

Abstract
Animal cells express heparan sulfate proteoglycans that perform many important cellular functions by way of heparan sulfate–protein interactions. The identification of membrane heparan sulfate–binding proteins is challenging because of their low abundance and the need for extensive enrichment. Here, we report a proteomics workflow for the identification and characterization of membrane-anchored and extracellular proteins that bind heparan sulfate. The technique is based on limited proteolysis of live cells in the absence of denaturation and fixation, heparin-affinity chromatography, and high-resolution LC-MS/MS, and we designate it LPHAMS. Application of LPHAMS to U937 monocytic and primary murine and human endothelial cells identified 55 plasma membrane, extracellular matrix, and soluble secreted proteins, including many previously unidentified heparin-binding proteins. The method also facilitated the mapping of the heparin-binding domains, making it possible to predict the location of the heparin-binding site. To validate the discovery feature of LPHAMS, we characterized one of the newly-discovered heparin-binding proteins, C-type lectin 14a (CLEC14A), a member of the C-type lectin family that modulates angiogenesis. We found that the C-type lectin domain of CLEC14A binds one-to-one to heparin with nanomolar affinity, and using molecular modeling and mutagenesis, we mapped its heparin-binding site. CLEC14A physically interacted with other glycosaminoglycans, including endothelial heparan sulfate and chondroitin sulfate E, but not with neutral or sialylated oligosaccharides. The LPHAMS technique should be applicable to other cells and glycans and provides a way to expand the repertoire of glycan-binding proteins for further study.
Keywords: proteomics, heparin-binding protein, heparin, heparan sulfate, endothelium, vascular biology, glycosaminoglycan, extracellular matrix, C-type lectin 14a (CLEC14A), LPHAMS
Introduction
All animal cells express heparan sulfate proteoglycans (HSPGs),2 either as transmembrane, secreted, and extracellular matrix proteins or as components of storage granules. HSPGs perform many functions in cells mediated to a large extent by the capacity of the heparan sulfate (HS) chains to interact with other proteins. HSPGs can tether and present cytokines, chemokines, growth factors, and morphogens (1). They can also act as a template to lower the concentration of growth factors required for signaling and to increase the duration of signaling. Binding of HS to proteases and protease inhibitors can lead to allosteric activation or inhibition of enzyme activity, for example in the activation of antithrombin and inhibition of Factors II and X in the coagulation cascade (2). HS also can induce oligomerization of soluble and membrane proteins (3). Notably, most angiogenic factors and vascular growth factor receptors interact with HS, emphasizing the role of HS–protein interactions in vascular development and angiogenesis (4, 5). Mice displaying undersulfated HS in endothelial tissues show altered angiogenesis in the diaphragm, due to diminished Slit-Robo signaling, and reduced tumor angiogenesis, due to dysregulated vascular endothelial cell growth factor (VEGF) and fibroblast growth factor (FGF) signaling, and altered chemokine and selectin-mediated responses to acute inflammation (6–9).
The size of the endothelial “HS-interactome,” i.e. the repertoire of heparan sulfate-binding proteins (HSBPs) on the surface and surrounding extracellular matrix of endothelial cells, is unknown in part due to technical challenges in working with membrane proteins and protein complexes. Previous attempts to elucidate the interactome using live cells and tissue extracts often enriched intracellular proteins, such as DNA- and RNA-binding proteins, which bind ligands that have charge characteristics similar to heparan sulfate. To circumvent these problems, preparative steps involving purification of plasma membranes have been applied to cells and tissues (10). Cell-surface biotinylation strategies coupled with streptavidin enrichment prior to affinity chromatography also have proven useful in studies of cultured cells (10–12). Often, the proteins identified were nuclear or cytoplasmic components. Heparan sulfate–binding membrane proteins were difficult to identify because of their low abundance and the need for detergents or prior enrichment techniques.
In this report, we describe a new simple proteomics strategy to identify plasma membrane and extracellular HSBPs that also permits the simultaneous determination of the binding domains that interact with heparin/heparan sulfate. The workflow combines Limited Proteolysis in the absence of denaturation, Heparin-Affinity chromatography, and high-resolution LC-MS/MS proteomics (LPHAMS). Application of LPHAMS to endothelial cells led to the identification of known HSBPs, including membrane receptors, secreted proteins, and extracellular matrix proteins, along with a set of previously unknown HSBPs. As a validation of LPHAMS, we characterized the heparin-binding properties of CLEC14A, a previously undocumented membrane HSBP involved in angiogenesis. This report lays the groundwork for screening and identifying not only HSBPs and their binding site, but other classes of glycan-binding proteins.
Results
Analysis of heparin-binding proteins on the surface of endothelial cells
Limited proteolysis is a powerful tool to map conformational features of proteins. By using suboptimal conditions for proteolysis (limiting enzyme, reduced temperature, and omission of reducing agents and denaturants), limited cleavage occurs at exposed hinges or loops resulting in the liberation of intact protein domains (13, 14). When applied to cells, limited proteolysis can be used to isolate and purify ectodomains of cell-surface transmembrane proteins, their subdomains, and subdomains of extracellular matrix and associated secreted proteins (15). We hypothesized that analysis of these liberated domains by chromatography on heparin-affinity resin would identify potential HSBPs and enrich for HS-binding domains, potentially indicating sites of contact between the protein and the ligand (Fig. 1A). To establish the feasibility of the approach, we treated confluent monolayers of human umbilical vein endothelial cells (HUVEC) with varying concentrations of proteinase K and chymotrypsin (see under “Experimental procedures”). The extent of proteolysis was monitored by SDS-PAGE and silver staining of released proteolytic fragments. Conditions were empirically adjusted to shift the pattern of bands on the gel from the pattern obtained for samples treated with buffer, but not to the extent that all of the material migrated as low molecular weight peptides. To enrich for HSBPs, we subjected the samples to heparin-affinity chromatography. Heparin is structurally related to HS, although it is more highly sulfated, enriched in iduronic acid, and more highly-negatively charged. Its commercial availability makes it an inexpensive surrogate for HS. Samples obtained after proteolytic digestion or mock digestions with PBS were bound to heparin-Sepharose, and weakly-bound proteins were washed out with low ionic strength buffer (0.3 m NaCl in 20 mm HEPES (pH 7.2)). Strongly-bound proteins were eluted with buffer containing 1 m NaCl. The eluted material was then analyzed by LC-MS/MS. As described above, the overall method was designated LPHAMS.
Figure 1.

Isolation and identification of heparin-binding proteins by LPHAMS. A, schematic representation of the analytical workflow. Living cells were subjected to limited proteolysis to liberate exposed domains. These domains were passed through a heparin-Sepharose column to enrich for heparin-binding domains and were subsequently analyzed by MS. B–D, alignments of peptides identified by MS to protein sequence (x axis). The colored bars represent peptides recovered in each step of heparin chromatography as indicated in the key. Domain structures for these three proteins are aligned above the peptide map, and the shaded region enclosed by broken lines indicate the known heparin binding site. E and F, frequency distribution of identified proteins (gray) in samples treated either with proteinase K or chymotrypsin and eluted from the heparin column with 1 m NaCl. Blue bars depict established heparin-binding proteins, and red bars indicate putative heparin-binding proteins.
To assess the feasibility and dynamic range of LPHAMS, HUVEC were first treated with proteinase K or chymotrypsin for 10 min at room temperature (see under “Experimental procedures”). Proteolytic fragments were collected and analyzed by the LPHAMS workflow. Proteomic characterization of the material displaying high affinity to heparin yielded numerous candidate HSBPs at 1% false discovery rate (FDR). All identified proteins were filtered based on the presence of at least two MS/MS counts for each identified peptide. Protein identifications were also filtered based on the presence of signal peptides (membrane and secreted proteins) and subcellular localization deduced via database searches and manual curation of the literature. Using these criteria, a total of 32 cell-surface or extracellular proteins were confidently identified in five independent experiments using proteinase K or chymotrypsin (Table S1). They included known HSBPs, such as thrombospondin 1 (THBS1) (16–18), hedgehog-interacting protein (HHIP) (19), and vascular endothelial growth factor receptor 1 (VEGFR1) (20). Previously unknown HSBPs were also detected, including C-type lectin domain family 14 member A (CLEC14A), tyrosine receptor phosphatase β (PTPRβ), lysyl oxidase–like protein 2 (LOXL2), transmembrane protein 132 (TMEM132A), growth/differentiation factor 15 (GDF15), adhesion G-protein–coupled receptor L2 (ADGRL2), and killer cell immunoglobulin-like receptor 3DL2 (KIR3DLR2).
We performed MS analysis at each step in the purification scheme and aligned identified peptides from known heparin-binding proteins to their respective protein sequence. Connective tissue growth factor (CTGF) is a 349-amino acid protein composed of four domains: an insulin-like growth factor–binding protein; a von Willebrand factor type C domain (VWFC); a thrombospondin type 1 repeat (TSP1); and a cystine-knot domain (CTCK) (Fig. 1B) (21). Peptides identified in the input and flow-through fractions originated from the VWFC domain, whereas peptides identified in the high-salt eluate were aligned to the heparin-binding CTCK domain (Fig. 1B). In the case of HHIP, peptides from the high-salt eluate aligned to the frizzled-like domain and two epidermal growth factor (EGF)-like domains (Fig. 1C), which is consistent with the observation that heparin binding occurs in the N-terminal frizzled-like domain (19). For THBS1, identified peptides from the input and flow-through aligned outside of the heparin-binding domain whereas peptides identified in the 0.3 m wash and high-salt eluate localized to the heparin-binding N-terminal thrombospondin domain (Fig. 1D) (17, 18). These findings show that the partial proteolysis released subdomains of accessible proteins and that fractionation of the released material by heparin-affinity chromatography enriched for subdomains that map to the heparin-binding site.
To examine the dynamic range of LPHAMS, we plotted the protein counts as a function of their normalized label-free intensities (Fig. 1, E and F). Interestingly, known HSBPs such as THBS1, HHIP, and CTGF displayed high intensities, whereas other low-abundant HSBPs, such as SDF1 and ADAMTS4, occurred at lower intensities. Many of the newly-identified, putative HSBPs also occurred at low intensities as well, which may explain why previous methods failed to detect them. Compared with chymotrypsin treatment, proteinase K treatment of HUVEC resulted in higher protein recovery based on protein intensities and consequently in more protein identifications (Fig. 1 F).
We next applied LPHAMS to mouse lung microvascular endothelial cells (MLEC), mouse brain microvascular endothelial cells, and U937 histiocytic lymphoma cells, which yielded 9, 12, and 20 HSBPs, respectively (Table 1 and Table S1). Of these proteins, unique HSBPs such as interleukin-1 receptor type 1 (IL1R1), transmembrane channel-like protein 5 (TMC5), and amyloid precursor like protein 2 (APLP2) were identified. Several proteins were shared between cell lines, whereas others were unique as might be expected given the diversity of protein expression across cell types (Table 1). In total, 55 HSBPs were identified, including 30 HSBPs not previously known to bind to heparin. About half of the proteins identified by LPHAMS were secreted soluble proteins or extracellular matrix proteins. Presumably, many of the soluble proteins were present in the extracellular matrix or bound to the cell surface, given that the cells were only gently rinsed with PBS prior to limited proteolysis. Twelve type I, one type II, two polytopic, and one glycosylphosphatidylinositol-anchored membrane proteins were identified in this way (Table 1). Thus, LPHAMS has the capacity to identify a broad range of membrane-associated and extracellular proteins, and the technique can be applied to different cell types.
Table 1.
Heparin-binding proteins identified by LPHAMS
| ID | Symbola | Protein name | Location | Cellular source | Heparin binding |
|---|---|---|---|---|---|
| Q80WJ7 | MTDHa,b | Protein LYRIC (metadherin) | Type II transmembrane | MLEC | ? |
| P13504 | IL1R1A | Interleukin-1 receptor type 1 | Type I transmembrane | BMEC | ? |
| Q86T13 | CLEC14A | C-type lectin domain family 14 member A | Type I transmembrane | HUVEC | ? |
| P43630 | KIR3DL2 | Killer cell immunoglobulin-like receptor 3DL2 | Type I transmembrane | HUVEC | ? |
| P23467–3 | PTPRB | Receptor-type tyrosine-protein phosphatase β | Type I transmembrane | HUVEC | ? |
| Q24JP5 | TMEM132A | Transmembrane protein 132A | Type I transmembrane | HUVEC | ? |
| P17948 | FLT1a | Vascular endothelial growth factor receptor 1 | Type I transmembrane | HUVEC/BMEC | Yes (20) |
| P07996 | THBS1a | Thrombospondin-1 | Type I transmembrane | HUVEC/BMEC | Yes (16–18, 79) |
| Q14956 | GPNMB | Transmembrane glycoprotein NMB | Type I transmembrane | U937 | Yes (80) |
| Q06481 | APLP2 | Amyloid-like protein 2 | Type I transmembrane | U937 | ? |
| Q92478 | CLEC2B | C-type lectin domain family 2 member B | Type I transmembrane | U937 | ? |
| P35613 | BSGa | Basigin | Type I transmembrane | U937 | ? |
| Q9NPY3 | CD93a | Complement component C1q receptor | Type I transmembrane | U937 | ? |
| Q32NZ6 | TMC5 | Transmembrane channel-like protein 5 | Polytopic membrane | BMEC | ? |
| O95490 | ADGRL2 | Adhesion G-protein-coupled receptor L2 | Polytopic membrane | HUVEC | ? |
| Q96QV1 | HHIPa | Hedgehog-interacting protein | GPI anchor | HUVEC/MLEC | Yes (19) |
| P20908 | COL5A1 | Collagen α1(V) chain | Extracellular matrix | HUVEC | ? |
| P05997 | COL5A2a | Collagen α2(V) chain | Extracellular matrix | HUVEC | ? |
| P39060 | COL18A1a | Collagen α1(XVIII) chain | Extracellular matrix | HUVEC/BMEC | Yes (32) |
| P02751 | FN1a | Fibronectin | Extracellular matrix | HUVEC/BMEC/MLEC | Yes (81) |
| P98160 | HSPG2 | Basement membrane-specific heparan sulfate proteoglycan core protein | Extracellular matrix | HUVEC/BMEC/MLEC | Yes (29) |
| P27658 | COL8A1 | Collagen α1(VIII) chain | Extracellular matrix | HUVEC/MLEC | ? |
| P01029 | C4Ba | Complement C4-B | Secreted | BMEC | ? |
| Q925I7 | PDGFD | Platelet-derived growth factor D | Secreted | BMEC | ? |
| Q9UHI8 | ADAMTS1a | A disintegrin and metalloproteinase with thrombospondin motifs 1 | Secreted | HUVEC | Yes (24, 25, 83) |
| O75173 | ADAMTS4a | A disintegrin and metalloproteinase with thrombospondin motifs 4 | Secreted | HUVEC | Yes (24, 25) |
| Q99988 | GDF15a | Growth/differentiation factor 15 | Secreted | HUVEC | ? |
| P01857 | IGHG1 | Immunoglobulin heavy constant γ1 | Secreted | HUVEC | ? |
| P04433 | IGKV3–11 | Immunoglobulin κ variable 3–11 | Secreted | HUVEC | ? |
| Q16363 | LAMA4a,b | Laminin subunit α-4 | Secreted | HUVEC | Yes (30, 31, 84) |
| Q9Y4K0 | LOXL2a | Lysyl oxidase homolog 2 | Secreted | HUVEC | ? |
| Q13201 | MMRN1 | Multimerin-1 | Secreted | HUVEC | ? |
| Q9HB63 | NTN4 | Netrin-4 | Secreted | HUVEC | ? |
| P05121 | SERPINE1 | Plasminogen activator inhibitor 1 | Secreted | HUVEC | Yes (85) |
| P50454 | SERPINH1 | Serpin H1 | Secreted | HUVEC | ? |
| Q8IWU5 | SULF2a | Extracellular sulfatase Sulf-2 | Secreted | HUVEC | Yes (86) |
| O00622 | CYR61 | CCN family member 1 | Secreted | HUVEC/BMEC | Yes (87, 88) |
| Q9H8L6 | MMRN2a | Multimerin-2 | Secreted | HUVEC/BMEC | ? |
| P9279 | CTGF | Connective tissue growth factor | Secreted | HUVEC/BMEC/MLEC | Yes (21) |
| P51858 | HDGFa | Hepatoma-derived growth factor | Secreted | HUVEC/MLEC/U937 | Yes (27, 28) |
| P07355 | ANXA2a | Annexin A2 | Secreted | HUVEC/U937 | Yes (33) |
| P14625 | HSP90B1a | Endoplasmin | Secreted | HUVEC/U937 | Yes (89–91) |
| P40224 | CXCL12 | Stromal cell-derived factor 1 | Secreted | MLEC | Yes (92) |
| Q9CXI5 | MANF | Mesencephalic astrocyte-derived neurotrophic factor | Secreted | MLEC/U937 | ? |
| P08311 | CTSGa | Cathepsin G | Secreted | U937 | Yes (93, 94) |
| P08246 | ELANE | Neutrophil elastase | Secreted | U937 | Yes (95) |
| Q07021 | C1QBP | Complement component 1 Q subcomponent-binding protein, mitochondrial | Secreted | U937 | ? |
| P06733 | ENO1 | α-Enolase | Secreted | U937 | ? |
| P61769 | B2M | β2-Microglobulin | Secreted | U937 | Yes (96) |
| P28799 | GRN | Progranulin | Secreted | U937 | Yes (97, 98) |
| P20160 | AZU1a | Azurocidin | Secreted | U937 | Yes (58) |
| P47929 | LGALS7 | Galectin-7 | Secreted | U937 | ? |
| Q9Y450 | HBS1La | HBS1-like protein | Secreted | U937 | ? |
| P07339 | CTSD | Cathepsin D | Secreted | U937 | Yes (59) |
| P08567 | PLEK | Pleckstrin | Intracellular | U937 | ? |
a Proteins with a predicted Cardin-Weintraub sequence are shown.
b Data contain a predicted turn-based motif.
LPHAMS facilitates mapping of heparin-binding domains in HSBPs
As indicated above, LPHAMS can enrich for protein subdomains that bind to heparin. Alignment of the peptides identified in the mass spectra to primary protein sequences in the UniProt database often mapped to specific subdomains in the known and putative HSBPs (Fig. 2A). For example, peptides derived from thrombospondin-1 (THBS1) were confined to the N-terminal laminin G (LamG)-like domain where the heparin-binding site was previously mapped by heparin-affinity chromatography of proteolytic fragments, molecular docking, and X-ray crystallography (Fig. 2A) (16–18). Several of the proteins (e.g. VEGFR1 (20), HHIP (19), and stromal cell–derived factor 1 (SDF1, CXCL12) (22, 23)) showed partial alignment of the recovered peptides with the putative heparin-binding domains. A disintegrin and metalloproteinase with thrombospondin repeats 4 (ADAMTS4) was identified in the screen as well, consistent with the observation that the protein can interact with heparan sulfate (24, 25). The location of the heparin-binding site in ADAMTS4 has not been established, but we found enrichment of peptides derived from a zinc-dependent metalloprotease domain, a thrombospondin type 1 repeat, and an ADAMTS spacer region, suggesting its location. In general, recovered peptides in secreted HSBPs aligned well with domains previously shown to bind heparin, for example in CTGF (21, 26), hepatic-derived growth factor (27, 28), perlecan (HSPG2) (29), and laminin α4 (LAMA4) (30, 31). In some reported HSBPs, we did not recover peptides corresponding to the location of the heparin-binding site, for example in the N-terminal domain of FN1, endostatin (the heparin-binding domain in collagen XVIII; COL18A1) (32), and annexin A2 (ANXA2) (33). However, peptides mapping to a heparin-binding site in the C-terminal domain of FN1 were detected (34).
Figure 2.

Limited proteolysis selects for ectodomains of cell-surface proteins and maps to putative sites. A and B, alignment and mass spectral counts of peptides detected by LPHAMS (black bars). Red highlighted areas represent documented heparin-binding sites, and blue highlighted areas represent putative heparin-binding sites based on LPHAMS and modeling studies. The subdomains comprising each protein are shown schematically in each panel. A, peptide alignments of known heparin-binding proteins. B, peptide alignments of previously unknown heparin-binding proteins.
We next inspected peptides derived from previously unidentified HSBPs (Fig. 2B) and examined their position in available crystal structures or in generated molecular models based on related structures to search for patches of positively-charged amino acids fitting the constraints described for heparin-binding domains (Fig. 3, A–H) (1, 35, 36). For reference, the crystal structure for THBS1 and SDF1 is shown with the electropositive surface previously documented to bind heparin (Fig. 3, A and B) (17, 22). Examination of the structure of THBS1 and SDF1, which have been co-crystallized with heparin, showed that peptide sequences retrieved by LPHAMS aligned with the heparin-binding site (17, 18, 22). For putative heparin-binding proteins, such as growth differentiation factor 15 (GDF15), LOXL2, and Netrin 4 (NTN4), we took advantage of previously existing crystal structures to search for regions of positive charge (37–39). The crystal structure of GDF15 is a dimer containing a large electropositive region (41 Å) spanning 108 amino acids of each monomer (Fig. 3C) (38). LOXL2 had peptides spanning the second to fourth scavenger receptor cysteine-rich (SRCR) domain and the lysyl oxidase-like domain (Fig. 2B). Inspection of the crystal structure (PDB 5ZE3) (39) revealed a large electropositive patch (45 × 22 Å) spanning the dimer interface of the SRCR4 domain, once again highly consistent with a putative heparin-binding site (Fig. 3D). NTN4 is an extracellular protein, consisting of lamin EGF-like domains, that functions in axon guidance (37). Inspection of the crystal structure revealed several linear stretches of positive charge spanning ∼25, 28, and 40 Å (Fig. 3E). We modeled the putative heparin-binding sites in the R-type lectin domain of PTPRβ and the C-type lectin domains of CLEC14A using Robetta (Fig. 3, F and G). PTPRβ consists of 1997 amino acids and a single ricin-like fold followed by 17 fibronectin repeats. All of the peptides recovered by LPHAMS mapped specifically to the N-terminal ricin domain (representing 5.1% of the total protein) (Fig. 2B). An area of positive charge spanning 20 × 20 Å was present in the model (Fig. 3F). In CLEC14A, recovered peptides predominantly localized to the C-type lectin and EGF domains (Fig. 2B). Interestingly, modeling of CLEC14A suggested a patch of positive charge stretching 10 × 30 Å, indicative of a putative heparin-binding site (Fig. 3G). Another C-type lectin 14 family member, CD93, also yielded peptides in its lectin domain that aligned with a putative heparin-binding site (Figs. 2B and 3H).
Figure 3.

Electropotential plots and ribbon diagrams of crystallized or modeled structures of candidate HSBPs. Blue represents regions of positive charge, and red represents regions of negative charge. A and B, reported three-dimensional structures of known heparin-binding proteins THSB1 (PDB 2OUJ) and SDF1 (PDB 2NWG). C–E, reported crystal structures of previously unknown heparin-binding proteins GDF15 (PDB 5VZ3), LOXL2 (PDB 5ZE3), and NTN4 (PDB 4WNX). F–H, structural models of protein domains of PTPRβ, CLEC14A, and CD93 enriched by heparin-affinity chromatography.
Some of the identified proteins lacked three-dimensional structures or were not of sufficient homology to previously crystallized proteins to allow molecular modeling. These proteins include transmembrane protein 132A (T132A) and the α1 and α2 chains of type V collagen (COL5A1 and COL5A2) (Fig. 2B). Interestingly, the recovered peptide sequences in type V collagen corresponded to the thrombospondin 1 TSP1/LamG domains and collagen EMF1a (COLF1) protein domains. TSP1 and LamG are protein modules known to interact with heparin (10), whereas COLF1 has not been previously associated with heparin binding.
CLEC14A binds heparan sulfate
To validate LPHAMS as a discovery tool for new HSBPs, we analyzed the glycosaminoglycan-binding properties of CLEC14A, a member of the C-type lectin family 14 (40). CLEC14A plays a role in physiological and pathological angiogenesis, but its identification as a heparin-binding protein and the structure and function of the heparin-binding domain have not been characterized (41, 42). CLEC14A is type I transmembrane protein containing a C-type lectin domain (CTLD), an EGF-module, and an endomucin domain rich in serine and threonine residues (Fig. S1). A 21-amino acid transmembrane peptide connects the ectodomain to a 71-amino acid cytoplasmic tail. C-type lectins in general bind calcium, and many bind glycans through a carbohydrate recognition domain in the CTLD. CLEC14A belongs to a subgroup of C-type lectins that includes CD93, THBS1, and CD248 (endosialin) (43).
Recombinant cDNA constructs spanning residues 1–325 (the entire ectodomain, CLEC14A-325) and residues 1–290 (the ectodomain lacking the endomucin domain, CLEC14A-290) were expressed in HEK293F cells (Fig. S1). Primary sequence analysis indicated the presence of a single consensus sequon (Asn–Leu–Ser) for N-linked glycosylation at Asn-189. SDS-PAGE before and after PNGase F digestion demonstrated that CLEC14A-325 contained an asparagine-linked glycan chain (Fig. S2A). Enzymatic deglycosylation of CLEC14–290 using PNGase F in the presence of 18O-labeled water (H218O) labeled the site of N-glycosylation (44, 45). The extent of glycosylation was estimated at ∼60% based on SDS-PAGE (Fig. S3) and the recovery of 18O-labeled peptides and peptides containing an asparagine residue at position 189 (“Experimental procedures” and Table S2, see supporting information). Glycan analysis showed that Asn-189 was occupied predominantly by either a complex-type biantennary, disialylated, and core-fucosylated glycan (HexNAc4Hex5Fuc1NeuAc) or a hybrid-type, core-fucosylated structure (HexNAc3Hex6Fuc1NeuAc1) (Fig. S2B). Removal of the N-glycans did not affect the ability of recombinant CLEC14A-325 to engage heparin (Fig. S2C).
CLEC14A contains a C-type lectin fold related to the fold present in E/L/P-selectins, which bind to glycans containing sialyl Lewis X (46). We tested whether CLEC14A could bind to sialyl Lewis X and other classes of acidic and neutral glycans through the Consortium for Functional Glycomics Protein–Glycan Interaction Core using a glycan array covering 609 different glycan structures unrelated to GAGs. CLEC14A-325 did not bind significantly to any of the glycans (Fig. S4). In contrast, surface plasmon resonance (SPR) confirmed that CLEC14A-325 bound to immobilized heparin in a dose-dependent manner (Fig. 4A). Binding of CLEC14A-290, lacking the endomucin domain, was reduced compared with CLEC14A-325 (Fig. 4, B and C). As shown below, CLEC14A-325 behaves as a trimer by size-exclusion chromatography, whereas CLEC14A-290 migrates as a monomer, most likely explaining the difference in binding of the two recombinant proteins to immobilized heparin (Fig. 4, A and B). To assess binding of different classes of GAGs, we immobilized recombinant CLEC14A-325 onto an SPR chip and tested different GAGs as the analyte (Fig. 4D and Fig. S5). Under these conditions, heparin exhibited similar binding characteristics, like the results obtained when heparin was immobilized on the chip. Chondroitin sulfate E, dermatan sulfate, and HS derived from Chinese hamster ovary cells (rHS01) also bound (Fig. 4D), whereas no binding was detected with hyaluronan (data not shown). Although the basic assumptions of single-site binding are violated due to the heterogeneous nature and valency of heparin and glycosaminoglycans, we calculated apparent binding constants using maximum response units (RU) (Fig. 4D). These data are summarized in Table 2.
Figure 4.

CLEC14A binds to several glycosaminoglycans. A, CLEC14A-325. B, CLEC14A-290 binding to immobilized heparin as measured by SPR fitted by a 1:1 site binding model (black lines). Vertical dotted lines indicate the start of the dissociation phase. C, maximum RU values for CLEC14A-325 and CLEC14A-290. D, binding curves for heparin, squid chondroitin sulfate E, dermatan sulfate, or CHO heparan sulfate to immobilized CLEC14A-325.
Table 2.
Binding values of CLEC14A-325 with glycosaminoglycans
| Analytea | KD | Bmax |
|---|---|---|
| μg/ml | RU | |
| Heparin | 0.45 ± 0.05 | 113 ± 4 |
| Chondroitin sulfate E | 0.51 ± 0.07 | 80 ± 3 |
| Dermatan sulfate | 0.53 ± 0.25 | 32 ± 6 |
| Heparan sulfate | 3.6 ± 0.2 | 28 ± 0.1 |
a The apparent KD and Bmax values were calculated from the data shown in Fig. 4D. The values for heparan sulfate include an additional data point obtained at 28 μg/ml. All data was analyzed using TraceDrawer version 1.6.1 (Ridgeview Instruments AB, Vänge, Sweden).
Binding of CLEC14A to endothelial HS was tested using 35S-labeled HS isolated from HUVEC. Samples were mixed with recombinant CLEC14A-325, and the solution was rapidly filtered through a nitrocellulose membrane. Free, uncomplexed GAGs do not bind to nitrocellulose, as demonstrated by the lack of counts bound to the filter when [35S]HS was incubated with BSA (Fig. 5) (47). In contrast, [35S]HS bound to CLEC14A-325 in a dose-dependent manner (3 μg of protein bound 10 ± 4% of the input [35S]HS counts) (Fig. 5). For comparison, 0.4 μg of FGF2, which binds to HS with high affinity (48–50), sequestered 35 ± 6% of input counts (Fig. 5).
Figure 5.
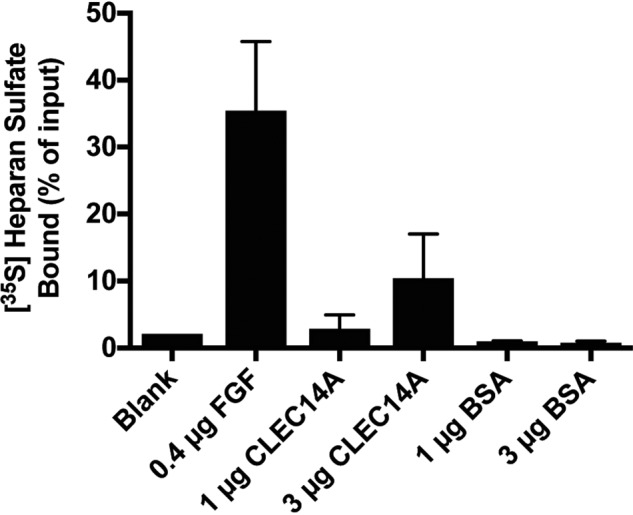
CLEC14A binds to endothelial cell heparan sulfate. Filter-binding assay of HUVEC-derived [35S]heparan sulfate to FGF2, CLEC14A-325, and BSA. The assay was performed in triplicate, and the average values ± S.D. were determined.
Heparin oligosaccharides bind to a single binding site in CLEC14A
Size-exclusion chromatography using globular protein standards showed that CLEC14A-290 ran as an ∼31-kDa monomer consistent with its predicted molecular mass of 31.6 kDa (Fig. 6A). In contrast, CLEC14A-325 migrated with an effective mass of ∼85 kDa, but the predicted molecular mass was only 35.6 kDa, suggesting that CLEC14–325 behaved as a dimer or trimer (Fig. 6B). When the experiment was repeated using multiangle light-scattering (MALS) to more accurately estimate molecular mass, CLEC14A-290 and CLEC14A-325 eluted with molecular masses of 35.4 ± 2.4 and 87.1 ± 18.1 kDa, respectively. Many C-type lectins behave as trimers, which increases their avidity for multivalent ligands (51). One function of the endomucin domain in CLEC14A may be to facilitate oligomerization, thus increasing its affinity for GAGs.
Figure 6.
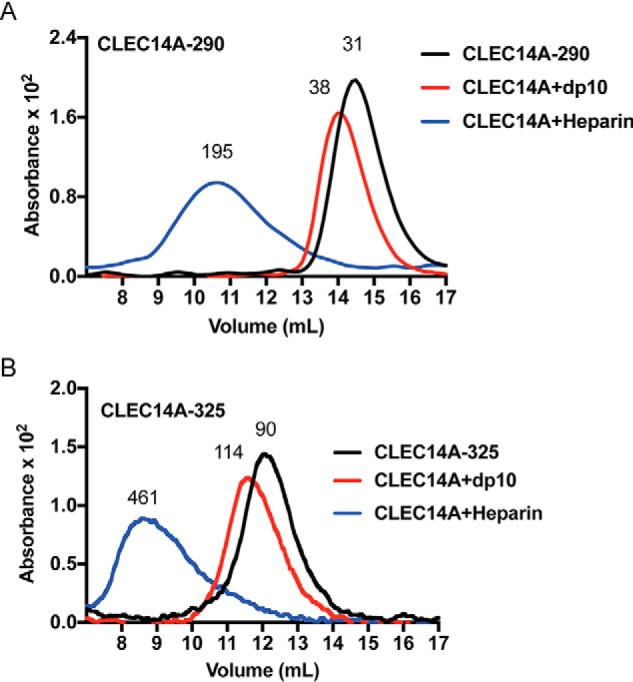
CLEC14A binds to heparin oligosaccharides. Recombinant CLEC14A-290 (A) and CLEC14A-325 (B) were analyzed by size-exclusion chromatography in the absence or presence of dp10 heparin oligosaccharides or heparin. CLEC14A-290 behaves as a monomer, and CLEC14A-325 behaves as an oligomer, most likely a trimer. Both recombinant proteins bind heparin oligosaccharides without affecting oligomerization.
As indicated above, modeling studies predicted a 22.3 × 29.2-Å electropositive surface embedded in the CTLD of CLEC14A, which could theoretically accommodate a dp10–12 heparin oligosaccharide (Fig. 3G). Incubation of CLEC14A-290 with dp10 heparin oligosaccharides shifted the elution pattern of the protein, increasing the apparent mass to ∼38 kDa (Fig. 6A). Incubation with heparin yielded a large complex of average relative mass of 195 kDa, suggesting a stoichiometry of 5:1 CLEC14A-290/heparin. The data also support the idea that CLEC14A accommodates oligosaccharides of ∼dp10 because heparin consists of a variety of chains with an average molecular mass of ∼14 kDa (∼dp50). Incubation of CLEC14A-325 oligomer with dp10 heparin oligosaccharides shifted its mass from 90 to 114 kDa; the difference of 24 kDa is close to the predicted value if CLEC14A-325 behaves like a trimer and the complex binds three dp10 oligosaccharides (Fig. 6B). Together, these findings suggest that CLEC14A monomers contain a single binding site for heparin and that these sites act independently in the trimer.
Binding of glycans to proteins can stabilize them against denaturation. To test the impact of heparin on CLEC14A stability, we examined the response of CLEC14A-325 to thermal denaturation using differential scanning fluorimetry (DSF). In this technique, denaturation is measured by binding of a hydrophobic dye to hydrophobic residues exposed by denaturation. An increase in melting temperature induced by ligand–protein binding reflects enhanced protein stability. CLEC14A-325 showed a typical biphasic melting curve, melting at 55 °C based on the first derivative spectrum. The addition of heparin increased thermostability by up to 9.8 °C (Fig. 7A). For reference, we examined the impact of heparin on stability of FGF2, a classical HSBP. The addition of heparin increased the stability of FGF2 by 33.7 ± 0.1 °C (Fig. 7A). Individual oligosaccharides dp6–18 added at a 9:1 molar ratio also increased the stability of CLEC14A-325 against thermal denaturation (Fig. 7B), with the effect reaching saturation with a dp12 oligosaccharide. Based on NMR structures for heparin oligosaccharides, a dp12 oligosaccharide has extended the length by ∼38–48 Å (52). This length corresponds well with the size of the modeled site shown in Fig. 3G. Chemically desulfating heparin at N- or C6 positions of glucosamine units or the C2 position of uronic acids reduced its ability to stabilize CLEC14A, indicating that binding involves contacts with multiple sulfate groups (Fig. 7C).
Figure 7.

Heparin stabilizes CLEC14A to thermal denaturation. CLEC14A-325 or FGF2 was thermally denatured, and the melting point (Tm) was determined by differential scanning fluorimetry (DSF). A, CLEC14A-325 (6 μm) was incubated with various concentrations of heparin to achieve the indicated molar ratios. FGF2 (24 μm) was incubated with heparin (9:1 molar ratio). B, heparin oligosaccharides (dp6–18) were incubated with CLEC-325 at a 9:1 molar ratio and analyzed by DSF. C, CLEC14A-325 was incubated with chemically de-sulfated forms of heparin. Each condition was performed in triplicate, and the data were analyzed by Prism (version 5.0).
Computational mapping of the site of CLEC14A
Because of the low-sequence homology of the lectin domain of CLEC14A with proteins available in the Protein Data Bank (53), we employed different homology modeling methods to predict 3D structural models of CLEC14A. Superimposition of the models showed that despite using different templates, the fold of the protein core is well-preserved, but the conformation of the various loops differs significantly (Fig. S6A). Molecular dynamics (MD) simulations of selected CLEC14A models in explicit solvent on the microsecond timescale confirmed that the fold of the protein core remained stable, but the loops were significantly flexible (Fig. S6B). This makes a reliable prediction of the binding site of heparin fragments using docking methods difficult. Therefore, a novel MD-based unbiased protocol was used to map the site of CLEC14A. Each predicted 3D model of CLEC14A was simulated together with a heparin fragment in explicit solvent on the microsecond timescale. In the starting assemblies, the heparin fragment was always positioned remotely, as shown in Fig. S6A. In this way any interaction with specific amino acids during the simulation was not biased by the starting orientation. The number of heavy atom contacts (<4.0 Å) between the heparin fragment and the individual amino acids was analyzed from the MD trajectories (accumulated 11.6 μs) for each frame in the corresponding model (Fig. 8, A–C). It was found that binding events occurred typically in less than 50 ns, but rearrangements in the exact binding pattern occurred even after 500 ns. From the interaction plots, it was obvious that Arg-161 reproducibly played a key role in most of the simulations, but other amino acids also contributed significantly. The statistics and a binding model derived from the simulations with the YASARA model are shown in Fig. 8.
Figure 8.

Molecular dynamics simulations of the models of the lectin domain of CLEC14A. A–C, accumulated trajectory plots of the number of heavy atom contacts (<4.0 Å) between the heparin fragment and the individual amino acids of CLEC14A. A vertical line is used to separate individual simulations. Amino acids that did not participate in any intermolecular interactions in the simulations are not shown in the plots. Red arrows indicate Arg-161. A, YASARA models; B, Swiss models; C, Robetta models. D, computational prediction of the heparin-binding site of the lectin domain of CLEC14A (YASARA model). Amino acids that participated in strong interactions with the heparin fragment during the MD simulations (accumulated 3.6 μs) are highlighted in green.
Genetic mapping of the site of CLEC14A
Arg-161 lies in a large positively-charged surface patch in the CTLD along with arginine and lysine residues Arg-141, Lys-158, and Arg-165 (Fig. 9A). To determine whether these residues are involved in binding, they were converted one–by–one to alanine residues, and recombinant protein was produced in HEK293F cells. Chromatography of the recombinant proteins on heparin-Sepharose showed that R141A, K158A, and R165A mutations had little effect on the salt concentration required for elution of the mutated proteins compared with the WT protein (488–528 mm NaCl for mutants R141A, K158A, and R165A versus 515 mm for the WT) (Fig. 9B). However, the R161A variant eluted at a much lower concentration of NaCl (373 mm) indicating impairment in its heparin-binding capacity (Fig. 9C). We validated this finding using an ELISA in which heparin was immobilized on a plate. WT CLEC14A-325 bound to immobilized heparin with an apparent affinity of ∼25 nm. In contrast, the R161A variant essentially lost its capacity to bind immobilized heparin under these conditions (Fig. 9D). Interestingly, the R161A mutant was more thermally stable compared with the WT protein, indicating that the decreased binding to heparin was not caused by unfolding of the mutant protein. As expected, heparin stabilized the WT protein to thermal denaturation, but it had a much reduced effect in the mutant (Fig. 9, E and F).
Figure 9.

Mapping the domain in CLEC14A. A, schematic representation of the CLEC14A lectin domain (CTLD, Robetta predictions). Arginine and lysine residues are shown as red stick representations on a ribbon diagram. B, elution of CLEC14A-325 mutants on heparin-Sepharose. WT CLEC14A-325 and the indicated mutants were chromatographed on heparin-Sepharose, and the concentration of NaCl required for elution was determined. C, heparin-Sepharose chromatography of WT CLEC14A-325 and mutant R161A. D, binding of CLEC14A-325 and mutant R161A to immobilized heparin. E, DSF of CLEC14A-325 and mutant R161A in the presence and absence of heparin. F, ΔTm (°C) of WT and mutant CLEC14A induced by heparin.
Discussion
In this report we describe the development and application of LPHAMS, a proteomic workflow integrating limited proteolysis, heparin-affinity chromatography, and high-resolution LC-MS/MS. Application of LPHAMS to human and murine endothelial cells led to the identification of large number of HSBPs, and in many examples, the method revealed the subdomains that facilitate binding to HS. We identified the endothelial transmembrane protein CLEC14A as a novel glycosaminoglycan-binding protein. CLEC14A, a C-type lectin, most likely exists as a trimer and does not bind typical N- and O-linked glycans but instead binds to glycosaminoglycans. In practice, LPHAMS is simple, does not require pre-fractionation methods or detergents, and can be applied to a variety of cell types. Types I and II, polytopic membrane proteins, as well as extracellular matrix and secreted proteins were discovered using this method.
Many investigators have used affinity chromatography coupled with MS to identify heparin-binding proteins, but typically, the source material consisted of a body fluid such as blood, serum, or cerebrospinal fluid (16, 26, 48, 49, 54–56). This approach led to the purification of soluble growth factors, plasma proteins of the coagulation cascade and complement systems, and RNA- and DNA-binding proteins, but few membrane proteins were identified. To enrich for membrane proteins, a technique was devised that involved isolation of a plasma membrane fraction, for example from liver (10, 57). Solubilization of the membranes and affinity purification of the material over heparin-Sepharose led to the identification of 148 HSBPs, including 79 membrane proteins. Although effective, this strategy involves homogenization of tissues, purification of plasma membranes, and detergents for solubilization of otherwise insoluble membrane proteins. Another approach employed cell-surface biotinylation of cultured cells, enrichment by streptavidin-affinity chromatography, and fractionation by heparin-Sepharose chromatography (10–12). Many HSBPs were discovered in this way, but few membrane proteins were enriched possibly because of their low abundance or limited access to the tagging reagents.
LPHAMS takes advantage of suboptimal proteolysis to selectively cleave proteins at exposed protein hinges or loops, and the method can be easily modified depending on the ultimate targets. When applied to cells, specific enrichment for protein ectodomains of cell-surface proteins occurred leading to the identification of previously unidentified heparin-binding proteins. One limitation of this method is the requirement that target proteins have accessible protease cleavage sites, but the use of a broad-spectrum serine protease (proteinase K) reduces this potential problem. In contrast, the use of a broad-spectrum protease could easily lead to the under-representation of certain proteins due to undesirable proteolysis. To mitigate this limitation, adjustments can be made to the duration of proteolytic treatment, concentration of the protease, the type of protease, and temperature. Other GAGs or glycans could be used as the affinity matrix as well.
LPHAMS also has the advantage of providing information about putative glycosaminoglycan-binding sites in the heparin-binding proteins. These predictions were often consistent with prior mapping studies in which the site of interaction had been deduced by crystallography, NMR, or modeling. Several of the binding sites in the HSBPs identified in this study mapped to larger domains that presumably depend on folding and approximation of subdomains to generate the positively-charged surfaces with affinity for heparin. Of the 55 identified proteins, 26 had at least one so-called Cardin-Weintraub sequence (Table 1): -XBBXBX- or -XBBBXXBX, where X is a hydrophilic amino acid and B is a basic amino acid (35). However, these Cardin-Weintraub sequences were not necessarily predictive, because many mapped outside of the established heparin-binding site or to domains not exposed at the cell surface. When examining secondary sequences for a heparin-binding turn based motifs (TXXBXXTBXXXTBB, where T denotes a turning amino acid (36)), only four proteins were predicted to contain a putative heparin-binding site.
Like any proteomic technique, LPHAMS may not detect all candidate proteins or subdomains fulfilling the criterion of binding heparin. For example, peptides from the heparin-binding domain in COL18A1 and ANXA2 were not recovered possibly because of proteolytic fragmentation or weak affinity for heparin. Although nearly all known heparin–protein interactions are driven by electrostatics, proteins that bind heparin via polar interactions and/or nonionic interactions may have been missed (60). Conceivably, some of the proteins identified by LPHAMS might not actually bind to heparin, but instead may form a complex with a bona fide heparin-binding protein, which then led to its co-purification. However, these interactions are of interest as well because they define possible complexes and biological functions.
In this study, we show for the first time that CLEC14A binds to GAGs and not to carbohydrates that typically associate with C-type lectins. CLEC14A is most likely a trimeric protein, typical of many C-type lectins. Binding to heparin oligosaccharides appeared to occur independently in each monomer. Interestingly, the heparin-binding site in CLEC14A does not map to the site typically associated with the carbohydrate recognition domain of this lectin subfamily (43), and binding does not depend on calcium. These observations suggest that the ability to bind heparin and other GAGs evolved independently of the C-type lectin fold. CLEC14A is an endothelial-specific gene up-regulated during tumor angiogenesis and regulates endothelial cell migration and adhesion in vitro and angiogenesis in vivo (41, 42, 61–67). The C-type lectin domain has been shown to engage other matrix proteins, such as multimerin 2 (MMRN2) and heat-shock protein 70-1A (61, 62). Antibodies blocking these interactions or targeting the C-type lectin domain have been shown to decrease cell migration and tumor angiogenesis in a manner dependent on MMRN2 (42, 62) or VEGFA (68). Whether these antibodies block the GAG-binding site is not known.
CLEC14A bound to specific glycosaminoglycans with fast on-rates and undetectable off-rates. Heparin and chondroitin sulfate E have high charge density and bound more avidly compared with heparan sulfate, chondroitin sulfate A, and dermatan sulfate, which have lower charge density (Fig. 4). There was no detectable binding to hyaluronan. CLEC14A also binds to heparan sulfate from HUVEC, but less so compared with FGF2 (Fig. 5). These findings suggest that CLEC14A prefers highly-charged polysaccharides, consistent with dramatically reduced binding to desulfated forms of heparin (Fig. 7). Although it is possible that CLEC14A prefers a specific arrangement of sulfated residues in the ligand, we think its most likely that the overall charge determines the affinity of the interaction, which would suggest that the GAG-binding site is somewhat promiscuous. The combination of unbiased molecular dynamics simulations to homology models and alanine mutagenesis indicates that binding of heparin to CLEC14A is highly dependent on Arg-161 (Figs. 8 and 9). Although we are confident in the location of the CLEC14A–heparin-binding pocket, we point out that the binding modes are approximate because it is not feasible to reach full convergence in the binding simulations. Attempts to crystallize the complex are currently underway.
In summary, we describe a facile method for discovery of heparin and other GAG-binding proteins using limited proteolysis. The technique can be readily adapted to other cell types by altering proteolytic conditions and in theory can be used to identify proteins that interact with other carbohydrate ligands by variation of the affinity matrix. The technique also aids in mapping the ligand-binding site. Finally, the characterization of CLEC14A as a heparin-binding protein validates the technology and suggests further studies of the function of CLEC14A–GAG complexes in vivo.
Experimental procedures
Limited proteolysis proteomics screening
Confluent HUVEC, grown in 100-mm diameter dishes in EGM-2 medium (Lonza), were washed twice with 5 ml of M199 medium (Life Technologies, Inc.). After testing different conditions, we typically treated cells with proteinase K (250 ng/ml) or chymotrypsin (1 μg/ml) in M199 medium for 10 min at room temperature. The supernatant was collected, centrifuged at 400 × g to remove cellular debris, and then placed on ice. The samples were applied to a 1-ml HiTrap heparin-Sepharose column (GE Healthcare) equilibrated in 150 mm NaCl in 25 mm HEPES buffer (pH 7.1). Columns were washed with 0.3 m NaCl in 25 mm HEPES buffer to remove low-affinity binding proteins and step-eluted with 1 m NaCl in 25 mm HEPES buffer. An in-solution digestion was performed on proteins in these fractions with mass spectrometry grade trypsin gold (Promega) at 37 °C. Peptides were desalted using C18 Tips (Pierce) and dried using a speed-vacuum centrifuge. Murine brain (mBMEC) and lung (mLEC) microvascular endothelial cells were isolated as described (6) and were cultured on gelatin (Sigma) in Dulbecco's modified Eagle's medium (DMEM; Lonza) containing 20% (v/v) fetal bovine serum (Atlanta Biologicals), heparin (100 μg/ml), and endothelial cell growth supplement (50 μg/ml; VWR International). Confluent mBMEC and mLEC were washed with serum-free DMEM and then treated with proteinase K (250 ng/ml) for 15 min. U937 cells were cultured in Roswell Park Memorial Institute 1640 (RPMI) medium containing 10% FBS (v/v). The cells were centrifuged, washed three times with PBS, and then digested with proteinase K (500 ng/ml) or trypsin (1 μg/ml) in PBS for 45 min with rotation. The supernatants from digestions of mBMEC, mLEC, and U937 cells underwent the same workflow for heparin purification, trypsinization and MS as HUVEC.
Liquid chromatography–mass spectrometry
Tryptic peptides were analyzed by ultra-HPLC (UPLC) coupled with tandem MS (LC-MS/MS) using nano-spray ionization. The experiments were performed using a TripleTOF 5600 hybrid mass spectrometer (ABSCIEX) interfaced with nano-scale reversed-phase UPLC (Waters®, nanoACQUITY) using a 20-cm × 75-μm, inner diameter, glass capillary packed with 2.5-μm C18 (130) CSHTM beads (Waters). Peptides were eluted from the C18 column using a linear gradient (5–80%) of acetonitrile at a flow rate of 250 μl/min for 1 h. Buffer A is 98% water, 2% acetonitrile, 0.1% formic acid, and 0.005% TFA, and buffer B is 100% acetonitrile, 0.1% formic acid, and 0.005% TFA. MS/MS data were acquired in a data-dependent manner. MS1 data were acquired for 250 ms at m/z of 400–1250 Da, and the MS/MS data were acquired from m/z of 50–2000 Da. MS1-TOF acquisition time was 250 ms, followed by 50 MS2 events with a 48-ms acquisition time for each event. The threshold to trigger the MS2 event was set to 150 counts when the ion had the charge state of +2, +3, and +4. The ion-exclusion time was set to 4 s.
In a few cases, the samples were also run on a Proxeon EASY nanoLC system (Thermo Fisher Scientific) coupled to a Q-Exactive Plus mass spectrometer (Thermo Fisher Scientific). Dried peptides were reconstituted with 2% acetonitrile, 0.1% formic acid, and quantified by modified BCA peptide assay (Thermo Fisher Scientific). Equal peptide amounts derived from each sample were injected and analyzed by LC-MS/MS using a Proxeon EASY nanoLC system (Thermo Fisher Scientific) coupled to a Q-Exactive Plus mass spectrometer (Thermo Fisher Scientific). Peptides were separated using an analytical C18 Acclaim PepMap column (0.075 × 500 mm, 2 μm; Thermo Fisher Scientific) equilibrated with buffer A (0.1% formic acid in water) and eluted in a 93-min linear gradient of 2–28% solvent B (100% acetonitrile) at a flow rate of 300 nl/min. The mass spectrometer was operated in positive data-dependent acquisition mode. MS1 spectra were measured with a resolution of 70,000, an automated gain control (AGC) target of 1e6, and a mass range from 350 to 1700 m/z. Up to 12 MS2 spectra per duty cycle were triggered, fragmented by higher-energy collisional dissociation, and acquired with a resolution of 17,500 and an AGC target of 5e4, an isolation window of 1.6 m/z, and a normalized collision energy of 25. Dynamic exclusion was enabled with duration of 20 s.
Protein and peptide identification and analysis
Bioinformatics and statistical analysis of proteomics results were conducted in the Perseus statistical suite (version 1.6.5.0) (69) and the MaxQuant computational platform. Raw data were searched by the Andromeda search engine (70) against the mouse UniProt FASTA database (17,009 entries, downloaded June 2, 2017), human Uniprot database (20,129 entries, downloaded June 3, 2017), and against a common contaminant database. Search parameters were set as follows: enzyme, trypsin with up to two potentially missed cleavages; fixed modifications, carbamidomethyl on cysteines; variable modifications, oxidation of methionine and acetylation of protein N terminus; minimum peptide length, 7 amino acids. The FDR for both peptide and protein identifications was set to 1% and was calculated by searching the MS/MS data against a reversed decoy database. Allowed mass deviation for precursor ions was set to 5 ppm and for peptide fragments was set to 20 ppm. Peptides having at least 95% confidence with multiple modified and cleaved states of the same underlying peptide sequence were considered distinct. Multiple spectra of the same peptide due to replicate acquisition or different charge states were counted once. Inclusion criteria required detection of ≥2 MS/MS counts in a given scan and/or detection in multiple samples. Several of the proteins from the U937 runs had low MS/MS counts, but three of these were established HSBPs or were detected in endothelial cells. The actual spectral counts and intensities for all the protein identifications are provided in Tables S1 and S3.
For peptide alignments, the number of distinct peptides identified from all limited proteolysis MS experiments was mapped to their respective UniProt protein sequence by using the multiple sequence alignment tool Clustal Omega (71, 72).
Expression and purification of CLEC14A
Mammalian codon-optimized DNA (Genewiz) encoding human CLEC14A-290 (residues 1–290) and CLEC14A-325 (residues 1–325) was cloned into pcDNA3.1A(+) (Invitrogen) with a C-terminal His6 tag. CLEC14A mutants were constructed by site-directed mutagenesis using QuikChange DNA mutagenesis kits (Agilent) and confirmed by sequencing (Genewiz). To produce recombinant protein, HEK293F cells (1.5–2 × 106 cells/ml) were transfected with 2.5 μg/ml plasmid DNA using polyethyleneimine (9 μg/ml) and cultured in FreeStyle 293 Expression Medium (Gibco). One day later, the cells were treated with 2 mm valproic acid, and 5 days after the initial transfection, the conditioned medium was mixed with cOmplete EDTA-free protease inhibitor (Roche Applied Science) and adjusted to 30 mm imidazole, 0.5 m NaCl, and 20 mm Tris-HCl (pH 7.4). Recombinant protein was purified by chromatography on a Ni2+-Sepharose 6 Fast Flow column (1 ml, GE Healthcare). The columns were washed with 30 mm imidazole, 0.5 m NaCl, 20 mm Tris buffer (pH 7.4), and recombinant protein was eluted using 0.5 m NaCl, 0.3 m imidazole in 20 mm Tris buffer (pH 7.4). The protein was further purified by size-exclusion chromatography (HiLoad 16/60 Superdex 200, preparation grade, GE Healthcare) in 0.3 m NaCl, 5% glycerol in 20 mm Tris buffer (pH 7.4). Mutant forms of CLEC14A were purified in the same manner as WT CLEC14A.
CLEC14A N-glycan site mapping and glycopeptide analysis
For studies involving N-linked glycans, CLEC14A was treated with PNGase F (New England Biolabs) for 16 h at 37 °C under nondenaturing conditions. Details are provided in the supporting information.
Heparin ELISA
Porcine mucosal heparin (SPL Scientific Protein Laboratories) was immobilized (50 μl at 1 mg/ml) on a 96-well Carbobind plate (Corning) in 0.1 m sodium acetate buffer (pH 5.5) for 2 h at room temperature. Wells were washed with PBST, blocked with 1% BSA (Sigma) for 2 h at 37 °C, and washed again with PBST. The wells were incubated with the indicated concentrations of recombinant CLEC14A at room temperature for 1 h. Bound protein was quantitated using THETM His tag antibody (GenScript) and anti-mouse horseradish peroxidase (Cell Signaling). The Kd value was calculated by fitting the binding data to a single-site binding model (Prism 8).
Endothelial heparan sulfate purification
HUVEC were grown on gelatin in EGM-2 medium until confluent. The cells were radiolabeled with 20 μCi of 35SO4 (9.25–37.0 GBq/mmol; PerkinElmer Life Sciences) in 5 ml of F12 medium (Gibco) supplemented with 10% FBS depleted of glycosaminoglycans by chromatography over DEAE-Sepharose. The culture medium was collected after 24 h, and the cell layer was treated with trypsin for 10 min at 37 °C. The trypsin solution was collected and centrifuged to remove cell debris. Secreted proteoglycans in the growth medium and cell surface were pooled and digested with 0.4 mg/ml Pronase (Sigma) overnight at 37 °C. Samples were diluted with 2 volumes of wash buffer and purified by anion-exchange chromatography. Briefly, columns were prepared by washing 1 ml of 50% slurry of DEAE-Sepharose beads (GE Healthcare) with 50 mm sodium acetate, 0.2 m NaCl, 0.1% Triton X-100 (pH 6.0). After applying the sample, the columns were rinsed with wash buffer, and proteoglycans and glycosaminoglycans were eluted with 2.5 ml of 50 mm sodium acetate buffer containing 2 m NaCl (pH 6.0). Samples were then desalted on a PD-10 column (GE Healthcare) equilibrated in 10% ethanol. Samples were lyophilized and resuspended in 50 mm Tris buffer containing 50 mm NaCl and 25 mm MgCl2 (pH 8.0). To remove DNA and chondroitin sulfate, samples were treated with 20 kilounits/ml DNase I (Sigma) and 20 milliunits of chondroitinase ABC (Amsbio) for 3 h at 37 °C. To liberate the [35S]heparan sulfate chains from residual peptides, the samples were β-eliminated with 0.4 m NaOH overnight at 4 °C. The heparan [35S]sulfate was then re-purified by anion-exchange chromatography and desalted.
Nitrocellulose filter binding assay
Recombinant CLEC14A, BSA, or FGF2 (PeproTech) were incubated with 10,000 counts of heparan [35S]sulfate for 30 min at room temperature. Samples were added to prewashed nitrocellulose membranes on a vacuum apparatus and rapidly filtered. The filters were added to 5 ml of Ultima Gold XR (PerkinElmer Life Sciences) scintillation fluid and counted by liquid scintillation.
Surface plasmon resonance
A Nicoya OpenSPR was used to generate binding curves for CLEC14A interaction with heparin (SPL Laboratories), porcine intestinal mucosal dermatan sulfate (Celsus Laboratories), chondroitin sulfate E (Sigma), Chinese hamster ovary cell heparan sulfate (rHS01, TEGA Therapeutics, Inc.), and umbilical cord hyaluronan (Sigma). Protein was immobilized on a Nicoya carboxyl sensor using amine coupling kit (Nicoya). Carboxyl sensors were functionalized using 0.2 ml of a 1:1 mix of N-hydroxysuccinimide (0.1 m) and 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (0.4 m) before coupling to recombinant CLEC14A under flow conditions. Ethanolamine was used to block remaining active sites on the chip. In other experiments, biotinylated heparin (Sigma) was immobilized onto a Nicoya Streptavidin Sensor chip. All surfaces were washed with SPR buffer (20 mm HEPES, 150 mm NaCl, 5 mm CaCl2, 17 mm NaN3, 5 mm MgCl2, 0.1% BSA, and 0.05% Tween 20 (pH 7.2)) and regenerated with 20 mm HEPES buffer (pH 7.2) containing 3 m NaCl. Ligands were allowed to associate with the chip at a flow rate of 20 μl/min in SPR buffer for 4 min and allowed to dissociate for 5 min. Regeneration buffer was used before each injection of ligand to clean the surface chip.
Analytical size-exclusion chromatography
Purified CLEC14A (150–200 μg) and size-defined heparin-derived oligosaccharides (Iduron) were incubated in 20 mm Tris buffer (pH 7.5) containing 0.2 m NaCl. Complexes were resolved on a Superdex200 column (size 10/300, GE Healthcare). The column was calibrated with gel-filtration standards (Bio-Rad) consisting of thyroglobulin (669 kDa), bovine γ-globulin (158 kDa), chicken ovalbumin (44 kDa), equine myoglobin (17 kDa), and vitamin B12 (1.35 kDa). For accurate molecular mass determinations, CLEC14A was resolved on a Superdex200 column (size 10/300), and the eluate was passed in-line to a miniDAWN TREOS MALS detector followed by an Optilab T-rEX refractive index detector (Wyatt Technology).
Analytical heparin-Sepharose chromatography
CLEC14A was applied to a 1-ml HiTrap heparin-Sepharose column (GE Healthcare) in PBS. Protein was eluted with a gradient of NaCl from 150 mm to 1 m.
Differential scanning fluorimetry
CLEC14A (6 μm) or FGF2 (24 μm) was incubated with 5X SYPRO Orange Protein Gel Stain (Thermo Fisher Scientific) in PBS. CLEC14A thermal denaturation was monitored on a CFx96 real-time PCR system (Bio-Rad) using a temperature gradient from 25 to 98 °C (1 °C/min). Heparin and heparin derivatives (Iduron) were added to the solution at a final concentration of 48 μm. Melting temperatures were calculated using first derivatives of the data assuming a Gaussian distribution (Prism 8).
Protein structures and molecular modeling
A model of the RICIN domain (residues 17–130) of a PTPRβ isoform was generated using the full-chain protein structure prediction server Robetta with the RosettaCM protocol (73). CD93 (residues 37–241) models were generated using Phyre2 (74). Because the sequence homology between the lectin domain (residues 33–173) of human CLEC14A and protein structures available in the PDB is rather low (e.g. <20% for CLEC4C, see PDB code 3wbq), three different homology modeling methods were used for generating 3D models of CLEC14A: Robetta (five models, template PDB code 3ALS); SwissModel (75) (three models based on templates PDB codes 1GZ2, 5K8Y, and 5AO6); and YASARA (76) (one model, template PDB code 4MTH). A 3D model of a heptasaccharide heparin fragment with sequence GlcNS6S-IdoUA2S(2SO)-GlcNS6S-IdoA2S (2SO)-GlcNS6S-IdoA2S (2SO)-GlcNS6S-OMe was built using the GAG builder (77). Molecular dynamics simulations were set up and performed in 0.9% saline solution (TIP3P water, AMBER15 force field, PBC box size 61 × 61 × 61 Å) using YASARA at 310 K (NPT ensemble) (78). To investigate the stability of selected models, 1-μs MD simulations of the apoprotein were performed and the per-residue RMSDs were analyzed. The dynamics of complex formation were sampled for each CLEC14A model in the presence of one heparin molecule for at least 1 μs leading to an accumulated simulated timescale of 11.6 μs. Analysis of the MD trajectories was performed using Conformational Analysis Tools (www.md-simulations.de/CAT/). Electrostatic potential maps were generated from TSP1 (PDB code 2OUJ), SDF1 (PDB code 2NWG), GDF15 (PDB code 5VZ3), LOXL2 (PDB code 5ZE3), and NTN4 (PDB code 4WNX) crystal structures (18, 22, 37–39). Structures were visualized using PyMOL (version 2.0.6 by Schrödinger).
Author contributions
D. R. S., D. X., and J. D. E. conceptualization; D. R. S. and A. G. T. data curation; D. R. S., M. O. S., and M. M. F. formal analysis; D. R. S., C. D. P., E. M. T., M. O. S., A. M. W., and M. M. F. investigation; D. R. S. and A. G. T. visualization; D. R. S. and M. O. S. methodology; D. R. S. writing-original draft; D. R. S., A. G. T., and J. D. E. writing-review and editing; L. W., R. B., K. D. C., and J. D. E. resources; L. W., D. X., K. D. C., and J. D. E. supervision; J. D. E. funding acquisition.
Supplementary Material
Acknowledgments
We thank Dr. David Smith (Emory University) for analysis of CLEC14A binding to the CFG glycan array, Benjamin Kellman for assistance on heparin-binding sequence analysis, the University of California at San Diego Biomolecular and Proteomics Mass Spectrometry Facility S10 OD021724, and the Sanford-Burnham-Prebys Proteomics core.
This work was supported in part by Program of Excellence in Glycoscience Grants P01 HL107150 and P01 HL131474 from the National Institutes of Health (to J. D. E.), a supplement to Grant P01 HL131474 (to D.R.S.), and National Institutes of Health Grants R01 AR070179 (to D. X.), R01 GM104141 (to K. D. C.), and P41 GM103490 (to L. W.). The University of California, San Diego, and J. D. E. have a financial interest in TEGA Therapeutics, Inc. J. D. E. and the University of California, San Diego, may financially benefit from this interest if the company is successful in marketing its products (recombinant heparan sulfate) that are related to this research. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Mass spectrometry data has been uploaded to MassIVE MSV000084782 and may be accessed online.
This article contains Figs. S1–S6 and Tables S1–S3.
- HSPG
- heparan sulfate proteoglycan
- HS
- heparan sulfate
- CTLD
- C-type lectin domain
- DSF
- differential scanning fluorimetry
- HSBP
- heparan sulfate-binding protein
- PDB
- Protein Data Bank
- SPR
- surface plasmon resonance
- RU
- response unit
- HUVEC
- human umbilical vein endothelial cell
- MLEC
- mouse lung microvascular endothelial cell
- MD
- molecular dynamics
- DMEM
- Dulbecco's modified Eagle's medium
- FBS
- fetal bovine serum
- FGF
- fibroblast growth factor
- VEGF
- vascular endothelial cell growth factor
- EGF
- epidermal growth factor
- AGC
- automated gain control
- PTPRβ
- tyrosine receptor phosphatase β
- LamG
- laminin G
- CTCK
- cystine-knot domain
- VWFC
- von Willebrand factor type C domain
- CTGF
- connective tissue growth factor
- MALS
- multiangle light scattering
- mBMEC
- murine brain microvascular endothelial cell
- PNGase F
- peptide:N-glycosidase F
- RMSD
- root mean square deviation
- UPLC
- ultra-HPLC
- FDR
- false discovery rate
- HHIP
- hedgehog-interacting protein
- GAG
- glycosaminoglycan.
References
- 1. Xu D., and Esko J. D. (2014) Demystifying heparan sulfate–protein interactions. Annu. Rev. Biochem. 83, 129–157 10.1146/annurev-biochem-060713-035314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Björk I., and Lindahl U. (1982) Mechanism of the anticoagulant action of heparin. Mol. Cell. Biochem. 48, 161–182 10.1007/BF00421226 [DOI] [PubMed] [Google Scholar]
- 3. Xu D., Young J. H., Krahn J. M., Song D., Corbett K. D., Chazin W. J., Pedersen L. C., and Esko J. D. (2013) Stable RAGE-heparan sulfate complexes are essential for signal transduction. ACS Chem. Biol. 8, 1611–1620 10.1021/cb4001553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chiodelli P., Bugatti A., Urbinati C., and Rusnati M. (2015) Heparin/heparan sulfate proteoglycans glycomic interactome in angiogenesis: biological implications and therapeutical use. Molecules 20, 6342–6388 10.3390/molecules20046342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. van Wijk X. M., and van Kuppevelt T. H. (2014) Heparan sulfate in angiogenesis: a target for therapy. Angiogenesis 17, 443–462 10.1007/s10456-013-9401-6 [DOI] [PubMed] [Google Scholar]
- 6. Fuster M. M., Wang L., Castagnola J., Sikora L., Reddi K., Lee P. H., Radek K. A., Schuksz M., Bishop J. R., Gallo R. L., Sriramarao P., and Esko J. D. (2007) Genetic alteration of endothelial heparan sulfate selectively inhibits tumor angiogenesis. J. Cell Biol. 177, 539–549 10.1083/jcb.200610086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang L., Fuster M., Sriramarao P., and Esko J. D. (2005) Endothelial heparan sulfate deficiency impairs L-selectin- and chemokine-mediated neutrophil trafficking during inflammatory responses. Nat. Immunol. 6, 902–910 10.1038/ni1233 [DOI] [PubMed] [Google Scholar]
- 8. Axelsson J., Xu D., Kang B. N., Nussbacher J. K., Handel T. M., Ley K., Sriramarao P., and Esko J. D. (2012) Inactivation of heparan sulfate 2-O-sulfotransferase accentuates neutrophil infiltration during acute inflammation in mice. Blood 120, 1742–1751 10.1182/blood-2012-03-417139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang B., Xiao W., Qiu H., Zhang F., Moniz H. A., Jaworski A., Condac E., Gutierrez-Sanchez G., Heiss C., Clugston R. D., Azadi P., Greer J. J., Bergmann C., Moremen K. W., Li D., et al. (2014) Heparan sulfate deficiency disrupts developmental angiogenesis and causes congenital diaphragmatic hernia. J. Clin. Invest. 124, 209–221 10.1172/JCI71090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ori A., Wilkinson M. C., and Fernig D. G. (2011) A systems biology approach for the investigation of the heparin/heparan sulfate interactome. J. Biol. Chem. 286, 19892–19904 10.1074/jbc.M111.228114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xu D., Young J., Song D., and Esko J. D. (2011) Heparan sulfate is essential for high mobility group protein 1 (HMGB1) signaling by the receptor for advanced glycation end products (RAGE). J. Biol. Chem. 286, 41736–41744 10.1074/jbc.M111.299685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Joo E. J., ten Dam G. B., van Kuppevelt T. H., Toida T., Linhardt R. J., and Kim Y. S. (2005) Nucleolin: acharan sulfate-binding protein on the surface of cancer cells. Glycobiology 15, 1–9 10.1093/glycob/cwh132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hubbard S. J. (1998) The structural aspects of limited proteolysis of native proteins. Biochim. Biophys. Acta 1382, 191–206 10.1016/S0167-4838(97)00175-1 [DOI] [PubMed] [Google Scholar]
- 14. Fontana A., de Laureto P. P., Spolaore B., Frare E., Picotti P., and Zambonin M. (2004) Probing protein structure by limited proteolysis. Acta Biochim. Pol. 51, 299–321 10.18388/abp.2004_3573 [DOI] [PubMed] [Google Scholar]
- 15. Olaya-Abril A., Jiménez-Munguía I., Gómez-Gascón L., and Rodríguez-Ortega M. J. (2014) Surfomics: shaving live organisms for a fast proteomic identification of surface proteins. J. Proteomics 97, 164–176 10.1016/j.jprot.2013.03.035 [DOI] [PubMed] [Google Scholar]
- 16. Dixit V. M., Grant G. A., Santoro S. A., and Frazier W. A. (1984) Isolation and characterization of a heparin-binding domain from the amino terminus of platelet thrombospondin. J. Biol. Chem. 259, 10100–10105 [PubMed] [Google Scholar]
- 17. Tan K., Duquette M., Liu J. H., Zhang R., Joachimiak A., Wang J. H., and Lawler J. (2006) The structures of the thrombospondin-1 N-terminal domain and its complex with a synthetic pentameric heparin. Structure 14, 33–42 10.1016/j.str.2005.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tan K., Duquette M., Liu J. H., Shanmugasundaram K., Joachimiak A., Gallagher J. T., Rigby A. C., Wang J. H., and Lawler J. (2008) Heparin-induced cis- and trans-dimerization modes of the thrombospondin-1 N-terminal domain. J. Biol. Chem. 283, 3932–3941 10.1074/jbc.M705203200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Holtz A. M., Griffiths S. C., Davis S. J., Bishop B., Siebold C., and Allen B. L. (2015) Secreted HHIP1 interacts with heparan sulfate and regulates Hedgehog ligand localization and function. J. Cell Biol. 209, 739–757 10.1083/jcb.201411024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Park M., and Lee S. T. (1999) The fourth immunoglobulin-like loop in the extracellular domain of FLT-1, a VEGF receptor, includes a major heparin-binding site. Biochem. Biophys. Res. Commun. 264, 730–734 10.1006/bbrc.1999.1580 [DOI] [PubMed] [Google Scholar]
- 21. Ball D. K., Rachfal A. W., Kemper S. A., and Brigstock D. R. (2003) The heparin-binding 10-kDa fragment of connective tissue growth factor (CTGF) containing module 4 alone stimulates cell adhesion. J. Endocrinol. 176, R1–R7 10.1677/joe.0.176r001 [DOI] [PubMed] [Google Scholar]
- 22. Murphy J. W., Cho Y., Sachpatzidis A., Fan C., Hodsdon M. E., and Lolis E. (2007) Structural and functional basis of CXCL12 (stromal cell-derived factor-1α) binding to heparin. J. Biol. Chem. 282, 10018–10027 10.1074/jbc.M608796200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bleul C. C., Farzan M., Choe H., Parolin C., Clark-Lewis I., Sodroski J., and Springer T. A. (1996) The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature 382, 829–833 10.1038/382829a0 [DOI] [PubMed] [Google Scholar]
- 24. Kashiwagi M., Enghild J. J., Gendron C., Hughes C., Caterson B., Itoh Y., and Nagase H. (2004) Altered proteolytic activities of ADAMTS-4 expressed by C-terminal processing. J. Biol. Chem. 279, 10109–10119 10.1074/jbc.M312123200 [DOI] [PubMed] [Google Scholar]
- 25. Fushimi K., Troeberg L., Nakamura H., Lim N. H., and Nagase H. (2008) Functional differences of the catalytic and non-catalytic domains in human ADAMTS-4 and ADAMTS-5 in aggrecanolytic activity. J. Biol. Chem. 283, 6706–6716 10.1074/jbc.M708647200 [DOI] [PubMed] [Google Scholar]
- 26. Brigstock D. R., Steffen C. L., Kim G. Y., Vegunta R. K., Diehl J. R., and Harding P. A. (1997) Purification and characterization of novel heparin-binding growth factors in uterine secretory fluids. Identification as heparin-regulated Mr 10,000 forms of connective tissue growth factor. J. Biol. Chem. 272, 20275–20282 10.1074/jbc.272.32.20275 [DOI] [PubMed] [Google Scholar]
- 27. Nakamura H., Izumoto Y., Kambe H., Kuroda T., Mori T., Kawamura K., Yamamoto H., and Kishimoto T. (1994) Molecular cloning of complementary DNA for a novel human hepatoma-derived growth factor. Its homology with high mobility group-1 protein. J. Biol. Chem. 269, 25143–25149 [PubMed] [Google Scholar]
- 28. Sue S. C., Chen J. Y., Lee S. C., Wu W. G., and Huang T. H. (2004) Solution structure and heparin interaction of human hepatoma-derived growth factor. J. Mol. Biol. 343, 1365–1377 10.1016/j.jmb.2004.09.014 [DOI] [PubMed] [Google Scholar]
- 29. Brown J. C., Sasaki T., Göhring W., Yamada Y., and Timpl R. (1997) The C-terminal domain V of perlecan promotes β1 integrin-mediated cell adhesion, binds heparin, nidogen and fibulin-2 and can be modified by glycosaminoglycans. Eur. J. Biochem. 250, 39–46 10.1111/j.1432-1033.1997.t01-1-00039.x [DOI] [PubMed] [Google Scholar]
- 30. Yamaguchi H., Yamashita H., Mori H., Okazaki I., Nomizu M., Beck K., and Kitagawa Y. (2000) High and low affinity heparin-binding sites in the G domain of the mouse laminin α4 chain. J. Biol. Chem. 275, 29458–29465 10.1074/jbc.M003103200 [DOI] [PubMed] [Google Scholar]
- 31. Yamashita H., Beck K., and Kitagawa Y. (2004) Heparin binds to the laminin α4 chain LG4 domain at a site different from that found for other laminins. J. Mol. Biol. 335, 1145–1149 10.1016/j.jmb.2003.11.047 [DOI] [PubMed] [Google Scholar]
- 32. O'Reilly M. S., Boehm T., Shing Y., Fukai N., Vasios G., Lane W. S., Flynn E., Birkhead J. R., Olsen B. R., and Folkman J. (1997) Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell 88, 277–285 10.1016/S0092-8674(00)81848-6 [DOI] [PubMed] [Google Scholar]
- 33. Shao C., Zhang F., Kemp M. M., Linhardt R. J., Waisman D. M., Head J. F., and Seaton B. A. (2006) Crystallographic analysis of calcium-dependent heparin binding to annexin A2. J. Biol. Chem. 281, 31689–31695 10.1074/jbc.M604502200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Benecky M. J., Kolvenbach C. G., Amrani D. L., and Mosesson M. W. (1988) Evidence that binding to the carboxyl-terminal heparin-binding domain (Hep II) dominates the interaction between plasma fibronectin and heparin. Biochemistry 27, 7565–7571 10.1021/bi00419a058 [DOI] [PubMed] [Google Scholar]
- 35. Cardin A. D., and Weintraub H. J. (1989) Molecular modeling of protein-glycosaminoglycan interactions. Arteriosclerosis 9, 21–32 10.1161/01.ATV.9.1.21 [DOI] [PubMed] [Google Scholar]
- 36. Hileman R. E., Fromm J. R., Weiler J. M., and Linhardt R. J. (1998) Glycosaminoglycan-protein interactions: definition of consensus sites in glycosaminoglycan binding proteins. BioEssays 20, 156–167 [DOI] [PubMed] [Google Scholar]
- 37. Reuten R., Patel T. R., McDougall M., Rama N., Nikodemus D., Gibert B., Delcros J. G., Prein C., Meier M., Metzger S., Zhou Z., Kaltenberg J., McKee K. K., Bald T., Tüting T., et al. (2016) Structural decoding of netrin-4 reveals a regulatory function towards mature basement membranes. Nat. Commun. 7, 13515 10.1038/ncomms13515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hsu J. Y., Crawley S., Chen M., Ayupova D. A., Lindhout D. A., Higbee J., Kutach A., Joo W., Gao Z., Fu D., To C., Mondal K., Li B., Kekatpure A., Wang M., et al. (2017) Non-homeostatic body weight regulation through a brainstem-restricted receptor for GDF15. Nature 550, 255–259 10.1038/nature24042 [DOI] [PubMed] [Google Scholar]
- 39. Zhang X., Wang Q., Wu J., Wang J., Shi Y., and Liu M. (2018) Crystal structure of human lysyl oxidase-like 2 (hLOXL2) in a precursor state. Proc. Natl. Acad. Sci. U.S.A. 115, 3828–3833 10.1073/pnas.1720859115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Khan K. A., McMurray J. L., Mohammed F., and Bicknell R. (2019) C-type lectin domain group 14 proteins in vascular biology, cancer and inflammation. FEBS J. 286, 3299–3332 10.1111/febs.14985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lee S., Rho S. S., Park H., Park J. A., Kim J., Lee I. K., Koh G. Y., Mochizuki N., Kim Y. M., and Kwon Y. G. (2017) Carbohydrate-binding protein CLEC14A regulates VEGFR-2- and VEGFR-3-dependent signals during angiogenesis and lymphangiogenesis. J. Clin. Invest. 127, 457–471 10.1172/JCI85145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Noy P. J., Lodhia P., Khan K., Zhuang X., Ward D. G., Verissimo A. R., Bacon A., and Bicknell R. (2015) Blocking CLEC14A-MMRN2 binding inhibits sprouting angiogenesis and tumour growth. Oncogene 34, 5821–5831 10.1038/onc.2015.34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zelensky A. N., and Gready J. E. (2005) The C-type lectin-like domain superfamily. FEBS J. 272, 6179–6217 10.1111/j.1742-4658.2005.05031.x [DOI] [PubMed] [Google Scholar]
- 44. Yu W. H., Zhao P., Draghi M., Arevalo C., Karsten C. B., Suscovich T. J., Gunn B., Streeck H., Brass A. L., Tiemeyer M., Seaman M., Mascola J. R., Wells L., Lauffenburger D. A., and Alter G. (2018) Exploiting glycan topography for computational design of Env glycoprotein antigenicity. PLoS Comput. Biol. 14, e1006093 10.1371/journal.pcbi.1006093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Angel P. M., Lim J. M., Wells L., Bergmann C., and Orlando R. (2007) A potential pitfall in 18O-based N-linked glycosylation site mapping. Rapid Commun. Mass Spectrom. 21, 674–682 10.1002/rcm.2874 [DOI] [PubMed] [Google Scholar]
- 46. Foxall C., Watson S. R., Dowbenko D., Fennie C., Lasky L. A., Kiso M., Hasegawa A., Asa D., and Brandley B. K. (1992) The three members of the selectin receptor family recognize a common carbohydrate epitope, the sialyl Lewis(x) oligosaccharide. J. Cell Biol. 117, 895–902 10.1083/jcb.117.4.895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kreuger J., Lindahl U., and Jemth P. (2003) Nitrocellulose filter binding to assess binding of glycosaminoglycans to proteins. Methods Enzymol. 363, 327–339 10.1016/S0076-6879(03)01062-0 [DOI] [PubMed] [Google Scholar]
- 48. Maciag T., Mehlman T., Friesel R., and Schreiber A. B. (1984) Heparin binds endothelial cell growth factor, the principal endothelial cell mitogen in bovine brain. Science 225, 932–935 10.1126/science.6382607 [DOI] [PubMed] [Google Scholar]
- 49. Shing Y., Folkman J., Sullivan R., Butterfield C., Murray J., and Klagsbrun M. (1984) Heparin affinity: purification of a tumor-derived capillary endothelial cell growth factor. Science 223, 1296–1299 10.1126/science.6199844 [DOI] [PubMed] [Google Scholar]
- 50. Mohammadi M., Olsen S. K., and Ibrahimi O. A. (2005) Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev. 16, 107–137 10.1016/j.cytogfr.2005.01.008 [DOI] [PubMed] [Google Scholar]
- 51. Cummings R. D., and McEver R. P. (2017) in Essentials of Glycobiology (Varki A., Cummings R. D., Esko J. D., Stanley P., Hart G. W., Aebi M., Darvill A. G., Kinoshita T., Packer N. H., Prestegard J. H., Schnaar R. L., and Seeberger P. H., eds), pp. 435–452, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 52. Khan S., Gor J., Mulloy B., and Perkins S. J. (2010) Semi-rigid solution structures of heparin by constrained X-ray scattering modelling: new insight into heparin–protein complexes. J. Mol. Biol. 395, 504–521 10.1016/j.jmb.2009.10.064 [DOI] [PubMed] [Google Scholar]
- 53. Burley S. K., Berman H. M., Bhikadiya C., Bi C., Chen L., Di Costanzo L., Christie C., Dalenberg K., Duarte J. M., Dutta S., Feng Z., Ghosh S., Goodsell D. S., Green R. K., Guranovic V., et al. (2019) RCSB Protein Data Bank: biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 47, D464–D474 10.1093/nar/gky1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Burgess W. H., Shaheen A. M., Ravera M., Jaye M., Donohue P. J., and Winkles J. A. (1990) Possible dissociation of the heparin-binding and mitogenic activities of heparin-binding (acidic fibroblast) growth factor-1 from its receptor-binding activities by site-directed mutagenesis of a single lysine residue. J. Cell Biol. 111, 2129–2138 10.1083/jcb.111.5.2129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hauschka P. V., Mavrakos A. E., Iafrati M. D., Doleman S. E., and Klagsbrun M. (1986) Growth factors in bone matrix. Isolation of multiple types by affinity chromatography on heparin-Sepharose. J. Biol. Chem. 261, 12665–12674 [PubMed] [Google Scholar]
- 56. Klagsbrun M., and Shing Y. (1985) Heparin affinity of anionic and cationic capillary endothelial cell growth factors: analysis of hypothalamus-derived growth factors and fibroblast growth factors. Proc. Natl. Acad. Sci. U.S.A. 82, 805–809 10.1073/pnas.82.3.805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nunes Q. M., Su D., Brownridge P. J., Simpson D. M., Sun C., Li Y., Bui T. P., Zhang X., Huang W., Rigden D. J., Beynon R. J., Sutton R., and Fernig D. G. (2019) The heparin-binding proteome in normal pancreas and murine experimental acute pancreatitis. PLoS ONE 14, e0217633 10.1371/journal.pone.0217633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Flodgaard H., Ostergaard E., Bayne S., Svendsen A., Thomsen J., Engels M., and Wollmer A. (1991) Covalent structure of two novel neutrophile leucocyte-derived proteins of porcine and human origin. Neutrophile elastase homologues with strong monocyte and fibroblast chemotactic activities. Eur. J. Biochem. 197, 535–547 10.1111/j.1432-1033.1991.tb15942.x [DOI] [PubMed] [Google Scholar]
- 59. Beckman M., Freeman C., Parish C. R., and Small D. H. (2009) Activation of cathepsin D by glycosaminoglycans. FEBS J. 276, 7343–7352 [DOI] [PubMed] [Google Scholar]
- 60. Hileman R. E., Jennings R. N., and Linhardt R. J. (1998) Thermodynamic analysis of the heparin interaction with a basic cyclic peptide using isothermal titration calorimetry. Biochemistry 37, 15231–15237 10.1021/bi980212x [DOI] [PubMed] [Google Scholar]
- 61. Jang J., Kim M. R., Kim T. K., Lee W. R., Kim J. H., Heo K., and Lee S. (2017) CLEC14a–HSP70–1A interaction regulates HSP70–1A–induced angiogenesis. Sci. Rep. 7, 10666 10.1038/s41598-017-11118-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Khan K. A., Naylor A. J., Khan A., Noy P. J., Mambretti M., Lodhia P., Athwal J., Korzystka A., Buckley C. D., Willcox B. E., Mohammed F., and Bicknell R. (2017) Multimerin-2 is a ligand for group 14 family C-type lectins CLEC14A, CD93 and CD248 spanning the endothelial pericyte interface. Oncogene 36, 6097–6108 10.1038/onc.2017.214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Noy P. J., Swain R. K., Khan K., Lodhia P., and Bicknell R. (2016) Sprouting angiogenesis is regulated by shedding of the C-type lectin family 14, member A (CLEC14A) ectodomain, catalyzed by rhomboid-like 2 protein (RHBDL2). FASEB J. 30, 2311–2323 10.1096/fj.201500122R [DOI] [PubMed] [Google Scholar]
- 64. Zanivan S., Maione F., Hein M. Y., Hernández-Fernaud J. R., Ostasiewicz P., Giraudo E., and Mann M. (2013) SILAC-based proteomics of human primary endothelial cell morphogenesis unveils tumor angiogenic markers. Mol. Cell. Proteomics 12, 3599–3611 10.1074/mcp.M113.031344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mura M., Swain R. K., Zhuang X., Vorschmitt H., Reynolds G., Durant S., Beesley J. F., Herbert J. M., Sheldon H., Andre M., Sanderson S., Glen K., Luu N. T., McGettrick H. M., Antczak P., et al. (2012) Identification and angiogenic role of the novel tumor endothelial marker CLEC14A. Oncogene 31, 293–305 10.1038/onc.2011.233 [DOI] [PubMed] [Google Scholar]
- 66. Zhuang X., Cross D., Heath V. L., and Bicknell R. (2011) Shear stress, tip cells and regulators of endothelial migration. Biochem. Soc. Trans. 39, 1571–1575 10.1042/BST20110746 [DOI] [PubMed] [Google Scholar]
- 67. Rho S. S., Choi H. J., Min J. K., Lee H. W., Park H., Park H., Kim Y. M., and Kwon Y. G. (2011) Clec14a is specifically expressed in endothelial cells and mediates cell to cell adhesion. Biochem. Biophys. Res. Commun. 404, 103–108 10.1016/j.bbrc.2010.11.075 [DOI] [PubMed] [Google Scholar]
- 68. Kim T. K., Park C. S., Jang J., Kim M. R., Na H. J., Lee K., Kim H. J., Heo K., Yoo B. C., Kim Y. M., Lee J. W., Kim S. J., Kim E. S., Kim D. Y., Cha K., Lee T. G., and Lee S. (2018) Inhibition of VEGF-dependent angiogenesis and tumor angiogenesis by an optimized antibody targeting CLEC14a. Mol. Oncol. 12, 356–372 10.1002/1878-0261.12169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M. Y., Geiger T., Mann M., and Cox J. (2016) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 10.1038/nmeth.3901 [DOI] [PubMed] [Google Scholar]
- 70. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., and Mann M. (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 10.1021/pr101065j [DOI] [PubMed] [Google Scholar]
- 71. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., and Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 10.1038/msb.2011.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sievers F., and Higgins D. G. (2018) Clustal Omega for making accurate alignments of many protein sequences. Protein Sci. 27, 135–145 10.1002/pro.3290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Raman S., Vernon R., Thompson J., Tyka M., Sadreyev R., Pei J., Kim D., Kellogg E., DiMaio F., Lange O., Kinch L., Sheffler W., Kim B. H., Das R., Grishin N. V., and Baker D. (2009) Structure prediction for CASP8 with all-atom refinement using Rosetta. Proteins 77, 89–99 10.1002/prot.22540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kelley L. A., Mezulis S., Yates C. M., Wass M. N., and Sternberg M. J. (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 10.1038/nprot.2015.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F. T., de Beer T. A. P., Rempfer C., Bordoli L., Lepore R., and Schwede T. (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303 10.1093/nar/gky427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Krieger E., Joo K., Lee J., Lee J., Raman S., Thompson J., Tyka M., Baker D., and Karplus K. (2009) Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: four approaches that performed well in CASP8. Proteins 77, Suppl. 9, 114–122 10.1002/prot.22570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Singh A., Montgomery D., Xue X., Foley B. L., and Woods R. J. (2019) GAG Builder: a web-tool for modeling 3D structures of glycosaminoglycans. Glycobiology 29, 515–518 10.1093/glycob/cwz027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Krieger E., and Vriend G. (2015) New ways to boost molecular dynamics simulations. J. Comput. Chem. 36, 996–1007 10.1002/jcc.23899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Lawler J. W., Slayter H. S., and Coligan J. E. (1978) Isolation and characterization of a high molecular weight glycoprotein from human blood platelets. J. Biol. Chem. 253, 8609–8616 [PubMed] [Google Scholar]
- 80. Shikano S., Bonkobara M., Zukas P. K., and Ariizumi K. (2001) Molecular cloning of a dendritic cell-associated transmembrane protein, DC-HIL, that promotes RGD-dependent adhesion of endothelial cells through recognition of heparan sulfate proteoglycans. J. Biol. Chem. 276, 8125–8134 10.1074/jbc.M008539200 [DOI] [PubMed] [Google Scholar]
- 81. Yamada K. M., Kennedy D. W., Kimata K., and Pratt R. M. (1980) Characterization of fibronectin interactions with glycosaminoglycans and identification of active proteolytic fragments. J. Biol. Chem. 255, 6055–6063 [PubMed] [Google Scholar]
- 82. Ishitsuka R., Kojima K., Utsumi H., Ogawa H., and Matsumoto I. (1998) Glycosaminoglycan binding properties of annexin IV, V, and VI. J. Biol. Chem. 273, 9935–9941 10.1074/jbc.273.16.9935 [DOI] [PubMed] [Google Scholar]
- 83. Kuno K., Kanada N., Nakashima E., Fujiki F., Ichimura F., and Matsushima K. (1997) Molecular cloning of a gene encoding a new type of metalloproteinase-disintegrin family protein with thrombospondin motifs as an inflammation associated gene. J. Biol. Chem. 272, 556–562 10.1074/jbc.272.1.556 [DOI] [PubMed] [Google Scholar]
- 84. Talts J. F., Sasaki T., Miosge N., Göhring W., Mann K., Mayne R., and Timpl R. (2000) Structural and functional analysis of the recombinant G domain of the laminin α4 chain and its proteolytic processing in tissues. J. Biol. Chem. 275, 35192–35199 10.1074/jbc.M003261200 [DOI] [PubMed] [Google Scholar]
- 85. Ehrlich H. J., Keijer J., Preissner K. T., Gebbink R. K., and Pannekoek H. (1991) Functional interaction of plasminogen activator inhibitor type 1 (PAI-1) and heparin. Biochemistry 30, 1021–1028 10.1021/bi00218a020 [DOI] [PubMed] [Google Scholar]
- 86. Morimoto-Tomita M., Uchimura K., Werb Z., Hemmerich S., and Rosen S. D. (2002) Cloning and characterization of two extracellular heparin-degrading endosulfatases in mice and humans. J. Biol. Chem. 277, 49175–49185 10.1074/jbc.M205131200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Yang G. P., and Lau L. F. (1991) Cyr61, product of a growth factor-inducible immediate early gene, is associated with the extracellular matrix and the cell surface. Cell Growth Differ. 2, 351–357 [PubMed] [Google Scholar]
- 88. Chen N., Chen C. C., and Lau L. F. (2000) Adhesion of human skin fibroblasts to Cyr61 is mediated through integrin α6β1 and cell surface heparan sulfate proteoglycans. J. Biol. Chem. 275, 24953–24961 10.1074/jbc.M003040200 [DOI] [PubMed] [Google Scholar]
- 89. Riera M., Roher N., Miró F., Gil C., Trujillo R., Aguilera J., Plana M., and Itarte E. (1999) Association of protein kinase CK2 with eukaryotic translation initiation factor eIF-2 and with grp94/endoplasmin. Mol. Cell. Biochem. 191, 97–104 10.1023/A:1006810311743 [DOI] [PubMed] [Google Scholar]
- 90. Ménoret A., and Bell G. (2000) Purification of multiple heat shock proteins from a single tumor sample. J. Immunol. Methods 237, 119–130 10.1016/S0022-1759(00)00137-X [DOI] [PubMed] [Google Scholar]
- 91. Reed R. C., Zheng T., and Nicchitta C. V. (2002) GRP94-associated enzymatic activities. Resolution by chromatographic fractionation. J. Biol. Chem. 277, 25082–25089 10.1074/jbc.M203195200 [DOI] [PubMed] [Google Scholar]
- 92. Bleul C. C., Fuhlbrigge R. C., Casasnovas J. M., Aiuti A., and Springer T. A. (1996) A highly efficacious lymphocyte chemoattractant, stromal cell-derived factor 1 (SDF-1). J. Exp. Med. 184, 1101–1109 10.1084/jem.184.3.1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kainulainen V., Wang H., Schick C., and Bernfield M. (1998) Syndecans, heparan sulfate proteoglycans, maintain the proteolytic balance of acute wound fluids. J. Biol. Chem. 273, 11563–11569 10.1074/jbc.273.19.11563 [DOI] [PubMed] [Google Scholar]
- 94. Ledoux D., Merciris D., Barritault D., and Caruelle J. P. (2003) Heparin-like dextran derivatives as well as glycosaminoglycans inhibit the enzymatic activity of human cathepsin G. FEBS Lett. 537, 23–29 10.1016/S0014-5793(03)00064-4 [DOI] [PubMed] [Google Scholar]
- 95. Spencer J. L., Stone P. J., and Nugent M. A. (2006) New insights into the inhibition of human neutrophil elastase by heparin. Biochemistry 45, 9104–9120 10.1021/bi060338r [DOI] [PubMed] [Google Scholar]
- 96. Ohashi K., Kisilevsky R., and Yanagishita M. (2002) Affinity binding of glycosaminoglycans with β(2)-microglobulin. Nephron 90, 158–168 10.1159/000049037 [DOI] [PubMed] [Google Scholar]
- 97. Yip C. W., Cheung P. F., Leung I. C., Wong N. C., Cheng C. K., Fan S. T., and Cheung S. T. (2014) Granulin-epithelin precursor interacts with heparan sulfate on liver cancer cells. Carcinogenesis 35, 2485–2494 10.1093/carcin/bgu164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Zhou J., Gao G., Crabb J. W., and Serrero G. (1993) Purification of an autocrine growth factor homologous with mouse epithelin precursor from a highly tumorigenic cell line. J. Biol. Chem. 268, 10863–10869 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.