α‐actinin binding to the IQ motif augments also the coupling between this gating movement and channel opening. This finding is reminiscent of the effect of CaV1.2 phosphorylation by PKA on S1700, which upregulates this coupling (Fuller et al, 2010, 2014). Because of the close proximity of S1700 to the IQ motif, it is tempting to speculate that phosphorylation of S1700 and α‐actinin binding to the IQ motif intersect to facilitate channel gating, possibly through similar effects on overall conformations of CaV1.2.
All three α‐actinin binding‐deficient IQ mutants of CaV1.2 reduced NPo by ~90% when channel density was reduced by only 35–40% as seen by surface labeling (Tseng et al, 2017). To further ascertain that a large portion of the reduction in NPo is due to a reduction in Po and not just N, i.e., not only due to a reduction in surface expression, we determined the 95% confidence intervals (CIs) for surface biotinylation and surface labeling with antibodies with subsequent analysis by fluorescence‐activated cell sorting (Bourdin et al, 2010; Yang et al, 2010), Po, and Qon (Appendix Tables S1–S3). The CIs for surface biotinylation and surface antibody labeling are remarkably similar (e.g., 52–69% and 51–71%, respectively, for the most relevant mutant K1647A, with WT being 100%) and overlap not at all with Qon and Po (26–44% and 6–10%, respectively, for K1647A, with WT again being 100%). Thus, the CIs for the 95% CIs for reductions are ~29–49% for surface stainings, 56–74% for Qon, and 90–94% for Po. Accordingly, it appears extremely unlikely that the reductions in surface expression can fully account for the reductions in Qon and Po for α‐actinin binding‐deficient Cav1.2; rather, loss of surface localization is only responsible for a fraction of the reductions in Qon and Po. Similarly, the reduction in Qon can most likely only partially account for the reduction in Po, supporting the notion that coupling between the movement of the voltage sensor and the channel gate is impaired in addition to the movement of this sensor.
If loss of α‐actinin binding to the IQ motif would not affect Po of individual channels, then NPo of all α‐actinin binding‐deficient IQ mutants should be ~60% of wild‐type CaV1.2. A 90% decrease in NPo means that 60% of the CaV1.2 channels that remain on the cell surface upon impairment of α‐actinin binding carry only ~10% of the current seen for the wild‐type CaV1.2 population. These effects translate into a sixfold reduction in Po of individual CaV1.2 channels upon loss of α‐actinin binding.
Together with earlier work that showed that α‐actinin increases surface localization and postsynaptic accumulation of CaV1.2 (Hall et al, 2013; Tseng et al, 2017), this new work now demonstrates that α‐actinin serves a dual role in regulating CaV1.2 function. It not only promotes CaV1.2 surface expression but, remarkably, also exerts a strong positive effect on Po. This mechanism allows CaV1.2 to be minimally active during secretory trafficking or when it is outside its target regions but to become functionally fully engaged when anchored at proper locations such as postsynaptic sites. Thus, coupling of α‐actinin binding to both localization and activity of CaV1.2 is perfectly fine‐tuned. This mechanism is so far unique. Whether analogous mechanisms apply to other channels and especially other Ca2+ channels, which may require potent mechanisms to prevent inappropriate and potentially harmful Ca2+ flux from secretory compartments or at the wrong locations on the cell surface, is now an intriguing premise that will inspire future work.
Materials and Methods
NMR spectroscopy
Xenopus calmodulin was expressed in E. coli strain BL21(DE3) grown in LB medium (unlabeled proteins) or M9 media supplemented with 15NH4Cl or 15NH4Cl/13C‐glucose for single‐ or double‐labeled proteins, respectively. Recombinant CaM was prepared as described previously (Zhang et al, 2012).
Human α‐actinin‐1_EF12 (residues 750–812) and α‐actinin‐1_EF34 (residues 822–893) were each subcloned into pET3b vector and expressed in E. coli strain BL21(DE3) grown in LB medium (unlabeled proteins) or M9 media supplemented with 15NH4Cl or 15NH4Cl/13C‐glucose for single‐ or double‐labeled proteins, respectively. α‐actinin‐1_EF12 and α‐actinin‐1_EF34 were each purified and prepared as described (Turner et al, 2016).
Unlabeled CaV1.2 IQ peptide (residues 1,644–1,664) was purchased from ChinaPeptides. The peptide was dissolved in d6‐DMSO to give a peptide concentration of 7.8 mM. An aliquot of peptide (1.5 equivalents) was added to a dilute solution of α‐actinin‐1_EF34 or apoCaM (50 μM protein dissolved in 20 mM 2‐amino‐2‐hydroxymethyl‐propane‐1,3‐diol‐d11 (Tris‐d 11) with 50 mM NaCl, 5 mM dithiothreitol‐d10 (DTT‐d10), and 95% H2O/5% D2O) and incubated at 15°C for 1 h to ensure complete binding of the peptide. The complex was then concentrated to a final concentration of 500 μM in a final volume of 500 μl for NMR experiments.
Production and use of isotope‐labeled peptides using pET‐31
We expressed 15N‐ or 15N/13C‐labeled IQ peptide in E. coli as a fusion protein with the ketosteroid isomerase (KSI) using the pET31 plasmid (Kuliopulos et al, 1994) (Novagen/EMD Biosciences). KSI fusion proteins are concentrated in inclusion bodies in E. coli, protecting the fused peptide from proteolysis. The fusion protein was purified by affinity chromatography under denaturing conditions via a 6x‐His tag at the C‐terminus. To release the peptide of interest, the purified fusion is cleaved using cyanogen bromide (CNBr). The CNBr cleaves at methionine residues that were engineered between the KSI and peptide, and between the peptide and 6x‐His tag. This cleavage mixture is then rotary evaporated to dryness and resuspended in an appropriate ratio of acetonitrile/water, which solubilizes primarily the peptide and not KSI. Finally, the peptide is purified by reverse‐phase HPLC.
Two complementary oligonucleotides that code for Cav1.2 IQ peptide (5′p‐TTT TAT GCG ACC TTT CTG ATT CAG GAA TAT TTT CGC AAA TTT AAA AAA CGC AAA ATG; 5′p‐TTT GCG TTT TTT AAA TTT GCG AAA ATA TTC CTG AAT CAG AAA GGT CGC ATA AAA CAT) were annealed in 40 mM Tris–HCl, pH 8.0, 10 mM MgCl2, and 50 mM NaCl by heating to 95°C and then cooling slowly (> 2 h) to room temperature (RT). The annealed, double‐stranded insert was then ligated into AlwNI‐digested and dephosphorylated pET31 using T4 DNA ligase. The 5′‐phosphorylation augments ligation, and the extra Met codons (underlined) provide compatible sticky ends for the AlwNI‐cut plasmid as well as the sites of CNBr cleavage. 2 μl of the ligation mixture was transformed into DH5 alpha cells and plasmids from individual ampicillin‐resistant colonies sequenced. Successful insertions were obtained, including some multiple insertions. We used one of the single‐insertion plasmids for expression in M9 minimal media. E. coli was lysed by sonication. The insoluble material was collected by ultracentrifugation and resuspended in 6M guanidine, insoluble material removed by ultracentrifugation, and the supernatant loaded onto a Ni‐NTA column equilibrated with 6M guanidine buffer. After elution with 300 mM imidazole, the purified fractions were pooled and then dialyzed against ultrapure water, causing the fusion protein to precipitate. Precipitate was collected by centrifugation and resuspended in 80% formic acid, transferred to a round‐bottom flask, and injected with N2 at ~3 psi. For a 2L scale expression, we used 60 ml of formic acid solution and added 2 g of solid CNBr to start the cleavage reaction. After overnight reaction, the flask was roto‐evaporated to dryness. The clear peptide film was resuspended in 40% acetonitrile/60% H2O and mixed for > 1 h. After centrifugation to remove any insoluble material, the supernatant was lyophilized, resuspended in H2O + 0.1% trifluoroacetic acid, and run over a C18 reverse‐phase HPLC column using a gradient of 9–28% acetonitrile. Peak fractions were collected, lyophilized, and analyzed by MALDI‐MS to identify the desired peptide product. Dry fractions of peptide were resolubilized in DMSO‐d6 to a concentration of > 5 mM.
To measure residual dipolar couplings (RDCs) of α‐actinin‐1_EF34 or apoCaM bound to IQ peptide, the filamentous bacteriophage Pf1 (Asla Biotech Ltd., Latvia) was used as an orienting medium. Pf1 (12 mg/ml) was added to 15N‐labeled α‐actinin‐1_EF34 or apoCaM bound to unlabeled IQ at pH 7.0, to produce weak alignment of the complex.
Haddock structure determination of α‐actinin‐1/IQ or apoCaM/IQ
The molecular docking of α‐actinin‐1 and apoCaM to the CaV1.2 IQ motif (residues 1,644–1,664) was performed using the Haddock d‐level 2.2 web server as described (van Zundert et al, 2016). Residual dipolar couplings, chemical shift perturbation, and mutagenesis data were used as structural restraints. For active restraints or ambiguous interaction restraints (AIRs), chemical shift perturbation was used, selecting residues whose chemical shift perturbation falls above the average perturbation.
The initial docking calculation used NMR‐derived structures of α‐actinin‐1 or apoCaM as determined in this study, which were each docked with the helical structure of Cav1.2 IQ peptide (residues 1,644–1,664) (Van Petegem et al, 2005) as input structures for Haddock. A total of 69 and 42 AIR restraints were used for α‐actinin‐1 and apoCaM, respectively, based on chemical shift perturbation data, and 16 active restraints were used for the IQ peptide from chemical shift perturbation. Unambiguous restraints were introduced to define key intermolecular interactions (IQ K1647–α‐actinin‐1_EF34 E847; IQ K1647–α‐actinin‐1_EF34 E851; IQ K1662–CaM E88; IQ I1654–α‐actinin‐1_EF3/4F833; and IQ I1654–apoCaM F90), which were each verified by mutagenesis (Tables 2 and 4).
Initial docking calculations used AIRs based on chemical shift perturbation data, and the top 200 structures were selected for simulated annealing and water refinement. The lowest energy structures were then run again, adding unambiguous restraints based on mutagenesis data. Rigid‐body docking, simulated annealing, and water refinement were run using the top 200 structures. RDC restraints assigned to α‐actinin‐1 and apoCaM were then added using the Sani statement, with tensor values Dr and Da calculated using the program PALES (Zweckstetter, 2008). A total of 74 RDC values were used from residues found in regions of regular secondary structure and as deemed reliable by the PALES calculation.
Fluorescence polarization (FP) assays
Fluorescein‐labeled peptides (100 nM; ChinaPeptides, Shanghai, China) were titrated with increasing concentrations of either purified α‐actinin‐1 or CaM in FP buffer (50 mM HEPES, pH 7.4, 100 mM KCl, 1 mM MgCl2, 0.05 mM EGTA, 5 mM nitrilotriacetic acid) and FP determined with a Synergy 2 plate reader (BioTek, Winooski, VT) as described (Zhang et al, 2014; Patriarchi et al, 2016; Tseng et al, 2017). FP was calculated as P = (Iv – g*Ih)/(Iv + g*Ih); Iv is vertical and Ih is horizontal fluorescence intensity, respectively, and g is the correction factor for fluorescein. To obtain binding curves and K d values, data were fitted in GraphPad Prism 5 to the equation Y = B*X/(K d + X); B is maximal FP value that would be reached at saturation as determined by extrapolation of the fitted curve.
Isothermal titration calorimetry
ITC experiments were performed using a VP‐ITC calorimeter (MicroCal) at 27°C, and data were acquired and processed with MicroCal software as described previously (Wingard et al, 2005). Samples of apoCaM (injectant) and IQ (titrant) were prepared by exchanging each into buffer containing 50 mM HEPES, pH 7.4, 100 mM NaCl, 0.05 mM EGTA, and 1 mM MgCl2. The IQ peptide in the sample cell (10 μM, 1.5 ml) was titrated with apoCaM (100 μM) using 35 injections of 10 μl each.
Expression of CaV1.2 and α‐actinin‐1 in HEK293 cells
HEK293T/17 cells were maintained in DMEM‐10 [Dulbecco's modified Eagle's medium (Life Technologies) supplemented with 10% fetal bovine serum (FBS, Atlanta Biologicals)] at 37°C in humidified incubators injected with 5% CO2 and 95% air (Tseng et al, 2017). For expression of CaV1.2, HEK293T/17 cells were transfected with rat α11.2 (GenBank ID: M67515.1) cDNA subcloned into pECFP‐C1 vector (Tseng et al, 2017) encoding an in‐frame N‐terminally fused eCFP tag and an HA tag in the S5‐H5 extracellular loop of domain II (Green et al, 2007), which does not affect channel properties (Altier et al, 2002). The point mutations in plasmids encoding single‐residue K1647A, K1647E, F1648A, Y1649A, I1654A, F1658A, and K1662E exchanges in α11.2 were generated via QuikChange II using the above pECFP‐C1 rat α11.2 plasmid DNA as template as described (Tseng et al, 2017) and the oligonucleotides described in Appendix Table S4. For full expression of CaV1.2, cells were also co‐transfected with pGWIH‐based plasmids encoding the auxiliary subunits rat β2A (Perez‐Reyes et al, 1992) and rabbit α2δ‐1 (Ellis et al, 1988). To assess the contributions of α‐actinin‐1 on CaV1.2, cells were transfected with pCMV plasmid DNAs encoding WT (Hall et al, 2013) or mutant α‐actinin‐1. The K847E/K851EE point mutations in α‐actinin‐1 were produced via QuikChange II mutagenesis as before (Tseng et al, 2017). The forward primer for making the K851E mutation, which was performed first, was 5′p‐CCA TGG ACA AAT TGC GCA GAA AGC TGC CAC CCG ACC AGG and reverse primer 5′p‐CCT GGT CGG GTG GCA GCT TTC TGC GCA ATT TGT CCA TGG. Forward primer for the K847E mutation was 5′p‐GAA CTA CAT TAC CAT GAA CAA ATT GCG CCG CGA GCT GCC ACC C and reverse primer 5′p‐GGG TGG CAG CTC GCG GCG CAA TTT GTC CAT GGT AAT GTA GTT C. Transfection of HEK293T/17 cells was accomplished using Ca2+ phosphate precipitation (Tseng et al, 2017) for single‐channel recording and surface labeling or Lipofectamine 2000 for the analysis of Qon.
Cell surface biotinylation assays
Seven hours after transfection, cells were washed once with 150 mM NaCl, 10 mM NaHCO2, pH 7.4 (PBS) and cultured for another 15–17 h in fresh medium. 22–24 h after transfection, cells were either harvested, monodispersed (> 95% viability), and then labeled with EZ‐link‐sulfo‐NHS‐LC‐biotin (Thermo Fisher Scientific) in solution essentially as described (Tseng et al, 2017) or directly labeled while adhered to the petri dish. In the latter case, adherent cells on the dish were washed twice with ice‐cold PBS containing 0.5 mM Mg2+ and 1 mM Ca2+ and incubated on ice with PBS containing 0.4 mg/ml EZ‐link‐sulfo‐NHS‐LC‐biotin for 15 min. The labeling reaction was quenched by three 5‐min washes with 40 mM glycine in PBS containing 0.5 mM Mg2+ and 1 mM Ca2+ on ice. Cells were harvested by scraping in PBS and collected by centrifugation. Cell pellets were lysed in ice‐cold RIPA buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 5 mM EGTA, 10 mM EDTA, 1% NP‐40, 0.05% SDS, 0.4% DOC, and 10% glycerol) supplemented with a cocktail of protease inhibitors (1 μg/ml leupeptin (Merck Millipore), 2 μg/ml aprotinin (Merck Millipore), 1 μg/ml pepstatin A (Merck Millipore), and 34 μg/ml phenylmethanesulfonyl fluoride (PMSF; Sigma)). The solubilized material was cleared of insoluble debris by centrifugation at 200,000× g for 30 min at 4°C. Biotinylated constituents in 600 μg of cell protein lysate were affinity purified by incubation with 30 μl of NeutrAvidin‐conjugated Sepharose beads (Thermo Fisher Scientific) for 2 h at 4°C. Bead‐bound material was collected by centrifugation and washed three times with ice‐cold buffer, and immobilized proteins were extracted in SDS sample buffer. Proteins were fractionated by SDS–PAGE in 7.5% acrylamide gels and transferred onto polyvinylidene difluoride (PVDF; Bio‐Rad) membranes. PVDF membranes were incubated in blocking buffer (BB) consisting of 150 mM NaCl, 10 mM Tris–HCl, pH 7.4 (TBS) with 0.10% Tween (TBST), and 2% bovine serum albumin (BSA; RPI Corp.) for 1 h at RT before incubation with primary antibodies in BB for 3 h at RT. α11.2 was detected by antibodies against the HA tag and, for confirmation, the intracellular loop II/III FP1 epitope (Buonarati et al, 2017) and the CNC2 epitope near the C‐terminus of α11.2 (Buonarati et al, 2017). Probing with antibodies against the cytosolic protein vinculin (Cell Signaling Technologies) and α‐tubulin (Santa Cruz Biotechnology) was used to correct for variation in protein content in lysates and as negative control for surface biotinylation. Membranes were washed for 40 min with at least five exchanges of TBST, incubated with horseradish peroxidase‐conjugated secondary goat anti‐mouse antibodies (HA, α‐tubulin; Jackson) or mouse anti‐rabbit antibodies (FP1, CNC2, vinculin; Jackson) for 1 h at RT, and washed again with TBST with at least five exchanges for 1.5 h. Immunosignals were detected using the horseradish peroxidase substrates Luminata Classico or Crescendo (Merck Millipore) or Femto (Thermo Fisher Scientific) by X‐ray film (Denville Scientific Inc.). Multiple exposures over increasing time periods were taken to ensure that all signals were in the linear range (Davare & Hell, 2003; Hall et al, 2006). Films were scanned and assessed via ImageJ to determine signal intensity for each band. Background signals in individual lanes were subtracted from the band signal before further analysis. To correct differences in immunosignal strengths due to potential differences during immunoblotting and film exposures between experiments, the individual immunosignals for each α11.2 pull‐down sample were divided by the sum of all immunosignals from one blot to obtain the relative signal fraction for each band (Degasperi et al, 2014). The mean of the WT control signals from all experiments was then calculated and all fractional values from all samples (WT and mutants) divided by this value (the WT mean transforms to 1 with this algorithm) (Degasperi et al, 2014). All values were then converted to percent, with the mean of WT control equaling 100%. The data were statistically analyzed applying either a Student's t‐test (two‐sample comparison) or ANOVA with Bonferroni's post hoc test as before (Tseng et al, 2017).
Cell‐attached Patch‐Clamp recording
Cell‐attached patch‐clamp recordings were performed as before (Davare et al, 2001; Patriarchi et al, 2016; Qian et al, 2017) on an Olympus IX70‐inverted microscope at room temperature (22°C). Recordings were obtained with an Axopatch 200B amplifier, and data were sampled at 10 kHz with a low‐pass filter at 2 kHz (3 dB, four‐pole Bessel) and digitalized with a Digidata 1440 digitizer. Recording electrodes were fabricated from borosilicate capillary glass (0.86 OD) with a Flaming micropipette puller (Model P‐97, Sutter Instruments) and polished (polisher from World Precision Instruments; 3.5–6.5 MΩ resistance). The extracellular solution contained (in mM) 145 KCl, 10 NaCl, and 10 HEPES, pH 7.4 (NaOH). The high K+ concentration was used for optimal control of the transmembrane potential under the patch during depolarizations to 0 mV. The pipette solution contained (in mM) 20 tetraethylammonium chloride (TEA‐Cl), 110 BaCl2 (as charge carrier), and 10 HEPES, pH 7.3 (TEA‐OH). Cells were depolarized from a holding potential of −80 mV to 0 mV every 5 s. Event lists were translated from raw Ba2+ currents after leak, and capacity transients were digitally subtracted. Data were analyzed based on the half‐height criterium (Sachs et al, 1982) with the single‐channel software provided by pClamp 10. The number of channels (k) in the patch was estimated based on the observed simultaneous and stacked openings over several minutes at the depolarizing test potential (Herzig et al, 2007; Bartels et al, 2018). The k‐value is then determined by the observed maximum current amplitude divided by the unitary current amplitude. Data with more than > 3 channels (k > 3) in the patch were not considered for statistical analysis in order to prevent overinterpretation of channel open probability. For statistical analysis, single‐channel parameters were corrected by the channel number as previously (Schroder et al, 1998; Bartels et al, 2009). For a sufficient statistical analysis, 50–100 Ba2+ current traces were recorded on average for each cell for each experimental condition.
Whole‐cell patch‐clamp recordings
Whole‐cell patch‐clamp recordings were performed as before (Bartels et al, 2018) on an Olympus IX70‐inverted microscope at room temperature (22°C). Macroscopic Ba2+ currents (IBa) of Cav1.2 were recorded in external solution containing (in mM) 75 CsCl, 40 TEA‐Cl, 20 BaCl2, 1 MgCl2, 10 HEPES, and 10 glucose with a pH adjusted to 7.2 (TEA‐OH) and an osmolarity of 300–310 (sucrose). The internal pipette solution contained (in mM) 110 CsCl, 30 TEA‐Cl, 1 MgCl2, 4 Mg‐ATP, and 10 HEPES, pH 7.2 (CsOH), mOsm 290–300 (sucrose). Pipette resistance was usually between 1.7 and 2.5 MΩ. The series resistance and the cell capacitance were taken from an Axopatch 200B amplifier (Molecular Devices) and compensated not more than < 40% in order to prevent current oscillation. On‐gating currents (Qon) were sampled at 50 kHz and low‐pass‐filtered at 5 kHz and further quantified through current integration over the first 2–3 ms of the beginning of the test pulse. Cells were clamped at a holding potential of −80 mV and depolarized by a 20 ms test pulse of a serious of activating potentials starting from −60 mV to +80 mV to determine the reversal potential (Erev). Tail currents (Itail) were then measured after repolarization to −50 mV for 10 ms. Recorded data were leak and capacity corrected with an online P/4 protocol. The liquid junction potential was not considered for correction in the experiment. Data acquisition and analysis were obtained with pClamp 10. Curve fitting was performed by using GraphPad Prism VIII software (San Diego).
Author contributions
MT, DEA, MN‐C, PB, KNMM, P‐YT, MFN, MCH, JBA, and JWH designed experiments; MT, DEA, MN‐C, PB, AMC, KNMM, PBH, P‐YT, MFN, and MCH performed experiments; MT, DEA, MN‐C, PB, AMC, PBH, VY‐Y, DMB, MFN, MCH, JBA, and JWH analyzed data; and MT, DEA, MN‐C, PB, VY‐Y, DMB, MFN, MCH, JBA, and JWH wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank Dr. Elza Kuzmenkina (University of Cologne, Germany) for providing the coding of the algorithm for calculating assembly averages. This work was supported by NIH grants T32 GM113770 (AMC), T32 GM099608 (PBH), R01 HL098200 (MFN), R01 HL121059 (MFN), R01 EY012347 (JBA), R01MH097887 (JWH), R01 AG017502 (JWH), R01 NS078792 (JWH), and the American Heart Association (AHA) Predoctoral Fellowship AHA 14PRE19900021 (PBH).
The EMBO Journal (2020) 39: e102622
Contributor Information
Mary C Horne, Email: mhorne@ucdavis.edu.
James B Ames, Email: jbames@ucdavis.edu.
Johannes W Hell, Email: jwhell@ucdavis.edu.
Data availability
The NMR assignments have been deposited in the BMRB (accession number 25902; http://www.bmrb.wisc.edu/). The atomic coordinates have been deposited into the Protein Databank (6COA and 6CTB).
References
- Adams PJ, Ben‐Johny M, Dick IE, Inoue T, Yue DT (2014) Apocalmodulin itself promotes ion channel opening and Ca(2+) regulation. Cell 159: 608–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander KA, Cimler BM, Meier KE, Storm DR (1987) Regulation of calmodulin binding to P‐57. A neurospecific calmodulin binding protein. J Biol Chem 262: 6108–6113 [PubMed] [Google Scholar]
- Altier C, Dubel SJ, Barrere C, Jarvis SE, Stotz SC, Spaetgens RL, Scott JD, Cornet V, De Waard M, Zamponi GW et al (2002) Trafficking of L‐type calcium channels mediated by the postsynaptic scaffolding protein AKAP79. J Biol Chem 277: 33598–33603 [DOI] [PubMed] [Google Scholar]
- Atkinson RA, Joseph C, Kelly G, Muskett FW, Frenkiel TA, Nietlispach D, Pastore A (2001) Ca2+‐independent binding of an EF‐hand domain to a novel motif in the alpha‐actinin‐titin complex. Nat Struct Biol 8: 853–857 [DOI] [PubMed] [Google Scholar]
- Backman L (2015) Calcium affinity of human alpha‐actinin 1. PeerJ 3: e944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartels P, Behnke K, Michels G, Groner F, Schneider T, Henry M, Barrett PQ, Kang HW, Lee JH, Wiesen MH et al (2009) Structural and biophysical determinants of single Ca(V)3.1 and Ca(V)3.2 T‐type calcium channel inhibition by N(2)O. Cell Calcium 46: 293–302 [DOI] [PubMed] [Google Scholar]
- Bartels P, Yu D, Huang H, Hu Z, Herzig S, Soong TW (2018) Alternative splicing at N terminus and domain I modulates CaV1.2 inactivation and surface expression. Biophys J 114: 2095–2106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Johny M, Yang PS, Bazzazi H, Yue DT (2013) Dynamic switching of calmodulin interactions underlies Ca2+ regulation of CaV1.3 channels. Nat Commun 4: 1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben‐Johny M, Yang PS, Niu J, Yang W, Joshi‐Mukherjee R, Yue DT (2014) Conservation of Ca(2+)/calmodulin regulation across Na and Ca(2+) channels. Cell 157: 1657–1670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkefeld H, Sailer CA, Bildl W, Rohde V, Thumfart JO, Eble S, Klugbauer N, Reisinger E, Bischofberger J, Oliver D et al (2006) BKCa‐Cav channel complexes mediate rapid and localized Ca2+‐activated K+ signaling. Science 314: 615–620 [DOI] [PubMed] [Google Scholar]
- Berman DE, Dudai Y (2001) Memory extinction, learning anew, and learning the new: dissociations in the molecular machinery of learning in cortex. Science 291: 2417–2419 [DOI] [PubMed] [Google Scholar]
- Bourdin B, Marger F, Wall‐Lacelle S, Schneider T, Klein H, Sauve R, Parent L (2010) Molecular determinants of the CaVbeta‐induced plasma membrane targeting of the CaV1.2 channel. J Biol Chem 285: 22853–22863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buonarati OR, Henderson PB, Murphy GG, Horne MC, Hell JW (2017) Proteolytic processing of the L‐type Ca 2+ channel alpha 11.2 subunit in neurons. F1000 Res 6: 1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill L, Prins B, Weber M, McGaugh JL (1994) Beta‐adrenergic activation and memory for emotional events. Nature 371: 702–704 [DOI] [PubMed] [Google Scholar]
- Carter ME, Yizhar O, Chikahisa S, Nguyen H, Adamantidis A, Nishino S, Deisseroth K, de Lecea L (2010) Tuning arousal with optogenetic modulation of locus coeruleus neurons. Nature Neurosci 13: 1526–1533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chagot B, Chazin WJ (2011) Solution NMR structure of Apo‐calmodulin in complex with the IQ motif of human cardiac sodium channel NaV1.5. J Mol Biol 406: 106–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimler BM, Andreasen TJ, Andreasen KI, Storm DR (1985) P‐57 is a neural specific calmodulin‐binding protein. J Biol Chem 260: 10784–10788 [PubMed] [Google Scholar]
- Cohen SM, Suutari B, He X, Wang Y, Sanchez S, Tirko NN, Mandelberg NJ, Mullins C, Zhou G, Wang S et al (2018) Calmodulin shuttling mediates cytonuclear signaling to trigger experience‐dependent transcription and memory. Nat Commun 9: 2451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai S, Hall DD, Hell JW (2009) Supramolecular assemblies and localized regulation of voltage‐gated ion channels. Physiological Rev 89: 411–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davare MA, Avdonin V, Hall DD, Peden EM, Burette A, Weinberg RJ, Horne MC, Hoshi T, Hell JW (2001) A beta2 adrenergic receptor signaling complex assembled with the Ca2+ channel Cav1.2. Science 293: 98–101 [DOI] [PubMed] [Google Scholar]
- Davare MA, Hell JW (2003) Increased phosphorylation of the neuronal L‐type Ca(2+) channel Ca(v)1.2 during aging. Proc Natl Acad Sci USA 100: 16018–16023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degasperi A, Birtwistle MR, Volinsky N, Rauch J, Kolch W, Kholodenko BN (2014) Evaluating strategies to normalise biological replicates of Western blot data. PLoS ONE 9: e87293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolmetsch RE, Pajvani U, Fife K, Spotts JM, Greenberg ME (2001) Signaling to the nucleus by an L‐type calcium channel‐calmodulin complex through the MAP kinase pathway. Science 294: 333–339 [DOI] [PubMed] [Google Scholar]
- Dolphin AC (2012) Calcium channel auxiliary alpha(2)delta and beta subunits: trafficking and one step beyond. Nature Rev Neurosci 13: 542–555 [DOI] [PubMed] [Google Scholar]
- Dolphin AC (2016) Voltage‐gated calcium channels and their auxiliary subunits: physiology and pathophysiology and pharmacology. J Physiol 594: 5369–5390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egrie JC, Campbell JA, Flangas AL, Siegel FL (1977) Regional, cellular and subcellular distribution of calcium‐activated cyclic nucleotide phosphodiesterase and calcium‐dependent regulator in porcine brain. J Neurochem 28: 1207–1213 [DOI] [PubMed] [Google Scholar]
- Ellis SB, Williams ME, Ways NR, Brenner R, Sharp AH, Leung AT, Campbell KP, McKenna E, Koch WJ, Hui A et al (1988) Sequence and expression of mRNAs encoding the a1 and a2 subunits of the DHP‐sensitive calcium channel. Science 241: 1661–1664 [DOI] [PubMed] [Google Scholar]
- Evans TI, Hell JW, Shea MA (2011) Thermodynamic linkage between calmodulin domains binding calcium and contiguous sites in the C‐terminal tail of Ca(V)1.2. Biophys Chem 159: 172–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldkamp MD, Yu L, Shea MA (2011) Structural and energetic determinants of apo calmodulin binding to the IQ motif of the Na(V)1.2 voltage‐dependent sodium channel. Structure 19: 733–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findeisen F, Rumpf CH, Minor DL Jr (2013) Apo states of calmodulin and CaBP1 control CaV1 voltage‐gated calcium channel function through direct competition for the IQ domain. J Mol Biol 425: 3217–3234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller MD, Emrick MA, Sadilek M, Scheuer T, Catterall WA (2010) Molecular mechanism of calcium channel regulation in the fight‐or‐flight response. Sci Signal 3: ra70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller MD, Fu Y, Scheuer T, Catterall WA (2014) Differential regulation of CaV1.2 channels by cAMP‐dependent protein kinase bound to A‐kinase anchoring proteins 15 and 79/150. J Gen Physiol 143: 315–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabelli SB, Boto A, Kuhns VH, Bianchet MA, Farinelli F, Aripirala S, Yoder J, Jakoncic J, Tomaselli GF, Amzel LM (2014) Regulation of the NaV1.5 cytoplasmic domain by calmodulin. Nat Commun 5: 5126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabelli SB, Yoder JB, Tomaselli GF, Amzel LM (2016) Calmodulin and Ca(2+) control of voltage gated Na(+) channels. Channels 10: 45–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh D, Syed AU, Prada MP, Nystoriak MA, Santana LF, Nieves‐Cintron M, Navedo MF (2017) Calcium channels in vascular smooth muscle. Adv Pharmacol 78: 49–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh D, Nieves‐Cintron M, Tajada S, Brust‐Mascher I, Horne MC, Hell JW, Dixon RE, Santana LF, Navedo MF (2018) Dynamic L‐type CaV1.2 channel trafficking facilitates CaV1.2 clustering and cooperative gating. Biochim Biophys Acta Mol Cell Res 1865: 1341–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green EM, Barrett CF, Bultynck G, Shamah SM, Dolmetsch RE (2007) The tumor suppressor eIF3e mediates calcium‐dependent internalization of the L‐type calcium channel CaV1.2. Neuron 55: 615–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grover LM, Teyler TJ (1990) Two components of long‐term potentiation induced by different patterns of afferent activation. Nature 347: 477–479 [DOI] [PubMed] [Google Scholar]
- Hall DD, Feekes JA, Arachchige Don AS, Shi M, Hamid J, Chen L, Strack S, Zamponi GW, Horne MC, Hell JW (2006) Binding of protein phosphatase 2A to the L‐type calcium channel Cav1.2 next to Ser 1928, its main PKA site, is critical for Ser1928 dephosphorylation. Biochemistry 45: 3448–3459 [DOI] [PubMed] [Google Scholar]
- Hall DD, Dai S, Tseng P‐Y, Malik ZA, Nguyen M, Matt L, Schnizler K, Shephard A, Mohapatra D, Tsuruta F et al (2013) Competition between α‐Actinin and Ca2+‐calmodulin controls surface retention of the L‐type Ca2+ channel CaV1.2. Neuron 78: 483–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell JW, Yokoyama CT, Wong ST, Warner C, Snutch TP, Catterall WA (1993) Differential phosphorylation of two size forms of the neuronal class C L‐type calcium channel a1 subunit. J Biol Chem 268: 19451–19457 [PubMed] [Google Scholar]
- Hell JW (2014) CaMKII: claiming center stage in postsynaptic function and organization. Neuron 81: 249–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig S, Khan IF, Grundemann D, Matthes J, Ludwig A, Michels G, Hoppe UC, Chaudhuri D, Schwartz A, Yue DT et al (2007) Mechanism of Ca(v)1.2 channel modulation by the amino terminus of cardiac beta2‐subunits. FASEB J 21: 1527–1538 [DOI] [PubMed] [Google Scholar]
- Houdusse A, Gaucher JF, Krementsova E, Mui S, Trybus KM, Cohen C (2006) Crystal structure of apo‐calmodulin bound to the first two IQ motifs of myosin V reveals essential recognition features. Proc Natl Acad Sci USA 103: 19326–19331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovey L, Fowler CA, Mahling R, Lin Z, Miller MS, Marx DC, Yoder JB, Kim EH, Tefft KM, Waite BC et al (2017) Calcium triggers reversal of calmodulin on nested anti‐parallel sites in the IQ motif of the neuronal voltage‐dependent sodium channel NaV1.2. Biophys Chem 224: 1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Real E, Takamiya K, Kang MG, Ledoux J, Huganir RL, Malinow R (2007) Emotion enhances learning via norepinephrine regulation of AMPA‐receptor trafficking. Cell 131: 160–173 [DOI] [PubMed] [Google Scholar]
- Huang KP, Huang FL, Li J, Schuck P, McPhie P (2000) Calcium‐sensitive interaction between calmodulin and modified forms of rat brain neurogranin/RC3. Biochemistry 39: 7291–7299 [DOI] [PubMed] [Google Scholar]
- Huang KP, Huang FL, Jager T, Li J, Reymann KG, Balschun D (2004) Neurogranin/RC3 enhances long‐term potentiation and learning by promoting calcium‐mediated signaling. J Neurosci 24: 10660–10669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacobucci GJ, Popescu GK (2017) Resident calmodulin primes NMDA receptors for Ca(2+)‐Dependent inactivation. Biophys J 113: 2236–2248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuliopulos A, Nelson NP, Yamada M, Walsh CT, Furie B, Furie BC, Roth DA (1994) Localization of the affinity peptide‐substrate inactivator site on recombinant vitamin K‐dependent carboxylase. J Biol Chem 269: 21364–21370 [PubMed] [Google Scholar]
- Li H, Pink MD, Murphy JG, Stein A, Dell'Acqua ML, Hogan PG (2012) Balanced interactions of calcineurin with AKAP79 regulate Ca2+‐calcineurin‐NFAT signaling. Nat Struct Mol Biol 19: 337–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian LY, Myatt D, Kitmitto A (2007) Apo calmodulin binding to the L‐type voltage‐gated calcium channel Cav1.2 IQ peptide. Biochem Biophys Res Commun 353: 565–570 [DOI] [PubMed] [Google Scholar]
- Ma H, Groth RD, Cohen SM, Emery JF, Li B, Hoedt E, Zhang G, Neubert TA, Tsien RW (2014) gammaCaMKII shuttles Ca(2+)/CaM to the nucleus to trigger CREB phosphorylation and gene expression. Cell 159: 281–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man KNM, Navedo MF, Horne MC, Hell JW (2020) β2 adrenergic receptor complexes with the L‐type Ca2+ channel Cav1.2 and AMPA‐type glutamate receptors: paradigms for pharmacological targeting of protein interactions. Annu Rev in Pharmacol and Toxicol 60: 29.1–29.20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrion NV, Tavalin ST (1998) Selective activation of Ca2+‐activated K+ channels by co‐localized Ca2+channels in hippocampal neurons. Nature 395: 900–905 [DOI] [PubMed] [Google Scholar]
- Matt L, Kim K, Hergarden AC, Patriarchi T, Malik ZA, Park DK, Chowdhury D, Buonarati OR, Henderson PB, Gokcek Sarac C et al (2018) alpha‐actinin anchors PSD‐95 at postsynaptic sites. Neuron 97: 1094–1109 e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minzenberg MJ, Watrous AJ, Yoon JH, Ursu S, Carter CS (2008) Modafinil shifts human locus coeruleus to low‐tonic, high‐phasic activity during functional MRI. Science 322: 1700–1702 [DOI] [PubMed] [Google Scholar]
- Patriarchi T, Qian H, Di Biase V, Malik ZA, Chowdhury D, Price JL, Hammes EA, Buonarati OR, Westenbroek RE, Catterall WA et al (2016) Phosphorylation of Cav1.2 on S1928 uncouples the L‐type Ca2+ channel from the β2 adrenergic receptor. EMBO J 35: 1330–1345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Reyes E, Castellano A, Kim HS, Bertrand P, Baggstrom E, Lacerda AE, Wei XY, Birnbaumer L (1992) Cloning and expression of a cardiac/brain beta subunit of the L‐type calcium channel. J Biol Chem 267: 1792–1797 [PubMed] [Google Scholar]
- Peterson BZ, DeMaria CD, Adelman JP, Yue DT (1999) Calmodulin is the Ca2+ sensor for Ca2+‐dependent inactivation of L‐ type calcium channels. Neuron 22: 549–558 [DOI] [PubMed] [Google Scholar]
- Qian H, Patriarchi T, Price JL, Matt L, Lee B, Nieves‐Cintron M, Buonarati OR, Chowdhury D, Nanou E, Nystoriak MA et al (2017) Phosphorylation of Ser1928 mediates the enhanced activity of the L‐type Ca2+ channel Cav1.2 by the beta2‐adrenergic receptor in neurons. Sci Signal 10: eaaf9659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran X, Miao HH, Sheu FS, Yang D (2003) Structural and dynamic characterization of a neuron‐specific protein kinase C substrate, neurogranin. Biochemistry 42: 5143–5150 [DOI] [PubMed] [Google Scholar]
- Ribeiro Ede A Jr, Pinotsis N, Ghisleni A, Salmazo A, Konarev PV, Kostan J, Sjoblom B, Schreiner C, Polyansky AA, Gkougkoulia EA et al (2014) The structure and regulation of human muscle alpha‐actinin. Cell 159: 1447–1460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs F, Neil J, Barkakati N (1982) The automated analysis of data from single ionic channels. Pflugers Arch 395: 331–340 [DOI] [PubMed] [Google Scholar]
- Schroder F, Handrock R, Beuckelmann DJ, Hirt S, Hullin R, Priebe L, Schwinger RH, Weil J, Herzig S (1998) Increased availability and open probability of single L‐type calcium channels from failing compared with nonfailing human ventricle. Circulation 98: 969–976 [DOI] [PubMed] [Google Scholar]
- Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM (2003) The Xplor‐NIH NMR molecular structure determination package. J Magn Reson 160: 65–73 [DOI] [PubMed] [Google Scholar]
- Seisenberger C, Specht V, Welling A, Platzer J, Pfeifer A, Kuhbandner S, Striessnig J, Klugbauer N, Feil R, Hofmann F (2000) Functional embryonic cardiomyocytes after disruption of the L‐type alpha1C (Cav1.2) calcium channel gene in the mouse. J Biol Chem 275: 39193–39199 [DOI] [PubMed] [Google Scholar]
- Sinnegger‐Brauns MJ, Hetzenauer A, Huber IG, Renstrom E, Wietzorrek G, Berjukov S, Cavalli M, Walter D, Koschak A, Waldschutz R et al (2004) Isoform‐specific regulation of mood behavior and pancreatic beta cell and cardiovascular function by L‐type Ca2+ channels. J Clin Invest 113: 1430–1439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K et al (2004) Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119: 19–31 [DOI] [PubMed] [Google Scholar]
- Tjandra N, Bax A (1997) Direct measurement of distances and angles in biomolecules by NMR in a dilute liquid crystalline medium. Science 278: 1111–1114 [DOI] [PubMed] [Google Scholar]
- Tseng PY, Henderson PB, Hergarden AC, Patriarchi T, Coleman AM, Lillya MW, Montagut‐Bordas C, Lee B, Hell JW, Horne MC (2017) alpha‐Actinin promotes surface localization and current density of the Ca2+ channel CaV1.2 by binding to the IQ region of the alpha1 subunit. Biochemistry 56: 3669–3681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuluc P, Molenda N, Schlick B, Obermair GJ, Flucher BE, Jurkat‐Rott K (2009) A CaV1.1 Ca2+ channel splice variant with high conductance and voltage‐sensitivity alters EC coupling in developing skeletal muscle. Biophys J 96: 35–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner M, Anderson DE, Rajan S, Hell JW, Ames JB (2016) Chemical shift assignments of the C‐terminal EF‐hand domain of alpha‐actinin‐1. Biomol NMR Assign 10: 219–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Petegem F, Chatelain FC, Minor DL Jr (2005) Insights into voltage‐gated calcium channel regulation from the structure of the CaV1.2 IQ domain‐Ca2+/calmodulin complex. Nat Struct Mol Biol 12: 1108–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingard JN, Chan J, Bosanac I, Haeseleer F, Palczewski K, Ikura M, Ames JB (2005) Structural analysis of Mg2+ and Ca2+ binding to CaBP1, a neuron‐specific regulator of calcium channels. J Biol Chem 280: 37461–37470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Bers DM (2007) Free and bound intracellular calmodulin measurements in cardiac myocytes. Cell Calcium 41: 353–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyszynski M, Lin J, Rao A, Nigh E, Beggs AH, Craig AM, Sheng M (1997) Competitive binding of a‐actinin and calmodulin to the NMDA receptor. Nature 385: 439–442 [DOI] [PubMed] [Google Scholar]
- Yang T, Xu X, Kernan T, Wu V, Colecraft HM (2010) Rem, a member of the RGK GTPases, inhibits recombinant CaV1.2 channels using multiple mechanisms that require distinct conformations of the GTPase. J Physiol 588: 1665–1681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamponi GW, Striessnig J, Koschak A, Dolphin AC (2015) The physiology, pathology, and pharmacology of voltage‐gated calcium channels and their future therapeutic potential. Pharmacol Rev 67: 821–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhabotinsky AM, Camp RN, Epstein IR, Lisman JE (2006) Role of the neurogranin concentrated in spines in the induction of long‐term potentiation. J Neurosci 26: 7337–7347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Li Z, Sacks DB, Ames JB (2012) Structural basis for Ca2+‐induced activation and dimerization of estrogen receptor alpha by calmodulin. J Biol Chem 287: 9336–9344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Matt L, Patriarchi T, Malik ZA, Chowdhury D, Park DK, Renieri A, Ames JB, Hell JW (2014) Capping of the N‐terminus of PSD‐95 by calmodulin triggers its postsynaptic release. EMBO J 33: 1341–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong L, Kaleka KS, Gerges NZ (2011) Neurogranin phosphorylation fine‐tunes long‐term potentiation. Eur J Neurosci 33: 244–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuhlke RD, Pitt GS, Deisseroth K, Tsien RW, Reuter H (1999) Calmodulin supports both inactivation and facilitation of L‐type calcium channels. Nature 399: 159–162 [DOI] [PubMed] [Google Scholar]
- van Zundert GC, Rodrigues JP, Trellet M, Schmitz C, Kastritis PL, Karaca E, Melquiond AS, van Dijk M, de Vries SJ, Bonvin AM (2016) The HADDOCK2.2 web server: user‐friendly integrative modeling of biomolecular complexes. J Mol Biol 428: 720–725 [DOI] [PubMed] [Google Scholar]
- Zweckstetter M (2008) NMR: prediction of molecular alignment from structure using the PALES software. Nat Protoc 3: 679–690 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Data Availability Statement
The NMR assignments have been deposited in the BMRB (accession number 25902; http://www.bmrb.wisc.edu/). The atomic coordinates have been deposited into the Protein Databank (6COA and 6CTB).