

Abstract
Duchenne muscular dystrophy (DMD) is a severe childhood muscle disease primarily caused by the lack of functional dystrophin at the muscle fiber membranes. Multiple therapeutic approaches are currently in (pre)clinical development, aimed at restoring expression of (truncated) dystrophin. Key questions in this phase relate to route of drug administration, dose regimen, and levels of dystrophin required to improve muscle function. A series of studies applying antisense oligonucleotides (AONs) in the mdx mouse model for DMD has been reported over the last two decades, claiming a variable range of exon skipping and increased dystrophin levels correlated to some functional improvement. The aim of this study was to compare the efficacy of subcutaneous (SC) versus intravenous (IV) dosing routes of an mdx-specific AON at both the molecular and functional level, using state-of-the-art quantitative technologies, including digital droplet polymerase chain reaction, capillary Western immunoassay, magnetic resonance imaging, and automated kinematic analysis. The majority of all readouts we quantified, both molecular and functional, showed that IV dosing of the AON had a more pronounced beneficial effect than SC dosing in mdx mice. Last, but not least, the more quantitative molecular and functional data obtained in this study suggest that low levels of dystrophin protein of at least 2.5% of wild type may already have a beneficial effect on muscle leakiness and may improve motor performance of mdx mice.
Keywords: exon skipping, mdx, kinematic gait analysis, digital droplet PCR, capillary Western immunoassay, MRI
Introduction
Duchenne muscular dystrophy (DMD) is a neuromuscular disease that affects 1 in 5,000–6,000 newborn boys and is characterized by progressive weakness of the skeletal and cardiac muscles, respiratory failure, and death in early adulthood [1,2]. This X-chromosome linked disorder is caused by mutations in the DMD gene, which codes for dystrophin, a large 427 kDa protein critical for sarcolemmal integrity and with an important role in intracellular signaling [3]. The mutations in DMD disrupt the open reading frame and therefore result in prematurely truncated and instable dystrophin variants lacking the C-terminus, giving rise to strongly reduced levels or complete absence of dystrophin. Mutations in the DMD gene that conserve the reading frame lead to a shorter, but (partially) functional, dystrophin protein typically lacking part of the central rod domain region and underlying the milder Becker muscular dystrophy (BMD) [4,5].
Several potential therapies are being explored and developed to restore dystrophin expression in DMD patients, including gene (construct) replacement, translational stop codon read-through, splice modification (exon skipping), and CRISPR/Cas9-mediated DNA correction [6,7]. To date, two drugs, Exondys 51 and Translarna, have been approved by the U.S. Food and Drug Administration (FDA) and European Medicines Agency (EMA), respectively, but their efficiency in restoring dystrophin in DMD patients appears modest [8,9].
Quantifying absolute levels of increased dystrophin expression in patient muscle biopsies following treatment has shown to be challenging, but remains an important biomarker readout to validate DMD drug efficacy. Besides the nonquantitative and less sensitive methods mostly applied over the last two decades, dystrophin quantification is further hampered by the limited availability of patient muscle biopsies. It therefore still is not clearly defined precisely how much dystrophin restoration is necessary to yield a functional improvement. Although levels of (lifelong expression of) dystrophin in BMD patients and transgenic mouse models may provide useful information [10–12], these are likely not obtainable when starting therapies in dystrophic muscle tissue later in life.
Multiple mdx mouse studies applying different dystrophin-restoring drug candidates have provided further insight. The mdx mouse is the most widely used model for DMD and carries a nonsense point mutation giving rise to a premature stop codon in exon 23 of mouse dystrophin [13]. Consequently, mdx mice lack functional dystrophin, although their phenotype is considered mild relative to the severity of the symptoms observed in DMD patients. The majority of these mdx studies applied antisense oligonucleotides (AON) to induce skipping of the mutated mouse exon 23 and thus restore the translational open reading frame.
A variable range of exon skipping and dystrophin levels has been reported and claimed to be associated with some functional improvement. However, these results were typically obtained using nonquantitative (nested) polymerase chain reaction (PCR) methodology (overestimating exon skip levels), nonquantitative Western blots to determine dystrophin levels and rather insensitive methods to measure functionality [14–16]. In recent years, the methods to quantify exon skip and dystrophin levels have significantly improved with, for instance, the availability of digital droplet PCR (ddPCR) [17], automated, sensitive and accurate immunofluorescence analysis (IFA) [18], and capillary Western immunoassay (Wes) [10]. In addition, more sensitive methods for quantification of functional endpoints have become available, including magnetic resonance imaging (MRI) [19] and fully automated kinematic analysis (ie, MotoRater analysis for mouse models), with which 95 different parameters related to fine motor capabilities and gait can be assessed [20–22].
Aim of this study was to apply these more quantitative and sensitive state-of-the-art technologies to mdx mice to compare the efficacy of subcutaneous (SC) versus intravenous (IV) administration routes and different dosing regimens of a 2′-O-methylphosphorothioate (2OMePS) AON targeting mdx exon 23, following its route from administration to tissue uptake, from exon skipping to production of dystrophin, and ultimately its therapeutic effect on muscle quality and motor performance (Fig. 1A).
FIG. 1.

Route of AONs from administration to therapeutic effects in mdx muscle. Upon dosing of M23D, it is taken up in muscle, where it induces exon 23 skipping of mouse dystrophin, resulting in production of dystrophin. The increase in dystrophin gives rise to an improvement of muscle integrity, which can be assessed by quantifying several markers of muscle leakiness and by MRI to assess hyperintense regions in muscle representing edemic damage. Ultimately, the improved muscle integrity results in an improvement of muscle performance. (A) The used methods for each step are indicated in italics below the corresponding arrow. (B) Study design. mdx mice (n = 15), 5 weeks of age at first dose, received either twice weekly SC injections or once weekly IV injections of 100 mg/kg with M23D for a total of 12 weeks. A scrambled M23D AON was taken along as a control for the SC dosing group. The total dose is indicated. Mice were subjected to MotoRater analysis at 14 weeks of age and to MRI at 15 weeks of age. AON, antisense oligonucleotide; IV, intravenous; MRI, magnetic resonance imaging; SC, subcutaneous.
Materials and Methods
Antisense oligonucleotides
2OMePS-modified AONs were synthesized by BioMarin Nederland B.V. (Leiden, The Netherlands). The previously published 20-mer M23D(+2–18), targeting the last 2 bases of mouse DMD exon 23 and the first 18 bases of intron 23 with the sequence 5′-GGCCAAACCUCGGCUUACCU-3′ [23], was used in this study as well as a scrambled (SCR) version of it: M23D-scr 5′-AGUCACCUGCUGACUCAGCC-3′.
Mouse husbandry and ethical approval
Male mdx mice (C57BL/10ScSn-Dmdmdx/J) and wild-type (WT) controls (C57BL/10ScSn) were obtained from Jax Labs. Animal experiments were carried out at Charles River Laboratories in Kuopio, Finland, according to the National Institute of Health (NIH) guidelines for the care and use of laboratory animals and were approved by the Animal Experiment Board in Finland. Animals were housed at a standard temperature (22 ± 1°C) and in a light-controlled environment (lights on from 7 a.m. to 8 p.m.) with ad libitum access to food and water.
Treatment of mice with different dosing regimens of M23D
mdx mice were randomized into groups so that whole litters of mice did not end up in a single testing group and taking the baseline T2 MRI data and body weight into account so that there were no group differences at the start of first treatment. Five -week-old male mdx mice were treated twice weekly subcutaneously with 100 mg/kg M23D (n = 15) or M23D-scr (n = 15). This SC dose of M23D and even higher doses have been previously used in other mdx studies [24,25]. In addition, a group of mdx mice was treated intravenously with M23D in the tail vein (n = 15). Since it is unfeasible to perform twice weekly IV injections in the tail vein for an extended period, we opted to dose once weekly with 100 mg/kg for a total of 12 weeks, resulting in a 50% lower total dose than the SC dose group (Fig. 1B).
A group of 10 male WT mice (C57BL/10ScSn) of the same age as the mdx mice was included as a control group for the serum biomarkers, the MRI, and motor function testing, and received twice weekly SC injections with vehicle (VEH) (20 mM sodium phosphate buffer pH 7 with 0.7% w/v NaCl). Before each dose, mice were weighed. Mice were sacrificed in week 13, 4 days after the final injection, and at the age of 17 weeks by deep anesthetization with sodium pentobarbital (60 mg/kg Mebunat; Orion Pharma, Finland), followed by cardiac puncture. A blood sample was collected to measure serum biomarkers, after which the mice were transcardially perfused with phosphate-buffered saline (PBS). Skeletal muscles (gastrocnemius and quadriceps), diaphragm, heart, intestine, spleen, kidney, and liver were isolated and snap-frozen by immersing in isopentane au bain marie in liquid nitrogen, placed in cryovials prechilled on dry ice, and stored at −80°C.
Quantification of hyperintense regions in gastrocnemius using MRI
To quantify muscle volume and muscle damage, 10 randomly selected mice per group were subjected to volumetric MRI and absolute T2 mapping of gastrocnemius muscle at baseline (5 weeks of age) and at 15 weeks of age. WT mice (n = 10) were included as a reference to check if treatment with M23D changed parameters in the expected direction, that is, toward WT. MRI analysis was performed using a horizontal 11.7T magnet with bore size 160 mm equipped with a gradient set capable of max. gradient strength 750 mT/m and interfaced to a Bruker Avance III console (Bruker Biospin GmbH, Ettlingen, Germany). A volume coil (Bruker Biospin GmbH) was used for transmission and a surface phased array coil for receiving (Rapid Biomedical GmbH, Rimpar, Germany). Isoflurane-anesthetized mice were fixed to a custom-built mouse lower body holder and positioned in the magnet bore in a standard orientation relative to gradient coils.
For determination of muscle volume, a 3D gradient echo sequence was used with repetition time (TR)/echo time (TE) ratio (TR/TE) = 25/5 ms, field-of-view (FOV) of 26 × 22 × 36 mm3, and matrix of 320 × 256 × 96. Absolute T2 mapping was achieved using multi-spin-multi-echo (MSME) sequence with TR of 2,300 ms, seven echo times in 10.5 ms intervals between range of 10.5–73.5 ms, 20 0.7 mm thick slices, and FOV/matrix of 25.6 × 19.2 mm2/256 × 192 providing 100 microns in-plane resolution. Manual region-of-interest analysis for muscle volumes and T2-hyperintensities was carried out in MATLAB environment (Mathworks, Natick, MA) utilizing aedes image-analysis toolbox (http://aedes.uef.fi/).
Fine motor and kinematic gait analysis
Fine motor skills and gait properties were assessed at the age of 14 weeks in 10 randomly selected mice per group using a high precision kinematic analysis method (MotoRater; TSE Systems, Homburg, Germany) using the walking mode. Before the test sessions, the mice were marked in appropriate anatomical landmark points to facilitate the data analysis process. The movement data were captured using a high-speed camera (300 frames/s) from three different views, from below and both sides. A trial was considered successful if the mouse performed five consecutive step cycles. The captured videos of each mouse were first analyzed using Simi Motion software (Simi Reality Motion Systems GmbH, Unterschleissheim, Germany) by tracking the anatomical landmark points from the three views.
Different gait patterns and movements were further analyzed using a custom-made automated analysis system. The analyzed parameters included, for example, (1) general spatiotemporal parameters (stride time and speed, step width, stance and swing time during a stride, and interlimb coordination), (2) body posture and balance (toe clearance, iliac crest and hip height, hind limb protraction and retraction, and tail position and movement), and (3) fine motor skills (swing speed during a stride, jerk metric during swing phase, angle ranges and deviations of different joints, and vertical and horizontal head movement). The analysis provided altogether 95 different parameters related to fine motor capabilities and gait.
The correlation structure between these distinctive parameters was further assessed using principal component analysis (PCA). A set of new uncorrelated parameters, the Principal Components (PCs), was determined. In this study, we transformed the PCs by using the orthogonality preserving Varimax rotation procedure to simplify the PCs into interpretable gait features such as “overall hip height” or “trotting step sequence.”
Finally, an overall gait analysis score, Gait Discriminant Score, based on Varimax-rotated PCs, was established and determined. The score is based on differences between mdx and WT groups using those PCs that demonstrate a large effect size (Cohen's d > 0.8). Essentially, the basis of the score, the discriminant vector, is a disease model-specific combination of original variables, which characterizes the model in the best possible way. The Gait Discriminant Score for each mouse was obtained by projecting the normalized parameter data onto the discriminant vector.
Clinical chemistry
Routine clinical chemistry was performed on unhemolyzed serum samples on a Thermofisher Konelab Xti 20 chemistry analyzer according to the manufacturer's instructions. Measured parameters included alanine aminotransferase (ALT), aspartate aminotransferase (AST), creatine kinase (CK), and lactate dehydrogenase (LDH).
ddPCR analysis of exon skip levels
Mouse dystrophin exon 23 skip was determined using ddPCR [17]. Total RNA was isolated from different mdx tissues homogenized in 1 mL RNA-Bee (no. CS-105B; Bio-Connect) by grinding in a MagNa Lyser (two to four cycles of 20 s at 7,000 rpm) using MagNA Lyser Green Beads (no. 03358941001; Roche). The RNA concentration was determined with Nanodrop. For cDNA synthesis, 1,000 ng of total RNA was used as input. cDNA was generated in 20 μL reactions using random hexamer primers (no. 11034731001; Roche) and Transcriptor Reverse transcriptase (no. 3531287001; Roche) according to the manufacturer's instructions, with the exception that incubation took place for 40 min at 50°C instead of 30 min at 55°C.
Specific TaqMan assays were designed to detect the dystrophin exon 22–24 boundary (transcript with exon 23 skipped) and the exon 22–23 boundary (transcript still containing exon 23, referred to as nonskipped) (Table 1). ddPCR was performed on 2 μL 2–10 × diluted cDNA in a 20 μL reaction volume containing 1 μL of the relevant TaqMan assay, 10 μL of ddPCR Supermix for probes (no. 186–3010; BioRad), and 7 μL of DNase/RNase-free H2O. Exon skip percentage was calculated with the following formula:
Table 1.
Primer and Probe Sequence TaqMan Assays for Exon Skip Quantification in mdx Mice
| Probe | Forward primer 5′ > 3′ | Reverse primer 5′ > 3′ | Probe 5′ > 3′ |
|---|---|---|---|
| mDMD_ex22–23 | ACTGAATATGAAATAATGGAGGAGAGACT | GCCATTTTGTTGCTCTTTCAAA | AAATTACAGGCTCTGCAAAG |
| mDMD_ex22–24 | AGCAGTCAGAAAGCAAACTCTCTG | TTCAGCCATCCATTTCTGTAAGGT | TGGAGGAGAGACTCGGGAAATTACAGAATCACAT |
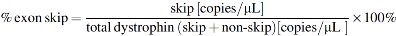
Protein lysate preparation from mdx tissues
Protein lysates were prepared from snap-frozen skeletal muscle biopsies, heart and diaphragm by sectioning or cutting off small biopsy pieces of in total ∼5–10 μg, placing them in a Magna Lyser vial (without beads), and adding 200 μL Protein Lysis Buffer (15% sodium dodecyl sulfate [SDS], 75 mM Tris-HCl pH 6.8, 1 Protease Inhibitor Cocktail tablet [no. 04693159001; Roche/Sigma]/8 mL; 5% β-Mercaptoethanol). After briefly spinning down, ∼15 ceramic beads (MagNA Lyser Green Beads; no. 03358941001; Roche) were added to each tube. The samples were then homogenized by two to four cycles (20 s; 7,000 rpm) in a MagNA Lyser (Roche) and spun down for 5 min at 13,000 rpm. The supernatant was supplemented with glycerol (final concentration 20%) and then samples were stored at −80°C until further use.
To measure total protein concentration, 20 × dilutions of the lysates in H2O were measured using the Pierce 660nm Protein assay (no. 226607; Thermo Scientific) with added Ionic Detergent Compatible Reagent (Thermo Scientific; no. 22663), according to the manufacturer's instructions.
Wes procedure to quantify dystrophin
Wes analysis was performed as previously described [10] on a Wes system (no. 004-600; ProteinSimple) according to the manufacturer's instructions using a 66–440 kDa Separation Module (no. SM-W006 or no. SM-W008; ProteinSimple) combined with the No secondary Detection Module (no. DM-003; ProteinSimple). For dystrophin detection, a rabbit monoclonal anti-dystrophin antibody (no. ab154168; Abcam; dilution 1/1,000) was used. In addition, an antibody targeting Vinculin (no. Vinculin E1E9V; Cell signaling 13901S; dilution 1/100) was used to control for sample loading. Both primary antibodies were raised in rabbit, so an anti-rabbit secondary antibody (no. 042-206; Protein Simple) was used. The lysed tissue samples were subsequently diluted to 250 μg/mL, resulting in a loading amount of 1.25 μg per well/capillary.
To control for differences in signal between experiments, a 6-point calibration curve of a WT C57BL6/10ScSn mouse muscle sample, ranging from 0.008 to 0.25 μg, was routinely included for dystrophin detection. This reference sample was selected from a panel of C57BL6/10ScSn mice based on it displaying average dystrophin levels. Wes runs were considered valid if the linearity of the calibration curve displayed an R2 > 0.99, based on 4–6 points in a relevant concentration range.
In addition, to correct for loading differences, vinculin levels were also determined by Wes in the diluted samples. Vinculin is commonly used for normalization and expressed in most tissues, with high levels in muscle tissue. A vinculin calibration curve of the same WT control mouse used for dystrophin analysis was therefore included, ranging from 0.25 to 4.0 μg. Since vinculin abundance was too high to get reliable chemiluminescence values (substrate depletion), we spiked/diluted the standard ProteinSimple secondary anti-rabbit-horseradish peroxidase antibody with 1/1,000 diluted unconjugated anti-rabbit antibody (no. ab6702; Abcam) to reduce HRP activity.
The final vinculin-corrected dystrophin values were generated by first expressing both the dystrophin and vinculin signals as percentage of control (% WT; using a calibration curve) using the formula below and then dividing the dystrophin % WT value by the Vinculin % WT value:
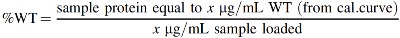
IFA of dystrophin at the muscle membrane
IFA was performed as previously described [18]. Briefly, cryosections (8 μm thick) prepared from quadriceps and gastrocnemius were mounted on Superfrost Ultra Plus microscopy slides (no. 10417002; Fisher Scientific) and incubated with primary antibody rabbit polyclonal anti-dystrophin (no ab15277; Abcam) in a dilution of 1/50 in a double staining with goat anti-spectrin (no. 7467; Santa Cruz biotechnology; dilution of 1/50). Slides were rinsed and washed twice for 5 min in PBS and subsequently incubated for 1 h with secondary antibodies donkey-anti-rabbit AlexaFluor488 (no. R37118; ThermoFisher) in a dilution of 1/150 (dystrophin) and donkey-anti-goat AlexaFluor594 (no. A-11058; ThermoFisher) in a dilution of 1/500 (spectrin).
Imaging of slides (five images per slide) was performed on the same day on a Zeiss LSM 710 confocal microscope using a 25 × objective. The 488 laser was set optimal on 1% beads (no. I-14785; ThermoFisher) for each individual experiment, which was typically gain 600 and transmission 5%. Images were analyzed using Definiens Architect software version 2.0 as previously described [18]. The average dystrophin intensity on the membrane of all analyzed cells was given in arbitrary units.
Hybridization-ligation assay for measuring M23D concentration
The concentration of M23D was determined using a hybridization/ligation enzyme-linked immunosorbent assay method based on an assay previously published [26] following adaptations previously described [27]. Briefly, tissues were homogenized in 100 mM Tris-HCl pH 8.5, 200 mM NaCl, 0.2% SDS, 5 mM ethylenediamine tetraaceticacid (all Sigma), and 2 mg/mL protK (ThermoFisher) using MagNA Lyser Green Beads (no. 03358941001; Roche) in a MagNA Lyser (Roche) (four cycles of 20 s at 7,000 rpm), followed by a 4-h rotation-incubation at 55°C. Tissue samples (60 mg/mL) were diluted 1/60 in PBS and further dilutions were made in tissue matrix (1/60 diluted pooled control tissue (60 mg/mL) in PBS). A signal probe (containing the peptide for antibody recognition) and a biotinylated template (complementary to M23D and the probe) were added to homogenized tissue samples and incubated in streptavidin-coated plates at 37°C, unbound AON and probe were washed off, and the bound fraction signal was developed with an antibody against the signal probe followed by an incubation with 3,3′,5,5′-Tetramethylbenzidine Liquid Substrate (TMB) (no. T4444, Sigma). Absorption at 450 nm was measured using a plate reader after stopping the reaction with 345 mM maleic acid (Sigma). Calibration curves of the analyzed M23D prepared in tissue matrix were included. All analyses were performed in duplicate.
Statistical analysis
All values are presented as mean ± standard error of mean, and differences were considered to be statistically significant at the P < 0.05 level. Significance between mdx groups was assessed by analysis of variance (ANOVA) and Tukey's multiple comparison test or by unpaired two-sided t-test (*P < 0.05; **P < 0.01; ***P < 0.001).
Results
Muscle uptake of the M23D AON
Tissue levels of M23D were quantified by ELISA in skeletal muscle and in several off-target tissues, including intestine, spleen, kidney, and liver. As anticipated, based on the differential total dose administered, M23D levels were ∼1.3-fold (P < 0.05) and 2-fold (P < 0.001) higher in quadriceps and gastrocnemius, respectively, in the SC dosing group compared to the IV group (Fig. 2A).
FIG. 2.

M23D tissue levels in SC and IV dosed mdx mice. (A). M23D levels (nmol) per gram in quadriceps and gastrocnemius tissue. (B) M23D levels (nmol) in several off-target tissues (per gram): liver (L), kidney (K), spleen (S), and intestine (I). (C) Muscle/kidney and muscle/liver ratios for quadriceps and gastrocnemius. Values are presented as mean ± SEM. Dots represent values obtained in individual mice (n = 14–15). Significance was assessed using unpaired two-tailed t-test (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). SEM, standard error of mean.
While absolute levels differed significantly between the off-target tissues (liver, kidney, spleen and intestine), the overall pattern was similar to what we observed in skeletal muscle, with 1.8–3-fold higher levels after SC compared to IV dosing depending on the tissue (Fig. 2B). Interestingly, the IV-dosed mice displayed a more favorable muscle-to-kidney ratio compared to mice dosed through the SC route (2.3-fold and 1.6-fold higher in quadriceps and gastrocnemius, respectively) (Fig. 2C). This effect was also observed, although less pronounced (1.5-fold), for muscle-to-liver ratios in quadriceps. It thus appears that, despite the lower total dose received, M23D uptake in muscle is more efficient through the IV dosing route, with relatively less uptake in off-target tissues. This confirms earlier observations with this AON in the mdx mouse [28].
IV dosing results in higher exon skip levels in skeletal muscle
Mean mouse exon 23 skip levels determined by ddPCR ranged between 2.5% and 5.0% of total dystrophin transcripts in skeletal muscle of M23D-treated mdx mice, depending on the muscle group and dosing regimen, and were close to zero in the scrambled control group (Fig. 3). In both quadriceps and gastrocnemius, mean exon skip levels were significantly higher in the IV dose group compared to the SC group (Fig. 3), despite the twofold lower dose received by mice in the IV group.
FIG. 3.

Exon 23 skip levels (% of total dystrophin transcripts) in quadriceps, gastrocnemius, heart, and diaphragm of SC and IV dosed mdx mice. Values are presented as mean ± SEM. Dots represent values obtained in individual mice (n = 14–15). Significance was assessed using ANOVA and Tukey's multiple comparison test (**P < 0.01, ****P < 0.0001). ANOVA, analysis of variance.
Exon skip levels were also determined in cardiac and diaphragm muscle, tissues affected in DMD that would likely positively contribute to overall functional improvement upon successful restoration of dystrophin expression. Mean exon skip levels in these tissues were lower compared to skeletal muscle, in particular, in heart (up to 0.5%). In contrast to skeletal muscle, in heart and diaphragm, exon skip levels were highest in the SC dose group. Since we did not measure tissue levels in heart and diaphragm, it is unclear whether this is due to lower M23D uptake in these tissues compared to skeletal muscle upon IV dosing (Fig. 3).
IV dosing results in higher dystrophin levels in skeletal muscle
Dystrophin protein levels were determined in quadriceps, gastrocnemius, diaphragm, and heart using Wes [10]. Mean dystrophin levels in the M23D-treated groups were 2.5 and 3.8% of WT levels in quadriceps and 1.6 and 2.5% of WT levels in gastrocnemius in the SC and IV groups, respectively (Fig. 4A). Comparison of dose routes revealed 1.5-fold higher mean dystrophin levels in both quadriceps and gastrocnemius in the IV group compared to the SC group, despite the twofold lower total dose received by mice in the IV group. No dystrophin was detectable by Wes in both diaphragm and heart (data not shown), consistent with the lower exon 23 skip in both these tissues.
FIG. 4.

Dystrophin levels (% of WT) in quadriceps and gastrocnemius of SC and IV dosed mdx mice, assessed using Wes (A) and IFA (B). For Wes, rabbit monoclonal anti-dystrophin antibody (no. ab154168; Abcam), and for IFA, rabbit polyclonal anti-dystrophin antibody (no. ab15277; Abcam) were used. Values are presented as mean ± SEM. Dots represent values obtained in individual mice (n = 14–15). Significance between the mdx groups was assessed using ANOVA and Tukey's multiple comparison test (*P < 0.05, **P < 0.01, ****P < 0.0001). (C) Representative IFA images are shown of quadriceps and gastrocnemius for M23D-treated mdx mice in the SC and IV groups in comparison to the scrambled control group. IFA, immunofluorescence analysis; Wes, capillary Western immunoassay.
We also performed IFA of dystrophin membrane signal on cross-sections of quadriceps and gastrocnemius muscle using Definiens Architect software for image analysis [18]. Mean dystrophin intensity was 1.2-fold higher in the IV group than in the SC group both in quadriceps and in gastrocnemius, consistent with the Wes results obtained in muscle homogenates (Fig. 4B, C).
A small increase in dystrophin expression is sufficient to reduce levels of biomarkers associated with muscle damage
CK, LDH, AST, and ALT are serum enzymes that are elevated in several muscular dystrophies, including DMD, and are widely used in the diagnosis of skeletal muscle injury and damage [29,30]. mdx mice are reported to also have elevated levels of these serum biomarkers compared to WT controls [31,32]. We here confirmed that all four biomarkers were indeed significantly increased in mdx mice treated with the scrambled control compared to WT controls (P < 0.0001) (Fig. 5). After treatment with M23D LDH, AST and ALT levels were significantly reduced in both the SC and IV M23D treatment groups compared to the scrambled control-treated mice, with the IV group displaying the strongest reduction for all three serum biomarkers (67% [LDH], 48% [AST], and 45% [ALT] reduction). Reductions in CK levels did not reach significance using ANOVA, but levels in the IV group were reduced by ∼40% when compared to the scrambled control group (two-tailed t-test, P < 0.05).
FIG. 5.

Levels of serum biomarkers CK, LDH, AST, and ALT in SC and IV dosed mdx mice compared to VEH-dosed WT mice. Values are presented as mean ± SEM. Dots represent values obtained in individual mice (n = 14–15). Significance between the scrambled control and WT group was assessed using an unpaired two-tailed t-test (####P < 0.0001). Significance between the mdx groups was assessed using ANOVA and Tukey's multiple comparison test (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). ALT, alanine aminotransferase; AST, aspartate aminotransferase; CK, creatine kinase; LDH, lactate dehydrogenase.
The collective lowering of all four serum biomarkers suggests that IV dosing for 12 weeks with M23D and a small increase in dystrophin levels from 2.5% to 3.8% of WT levels were sufficient to reduce the leakiness of muscle fibers in mdx mice.
A small increase in dystrophin expression is sufficient to reduce edemic damage in gastrocnemius muscle
To further confirm that M23D treatment improved muscle quality, 10 randomly selected mdx mice from each group were subjected to volumetric MRI and absolute T2 mapping of gastrocnemius at baseline (5 weeks of age) and at 15 weeks of age. While no differences in gastrocnemius volume were observed with MRI at baseline or at 15 weeks in any of the M23D treatment groups compared to the scrambled control (data not shown), differences were revealed with the quantitative T2-MRI mapping of gastrocnemius muscle by visualizing edemic damage as areas of hyperintensity [33] 2017).
At 5 weeks of age, before any treatment, mdx mice showed a significantly higher % of hyperintensities (P < 0.0001) and hyperintensity volume (P < 0.001) compared to WT controls, in which these hyperintensity scores were close to zero (Fig. 6). This baseline measurement also revealed that there were no significant differences in hyperintensity % or volume between any of the mdx treatment groups before treatment.
FIG. 6.

Hyperintensities identified by MRI in gastrocnemius in SC and IV dosed mdx mice compared to VEH-dosed WT mice. (A)Representative T2-weighted MRI images of gastrocnemius muscles of mice in the different mdx and WT treatment groups at baseline before treatment at 5 weeks of age obtained by MRI showing hyperintense regions as white areas. The arrows indicate some hyperintense regions. (B)Measurements of % and volume of hyperintense tissue at baseline (5 weeks of age) before any treatment and after 10 weeks of treatment (15 weeks of age) in mdx mice treated with M23D or the scrambled control compared to VEH-dosed WT mice. Values are presented as mean ± SEM. Dots represent values obtained in individual mice (n = 10). Significance between the scrambled control and WT group was assessed using an unpaired two-tailed t-test (###P < 0.001; ####P < 0.0001). Significance between the mdx groups was assessed using ANOVA and Tukey's multiple comparison test or unpaired two-tailed t-test (*P < 0.05, **P < 0.01, ***P < 0.001).
At the age of 15 weeks, both hyperintensity volume and % in mdx mice were higher than observed at 5 weeks of age and levels in the M23D SC group were very similar to those observed in the scrambled control group. However, in the M23D IV group, hyperintensity volume did not show a rise at 15 weeks of age and % hyperintensities even showed a significant decline compared to the levels observed at 5 weeks (Fig. 6). Moreover, mice in the IV group displayed a significant reduction of hyperintensity volume compared to the M23D SC group, despite the twofold lower dose they received compared to the M23D SC group. The difference in hyperintensity volume compared to the scrambled control group was just above the statistical threshold of 0.05 when using ANOVA, but did reach statistical significance by t-test (Fig. 6B). In the IV group, the mean hyperintensity volume was reduced by 45% (t-test, P < 0.001) and 49% (t-test, P < 0.05) relative to the scrambled control group and M23D SC group, respectively. Similarly, a reduction of mean hyperintensity percentage of 47% (t-test, P < 0.01) and 49% (t-test, P < 0.05) was observed relative to the scrambled control group and M23D SC group, respectively (Fig. 6B), This suggests a positive treatment effect leading to reduction of edemic damage in gastrocnemius muscle upon IV administration of M23D.
A small increase in dystrophin levels may already have a beneficial effect on motor performance
mdx mice, in particular in the original C57BL/10ScSn genetic background, are known to have a relatively mild motor phenotype [34]. In this study, we used the highly sensitive MotoRater system to evaluate gait characteristics and fine motor skills in walking mode of all treatment groups at 14 weeks of age. Data were generated per individual parameter as well as by using PCA. Using this approach, a clear phenotype of the mdx mice was apparent, with 37 motor and gait parameters significantly differing between mdx mice treated with the scrambled control compared to vehicle (VEH)-treated WT mice (P < 0.05; Table 2).
Table 2.
List of 37 Motor and Gait Parameters That Are Significantly Different Between mdx and Wild-Type Mice
| Parameter | P |
|---|---|
| strideTime | 0.0152 |
| strideSpeed | 0.0171 |
| strideDistance | 0.0281 |
| stepWidhHind | 0.00608 |
| stanceTimeHind | 0.0305 |
| swingTimeHind | 0.00295 |
| stanceTimeFore | 0.0213 |
| swingTimeFore | 0.0179 |
| foreCoupling | 0.0358 |
| iliacCrestHeight | 0.000443 |
| tailBaseMean | 0.0164 |
| tailBaseMax | 0.00431 |
| tailBaseRange | 0.0422 |
| tailTipMin | 0.00939 |
| tailTipMean | 0.0206 |
| swingMeanSpeedHind | 0.0095 |
| swingMeanSpeedFore | 0.018 |
| swingPeakSpeedHind | 0.00558 |
| swingPeakSpeedFore | 0.00515 |
| swingMeanJerkFore | 0.0325 |
| angleRangeHip | 0.00106 |
| lengthNoseTail | 0.000189 |
| hipHeight | 0.00148 |
| tailTouchesGround | 0.0244 |
| tailTipOverHip | 0.0222 |
| tailTipDist2d | 0.00617 |
| tailTipDist3d | 0.00117 |
| toeLiftoffAngleHind | 0.036 |
| dutyCycleFore | 0.0324 |
| singleSupportFore | 0.0426 |
| doubleSupportFore | 0.0318 |
| supportLateral | 0.00199 |
| supportFour | 0.0132 |
| minAngleHip | 0.0432 |
| minAngleKnee | 4.98e-06 |
| maxAngleKnee | 0.0156 |
| minAngleAnkle | 0.0453 |
The P-value (two-tailed t-test)is indicated.
A discriminant vector was established based on those gait features, which demonstrated a large effect size (Fig. 7A). The vector can be seen as overall kinematic fingerprint of mdx mice, characterizing all relevant gait changes, including lower overall speed and swing speed, increased overall hip height, slightly decreased tail tip position, decreased hip range of motion (ROM), increased hip, knee, and ankle angles, decreased retraction (more forward direction) and increased nose height and ROM. The corresponding gait discriminant score displayed a highly significant (P < 0.0001) differentiation between the mdx mice and WT controls and a significant shift toward WT in the SC and IV M23D treatment groups compared to the scrambled control group (P < 0.05), indicating an M23D treatment effect on overall motor performance (Fig. 7B).
FIG. 7.

MotoRater analysis of VEH-treated WT mice compared to SC and IV dosed mdx mice at 14 weeks of age. (A) Discriminant vector representing an overall kinematic fingerprint of and the differences between VEH-dosed WT mice and scrambled control-dosed mdx mice. (B) Overall gait discriminant score, based on the established discriminant vector. (C) Heatmap showing three PC clusters reflecting different gait features that differ between VEH-dosed WT mice and scrambled M23D-dosed mdx mice. Per cluster, the corresponding gait score is represented in a graph (PC1: speed, PC2: hip height, and PC3: angles, tail, and nose height). The % of total variation of the data explained per cluster is indicated above each graph. Values are presented as mean ± SEM (n = 10). Significance between the scrambled control and WT group was assessed using an unpaired two-tailed t-test (#P < 0.05; ###P < 0.001; ####P < 0.0001). Significance between the mdx groups was assessed using ANOVA and Tukey's multiple comparison test (*P < 0.05, **P < 0.01).
Principle component analysis revealed three principle components (PCs), each reflecting different gait features (see heatmap, Fig. 7C). PC1 reflected overall speed, PC2 overall hip height and tail base, and PC3 ankle angles, tail tip height, nose height, and trajectory profiles. The difference between mdx mice treated with scrambled (SCR) M23D and WT controls was significant for PCs 1 and 2 and displayed a trend toward significance (P = 0.07) for PC3. The most characteristic mdx phenotype was captured by PC2 (Hip height, consisting of hip and tail base heights, explaining 8.2% of the total variation of data). A significant treatment effect was seen in PC2 for both the SC and IV groups compared to the scrambled control group (P < 0.05), with a more pronounced shift toward WT observed in the IV group, suggesting recovery in overall hip height toward WT (Fig. 7C).
Discussion
The mdx mouse is the most widely used animal model in the preclinical phase of dystrophin-restoring therapeutics for DMD, and numerous mdx studies have been performed to explore different routes of drug administration and dose regimens, but also to define what increase and/or absolute level of dystrophin is required to obtain functional improvement of muscle. In BMD patients, dystrophin levels have been reported to range between ∼5% and 90% of healthy control, with levels below 10% typically associated with a more severe disease progression [35,36,10]. A study on transgenic mdx/utrn(-/-)/Xist(Δhs) mice has further suggested that with dystrophin levels up to 4%, survival and motor function may be improved, but that levels higher than 4% are required to ameliorate histopathology as well [12].
Although informative, data derived from BMD patients and transgenic mouse studies are based on lifelong expression of (truncated) dystrophin and difficult to compare with DMD patients starting (truncated) dystrophin expression upon treatment later in life. Thus, although phenotypically quite distinct from DMD patients, studies in DMD animal models like the mdx mouse remain important to explore and define the minimal requirements for therapeutic dystrophin expression in a (mouse) dystrophic background. A series of mdx studies applying AONs to correct dystrophin expression has been published over the last two decades, claiming a variable range of exon skipping and dystrophin levels correlated to some functional improvement.
More recently, several improved and more quantitative methods for quantifying RNA and protein levels and technologies to measure functionality have become available. Therefore, one of the aims of this study was to perform an mdx AON study using a state-of-the-art toolbox applying the best quantitative technologies available today. We applied a relatively large sample size per group (n = 10–15, depending on the readout) to have sufficient statistical power, given the natural variation between mice.
While the first clinical studies on AONs with a PS backbone applied SC administration to DMD patients due to the chronic nature of the treatment, more recently it has become clear that this route may induce local injection site reactions [37,38]. To explore the possibility of future IV dosing in clinical trials, a second aim of this study was to perform a side-to-side comparison of the efficacy of SC versus IV dosing routes of the mdx-specific M23D (with a PS backbone) at both the molecular and functional level, using the latest methodologies.
Previous studies in mdx indicated that twice weekly SC administration of 100 mg/kg M23D gave rise to optimal tissue levels and exon skipping [24,25]. Therefore, this dosing regimen was the starting point of this study, including both an M23D and a scrambled control group, applying the same SC dosing route and twice weekly dosing regimen. Ideally, to allow comparison of SC and IV dosing routes, the IV group should also have received a twice weekly administration of 100 mg/kg M23D. However, due to practical reasons, performing twice weekly IV injections in the tail vein of mdx mice turned out to not be practically feasible for a 12-week study. Mouse tail veins are fragile and multiple IV injections result in an increased risk of tail necrosis and/or scarring around the puncture site, making repeated injections difficult and limiting the total number of IV injections that can be applied. Therefore, we chose to perform the IV dosing once weekly, thus resulting in a 50% lower total dose. While this is not an optimal experimental design for comparison of SC and IV dosing routes, the majority of readouts we quantified, both molecular and functional, showed that IV dosing of M23D had a more pronounced beneficial effect than SC dosing, despite the fact that dose levels were only half of those applied in the SC group.
Muscle/kidney and muscle/liver ratios were higher in the IV group, indicating a more favorable target-to-off-target ratio, which may be related to higher plasma Cmax levels obtained upon IV dosing [39]. Exon skip levels were significantly higher in skeletal muscle of the IV compared to the SC group, despite that absolute tissue levels were lower in skeletal muscle of the IV group. It must be noted that M23D tissue levels were quantified in tissue homogenates, so do not reflect potential differences in intracellular uptake or subcellular distribution. Therefore, the higher exon skip levels in the IV group may be caused by a better Cmax-driven uptake into the nucleus, the site of action for splicing modulation by AONs, which is not evident when assessing absolute tissue levels.
Consistent with the higher exon skip levels observed in the IV group, dystrophin levels were also higher as determined by both Wes and IFA analysis. Mean absolute levels of dystrophin were slightly lower in gastrocnemius (1.6% [SC] and 2.5% [IV] of WT levels) than in quadriceps (2.5% [SC] and 3.8% [IV] of WT levels). While dystrophin levels were relatively low, some beneficial effects were observed on markers of muscle leakiness, muscle quality, and motor performance. In particular, in the IV-treated mice, there was a 40–67% reduction of serum biomarkers CK, LDH, AST, and ALT, pointing toward a reduced muscle leakiness. In line with this, IV-dosed mice displayed a significant reduction of both hyperintensity percentage and volume in gastrocnemius. In contrast, the lower mean dystrophin levels in the SC group (1.6% of WT levels) were not sufficient to reduce these hyperintensity parameters.
Finally, motor performance was assessed using kinematic gait analysis. Mice in both the SC and IV group displayed a significant reduction of gait discriminant score and a significant shift toward WT for overall hip height and tail base properties captured in PC2, with the shift being larger in the IV group. Taking all these data together, the overall response was stronger and more significant upon IV dosing at a twofold lower dose than in the SC dose group. This supports implementation of IV dosing of 2OMePS AONs in future clinical trials.
A second aim of this study was to make use of modern cutting-edge quantitative and sensitive methods, some of which can also be implemented in clinical trials. Since the initial studies on induction of exon 23 skipping in mdx myoblasts published over two decades ago [40,41], there have been several technological advances allowing more accurate quantification of the main molecular readouts of efficacy: exon 23 skipping and dystrophin restoration. In addition, there have been several innovations in imaging and kinematic gait analysis, providing novel tools to better assess on the one hand muscle condition and on the other hand motor performance.
Some of the exon skip studies in mdx mice, including recent publications, have reported on AON-induced exon 23 skipping data assessed using nonquantitative, sometimes even nested, PCR with gel-based or densitometric assessment of exon skip levels [42–45,25]. A disadvantage of this approach is that it has the propensity to overestimate exon skip levels due to the PCR bias, favoring amplification of the smaller exon skip product over the exon 23-containing nonskipped amplicon, in particular, when using a large number of PCR cycles or nested PCR [46,17]. A more valid way of quantifying exon skip is quantitative PCR (qPCR), which is an improvement compared to non-qPCR, but may require preamplification of cDNA if exon skip levels are low [47,48]. The gold standard, however, is absolute quantification of exon skip levels by ddPCR, which has been validated for its linearity and range, repeatability, accuracy, detection, and quantitation limits [17]. A recent multicenter comparison of quantification methods for AON-induced DMD exon 51 skipping in DMD patient concluded that ddPCR was the most precise and quantitative method and did not overestimate exon skip levels [49]. Despite its advantages, ddPCR is still not widely used and this study is one of the first to apply this accurate and highly sensitive methodology to quantify AON-induced exon skip in muscle tissue.
Western blotting (WB) is still the most widely used method in the DMD field for quantification of dystrophin levels. Limitations of WB are the relatively large amount of protein required and the technical challenges related to the large size of dystrophin, which make it notoriously difficult to transfer efficiently onto membrane during blotting. Consequently, reproducibility is poor and the procedure is not very quantitative.
Recently, we demonstrated the use of Wes for quantification of dystrophin in skeletal muscle biopsies of healthy controls and individuals with BMD or DMD [10]. In Wes, proteins are size separated in capillaries, where they are incubated with primary and (HRP conjugated) secondary antibodies and finally with Luminol/peroxidase. The produced chemiluminescent signal is automatically quantified and is displayed as an electropherogram showing automatically detected intensity peaks that can be quantified by calculation of the area under the curve. Besides quantification of the full-length Dp427 dystrophin isoform, Wes can also be used to quantify levels of shorter dystrophin isoforms, such as, for example, the Dp71 isoform [10,50]. We validated this method by assessing linearity of quantification for different antibodies, reproducibility (interassay and intra-assay, interoperator), and found the assay to be highly sensitive and linear over a wide dynamic range and with a lower limit of quantification (LLOQ) low enough to quantify trace dystrophin levels present in skeletal muscle of DMD patients. The reproducibility of dystrophin quantification by Wes was in line with the FDA/EMA regulations for quantitative biochemical analysis [10]. Since it is a relatively new method, so far only a few studies have been published making use of this methodology [10,50]. In this study, we applied Wes, allowing accurate quantification of the amount of dystrophin produced by exon 23 skipping in the mdx mouse.
The Wes results were confirmed by quantifying dystrophin signal intensity at the muscle membrane in muscle cross-sections using IFA. We applied customized Definiens software, which automatically identifies individual muscle fiber membranes using their spectrin signal and measures the membrane dystrophin intensity per muscle fiber [18]. This method was previously shown to be an objective, sensitive, reproducible, and standardized IFA method to assess dystrophin intensity levels per individual fiber in an entire fiber population in a semiautomated manner [18].
The downstream effects of the observed dystrophin restoration on muscle quality were assessed using MRI of gastrocnemius muscle. mdx mice displayed hyperintense regions in their skeletal muscle at both the baseline (5 weeks of age) and endpoint measurements (15 weeks of age), indicating that MRI is sensitive enough to clearly detect the dystrophic phenotype in mdx from an early age onward. This is striking, given that mdx mice on a C57BL/10ScSn genetic background are known to have a mild skeletal muscle phenotype [13,34]. The hyperintense regions are thought to represent edemic damage due to impaired membrane integrity arising from the lack of dystrophin and were absent in WT controls. The IV-dosed mdx mice showed a significant reduction of hyperintensities, suggesting a decrease of muscle leakiness. This was backed up by the reduction of CK, LDH, AST, and ALT, which are serum biomarkers of muscle damage. Similar to the MRI analysis, a clear phenotype of mdx mice was evident from the fine motor and kinematic gait analysis using the MotoRater system in 15-week mdx mice. A total of 37 motor and gait parameters significantly differed between mdx and WT mice, resulting in an overall gait discriminant score that significantly differed from WT. Together, these changes signify a more upright gait and posture of mdx mice, suggesting physiological changes in hind limb muscles potentially due to increased stiffness/spasticity/rigidity in knee and hip extensor muscles.
Mice in both the SC and IV group showed a clear shift in normalized gait discriminant score toward WT. In addition, overall hip height was significantly affected and displayed a shift toward WT in both the SC and IV M23D treatment groups, indicated by the gait PC score in PC2, with the largest effect being observed in the IV-treated mice. Strikingly, while the MotoRater analysis was sensitive enough to clearly detect a muscle and motor phenotype with a sufficient window to allow accurate monitoring of therapeutic effects in mdx mice, in contrast to the majority of the other readouts, it did not detect much difference between the SC and IV dosing groups.
Last, but not least, the more quantitative molecular and functional data obtained in this study suggest that low levels of dystrophin of at least 2.5–3.8% of WT levels may already be sufficient to reduce muscle leakiness and improve motor performance in the mdx mouse model. This is considerably lower than the 15–20% of normal levels of dystrophin that have been postulated by others to be the lower threshold required for restoration of functional deficits in dystrophic mouse models [11, 51–54]. It cannot be excluded that dystrophin levels in these studies were overestimated, since nonquantitative methods were used. In addition, the functional improvement observed in this study should not be overinterpreted, since we only applied a limited set of functional tests and did not include electrophysiology or force tests to directly quantify muscle function.
While it is encouraging that a relatively low amount of dystrophin may already be sufficient to restore muscle leakiness and improve motor function in mdx mice, it remains to be seen to what extent this can be extrapolated to the human situation in DMD. So far, in clinical trials applying exon skipping AONs, reported dystrophin levels have been low, but in some patients a functional improvement was observed [55,56]. However, in the absence of more robust dystrophin levels and functional outcome, it is not justified to make an accurate statement on how much dystrophin restoration in DMD patients is necessary for a specific functional benefit. Nonetheless, several lessons from this study can be implemented in a clinical trial setting, including the better efficacy of IV compared to SC dosing and the use of innovative methods to more sensitively and reliably quantify exon skip, dystrophin, muscle quality, and motor function.
Author Disclosure Statement
N.A.D., S.B., A.J., J.T., R.v.d.E., R.W., and J.C.v.D. are employees of (former) BioMarin Nederland BV (formerly Prosensa Therapeutics BV) and performed the work with company budget in the form of salaries, equipment, and facilities. J.C.v.D. discloses being co-inventor on patents on exon skipping technology, some of which have been licensed to BioMarin and subsequently sublicensed to Sarepta. As co-inventor of some of these patents, J.C.v.D. is entitled to a share of royalties. J.P., K.L., T.B., and T.A. are employees of Charles River Research Discovery Services in Finland and have no financial conflict of interests related to the submitted article.
Funding Information
No external funding was received for this study. All experiments were performed with company budget.
References
- 1. Emery AEH. (1993). Duchenne Muscular Dystrophy. Oxford University Press, Oxford [Google Scholar]
- 2. Muntoni F, Torelli S and Ferlini A (2003). Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol 2:731–740 [DOI] [PubMed] [Google Scholar]
- 3. Allen DG, Whitehead NP and Froehner SC (2016). Absence of dystrophin disrupts skeletal muscle signaling: roles of Ca2+, reactive oxygen species, and nitric oxide in the development of muscular dystrophy. Physiol Rev 96:253–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Koenig M, Beggs AH, Moyer M, Scherpf S, Heindrich K, Bettecken T, Meng G, Müller CR, Lindlöf M, et al. (1989). The molecular basis for Duchenne versus Becker muscular dystrophy: correlation of severity with type of deletion. Am J Hum Genet 45:498–506 [PMC free article] [PubMed] [Google Scholar]
- 5. Monaco AP, Bertelson CJ, Liechti-Gallati S, Moser H and Kunkel LM (1988). An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 2:90–95 [DOI] [PubMed] [Google Scholar]
- 6. Salmaninejad A, Valilou SF, Bayat H, Ebadi N, Daraei A, Yousefi M, Nesaei A and Mojarrad M (2018). Duchenne muscular dystrophy: an updated review of common available therapies. Int J Neurosci 128:854–864 [DOI] [PubMed] [Google Scholar]
- 7. Shimizu-Motohashi Y, Komaki H, Motohashi N, Takeda S, Yokota T and Aoki Y (2019). Restoring dystrophin expression in Duchenne muscular dystrophy: current status of therapeutic approaches. J Pers Med 9:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Aartsma-Rus A and Krieg AM (2017). FDA approves eteplirsen for Duchenne muscular dystrophy: the next chapter in the eteplirsen saga. Nucleic Acid Ther 27:1–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ebrahimi-Fakhari D, Dillmann U, Flotats-Bastardas M, Poryo M, Abdul-Khaliq H, Shamdeen MG, Mischo B, Zemlin M and Meyer S (2018). Off-label use of ataluren in four non-ambulatory patients with nonsense mutation Duchenne muscular dystrophy: effects on cardiac and pulmonary function and muscle strength. Front Pediatr 6:316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Beekman C, Janson AA, Baghat A, van Deutekom JC and Datson NA (2018). Use of capillary Western immunoassay (Wes) for quantification of dystrophin levels in skeletal muscle of healthy controls and individuals with Becker and Duchenne muscular dystrophy. PLoS One 13:e0195850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. van Putten M, Hulsker M, Nadarajah VD, van Heiningen SH, van Huizen E, van Iterson M, Admiraal P, Messemaker T, den Dunnen JT, et al. (2012). The effects of low levels of dystrophin on mouse muscle function and pathology. PLoS One 7:e31937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. van Putten M, Hulsker M, Young C, Nadarajah VD, Heemskerk H, van der Weerd L, Hoen PAC't, van Ommen G-J and Aartsma-Rus AM (2013). Low dystrophin levels increase survival and improve muscle pathology and function in dystrophin/utrophin double-knockout mice. FASEB J 27:2484–2495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Banks GB and Chamberlain JS (2008). The value of mammalian models for Duchenne muscular dystrophy in developing therapeutic strategies. Curr Top Dev Biol 84:431–453 [DOI] [PubMed] [Google Scholar]
- 14. Vitiello L, Bassi N, Campagnolo P, Zaccariotto E, Occhi G, Malerba A, Pigozzo S, Reggiani C, Ausoni S, et al. (2008). In vivo delivery of naked antisense oligos in aged mdx mice: analysis of dystrophin restoration in skeletal and cardiac muscle. Neuromuscul Disord 18:597–605 [DOI] [PubMed] [Google Scholar]
- 15. Yin H, Betts C, Saleh AF, Ivanova GD, Lee H, Seow Y, Kim D, Gait MJ and Wood MJ (2010). Optimization of peptide nucleic acid antisense oligonucleotides for local and systemic dystrophin splice correction in the mdx mouse. Mol Ther 18:819–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yang L, Niu H, Gao X, Wang Q, Han G, Cao L, Cai C, Weiler J and Yin H (2013). Effective exon skipping and dystrophin restoration by 2′-O-methoxyethyl antisense oligonucleotide in dystrophin-deficient mice. PLoS One 8:e61584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Verheul RC, van Deutekom JCT and Datson NA (2016). Digital droplet PCR for the absolute quantification of exon skipping induced by antisense oligonucleotides in (pre-)clinical development for Duchenne muscular dystrophy. PLoS One 11:e0162467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Beekman C, Sipkens JA, Testerink J, Giannakopoulos S, Kreuger D, van Deutekom JC, Campion GV, de Kimpe SJ and Lourbakos A (2014). A sensitive, reproducible and objective immunofluorescence analysis method of dystrophin in individual fibers in samples from patients with Duchenne muscular dystrophy. PLoS One 9:e107494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Walter G, Cordier L, Bloy D and Sweeney HL (2005). Noninvasive monitoring of gene correction in dystrophic muscle. Magn Reson Med 54:1369–1376 [DOI] [PubMed] [Google Scholar]
- 20. Datson NA, González-Barriga A, Kourkouta E, Weij R, van de Giessen J, Mulders S, Kontkanen O, Heikkinen T, Lehtimäki K and van Deutekom JCT (2017). The expanded CAG repeat in the huntingtin gene as target for therapeutic RNA modulation throughout the HD mouse brain. PLoS One 12:e0171127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hou Y, Lautrup S, Cordonnier S, Wang Y, Croteau DL, Zavala E, Zhang Y, Moritoh K, O'Connell JF, et al. (2018). NAD+ supplementation normalizes key Alzheimer's features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc Natl Acad Sci U S A 115:E1876–E1885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Johnson TB, Brudvig JJ, Lehtimäki KK, Cain JT, White KA, Bragge T, Rytkönen J, Huhtala T, Timm D, et al. (2019). A multimodal approach to identify clinically relevant parameters to monitor disease progression in a preclinical model of neuropediatric disease. bioRxiv; DOI: 10.1101/522011 [DOI] [PubMed] [Google Scholar]
- 23. Mann CJ, Honeyman K, McClorey G, Fletcher S and Wilton SD (2002). Improved antisense oligonucleotide induced exon skipping in the mdx mouse model of muscular dystrophy. J Gene Med 4:644–654 [DOI] [PubMed] [Google Scholar]
- 24. Verhaart IEC, Tanganyika-de Winter CL, Karnaoukh TG, Kolfschoten IGM, de Kimpe SJ, van Deutekom JCT and Aartsma-Rus A (2013). Dose-dependent pharmacokinetic profiles of 2′-O-methyl phosphorothioate antisense oligonucleotidesin mdx mice. Nucleic Acid Ther 23:228–237 [DOI] [PubMed] [Google Scholar]
- 25. van Putten M, Tanganyika-de Winter C, Bosgra S and Aartsma-Rus A (2019). Nonclinical exon skipping studies with 2′-O-methyl phosphorothioate antisense oligonucleotides in mdx and mdx-utrn-/- mice inspired by clinical trial results. Nucleic Acid Ther 29:92–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yu RZ, Baker B, Chappell A, Geary RS, Cheung E and Levin AA (2002). Development of an ultrasensitive noncompetitive hybridization-ligation enzyme-linked immunosorbent assay for the determination of phosphorothioate oligodeoxynucleotide in plasma. Anal Biochem 304:19–25 [DOI] [PubMed] [Google Scholar]
- 27. Jirka SMG, Tanganyika-de Winter CL, Boertje-van der Meulen JW, van Putten M, Hiller M, Vermue R, de Visser PC and Aartsma-Rus A (2015). Evaluation of 2′-deoxy-2′-fluoro antisense oligonucleotides for exon skipping in Duchenne muscular dystrophy. Mol Ther Nucleic Acids 4:e265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Heemskerk H, de Winter C, van Kuik P, Heuvelmans N, Sabatelli P, Rimessi P, Braghetta P, van Ommen G-JB, de Kimpe S, et al. (2010). Preclinical PK and PD studies on 2′-O-methyl-phosphorothioate RNA antisense oligonucleotides in the mdx mouse model. Mol Ther 18:1210–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhu Y, Zhang H, Sun Y, Li Y, Deng L, Wen X, Wang H and Zhang C (2015). Serum enzyme profiles differentiate five types of muscular dystrophy. Dis Markers 2015:543282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McMillan HJ, Gregas M, Darras BT and Kang PB (2011). Serum transaminase levels in boys with Duchenne and Becker muscular dystrophy. Pediatrics 127:e132–e136 [DOI] [PubMed] [Google Scholar]
- 31. González-Sánchez J, Sánchez-Temprano A, Cid-Díaz T, Pabst-Fernández R, Mosteiro CS, Gallego R, Nogueiras R, Casabiell X, Butler-Browne GS, et al. (2018). Improvement of Duchenne muscular dystrophy phenotype following obestatin treatment. J Cachexia Sarcopenia Muscle 9:1063–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang L, Chen M, Xu M, Li J, Feng P, He R, Zhu Y, Li H, Lin J and Zhang C (2018). Ratio of creatine kinase to alanine aminotransferase as a biomarker of acute liver injury in dystrophinopathy. Dis Markers 2018:6484610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vohra R, Batra A, Forbes SC, Vandenborne K and Walter GA (2017). Magnetic resonance monitoring of disease progression in mdx mice on different genetic backgrounds. Am J Pathol 187:2060–2070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Coley WD, Bogdanik L, Vila MC, Yu Q, Van Der Meulen JH, Rayavarapu S, Novak JS, Nearing M, Quinn JL, et al. (2016). Effect of genetic background on the dystrophic phenotype in mdx mice. Hum Mol Genet 25:130–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Neri M, Torelli S, Brown S, Ugo I, Sabatelli P, Merlini L, Spitali P, Rimessi P, Gualandi F, et al. (2007). Dystrophin levels as low as 30% are sufficient to avoid muscular dystrophy in the human. Neuromuscul Disord 17:913–918 [DOI] [PubMed] [Google Scholar]
- 36. van den Bergen JC, Wokke BH, Janson AA, van Duinen SG, Hulsker MA, Ginjaar HB, van Deutekom JC, Aartsma-Rus A, Kan HE and Verschuuren JJ (2014). Dystrophin levels and clinical severity in Becker muscular dystrophy patients. J Neurol Neurosurg Psychiatry 85:747–753 [DOI] [PubMed] [Google Scholar]
- 37. van Meer L, Moerland M, Gallagher J, van Doorn MBA, Prens EP, Cohen AF, Rissmann R and Burggraaf J (2016). Injection site reactions after subcutaneous oligonucleotide therapy. Br J Clin Pharmacol 82:340–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hilhorst N, Spanoudi-Kitrimi I, Goemans N and Morren M-A (2019). Injection site reactions after long-term subcutaneous delivery of drisapersen: a retrospective study. Eur J Pediatr 178:253–258 [DOI] [PubMed] [Google Scholar]
- 39. Bosgra S, Sipkens J J, de Kimpe S, den Besten C, Datson N and van Deutekom J (2019). The pharmacokinetics of 2′-O-methyl phosphorothioate antisense oligonucleotides: experiences from developing exon skipping therapies for Duchenne muscular dystrophy. Nucleic Acid Ther; DOI: 10.1089/nat.2019.0805 [DOI] [PubMed] [Google Scholar]
- 40. Dunckley MG, Manoharan M, Villiet P, Eperon IC and Dickson G (1998). Modification of splicing in the dystrophin gene in cultured Mdx muscle cells by antisense oligoribonucleotides. Hum Mol Genet 7:1083–1090 [DOI] [PubMed] [Google Scholar]
- 41. Wilton SD, Lloyd F, Carville K, Fletcher S, Honeyman K, Agrawal S and Kole R (1999). Specific removal of the nonsense mutation from the mdx dystrophin mRNA using antisense oligonucleotides. Neuromuscul Disord 9:330–338 [DOI] [PubMed] [Google Scholar]
- 42. Le BT, Adams AM, Fletcher S, Wilton SD and Veedu RN (2017). Rational Design of short locked nucleic acid-modified 2′-O-methyl antisense oligonucleotides for efficient exon-skipping in vitro. Mol Ther Nucleic Acids 9:155–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Han G, Lin C, Ning H, Gao X and Yin H (2018). Long-term morpholino oligomers in hexose elicit long-lasting therapeutic improvements in mdx mice. Mol Ther Nucleic Acids 12:478–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lu-Nguyen N, Ferry A, Schnell FJ, Hanson GJ, Popplewell L, Dickson G and Malerba A (2019). Functional muscle recovery following dystrophin and myostatin exon splice modulation in aged mdx mice. Hum Mol Genet; DOI: 10.1093/hmg/ddz125 [DOI] [PubMed] [Google Scholar]
- 45. Chen S, Le BT, Chakravarthy M, Kosbar TR and Veedu RN (2019). Systematic evaluation of 2′-Fluoro modified chimeric antisense oligonucleotide-mediated exon skipping in vitro. Sci Rep 9:6078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Spitali P, Heemskerk H, Vossen RHAM, Ferlini A, den Dunnen JT, Hoen PAC't and Aartsma-Rus A (2010). Accurate quantification of dystrophin mRNA and exon skipping levels in Duchenne muscular dystrophy. Lab Invest 90:1396–1402 [DOI] [PubMed] [Google Scholar]
- 47. Anthony K, Feng L, Arechavala-Gomeza V, Guglieri M, Straub V, Bushby K, Cirak S, Morgan J and Muntoni F (2012). Exon skipping quantification by quantitative reverse-transcription polymerase chain reaction in Duchenne muscular dystrophy patients treated with the antisense oligomer eteplirsen. Hum Gene Ther Methods 23:336–345 [DOI] [PubMed] [Google Scholar]
- 48. Echevarría L, Aupy P, Relizani K, Bestetti T, Griffith G, Blandel F, Komisarski M, Haeberli A, Svinartchouk F, et al. (2019). Evaluating the impact of variable phosphorothioate content in tricyclo-DNA antisense oligonucleotides in a Duchenne muscular dystrophy mouse model. Nucleic Acid Ther 29:148–160 [DOI] [PubMed] [Google Scholar]
- 49. Hiller M, Falzarano MS, Garcia-Jimenez I, Sardone V, Verheul RC, Popplewell L, Anthony K, Ruiz-Del-Yerro E, Osman H, et al. (2018). A multicenter comparison of quantification methods for antisense oligonucleotide-induced DMD exon 51 skipping in Duchenne muscular dystrophy cell cultures. PLoS One 13:e0204485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kawaguchi T, Niba ETE, Rani AQM, Onishi Y, Koizumi M, Awano H, Matsumoto M, Nagai M, Yoshida S, et al. (2018). Detection of dystrophin Dp71 in human skeletal muscle using an automated capillary western assay system. Int J Mol Sci 19:E1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wells DJ. (2019). What is the level of dystrophin expression required for effective therapy of Duchenne muscular dystrophy? J Muscle Res Cell Motil 40:141–150 [DOI] [PubMed] [Google Scholar]
- 52. Barton-Davis ER, Cordier L, Shoturma DI, Leland SE and Sweeney HL (1999). Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J Clin Invest 104:375–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sharp PS, Bye-a-Jee H and Wells DJ (2011). Physiological characterization of muscle strength with variable levels of dystrophin restoration in mdx mice following local antisense therapy. Mol Ther 19:165–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Godfrey C, Muses S, McClorey G, Wells KE, Coursindel T, Terry RL, Betts C, Hammond S, O'Donovan L, et al. (2015). How much dystrophin is enough: the physiological consequences of different levels of dystrophin in the mdx mouse. Hum Mol Genet 24:4225–4237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Alfano LN, Charleston JS, Connolly AM, Cripe L, Donoghue C, Dracker R, Dworzak J, Eliopoulos H, Frank DE, et al. (2019). Long-term treatment with eteplirsen in nonambulatory patients with Duchenne muscular dystrophy. Medicine 98:e15858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Voit T, Topaloglu H, Straub V, Muntoni F, Deconinck N, Campion G, De Kimpe SJ, Eagle M, Guglieri M, et al. (2014). Safety and efficacy of drisapersen for the treatment of Duchenne muscular dystrophy (DEMAND II): an exploratory, randomised, placebo-controlled phase 2 study. Lancet Neurol 13:987–996 [DOI] [PubMed] [Google Scholar]