

Abstract
Serum amyloid A (SAA), one of the major highly conserved acute-phase proteins in most mammals, is predominantly produced by hepatocytes and also by a variety of cells in extrahepatic tissues. It is well-known that the expression of SAA is sharply increased in bacterial infections. However, the exact physiological function of SAA during bacterial infection remains unclear. Herein, we showed that SAA expression significantly increased in abscesses of Staphylococcus aureus cutaneous infected mice, which exert direct antibacterial effects by binding to the bacterial cell surface and disrupting the cell membrane in acidic conditions. Mechanically, SAA disrupts anionic liposomes by spontaneously forming small vesicles or micelles under acidic conditions. Especially, the N-terminal region of SAA is necessary for membrane disruption and bactericidal activity. Furthermore, we found that mice deficient in SAA1/2 were more susceptible to infection by S. aureus. In addition, the expression of SAA in infected skin was regulated by interleukin-6. Taken together, these findings support a key role of the SAA in host defense and may provide a novel therapeutic strategy for cutaneous bacterial infection.
Keywords: antimicrobial peptide (AMP), lipid-binding protein, Staphylococcus aureus (S. aureus), host defense, phospholipid vesicle, acute-phase proteins, bactericidal mechanism, cutaneous infection, serum amyloid A
Introduction
Serum amyloid A (SAA)2 is a major acute-phase protein and highly conserved from sea cucumber to humans. SAA increases rapidly in serum during the acute-phase response and is thought to be involved in innate immunity and lipid homeostasis during inflammation. Three isoforms of acute-phase SAA have been reported in mice: Saa1, Saa2, and Saa3. However, the third gene, SAA3, is a pseudogene in humans. The acute-phase SAAs in humans and mice are synthesized largely by hepatocytes under inflammatory conditions (1, 2). SAAs are also widely expressed in normal extrahepatic tissues (3) and significantly increased in inflammatory tissue (4–6). Furthermore, the plasma extravasation may enable delivery of hepatocyte-derived SAA to the infection site. On-site SAA may play a role in protecting the blood and other organs from disseminating microbes (7, 8). SAA has long been used as a clinical biomarker for acute infections and also has been reported to be expressed in keratinocytes and sebocytes and up-regulated in some inflammatory cutaneous diseases (3, 9, 10), such as psoriasis, papulopustular acne, and papulopustular rosacea. Although SAA has a similar expression pattern with typical cutaneous antimicrobial proteins and peptides (AMPs) (11, 12), its role in skin surface defense remains unclear.
The acidic condition (pH 4–6) is an important character of skin surface. Because the bactericidal efficacy of many skin AMPs depends on the acidic pH of the skin, acidity of the stratum corneum and sweat is important for antimicrobial activity (13) Local acidosis also occurs during inflammation due to massive infiltration of neutrophils and macrophages at the site of inflammation, which subsequently activates respiratory burst, and hypoxia at sites of inflammation leading to local accumulation of lactic acid and a significant decrease in pH (14, 15).
Many members of the SAA protein family are amphipathic and positively charged, which are typical characteristics of AMPs. In some species, such as goldfish and common carp, SAAs have a highly alkaline pI and present antimicrobial activity in vitro (16, 17). In vitro antibacterial assays at neutral pH do not reveal that human or mouse SAAs have bactericidal properties (6). However, overexpression of mouse SAA1 and SAA2 (mSAA1 and mSAA2) in cultured epithelial cell lines reduces co-cultured Escherichia coli via an undefined mechanism (18). These works suggest that the local microenvironment of inflammatory foci may affect the bactericidal properties of SAA.
In this study, we demonstrate that SAA exhibits direct antibacterial activity at low pH conditions through targeted disruption of bacterial membrane structures, which are negatively charged phospholipids. Intriguingly, SAA rapidly self-assembles with anionic phospholipids, forming small vesicles or micelles under mildly acidic pH conditions. Moreover, SAA was strongly induced in Staphylococcus aureus–infected skin and reduced bacteria burden in vivo.
Results
SAA is strongly induced in S. aureus–infected skin and presents antimicrobial properties by disrupting bacterial membranes
We first evaluated the expressions of SAA1/2/3 in an S. aureus cutaneous infected mouse model and found that mRNA levels of SAAs were significantly increased both in the skin and in the liver of the infected mice (Fig. 1A and Fig. S1). The expression of SAA was also detected by immunohistochemical staining. We found that all three mice SAAs were abundantly expressed in cutaneous abscesses (Fig. 1B). To investigate whether SAA has a direct bactericidal effect, we tested the activity of SAA against E. coli and S. aureus, which are representatives of Gram-negative and Gram-positive bacterial strains, respectively. We found that bactericidal activity of mSAA1 was pH-dependent (Fig. 1C), with higher activity at acidic pH. The survival rates of both E. coli and S. aureus declined by 90% after incubation with 2.5 μm mSAA1 at pH 5.5 (Fig. 1D). We also tested the antibacterial activity of hSAA1, hSAA2, mSAA2, and mSAA3, and the bactericidal activity of all of these proteins increased significantly at pH 5.5 (Fig. 1, E and F). To further validate the interaction between SAA and bacteria, we used serum from AgNO3-injected mice (endogenous mSAA) and the purified recombinant mSAA1 (exogenous) to incubate with E. coli and S. aureus at different pH conditions. The binding abilities of endogenous mSAA and the purified recombinant mSAA1 to E. coli and S. aureus at acidic pH (pH 5.5) were strongly enhanced (Fig. 2, A and B). The induced expression of recombinant SAA in bacteria causes bacterial cell lysis, suggesting the destructive effect of SAA on bacterial cell membranes (19). To investigate the direct interaction between SAA and bacteria, membrane integrity of mSAA1-treated bacterial cells was observed by using transmission EM. Drastic morphological changes in the bacterial cells were observed after incubating mSAA1 at low pH, and cell membrane damage and component leakage in both S. aureus and E. coli cells and cytoplasm condensation in E. coli cells were evident (Fig. 2, C–E).
Figure 1.

The expression of SAA in the S. aureus cutaneous infection model and the antibacterial activity of SAA to E. coli and S. aureus in a pH-dependent manner. A, SAA mRNA expression in skin and liver after mouse back skin infected with 3 × 106 cfu of S. aureus (n = 5) on day 3. B, immunohistochemistry analysis of SAA in mouse skin after S. aureus infection on day 3 (scale bar, 200 μm). C, bactericidal activity of mSAA1 at different pH values was measured against E. coli and S. aureus using a microdilution susceptibility assay (n = 3). 100% survival was defined as total survival of bacteria in the same buffer and under the same conditions as in the absence of proteins. D, percentage of cfu remaining after exposure to purified mSAA1. S. aureus and E. coli were grown to midlog phase and incubated with purified proteins. After incubation for 2 h at pH 5.5 at 37 °C, the viability of bacteria was quantified by dilution plating (n = 3). E and F, bactericidal activity of different SAA proteins. The viability of bacteria was quantified by dilution plating (n = 3). All data are representative of three independent experiments. Significant differences versus control group are indicated by asterisks: *, p < 0.05; **, p < 0.01; ***, p < 0.001. Error bars, S.E.M.
Figure 2.

SAA binds bacteria and disrupts its cell membrane at low pH. A, binding of mSAA1 to E. coli or S. aureus was incubated with 5 μm recombinant mSAA1 in 10 mm sodium phosphate, 150 mm NaCl buffer, pH 5.5 or pH 7.4. After centrifugation, the supernatant (S) and the pellet (P) were analyzed by SDS-PAGE and Coomassie Blue staining. B, binding of mSAA1 to E. coli or S. aureus was incubated with 10% AgNO3-injected mice serum in 10 mm sodium phosphate, 150 mm NaCl buffer, pH 5.5 or pH 7.4. After centrifugation, SAA in the pellet was detected by immunoblotting. C, transmission EM micrographs of E. coli and S. aureus cells were incubated with 10 μm mSAA1 or buffer. (scale bar, 500 nm). D and E, cell component leakage areas of S. aureus (D) and E. coli (E) cells (n = 30) were incubated with 10 μm mSAA1 or buffer. Significant differences versus control group are indicated by asterisks: ***, p < 0.001. Error bars, S.E.M.
SAA induces anionic liposome leakage by forming micelles under acidic conditions.
The phospholipid-binding specificity of SAA was examined by measuring releasing entrapped terbium (Tb3+) from liposomes of defined composition (20). Negatively charged phospholipids, such as phosphatidylglycerol (PG) and cardiolipin, are abundantly present in bacterial cell membranes (21, 22). To further understand the interaction between SAA and bacterial cell membranes, unilamellar liposomes were used as an in vitro model. No phosphatidylcholine (PC)-only liposome leakage was observed with 5 μm mSAA1 and hSAA2 under neutral or acidic conditions, whereas 100% leakage in liposomes containing 20% cardiolipin (cardiolipin liposomes) and weaker leakage in liposomes containing 20% PG (PG liposomes) were observed with 5 μm mSAA1 and hSAA2 (Fig. 3, A and B). Moreover, mSAA1, mSAA2, hSAA1, and hSAA2 also induced negatively charged liposome disruption under acidic conditions. Moreover, 100% PG liposome leakage with a higher concentration of SAA proteins was evident (Fig. 3 (C and D) and Fig. S2A). To investigate the interaction between SAA and cardiolipin, we performed biolayer interferometry to explore the binding affinity of proteins and liposomes. The biotinylated SAA protein was immobilized on a streptavidin (SA) sensor, which was then flown through with the different liposomes. There was no observable binding to PC liposomes, but both mSAA1 and hSAA2 bound to cardiolipin liposomes were prominent (Fig. 3, E and F). As the cardiolipin could not dissociate from SAA proteins, we speculated that SAA formed a stable complex with cardiolipin at acidic pH. Consistent with the liposome leakage assays, no interactions between SAA and PC or cardiolipin at neutral pH were observed.
Figure 3.

SAA binds to negatively charged lipids and induces liposome leakage. A and B, SAA disrupted terbium (Tb3+)-loaded unilamellar liposomes containing the negatively charged lipid phosphatidylglycerol (PG) or cardiolipin (Card), but not liposomes composed of the zwitterionic lipid PC. Liposomes were treated with 5 μm mSAA1 (A) or hSAA2 (B) after incubation with SAA proteins. Detergent was added after 20 min. C and D, PG (C) and cardiolipin (D) liposome leakage–inducing activity of mSAA1, mSAA2, hSAA1, and hSAA2. E, Octet binding response of PC liposomes and cardiolipin liposomes to mSAA1 or hSAA2 at pH 5.5. F, Octet binding response of PC liposomes and cardiolipin liposomes to mSAA1 or hSAA2 at pH 7.4. The proteins were immobilized onto an SA sensor followed by dipping these biosensors into the liposome solution, and the protein liposome interaction was represented as binding curves showing the association. All data are representative of three independent experiments.
To determine the exact interaction between SAA and liposomes, we performed transmission EM on negatively stained liposomes treated with mSAA1 at different pH conditions. We observed that SAA induced PG or cardiolipin liposome collapse at acidic pH. Furthermore, the SAAs rapidly formed small vesicles or micelles with large-scale disruption of the cardiolipin liposomes and PG liposomes under acidic conditions, and no significant morphological changes were observed in PG or cardiolipin liposomes at neutral pH and in PC liposome under both pH conditions (Fig. 4 and Fig. S2B). This indicated that SAA binds anionic phospholipids to form small vesicles or micelles and induce liposome leakage under acidic pH conditions.
Figure 4.

Membrane disruption and micelle nanoparticle formation activity of SAA. PG liposomes (left), cardiolipin liposomes (middle), and PC liposomes (right) after 1 h of incubation with mSAA1 at pH 7.4 (bottom) or pH 5.5 (top) at 37 °C. The protein/lipid molar ratio was 1:100. Shown are representative negative-stain EM micrographs of the liposomes (scale bar, 100 nm). Black arrows, small vesicles or micelles. All data are representative of three independent experiments.
The N-terminal region of SAA is necessary for liposome disruption and bactericidal activity
The crystal structures of hSAA1 and mSAA3 had been determined, and the known monomer structures of hSAA1 and mSAA3 are very similar, containing a cone with four α-helices and a tail (23, 24). By analyzing the surface hydrophobicity of SAA, a hypothetical model of SAA binding to high-density lipoprotein (HDL) suggested that helix 1 and helix 3 of SAA form a hydrophobic surface that binds to HDL (25). Because the N terminus of SAA is important for HDL binding, we speculated that the N-terminal region of SAA was also essential for its membrane disruption activity. A mutation with a 20-amino acid truncation of mSAA1 (ΔN20-mSAA1) was constructed. We found that the ΔN20-mSAA1 mutation did not induce PG and cardiolipin liposome leakage (Fig. 5, A–D). The negative stain EM also showed that the truncation mutation did not induce cardiolipin liposome disruption (Fig. 5E). Furthermore, the interaction with cardiolipin liposomes was detected by biolayer interferometry. It was also found that under acidic conditions, ΔN20-mSAA1 could not bind to cardiolipin liposomes (Fig. 5F). Consequently, the bactericidal activity of the truncation mutation was also significantly impaired (Fig. 5G).
Figure 5.

Structure analysis of SAA shows that the N terminus of SAA is necessary for liposome disruption and bactericidal activity. A–D, effects of ΔN20-mSAA1 mutation on mSAA1 liposome leakage–inducing activities. A, purified mSAA1 and ΔN20-mSAA1 (1 μm) proteins were incubated with cardiolipin liposomes. B, means ± S.E.M. (error bars) from three independent replicates of the experiment shown in A. C, mSAA1 and ΔN20-mSAA1 (10 μm) were incubated with PG liposomes at pH 5.5. D, means ± S.E.M. from three independent replicates of the experiment shown in C. E, negative-stain EM micrographs of cardiolipin liposomes incubated with mSAA1 and ΔN20-mSAA1 mutation at pH 5.5 (scale bar, 100 nm). Protein/lipid molar ratio was 1:100. F, Octet binding response of cardiolipin liposomes to ΔN20-mSAA1 at pH 5.5. G, effects of ΔN20-mSAA1 mutation on mSAA1 antibacterial activity (n = 3). All data are representative of three independent experiments. Significant differences versus control group are indicated by asterisks: **, p < 0.01.
Deletion of Saa1/2 in mice impaired the clearance of S. aureus during cutaneous infections
To further explore the role of SAA in host defense, we generated Saa1/2 double knockout mice (SAA1/2 DKO) and mice to test the function for endogenous SAAs in host defense against cutaneous bacterial infections. We verified that SAA1/2 was absent in the skin of SAA1/2 DKO mice (Fig. 6A) and showed that S. aureus infection led to higher numbers of cutaneous bacteria and a larger abscess area in the absence of SAA1/2 (Fig. 6, B and C). Because IL-6 is critical for the acute phase response and the production of acute reactive proteins including SAA in mice liver during bacterial infections (26). Real-time PCR analysis of IL-6 also showed that mRNA levels for IL-6 were markedly increased in the infected skin (Fig. 6D). We assumed that the production of SAA in skin induced by S. aureus was IL-6–dependent. To test this hypothesis, we infected the WT and Il6−/− mice with S. aureus. Compared with the WT mice, mRNA levels of SAAs were significantly decreased in the skin of the infected Il6−/− mice (Fig. 6E). Taking these results together, we demonstrated that SAA has antibacterial activity both in vitro and in vivo and regulated by IL-6.
Figure 6.

Deletion of Saa1/2 in mice impaired the clearance of S. aureus in the skin. A, WT and SAA1/2DKO littermates were analyzed at 8 weeks of age by quantitative PCR for Saa1/2 gene expression in S. aureus-infected skin (n = 5). B, bacterial counts from S. aureus–infected WT and SAA1/2 DKO mouse cutaneous abscesses. C, abscesses areas of S. aureus–infected WT and SAA1/2 DKO mice. Mice were subcutaneously infected for 4 days (n = 4–5). D, IL-6 mRNA levels in the infected skin were determined by quantitative PCR and are expressed relative to the levels in uninfected WT mice (n = 5). E, SAA mRNA expression in skin of WT and Il6−/− mice infected with S. aureus (n = 3–5). Significant differences versus control group are indicated by asterisks: *, p < 0.05; **, p < 0.01. Error bars, S.E.M.
Discussion
SAA has long been recognized as a major acute-phase response protein in inflammatory diseases, but its molecular actions of innate immunity during bacterial infection remain largely unclear. In the current study, we demonstrated that SAA was abundantly expressed in cutaneous abscesses when infected with S. aureus. SAA not only binds to phospholipids of bacterial membranes through its hydrophobic N-terminal region, but exerts direct bactericidal by disrupting the bacterial cell membrane under acidic conditions. Furthermore, we demonstrated the antimicrobial effects of SAA against skin bacterial infections in vivo, which may provide a novel therapeutic strategy for cutaneous bacterial infection.
The SAA protein family is highly conserved, and the crystal structure reveals that SAA contains a four-alpha-helix bundle in which helix 1 and helix 3 form a hydrophobic surface that binds hydrophobic ligands, such as the phospholipids cholesterol and retinol (24). Some conserved charged amino acid residues interact with polysaccharides and cell receptors (25), suggesting that SAA exerts multiple functions at the inflammatory site, where the concentration of SAA rises sharply during the acute response. SAA, during the acute phase, is primarily associated with HDL (27). Under mild acidic conditions, heparan sulfate binds to SAA and causes differential dissociation of SAA from the HDL particle, but it has no effect on the HDL-associated ApoA-I (28). A recent study also showed that SAA solubilized phospholipid bilayers to form lipoproteins that provided substrates for sPLA2 and effectively removed free fatty acids under acidic conditions (29). Inflammatory foci are characterized by low pH levels reaching as low as pH 5 (30), indicating that the microenvironment at inflammatory foci may influence the biochemical properties and physiological function of SAA. Increased SAA expression was observed in biopsies from patients with Crohn's disease and other inflammatory diseases (4, 18), and such increased SAA was co-localized with bacteria in an animal model (6, 31), which has been speculated to play a role in bacterial sensing and killing mechanisms to protect tissues of the host.
It has been reported that SAA is able to bind to Gram-negative but not Gram-positive bacteria under neutral conditions and acts as an opsonin (32). In this study, we found that SAAs bind both Gram-positive and Gram-negative bacteria and disrupt the bacterial cell membrane, which in turn causes bacterial cell death under the mild acidic pH of the local inflammatory site. AMPs have three different strategies to insert and disrupt the cell membrane: by forming a barrel stave or toroidal pore to make the membrane permeable or by forming a “carpet model,” which causes the accumulation of peptides or proteins on the bilayer surface, leading to disruption of the bilayer in a detergent-like manner, eventually leading to the formation of micelles (33). We demonstrated that SAA was electrostatically attracted to the anionic phospholipid headgroups at the surface of the membrane and disrupted the cell membrane in a carpet-like manner by its hydrophobic surface under acidic pH conditions. These results were consistent with the fact that a group of AMPs present optimal antimicrobial activity at low pH (30, 34, 35).
In this study, we also found that the N-terminal region of mSAA1 is necessary for phospholipid binding and bactericidal activity. Consist with our results, Patel et al. (36) found that deletion of the first 11 amino acid residues at the N terminus of recombinant human SAA diminishes its capacity to bind to HDL and decreases amyloid Fibril formation. Also, another group had reported that the N-terminal region (residues 1–27) of human SAA1 is important for lipid interaction and sufficient for phospholipid binding (37). Previous studies have reported that oligomerized SAA forms a channel on the planar membrane and is associated with toxic amyloid oligomers (19, 33, 38). However, we did not observe channel formation on PG liposomes incubated with SAAs. In contrast, SAAs bound PG liposomes and cardiolipin liposomes, but not PC liposomes, and spontaneously formed small vesicles or micelles under acidic conditions, like a typical apolipoprotein. A reasonable explanation is that SAAs bind to anionic phospholipids with a higher affinity than a zwitterionic phospholipid under acidic conditions. Like other lipoproteins, the amphipathic SAA helix bundle forms a stable HDL-like nanoparticle with the zwitterionic phospholipids; however, this nanoparticle requires a high protein/phospholipid ratio and longer incubation time under neutral conditions (39); a low protein/lipid ratio and short time does not result in morphological changes in vesicles (40). In addition, other studies have reported that some HDL components can be involved in the resistance to microbial infections, in which apoL-1 can selectively bind anionic phospholipids under acidic conditions to disrupt cell membranes and kill trypanosomes (41), whereas apoA-1 also has a higher affinity to anionic phospholipids and inhibits bacterial growth (22). The results of this study indicate that SAA has similar biochemical and biological functions to these apolipoproteins and provide new evidence for the important role of HDL in host defense.
S. aureus is a major cause of skin and soft-tissue infections in humans, which cause both local and systemic diseases (42, 43). In this study, we found that SAA was strongly induced in infected skin of an S. aureus cutaneous infected mouse model. We also found that both SAA1/2 DKO and mice have higher bacterial loads in cutaneous abscesses after subcutaneous S. aureus infection. Furthermore, the production of SAA in infected skin was IL-6–dependent. Consistent with our results, many studies have shown that IL-6 is critical for acute-phase response and acute-phase protein production (26, 44). Previous studies had shown that SAA plays a role in host defense of Salmonella typhimurium and Klebsiella pneumoniae infection (24, 45). Our data demonstrate that SAA also participates in host defense of S. aureus cutaneous infection in vivo. SAA has also been reported to regulate immune cells, such as promoting the secretion of cytokines, such as IL-17 and IL-22 (46, 47), and recruiting neutrophils and monocytes (48). Whether the immunomodulatory function of SAA is also involved in the resistance against S. aureus infection remains to be confirmed by further studies.
Although no case report has identified a genetic human SAA deficiency, evidence suggests that many patients with liver dysfunction have defective acute-phase responses to bacterial infection, contributing to an increased susceptibility to bacterial infection (49). SAA plays a pivotal role in regulating inflammatory responses, suggesting that SAA may be not only the major source of acute-phase proteins and a clinical marker, but a functional immune response player that provides a novel therapeutic strategy for inflammation during the innate immune response to infection. However, its direct innate immune effect in humans needs to be confirmed by large-scale clinical trials.
In summary (Fig. 7), we demonstrated that SAA not only bound to anionic phospholipids of the bacterial cell membrane and self-assembled to form micelles, but elicited direct bactericidal effects through a hydrophobic N-terminal region as well. Our study also confirmed that SAA is abundantly expressed in S. aureus infection sites and protects from S. aureus cutaneous infection in vivo.
Figure 7.
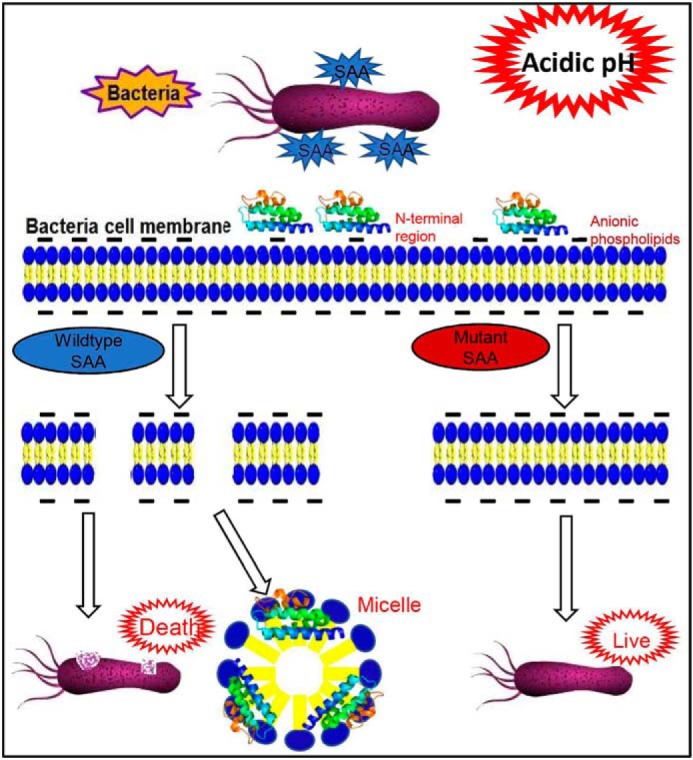
Graphic summary. The bacterial killing mechanism of SAA under acidic conditions. SAA bounds to anionic phospholipid of the bacterial membrane and disrupts cell membrane by self-assembling to form micelles through the hydrophobic N-terminal region.
Materials and methods
Animals
All mice were housed and bred in the specific pathogen–free facility at China Agricultural University. 8–12-Week-old male mice were used for all experiments. All animal experiments were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee of China Agricultural University (approval number SKLAB-2017-01-05). Saa1 and Saa2 double knockout mice (C57BL/6 background) were generated by the Nanjing biomedical research institute of Nanjing University. SAA1/2 double knockout mice were generated by a 15-kb chromosomal deletion at the Saa1 and Saa2 locus in the mouse genome using CRISPR/Cas9 technology. IL-6–deficient (Il6−/−) mice (C57BL/6 background) were obtained from Dr. Zhinan Yin (Jinan University, Guangzhou, China).
Microorganisms
Bacterial isolates E. coli BL21 and S. aureus ATCC 25923 were obtained from the China General Microbiological Culture Collection Center (CGMCC, Beijing, China).
Antibodies and reagents
All lipids were purchased from Avanti Polar Lipids Inc. (Alabaster, AL). The polyclonal antibody for mSAA1/2 was purchased from R&D Systems (Minneapolis, MN). The polyclonal antibodies for human SAA1/2 (hSAA1/2) and mouse SAA3 (mSAA3) were purchased from ABclonal (Wuhan, China). Terbium chloride (TbCl3) and DPA were purchased from Sigma-Aldrich, and all other chemicals were from Sigma-Aldrich.
Mouse serum sample preparation
Ten-week-old male C57BL/6 mice were intraperitoneally injected with AgNO3 (0.5 ml, 0.01 g/ml). After 24 h, serum was obtained by centrifugation (50).
Gene cloning, protein expression, and purification
The human and mouse SAA cDNAs encoding 103 amino acid residues were cloned into pET21a with an N-terminal His6 tag followed by a tobacco etch virus protease cleavage site and a C-terminal stop codon. Recombinant pET-SAA plasmids were transformed into E. coli strain BL21 (DE3) pLysS (Tiangen, China). The cells were grown at 37 °C in Luria–Bertani (LB) medium supplemented with 50 μg/ml ampicillin and 34 μg/ml chloramphenicol until OD600 reached about 0.6. Protein expression was induced with 0.5 mm isopropyl-β-d-galactoside and incubated at 25 °C; after 30 min, 100 μg/ml rifampicin was added to inhibit E. coli RNA polymerase. After an additional 2.5-h incubation, the cells were harvested and resuspended in lysis buffer (50 mm NaH2PO4, 500 mm NaCl, pH 8.0, for mSAA1, mSAA2, mSAA3, hSAA1, and hSAA2), and then a 0.5% final concentration of n-dodecyl-β-d-maltoside was added to the lysis buffer and incubated for 3 h at 4 °C after sonication. The lysate was clarified by centrifugation at 42,000 × g for 30 min at 4 °C. The supernatant was loaded onto a nickel-SepharoseTM (GE Healthcare) column pre-equilibrated with lysis buffer containing 0.05% n-dodecyl-β-d-maltoside. The column was washed at least 40 times with 25 mm imidazole in lysis buffer to completely remove nonspecific contaminants and detergent (21), and the proteins were eluted in lysis buffer containing 300 mm imidazole. The eluted product was pooled and buffer-exchanged into 20 mm Tris-HCl, pH 8, 200 mm NaCl and then digested with tobacco etch virus protease at 18 °C for 12 h. Undigested protein was removed by passing the protein through a nickel-affinity matrix and collected only the flow through. The eluate was concentrated and further purified with a Resource Q column (GE Healthcare) and a HiLoad 16/600 SuperdexTM200pg column (GE Healthcare) in 100 mm NaCl, 20 mm Tris, pH 8, 1 mm tris(2-carboxyethyl)phosphine, and 5% glycerol. Finally, the purified protein was >95% pure according to SDS-PAGE analysis.
Mutagenesis
Deletion mutant was generated using standard PCR methods. Protein expression and purification were done as for WT SAA
Viable count analysis
Bacteria were grown overnight in LB medium. The microbes were washed twice with 10 mm sodium phosphate, 150 mm NaCl buffer, pH 7.4, and diluted in the same buffer at a pH range of 5.2–7.4. Then about 107 cfu of microbes were incubated at 37 °C for 2 h at the indicated concentrations with SAA. To quantify bactericidal activity, serial dilutions of the incubated mixtures were plated on LB (for bacteria), followed by an incubation at 37 °C overnight, and the number of cfu was determined. Then 100% survival was defined as total survival of bacteria in the same buffer and under the same conditions as in the absence of proteins.
Binding assay
E. coli and S. aureus (108 cfu) were incubated with 5 μm mSAA1 in 500 μl of 10 mm sodium phosphate, 150 mm NaCl, pH 5.5 or pH 7.4 for 1 h at 37 °C and centrifuged, and the pellets were washed three times in the same buffer. The pellet and the supernatant were resuspended in SDS sample buffer, electrophoresed (15% SDS-PAGE), and then transferred to a polyvinylidene difluoride membrane with a transfer buffer (25 mm Tris, 192 mm glycine, and 20% (v/v) methanol). The membranes were blocked with 5% dried milk in TBS-T (20 mm Tris, 500 mm NaCl, 0.05% Tween 20) and incubated with mSAA1 antibody for 1 h at room temperature. Then secondary antibody (rabbit anti-goat horseradish peroxidase; 1:5,000; Santa Cruz Biotechnology). was diluted into hybridization buffer and incubated at room temperature for 1 h. The blot was then washed five times for 5 min with wash buffer, and horseradish peroxidase was detected with ECL reagent (GE Healthcare).
Liposome preparation
The method for liposome preparation in this study was modified from Ding et al. (20). In brief, different components of phospholipids were separately dissolved in chloroform. The lipid of the specified fraction (0.5 μmol) was mixed in a glass vial. The solvent was evaporated under a stream of nitrogen, and the dried lipid film was hydrated at room temperature and mixed in 500 μl of buffer 1 (20 mm HEPES (pH 7.4) or 20 mm MES (pH 5.5) and 150 mm NaCl). The Tb3+-packaged liposomes were hydrated in 500 μl of buffer 2 (20 mm HEPES (pH 7.4) or 20 mm MES (pH 5.5)), 100 mm NaCl, 50 mm sodium citrate, and 15 mm TbCl3. Liposomes were generated by repeated pushes of hydrated lipids through a 100-nm polycarbonate filter (Whatman, UK) 35 times using a Mini-Extruder apparatus (Avanti Polar Lipids). After the push process, the liposomes were added to a centrifugal filter tube (Amicon Ultra-4, 100,000 molecular weight cutoff; Millipore), and the Tb3+ ions outside the liposome were removed by repeated washings five times with Tb3+-free buffer 2. The liposomes were replaced with buffer 1 for use.
Lipid leakage assay
The lipid leakage assay implemented for this study was modified from Ding et al. (20). In brief, a 30-μl aliquot of Tb3+-packaged liposomes was mixed with 60 μl of DPA-containing buffer 1, and 10 μl of the specified concentration of SAA recombinant protein was added before the assay. The final concentration of phospholipid was 300 μm, and the concentration of DPA was 15 μm. The Tb3+/DPA chelate was examined using excitation and emission wavelengths of 270 and 490 nm, respectively, in a full-wavelength fluorescence microplate reader. The emission fluorescence before the addition of the SAA protein was regarded as Ft0. A 10 μl aliquot of the protein was added to the designated final concentration, and the emission fluorescence was continuously recorded as Ft at 30-s intervals. After 20 min, 10 μl of 1% Triton X-100 was added to achieve complete release of Tb3+, and the average of the first three fluorescence readings was defined as Ft100, if the fluorescence value of the protein treatment group was greater than after adding 1% Triton X-100 The fluorescence value takes three maximum fluorescence values of the protein treatment group as Ft100. The percentage of liposome leakage at each time point is defined as follows: Leakage (t) (%) = (Ft − Ft0) × 100/(Ft100 − Ft0).
Transmission EM
Midlogarithmic phase E. coli and S. aureus cells (OD600 at 0.4) were incubated with 10 μm mSAA1 at pH 5.5 for 2 h. After treatment, the bacterial pellets were pre-fixed with 2.5% glutaraldehyde at pH 7.4 for 2 h at 4 °C and post-fixed in 1% osmium tetroxide buffered at pH 7.4 for 2 h at 4 °C. The samples were dehydrated in acetone (50, 70, 90, 95, and 100%). The cells were immersed in EPON resin, and ultrathin sections were examined under a JEOL JEM-1230 (Jeol Ltd., Tokyo, Japan). The leakage area of bacterial cell component was quantified by Image J (National Institutes of Health).
Negative staining EM of the SAA-lipid micelles
SAA proteins (5 μm) were incubated with the indicated liposomes (500 μm lipids) at room temperature for 30 min. Aliquots of the mixture (5 μl) were transferred to a glow-discharged carbon-coated copper grid for EM and negatively stained with 2% uranyl acetate. Samples were imaged on a JEM-1400 electron microscope (Jeol Ltd.) at 120 kV.
Biolayer interferometry
Biolayer interferometry analysis was performed using an Octet Red 96 system (Molecular Devices, Fremont, CA) with SA chips at room temperature (25 °C). All assays were performed in 10 mm sodium phosphate, 150 mm NaCl, pH 5.5 or 7.4. SAA proteins were biotinylated and then flowed through with 100 μm PC liposome or cardiolipin liposome in different buffers.
Immunohistochemistry
Immunohistochemistry was performed as described previously (51). Briefly, 4-μm tissue sections were deparaffinized in xylene and rehydrated through a series of decreasing ethanol concentrations. The slides were pretreated with hydrogen peroxide (3%) for 10 min to remove endogenous peroxidase, followed by antigen retrieval in a microwave for 15 min in 10 mm citrate buffer (pH 6.0). The primary antibodies were applied, followed by washing and incubation with the biotinylated secondary antibody for 30 min at room temperature. The slides were counterstained with hematoxylin and dehydrated in alcohol and xylene before mounting.
RNA extraction and real-time RT-PCR
Total RNA and quantitative real-time PCR were performed as described previously (51). Data were analyzed with LightCycler® 480 software, version 1.5 (Roche Applied Science). Relative quantification of gene expression was performed using the standard curves and normalized to the value for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in each sample. Primers used for Saa1/2, Saa3, Il6, and Gapdh are listed in Table S1.
Cutaneous infection in vivo
The mouse model of cutaneous infection was modified from Li et al. (52) and Zhang et al. (43). In brief, the backs of age-matched adults were shaved, and hair was removed by using chemical depilation. On the same day, S. aureus was grown in LB medium overnight. The next day, 1 ml of overnight culture of S. aureus was re-inoculated into 30 ml of fresh LB and grew to logarithmic phase (OD600 = 0.7–0.8). Then the bacterium was centrifuged, and the pellets were washed and resuspended in sterile PBS. 3 × 106 cfu of live S. aureus was subcutaneously injected into mouse back skin (n = 5/group). Mice were euthanized after 4 days. In some experiments, skin around the abscess was collected and homogenized in PBS to determine the number of surviving S. aureus. In other experiments, RNA from normal or infected skin was either collected for real-time RT-PCR or stored in 4% paraformaldehyde for immunostaining. Samples were randomized during data collection. Investigators were not blinded to the group allocation during data acquisition.
Statistics
Data were analyzed for statistical significance with SPSS 12.0.1. All numerical data are presented as mean ± S.E.M. p < 0.05 was considered significant. All graphs were generated with GraphPad Prism 6.0 (GraphPad Software, Inc.)
Author contributions
H. Z. and X. L. conceptualization; H. Z. and X. L. data curation; H. Z. and X. L. formal analysis; H. Z., H. L., J. Z., H. F., L. J., W. M., S. M., and S. W. investigation; H. Z. visualization; H. Z. methodology; H. Z. writing-original draft; H. Y. and X. L. funding acquisition; Z. Y. resources; X. L. supervision; X. L. validation; X. L. project administration; X. L. writing-review and editing.
Supplementary Material
Acknowledgments
We thank Prof. Feng Shao and Xuyan Shi (National Institute of Biological Sciences, Beijing) for providing protocols of liposome preparation and the liposome leakage assay. We also thank Dr. Yuanyuan Chen and Dr. Zhenwei Yang (Institute of Biophysics, Chinese Academy of Sciences) for expert technical assistance.
This work was supported by National Science and Technology Major Project Grant 2013ZX10004608; National Key Research and Development Grant 2018YFC1004702; National Natural Science Foundation of China Grants NSFC31970802, NSFC31071316 and NSFC81261130024; Natural Science Foundation of Beijing Grant 20G10613; State Key Laboratory of Agrobiotechnology, China Agricultural University, Grant 2018SKLAB6-18; and project for Extramural Scientists of State Key Laboratory of Agrobiotechnology Grant 2015SKLAB6-2. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Table S1 and Figs. S1 and S2.
- SAA
- serum amyloid A
- AMP
- antimicrobial peptide
- PG
- phosphatidylglycerol
- PC
- phosphatidylcholine
- SA
- streptavidin
- HDL
- high-density lipoprotein
- DKO
- double knockout
- LB
- Luria–Bertani
- OD
- optical density
- DPA
- dipicolinic acid
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- IL
- interleukin.
References
- 1. Uhlar C. M., and Whitehead A. S. (1999) Serum amyloid A, the major vertebrate acute-phase reactant. Eur. J. Biochem. 265, 501–523 10.1046/j.1432-1327.1999.00657.x [DOI] [PubMed] [Google Scholar]
- 2. Tannock L. R., De Beer M. C., Ji A., Shridas P., Noffsinger V. P., den Hartigh L., Chait A., De Beer F. C., and Webb N. R. (2018) Serum amyloid A3 is a high density lipoprotein-associated acute-phase protein. J. Lipid Res. 59, 339–347 10.1194/jlr.M080887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Urieli-Shoval S., Cohen P., Eisenberg S., and Matzner Y. (1998) Widespread expression of serum amyloid A in histologically normal human tissues: predominant localization to the epithelium. J. Histochem. Cytochem. 46, 1377–1384 10.1177/002215549804601206 [DOI] [PubMed] [Google Scholar]
- 4. Bozinovski S., Uddin M., Vlahos R., Thompson M., McQualter J. L., Merritt A. S., Wark P. A., Hutchinson A., Irving L. B., Levy B. D., and Anderson G. P. (2012) Serum amyloid A opposes lipoxin A(4) to mediate glucocorticoid refractory lung inflammation in chronic obstructive pulmonary disease. Proc. Natl. Acad. Sci. U.S.A. 109, 935–940 10.1073/pnas.1109382109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cleaver J. O., You D., Michaud D. R., Pruneda F. A., Juarez M. M., Zhang J., Weill P. M., Adachi R., Gong L., Moghaddam S. J., Poynter M. E., Tuvim M. J., and Evans S. E. (2014) Lung epithelial cells are essential effectors of inducible resistance to pneumonia. Mucosal Immunol. 7, 78–88 10.1038/mi.2013.26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Erman A., Lakota K., Mrak-Poljsak K., Blango M. G., Krizan-Hergouth V., Mulvey M. A., Sodin-Semrl S., and Veranic P. (2012) Uropathogenic Escherichia coli induces serum amyloid a in mice following urinary tract and systemic inoculation. PLoS One 7, e32933 10.1371/journal.pone.0032933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Quinton L. J., Blahna M. T., Jones M. R., Allen E., Ferrari J. D., Hilliard K. L., Zhang X., Sabharwal V., Algül H., Akira S., Schmid R. M., Pelton S. I., Spira A., and Mizgerd J. P. (2012) Hepatocyte-specific mutation of both NF-κB RelA and STAT3 abrogates the acute phase response in mice. J. Clin. Invest. 122, 1758–1763 10.1172/JCI59408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hilliard K. L., Allen E., Traber K. E., Yamamoto K., Stauffer N. M., Wasserman G. A., Jones M. R., Mizgerd J. P., and Quinton L. J. (2015) The lung-liver axis: a requirement for maximal innate immunity and hepatoprotection during pneumonia. Am. J. Respir. Cell Mol. Biol. 53, 378–390 10.1165/rcmb.2014-0195OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Morizane S., Mizuno K., Takiguchi T., Sugimoto S., and Iwatsuki K. (2017) The involvement of serum amyloid A in psoriatic inflammation. J. Invest. Dermatol. 137, 757–760 10.1016/j.jid.2016.10.016 [DOI] [PubMed] [Google Scholar]
- 10. Törcsik D., Kovács D., Póliska S., Szentkereszty-Kovács Z., Lovászi M., Hegyi K., Szegedi A., Zouboulis C. C., and Ståhle M. (2018) Genome wide analysis of TLR1/2- and TLR4-activated SZ95 sebocytes reveals a complex immune-competence and identifies serum amyloid A as a marker for activated sebaceous glands. PLoS One 13, e0198323 10.1371/journal.pone.0198323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gallo R. L., and Hooper L. V. (2012) Epithelial antimicrobial defence of the skin and intestine. Nat. Rev. Immunol. 12, 503–516 10.1038/nri3228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang L. J., and Gallo R. L. (2016) Antimicrobial peptides. Curr. Biol. 26, R14–R19 10.1016/j.cub.2015.11.017 [DOI] [PubMed] [Google Scholar]
- 13. Eyerich S., Eyerich K., Traidl-Hoffmann C., and Biedermann T. (2018) Cutaneous barriers and skin immunity: differentiating a connected network. Trends Immunol. 39, 315–327 10.1016/j.it.2018.02.004 [DOI] [PubMed] [Google Scholar]
- 14. Zhang J., Koh J., Lu J., Thiel S., Leong B. S., Sethi S., He C. Y., Ho B., and Ding J. L. (2009) Local inflammation induces complement crosstalk which amplifies the antimicrobial response. PLoS Pathog. 5, e1000282 10.1371/journal.ppat.1000282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lardner A. (2001) The effects of extracellular pH on immune function. J. Leukoc. Biol. 69, 522–530 [PubMed] [Google Scholar]
- 16. Kovacevic N., and Belosevic M. (2015) Molecular and functional characterization of goldfish (Carassius auratus L.) serum amyloid A. Fish Shellfish Immunol. 47, 942–953 10.1016/j.fsi.2015.10.041 [DOI] [PubMed] [Google Scholar]
- 17. Yu J., Tang Y., Li J., Li H., Yu F., Yu W., He F., Fu C., and Mao S. (2017) Cloning, expression analysis, and antibacterial propertiesof three serum amyloid A in common carp (Cyprinus carpio). Fish Shellfish Immunol. 65, 267–277 10.1016/j.fsi.2017.04.021 [DOI] [PubMed] [Google Scholar]
- 18. Eckhardt E. R., Witta J., Zhong J., Arsenescu R., Arsenescu V., Wang Y., Ghoshal S., de Beer M. C., de Beer F. C., and de Villiers W. J. (2010) Intestinal epithelial serum amyloid A modulates bacterial growth in vitro and pro-inflammatory responses in mouse experimental colitis. BMC Gastroenterol. 10, 133 10.1186/1471-230X-10-133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hirakura Y., Carreras I., Sipe J. D., and Kagan B. L. (2002) Channel formation by serum amyloid A: a potential mechanism for amyloid pathogenesis and host defense. Amyloid 9, 13–23 10.3109/13506120209072440 [DOI] [PubMed] [Google Scholar]
- 20. Ding J., Wang K., Liu W., She Y., Sun Q., Shi J., Sun H., Wang D. C., and Shao F. (2016) Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535, 111–116 10.1038/nature18590 [DOI] [PubMed] [Google Scholar]
- 21. Liu X., Zhang Z., Ruan J., Pan Y., Magupalli V. G., Wu H., and Lieberman J. (2016) Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158 10.1038/nature18629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Beck W. H., Adams C. P., Biglang-Awa I. M., Patel A. B., Vincent H., Haas-Stapleton E. J., and Weers P. M. (2013) Apolipoprotein A-I binding to anionic vesicles and lipopolysaccharides: role for lysine residues in antimicrobial properties. Biochim. Biophys. Acta 1828, 1503–1510 10.1016/j.bbamem.2013.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lu J., Yu Y., Zhu I., Cheng Y., and Sun P. D. (2014) Structural mechanism of serum amyloid A-mediated inflammatory amyloidosis. Proc. Natl. Acad. Sci. U.S.A. 111, 5189–5194 10.1073/pnas.1322357111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Derebe M. G., Zlatkov C. M., Gattu S., Ruhn K. A., Vaishnava S., Diehl G. E., MacMillan J. B., Williams N. S., and Hooper L. V. (2014) Serum amyloid A is a retinol binding protein that transports retinol during bacterial infection. eLife 3, e03206 10.7554/eLife.03206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Frame N. M., and Gursky O. (2016) Structure of serum amyloid A suggests a mechanism for selective lipoprotein binding and functions: SAA as a hub in macromolecular interaction networks. FEBS Lett. 590, 866–879 10.1002/1873-3468.12116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kopf M., Baumann H., Freer G., Freudenberg M., Lamers M., Kishimoto T., Zinkernagel R., Bluethmann H., and Köhler G. (1994) Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature 368, 339–342 10.1038/368339a0 [DOI] [PubMed] [Google Scholar]
- 27. Coetzee G. A., Strachan A. F., van der Westhuyzen D. R., Hoppe H. C., Jeenah M. S., and de Beer F. C. (1986) Serum amyloid A-containing human high density lipoprotein 3: density, size, and apolipoprotein composition. J. Biol. Chem. 261, 9644–9651 [PubMed] [Google Scholar]
- 28. Noborn F., Ancsin J. B., Ubhayasekera W., Kisilevsky R., and Li J. P. (2012) Heparan sulfate dissociates serum amyloid A (SAA) from acute-phase high-density lipoprotein, promoting SAA aggregation. J. Biol. Chem. 287, 25669–25677 10.1074/jbc.M112.363895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jayaraman S., Fändrich M., and Gursky O. (2019) Synergy between serum amyloid A and secretory phospholipase A2 suggests a vital role for an ancient protein in lipid clearance. eLife 8, e46630 10.7554/eLife.46630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rydengård V., Shannon O., Lundqvist K., Kacprzyk L., Chalupka A., Olsson A. K., Mörgelin M., Jahnen-Dechent W., Malmsten M., and Schmidtchen A. (2008) Histidine-rich glycoprotein protects from systemic Candida infection. PLoS Pathog. 4, e1000116 10.1371/journal.ppat.1000116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Reigstad C. S., and Bäckhed F. (2010) Microbial regulation of SAA3 expression in mouse colon and adipose tissue. Gut Microbes 1, 55–57 10.4161/gmic.1.1.10514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shah C., Hari-Dass R., and Raynes J. G. (2006) Serum amyloid A is an innate immune opsonin for Gram-negative bacteria. Blood 108, 1751–1757 10.1182/blood-2005-11-011932 [DOI] [PubMed] [Google Scholar]
- 33. Brogden K. A. (2005) Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 3, 238–250 10.1038/nrmicro1098 [DOI] [PubMed] [Google Scholar]
- 34. Malik E., Dennison S. R., Harris F., and Phoenix D. A. (2016) pH-dependent antimicrobial peptides and proteins, their mechanisms of action and potential as therapeutic agents. Pharmaceuticals (Basel) 9, E67 10.3390/ph9040067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Propheter D. C., Chara A. L., Harris T. A., Ruhn K. A., and Hooper L. V. (2017) Resistin-like molecule β is a bactericidal protein that promotes spatial segregation of the microbiota and the colonic epithelium. Proc. Natl. Acad. Sci. U.S.A. 114, 11027–11033 10.1073/pnas.1711395114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Patel H., Bramall J., Waters H., De Beer M. C., and Woo P. (1996) Expression of recombinant human serum amyloid A in mammalian cells and demonstration of the region necessary for high-density lipoprotein binding and amyloid fibril formation by site-directed mutagenesis. Biochem. J. 318, 1041–1049 10.1042/bj3181041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ohta S., Tanaka M., Sakakura K., Kawakami T., Aimoto S., and Saito H. (2009) Defining lipid-binding regions of human serum amyloid A using its fragment peptides. Chem. Phys. Lipids 162, 62–68 10.1016/j.chemphyslip.2009.07.008 [DOI] [PubMed] [Google Scholar]
- 38. Quist A., Doudevski I., Lin H., Azimova R., Ng D., Frangione B., Kagan B., Ghiso J., and Lal R. (2005) Amyloid ion channels: a common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. U.S.A. 102, 10427–10432 10.1073/pnas.0502066102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Frame N. M., Jayaraman S., Gantz D. L., and Gursky O. (2017) Serum amyloid A self-assembles with phospholipids to form stable protein-rich nanoparticles with a distinct structure: a hypothetical function of SAA as a “molecular mop” in immune response. J. Struct. Biol. 200, 293–302 10.1016/j.jsb.2017.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jayaraman S., Gantz D. L., Haupt C., and Gursky O. (2017) Serum amyloid A forms stable oligomers that disrupt vesicles at lysosomal pH and contribute to the pathogenesis of reactive amyloidosis. Proc. Natl. Acad. Sci. U.S.A. 114, E6507–E6515 10.1073/pnas.1707120114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Harrington J. M., Howell S., and Hajduk S. L. (2009) Membrane permeabilization by trypanosome lytic factor, a cytolytic human high density lipoprotein. J. Biol. Chem. 284, 13505–13512 10.1074/jbc.M900151200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Miller L. S., and Cho J. S. (2011) Immunity against Staphylococcus aureus cutaneous infections. Nat. Rev. Immunol. 11, 505–518 10.1038/nri3010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang L. J., Guerrero-Juarez C. F., Hata T., Bapat S. P., Ramos R., Plikus M. V., and Gallo R. L. (2015) Innate immunity: dermal adipocytes protect against invasive Staphylococcus aureus skin infection. Science 347, 67–71 10.1126/science.1260972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McFarland-Mancini M. M., Funk H. M., Paluch A. M., Zhou M., Giridhar P. V., Mercer C. A., Kozma S. C., and Drew A. F. (2010) Differences in wound healing in mice with deficiency of IL-6 versus IL-6 receptor. J. Immunol. 184, 7219–7228 10.4049/jimmunol.0901929 [DOI] [PubMed] [Google Scholar]
- 45. Zheng M., Horne W., McAleer J. P., Pociask D., Eddens T., Good M., Gao B., and Kolls J. K. (2016) Therapeutic role of interleukin 22 in experimental intra-abdominal Klebsiella pneumoniae infection in mice. Infect. Immun. 84, 782–789 10.1128/IAI.01268-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sano T., Huang W., Hall J. A., Yang Y., Chen A., Gavzy S. J., Lee J. Y., Ziel J. W., Miraldi E. R., Domingos A. I., Bonneau R., and Littman D. R. (2015) An IL-23R/IL-22 circuit regulates epithelial serum amyloid A to promote local effector Th17 responses. Cell 163, 381–393 10.1016/j.cell.2015.08.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang G., Liu J., Wu L., Fan Y., Sun L., Qian F., Chen D., and Ye R. D. (2018) Elevated expression of serum amyloid A3 protects colon epithelium against acute injury through TLR2-dependent induction of neutrophil IL-22 expression in a mouse model of colitis. Front. Immunol. 9, 1503 10.3389/fimmu.2018.01503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. De Buck M., Gouwy M., Wang J. M., Van Snick J., Opdenakker G., Struyf S., and Van Damme J. (2016) Structure and expression of different serum amyloid A (SAA) variants and their concentration-dependent functions during host insults. Curr. Med. Chem. 23, 1725–1755 10.2174/0929867323666160418114600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhou Z., Xu M. J., and Gao B. (2016) Hepatocytes: a key cell type for innate immunity. Cell. Mol. Immunol. 13, 301–315 10.1038/cmi.2015.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Han C. Y., Tang C., Guevara M. E., Wei H., Wietecha T., Shao B., Subramanian S., Omer M., Wang S., O'Brien K. D., Marcovina S. M., Wight T. N., Vaisar T., de Beer M. C., de Beer F. C., Osborne W. R., Elkon K. B., and Chait A. (2016) Serum amyloid A impairs the antiinflammatory properties of HDL. J. Clin. Invest. 126, 266–281 10.1172/JCI83475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yu W., Zheng H., Lin W., Tajima A., Zhang Y., Zhang X., Zhang H., Wu J., Han D., Rahman N. A., Korach K. S., Gao G. F., Inoue I., and Li X. (2014) Estrogen promotes Leydig cell engulfment by macrophages in male infertility. J. Clin. Invest. 124, 2709–2721 10.1172/JCI59901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Li C., Li H., Jiang Z., Zhang T., Wang Y., Li Z., Wu Y., Ji S., Xiao S., Ryffel B., Radek K. A., Xia Z., and Lai Y. (2014) Interleukin-33 increases antibacterial defense by activation of inducible nitric oxide synthase in skin. PLoS Pathog. 10, e1003918 10.1371/journal.ppat.1003918 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.