

Abstract
Fatty acid–binding protein 4 (FABP4) is predominantly expressed in adipocytes and macrophages and regulates metabolic and inflammatory pathways. FABP4 is secreted from adipocytes during lipolysis, and elevated circulating FABP4 levels are associated with obesity, metabolic disease, and cardiac dysfunction. We previously reported that the bacterial respiratory pathogen Chlamydia pneumoniae infects murine adipocytes and exploits host FABP4 to mobilize fat and replicate within adipocytes. However, whether C. pneumoniae induces FABP4 secretion from adipocytes has not been determined. Here, we show that FABP4 is actively secreted by murine adipocytes upon C. pneumoniae infection. Chemical inhibition of lipase activity and genetic deficiency of hormone-sensitive lipase blocked FABP4 secretion from C. pneumoniae–infected adipocytes. Mechanistically, C. pneumoniae infection induced endoplasmic reticulum (ER) stress and the unfolded protein response (UPR), resulting in elevated levels of mitochondrial reactive oxygen species and cytosolic Ca2+. Of note, exposure to a mitochondrial reactive oxygen species–specific scavenger, MitoTEMPO, reduced FABP4 release from C. pneumoniae–infected adipocytes. Furthermore, treatment with azoramide, which protects cells against ER stress, decreased FABP4 release from C. pneumoniae–infected adipocytes. Using gene silencing of CHOP (C/EBP homologous protein), a central regulator of ER stress, we further validated the role of C. pneumoniae infection–induced ER stress/UPR in promoting FABP4 secretion. Overall, these results indicate that C. pneumoniae infection robustly induces FABP4 secretion from adipocytes by stimulating ER stress/UPR. Our findings shed additional light on the etiological link between C. pneumoniae infection and metabolic syndrome.
Keywords: Chlamydia, bacteria, infection, adipocyte, fatty acid binding protein, endoplasmic reticulum stress (ER stress), lipolysis, unfolded protein response (UPR), oxidative stress, metabolic disorder
Introduction
Adipocyte fatty acid–binding protein (FABP4),3 also known as adipocyte protein 2 (aP2), is abundantly expressed in adipocytes and functions as an intracellular lipid chaperone that can affect the uptake, transportation, esterification, and β-oxidation of fatty acids and that regulates energy balance and lipid signal transduction within cells (1, 2). Under fasting and lipolysis stimuli, FABP4 has been shown to be actively secreted from adipocytes (3–5) and acts on the liver to stimulate glucose production (6). In humans, elevated circulating FABP4 levels are associated with obesity and metabolic diseases. FABP4 acts on multiple integrated pathways to regulate lipid metabolism and inflammation, impairs insulin action, promotes glucose production, and contributes to the pathogenesis of immune–metabolic diseases such as diabetes mellitus and atherosclerosis (7–11). However, the role of infectious agents on FABP4 secretion or the role of FABP4 on the bacterial pathogenesis has not been studied.
The association of Chlamydia pneumoniae infection with metabolic syndrome has been intensively studied (12–15); however, whether C. pneumoniae has a causal role in metabolic syndrome remains undetermined (16–19). We have recently demonstrated that C. pneumoniae proliferates in mature adipocytes by inducing lipolysis and unveils a new mechanism of host lipid metabolism modulation by C. pneumoniae infection (20). We reported that liberated free fatty acids are utilized to generate ATP via β-oxidation, which C. pneumoniae usurps for its replication. C. pneumoniae exploits host FABP4 to facilitate fat mobilization and intracellular replication in adipocytes. However, whether C. pneumoniae infection causes FABP4 secretion from adipocytes has not been clarified.
The endoplasmic reticulum (ER) is an important intracellular compartment for regulation of protein synthesis and lipid metabolism. Perturbations of ER functions, referred to as “ER stress,” leads to the activation of the unfolded protein response (UPR) (21) and has been linked to numerous pathological conditions, including inflammation, cardiovascular diseases, and metabolic disorders (22). The UPR relies on ER membrane-localized sensors, including ATF6 (activating transcription factor 6), IRE1 (inositol-requiring enzyme 1), and dsRNA-dependent protein kinase R–like ER kinase (PERK), which at steady state are bound to the ER chaperone immunoglobulin protein (BiP), also known as GRP78 (78-kDa glucose-related protein). The UPR has emerged as a key target for host cells and viruses to control infection outcomes (23). However, the connection between bacterial pathogens and the UPR has been poorly explored (21, 24)
Recently, George et al. (25) have demonstrated that the three transducers of UPR (PERK, IRE1α, and ATF6α) are activated during Chlamydia muridarum infection of murine oviduct epithelial cells and suggested that UPR increases host-cell glucose utilization, ATP synthesis by substrate level phosphorylation, and phospholipid production, resulting in bacterial replication. Little, however, is known about the role of ER stress/UPR on C. pneumoniae infection or the possible linkage to C. pneumoniae infection–induced pathogenesis.
In this study, we found that FABP4 is secreted from adipocytes by C. pneumoniae infection via ER stress/UPR. Our data indicate that C. pneumoniae infection–induced ER stress/UPR causes the elevation of mitochondrial reactive oxygen species (ROS) and cytoplasmic calcium in adipocytes, resulting in robust FABP4 secretion associated with lipolysis. These results demonstrate that C. pneumoniae infection–induced ER stress/UPR causes robust secretion of FABP4 from adipocytes and provide new insights into the etiological link between C. pneumoniae infection and metabolic syndrome.
Results
C. pneumoniae infection induces FABP4 secretion from murine adipocytes
We previously reported that C. pneumoniae successfully infects and proliferates in differentiated 3T3-L1 mouse adipocytes by inducing vigorous lipolysis (20). Because FABP4 is known to be secreted by adipocytes subjected to lipolytic agonists, we examined whether C. pneumoniae infection–induced lipolysis causes FABP4 secretion from adipocytes. Immunoblot analyses of cultured medium of adipocytes revealed that FABP4 secretion was robustly induced by infection with C. pneumoniae when compared with mock infections (Fig. 1A and Fig. S1). Increased FABP4 secretion from adipocytes upon C. pneumoniae infection was also confirmed by ELISA (Fig. 1B). Expression of Fabp4 mRNA was significantly induced by C. pneumoniae infection (Fig. 1C). A lactate dehydrogenase (LDH) release assay revealed similar cell death rates among C. pneumoniae– and mock-infected adipocytes (Fig. 1D), suggesting that FABP4 is actively secreted from live cells. Taken together, these results clearly demonstrate that C. pneumoniae infection in adipocytes induces FABP4 secretion associated with lipolysis.
Figure 1.

C. pneumoniae infection induces the secretion of FABP4 from murine adipocytes. A, immunoblot analysis of FABP4 in cultured medium (CM) and cell lysates (CL) of 3T3-L1 adipocytes after mock or C. pneumoniae (Cpn) infection for 2–24 h. β-Actin served as the standard. B, secretion of FABP4 was measured in the cultured medium of 3T3-L1 adipocytes after mock or Cpn infection for 2–24 h. A 4-h incubation with forskolin (20 μm) served as the positive control for lipolysis. C, relative levels of Fabp4 mRNA in 3T3-L1 adipocytes after mock or Cpn infection for 4–24 h, as determined by real-time PCR. Gus mRNA served as the internal control. D, LDH assay using the supernatant of 3T3-L1 adipocytes at 2–24 h after mock or Cpn infection (n = 3/group; B–D). **, p < 0.01 by two-way ANOVA (B and C). The data are shown as the means ± S.E. and are representative of at least three experiments.
Chemical inhibition or genetic manipulation of HSL abrogates C. pneumoniae infection–induced FABP4 secretion from adipocytes
Next, we examined the mechanism underlying C. pneumoniae infection–induced FABP4 secretion from adipocytes. FABP4 secretion is responsive to signals that induce lipolysis, including β-adrenergic receptor agonists and forskolin (an adenylyl cyclase activator); furthermore, chemical inhibition or genetic deficiency of hormone-sensitive lipase (HSL) and adipose triglyceride lipase (ATGL) abrogates β-adrenergic–induced FABP4 secretion (5). It is well-known that after lipolytic stimulation, HSL is phosphorylated and translocated to lipid droplet surfaces via the cAMP–PKA–HSL signaling pathway (Fig. 2A) (26, 27). We previously demonstrated that C. pneumoniae infection induces HSL activation in adipocytes (20). Thus we examined the relative importance of lipase in C. pneumoniae infection–induced FABP4 secretion from adipocytes. Strikingly, FABP4 secretion from C. pneumoniae–infected adipocytes was almost completely blocked by an HSL inhibitor (CAY10499) (Fig. 2B), whereas a monoacylglycerol lipase inhibitor (JZL184) partially decreased FABP4 secretion, and the inhibitory effect of an ATGL inhibitor (atglistatin) (28) was negligible. Treatment with KH7 (adenylyl cyclase inhibitor) or H89 (PKA inhibitor) significantly abrogated FABP4 secretion from C. pneumoniae–infected adipocytes (Fig. 2, C and D), indicating that C. pneumoniae infection–induced FABP4 secretion depends on the cAMP–PKA–HSL axis. Furthermore, the importance of HSL in C. pneumoniae-induced FABP4 secretion was confirmed by data acquired using 3T3-L1 cells stably expressing a short hairpin RNA (shRNA) against mRNA encoding either murine EGFP (control) or HSL. The C. pneumoniae infection–induced FABP4 secretion from 3T3-L1 adipocytes expressing shRNA Lipe encoding HSL was severely abrogated (Fig. 2, F and G). We previously demonstrated that chemical inhibition or genetic manipulation of HSL abrogates the bacterial growth of C. pneumoniae in adipocytes (20). The importance of cAMP–PKA–HSL signaling pathway in the intracellular bacterial growth of C. pneumoniae was further confirmed by the treatment with KH7 or H89 (Fig. S2).
Figure 2.

C. pneumoniae infection–induced FABP4 secretion is regulated by cAMP–PKA–HSL pathway. A, the lipolytic pathway inhibitors used in this experiment. B–E, FABP4 levels were measured in the cultured medium of 3T3-L1 adipocytes at 24 h after Cpn infection at a MOI of 5 in the presence or absence of atglistatin (50 μm), CAY10499 (50 μm) or JZL184 (1 μm) (B), KH7 (50 μm) (C), H89 (50 μm) (D), or DMSO 1% solvent control (E). F and G, 3T3-L1 adipocytes differentiated from 3T3-L1 preadipocyte lines each stably expressing a short hairpin RNA (shRNA) against mRNAs encoding either murine EGFP (control) or HSL were infected with Cpn MOI 5 for 24 h. F, immunoblot analysis of FABP4 in the cultured medium (CM) and cell lysates (CL) of 3T3-L1 adipocytes. β-Actin served as the standard. G, secretion of FABP4 in cultured medium of these 3T3-L1 adipocytes was examined by ELISA (n = 3/group; B–E and G). **, p < 0.01, one-way ANOVA (B–E); two-way ANOVA (G). The data are shown as the means ± S.E. and are representative of at least three experiments. AC, adenylyl cyclase; PKA, protein kinase A; MAGL, monoacylglycerol lipase; TAG, triacylglycerol; DAG, diacylglycerol; MAG, monoacylglycerol; FFA, free fatty acid.
C. pneumoniae infection–induced FABP4 secretion depends on mitochondrial ROS and cytoplasmic Ca2+ elevation
Considering the importance of HSL in C. pneumoniae infection–induced FABP4 secretion, we further examined the conditions required for HSL activation. It was previously reported that ROS facilitate HSL translocation to lipid droplets during lipolysis in adipocytes (26). We found that the increase in mitochondrial ROS generation occurs after C. pneumoniae infection in adipocytes (Fig. 3A). Importantly, MitoTEMPO, a mitochondrial ROS-specific scavenger, markedly inhibited HSL phosphorylation (Fig. 3B), lipolysis (Fig. 3C), and FABP4 secretion (Fig. 3D). Mitochondrial ROS-dependent HSL activation and FABP4 secretion in C. pneumoniae–infected adipocytes were further confirmed by using another mitochondrial ROS scavenger, MitoQ (Fig. 3, E and F) or N-acetyl cysteine (data not shown). We also observed that intracellular Ca2+ levels increased at 12 and 24 h after infection with C. pneumoniae in adipocytes (Fig. 4A). Treatment with BAPTA-AM, an intracellular calcium chelator, attenuated FABP4 secretion in C. pneumoniae–infected adipocytes (Fig. 4B), which is consistent with the recent reports showing the calcium-dependent release of FABP4 from adipocytes (3, 4). Although C. pneumoniae infection–induced lipolysis was not inhibited by BAPTA-AM treatment (Fig. 4C), this treatment decreased the intracellular growth of C. pneumoniae in adipocytes (Fig. 4D). Taken together, these results strongly indicate that elevation of mitochondrial ROS and intracellular calcium play an important role in infection-induced FABP4 secretion in adipocytes.
Figure 3.

C. pneumoniae infection–induced FABP4 secretion depends on mitochondrial ROS. A, flow cytometry (left panel) and quantification (right panel) of MitoSOX-stained 3T3-L1 adipocytes at 24 h after Cpn infection at a MOI of 5 in the presence or absence of MitoTEMPO (100 μm). B and E, immunoblot analysis of p-HSL and HSL in cell lysates of 3T3-L1 adipocytes at 24 h after Cpn infection in the presence or absence of MitoTEMPO (100 μm) (B) or increasing doses of MitoQ (E). A 2-h incubation with forskolin (20 μm) served as the positive control for lipolysis. β-Actin served as the standard. C, glycerol levels were measured in cultured medium of 3T3-L1 adipocytes at 24 h after Cpn infection in the presence or absence of MitoTEMPO. D and F, FABP4 levels in cultured medium of 3T3-L1 adipocytes at 24 h after Cpn infection at a MOI of 5 in the presence or absence of MitoTEMPO (100 μm) (D), increasing doses of MitoQ (Mitoquinone) (F). In A, C, D, and F, n = 3/group. *, p < 0.05; **, p < 0.01, one-way ANOVA (A, C, D, and F). The data are shown as the means ± S.E. and are representative of at least three experiments.
Figure 4.

C. pneumoniae infection–induced FABP4 secretion is regulated by cytoplasmic calcium elevation. A, intracellular Ca2+ was assessed by Fluo-4 AM fluorescence in 3T3-L1 adipocytes after mock or Cpn infection for 2, 4, 12, and 24 h. Thapsigargin (10 μm) for 6 h served as a positive control. B and C, FABP4 (B) and glycerol (C) levels in cultured medium of 3T3-L1 adipocytes at 24 h after Cpn infection at a MOI of 5 in the presence or absence of BAPTA-AM (20 μm). D, the number of infectious EB progeny of 3T3-L1 adipocytes at 24 h after Cpn infection in the presence or absence of BAPTA-AM (20 μm) or BAPTA (50 μm) was determined using an IFU assay (n = 3/group; B–D). *, p < 0.05; **, p < 0.01, one-way ANOVA (B–D). The data are shown as the means ± S.E. and are representative of at least three experiments.
C. pneumoniae infection induces ER stress and the UPR in adipocytes, which leads to lipolysis and FABP4 secretion
Next, we determined what triggers the elevation of mitochondrial ROS and cytosolic Ca2+ after infection with C. pneumoniae. ER stress and the subsequent UPR, especially CHOP expression, induces the elevation of cytoplasmic calcium (29). Furthermore, it has been shown that ER stress induces lipolysis in adipocytes (30). Thus we hypothesized that C. pneumoniae infection in adipocytes might induce ER stress/UPR, which then causes lipolysis and FABP4 secretion via elevating cytoplasmic Ca2+ and mitochondrial ROS. We found that the mRNA expression of Chop, Grp78/Bip (glucose regulated 78-kDa protein), Atf4, and sXbp1 were robustly induced by infection with C. pneumoniae when compared with a mock infection (Fig. 5A). Immunoblot analyses revealed that the central regulator of ER stress, CHOP, as well as Bip and phospho-eIF2α showed time-dependent elevation after C. pneumoniae infection in adipocytes (Fig. 5B and Fig. S3), clearly demonstrating that C. pneumoniae infection induces ER stress and the UPR.
Figure 5.

C. pneumoniae infection induces ER stress and the UPR in adipocytes, which leads to lipolysis and FABP4 secretion. A, relative levels of Chop, Bip, Atf4, or sXbp1 mRNA, as determined by real-time PCR, in 3T3-L1 adipocytes after mock or Cpn infection for 24 h and treatment with thapsigargin (1 μm) for 6 h. Gus mRNA served as the internal control. B, immunoblot analysis of CHOP, BIP, and p-eif2α in cell lysates of 3T3-L1 adipocytes after mock or Cpn infection at 4, 6, and 24 h. A 6-h incubation with thapsigargin (1 μm) served as the positive control for ER stress. β-Actin served as the standard. C, flow cytometry (left panel) and quantification (right panel) of MitoSOX-stained 3T3-L1 adipocytes at 24 h after Cpn infection in the presence or absence of azoramide (30 μm). D–G, glycerol (D and F) and FABP4 (E and G) levels in cultured medium of 3T3-L1 adipocytes at 24 h after Cpn infection at a MOI of 5 in the presence or absence of increasing doses of azoramide (D and E) or in the presence of GSK2606414) (PERK inhibitor) or STF-083010 (IRE1α RNase-specific inhibitor) (F and G). H, secretion of FABP4 in cultured medium of 3T3-L1 adipocytes at 6 h after treatment with thapsigardin (TG, 1 μm) and tunicamycin (TU, 5 μg/ml) was measured by ELISA. LDH assay using the supernatant was performed. As positive control, the cells were lysed with 2% Triton X-100 containing culture medium (n = 3/group; A and C–H). *, p < 0.05; **, p < 0.01, two-way ANOVA (A); one-way ANOVA (C–H). The data are shown as the means ± S.E. and are representative of at least three experiments.
C. pneumoniae infection–induced elevation of mitochondrial ROS was significantly inhibited by the treatment with the chemical chaperone azoramide, which has been shown to reduce ER stress (31, 32) (Fig. 5C). Notably, treatment with azoramide greatly attenuated C. pneumoniae infection–induced lipolysis and FABP4 secretion (Fig. 5, D and E). ER stress/UPR-dependent lipolysis and FABP4 secretion were confirmed using tauroursodeoxycholic acid, another pharmacological chaperone that ameliorates the UPR (data not shown). An IRE1α RNase-specific inhibitor (STF-083010) (33) and PERK inhibitor (GSK2606414) (34) also decreased C. pneumoniae infection–induced lipolysis and FABP4 secretion (Fig. 5, F and G). Furthermore, we found that gene silencing of CHOP or PERK abrogates C. pneumoniae infection–induced FABP4 secretion (Fig. 6, A and B). It is worth mentioning that ER stress/UPR induced by thapsigardin or tunicamycin in 3T3-L1 adipocytes causes FABP4 secretion (Fig. 5H), strongly supporting the ER stress/UPR–mediated FABP4 secretion. Taken together, these results clearly demonstrate that C. pneumoniae infection induces ER stress/UPR in adipocytes and causes FABP4 secretion in murine adipocytes.
Figure 6.
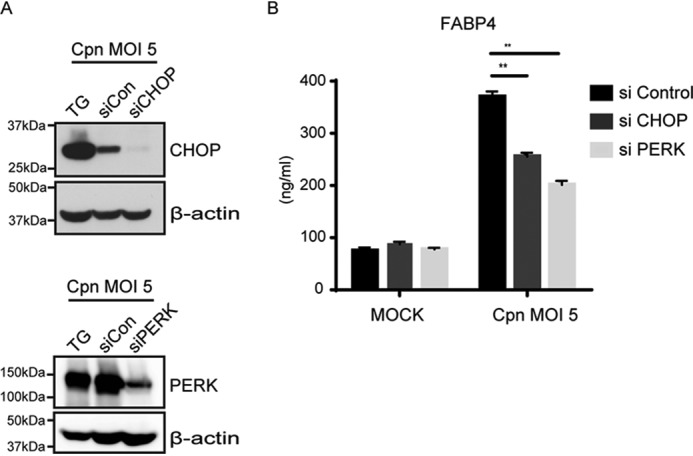
Gene silencing of CHOP or PERK abolishes C. pneumoniae infection–induced FABP4 secretion. A, at 48 h after transfection (siCHOP, siPERK, or siControl), 3T3-L1 adipocytes were infected with Cpn MOI 5 for 24 h, and immunoblot analysis was done to confirm the effectiveness of CHOP or PERK gene silencing. B, FABP4 levels were measured in the cultured medium of 3T3-L1 siControl, siCHOP, or siPERK adipocytes at 24 h after Cpn infection at a MOI of 5 (n = 3/group). **, p < 0.01, two-way ANOVA (B). The data are shown as the means ± S.E. and are representative of at least three experiments.
C. pneumoniae usurps infection-induced ER stress/UPR and subsequent elevation of mitochondrial ROS for intracellular replication
We previously reported the infectivity of adipocytes by C. pneumoniae and the subsequent lipolysis to facilitate bacterial growth (20). We reported that liberated free fatty acids are utilized to generate ATP via β-oxidation, which C. pneumoniae usurps for its replication (20). In this study we have demonstrated that C. pneumoniae infection–induced ER stress/UPR and mitochondrial ROS are the central mechanisms promoting lipolysis and FAPB4 secretion. Finally, we asked the role of ER stress/UPR and subsequent elevation of mitochondrial ROS on bacterial growth. The mitochondrial ROS-specific scavenger, MitoTEMPO and MitoQ, markedly inhibited the intracellular growth of C. pneumoniae in adipocytes (Fig. 7, A and B). Treatment with the chemical chaperone azoramide significantly attenuated intracellular bacterial growth of C. pneumoniae (Fig. 7C). An IRE1α RNase-specific inhibitor (STF-083010) and PERK inhibitor (GSK2606414) also decreased intracellular growth of C. pneumoniae infection–induced lipolysis and FABP4 secretion (Fig. 7D). We also confirmed that gene silencing of CHOP or PERK abrogates intracellular bacterial growth in adipocytes (Fig. 7E). Taken together, these results clearly indicate that infection-induced ER stress/UPR and the subsequent elevation of mitochondrial ROS play an important role not only on FABP4 secretion but also on the bacterial proliferation.
Figure 7.
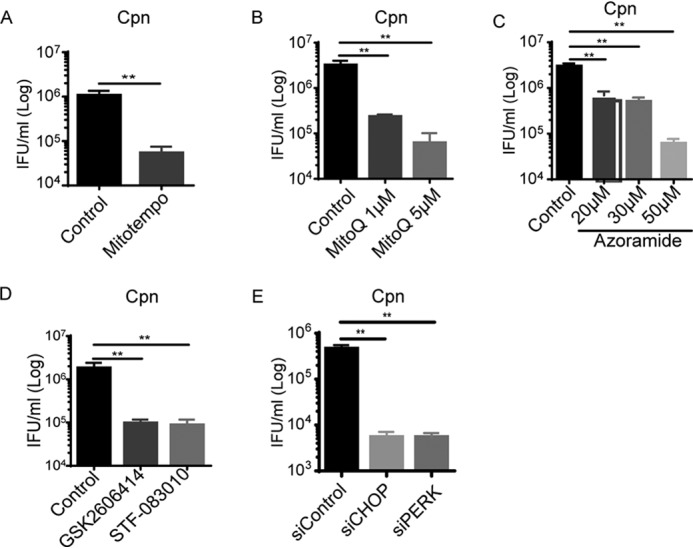
C. pneumoniae usurps infection-induced ER stress/UPR and subsequent elevation of mitochondrial ROS for its replication. A–D, the number of infectious EB progeny of 3T3-L1 adipocytes at 24 h after Cpn infection at a MOI of 5 in the presence or absence of MitoTEMPO (A, 100 μm), MitoQ (B, 1 or 5 μm), azoramide (C, 20 μm to 50 μm), GSK2606414 (D, PERK inhibitor, 2 μm), or STF-083010 (IRE1α RNase-specific inhibitor, 50 μm) was determined using an IFU assay. E, at 48 h after transfection (siCHOP, siPERK, or siControl), 3T3-L1 adipocytes were infected with Cpn MOI 5 for 24 h, and infectious EB progeny was determined using an IFU assay (n = 3/group). *, p < 0.05; **, p < 0.01, Student's t test (A); one-way ANOVA (B–E). The data are shown as the means ± S.E. and are representative of at least three experiments.
Discussion
Here we clearly showed the effect of infectivity of C. pneumoniae on FABP4 secretion. Our data suggest that bacterially induced ER stress/UPR, lipolysis, FABP4 secretion, and intracellular growth of C. pneumoniae are functionally related. We demonstrated that C. pneumoniae infection–induced ER stress/UPR in adipocytes causes the elevation of mitochondrial ROS and cytoplasmic calcium levels, followed by HSL-mediated lipolysis and FABP4 secretion. Treatment with an ER chemical chaperone, gene silencing of CHOP, and mitochondrial ROS scavenging by MitoTEMPO inhibited the HSL mediated lipolysis and FABP4 secretion and reduced the intracellular replication of C. pneumoniae in adipocytes. These findings are depicted in Fig. 8, which represents a model based upon the current study and our previous report showing that C. pneumoniae grows intracellularly exploiting lipolysis and FABP4 (20). The current study highlights the previously unrecognized mechanism of FABP4 secretion induced by C. pneumoniae infection in adipocytes, although the precise molecular mechanisms underlying how C. pneumoniae infection induces FABP4 secretion largely remain to be resolved.
Figure 8.
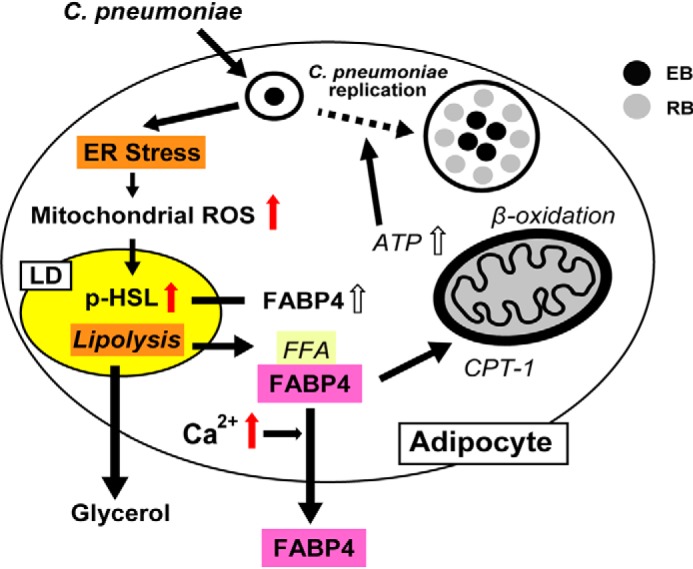
The possible model of FABP4 secretion in C. pneumoniae–infected adipocytes. C. pneumoniae infection–induced ER stress/UPR in murine adipocytes causes the elevation of mitochondrial ROS and cytoplasmic calcium levels, followed by HSL-mediated lipolysis and FABP4 secretion. The right part of this illustration (italics) is based on our previous report (20). CPT-1, carnitine palmitoyltransferase 1; FFA, free fatty acid.
Because FABP4 lacks a signal peptide, it has been considered that FABP4 secretion occurs via a unconventional secretion pathway (2). FABP4 secretion is stimulated by an increase in intracellular calcium (3, 4), and it has been suggested that multivesicular bodies and exosomes contribute to FABP4 secretion (5). More recently, Villeneuve et al. (35) reported that FABP4 secretion in adipocytes upon lipolysis stimulation involves endosomes and secretory lysosomes, which are promoted by an increase in intracellular calcium. They demonstrated that an increase in FABP4 secretion in plasma is inhibited by chloroquine treatment of mice (35). In this study, we showed that C. pneumoniae infection–induced FABP4 secretion is also dependent on cytoplasmic Ca2+ elevation. Further studies will be needed to determine whether endosomes and secretory lysosome are involved in C. pneumoniae infection–induced FABP4 secretion in adipocytes.
One can raise a question such as the possibility of FABP4 release by other pathogens. In this study we found that Listeria monocytogenes or Staphylococcus aureus did not augment the secretion of FABP4 from murine adipocytes (Fig. S4). Recently adipocytes were acknowledged as prime targets of a number of intracellular parasites, such as Mycobacterium tuberculosis (36), Rickettsia prowazekii (37), and Trypanosoma cruzi (38). Therefore, further study is required to address this possibility and its significance.
Suzuki et al. (39) recently reported the importance of an ER stress protein, CHOP, in determining adipose tissue macrophage polarity (M1 versus M2 macrophages) and systemic insulin sensitivity. The ER stress protein CHOP mediates insulin resistance by modulating adipose tissue macrophage polarity (39). This molecular mechanism may link adipose ER stress with systemic insulin resistance. In this study we demonstrated that C. pneumoniae infection induces ER stress/UPR followed by lipolysis and FABP4 secretion. Increase of CHOP protein expression after C. pneumoniae infection in adipocytes (Fig. 5B) and FABP4 secretion provides the clue for the causal relationship between C. pneumoniae infection and metabolic syndrome.
We previously reported the infectivity of adipocytes by C. pneumoniae and the subsequent lipolysis to facilitate bacterial growth (20). Here we extended the previous study and found that C. pneumoniae infection–induced ER stress/UPR and mitochondrial ROS are the central mechanisms promoting lipolysis and FAPB4 secretion. Considering the important pathogenic role of secreted FABP4 in obesity-induced type 2 diabetes and atherosclerosis (9), C. pneumoniae infection–induced FABP4 secretion may have an important in vivo relevance. Interestingly, infection-induced FABP4 secretion was not obvious after infection with C. muridarum (the mouse pneumonitis strain of Chlamydia trachomatis), which does not show the atherogenic effect in mice (40) (data not shown). The association of C. pneumoniae infection with cardiovascular disease has been extensively studied (12, 15, 16); however, whether C. pneumoniae has a causal role in metabolic syndrome remains undetermined (18, 19, 41). Therefore, our current results warrant the examination of the hypothesis that FABP4 released from C. pneumoniae–infected adipocytes might play a role in C. pneumoniae-induced metabolic pathologies, such as glucose intolerance, hepatic steatosis, or atherosclerotic pathogenic changes. In summary, the present study demonstrates that C. pneumoniae infection–induced ER stress/UPR causes robust secretion of FABP4 from adipocytes and sheds new light on the etiological link between C. pneumoniae infection and metabolic syndrome.
Experimental procedures
Reagents and antibodies
Reagents were obtained from the following sources: forskolin (F6886, Sigma–Aldrich), polyoxyethylene octylphenyl ether/Triton X-100 (168-11805, Wako), CAY10499 (10007875, Cayman), atglistatin (M60150-2s, Xcess Biosciences), JZL184 (S4904, Selleckchem), KH7 (13243, Cayman), H89 (10010556, Cayman), MitoTEMPO (ALX-430-150-M005, Enzo Life Sciences), BAPTA-AM (BML-CA411-0025, Enzo Life Sciences), BAPTA (2786, Tocris Bioscience), azoramide (HY-18705, MedChem Express), GSK2606414 (516535, Calbiochem), STF-083010 (SML0409, Sigma–Aldrich), thapsigargin (T9033, Sigma), and tunicamycin (T8153, LKT Laboratories). The following antibodies were used: polyclonal goat anti-FABP4 (AF1443, R&D Systems), monoclonal mouse anti-β-actin (SC-47778, Santa Cruz Biotechnology), polyclonal rabbit anti–phospho-HSL (Ser-660) (4126, Cell Signaling Technology), polyclonal rabbit anti-HSL (4107, Cell Signaling Technology), monoclonal mouse anti-CHOP (L63F7) (2895, Cell Signaling Technology), polyclonal rabbit anti-BiP (C50B12) (3177, Cell Signaling Technology), polyclonal rabbit and anti–phospho-eIF2α (Ser-51) (9721, Cell Signaling Technology).
Microbes
C. pneumoniae (strain AR39, ATCC53592) was obtained from the ATCC and propagated as previously described (42). Chlamydial EBs were purified using Urografin (Bayer) density gradient centrifugation, resuspended in sucrose–phosphate–glutamate (SPG) buffer, and stored at −80 °C. All Chlamydia stocks were confirmed as negative for Mycoplasma contamination using a MycoAlert Mycoplasma detection kit (Lonza). S. aureus (NCTC 10442) was obtained from the NCTC (The National Collection of Type Cultures of Public Health England). S. aureus was propagated in brain–heart infusion (BHI) and stocked in 30% glycerol containing BHI at −80 °C. L. monocytogenes (VTU206) was obtained from Japanese Society for Bacteriology. L. monocytogenes was grown in BHI and stocked in 30% glycerol containing BHI at −80 °C.
3T3-L1 adipocyte differentiation
3T3-L1 preadipocytes (ATCC CL-173) or stable shRNA knockdown (control EGFP, HSL) were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal calf serum (FCS) and streptomycin (100 μg/ml). For differentiation, the cells were seeded in 24-well plates (2 × 105 cells/well) and allowed to reach confluence for 2 days (day −2). On day 0, adipocyte differentiation was induced by adding 2.5 μm dexamethasone, 0.5 mm 3-isobutyl-1-methylexanthine, and 10 μg/ml insulin. On day 2 and thereafter, the medium was replaced with DMEM with 10% FCS containing only 10 μg/ml insulin. Differentiated adipocytes were infected with C. pneumoniae after an additional 2–4 days.
In vitro infection of adipocytes with C. pneumoniae
Cultured adipocytes were infected at days 4–6 of differentiation. The cells were infected with C. pneumoniae at a multiplicity of infection (MOI) of 5 IFU/cell or mock-infected with SPG buffer. To initiate infection, C. pneumoniae was added to plates at a MOI of 5, and the plates were centrifuged at 900 × g and 25 °C for 1 h. Next, the inoculum was removed, and the cells were cultured in DMEM containing 10% FCS and streptomycin (100 μg/ml). Inhibitors were added 60 min after inoculation with C. pneumoniae.
IFU assay
All Chlamydia-infected cells were collected, frozen, thawed, serially diluted 10-fold in SPG medium, and reseeded into 24-well plates containing a HeLa-cell monolayer (2 × 105 cells/well). After centrifugation at 900 × g and 25 °C for 1 h, the inoculum was removed, and the cells were cultured in DMEM containing 10% FCS and 1 μg/ml cycloheximide. After 24–48 h, the cells were fixed with ice-cold methanol for 10 min and incubated with a FITC-conjugated anti-Chlamydia LPS-specific mAb (FR97457, PROGEN). After being washed in PBS, the slides were mounted with coverslips, and the cells were imaged and quantified with an Axioskop fluorescence microscope (Zeiss).
In vitro treatment of adipocytes with heat-killed S. aureus
Cultured adipocytes were treated at days 8–10 of differentiation. S. aureus were killed at 80 °C for 30 min, and the cells were treated with heat-killed S. aureus at a MOI of 5.
In vitro infection of adipocytes with L. monocytogenes
Cultured adipocytes were infected at day 8–10 of differentiation. The cells were infected with L. monocytogenes at a MOI of 10 for 30 min and replaced with fresh culture medium.
Generation of 3T3-L1 preadipocyte cell lines stably expressing shRNAs
Retroviruses encoding shRNAs targeting EGFP mRNA were generated as previously described (43). Lentiviruses expressing shRNAs targeting HSL (sc-77404-V) were obtained from Santa Cruz Biotechnology. To generate 3T3-L1 cells constitutively expressing shRNAs targeting EGFP or HSL mRNA, 3T3-L1 cells were infected with the respective shRNA-expressing retrovirus/lentivirus in the presence of Polybrene (8 μg/ml) at 37 °C overnight. The cells were washed with PBS and cultured for 48 h in complete DMEM in the presence of Polybrene. The cells were cultured for 2–3 weeks in complete DMEM containing puromycin (4 μg/ml) to kill untransduced cells.
Density-based separation followed by replating of enriched adipocytes in monolayer (DREAM)
On days 6–7, the differentiated 3T3-L1 cells were replated as previously described (44). The cells were washed once with PBS and treated with 0.05% trypsin EDTA in PBS at 37 °C for 5 min until most cells were detached from the culture dish. The detached cells were centrifuged at 400 × g for 5 min and resuspended in the medium mixed with 1:1 ratio of DMEM containing 10% FCS and Histopaque-1077 (Sigma–Aldrich). Next, the cell suspension was filtered through a 100-μm cell strainer (BD Falcon) and then centrifuged at 400 × g for 10 min. Floating cells containing the differentiated adipocytes were collected into a new tube, washed once with DMEM containing 10% FCS, and then replated onto 24-well plates (5 × 104 cells/well) for transfection.
Transfection of the differentiated 3T3-L1 cells with siRNA
Mouse Ddit3/Chop siRNA (4390771, siRNA ID: s64889, Ambion) or mouse Eif2ak3/Perk siRNA (4390771, siRNA ID: s65405, Ambion) was used for transient knockdown of Ddit3/Chop or Eif2ak3/Perk, respectively. 3T3-L1 adipocytes were replated in 24-well plates and transfected with siRNA for mouse Ddit3/Chop, Eif2ak3/Perk, or siRNA universal negative control (SIC001, Sigma–Aldrich) using Lipofectamine RNAiMAX reagent (13778-075, Invitrogen) according to the manufacturer's instructions.
ELISA
Cell culture supernatants and plasma were measured for mouse FABP4 with an adipocyte FABP mouse ELISA (Biovendor) or Circulex mouse FABP4/A-FABP ELISA kit according to the manufacturer's instructions. ELISA plates were read using a model 680 microplate reader (Bio-Rad).
Cytotoxicity assay
LDH released from dead cells was measured using a CytoTox 96 nonradioactive cytotoxic assay kit (Promega).
Glycerol assay
Glycerol levels in the cultured supernatants were measured using a glycerol cell–based assay kit (Cayman Chemical Company) according to the manufacturer's instructions.
Quantitative real-time PCR
Total RNA was isolated from cultured cells using ISOGEN II (Nippon Gene). cDNA synthesis was performed using a PrimeScriptTM RT reagent kit with gDNA Eraser (Perfect Real Time) (TaKaRa Bio) and 1 μg of total RNA as a template. Relative gene expression levels were determined in 96-well plates using the SYBR Premix Dimer Eraser (Perfect Real Time) (TaKaRa Bio) with an Applied Biosystems 7500 Real-Time PCR system (Applied Biosystems). All primer sequences used in the present study are listed in Table 1. The specific thermal cycling parameters were as follows: 30 s at 95 °C, 40 cycles of denaturation at 95 °C for 5 s, annealing at 55 °C for 30 s, and extension at 72 °C for 34 s.
Table 1.
Quantitative PCR primers used in this study
| Gene | Forward primer | Reverse primer |
|---|---|---|
| Fabp4 | TTTGGTCACCATCCGGTCAG | TGTCGTCTGCGGTGATTTCA |
| Chop | CACATCCCAAAGCCCTCGCTCTC | TCATGCTTGGTGCAGGCTGACCAT |
| Bip | CTGGGTACATTTGATCTGACTGG | GCATCCTGGTGGCTTTCCAGCCATTC |
| Atf4 | GGGTTCTGTCTTCCACTCCA | AAGCAGCAGAGTCAGGCTTTC |
| sXbp1 | CTGAGTCCGAATCAGGTGCAG | GTCCATGGGAAGATGTTCTGG |
| Gus | ATGACGAACCAGTCACC | CCTCCAGTATCTCTCTCGCAAA |
Immunoblot analysis
The cells were washed once in ice-cold PBS, lysed in radioimmune precipitation assay buffer (50 mm Tris, pH 7.4, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 150 mm NaCl, 2 mm EDTA, and 50 mm NaF) with a protease inhibitor mixture (Nacalai Tesque) and PhosSTOP phosphatase inhibitor mixture (Roche) and then briefly sonicated. Protein concentration was determined with a BCA protein assay kit (Thermo Fisher Scientific), and equal amounts of proteins were loaded onto SDS-polyacrylamide gels. The proteins were transferred to membranes using a Trans Blot SD Semi-Dry Transfer Cell (Bio-Rad) following the manufacturer's instructions. The membranes were blocked in Blocking One (Nacalai Tesque) for 30 min. The membranes were incubated with primary antibody diluted in Can Get Signal solution 1 (TOYOBO) overnight at 4 °C and then with horseradish peroxidase–conjugated secondary antibodies to rabbit/mouse IgGs (GE Healthcare) or goat IgG (SeraCare) diluted in Can Get Signal solution 2 (TOYOBO) for 1 h at room temperature. Immunoreactive bands were detected by ECL blotting reagents (GE Healthcare, RPN2109) or EzWestLumi plus (ATTO). The densitometric analysis was performed using the Image Studio Lite software (Licor).
Mitochondrial ROS production assay
Mitochondrial ROS levels were measured using MitoSOX (M36008, Molecular Probes) staining. 3T3-L1 adipocytes after mock or C. pneumoniae infection were incubated with a mitochondrial-superoxide-specific stain MitoSOX (5 μm) for 15 min at 37 °C. 3T3-L1 adipocytes were washed once with PBS, treated with 0.05% trypsin EDTA, and resuspended in PBS. Flow cytometry was performed using a BD FACS Canto II (BD Bioscience), and the data were analyzed using the FlowJo software.
Intracellular Ca2+ assay
Intracellular Ca2+ levels were measured using Fluo-4 AM (F311, Dojindo Laboratories) staining. 3T3-L1 adipocytes after mock or C. pneumoniae infection were incubated with Fluo-4 AM (1 μm) for 45 min at 37 °C. 3T3-L1 adipocytes were washed once with PBS, treated with 0.05% trypsin EDTA, and resuspended in PBS. Fluo-4 AM fluorescence analysis was completed using a Tristar LB 941 (Berthold Technologies).
Statistics
The results are expressed as the means ± S.E. The statistical significance of the differences between various treatments was measured by either a two-tailed Student's t test for comparison of two groups or a one-way or two-way ANOVA with Dunnett's multiple comparisons for comparison of multiple groups. The data analyses were performed using the GraphPad Prism software version 6.0. All p < 0.05 were considered statistically significant.
Author contributions
N. F. W., Y. K., B. C., K. I., and T. S. formal analysis; N. F. W., Y. K., B. C., and K. I. investigation; N. F. W., Y. K., and K. H. visualization; Y. K., B. C., K. I., T. S., and K. H. supervision; Y. K. funding acquisition; Y. K., B. C., K. I., and T. S. methodology; Y. K. and K. H. writing-review and editing; B. C., K. I., and T. S. validation; T. S. resources; K. H. conceptualization; K. H. writing-original draft; K. H. project administration.
Supplementary Material
Acknowledgment
We thank Tetsuya Hayashi (Department of Bacteriology, Kyushu University) for critical discussion.
This work was supported in part by Japan Society for the Promotion of Science Grant-in-Aid for Scientific Research 19K166590002. This work was also supported in part by an Indonesia Endowment Fund for Education scholarship from Ministry of Finance of the Republic of Indonesia (to N. F. W.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S4.
- FABP4
- fatty acid–binding protein 4
- Cpn
- C. pneumoniae
- ER
- endoplasmic reticulum
- HSL
- hormone-sensitive lipase
- MOI
- multiplicity of infection
- ROS
- reactive oxygen species
- PERK
- double-stranded RNA-dependent protein kinase R–like ER kinase
- UPR
- unfolded protein response
- LDH
- lactate dehydrogenase
- ATGL
- adipose triglyceride lipase
- shRNA
- short hairpin RNA
- BAPTA-AM
- 1,2-bis(2-aminophenoxyl)ethane-N,N,N,N-tetraacetic acid
- SPG
- sucrose–phosphate–glutamate
- BHI
- brain–heart infusion
- EGFP
- enhanced GFP
- DMEM
- Dulbecco's modified Eagle's medium
- FCS
- fetal calf serum
- ANOVA
- analysis of variance
- IFU
- inclusion-forming unit
- EB
- elementary body.
References
- 1. Furuhashi M., and Hotamisligil G. S. (2008) Fatty acid–binding proteins: role in metabolic diseases and potential as drug targets. Nat. Rev. Drug Discov. 7, 489–503 10.1038/nrd2589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hotamisligil G. S., and Bernlohr D. A. (2015) Metabolic functions of FABPs: mechanisms and therapeutic implications. Nat. Rev. Endocrinol. 11, 592–605 10.1038/nrendo.2015.122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kralisch S., Ebert T., Lossner U., Jessnitzer B., Stumvoll M., and Fasshauer M. (2014) Adipocyte fatty acid–binding protein is released from adipocytes by a non-conventional mechanism. Int. J. Obes. (Lond.) 38, 1251–1254 10.1038/ijo.2013.232 [DOI] [PubMed] [Google Scholar]
- 4. Schlottmann I., Ehrhart-Bornstein M., Wabitsch M., Bornstein S. R., and Lamounier-Zepter V. (2014) Calcium-dependent release of adipocyte fatty acid binding protein from human adipocytes. Int. J. Obes. (Lond.) 38, 1221–1227 10.1038/ijo.2013.241 [DOI] [PubMed] [Google Scholar]
- 5. Ertunc M. E., Sikkeland J., Fenaroli F., Griffiths G., Daniels M. P., Cao H., Saatcioglu F., and Hotamisligil G. S. (2015) Secretion of fatty acid binding protein aP2 from adipocytes through a nonclassical pathway in response to adipocyte lipase activity. J. Lipid Res. 56, 423–434 10.1194/jlr.M055798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cao H., Sekiya M., Ertunc M. E., Burak M. F., Mayers J. R., White A., Inouye K., Rickey L. M., Ercal B. C., Furuhashi M., Tuncman G., and Hotamisligil G. S. (2013) Adipocyte lipid chaperone AP2 is a secreted adipokine regulating hepatic glucose production. Cell Metab. 17, 768–778 10.1016/j.cmet.2013.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Maeda K., Cao H., Kono K., Gorgun C. Z., Furuhashi M., Uysal K. T., Cao Q., Atsumi G., Malone H., Krishnan B., Minokoshi Y., Kahn B. B., Parker R. A., and Hotamisligil G. S. (2005) Adipocyte/macrophage fatty acid binding proteins control integrated metabolic responses in obesity and diabetes. Cell Metab. 1, 107–119 10.1016/j.cmet.2004.12.008 [DOI] [PubMed] [Google Scholar]
- 8. Erbay E., Babaev V. R., Mayers J. R., Makowski L., Charles K. N., Snitow M. E., Fazio S., Wiest M. M., Watkins S. M., Linton M. F., and Hotamisligil G. S. (2009) Reducing endoplasmic reticulum stress through a macrophage lipid chaperone alleviates atherosclerosis. Nat. Med. 15, 1383–1391 10.1038/nm.2067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Furuhashi M., Tuncman G., Görgün C. Z., Makowski L., Atsumi G., Vaillancourt E., Kono K., Babaev V. R., Fazio S., Linton M. F., Sulsky R., Robl J. A., Parker R. A., and Hotamisligil G. S. (2007) Treatment of diabetes and atherosclerosis by inhibiting fatty-acid-binding protein aP2. Nature 447, 959–965 10.1038/nature05844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xu H., Hertzel A. V., Steen K. A., Wang Q., Suttles J., and Bernlohr D. A. (2015) Uncoupling lipid metabolism from inflammation through fatty acid binding protein–dependent expression of UCP2. Mol. Cell. Biol. 35, 1055–1065 10.1128/MCB.01122-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Furuhashi M., Fucho R., Görgün C. Z., Tuncman G., Cao H., and Hotamisligil G. S. (2008) Adipocyte/macrophage fatty acid–binding proteins contribute to metabolic deterioration through actions in both macrophages and adipocytes in mice. J. Clin. Invest. 118, 2640–2650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kalayoglu M. V., Libby P., and Byrne G. I. (2002) Chlamydia pneumoniae as an emerging risk factor in cardiovascular disease. JAMA 288, 2724–2731 10.1001/jama.288.21.2724 [DOI] [PubMed] [Google Scholar]
- 13. Fernández-Real J. M., López-Bermejo A., Vendrell J., Ferri M. J., Recasens M., and Ricart W. (2006) Burden of infection and insulin resistance in healthy middle-aged men. Diabetes Care 29, 1058–1064 10.2337/dc05-2068 [DOI] [PubMed] [Google Scholar]
- 14. Wang C., Gao D., and Kaltenboeck B. (2009) Acute Chlamydia pneumoniae reinfection accelerates the development of insulin resistance and diabetes in obese C57BL/6 mice. J. Infect. Dis. 200, 279–287 10.1086/599796 [DOI] [PubMed] [Google Scholar]
- 15. Campbell L. A., and Kuo C. C. (2004) Chlamydia pneumoniae: an infectious risk factor for atherosclerosis? Nat. Rev. Microbiol. 2, 23–32 10.1038/nrmicro796 [DOI] [PubMed] [Google Scholar]
- 16. Grayston J. T., Kronmal R. A., Jackson L. A., Parisi A. F., Muhlestein J. B., Cohen J. D., Rogers W. J., Crouse J. R., Borrowdale S. L., Schron E., Knirsch C., and ACES Investigators (2005) Azithromycin for the secondary prevention of coronary events. N. Engl. J. Med. 352, 1637–1645 10.1056/NEJMoa043526 [DOI] [PubMed] [Google Scholar]
- 17. Cannon C. P., Braunwald E., McCabe C. H., Grayston J. T., Muhlestein B., Giugliano R. P., Cairns R., and Skene A. M. (2005) Antibiotic treatment of Chlamydia pneumoniae after acute coronary syndrome. N. Engl. J. Med. 352, 1646–1654 10.1056/NEJMoa043528 [DOI] [PubMed] [Google Scholar]
- 18. Anderson J. L. (2005) Infection, antibiotics, and atherothrombosis: end of the road or new beginnings? N. Engl. J. Med. 352, 1706–1709 10.1056/NEJMe058019 [DOI] [PubMed] [Google Scholar]
- 19. Campbell L. A., and Rosenfeld M. E. (2014) Persistent C. pneumoniae infection in atherosclerotic lesions: rethinking the clinical trials. Front. Cell. Infect. Microbiol. 4, 34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Walenna N. F., Kurihara Y., Chou B., Ishii K., Soejima T., Itoh R., Shimizu A., Ichinohe T., and Hiromatsu K. (2018) Chlamydia pneumoniae exploits adipocyte lipid chaperone FABP4 to facilitate fat mobilization and intracellular growth in murine adipocytes. Biochem. Biophys. Res. Commun. 495, 353–359 10.1016/j.bbrc.2017.11.005 [DOI] [PubMed] [Google Scholar]
- 21. Celli J., and Tsolis R. M. (2015) Bacteria, the endoplasmic reticulum and the unfolded protein response: friends or foes? Nat. Rev. Microbiol. 13, 71–82 10.1038/nrmicro3393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhou H., and Liu R. (2014) ER stress and hepatic lipid metabolism. Front. Genet. 5, 112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li S., Kong L., and Yu X. (2015) The expanding roles of endoplasmic reticulum stress in virus replication and pathogenesis. Crit. Rev. Microbiol. 41, 150–164 10.3109/1040841X.2013.813899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pillich H., Loose M., Zimmer K. P., and Chakraborty T. (2016) Diverse roles of endoplasmic reticulum stress sensors in bacterial infection. Mol. Cell. Pediatr. 3, 9 10.1186/s40348-016-0037-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. George Z., Omosun Y., Azenabor A. A., Partin J., Joseph K., Ellerson D., He Q., Eko F., Bandea C., Svoboda P., Pohl J., Black C. M., and Igietseme J. U. (2017) The roles of unfolded protein response pathways in Chlamydia pathogenesis. J. Infect. Dis. 215, 456–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Krawczyk S. A., Haller J. F., Ferrante T., Zoeller R. A., and Corkey B. E. (2012) Reactive oxygen species facilitate translocation of hormone sensitive lipase to the lipid droplet during lipolysis in human differentiated adipocytes. PLoS One 7, e34904 10.1371/journal.pone.0034904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Carmen G. Y., and Víctor S. M. (2006) Signalling mechanisms regulating lipolysis. Cell. Signal. 18, 401–408 10.1016/j.cellsig.2005.08.009 [DOI] [PubMed] [Google Scholar]
- 28. Mayer N., Schweiger M., Romauch M., Grabner G. F., Eichmann T. O., Fuchs E., Ivkovic J., Heier C., Mrak I., Lass A., Höfler G., Fledelius C., Zechner R., Zimmermann R., and Breinbauer R. (2013) Development of small-molecule inhibitors targeting adipose triglyceride lipase. Nat. Chem. Biol. 9, 785–787 10.1038/nchembio.1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang K., and Kaufman R. J. (2008) From endoplasmic-reticulum stress to the inflammatory response. Nature 454, 455–462 10.1038/nature07203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Deng J., Liu S., Zou L., Xu C., Geng B., and Xu G. (2012) Lipolysis response to endoplasmic reticulum stress in adipose cells. J. Biol. Chem. 287, 6240–6249 10.1074/jbc.M111.299115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fu S., Yalcin A., Lee G. Y., Li P., Fan J., Arruda A. P., Pers B. M., Yilmaz M., Eguchi K., and Hotamisligil G. S. (2015) Phenotypic assays identify azoramide as a small-molecule modulator of the unfolded protein response with antidiabetic activity. Sci. Transl. Med. 7, 292ra298 10.1126/scitranslmed.aaa9134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang M., and Kaufman R. J. (2016) Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 529, 326–335 10.1038/nature17041 [DOI] [PubMed] [Google Scholar]
- 33. Papandreou I., Denko N. C., Olson M., Van Melckebeke H., Lust S., Tam A., Solow-Cordero D. E., Bouley D. M., Offner F., Niwa M., and Koong A. C. (2011) Identification of an Ire1α endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood 117, 1311–1314 10.1182/blood-2010-08-303099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Axten J. M., Medina J. R., Feng Y., Shu A., Romeril S. P., Grant S. W., Li W. H., Heerding D. A., Minthorn E., Mencken T., Atkins C., Liu Q., Rabindran S., Kumar R., Hong X., et al. (2012) Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-p yrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J. Med. Chem. 55, 7193–7207 10.1021/jm300713s [DOI] [PubMed] [Google Scholar]
- 35. Villeneuve J., Bassaganyas L., Lepreux S., Chiritoiu M., Costet P., Ripoche J., Malhotra V., and Schekman R. (2018) Unconventional secretion of FABP4 by endosomes and secretory lysosomes. J. Cell Biol. 217, 649–665 10.1083/jcb.201705047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Neyrolles O., Hernández-Pando R., Pietri-Rouxel F., Fornès P., Tailleux L., Barrios Payán J. A., Pivert E., Bordat Y., Aguilar D., Prévost M. C., Petit C., and Gicquel B. (2006) Is adipose tissue a place for Mycobacterium tuberculosis persistence? PLoS One 1, e43 10.1371/journal.pone.0000043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bechah Y., Paddock C. D., Capo C., Mege J. L., and Raoult D. (2010) Adipose tissue serves as a reservoir for recrudescent Rickettsia prowazekii infection in a mouse model. PLoS One 5, e8547 10.1371/journal.pone.0008547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Combs T. P., Nagajyothi Mukherjee S., de Almeida C. J., Jelicks L. A., Schubert W., Lin Y., Jayabalan D. S., Zhao D., Braunstein V. L., Landskroner-Eiger S., Cordero A., Factor S. M., Weiss L. M., Lisanti M. P., Tanowitz H. B., et al. (2005) The adipocyte as an important target cell for Trypanosoma cruzi infection. J. Biol. Chem. 280, 24085–24094 10.1074/jbc.M412802200 [DOI] [PubMed] [Google Scholar]
- 39. Suzuki T., Gao J., Ishigaki Y., Kondo K., Sawada S., Izumi T., Uno K., Kaneko K., Tsukita S., Takahashi K., Asao A., Ishii N., Imai J., Yamada T., Oyadomari S., and Katagiri H. (2017) ER stress protein CHOP mediates insulin resistance by modulating adipose tissue macrophage polarity. Cell Reports 18, 2045–2057 10.1016/j.celrep.2017.01.076 [DOI] [PubMed] [Google Scholar]
- 40. Blessing E., Nagano S., Campbell L. A., Rosenfeld M. E., and Kuo C. C. (2000) Effect of Chlamydia trachomatis infection on atherosclerosis in apolipoprotein E-deficient mice. Infect. Immun. 68, 7195–7197 10.1128/IAI.68.12.7195-7197.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Grayston J. T., Belland R. J., Byrne G. I., Kuo C. C., Schachter J., Stamm W. E., and Zhong G. (2015) Infection with Chlamydia pneumoniae as a cause of coronary heart disease: the hypothesis is still untested. Pathog. Dis. 73, 1–9 10.1093/femspd/ftu015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Itoh R., Murakami I., Chou B., Ishii K., Soejima T., Suzuki T., and Hiromatsu K. (2014) Chlamydia pneumoniae harness host NLRP3 inflammasome-mediated caspase-1 activation for optimal intracellular growth in murine macrophages. Biochem. Biophys. Res. Commun. 452, 689–694 10.1016/j.bbrc.2014.08.128 [DOI] [PubMed] [Google Scholar]
- 43. Ito M., Yanagi Y., and Ichinohe T. (2012) Encephalomyocarditis virus viroporin 2B activates NLRP3 inflammasome. PLoS Pathogens 8, e1002857 10.1371/journal.ppat.1002857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kajimoto K., Takayanagi S., Sasaki S., Akita H., and Harashima H. (2012) RNA interference-based silencing reveals the regulatory role of fatty acid–binding protein 4 in the production of IL-6 and vascular endothelial growth factor in 3T3-L1 adipocytes. Endocrinology 153, 5629–5636 10.1210/en.2012-1456 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.