

Abstract
The expression of the extracellular sulfatase SULF2 has been associated with increased hepatocellular carcinoma (HCC) growth and poor patient survival. However, the molecular mechanisms underlying SULF2-associated tumor growth remain unclear. To address this gap, here we developed a transgenic mouse overexpressing Sulf2 in hepatocytes under the control of the transthyretin promoter. In this model, Sulf2 overexpression potentiated diethylnitrosamine-induced HCC. Further analysis indicated that the transcription factor GLI family zinc finger 1 (GLI1) mediates Sulf2 expression during HCC development. A cross of the Sulf2-overexpressing with Gli1-knockout mice revealed that Gli1 inactivation impairs SULF2-induced HCC. Transcriptomic analysis revealed that Sulf2 overexpression is associated with signal transducer and activator of transcription 3 (STAT3)-specific gene signatures. Interestingly, the Gli1 knockout abrogated SULF2-mediated induction of several STAT3 target genes, including suppressor of cytokine signaling 2/3 (Socs2/3); Pim-1 proto-oncogene, Ser/Thr kinase (Pim1); and Fms-related tyrosine kinase 4 (Flt4). Human orthologs were similarly regulated by SULF2, dependent on intact GLI1 and STAT3 functions in HCC cells. SULF2 overexpression promoted a GLI1-STAT3 interaction and increased GLI1 and STAT3 enrichment at the promoters of their target genes. Interestingly, the SULF2 overexpression resulted in GLI1 enrichment at select STAT3 consensus sites, and vice versa. siRNA-mediated STAT3 or GLI1 knockdown reduced promoter binding of GLI1 and STAT3, respectively. Finally, chromatin-capture PCR confirmed long-range co-regulation of SOCS2 and FLT3 through changes in promoter conformation. These findings define a mechanism whereby SULF2 drives HCC by stimulating formation of a GLI1-STAT3 transcriptional complex.
Keywords: hepatocellular carcinoma, carcinogenesis, STAT transcription factor, gene transcription, gene regulation, GLI1, promoter conformation, STAT3, SULF2, tumorigenesis
Introduction
Sulfatase 2 (SULF2)3 has been identified as an important prognostic marker and regulator of HCC tumorigenesis. Elevated SULF2 expression has been identified in ∼60% of human HCC. Increased expression of SULF2 in HCC is associated with increased tumor growth, migration, microvascular density, hepatoblast phenotype, and poorer patient survival, with higher rates of tumor recurrence after surgical resection (1). Furthermore, a Sulf2 knockout (KO) mouse was found to have diminished diethylnitrosamine (DEN)-induced liver tumorigenesis (2). SULF2 is an extracellular enzyme with 6-O-desulfatase activity acting on heparan sulfate proteoglycans. Heparan sulfate proteoglycans function as storage sites, harboring various cytokines and growth factors that are released via SULF2 enzymatic activity (3). Given the ability of SULF2 to regulate release of many signaling factors, it is likely that SULF2-mediated tumorigenesis is facilitated through a number of signaling pathways, including ligands of Hedgehog, WNT, and TGFβ pathways (4). Interestingly, these cascades all have transcription factor GLI1 as a common downstream effector (4–6). Furthermore, SULF2-mediated WNT signaling, critical for liver regeneration, is GLI1-dependent (6).
We therefore hypothesized that the SULF2-Gli1 signaling axis is a key regulator of HCC tumorigenesis in vivo. Here, we demonstrate that GLI1 is critical for SULF2-potentiated HCC development. Not only is GLI1 important, but it promotes tumor growth via heterodimerization with STAT3 to cooperatively regulate transcription of JAK/STAT-signaling target genes. These findings define a novel mechanism controlling HCC pathobiology and identified this new GLI1-STAT3 complex as a central mediator of this phenomenon.
Results
Genetically engineered mouse model with transthyretin promoter (TTR)-driven Sulf2 overexpression potentiates liver tumorigenesis
To determine the pathophysiologic role of Sulf2 in HCC, we developed a transgenic mouse expressing murine Sulf2 under control of the hepatocyte-specific TTR (Fig. 1A). Nontransgenic controls are referred to as Sulf2-WT and have physiologic expression of Sulf2. Histopathologic assessment of liver tissue from 8-month-old Sulf2-WT and Sulf2-TTR mice did not reveal any major phenotypic differences. Analysis revealed normal liver architecture without evidence of cellular atypia or inflammation (Fig. 1B). Overexpression of Sulf2 was confirmed by quantitative PCR (Fig. 1C). A cohort of 16 mice was observed long-term, consisting of six Sulf2-TTR and 10 Sulf2-WT mice. One of the six Sulf2-TTR mice developed spontaneous liver tumorigenesis at 19 months, demonstrating typical hepatocellular carcinoma with enlarged round hyperchromatic nuclei, high nuclear/cytoplasmic ratio, and moderate microvesicular fat globules in the cytoplasm. Tumor cells were arranged in thick trabecular, solid, and pseudoglandular growth patterns (Fig. 1D).
Figure 1.
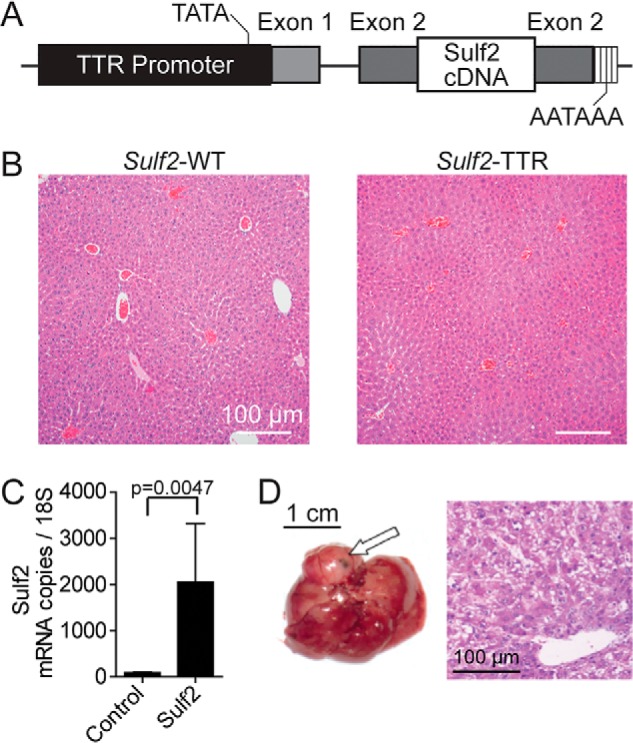
Genetically engineered mouse model with TTR-driven Sulf2 expression. A, schematic demonstrating design of Sulf2-overexpressing genetically engineered mouse model. Sulf2 cDNA is inserted into exon 2 of the TTR promoter, resulting in hepatocyte-specific expression. Relative locations of the TATA box and poly(A) tail are indicated. B, histopathologic analysis of liver tissue from WT (Sulf2-WT) and Sulf2-overexpressing (Sulf2-TTR) mice at 8 months of age. Normal liver morphology is demonstrated with both phenotypes. C, validation of Sulf2 overexpression in the transgenic mouse model by qPCR in control (Sulf2-WT, n = 3) and transgenic (Sulf2-TTR, n = 3) mice at 8 months of age. Results are expressed as mean ± S.E. (error bars). D, hepatic tissue (left) and corresponding histopathologic analysis (right) of spontaneous liver carcinogenesis from a single mouse (one of six) with the Sulf2-TTR genotype with no liver tumors spontaneously developing in the 10 Sulf2-WT mice. The tumor is indicated by an arrow.
Given the low rate of spontaneous liver tumorigenesis, DEN was used to induce HCC. Briefly, at 14 days of age, male mice were treated with intraperitoneal DEN and sacrificed at 8 months (Fig. 2A). Gross examination of murine livers demonstrated substantial tumor burden in Sulf2-TTR mice relative to Sulf2-WT mice (Fig. 2C). Histopathologic analysis revealed no obvious morphologic differences in tumors between mouse genotypes. Pathology consistently demonstrated well-differentiated HCC (Fig. 2D). Interestingly, there was no significant difference in overall tumor number between the two mouse genotypes. However, in line with our gross observations, Sulf2-TTR mice had significantly larger tumor volume, weight, and tumor/liver weight ratio relative to Sulf2-WT (Fig. 2E). Finally, the number of mice with metastatic disease in the lung was not clearly different between the two mice (Fig. 2F). Taken together, hepatocyte-specific Sulf2 overexpression potentiates DEN-induced liver tumorigenesis.
Figure 2.

Gli1 loss impairs Sulf2-potentiated, DEN-induced liver tumorigenesis in transgenic mice. A, schematic depicting the experimental design of DEN-induced liver tumorigenesis studies. DEN is administered intraperitoneally on day 14, and the mouse is sacrificed at 8 months of age. B, validation of Gli1 knockout by qPCR of liver tissue from Gli1 HET (n = 3) and KO (n = 3) mice relative to WT (n = 3). Results are expressed as mean ± S.E. (error bars). C, three representative examples of liver tissue from DEN-treated mice with the indicated genotypes. Individual tumors are outlined in white. A 1-cm scale for each image is depicted in the bottom left corner. D, representative images of histopathological analysis of tumors derived from DEN-treated mice with the indicated genotypes. E, quantitative assessments of tumor burden (number, volume, weight, and tumor/liver weight ratio) with data points representing individual mice. Bars, mean ± S.E. F, percentage of mice in each genotype determined to have metastatic disease (lesions outside the liver).
Gli1 loss impairs Sulf2-potentiated liver tumorigenesis
Consistent with prior studies between SULF2 and GLI1 in vitro, overexpression of Sulf2 correlated with increased Gli1 expression in Sulf2-TTR mice relative to Sulf2-WT mice (6, 7) (Fig. S1A). Furthermore, as Sulf2-WT and Sulf2-TTR mice age, relative expression levels of Sulf2 and Gli1 remain stable without statistical differences. With exposure to DEN, there appears to be a trend for increased expression of both Sulf2 and Gli1 with increased variability of expression relative to baseline. However, these findings were also not significantly different (Fig. S1B).
To further delineate the interplay between Sulf2 and Gli1 in liver tumorigenesis in vivo, Sulf2-TTR mice were crossed with Gli1 knockout mice (8) (Fig. S1C). Resultant genotypes of interest included Gli1 WT, heterozygous (HET), and homozygous KO mice, all with TTR-driven Sulf2 overexpression. Gli1 knockout did not significantly alter Sulf2 expression levels (Fig. 2B). As described above, male mice were treated with intraperitoneal DEN to induce liver tumorigenesis. Gross analysis revealed marked differences between genotypes. As noted above, Sulf2-TTR, Gli1 WT mice demonstrated significant liver tumor burden compared with Sulf2-WT, GLI1 WT controls; however, knockout of Gli1 impaired DEN-induced tumorigenesis in a Gli1 allele dose-dependent manner, with homozygous knockouts having significantly lower gross tumor burden (Fig. 2C). Although significant differences in tumor burden were observed, once again histopathologic analysis of tumors across genotypes did not differ significantly, demonstrating well-differentiated HCC (Fig. 2D).
Further quantitative analyses reinforced gross observations of tumor burden. With knockout of Gli1, there was an allele dose-dependent decrease in tumor number, tumor volume, tumor weight, and tumor/liver weight ratio (Fig. 2E). Further, several Gli1 WT mice developed metastatic disease, Gli1 KO mice lacked visible metastases, and Gli1 HET mice demonstrated an intermediate phenotype (Fig. 2F). Thus, intact Gli1 is critical for Sulf2-potentiated liver tumorigenesis.
Gli1 abrogation down-regulates Stat3 target genes
To elucidate transcriptional target genes mediating HCC tumorigenesis via the Sulf2-Gli1 signaling axis, we determined the differential gene expression between tumors derived from Sulf2-TTR, Gli1 WT, and Sulf2-TTR, Gli1 KO mice. RNA-Seq was performed on these tumors in triplicate. Unsupervised clustering of transcriptomic profiles resulted in appropriate segregation between Gli1 WT and Gli1 KO tumors (Fig. S2A). Relative to Gli1 WT tumors, in Gli1 KO tumors, 11 genes were significantly up-regulated, whereas 34 genes were significantly down-regulated (Fig. 3A). Ingenuity pathway analysis revealed the canonical Stat3 pathway as one of the most differentially regulated. Similarly, KEGG pathway analysis revealing significant down-regulation of Jak-Stat signaling (Fig. S2B), including known canonical targets of Stat3 such as Socs2, Socs3, Pim1, and Flt4 (Fig. 3A).
Figure 3.

Gli1 abrogation down-regulates Stat3 target genes. A, volcano plot demonstrating significantly differentially up-regulated (red) and down-regulated (green) genes in Sulf2-TTR Gli1 KO tumors relative to Sulf2-TTR Gli1 WT tumors. Each data point represents a single transcript. Those four transcripts highlighted are involved in canonical JAK/STAT signaling. B, validation of differential expression of Socs2, Socs3, Pim1, and Flt4 by qPCR on tumor-derived RNA from mice with Gli WT (n = 3), HET (n = 3), and KO (n = 3). Results are expressed as means ± S.E. (error bars). C, validation of expression regulation of human orthologues SOCS2, SOCS3, PIM1, and FLT3. Huh-7 cells were transduced with either a null adenoviral construct (Control, white, n = 3) or an adenovirus vector expressing SULF2 (SULF2, black, n = 3). The effect of siRNA-mediated knockdown of GLI1 (siGLI1) was tested with (n = 3) and without (n = 3) SULF2 transduction. Expression was measured by qPCR. Results are expressed as means ± S.E. D, promoter diagrams are depicted for SOCS2 (top left), SOCS3 (top right), PIM1 (bottom left), and FLT3 (bottom right), consisting of the 3000 nucleotides upstream of the transcriptional start site. The relative positions of putative GLI1 consensus binding sites are indicated in red. Graphs for each binding site represent ChIP experiments expressed as GLI1 -fold enrichment at these binding sites in Huh-7 cells under conditions of Adv-Null (n = 3) infection and transduction of SULF2 (Adv-SULF2; n = 3). Results are expressed as means ± S.E.
In addition to these well-characterized Stat3 target genes, several additional significantly down-regulated genes in Gli KO mice were identified as possible targets of Stat3 transcriptional regulation. For example, 12 of the top 34 down-regulated genes relative to Gli1 WT tumors were major urinary proteins (Fig. S2C). These proteins are synthesized in hepatocytes of rodents, but not humans, and their expression is regulated by Jak/Stat signaling (9). Additional genes of interest with prior evidence of regulation by Stat3 included Abcb1 (10–14), Capn1 (15), Cxcl1 (16), Derl3 (17), Gadd45g (18, 19), Itgb3 (20), Nnmt (21), Ptges2 (22), S100a8 (23, 24), Serinc2 (25), and Thbs1 (26, 27).
Validation was then carried out in homogenized tumor tissue derived from three mice of each Gli1 genotype. Quantitative PCR confirmed an allele dose-dependent reduction in expression of the canonical Jak/Stat signaling components Socs2, Socs3, Pim1, and Flt4 (Fig. 3B). Additional differentially expressed genes with evidence of Stat3 regulation in the literature (listed above), were also validated. This revealed several candidates that have reduced expression in Gli1 KO relative to Gli1 WT tumors (Fig. S2D). Thus, these findings suggest that increased Stat3-regulated gene expression underlies Sulf2-Gli1 axis mediated liver tumorigenesis.
SULF2-mediated up-regulation of STAT3 target genes depends on both GLI1 and STAT3
The significance of the observed changes for human HCC was first investigated using the Cancer Genome Atlas. Assessing liver cancer Cancer Genome Atlas transcriptomic data (n = 438 cases) revealed positive correlations between SULF2 expression and not only GLI1, but also JAK/STAT3 signaling components, including SOCS2, SOCS3, FLT3, FLT4, and STAT3. The only exception was PIM1, which did not seem to be related to SULF2 expression (Fig. S3).
Thus, the role of SULF2, GLI1, and STAT3 in regulation of these human orthologues was investigated in the HCC cell lines Huh-7 and Hep3B. HCC cells were transfected with either nontargeting siRNA (siNT) or siRNA against GLI1 (siGLI1). Knockdown efficiency was evaluated by Western blotting (Fig. S4A). 24 h after transfection, cells were transduced with either control adenovirus or viral particles expressing SULF2. Expression of SOCS2, SOCS3, PIM1, and FLT3 was assessed by qPCR 72 h after transfection. The expression patterns of all four genes were similar. As seen in murine tissue, SULF2 transduction increased expression of the above STAT3 targets, and GLI1 knockdown abrogated this induction (Fig. 3C).
Using both the in silico tool INSECT 2.0 (28) and manual analysis of the 3000 base pairs upstream of each gene's transcriptional start site (TSS), putative GLI1 consensus sites were identified in promoters of SOCS2 (1 site; −2737), SOCS3 (4 sites; −2871, −1820, −1314, −399), PIM1 (1 site; −669), and FLT3 (3 sites; −2173, −1589, −374). These sites are graphically depicted in Fig. 3D. Binding was confirmed by ChIP studies. Relative to cells transduced with control adenoviral particles, SULF2 transduction resulted in GLI1 enrichment within at least one of the putative binding sites identified (Fig. 3D). The exception was PIM1, where GLI1 was not significantly enriched at its only putative binding site as identified by our methodology (Fig. 3D). This finding suggests either a more indirect regulatory mechanism or a more distant GLI1-binding site. Thus, transcriptomic analysis revealed a STAT3 signature among a cohort of genes positively regulated by SULF2 and dependent on intact GLI1. SULF2 expression results in GLI1 enrichment at the promoters of these genes.
Given the numerous STAT3-regulated genes that appear to be important in GLI1-dependent liver tumorigenesis, the requirement for STAT3 in gene expression was also interrogated. SULF2-mediated increased expression of SOCS2, SOCS3, PIM1, and FLT3 was abrogated by siRNA-mediated STAT3 knockdown (Fig. 4A). Knockdown efficiency was evaluated by Western blotting (Fig. S4A). Expression patterns of additional human orthologues of genes of interest discovered during RNA-Seq of mouse tissues (Fig. 3 and Fig. S2) were also assessed under these experimental conditions. A similar pattern was observed to the canonical JAK/STAT signaling genes for most of these. Exceptions included DERL3 and SERINC2. DERL3 expression was unaffected by GLI1 or STAT3 knockdown or SULF2 overexpression, whereas SERINC2 expression was decreased by not only GLI1 and STAT3 knockdown but also SULF2 overexpression (Fig. S4B).
Figure 4.

SULF2-mediated up-regulation of STAT3 target genes depends on both GLI1 and STAT3. A, assessing the role of STAT3 in regulating SOCS2, SOCS3, PIM1, and FLT3 expression in Huh-7 cells by qPCR. Cells were transduced with either Adv-Null (Control, n = 3) or Adv-SULF2 (SULF2, n = 3) and transfected with either nontargeting siRNA (n = 3) or siRNA against STAT3 (siSTAT3, n = 3). Results are expressed as means ± S.E. (error bars). B, promoter diagrams are reproduced for SOCS2, SOCS3, PIM1, and FLT3. Relative position of putative STAT3 consensus binding sites are indicated in blue. ChIP-PCR experiments are expressed as STAT3 -fold enrichment at these binding sites in Huh-7 cells under conditions of Adv-Null (n = 3) and Adv-SULF2 (n = 3) transduction. Results are expressed as means ± S.E. C, representative co-immunoprecipitation assays in Huh-7 cells after co-transfection of STAT3-FLAG and GLI-His constructs under conditions of transduction with Adv-Null (left) and Adv-SULF2 (right) transduction. MW, molecular weight presented in kilodaltons.
Furthermore, putative STAT3 consensus binding sites were identified. Several sites were found in SOCS2 (1 site; −1043), SOCS3 (5 sites; −2766, −1053, −441, −263, −54), PIM1 (1 site; −968), and FLT3 (1 site; −857), all graphically depicted in Fig. 4B. Interestingly, much as with GLI1, STAT3 occupancy was enriched at all gene promoters tested with SULF2 transduction relative to controls (Fig. 4B).
In addition to these cis regulatory elements within promoter regions, we also used in silico analyses to identify enhancer elements for the canonical JAK/STAT signaling genes. The genomic coordinates for enhancer and double-elite regions annotated to regulate the JAK/STAT genes were identified using the GeneHancer data (version 4.4) from GeneCards or UCSC Genome Browser. Using the Homer tool findMotifsGenome, these genomic coordinates were searched for transcription factor enrichment. Ultimately, this exploratory search of a small gene set (n = 6) did not reveal an enrichment of GLI1 or STAT3 motifs associated with enhancer regulation of SOCS2, SOCS3, PIM1, ITGB3, PTGES2, or FLT3. Taken together, these findings support a promoter-based co-regulation by GLI1 and STAT3. This is further supported by a nonsignificant enrichment of GLI motifs in the promoters (−2000 to +100 bp around the TSS) of these six genes using the Homer tool findMotifs.
Given expression of SOCS2, SOCS3, PIM1, and FLT3 are dependent on both GLI1 and STAT3; co-regulation was hypothesized to be mediated by GLI1 and STAT3 heterodimerization. Therefore, HCC cell lines were co-transfected with FLAG-tagged STAT3 and His-tagged GLI1. Immunoprecipitation of either FLAG or His did not result in detectable levels of GLI1 or STAT3, respectively (Fig. 4C). However, transduction with adenovirus expressing SULF2 after co-transfection of transcription factors resulted in increased co-immunoprecipitation (Fig. 4C). Together, these findings confirmed interaction between STAT3 and GLI1 and define these factors as common effectors of SULF2.
SULF2 expression induces GLI1 and STAT3 enrichment at reciprocal consensus binding sites
The relative location of GLI1 and STAT3 consensus sites, where enrichment was confirmed with SULF2 expression, was mapped on promoters of SOCS2 (Fig. 5A), SOCS3 (Fig. 5B), PIM1 (Fig. 5C), and FLT3 (Fig. 5D). As depicted, the relative distribution of the confirmed binding sites is significantly dispersed. This led to the hypothesis that either GLI1 or STAT3 could bind to its respective consensus site with the other transcription factor in complex with it to promote transcription. Alternatively, GLI1 and STAT3, bound at disparate sites, could physically augment the promoter to come into close proximity and regulate gene expression.
Figure 5.

SULF2 expression induces GLI1 and STAT3 enrichment at reciprocal consensus binding sites. Depicted in this figure are reproductions of the 3000 nucleotide promoter maps depicted previously for SOCS2 (A), SOCS3 (B), PIM1 (C), and FLT3 (D). Relative positions of consensus binding sites for GLI1 (red) and STAT3 (blue) are depicted on the promoter maps. Four separate ChIP-PCR assays were performed for each depicted consensus binding site represented as the bar graphs below each site. The top two graphs depict either STAT3 or GLI1 -fold enrichment at that site in Huh-7 cells transduced with either Adv-Null (n = 3) or Adv-SULF2 (n = 3). The bottom pair of graphs represent ChIP-PCR assays in Huh-7 cells transfected with either siNT (gray bars, n = 3) or siRNA against GLI1 (red bars, n = 3) or STAT3 (blue bars, n = 3) prior to transduction of Adv-SULF2. Results are expressed as means ± S.E. (error bars).
To address the former hypothesis, ChIP-PCR was carried out to determine whether enrichment of GLI1 or STAT3 was observed at reciprocal transcription factor–binding sites. Enrichment of GLI1 at STAT3-binding sites and STAT3 at GLI1 sites was assessed both with transduction of control adenovirus and a vector expressing SULF2. Interestingly, depicted in the top row of bar graphs for each panel, GLI1 was enriched at all STAT3 consensus sites with SULF2 expression relative to controls. STAT3 occupancy was also enriched at all GLI1-binding sites (Fig. 5, A–D). Further, the same patterns were seen in a majority of other putative STAT3 target genes (Fig. S5).
Knockdown experiments were then performed with transfection of siNT, siGLI1, or siSTAT3 prior to SULF2 transduction. Results are shown in the bottom row of each panel of Fig. 6. A common pattern across promoters was observed. If studying a GLI1 consensus site, STAT3 knockdown did not result in significantly reduced GLI1 enrichment. However, GLI1 knockdown resulted in depletion of STAT3 at the binding site. The opposite was true at STAT3-binding sites. GLI1 knockdown had no significant effect on STAT3 binding, whereas STAT3 knockdown reduced GLI1 enrichment (Fig. 5, A–D). Notable exceptions were the GLI1 and second (from the TSS) STAT3-binding sites within the SOCS3 promoter. At these sites, GLI1 or STAT3 knockdown had no significant effect on STAT3 or GLI1 occupancy, respectively (Fig. 5B). This can be explained by the close proximity of the consensus sites, prohibiting the design of primer sets to interrogate them individually.
Figure 6.

GLI1 and STAT3 coordinately control promoter conformation in HCC cells. A, schematic of experimental design for chromatin conformation capture assays not presented at scale. Forward and reverse primers are arrows indicated in red and blue, respectively. Perpendicular lines indicate the site of restriction digestion with the indicated enzyme. Green lines, location of the TaqMan probe. The presented base pair length indicates the distance between restriction digestion sites in the SOCS2 and FLT3 promoters. Below each loop diagram is a representation of the ligation product with predicted amplicon length after digestion and ligation. B, results of qPCR using primers depicted in A. Because the ligation product of interest is rare, data are presented as -fold change in amplification normalized to the undigested control. Huh-7 cells were transfected with siNT (n = 6), siGLI1 (n = 6), or siSTAT3 (n = 6). Afterward, they were transduced with either Adv-Null (n = 6) or Adv-SULF2 (n = 18). DNA was either undigested as a negative control or digested with the restriction enzyme depicted. Results are expressed as means ± S.E. (error bars). C, diagram depicting a working model of SULF2-potentiated liver tumorigenesis. SULF2 overexpression results in increased GLI1 expression and enrichment at target genes. Co-regulation of genes occurs with STAT3 through heterodimerization and either co-enrichment or promoter DNA looping to mediate gene expression.
These results support the hypothesis that enrichment of GLI1 at a STAT3 consensus site depends on intact STAT3 and vice versa. However, this does not rule out the possibility of distant, transcription factor–bound sites being structurally brought into close proximity to regulate gene expression.
GLI1 and STAT3 coordinately control promoter DNA looping in HCC cells
The relative positions of confirmed GLI1 and STAT3 consensus binding sites, as depicted in Fig. 5, prohibit direct interaction of DNA-bound transcription factors in a number of cases. This suggested two possibilities. First, one of the two transcription factors may simply recruit the other, as supported by enrichment of one transcription factor at reciprocal consensus sites. Alternatively, GLI1 and STAT3 may augment promoter structure to bring the factors into close proximity, forming heterodimers. The latter hypothesis was tested with chromatin conformation capture (3C) qPCR. Among the four genes of interest, we chose to investigate SOCS2 and FLT3 loci, given the more feasible transcription factor-binding orientation. Specifically, each gene promoter contains a single GLI1-binding site and STAT3-binding site separated by >1000 nucleotides (Fig. 5, A and D). The schematics for SOCS2 and FLT3 are depicted in Fig. 6A. In the SOCS2 promoter, the GLI1-binding site is located 1740 bp upstream of the STAT3-binding site. Primers designed to anneal between these sites were located 1097 nucleotides apart. Two BsrI digestion sites were identified between this primer set. Successful promoter digestion and ligation between these sites would result in reduction of a 1222-bp amplicon to 145 bp (Fig. 6A, left). Similarly, the GLI1 site in the FLT3 promoter is located 1305 bp upstream of the STAT3-binding site. Primers designed to amplify the region between the sites would result in a 1097-bp amplicon. Using BsrGI digestion and subsequent ligation, this would reduce the amplicon to 500 bp (Fig. 6A, right).
To determine whether GLI1 and STAT3 were interacting at these distances in each promoter, 3C-qPCR was performed on genomic DNA isolated from Huh-7 cells under various experimental conditions. First, cells were transfected with siNT, siGLI1, or siSTAT3. 24 h after transfection, cells were transduced with either control adenoviral particles or a vector expressing SULF2. PCR was performed 48 h after transduction. Isolated genomic DNA was either left undigested or incubated with BsrI (for SOCS2 studies) or BsrGI (for FLT3 studies) with subsequent ligation and qPCR. Under conditions of transduction with control adenovirus, effectively no amplification was detected. However, transduction with SULF2 resulted in a significant increase in amplification under conditions of BsrI or BsrGI digestion with minimal detection without digestion. Furthermore, siRNA-mediated knockdown of both GLI1 and STAT3 abrogated this signal, supporting their importance in co-regulation (Fig. 6B). Confirmation of these results was conducted by gel electrophoresis of PCR products (data not shown). These findings indicated likely physical interaction between GLI1- and STAT3-binding sites of the SOCS2 and FLT3 promoters in the context of SULF2 expression.
Discussion
Taken together, in HCC, we have previously described the role of SULF2 in releasing growth factors such as Hedgehog (SHH) ligands, TGFβ, and Wnt3A, which can increase expression of GLI1 (4, 6, 7). Here, using a novel Sulf2 transgenic mouse model, we found that Sulf2 potentiates HCC tumorigenesis. Furthermore, this process depends on intact Gli1. Interestingly, Gli1 promotes a gene expression profile including many well-characterized Stat3 target genes both in the mouse model and in human orthologues. Regulatory interaction between GLI1 and STAT3 is not without precedence (33–35), but out study demonstrates that SULF2 expression results in increased heterodimerization with GLI1. Furthermore, GLI1 and STAT3 are both found to be enriched not only at their respective consensus promoter-binding sites, but also at a number of reciprocal binding sites for many target genes. Thus, co-regulation of this gene cohort is likely mediated by multiple mechanisms. GLI1 and STAT3 may directly interact at a single consensus binding site. Alternatively, we show that spatially distant STAT3 and GLI1 sites can be structurally brought into close proximity to promote gene expression (Fig. 6C).
This work interweaves a number of critical signaling pathways in HCC pathogenesis. GLI1 is a common downstream effector of both Hedgehog and WNT signaling, whereas STAT3 is a key downstream mediator of inflammatory signaling via the IL-6/JAK/STAT axis. In hepatocellular adenomas, benign lesions with potential for malignant transformation, multiple molecular subtypes have been defined. Interestingly, mutations within the aforementioned pathways define some of these subtypes: activating mutations in β-catenin, activating mutations within JAK2/STAT3 signaling, and a more recently described subtype characterized by increased GLI1 expression as a consequence of INHBE and GLI1 fusion (29). Not only do aberrations in these pathways play a role in defining adenoma subtypes, but they are also found in a large proportion of HCC.
In fact, adenomas harboring mutations in β-catenin carry a higher risk of malignant transformation (29). It follows that WNT/β-catenin signaling activation, most commonly caused by mutations in CTNNB1 (β-catenin), has been observed in approximately half of HCC cases (30–32). Inactivating mutations of TP53 are also abundant in HCC (30, 31). Recently, multi-omics characterization of HCC by the Cancer Genome Atlas Research Group identified an association between TP53 mutation status and SHH signaling activity. In the context of inactive TP53, the known target gene and negative regulator of SHH, PTCHD4, had markedly reduced expression. Furthermore, gene set enrichment analysis revealed up-regulated SHH expression (32). Finally, although not as common as in hepatocellular adenomas, activating mutations in IL-6 signal transducer (IL6ST), which lead to increased JAK/STAT signaling, are among the top 15 recurrently mutated genes in HCC (32). Consequently, recurrent mutations in HCC resulting in aberrantly activated WNT/β-catenin, SHH, and JAK/STAT signaling may converge on transcriptional effectors GLI1 and STAT3, which co-regulate genes.
Unfortunately, current therapeutic modalities for HCC have advanced only incrementally over the last decade (36–40). However, as molecular pathogenesis of HCC is better understood, novel treatment approaches have been developed. Given the relevance of SULF2 in pathogenesis and prognosis of HCC (1–4, 42, 43), targeting SULF2 has been investigated and seems promising in preclinical studies (7). In addition, small-molecule inhibitors for WNT signaling are also being investigated (44). However, whether WNT, JAK/STAT, or SHH signaling is clinically targeted in HCC, the current study suggests the possibility of cross-talk limiting the efficacy of such approaches. Instead, given that these frequently aberrantly activated pathways appear to have the common transcriptional complex of GLI1 and STAT3 as a downstream mediator of pathogenesis, novel approaches disrupting these effectors may be less susceptible to treatment resistance.
Experimental procedures
Animals
FVB strains overexpressing Sulf2 under control of the hepatocyte-specific transthyretin promoter (Sulf2-TTR mice) were developed by the Mayo Clinic transgenic and knockout core facility. Mice were created with the −3 kb TTR promoter region driving the expression of Sulf2 cDNA, which was cloned into the TTR second exon. Cloning was achieved by amplifying the coding sequence of murine Sulf2 with restriction sites for BamHI on the 5′ end and XbaI on the 3′ end using mouse kidney cDNA. The PCR product was digested with BamHI and XbaI and ligated into vector pcDNA3.1. Sulf2 was then cut out of the pcDNA3.1 vector using BamHI and XbaI, and 5′ overhangs were filled using T4 DNA polymerase and dNTPs and subsequently ligated into the pTTR exV3 vector at the StuI blunt cut site. Insertion and orientation were confirmed by sequencing (45).
FVB founders were back-crossed into the C57/BL6 background to the N6 generation. This was done to ensure uniform susceptibility to carcinogenesis, as C57/BL6 mice are known to have a higher propensity for developing tumors with DEN treatment when compared with FVB mice. The care and use of the animals for these studies were reviewed and approved by the Mayo Clinic Institutional Animal Care and Use Committee.
The Sulf2 transgenic mice were not found to have any major phenotypic changes when compared with WT. Mice did not show any signs of liver injury or inflammation. WT littermates were used as controls. Mice were maintained in a temperature-controlled (22 °C), pathogen-free environment and fed a standard rodent chow diet and water ad libitum. The care and use of the animals for these studies were reviewed and approved by the Mayo Clinic Institutional Animal Care and Use Committee.
Gli1 knockout mice have a C57/BL6 background (8) and were raised and maintained in the animal facilities of Mayo Clinic. Gli1 heterozygous null and Sulf2-TTR mice were crossed, ultimately producing three groups of mice: Sulf2-TTR, Gli1 WT; Sulf2-TTR, Gli1 HET; Sulf2-TTR, Gli1 KO. Primers used for genotyping of Sulf2 and Gli1 status are listed in Table 1.
Table 1.
Primers used for genotyping of Sulf2 and Gli1 status
| Primer | Sequence |
|---|---|
| Gli1 WT | |
| Sense | CCAGTTTCTGAGATGAGGGTTAGAGGC |
| Antisense | TTGAATGGGGAATACAGGGGCTTAC |
| Gli1 KO | |
| Sense | GCATCGAGCTGGGTAATAAGCGTTGGCAAT |
| Antisense | GACACCAGACCAACTGGTAATGGTAGCGAC |
| Sulf2 | |
| Sense | CCATATCCCTGGTGCACAGCAG |
| Antisense | GAAGGTGCGGCTCTCGTGCTG |
DEN-induced liver tumors in mice
DEN was used as a carcinogen to induce primary liver tumors. DEN is a genotoxic drug, and its use has been well-established in mouse models for hepatocarcinogenesis (46). At 14 days of age, mice received a single intraperitoneal injection of DEN (15 mg/kg body weight). At 21 days of age, mice were separated by sex and genotyped. Littermates with negative genotypes were used as WT controls. Only male mice were included in the analysis, as female mice have a substantially lower rate of tumor development after DEN treatment. All mice were sacrificed at 8 months, and their liver and lungs were examined for tumors. The liver weight, number of visible tumors, and size of visible tumors were recorded.
Histopathology
Liver tissue from mouse models were removed and fixed in 10% neutral buffered formalin. Tissues were sent to Mayo Clinic Histology Core Laboratory for paraffin embedding and sectioning for hematoxylin and eosin staining. Histopathology was independently reviewed by two pathologists, R. G. and M. V., who were blinded to genotype.
RNA-Seq
S. W. provided histopathologic assessment of murine liver tissue prior to RNA extraction for RNA-Seq. Replicates submitted were considered morphologically similar. cDNA libraries were generated in accord with the Illumina RNA-Seq preparation protocol. The cDNA fragments were amplified by PCR and sequenced at both ends using an Illumina Genome Analyzer IIx. All samples had over 90 million reads with over 75% of the reads mapping to the mouse genome (obtained from the UCSC Genome Browser assembly ID: mm10). Bioinformatics analysis was done using Qiagen Ingenuity Pathway Analysis. Reads were analyzed using MAPRSeq version 2.0.0 against the mouse reference for calculating gene expression counts. Differential expression using edgeR was performed between WT and Gli1 KO mouse liver tumors. Samples were sequenced in triplicate. Gene expression levels were filtered using a false discovery rate of <0.1 and a -fold change whose magnitude is greater than 1 and log counts per million >−1. Data have been deposited in the GEO database, GSE139374 (“Investigating the role of GLI1 in SULF2-potentiated liver tumorigenesis”) (Table 2).
Table 2.
GEO accession numbers
| Accession no. | Sample name |
|---|---|
| GSM4138971 | Sulf2-TTR Gli1 WT replicate 1 |
| GSM4138972 | Sulf2-TTR Gli1 WT replicate 2 |
| GSM4138973 | Sulf2-TTR Gli1 WT replicate 3 |
| GSM4138974 | Sulf2-TTR Gli1 KO replicate 1 |
| GSM4138975 | Sulf2-TTR Gli1 KO replicate 2 |
| GSM4138976 | Sulf2-TTR Gli1 KO replicate 3 |
Quantitative PCR
Total RNA was extracted from cultured cells or primary murine tissue using TRIzol reagent (Invitrogen) following the manufacturer's protocol. 2 μg of total RNA was reverse-transcribed using a high-capacity cDNA synthesis kit (Applied Biosystems). A portion of the total cDNA was amplified by quantitative PCR. Samples were prepared with 1× SYBR Green Supermix (Bio-Rad). Primers used for validation of RNA-Seq findings in mouse model-derived liver tumors are listed in Table 3. Primers used to assess levels of human orthologues in HCC cell lines are listed in Table 4.
Table 3.
Primers used for validation of RNA-Seq findings
| Primer | Sequence |
|---|---|
| Gapdh | |
| Sense | TCCATGACAACTTTGGCATTG |
| Antisense | CAGTCTTCTGGGTGGCAGTGA |
| Tbp | |
| Sense | GAAGTTCCCTATAAGGCTGGAAG |
| Antisense | AGGAGAACAATTCTGGGTTTGA |
| Flt4 | |
| Sense | AGGGAGACGCCCTTTCATG |
| Antisense | GAGGGCTCTTTGGTCAAGCA |
| Pim1 | |
| Sense | CGACATCAAGGACGAGAACAT |
| Antisense | GTCCCATCAAAGTCCGTGTAG |
| Socs2 | |
| Sense | GTTGCCGGAGGAACAGTCCC |
| Antisense | TCGGTCCAGCTGACGTCTTAA |
| Socs3 | |
| Sense | TGCAGGAGAGCGGATTCTA |
| Antisense | TGACGCTCAACGTGAAGAAG |
| Abcb1 | |
| Sense | GCTGTCTGGGCAAAGATACT |
| Antisense | CACAGTTCTAATTGCTGCCAAG |
| Capn1 | |
| Sense | TTCTTCCATTCTTCCTCTG |
| Antisense | CTTCCTTCCTGTTCTGAG |
| Cxcl1 | |
| Sense | TGTTGTGCGAAAAGAAGTGC |
| Antisense | ACACGTGCGTGTTGACCATA |
| Derl3 | |
| Sense | CATCACCACCTTCCTCTTCTTC |
| Antisense | AGAACACCACCGAAGAGAAAC |
| Gadd45g | |
| Sense | AAGCACTGCACGAACTTCT |
| Antisense | CTATGTCGCCCTCATCTTCTTC |
| Itgb3 | |
| Sense | GTGAAAGAGCTGACGGATAC |
| Antisense | TCTTCCACCACATAGAGGAC |
| Nnmt | |
| Sense | TGGAGAAGTGGCTGAAGAAAG |
| Antisense | TTCTCTGGACCCTTGACTCT |
| Ptges2 | |
| Sense | CTCTATGAGGCTGCTGACAAG |
| Antisense | GCCATACACCGCCAAATCA |
| S100a8 | |
| Sense | GAATTTCCATGCCGTCTACAG |
| Antisense | AGACGTCTGCACCCTTT |
| Serinc2 | |
| Sense | TCTTTAAGTTCCTGATCCTGGTG |
| Antisense | GCCGAAGTAGAACCAGATGTT |
| Thbs1 | |
| Sense | GAGTGTCACTGTCAGAACTCAG |
| Antisense | GAGGACAGCATTCTCCATCAG |
Table 4.
Primers used to assess levels of human orthologues in HCC cell lines
| Primer | Sequence |
|---|---|
| GAPDH | |
| Sense | GACCTGACCTGCCGTCTAGAAAAA |
| Antisense | ACCACCCTGTTGCTGTAGCCAAAT |
| TBP | |
| Sense | TATAATCCCAAGCGGTTTGC |
| Antisense | CCCAACTTCTGTACAACTCTAGCA |
| FLT3 | |
| Sense | CCGCCAGGAACGTGCTTG |
| Antisense | ATGCCAGGGTAAGGATTCACACC |
| PIM1 | |
| Sense | CGAGCATGACGAAGAGATCAT |
| Antisense | TCGAAGGTTGGCCTATCTGA |
| SOCS2 | |
| Sense | GGATGGTACTGGGGAAGTATGACTG |
| Antisense | AGTCGATCAGATGAACCACACTGTC |
| SOCS3 | |
| Sense | AGTCTGGGACCAAGAACCT |
| Antisense | TTGAGCACGCAGTCGAAG |
| ABCB1 | |
| Sense | GCTGTCTGGGCAAAGATACT |
| Antisense | CACAGTTCTAATTGCTGCCAAG |
| CAPN1 | |
| Sense | ACATGGAGGCCATCACTTTC |
| Antisense | CTCGGTAGTTCACCTGCTTG |
| CXCL1 | |
| Sense | CGAAGTCATAGCCACACTCAA |
| Antisense | GATTTGTCACTGTTCAGCATCTT |
| DERL3 | |
| Sense | CGGTGGGCCATATCTACTACT |
| Antisense | TGCTCTGAAGTCCCAGGAA |
| GADD45G | |
| Sense | GGAAAGCACAGCCAGGAT |
| Antisense | CAGTGAGGCAGCCCTGA |
| ITGB3 | |
| Sense | ACCATGGATTATCCCTCTTTGG |
| Antisense | GGGATGAGCTCACTATAGTTCTG |
| NNMT | |
| Sense | TGGAGAAGTGGCTGAAGAAAG |
| Antisense | TTCTCTGGACCCTTGACTCT |
| PTGES2 | |
| Sense | CTCTATGAGGCTGCTGACAAG |
| Antisense | GCCATACACCGCCAAATCA |
| S100A8 | |
| Sense | GAATTTCCATGCCGTCTACAG |
| Antisense | AGACGTCTGCACCCTTT |
| SERINC2 | |
| Sense | TCTTTAAGTTCCTGATCCTGGTG |
| Antisense | GCCGAAGTAGAACCAGATGTT |
| THBS1 | |
| Sense | GAGTGTCACTGTCAGAACTCAG |
| Antisense | GAGGACAGCATTCTCCATCAG |
Amplification was performed using the C1000 thermal cycler (Bio-Rad) under the following reaction conditions: 95 °C for 3 min followed by 40 cycles of 30 s at 95 °C, 30 s at 60 °C, and 20 s at 72 °C. Each mRNA level was normalized by comparison with glyceraldehyde-3-phosphate dehydrogenase and TATA-binding protein RNA levels in the same sample. The results were calculated following the 2ΔCp method.
Tissue culture
The Hep3B cell line was obtained from ATCC and cultured in complete minimum essential medium with 10% FBS. The Huh-7 cell line was obtained from the Japan Health Science Research Resources Bank (HSRRB, Osaka, Japan) and grown in Dulbecco's modified Eagle's medium with 10% FBS.
Adenoviral transduction
Adenoviral transduction was performed using null adenovirus (Adv-Null) (1060, Vector Biolabs) or Adv-SULF2 (ADV-224703, Vector Biolabs). Viral particles were applied to culture medium at a multiplicity of infection of 50 based on optimization experiments. Transduction efficiency was confirmed by qPCR and Western blotting.
siRNA knockdown studies
Knockdown experiments were performed using transfection reagent DharmaFECT 3 (T-2003-04, Dharmacon). Briefly, on day 0 of transfection, 1 × 106 cells were plated in a 10-cm dish. On day 1 of transfection, two 500-μl aliquots of Opti-MEM were prepared in 1.7-ml microcentrifuge tubes. 6.5 μl of each siRNA was added to one tube, whereas 10 μl of transfection reagent was added to the other. Medium aliquots were mixed and incubated at room temperature for 20 min prior to dropwise application to a single 10-cm dish. Knockdown of GLI1 was achieved using two ON-TARGETplus siRNA constructs (J-003896-05-0002 and J-003896-06-0002, Dharmacon), whereas reagents used against STAT3 included two ON-TARGETplus siRNA constructs (J-003544-08-0002 and J-00354-10-0002, Dharmacon). Knockdown efficiency was confirmed by Western blotting and qPCR as described.
ChIP PCR
ChIP was conducted as described previously (5). Primers used for assays are listed in Table 5.
Table 5.
Primers used for ChIP assays
| Primer | Sequence |
|---|---|
| FLT3 GLI1 site 1 | |
| Sense | CCTCCCAAGTAGCTACGACTA |
| Antisense | GCCAAGACCTAACACACTACC |
| FLT3 GLI1 site 2 | |
| Sense | GAGTGCAGTGGTGTGATCT |
| Antisense | TGCCTGTAATCCTAGCACTTT |
| FLT3 GLI1 site 3 | |
| Sense | CTGACTGGGTCCTGAGCATC |
| Antisense | TCCCCGAACTCTGTCGTTTG |
| FLT3 STAT3 site | |
| Sense | GCCATACCCTAGGGGAAAGAATTA |
| Antisense | CGTTTGACCTCCACAAACCT |
| PIM1 GLI1 site | |
| Sense | CTCATGTCCTGCGGATCCTT |
| Antisense | TCGAGCCGGAGCATAACAAG |
| PIM1 STAT3 site | |
| Sense | CAGCATCTGGCATCACAACAC |
| Antisense | GCAAAAGCACCAGCGAATCC |
| SOCS2 GLI1 site | |
| Sense | AGAGCCTGCTTCTCTTGTAGC |
| Antisense | CCTGGGATGCACTCAACGA |
| SOCS2 STAT3 site | |
| Sense | TCAAGTATGATGCAGCCATCAGGG |
| Antisense | TGAAATGAATCTTCCAGCAACTCCT |
| SOCS3 GLI1 site 1 | |
| Sense | CTGCTGAGATGAGCAGGCAG |
| Antisense | GAATCCTGCAGGGGGAGATG |
| SOCS3 GLI1 site 2 | |
| Sense | GACTCAAGCGATCCTTCCACCTT |
| Antisense | GTGAGATTTGGACAAGACACACAG |
| SOCS3 GLI1 site 3 | |
| Sense | GTCGGCCTCCTAGAACTGCC |
| Antisense | ACTCGCAGCAGACAAAGGCT |
| SOCS3 GLI1 site 4 | |
| Sense | CCTTCCCTTTCAGCACCTCA |
| Antisense | GTCACGTTGGCACTGACGTA |
| SOCS3 STAT3 site 1 | |
| Sense | TAGCCTGAACCCCCGAAAAC |
| Antisense | CTGTCTCCGGAGCTGGAAAG |
| SOCS3 STAT3 site 2 | |
| Sense | GTCGGCCTCCTAGAACTGCC |
| Antisense | ACTCGCAGCAGAGAAAGGCT |
| SOCS3 STAT3 site 3 | |
| Sense | AGCCTTTCTCTGCTGCGAGT |
| Antisense | CCGCGCTCGCGGGTAT |
| SOCS3 STAT3 site 4 | |
| Sense | CACCTCACTCATCACCTGGG |
| Antisense | ATAAACCTCTGCCGAGAGCC |
| SOCS3 STAT3 site 5 | |
| Sense | GACTGTCGCACGTCTCCAA |
| Antisense | GGCAGTTCTAGGAGGCCGAC |
| CAPN1 GLI1 site | |
| Sense | TCAACAATTCTCCTGCCTCA |
| Antisense | GTGGTGGCTAACACCTGTAAT |
| CAPN1 STAT3 site | |
| Sense | CCTGGCAACTGCTGATCTT |
| Antisense | TCACCACCACTTTGGAAGAC |
| GADD45G GLI1 site 1 | |
| Sense | AGGGAATTACTTGTAGCAGGGC |
| Antisense | TTGCTGTAGGGACAATCGAGG |
| GADD45G GLI1 site 2 | |
| Sense | CCTGGCATGTAGGCAGATT |
| Antisense | CAAACTCCTGACCTCGTGAT |
| GADD45G GLI1 site 3 | |
| Sense | GCAAGCGCAGAATGGAAACT |
| Antisense | GGTGAAGCCTTGGAGACTGG |
| GADD45G STAT3 site 1 | |
| Sense | ATTGAGATGGGGGCTTTGGG |
| Antisense | AGCTAGCCTGGACCAGAAGA |
| GADD45G STAT3 site 2 | |
| Sense | ACTAGCGAACAAACGGGGC |
| Antisense | GGTGAGAAGCTGGCGTCTTG |
| GADD45G STAT3 site 3 | |
| Sense | CGTGTATGGTCAAACGGCAG |
| Antisense | AAGGGCTGCAAAACGCATTC |
| GADD45G STAT3 site 4 | |
| Sense | GGGATCTTCCAGAGACGGC |
| Antisense | GGTAAAAACAGGTAGCGCCAG |
| ITGB3 GLI1 site | |
| Sense | CCCAAACCTGCTTCACATCA |
| Antisense | CCTCCTTAACTTATCATACAGTAGCC |
| PTGES2 GLI1 site 1 | |
| Sense | AGGCAGGCCAATAATGGGAG |
| Antisense | AGCCTATGCTCCCCAAGGTA |
| PTGES2 GLI1 site 2 | |
| Sense | AAGATCTTCATCACCTCAAACA |
| Antisense | TATACTGGAAGAGCTCGTGT |
| PTGES2 GLI1 site 3 | |
| Sense | CTGGTCTGTAAAGTGGCTGAA |
| Antisense | GAGTGGGTTTGGAGTTGTAGG |
| PTGES2 STAT3 site 1 | |
| Sense | TTGGCACACTGTAGGCATGT |
| Antisense | CAAAGCCACCCTCTTCCTCC |
| PTGES2 STAT3 site 2 | |
| Sense | GCCTTCCAGATAGCAGTAGTC |
| Antisense | TGTCAGTCACCTCCAATGATAG |
| PTGES2 STAT3 site 3 | |
| Sense | TTCGAAGCCAACATCCCGATT |
| Antisense | TTCCTTCCAGTCCTTGCGG |
| S100A8 GLI1 site | |
| Sense | CAGGGATGTATGGCCTGACC |
| Antisense | TTTTGGGTGGGGAGGTAGGA |
| S100A8 STAT3 site 1 | |
| Sense | GACTGAGCCCTTTCCTGTAAAC |
| Antisense | CTGACCAGCAAGATCGTGAAA |
| S100A8 STAT3 site 2 | |
| Sense | CTCAGGACTCCCTCACTGGA |
| Antisense | AGCATAACAGCACCTGCCTC |
| S100A8 STAT3 site 3 | |
| Sense | AAGGGTGGATCCTTGGTTG |
| Antisense | TAAGTTTGACCCTTCCCTTATCC |
| S100A8 STAT3 site 4 | |
| Sense | GGAATGGTGAGGAGGACATTA |
| Antisense | CCATGTCCGGGAACTATTCTAAG |
| THBS1 GLI1 site 1 | |
| Sense | GACCAGCTCCCTGAAAGGTC |
| Antisense | GCAGGGATGGGAAAAGGGAA |
| THBS1 GLI1 site 2 | |
| Sense | CATTCCGGGAGATCAGCTCG |
| Antisense | AAGCATCCCGAAAAGGGACG |
| THBS1 STAT3 site 1 | |
| Sense | TCCTGCCACCGTATAGGTCA |
| Antisense | GGCTTAGCTGTGCAGACAGA |
| THBS1 STAT3 site 2 | |
| Sense | TTGGTCAAGCTTCTACCTATGC |
| Antisense | CTGTCATTGAAGTCTGGTCTCC |
Western blotting
Polyvinylidene difluoride membranes were blocked in TBS and 0.3% Tween 20 (TBST) with 5% skim milk overnight at 4 °C. Primary antibodies were prepared in a solution of TBST with 5% skin milk at a working concentration of 1:1000. Anti-rabbit secondary antibody was prepared at a working concentration of 1:5000, whereas anti-mouse secondary was at 1:3000. Antibodies used include GLI1 (C68H3) rabbit mAb (3538S, Cell Signaling), STAT3 (D3Z2G) rabbit mAb (12640S, Cell Signaling), SULF2 mouse mAb (MCA5692T, Bio-Rad), vinculin mouse mAb (05-386, Millipore), goat anti-rabbit IgG horseradish peroxidase (AP132P, Millipore), and goat anti-mouse IgG horseradish peroxidase (AP124P, Millipore).
Co-immunoprecipitation
Huh-7 and Hep3B cells were transfected with both GLI1-His and STAT3-FLAG on day 1 followed by transduction with Adv-Null or Adv-SULF2 on day 2. Cells were then harvested and lysed in 50 mm Tris, pH 7.5, 300 mm NaCl, 0.5% Nonidet P-40 with Complete inhibitor. Lysates were passed five times through a 27½-gauge needle and then diluted with 50 mm Tris, pH 7.5, 0.5% Nonidet P-40 to 150 mm NaCl. Samples were centrifuged at 17,000 × g for 10 min. Supernatants were subjected to immunoprecipitation following the Dynabeads Protein G immunoprecipitation kit protocol (Invitrogen). The following antibodies were cross-linked to Dynabeads Protein G for 1 h at room temperature: anti-GLI1 (as above), anti-STAT3 (as above), and normal goat and rabbit IgGs (Santa Cruz Biotechnology, Inc.). Dynabeads-antibody complexes and lysates were incubated overnight at 4 °C with rotation. Proteins were eluted by the addition of SDS sample buffer and incubation at 95 °C for 5 min. These eluates were subjected to Western blot analysis using primary antibodies as mentioned above.
3C PCR
Experiments were conducted as described previously (41). Briefly, single-cell suspensions of 1 × 107 cells under each of the experimental conditions described were prepared. Cross-linking with 2% formaldehyde, 10% fetal bovine serum/PBS was performed for 10 min at room temperature while tumbling. On-ice quenching was achieved by adding 1 m ice-cold glycine. After isolation of the cell pellet from the supernatant, cell lysis was conducted on ice for 10 min. Lysis buffer consisted of 10 mm Tris-HCl, pH 7.5, 10 mm NaCl, 5 mm MgCl2, 0.1 mm EGTA, 1× complete protease inhibitor (11836145001, Roche Applied Science). After centrifugation and removal of supernatant, the pellet was resuspended in restriction enzyme buffer using NEBuffer 3.1 (B7203S, NE BioLabs) or Cut Smart Buffer (B7204S, NE BioLabs) and incubated at 37 °C with added SDS to a concentration of 0.3% while shaking for 1 h. Then Triton X-100 was added to a concentration of 2%, and the reaction was incubated again for 1 h at 37 °C while shaking. Selected restriction enzyme, BsrI (R0527S, NE BioLabs) or BsrGI-HF (R3575S, NE BioLabs), was then added at 400 units/ml and incubated overnight at 37 °C while shaking. SDS was added to a concentration of 1.6% and incubated for 25 min at 65 °C while shaking. Ligation buffer was then added with Triton X-100 to a concentration of 1%, followed by incubation for 1 h at 37 °C shaking. 100 units of ligase was added and incubated for 4 h at 16 °C followed by 30 min at room temperature. 300 μg of proteinase K was then added, and the mixture was incubated at 65 °C overnight to de-cross-link. We then added 300 μg of RNase and incubated for 45 min at 37 °C. Phenol-chloroform was added and mixed vigorously. The supernatant was saved, and two washes were performed. The first wash was with 2 m sodium acetate, pH 5.6, distilled water, and ethanol followed by incubation at −80 °C for 1 h. The pellet was then washed again with 70% ethanol. The pellet was dried and resuspended in 10 mm Tris, pH 7.5.
qPCRs were set up using Prime Time Gene Expression 2× Master Mix (1055772, IDT) and the primers and TaqMan probes listed in Table 6.
Table 6.
Primers and TaqMan probes used for qPCR
| Primer/Probe | Sequence |
|---|---|
| SOCS2 experimental | |
| Sense | CAATCTCCAAAGGCCATCCTAT |
| Antisense | CATTTCCTCCTGCTCTGTTCT |
| SOCS2 control-1 R | GTTATTCTCCATGAGGAGCGAA |
| SOCS2 control-2 R | TCTATTCAGTGTGGCAGATTCA |
| SOCS2 control-3 R | GATGATCATGGTCCCTACCTTAC |
| SOCS2 probe | AATGCTCCACACTGAGCTAC |
| FLT3 experimental | |
| Sense | GAGTGCAGTGGTGTGATCT |
| Antisense | TAATTCTTTCCCCTAGGGTATGGC |
| FLT3 control-1 R | CAAGATGAGCAGAGTTCTGGAG |
| FLT3 control-2 R | TAAATGGTGGTTACCATGGGC |
| FLT3 control-3 R | CCAGTGTTTAATGGATACAGAGTTTC |
| FLT3 probe | TGCCTGTAATCCTAGCACTTT |
Statistical analysis
Results are expressed as means ± S.E. Comparisons between means were evaluated by Student's t test. A one-way analysis of variance was used for multiple comparisons. A value of p < 0.05 was considered significant.
Author contributions
R. M. C., P. A. R. D., E. J. T., C. M., F. A., R. D., R. P. G., M. D. T., S. L. S., L. L. A., S. W., M. M. P., L. R. R., and M. E. F.-Z. conceptualization; R. M. C., P. A. R. D., E. J. T., A. M. O., C. D. M., B. A. B., J. H., F. A., R. D., R. P. G., M. D. T., S. L. S., L. L. A., S. W., M. M. P., L. R. R., and M. E. F.-Z. data curation; R. M. C., P. A. R. D., E. J. T., C. M., A. M. O., C. D. M., B. A. B., J. H., F. A., R. D., R. P. G., M. D. T., S. L. S., L. L. A., S. W., M. M. P., L. R. R., and M. E. F.-Z. formal analysis; R. M. C., P. A. R. D., E. J. T., C. M., A. M. O., C. D. M., B. A. B., J. H., F. A., R. D., R. P. G., M. D. T., S. L. S., L. L. A., S. W., M. M. P., L. R. R., and M. E. F.-Z. investigation; R. M. C., E. J. T., C. D. M., B. A. B., J. H., F. A., R. D., R. P. G., M. D. T., S. L. S., L. L. A., S. W., M. M. P., L. R. R., and M. E. F.-Z. methodology; R. M. C., M. D. T., S. L. S., L. L. A., S. W., M. M. P., L. R. R., and M. E. F.-Z. writing-original draft; R. M. C., P. A. R. D., E. J. T., A. M. O., C. D. M., B. A. B., J. H., F. A., R. D., R. P. G., M. D. T., S. L. S., L. L. A., S. W., M. M. P., L. R. R., and M. E. F.-Z. writing-review and editing; F. A., R. D., R. P. G., M. D. T., S. L. S., L. L. A., S. W., M. M. P., L. R. R., and M. E. F.-Z. funding acquisition.
Supplementary Material
Acknowledgments
We thank Terry Stephenson and Emily Porcher for secretarial assistance, Drs. Jan M. van Deursen and Wei Zhou from the Mayo Clinic Transgenic and Knockout Mouse Core for help with the development of the Sulf2 transgenic animal, and Dr. Stephen Duncan for providing the TTR construct.
This work was supported by NCI, National Institutes of Health, Grant CA136526, Mayo Clinic Pancreatic SPORE P50 CA102701, and Mayo Clinic Center for Cell Signaling in Gastroenterology P30 DK84567 (to M. E. F.-Z.); NCI, National Institutes of Health, Grant CA165076 (to M. E. F.-Z. and L. R. R.), and Mayo Clinic Hepatobiliary SPORE P50 CA210964 (to L.R. R.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S5.
Data have been deposited in the GEO database under accession number GSE139374.
- SULF2
- sulfatase 2
- HCC
- hepatocellular carcinoma
- KO
- knockout
- DEN
- diethylnitrosamine
- TGF
- transforming growth factor
- JAK
- Janus kinase
- STAT
- signal transducers and activators of transcription
- TTR
- transthyretin promoter
- HET
- heterozygous
- siNT
- nontargeting siRNA
- siGLI1 and siSTAT3
- siRNA against GLI1 and STAT3, respectively
- qPCR
- quantitative PCR
- TSS
- transcription start site
- SHH
- Sonic Hedgehog
- 3C
- chromatin conformation capture
- Adv-Null
- null adenovirus
- Adv-SULF2
- SULF2 adenovirus.
References
- 1. Yang J. D., Sun Z., Hu C., Lai J., Dove R., Nakamura I., Lee J. S., Thorgeirsson S. S., Kang K. J., Chu I. S., and Roberts L. R. (2011) Sulfatase 1 and sulfatase 2 in hepatocellular carcinoma: associated signaling pathways, tumor phenotypes, and survival. Genes Chromosomes Cancer 50, 122–135 10.1002/gcc.20838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chen G., Nakamura I., Dhanasekaran R., Iguchi E., Tolosa E. J., Romecin P. A., Vera R. E., Almada L. L., Miamen A. G., Chaiteerakij R., Zhou M., Asiedu M. K., Moser C. D., Han S., Hu C., et al. (2017) Transcriptional induction of periostin by a sulfatase 2-TGFB1-SMAD signaling axis mediates tumor angiogenesis in hepatocellular carcinoma. Cancer Res. 77, 632–645 10.1158/0008-5472.CAN-15-2556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lai J. P., Thompson J. R., Sandhu D. S., and Roberts L. R. (2008) Heparin-degrading sulfatases in hepatocellular carcinoma: roles in pathogenesis and therapy targets. Future Oncol. 4, 803–814 10.2217/14796694.4.6.803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zheng X., Vittar N. B., Gai X., Fernandez-Barrena M. G., Moser C. D., Hu C., Almada L. L., McCleary-Wheeler A. L., Elsawa S. F., Vrabel A. M., Shire A. M., Comba A., Thorgeirsson S. S., Kim Y., Liu Q., Fernandez-Zapico M. E., et al. (2012) The transcription factor GLI1 mediates TGFB1 driven EMT in hepatocellular carcinoma via a SNAI1-dependent mechanism. PLoS ONE 7, e49581 10.1371/journal.pone.0049581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nye M. D., Almada L. L., Fernandez-Barrena M. G., Marks D. L., Elsawa S. F., Vrabel A., Tolosa E. J., Ellenrieder V., and Fernandez-Zapico M. E. (2014) The transcription factor GLI1 interacts with SMAD proteins to modulate transforming growth factor B-induced gene expression in a p300/CREB-binding protein-associated factor (PCAF)-dependent manner. J. Biol. Chem. 289, 15495–15506 10.1074/jbc.M113.545194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nakamura I., Fernandez-Barrena M. G., Ortiz-Ruiz M. C., Almada L. L., Hu C., Elsawa S. F., Mills L. D., Romecin P. A., Gulaid K. H., Moser C. D., Han J. J., Vrabel A., Hanse E. A., Akogyeram N. A., Albrecht J. H., et al. (2013) Activation of the transcription factor GLI1 by WNT signaling underlies the role of sulfatase 2 as a regulator of tissue regeneration. J. Biol. Chem. 288, 21389–21398 10.1074/jbc.M112.443440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zheng X., Gai X., Han S., Moser C. D., Hu C., Shire A. M., Floyd R. A., and Roberts L. R. (2013) The human sulfatase 2 inhibitor 2,4-disufonylphenyl-tert-butylnitrone (OKN-007) has an antitumor effect in hepatocellular carcinoma mediated via suppression of TGFB1/SMAD2 and Hedgehog/GLI1 signaling. Genes Chromosomes Cancer 52, 225–236 10.1002/gcc.22022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mills L. D., Zhang Y., Marler R. J., Herreros-Villanueva M., Zhang L., Almada L. L., Couch F., Wetmore C., Pasca di Magliano M., and Fernandez-Zapico M. E. (2013) Loss of the transcription factor GLI1 identifies a signaling network in the tumor microenvironment mediating KRAS oncogene-induced transformation. J. Biol. Chem. 288, 11786–11794 10.1074/jbc.M112.438846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Davey H. W., Park S. H., Grattan D. R., McLachlan M. J., and Waxman D. J. (1999) STAT5b-deficieint mice are growth hormone pulse-resistant: role of STAT5b in sex-specific liver p450 expression. J. Biol. Chem. 274, 35331–35336 10.1074/jbc.274.50.35331 [DOI] [PubMed] [Google Scholar]
- 10. Wu K., Wang B., Chen Y., Zhou J., Huang J., Hui K., Zeng J., Zhu J., Zhang K., Li L., Guo P., Wang X., Hsieh J. T., He D., and Fan J. (2015) DAB2IP regulates the chemoresistance to piarubicin and tumor recurrence of non-muscle invasive bladder cancer through STAT3/Twist1/P-glycoprotein signaling. Cell. Signal. 27, 2515–2523 10.1016/j.cellsig.2015.09.014 [DOI] [PubMed] [Google Scholar]
- 11. Yun M., Lee D., Park M. N., Kim E. O., Sohn E. J., Kwon B. M., and Kim S. H. (2015) Cinnamaldehyde derivative (CB-PIC) sensitizes chemo-resistant cancer cells to drug-induced apoptosis via suppression of MDR1 and its upstream STAT3 and AKT signaling. Cell Physiol. Biochem. 35, 1821–1830 10.1159/000373993 [DOI] [PubMed] [Google Scholar]
- 12. Bourguignon L. Y., Peyrollier K., Xia W., and Gilad E. (2008) Hyaluronan-CD44 interaction activates stem cell marker Nanog, Stat-3-mediated MDR1 gene expression, and Ankyrin-regulated multidrug efflux in breast and ovarian tumor cells. J. Biol. Chem. 283, 17635–17651 10.1074/jbc.M800109200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang Y., and Wang Q. (2013) Sunitinib reverse multidrug resistance in gastric cancer cells by modulating Stat3 and inhibiting P-gp function. Cell Biochem. Biophys. 67, 575–581 10.1007/s12013-013-9544-5 [DOI] [PubMed] [Google Scholar]
- 14. Brault L., Menter T., Obermann E. C., Knapp S., Thommen S., Schwaller J., and Tzankov A. (2012) PIM kinases are progression markers and emerging therapeutic targets in diffuse large B-cell lymphoma. Br. J. Cancer 107, 491–500 10.1038/bjc.2012.272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lin Y. W., Lee B., Liu P. S., and Wei L. N. (2016) Receptor-interacting protein 140 orchestrates the dynamics of macrophage M1/M2 polarization. J. Innate Immun. 8, 97–107 10.1159/000433539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tang A. C., Saferali A., He G., Sandford A. J., Strug L. J., and Turvey S. E. (2017) Endoplasmic reticulum stress and chemokine production in cystic fibrosis airway cells: regulation by STAT3 modulation. J. Infect. Dis. 215, 293–302 [DOI] [PubMed] [Google Scholar]
- 17. Han H., Hu J., Lau M. Y., Feng M., Petrovic L. M., and Ji C. (2013) Altered methylation and expression of ER-associated degradation factors in long-term alcohol and constitutive ER stress-induced murine hepatic tumors. Front. Genet. 4, 224 10.3389/fgene.2013.00224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang L., Yang Z., Ma A., Qu Y., Xia S., Xu D., Ge C., Qiu B., Xia Q., Li J., and Liu Y. (2014) Growth arrest and DNA damage 45G down-regulation contributes to Janus kinase/signal transducer and activator of transcription 3 activation and cellular senescence evasion in hepatocellular carcinoma. Hepatology 59, 178–189 10.1002/hep.26628 [DOI] [PubMed] [Google Scholar]
- 19. Scuto A., Kirschbaum M., Buettner R., Kujawski M., Cermak J. M., Atadja P., and Jove R. (2013) SIRT1 activation enhances HDAC inhibition-mediated upregulation of GADD45G by repressing the binding of NFkB/STAT3 complex to its promoter in malignant lymphoid cells. Cell Death Dis. 4, e635 10.1038/cddis.2013.159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lau A. H., Lai H. K., Yeung B. H., Leung S. L., Tsang S. Y., Wong Y. H., and Wise H. (2012) Prostacyclin receptor-dependent inhibition of human erythroleukemia cell differentiation is STAT3-dependent. Prostaglandins Leukot. Essent. Fatty Acids 86, 119–126 10.1016/j.plefa.2011.12.002 [DOI] [PubMed] [Google Scholar]
- 21. Tomida M., Ohtake H., Yokota T., Kobayashi Y., and Kurosumi M. (2008) Stat3 up-regulates expression of nicotinamide N-methyltransferase in human cancer cells. J. Cancer Res. Clin. Oncol. 134, 551–559 10.1007/s00432-007-0318-6 [DOI] [PubMed] [Google Scholar]
- 22. Ke J., Shen Z., Li M., Peng C., Xu P., Wang M., Zhu Y., Zhang X., and Wu D. (2018) Prostaglandin E2 triggers cytochrome P450 17a hydroxylase overexpression via signal transducer and activator of transcription 3 phosphorylation and promotes invasion in endometrial cancer. Oncol. Lett. 16, 4577–4585 10.3892/ol.2018.9165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rodriguez-Barrueco R., Yu J., Saucedo-Cuevas L. P., Olivan M., Llobet-Navas D., Putcha P., Castro V., Murga-Penas E. M., Collazo-Lorduy A., Castillo-Martin M., Alvarez M., Cordon-Cardo C., Kalinsky K., Maurer M., Califano A., and Silva J. M. (2015) Inhibition of the autocrine IL-6-JAK2-STAT3-calprotectin axis as targeted therapy for HR−/HER2+ breast cancers. Genes Dev. 29, 1631–1648 10.1101/gad.262642.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hsu K., Chung Y. M., Endoh Y., and Geczy C. L. (2014) TLR9 ligands induce S100A8 in macrophages via a STAT3-dependent pathway which requires IL-10 and PGE2. PLoS ONE 9, e103629 10.1371/journal.pone.0103629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Qin H., Buckley J. A., Li X., Liu Y., Fox T. H. 3rd, Meares G. P., Yu H., Yan Z., Harms A. S., Li Y., Standaert D. G., and Benveniste E. N. (2016) Inhibition of the JAK/STAT pathway protects against alpha-synuclein-induced neuroinflammation and dopaminergic neurodegeneration. J. Neurosci. 36, 5144–5159 10.1523/JNEUROSCI.4658-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li S. W., Wang C. Y., Jou Y. J., Yang T. C., Huang S. H., Wan L., Yin Y. J., and Lin C. W. (2016) SARS coronavirus papain-like protease induces Egr-1-dependent up-regulation of TGF-B1 via ROS/p38 MAPK/STAT3 pathway. Sci. Rep. 6, 25754 10.1038/srep25754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tyzack G. E., Sitnikov S., Barson D., Adams-Carr K. L., Lau N. K., Kwok J. C., Zhao C., Franklin R. J., Karadottir R. T., Fawcett J. W., and Lakatos A. (2014) Astrocyte response to motor neuron injury promotes structural synaptic plasticity via STAT3-regulated TSP-1 expression. Nat. Commun. 5, 4294 10.1038/ncomms5294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Parra R. G., Rohr C. O., Koile D., Perez-Castro C., and Yankilevich P. (2016) INSECT 2.0: a web-server for genome-wide cis-regulatory modules prediction. Bioinformatics 32, 1229–1231 10.1093/bioinformatics/btv726 [DOI] [PubMed] [Google Scholar]
- 29. Nault J. C., Couchy G., Balabaud C., Morcrette G., Caruso S., Blanc J. F., Bacq Y., Calderaro J., Paradis V., Ramos J., Scoazec J. Y., Gnemmi V., Sturm N., Guettier C., Fabre M., et al. (2017) Molecular classification of hepatocellular adenoma associates with risk factors, bleeding and malignant transformation. Gastroenterology 152, 880–894 10.1053/j.gastro.2016.11.042 [DOI] [PubMed] [Google Scholar]
- 30. Totoki Y., Tatsuno K., Covington K. R., Ueda H., Creighton C. J., Kato M., Tsuji S., Donehower L. A., Slagle B. L., Nakamura H., Yamamoto S., Shinbrot E., Hama N., Lehkuhl M., Hosoda F., et al. (2014) Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat. Genet. 46, 1267–1273 10.1038/ng.3126 [DOI] [PubMed] [Google Scholar]
- 31. Schulze K., Imbeaud S., Letouzé E., Alexandrov L. B., Calderaro J., Rebouissou S., Couchy G., Meiller C., Shinde J., Soysouvanh F., Calatayud A. L., Pinyol R., Pelletier L., Balabaud C., Laurent A., et al. (2015) Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 47, 505–511 10.1038/ng.3252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. The Cancer Genome Atlas Research Network. (2017) Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell 169, 1327–1341.e23 10.1016/j.cell.2017.05.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. El-Zaatari M., Kao J. Y., Tessier A., Bai L., Hayes M. M., Fontaine C., Eaton K. A., and Merchant J. L. (2013) Gli1 deletion prevents Helicobacter-induced gastric metaplasia and expansion of myeloid cell subsets. PLoS ONE 8, e58935 10.1371/journal.pone.0058935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Geng L., Lu K., Li P., Li X., Zhou X., Li Y., and Wang X. (2017) GLI1 inhibitor GANT61 exhibits antitumor efficacy in T-cell lymphoma cells through down-regulation of p-STAT3 and SOCS3. Oncotarget 8, 48701–48710 10.18632/oncotarget.9792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sirkisoon S. R., Carpenter R. L., Rimkus T., Anderson A., Harrison A., Lange A. M., Jin G., Watabe K., and Lo H. W. (2018) Interaction between STAT3 and GLI1/tGLI1 oncogenic transcription factors promotes the aggressiveness of triple-negative breast cancers and HER2-enriched breast cancer. Oncogene 37, 2502–2514 10.1038/s41388-018-0132-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Llovet J. M., Ricci S., Mazzaferro V., Hilgard P., Gane E., Blank J. F., de Oliveira A. C., Santoro A., Raoul J. L., Forner A., Schwartz M., Porta C., Zeuzem S., Bolondi L., Greten T. F., et al. (2008) Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 359, 378–390 10.1056/NEJMoa0708857 [DOI] [PubMed] [Google Scholar]
- 37. Zhu A. X., Park J. O., Ryoo B.-Y., Yen C.-J., Poon R., Pastorelli D., Blanc J. F., Chung H. C., Baron A. D., Pfiffer T. E., Okusaka T., Kubackova K., Trojan J., Sastre J., Chau I., et al. (2015) Ramucirumab versus placebo as second-line treatment in patients with advanced hepatocellular carcinoma following first-line therapy with sorafenib (REACH): a randomized, double-blind, multicenter, phase 3 trial. Lancet Oncol. 16, 859–870 10.1016/S1470-2045(15)00050-9 [DOI] [PubMed] [Google Scholar]
- 38. Bruix J., Qin S., Merle P., Granito A., Huang Y.-H., Bodoky G., Pracht M., Yokosuka O., Rosmorduc O., Breder V., Gerolami R., Masi G., Ross P. J., Song T., Bronowicki J. P., et al. (2017) Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESOURCE): a randomized, double-blind, placebo-controlled, phase 3 trial. Lancet 389, 56–66 10.1016/S0140-6736(16)32453-9 [DOI] [PubMed] [Google Scholar]
- 39. Kudo M., Finn R. S., Qin S., Han K.-H., Ikeda K., Piscaglia F., Baron A., Park J. W., Han G., Jassem J., Blanc J. F., Vogel A., Komov D., Evans T. R. J., Lopez C., et al. (2018) Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomized phase 3 non-inferiority trial. Lancet 391, 1163–1173 10.1016/S0140-6736(18)30207-1 [DOI] [PubMed] [Google Scholar]
- 40. Abou-Alfa G. K., Meyer T., Cheng A.-L., El-Khoueiry A. B., Rimassa L., Ryoo B. Y., Cicin I., Merle P., Chen Y., Park J. W., Blanc J. F., Bolondi L., Klümpen H. J., Chan S. L., Zagonel V., et al. (2018) Cabozantinib in patients with advanced and progressing hepatocellular carcinoma. N. Engl. J. Med. 379, 54–63 10.1056/NEJMoa1717002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hagège H., Klous P., Braem C., Splinter E., Dekker J., Cathala G., de Laat W., and Forné T. (2007) Quantitative analysis of chromosome conformation capture assays (3C-qPCR). Nat. Protoc. 2, 1722–1733 10.1038/nprot.2007.243 [DOI] [PubMed] [Google Scholar]
- 42. Lai J. P., Oseini A. M., Moser C. D., Yu C., Elsawa S. F., Hu C., Nakamura I., Han T., Aderca I., Isomoto H., Garrity-Park M. M., Shire A. M., Li J., Sanderson S. O., Adjei A. A., et al. (2010) The oncogenic effect of sulfatase 2 in human hepatocellular carcinoma is mediated in part by glypican 3-dependent Wnt activation. Hepatology 52, 1680–1689 10.1002/hep.23848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lai J. P., Sandhu D. S., Yu C., Han T., Moser C. D., Jackson K. K., Guerrero R. B., Aderca I., Isomoto H., Garrity-Park M. M., Zhou H., Shire A. M., Nagorney D. M., Sanderson S. O., Adjei A. A., et al. (2008) Sulfatase 2 up-regulates glypican 3, promotes fibroblast growth factor signaling, and decreases survival in hepatocellular carcinoma. Hepatology 47, 1211–1222 10.1002/hep.22202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pez F., Lopez A., Kim M., Wands J. R., Caron de Fromentel C., and Merle P. (2013) Wnt signaling and hepatocarcinogenesis: molecular targets for the development of innovative anticancer drugs. J. Hepatol. 59, 1107–1117 10.1016/j.jhep.2013.07.001 [DOI] [PubMed] [Google Scholar]
- 45. Rausa F. M., Tan Y., Zhou H., Yoo K. W., Stolz D. B., Watkins S. C., Franks R. R., Unterman T. G., and Costa R. H. (2000) Elevated levels of hepatocyte nuclear factor 3β in mouse hepatocytes influence expression of genes involved in bile acid and glucose homeostasis. Mol. Cell. Biol. 20, 8264–8282 10.1128/MCB.20.21.8264-8282.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dhanasekaran R., Nakamura I., Hu C., Chen G., Oseini A. M., Seven E. S., Miamen A. G., Moser C. D., Zhou W., van Kuppevelt T. H., van Deursen J. M., Mounajjed T., Fernandez-Zapico M. E., and Roberts L. R. (2015) Activation of the transforming growth factor-β/SMAD transcriptional pathway underlies a novel tumor-promoting role of sulfatase 1 in hepatocellular carcinoma. Hepatology 61, 1269–1283 10.1002/hep.27658 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.