

Abstract
Cholesterol synthesis is a tightly regulated process, both transcriptionally and post-translationally. Transcriptional control of cholesterol synthesis is relatively well-understood. However, of the ∼20 enzymes in cholesterol biosynthesis, post-translational regulation has only been examined for a small number. Three of the four sterol reductases in cholesterol production, 7-dehydrocholesterol reductase (DHCR7), 14-dehydrocholesterol reductase (DHCR14), and lamin-B receptor (LBR), share evolutionary ties with a high level of sequence homology and predicted structural homology. DHCR14 and LBR uniquely share the same Δ-14 reductase activity in cholesterol biosynthesis, yet little is known about their post-translational regulation. We have previously identified specific modes of post-translational control of DHCR7, but it is unknown whether these regulatory mechanisms are shared by DHCR14 and LBR. Using CHO-7 cells stably expressing epitope-tagged DHCR14 or LBR, we investigated the post-translational regulation of these enzymes. We found that DHCR14 and LBR undergo differential post-translational regulation, with DHCR14 being rapidly turned over, triggered by cholesterol and other sterol intermediates, whereas LBR remained stable. DHCR14 is degraded via the ubiquitin-proteasome system, and we identified several DHCR14 and DHCR7 putative interaction partners, including a number of E3 ligases that modulate DHCR14 levels. Interestingly, we found that gene expression across an array of human tissues showed a negative relationship between the C14-sterol reductases; one enzyme or the other tends to be predominantly expressed in each tissue. Overall, our findings indicate that whereas LBR tends to be the constitutively active C14-sterol reductase, DHCR14 levels are tunable, responding to the local cellular demands for cholesterol.
Keywords: cholesterol, cholesterol regulation, endoplasmic reticulum-associated protein degradation (ERAD), protein degradation, enzyme degradation, DHCR14, LBR, TM7SF2
Introduction
Cholesterol is a vital lipid in the human body, and its deficiency can result in a wide array of health problems (1). It plays an essential role in membrane fluidity and the formation of lipid rafts (2), in the production of bile acids (3) and steroid hormones (4), and in embryonic development (5). However, an excess of cholesterol also leads to disease (6).
In the cholesterol synthesis pathway, there are four sterol reductases that catalyze three distinct reductive steps on sterol intermediates (7) (Fig. 1). Two of these reductases, 7-dehydrocholesterol reductase (DHCR7)3 and 24-dehydrocholesterol reductase (DHCR24) (the terminal enzymes of cholesterol biosynthesis), have been relatively well-characterized, and their transcriptional (8, 9) and post-translational regulation has been studied (10–14). Whereas their transcriptional regulation is generally understood (15–17), little is known about the post-translational regulation of the two remaining sterol reductases, lamin-B receptor (LBR) and 14-dehydrocholesterol reductase (DHCR14).
Figure 1.

Sterol reductases in cholesterol biosynthesis. Four sterol reductases, DHCR24, DHCR7, DHCR14, and LBR, catalyze three distinct reactions in cholesterol synthesis. In the Bloch pathway, LBR or DHCR14 first reduces the C(14–15) double bond. Then DHCR7 and DHCR24 act consecutively to form cholesterol. In the Kandutsch–Russell pathway, DHCR24 acts first, followed by LBR or DHCR14 and DHCR7 as the terminal enzyme. In addition to these two defined pathways, DHCR24 can shunt intermediates from the Bloch pathway into the Kandutsch–Russell pathway at any point. Dashed arrows represent multiple enzymatic steps. The purple box indicates the C(14–15) bond on FF-MAS and T-MAS, the reaction catalyzed by DHCR14 and LBR. The green box indicates the C(24–25) bond on sterol intermediates, the reaction catalyzed by DHCR24 at numerous steps in the pathway. The red box indicates the C(7–8) bond on 7DHC and cholesterol, the reaction catalyzed by DHCR7.
LBR and DHCR14 share the same C14-sterol reductase (C14-SR) activity, reducing the carbon 14–15 double bond in sterol intermediates. The substrates and products of C14-SRs were initially ascribed meiotic activities (18) and were collectively called meiosis-activating sterols (MASs), consisting of FF-MAS (follicular fluid MAS) and T-MAS (testis MAS) (19). DHCR24 can act on these sterols to produce dihydro-FF-MAS or dihydro-T-MAS.
DHCR14 and LBR emerged as a result of a gene duplication event (20) and share a high level of sequence homology with 58% protein similarity over their shared sterol reductase domain in humans. Whether DHCR14 or LBR is responsible for the majority of C14-SR activity in mammals has been contentious (15, 20–24), with evidence presented for each being the primary C14-SR.
DHCR14 localizes to the ER (20), the site of post-squalene cholesterol biosynthesis, suggesting that it is well-positioned to participate in C14-SR activity. Previous work has also highlighted that human DHCR14 has higher C14-SR activity than human LBR in microsomal preparations, suggesting that it is the major C14-SR in cholesterol biosynthesis (15). Despite the localization and higher activity of DHCR14, mice deficient in this enzyme have normal sterol profiles and an average life expectancy (25), indicating that LBR may compensate for the absence of DHCR14. Moreover, LBR knockout cells fail to thrive in sterol depleted conditions, whereas DHCR14 knockout cells were not challenged by the sterol-deficient environment (24).
LBR is a multidomain protein, localized at the inner nuclear membrane, consisting of the sterol reductase domain, which is homologous with DHCR14 and DHCR7, and an N-terminal Tudor domain that binds lamin-B, chromatin, and both assembled and unassembled histones H3 and H4 (26, 27). Despite the role of LBR in binding lamin-B, LBR knockout cells do not have altered nuclear morphology (24). In mouse models, LBR-null knockouts proved to be embryonically lethal (23), and whereas the deleterious impact of LBR loss was initially presumed to only be a laminopathy, later work has indicated that knockout mice and cells have altered sterol profiles indicative of reduced C14-SR activity (24). Furthermore, in Saccharomyces cerevisiae, LBR can complement ERG24 (the yeast homologue of the sterol reductase domain of LBR and DHCR14) knockouts, whereas DHCR14 is unable to compensate for this deficiency (21, 28).
Whereas the necessity of LBR cannot be disputed, it appears that both LBR and DHCR14 are used for C14-SR activity in mammalian cells. The enzymatic redundancy of these two C14-SRs may have an elusive functional role, and understanding the post-translational regulation of LBR and DHCR14 could help to clarify this conundrum.
Here, we show that DHCR14 undergoes sterol-dependent turnover, whereas LBR does not. We elucidate the mechanism of post-translational regulation of DHCR14 and identify E3 ligases that modulate basal DHCR14 levels. We further identify a tissue-specific RNA expression pattern between TM7SF2 (the gene encoding DHCR14) and LBR, suggesting preferential use of particular C14-SRs in different tissues.
Results
Human TM7SF2, but not LBR, responds to changing sterol status
Many cholesterol synthesis enzymes are transcriptionally controlled by sterol regulatory element–binding protein 2 (SREBP-2) (29). This was thought to be the case for TM7SF2 (the gene encoding DHCR14) (15, 16), but not for LBR. Despite the characterized SREBP-2–binding motif in the TM7SF2 promoter (15, 16), recent literature has yielded conflicting data about the sterol responsiveness of human TM7SF2 in some cell lines (24).
We employed previously generated cDNA sets derived from CHO cells with sterol-related mutations, CHO-7 and SRD-1 (30). There is an increase in gene expression of SREBP-2 target genes in CHO-7 cells with statins and a decrease with oxysterol treatment. SRD-1 cells have constitutively active SREBP-2 and therefore high levels of SREBP-2 target genes, irrespective of cellular sterol status (31). We found that 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR), a canonical SREBP-2 target, follows this trend in expression. However, neither TM7SF2 nor LBR follow this pattern, showing no response to variable SREBP-2 levels in these cell lines (Fig. 2A).
Figure 2.
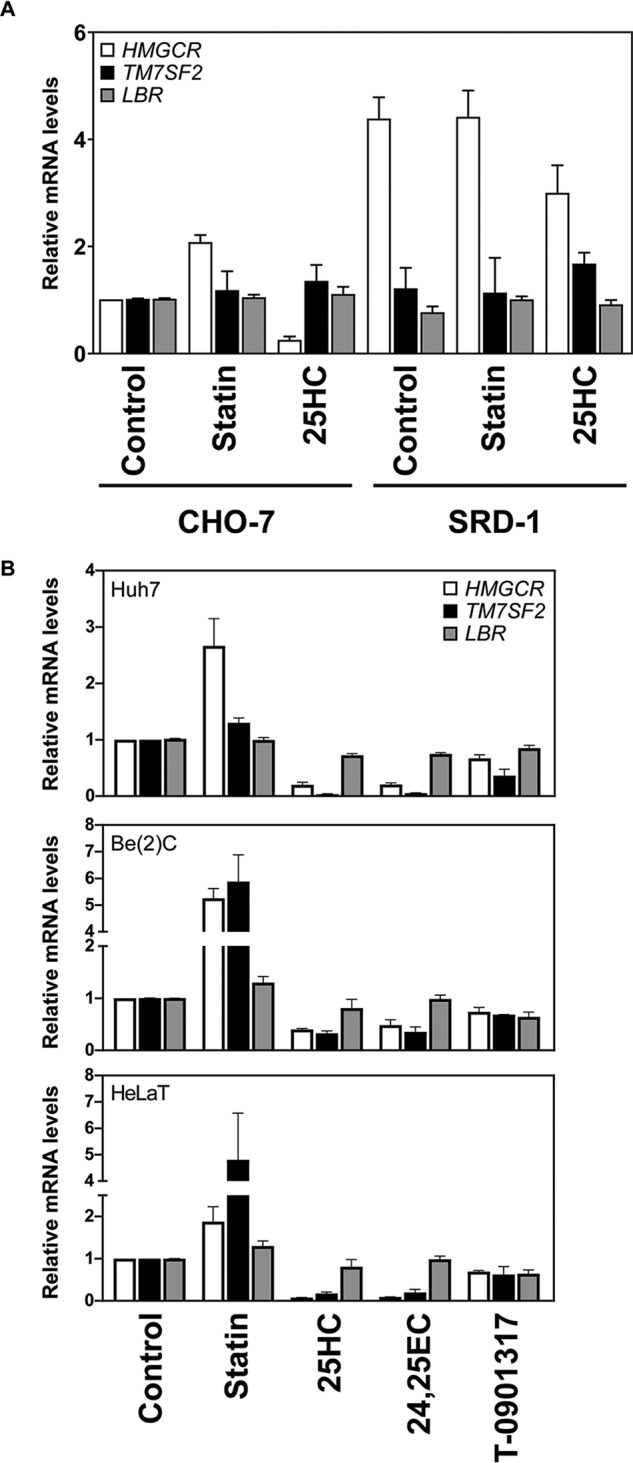
TM7SF2 mRNA is responsive to sterols in human but not hamster cell lines. A, cDNA samples were prepared from CHO-7 or SRD-1 cells treated for 24 h with 5 μm compactin (statin) or 10 μm 25-hydroxycholesterol (25HC) (30). B, Huh7, Be(2)C, and HeLaT cells were treated for 24 h with the following: 5 μm compactin (statin), 10 μm 25-hydroxycholesterol, 10 μm 24(S),25-epoxycholesterol (24,25EC), or 10 μm T0-901317 (60). Total RNA was harvested, and cDNA was prepared. A and B, mRNA levels for TM7SF2 and LBR were measured using qRT-PCR and normalized to PBGD. mRNA levels are relative to the control condition (for A, the CHO-7 control), which was set to 1. Data are presented as mean ± S.E. (error bars) from three independent experiments, each performed in triplicate.
We next investigated the sterol-dependent transcriptional regulation of human TM7SF2 under different sterol conditions in three human-derived cell lines: Huh7 liver cells, Be(2)C brain cells, and HeLaT cervical cancer cells. Consistent with previous studies (15, 16), human TM7SF2 was affected by altered sterol levels, with mRNA increasing with sterol depletion (statin treatment) and decreasing when sterol levels were high (Fig. 2B). The liver X receptor agonist, T0901317, did not increase TM7SF2 levels. The HeLaT, Be(2)C, and Huh7 TM7SF2 expression profiles are in line with our previously published data for other established SREBP-2 targets, including HMGCR (8, 30). In contrast, LBR was not responsive to cellular sterol levels, which is also in line with previously published studies (15, 24).
Epitope-tagged DHCR14 and LBR localize as expected
We next generated CHO-7 cells stably expressing CMV-driven, V5-tagged human LBR or DHCR14. After generation of these cell lines, we confirmed the proper localization of our ectopic V5-tagged proteins. Endogenous DHCR14 is reported to localize to the ER (32), and the majority of LBR is localized to the inner nuclear membrane (20). However, a small portion of LBR can also be found in the ER (33). V5-tagged DHCR14 localized to the ER, and the majority of LBR-V5 was observed around the nucleus (Fig. 3), indicating that the ectopically V5-tagged DHCR14 and LBR are located similarly to their endogenous counterparts.
Figure 3.

DHCR14-V5 localizes to the ER, whereas LBR-V5 localizes to the perinuclear region. CHO-EV, CHO-DHCR14-V5, and CHO-LBR-V5 cells were transfected with dsRedER plasmid (an ER marker), prior to staining with anti-V5 and DAPI. Slides were imaged with a Nikon C1 confocal microscope at ×100 for DAPI, dsRedER, and Alexa Fluor® 488.
LBR protein is stable, whereas DHCR14 is rapidly turned over and triggered by cholesterol
We investigated the post-translational regulation of both LBR and DHCR14 by comparing the turnover of these two C14-SR enzymes using a stably expressing system. This system, consisting of the human protein-coding sequence with a V5 epitope tag driven by the CMV promoter, was necessary to remove the normal transcriptional control by sterols.
CHO-7 cells stably expressing V5-tagged human LBR or DHCR14 were treated with or without the protein synthesis inhibitor cycloheximide for up to 8 h. In line with previous work (24), we found that LBR was stable over 8 h (Fig. 4A). However, DHCR14 was rapidly turned over, with less than 50% of the protein remaining after 1 h of treatment with cycloheximide (Fig. 4A). This was further reduced throughout the time course, with only ∼20% of DHCR14 remaining after 8 h of protein synthesis inhibition, similar to the rapid turnover we previously observed for the homologous DHCR7 (10).
Figure 4.

DHCR14 is rapidly turned over in response to cholesterol and is stabilized by statin treatment. CHO-DHCR14 and CHO-LBR cells were treated with 10 μg/ml cycloheximide (CHX) (A) or 20 μg/ml cholesterol complexed to cyclodextrin (B) for 0–8 h. C, CHO-DHCR14 cells were treated with 10 μg/ml cycloheximide and/or 20 μg/ml cholesterol complexed to cyclodextrin for 8 h. All conditions are significant; p < 0.05 compared with the solvent control. D, CHO-DHCR14 cells were pretreated with or without 5 μm compactin (statin) for 16 h and then refreshed with or without 5 μm compactin for an additional 8 h. Cell lysates were separated by SDS-PAGE, and protein levels were quantified by Western blotting using V5 (LBR and DHCR14) and GAPDH. Protein levels are relative to the paired control for each time point, where 0 h has been set to 1 for each protein. Data are presented as mean ± S.E. (error bars) of n > 3 independent experiments; Western blots are representative. A paired Student's two-tailed t test was used for statistical analysis. *, p < 0.05.
Considering that DHCR7 turnover is triggered by cholesterol (10), we investigated cholesterol as a trigger for DHCR14 turnover. Approximately 45% of DHCR14 remained after 8 h of cholesterol treatment (Fig. 4B). In contrast, LBR-V5 protein levels were unchanged by cholesterol treatment (Fig. 4B). Together, this indicated that DHCR14 protein, unlike LBR protein, is highly susceptible to basal and cholesterol-mediated turnover.
Co-treatment of cycloheximide and cholesterol led to a further reduction in DHCR14-V5 levels compared with cycloheximide or cholesterol alone. This shows that the cholesterol-mediated reduction of DHCR14-V5 protein levels is independent of protein synthesis (Fig. 4C). Conversely, decreasing cellular cholesterol status with statin treatment increased DHCR14-V5 protein levels (Fig. 4D).
C14-demethylated sterol intermediates, but not oxysterols, mediate turnover of DHCR14
The selectivity of different sterols as degradation triggers varies between cholesterogenic enzymes, with HMGCR degradation primarily being induced by lanosterol (34) and side-chain oxysterols like 25-hydroxycholesterol, whereas squalene monooxygenase (SM) and DHCR7 degradation is induced by cholesterol and certain other sterol intermediates (10, 35). We anticipated that the sterol degradation profile of DHCR14 would align closely with that of DHCR7, considering their homology.
A large number of mevalonate pathway sterols, including the substrate and product of C14-SRs, MASs, triggered the turnover of DHCR14 (Fig. 5A). Lanosterol, an early precursor to cholesterol, was the only sterol tested that failed to trigger this turnover, with all tested C14-demethylated sterols (post-lanosterol) triggering the degradation of DHCR14. There was no significant reduction in DHCR14 levels in response to any of the tested oxysterols (Fig. 5B). This low level of sterol specificity hints that DHCR14 is highly susceptible to variations in cholesterol production in the cell and that accumulation of any of the tested post-lanosterol intermediates rapidly triggers DHCR14 turnover.
Figure 5.

DHCR14 is degraded in response to numerous sterols. CHO-DHCR14 cells were treated for 8 h with or without 20 μg/ml of the indicated sterols complexed to cyclodextrin (A) or 1 μg/ml of the indicated oxysterols (B). Cell lysates were separated by SDS-PAGE, and protein levels were quantified by Western blotting using V5 (DHCR14), α-tubulin, and GAPDH. Data are presented as mean ± S.E. (error bars) of n = 4 (A) or n = 3 (B) independent experiments. Western blots are representative. A paired Student's two-tailed t test was used for statistical analysis. *, p < 0.05. C, sterol specificity of DHCR14 versus DHCR7. The reciprocals of data from Fig. 5A (DHCR14) or extracted from Ref. 10 (DHCR7) were plotted against the listed sterols presented in pathway order. #, no data available for this sterol.
Intriguingly, plotting the reciprocal of these data (Fig. 5A) to reflect degradative potency of the sterols, along with previously published data on the sterol-driven turnover of DHCR7 (10), suggests that mid-post-lanosterol intermediates (such as zymosterol and lathosterol) trigger the turnover of DHCR14 to a greater extent than later intermediates and cholesterol itself (Fig. 5C). The opposite is observed for the turnover of DHCR7, with later post-lanosterol intermediates (including 7DHC and desmosterol) inducing higher turnover of DHCR7 (Fig. 5C).
The sterol-responsive turnover of DHCR14 is via the ubiquitin-proteasome system
Many well-characterized cholesterol biosynthetic enzymes are turned over via the ubiquitin-proteasome system, including HMGCR (36), SM (37), and DHCR7 (10).
To evaluate which degradation pathway regulates the cholesterol-mediated turnover of DHCR14, we treated CHO-DHCR14 cells with proteasomal or lysosomal inhibitors. In the presence of the general proteasomal inhibitors MG132 and ALLN, DHCR14 was rescued from cholesterol-driven turnover (Fig. 6A). DHCR14 protein levels were not rescued when cells were treated with the lysosomal inhibitor ammonium chloride (Fig. 6A), indicating that the degradation of DHCR14 occurs primarily through the proteasome.
Figure 6.

DHCR14 is degraded by the proteasome and is ubiquitinated, although the published ubiquitination site is not responsible for DHCR14 sterol-mediated turnover. A, CHO-DHCR14 cells were treated for 8 h with or without 20 μg/ml Chol/CD, with either MG132 (10 μm), ALLN (25 μg/ml), or NH4Cl (20 mm). B, CHO-DHCR14 cells were treated for 8 h with or without 10 μm MG132 and with or without 20 μg/ml Chol/CD, and cell lysates were harvested. C, CHO-EV and CHO-DHCR14 cells were transfected with empty vector or HA-tagged ubiquitin plasmids for 24 h. Cells were treated for 2 h with 10 μm MG132 to block proteasomal degradation and subsequently treated with 10 μm MG132 with or without 20 μg/ml Chol/CD for 4 h. Cell lysate was immunoprecipitated with V5 antibody conjugated to magnetic dynabeads. D, HEK293-SM N100-GFP-V5, CHO-LBR, and CHO-DHCR14 cells were treated for 8 h with or without the VCP inhibitor CB-5083. E, CMV-DHCR14-V5 or CMV-DHCR14-K397R-V5 was transiently transfected into CHO-7 cells for 24 h. Transfected cells were treated for 8 h with or without 20 μg/ml cholesterol complexed to cyclodextrin. Cell lysates or IP products were separated by SDS-PAGE, and protein levels were quantified by Western blotting using V5 (DHCR14), HA (HA-ubiquitin), and α-tubulin or GAPDH. Data are presented as mean ± S.E. (error bars) of n = 5 (A) or n = 6 (E) independent experiments or presented as representative blots of n = 4 (B), n = 2 (C), and n = 1 (D) for previously published SM-N100 (40), n = 3 for LBR-V5, and n = 6 for DHCR14-V5. A paired Student's two-tailed t test was used for statistical analysis. *, p < 0.05; **, p < 0.01; ns, nonsignificant.
To further investigate the role of the proteasome in the sterol-mediated turnover of DHCR14, we treated CHO-DHCR14 cells with or without MG132, in the absence or presence of cholesterol (Fig. 6B). DHCR14 protein levels were increased with MG132, irrespective of cellular cholesterol status. Moreover, higher-molecular weight laddering occurred with proteasomal inhibition, consistent with polyubiquitination of DHCR14-V5.
After establishing that DHCR14 is degraded by the proteasome, we sought to investigate other proteasome-related factors that may play a role in the turnover of DHCR14. For a substrate to be degraded by the proteasome, it is typically ubiquitinated on a lysine residue. We transfected HA ectopically tagged ubiquitin (HA-Ub) into CHO-DHCR14 stable cells and immunoprecipitated DHCR14 using an anti-V5 antibody. When DHCR14 was pulled down, there was an enrichment of HA-Ub in the immunoprecipitation product, indicative of DHCR14 being ubiquitinated in the cell (Fig. 6C).
Valosin-containing protein (VCP), an ATPase involved in the destruction of ubiquitinated substrates (38), plays a role in the degradation of cholesterol-regulated proteins DHCR7 (39), SM (and its truncated regulatory domain SM N100) (40), and MARCH6 (41). We inhibited VCP using CB-5083 (39) and found that DHCR14-V5 accumulated (similarly to SM N100), whereas LBR-V5 did not (Fig. 6D). Analogous to proteasomal inhibition (Fig. 6B), far more marked higher-molecular weight laddering was observed with VCP inhibition for DHCR14-V5, compared with either SM N100-V5 or LBR-V5 (Fig. 6D).
Next, we investigated the residue responsible for sterol-mediated turnover of DHCR14. Although there are 10 lysines in DHCR14, there is only experimental evidence for the ubiquitination of Lys-397 (42). We mutated Lys-397 to arginine (K397R) to prevent ubiquitination and found no blunting of sterol-mediated turnover compared with the WT plasmid (Fig. 6E).
Multiple E3 ligases may interact with DHCR14 and its homologous enzyme, DHCR7
We set out to identify the E3 ligase(s) that mediate the degradation of DHCR14 and DHCR7. In our previous work, knockdown of MARCH6, an E3 ligase that can target the two rate-limiting enzymes in cholesterol biosynthesis, HMGCR and SM (37), failed to rescue the sterol-driven turnover of DHCR7 (10). In the present study, we knocked down MARCH6 using siRNA (Fig. 7A) and found that it was also not responsible for the turnover of DHCR14 (Fig. 7, B and C).
Figure 7.

Numerous E3 ligases affect basal protein levels of DHCR14, but none of the tested E3 ligases are solely responsible for the sterol-mediated turnover of DHCR14. A–C, CHO-DHCR14 cells were transfected for 24 h with a 25 nm concentration of the indicated siRNA and pretreated for 16 h in sterol-deficient medium. A, total RNA was harvested, cDNA was prepared, and mRNA levels of the siRNA targets were measured using qRT-PCR and normalized to PBGD. mRNA levels are relative to the negative siRNA control, which was set to 1. B and C, cells were treated for 8 h with or without 20 μg/ml Chol/CD, and cell lysates were harvested. Cell lysates were separated by SDS-PAGE, and protein levels were quantified by Western blotting using V5 (DHCR14) and GAPDH. Data are presented as mean ± S.E. (error bars) of n = 3 (A) or n = 5–8 (C) independent experiments. Western blots are representative. A paired Student's two-tailed t test was used for statistical analysis. *, p < 0.05; **, p < 0.01. D, Relative cholesterol driven degradation of DHCR14-V5 with respect to the control siRNA condition was calculated using values from figure 7C.
To identify new candidate E3 ligases for the degradation of DHCR14 and DHCR7, we employed a proteomics-based approach. Utilizing CHO-DHCR14 cells and HEK293 cells transiently expressing DHCR7-Myc with appropriate empty vector controls, interaction partners were co-immunoprecipitated and separated via SDS-PAGE. After pulldown of target proteins, MS was performed to identify E3 ligases with peptides present in the DHCR14 or DHCR7 samples, and absent in the empty vector controls, as potential interacting partners (Table 1). The full list of identified proteins can be found in Table S1 and the MS/MS fragmentation patterns for single peptide identifications can be found in figures S2–S9.
Table 1.
E3 ligases identified in DHCR14 and DHCR7 immunoprecipitation
Scores were assigned by MASCOT, and MASCOT scores >30 were used as a threshold for identification. Coverage represents the percentage of the protein that was mapped by found peptides.
| Accession | Gene name | Description | Immunoprecipitation experiment | Found? | Score | Coverage % |
|---|---|---|---|---|---|---|
| Q86TM6 | HRD1 | E3 ubiquitin-protein ligase synoviolin1 (SYVN1) | DHCR7 | Yes | 41.01 | 1.46 |
| DHCR14 | No | |||||
| Q9UKV5 | GP78 | E3 ubiquitin-protein ligase AMFR | DHCR7 | Yes | 266.07 | 7.15 |
| DHCR14 | Yes | 47.84 | 2.80 | |||
| Q9UNE7 | CHIP | E3 ubiquitin-protein ligase CHIP | DHCR7 | Yes | 54.8 | 3.96 |
| DHCR14 | No | |||||
| Q5T447 | HECTD3 | E3 ubiquitin-protein ligase HECTD3 | DHCR7 | Yes | 53.42 | 1.63 |
| DHCR14 | No | |||||
| Q15034 | HERC3 | Probable E3 ubiquitin-protein ligase HERC3 | DHCR7 | Yes | 47.42 | 1.33 |
| DHCR14 | No | |||||
| Q7Z6Z7 | HUWE1 | E3 ubiquitin-protein ligase HUWE1 | DHCR7 | Yes | 137.07 | 0.87 |
| DHCR14 | No | |||||
| 884908087 | Itch | E3 ubiquitin-protein ligase Itchy | DHCR7 | No | ||
| DHCR14 | Yes | 57.93 | 1.73 | |||
| O43164 | PRAJA2 | E3 ubiquitin-protein ligase Praja2 | DHCR7 | Yes | 31.69 | 2.54 |
| DHCR14 | No | |||||
| O00308 | WWP2 | NEDD4-like E3 ubiquitin-protein ligase WWP2 | DHCR7 | Yes | 53.91 | 1.49 |
| DHCR14 | No |
We next determined whether the identified E3 ligases played a role in the sterol-dependent turnover of DHCR14. Using siRNA to knock down the candidate E3 ligases (Fig. 7A and Fig. S1A), we observed a significant increase in the basal protein levels of DHCR14-V5 when WW domain-containing E3 ubiquitin protein ligase 2 (WWP2) was knocked down (Fig. 7, B and C). All other E3 ligases identified from our interaction screen, including glycoprotein 78 and Hrd1, which are implicated in the turnover of HMGCR (43), failed to increase basal DHCR14 levels and/or rescue the sterol-mediated turnover of DHCR14 (Fig. 7 (B and C) and Fig. S1B).
WWP2 knockdown additionally resulted in a significant decrease of cholesterol-induced degradation, with WWP2 siRNA knockdown leading to a 45% reduction in the sterol-driven turnover of DHCR14 (Fig. 7, B and D). This suggests that WWP2 may play a role in the cholesterol-mediated turnover of DHCR14.
DHCR14 and LBR have differential expression between tissues
Finally, we hypothesized that the expression of DHCR14 and LBR may differ between tissues to help explain the apparent redundancy of these two enzymes. Using the publicly accessible median gene expression of human tissues from the GTEx expression database (dbGaP accession code phs000424.v7.p2), we investigated the correlation of TM7SF2 and LBR transcript levels in six tissues and noted both tissue-specific expression patterns (unlike for porphobilinogen deaminase (PBGD)) and a negative relationship between expression of TM7SF2 and LBR (Fig. 8A). Expanding this analysis to 51 human tissues highlighted a moderate negative correlation of expression between TM7SF2 and LBR, with the linear correlation producing a significantly nonzero line of best fit (Pearson correlation coefficient, r = −0.37, p = 0.01). Various attempts at curve fitting were made on the data to elucidate the relationship; however, the “best-fitting” models were of questionable biological relevance. The expression data were moderately well-fitted (r = −0.48), with no significant deviation (p = 0.14), to a semi-log model (Fig. 8B).
Figure 8.

Human tissue gene expression of TM7SF2 versus LBR. Analysis of gene expression suggests a reciprocal, nonlinear relationship between TM7SF2 and LBR gene expression. A, bar chart of PBGD, TM7SF2, and LBR expression, using the publicly accessible data for six listed human tissues from the GTEx expression database (dbGaP accession code phs000424.v7.p2). B, TM7SF2 and LBR expression (expressed as transcripts per million (TPM)), using data for 51 human tissues from the GTEx expression database (dbGaP accession code phs000424.v7.p2). Pearson's correlation coefficients were as follows: p < 0.01, r = −0.37. Semi-log relationship statistics were as follows: p = 0.14, r = −0.48 with data not significantly deviating (#) from the semi-log model. C, TM7SF2 and LBR gene expression versus HMGCR or DHCR7 gene expression (expressed as transcripts per million), using data for 51 human tissues from the GTEx expression database (dbGaP accession code phs000424.v7.p2). All statistical analysis was performed in GraphPad Prism 8.
This negative correlation of expression was specific to the TM7SF2 and LBR pairing; there were no discernible relationships between the median tissue expression of LBR and two other examined cholesterol synthesis enzymes (HMGCR, r = 0.07, p = 0.62; DHCR7, r = −0.13, p = 0.36). Strong linear relationships were observed between TM7SF2 and HMGCR (r = 0.57, p < 0.001) or DHCR7 (r = 0.77, p < 0.001) expression in tissues (Fig. 8C); these correlations are in line with all of these enzymes being predominantly controlled by the same transcription factor, SREBP-2 (29).
Discussion
Cell cholesterol synthesis is tightly controlled by both transcriptional and post-translational regulation. In this study, we showed differential regulation of two homologous cholesterol biosynthesis enzymes, DHCR14 and LBR, elucidated the degradation pathway for DHCR14, and proposed a tunable model for the regulation of C14-SR catalytic activity (Fig. 9). We found that DHCR14 overall follows a regulatory pattern similar to many other cholesterol biosynthesis enzymes, particularly its homolog DHCR7 and the well-studied second rate-limiting enzyme of cholesterol biosynthesis, SM (8, 10, 35, 44).
Figure 9.
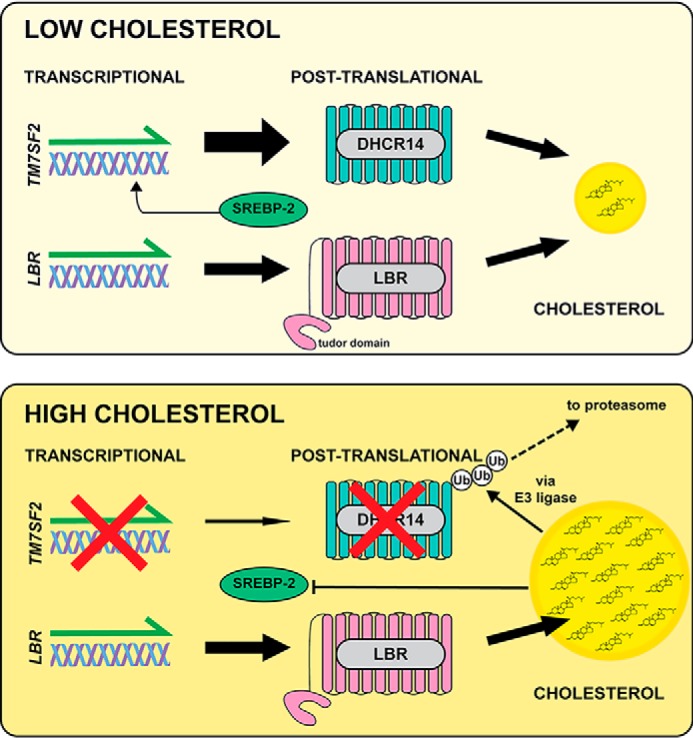
Proposed model of DHCR14 and LBR expression linked to cellular cholesterol status. Under conditions of low cholesterol (top), TM7SF2 gene expression is up-regulated by SREBP-2, and LBR is constitutively expressed. In this scenario, both DHCR14 and LBR protein are active and contribute to cholesterol synthesis. When cholesterol levels are high (bottom), TM7SF2 gene expression is inhibited, and DHCR14 protein is degraded by the proteasome. LBR gene and protein remain at basal levels, and LBR becomes the main contributor to Δ14 reductase activity.
The observed degradation of DHCR14 in the presence of sterols (Figs. 4 and 5), coupled with it being an SREBP-2 target gene (Fig. 2B) (16), suggests that levels of this enzyme are highly susceptible to cellular cholesterol status. This was observed of DHCR14 as early as its initial isolation studies (45), where the treatment of excess cholesterol reduced activity of the enzyme in isolated microsomes.
HMGCR, SM, and DHCR7 are all known to be regulated by changing sterol levels transcriptionally and post-translationally (8, 10, 29, 35, 44, 46). The addition of DHCR14 into this suite of enzymes, despite the enzymatic redundancy of C14-SRs, forges a strong case for its biological relevance and importance in cholesterol biosynthesis. Intriguingly, we show that TM7SF2 in CHO-7–derived cell lines does not respond like a canonical SREBP-2 target. We have previously found that SREs are not necessarily conserved between rodents and human cell models (8). The primary work in mapping the SREs of TM7SF2 has been carried out in human cell lines (15, 16), and further work is required to explain the lack of sterol response in hamster cell models.
In agreement with a previous study that found that the Tudor domain of LBR confers its stability (24), we show that LBR is resistant to cholesterol-mediated turnover. It is likely that this stability is necessary for its additional role at the inner nuclear membrane of binding lamin B and interacting with H3/H4 histones (24).
We found that DHCR14 turnover, similarly to DHCR7 (10), is triggered by a wide variety of sterols (Fig. 5A), including both its substrate and product. Comparing the turnover of DHCR14 and DHCR7 revealed that sterols appearing earlier in the post-lanosterol pathway preferentially degrade DHCR14. Comparison of this turnover with previously published data on the sterol-driven turnover of DHCR7 (10) revealed that DHCR7 is degraded to a greater extent by sterols later in the sterol synthesis pathway (Fig. 5C). Recently, published work on HMGCR has shown that lanosterol and post-lanosterol C4-dimethylated sterol intermediates, including T-MAS and FF-MAS, induce rapid degradation of HMGCR (34). Within the large feedback loops of cholesterol biosynthesis, it seems likely that there are smaller feedback loops that regulate the degradation of crucial enzymes and help to control flux through the pathway. For instance, early pathway sterols affect early pathway enzymes (e.g. DHCR14), with late-pathway sterols including cholesterol controlling later-pathway enzymes (e.g. DHCR7).
Furthermore, we identified that the predominant degradation pathway of DHCR14 is via the proteasome (Fig. 6A), with proteasomal inhibition leading to a 2–3-fold increase in DHCR14 levels. The increase in DHCR14-V5 levels with the addition of MG132 occurred irrespective of the addition of cholesterol, but cholesterol-mediated turnover of DHCR14 could be fully rescued by MG132 addition (Fig. 6, B and C). We additionally showed that DHCR14 is ubiquitinated with or without added cholesterol (Fig. 6C). Inhibition of VCP, an ATPase involved in extracting ubiquitinated substrates from the endoplasmic reticulum, led to the accumulation of DHCR14. Additionally, high-molecular weight banding was prominent with VCP inhibition, a phenomenon that was not observed for SM N100 GFP, another VCP substrate that our laboratory has reported (40). However, high-molecular weight banding of DHCR14 occurs after MG132 treatment, irrespective of cholesterol treatment (Fig. 6B), as does ubiquitination of DHCR14 (Fig. 6C). These findings, together with the failure of mutating the only published ubiquitination site to rescue cholesterol-mediated turnover of DHCR14, suggest that cholesterol is not the only trigger for the ubiquitination of DHCR14.
We previously found that MARCH6, an E3 ligase for SM and HMGCR (37), is not responsible for the cholesterol-mediated turnover of DHCR7 (10) and here show that this E3 ligase also does not affect the levels of ectopic DHCR14 (Fig. 7, B and C). We identified nine E3 ligases that potentially interact with DHCR14 and/or DHCR7 (Table 1). The different effects of a wide variety of E3 ligase knockdowns on basal DHCR14 protein levels (Fig. 7 (B and C) and Fig. S1B) suggest that many of these identified and tested E3 ligases are altering cellular proteostasis in poorly understood ways and indicate that the role of the ubiquitin-proteasome system in the regulation of cholesterol metabolism is far more complex than initially envisaged.
From our results, WWP2, a member of the NEDD4-1 family of E3 ligases, modulates DHCR14 levels and may blunt the sterol-mediated degradation of DHCR14 (Fig. 7, C and D). However, considering that WWP2 knockdown does not completely rescue sterol-mediated turnover of DHCR14 and only increases basal DHCR14 levels by 2-fold (compared with an order of magnitude increase in basal SM levels with MARCH6 knockdown (37)), we speculate that WWP2 is not the only E3 ligase responsible for the degradation of DHCR14. WWP2 interacts with and targets substrates that contain a PPXY motif (47) that is not found in the protein-coding sequence of DHCR14 and as such may indirectly mediate DHCR14 levels through another substrate.
Our experiments have focused on an ectopic model of DHCR14 expression under the control of a CMV promoter to avoid sterol-mediated transcriptional control. Future work should confirm that endogenous DHCR14 behaves similarly to ectopic DHCR14 post-translationally when a suitable antibody becomes available.
We have observed via the negative relationship of TM7SF2 and LBR median tissue transcript levels (Fig. 8, A and B) that tissues preferentially express one or the other C14-SR. Blood and blood peripheral tissue (the spleen) had overall high levels of LBR and low TM7SF2 levels. Conversely, tissues with high cholesterol synthetic capacity showed enrichment of TM7SF2 transcripts. Skin samples showed a ∼6-fold difference between the two C14-SRs, and the liver, the primary site of cholesterol synthesis, had TM7SF2 levels ∼4-fold higher. The brain, while having a moderate expression of TM7SF2, also had TM7SF2 levels ∼4-fold higher than that of LBR. No tissue listed in the database had high levels of both TM7SF2 and LBR. This tissue-specific switching appears to be specific to C14-SR expression; other SREBP-2 target genes, HMGCR and DHCR7, while having strong positive linear relationships with TM7SF2 expression, shared no discernible pattern of expression with LBR, indicating that there is another factor controlling the TM7SF2 and LBR expression axis.
From our work and that of others, LBR is constitutively active independent of sterol levels, with LBR protein levels remaining steady. On the other hand, we have shown that DHCR14 is a highly tunable and inducible C14-SR, with an increase in mRNA levels in times of low sterols and a rapid reduction in protein level when cellular sterol status is high—a trait mirrored in several other sterol synthesis enzymes. We propose that DHCR14, being transcriptionally and post-translationally sensitive to sterol levels, provides extra C14-SR capacity in times of sterol depletion. However, LBR is constitutively active as a C14-SR independent of cellular cholesterol status (Fig. 9).
In conclusion, DHCR14 and LBR are differentially regulated at both the transcriptional and post-translational levels. We identified triggers for the degradation of DHCR14 and screened a panel of putative E3 ligases responsible for its sterol-driven turnover. We propose a model for the duality of C14-SR enzymes, which further highlights the exquisite regulation of cholesterol biosynthesis.
Experimental procedures
Cell culture
Cell lines stably expressing DHCR14-V5 (CHO-DHCR14) or LBR-V5 (CHO-LBR) under the control of a CMV promoter were generated for this study from CHO-7 Flp-In cells as described previously (14). Briefly, plasmids containing an FRT recombination site and either the DHCR14 or LBR coding sequence (pcDNA5-FRT-CMV-DHCR14-V5 or pcDNA5-FRT-CMV-LBR-V5) were transfected into CHO-7 stable cells generated in-house with the Flp-In system (Invitrogen). Single colonies were selected for by the addition of hygromycin B (400 μg/ml). Clones were screened for expression via Western blotting, and clones that had growth rates similar to the parental cell lines and expressed moderate levels of DHCR14-V5 or LBR-V5 were selected for further study.
CHO-7 cells stably expressing DHCR7-Myc (CHO-DHCR7) (10) or pcDNA5/FRT empty vector (EV) (CHO-EV) (48) and HEK293-SM N100-GFP-V5 (37) were previously generated in-house.
CHO-derived cell lines were maintained in DMEM/F-12 medium with 5% (v/v) lipoprotein-deficient serum (LPDS) (50 mg/ml protein) and supplemented with penicillin (100 units/ml) and streptomycin (100 μg/ml). CHO-DHCR14, CHO-LBR, and CHO-DHCR7 cell lines were supplemented with 400 μg/ml hygromycin B, and CHO-EV cell lines were supplemented with 150 μg/ml hygromycin B.
HeLaT and Be(2)C cells were gifts from Drs. Noel Whitaker and Louise Lutze-Mann, respectively (University of New South Wales, Sydney, Australia), Huh7 cells were gifts from the Centre for Vascular Research (University of New South Wales, Sydney, Australia), and HEK293 cells were gifts of Drs. Goldstein and Brown (University of Texas Southwestern Medical Center, Dallas, TX).
HeLaT cells were maintained in RPMI medium, and Huh7, Be(2)C, and HEK293 cells were maintained in DMEM:HG medium. HEK293-SM N100-GFP-V5 cells were maintained in DMEM:HG medium supplemented with 200 μg/ml hygromycin B. All human-derived cell lines were grown with 10% (v/v) fetal calf serum (FCS) and supplemented with penicillin (100 units/ml) and streptomycin (100 μg/ml). Lipoprotein-deficient serum was prepared in-house from newborn calf serum (LPDS) or FCS (FCLPDS) as described previously (49, 50).
Immunofluorescent microscopy
Cells were grown on coverslips and transfected for 24 h with an ER marker plasmid (pDsRed-ER) using Lipofectamine LTX reagent. Cells were fixed in 4% (v/v) paraformaldehyde, rinsed in PBS, and permeabilized with 0.3% (v/v) Triton X-100. Fixed cells were washed with PBS and blocked for 1 h with 10% FCS (v/v) in PBS. Cells were incubated with anti-V5 antibody in 10% FCS (v/v) in PBS and 0.1% (w/v) saponin in the dark at 4 °C overnight.
Samples were washed and incubated with Alexa Fluor 488 anti-mouse in 10% FCS (v/v) in PBS and 0.1% (w/v) saponin in the dark for 1 h. Cells were washed, and coverslips were mounted onto glass slides with ProLong Gold AntiFade Reagent with 4′,6-diamidino-2-phenylindole (DAPI). Slides were dried, sealed, and imaged. Images were obtained using a Nikon C1 confocal microscope with laser excitation at 408 nm (DAPI), 488 nm (Alexa Fluor 488), and 561 nm (DsRed-ER).
Treatments
Pretreatment and all experiments (unless stated otherwise in the figure legends) were carried out in lipoprotein-deficient media (LPDS for CHO-derived cell lines and FCLPDS for human-derived cell lines) supplemented with the statin compactin (5 μm) and mevalonate (50 μm).
T0901317 (Sigma), compactin (Sigma), cycloheximide (Sigma), CB-5083 (Sigma), ALLN (Sigma), and MG132 (carbobenzoxy-Leu-Leu-leucinal, Sigma) were dissolved in DMSO. 25-Hydroxycholesterol, 24,25-epoxycholesterol, and mevalonate were dissolved in 100% ethanol. Complexed sterols (cholesterol-cyclodextrin purchased from Sigma-Aldrich, remaining sterols purchased from Steraloids and complexed in-house as described previously (51)) were dissolved in filtered MilliQ water. Appropriate solvent controls were used.
Western blotting
CHO-DHCR14 and CHO-LBR protein samples were harvested in 3% (w/v) SDS supplemented with 2% (v/v) protease inhibitor mixture (Sigma-Aldrich). Protein concentrations were measured by a BCA protein assay (Thermo Fisher Scientific), normalized, and separated by 10% SDS-PAGE. Samples were transferred onto nitrocellulose membranes. Membranes were blocked in 5% (w/v) skim milk/PBS with 0.1% (v/v) Tween 20 (PBST) and probed with the following primary antibodies: anti-V5 (1:5000 in PBST; Invitrogen, R960-25), anti-GAPDH (1:2000 in 5% (w/v) BSA/PBST; Cell Signaling Technologies, 2118L), anti-HA (1:2000 in 5% (w/v) BSA/PBST; Cell Signaling Technologies, C29F4), Anti-Myc (1:2000 in 0.5% (w/v) BSA/PBST; Santa Cruz Biotechnology, Inc., sc-40).
Secondary antibodies were diluted 1:10,000 in 5% (w/v) skim milk/PBST (Jackson Immunoresearch Laboratories; HRP-anti-mouse (715-035-150) and HRP-anti-rabbit (715-035-150 711-035-152) or 5% (w/v) BSA/PBST (LI-COR anti-mouse (926-68073) and anti-rabbit (926-32212)). HRP-conjugated antibodies were imaged using Immobilon Western HRP substrate (Merck, WBKLS0500) on an ImageQuant LAS 500. LI-COR antibodies were imaged on a LI-COR Odyssey CLx.
Quantitative real-time PCR
Total RNA was harvested from cells using TRI Reagent. cDNA was synthesized from RNA using the SuperScript III First Strand cDNA synthesis kit (Thermo Fisher Scientific).
Gene expression was determined using the SensiMix SYBR® kit (Bioline), and expression was normalized to the housekeeping gene coding for PBGD. Primer sequences used for qRT-PCR can be found in Table S2. Further normalization was carried out as described in the figure legends.
Protein immunoprecipitation
Cells were lysed in radioimmunoprecipitation assay buffer (20 mm Tris-HCl, 150 mm NaCl, 0.1% SDS, 1% Nonidet P-40, 1 mm sodium orthovandate, 5 mm EDTA). Cell lysate was rotated overnight with anti-V5 (Invitrogen, R960-25) or anti-Myc (Santa Cruz Biotechnology, sc-40) conjugated to DynaBeads (Thermo Fisher Scientific). Proteins were separated by 10% SDS-PAGE for either Western blotting or band excision for MS.
Liquid chromatography mass spectrometry
Gel bands were excised, destained, reduced, and alkylated following the methodology outlined by Shevchenko et al. (52). In-gel tryptic digestions and peptide extractions were performed following procedures described by Luu et al. (11). Peptide solutions were dried and resuspended in 0.1% (v/v) formic acid.
For DHCR14 samples, peptides were subjected to LC-MS/MS analysis on an LTQ Orbitrap Velos Pro (Thermo Scientific, Bremen, Germany) interfaced with an UltiMate 3000 HPLC and autosampler system (Dionex, Amsterdam, The Netherlands). Peptides were separated by nano-LC and ionized using positive ion mode nano-ESI as described by Hart-Smith and Raftery (53). MS survey scans were performed using parameters described in Luu et al. (11). Up to the 10 most abundant ions (>5000 counts) with charge state >2 were sequentially isolated and fragmented via collision-induced dissociation using parameters described by Luu et al. (11).
For DHCR7 samples, peptides were subjected to an LC-MS/MS analysis on a Tribrid Fusion Lumos (Thermo Scientific) interfaced with an UltiMate 3000 HPLC and autosampler system (Dionex, Amsterdam, The Netherlands). Peptides were separated by nano-LC following conditions described by Smith et al. (54), and eluting peptides were ionized using positive ion mode nano-ESI as described by Hart-Smith and Raftery (53). MS survey scans were performed using parameters described by Chua et al. (55), and ions (>2500 counts) with charge state +2 to +5 were sequentially isolated and fragmented via higher-energy collision dissociation using parameters described by Chua et al. (55).
Sequence database searches were performed using the Proteome Discoverer mass informatics platform (version 1.4, Thermo Scientific), using the search program Mascot (version 2.3, Matrix Science). Peak lists derived from LC-MS/MS were searched using the following parameters: instrument type was ESI-TRAP; precursor ion and peptide fragment mass tolerances were ±5 ppm and ±0.4 Da, respectively, for DHCR14 samples and ±5 ppm and ±0.02 Da, respectively, for DHCR7 samples; variable modifications included in each search were carbamidomethyl (C) and oxidation (M); and enzyme specificity was trypsin with up to two missed cleavages. For DHCR14 samples and empty vector controls, the NCBI protein database (56) was searched using rodentia taxonomies only. For DHCR7 samples and empty vector controls, the UniProt database (July 2013 release, 540,732 sequence entries) was searched using human sequences only. E3 ligases putatively identified from one or more statistically significant (p < 0.05 according to the Mascot expect metric) peptides and absent in empty vector controls were subjected to downstream analysis. The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (57) partner repository with the data set identifier PXD016417.
Site-directed mutagenesis
The CMV-DHCR14-V5 plasmid construct was generated using methods described previously (14). We used megaprimer site-directed mutagenesis (58) to mutate CMV-DHCR14-V5 to generate the K397R mutant plasmid. Megaprimers were generated using the forward mutagenic primer GCCTGCAGAGGTACGGCCTG and a BGH reverse primer GCGATGCAATTTCCTCATTT.
Transient transfection
To express DHCR14 WT and DHCR14 K397R plasmids, CHO-7 cells were seeded in 6-well plates and transfected with 0.25 μg of expression plasmid, 0.75 μg of pTK-empty vector DNA, and 4 μl of Lipofectamine-LTX for 24 h. To express HA-tagged ubiquitin, CHO-EV or CHO-DHCR14 cells were seeded in 6-cm dishes and transfected with either 2 μg of pTK-empty vector DNA and 4 μl of Lipofectamine-LTX or 1.8 μg of pTK-empty vector DNA, 0.2 μg of pEF1a-HA-ubiquitin (HA-Ub) (a kind gift from Dr. Bao-Liang Song (Wuhan University)), and 4 μl of Lipofectamine-LTX for 24 h. Cells were then treated as described in the figure legends.
siRNA transfection
To knock down candidate E3 ligase genes, CHO-DHCR14 cells were transfected with 25 nm siRNA using Lipofectamine RNAiMAX (Thermo Fisher Scientific) for 24 h followed by pretreatment and treatment as listed in figure legends. Nonproprietary siRNA sequences are available in Table S3.
Calculation of cholesterol-induced degradation
To account for changes in basal protein expression with transient transfections and E3 ligases knockdowns, we calculated the cholesterol-induced degradation of DHCR14 with respect to each transfected plasmid or siRNA. This is a measure of the ratio of protein degraded in the plasmid/siRNA condition compared with the control.
A ratio smaller than 1 indicates that there is reduced cholesterol-mediated degradation, and a ratio greater than 1 indicates increased cholesterol-driven degradation. This method has been described in previous work (59).
GTEx database
The data used for the analyses described in this paper were obtained from the GTEx Portal on July 12, 2019, dbGaP accession number phs000424.v7.p2. Median tissue expression data were extracted from the GTEx Portal for 51 tissues. Correlations were generated by GraphPad Prism 8 (GraphPad Software Inc.).
Data presentation and statistical analysis
Image analysis was carried out in Image Studio Lite (version 5.2, LI-COR Biosciences). Statistical analysis was conducted as described in the respective figure legends. Pearson correlation coefficients and the fitting of curves were calculated in GraphPad Prism 8 (GraphPad Software). Graphs were generated in GraphPad Prism 8 (GraphPad Software).
Author contributions
I. M. C.-H. and A. J. B. conceptualization; I. M. C.-H. data curation; I. M. C.-H. and L. J. S. formal analysis; I. M. C.-H., L. J. S., L. Q., G. H.-S., and A. V. P. investigation; I. M. C.-H., L. J. S., G. H.-S., and A. J. B. methodology; I. M. C.-H. and A. J. B. writing-original draft; I. M. C.-H., L. J. S., G. H.-S., A. V. P., and A. J. B. writing-review and editing; L. J. S., A. V. P., and A. J. B. supervision; A. J. B. funding acquisition; A. J. B. project administration.
Supplementary Material
Acknowledgments
We thank members of the Brown laboratory for reviewing the manuscript, the Sytnyk laboratory (University of New South Wales) for technical assistance with microscopy, Dr. Winnie Luu for cloning expression plasmids, and Dr. Vicky Howe for assistance with quantitative PCR.
This work was supported by Australian Research Council Grant DP170101178 to (A. J. B.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Tables S1–S3 and Figs. S1–S9.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (57) partner repository with the data set identifier PXD016417.
- DHCR7
- 7-dehydrocholesterol reductase
- 7DHC
- 7-dehydrocholesterol
- ALLN
- N-acetyl-Leu-Leu-Norleu-al
- C14-SR
- 3β-hydroxysterol Δ14-reductases
- CHO
- Chinese hamster ovary
- Chol/CD
- cholesterol/methyl-β-cyclodextrin
- CMV
- cytomegalovirus
- DAPI
- 4′,6-diamidino-2-phenylindole
- DHCR14
- 14-dehydrocholesterol reductase
- DHCR24
- 24-dehydrocholesterol reductase
- DMEM
- Dulbecco's modified Eagle's medium
- ER
- endoplasmic reticulum
- ESI
- electrospray ionization
- EV
- empty vector
- FCLPDS
- lipoprotein-deficient serum prepared from FCS
- FCS
- fetal calf serum
- FF-MAS
- follicular fluid MAS
- FRT
- Flp-In recombination target
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- HA-Ub
- HA ectopically tagged ubiquitin
- HMGCR
- 3-hydroxy-3-methylglutaryl-CoA reductase
- Hrd1
- hydroxymethyl glutaryl-coenzyme A reductase degradation protein 1
- HRP
- horseradish peroxidase
- LBR
- lamin-B receptor
- LPDS
- lipoprotein-deficient serum
- MARCH6
- membrane-associated Ring-CH-type finger 6
- MAS
- meiosis-activating sterol
- MG132
- carbobenzoxy-Leu-Leu-leucinal
- PBGD
- porphobilinogen deaminase
- qRT-PCR
- quantitative RT-PCR
- SM
- squalene monooxygenase
- SRE
- sterol regulatory element
- SREBP-2
- SRE-binding protein 2
- TM7SF2
- transmembrane 7 superfamily member 2
- T-MAS
- testis MAS
- V5
- V5 epitope tag
- VCP
- valosin-containing protein
- WWP2
- WW domain-containing protein 2.
References
- 1. Platt F. M., Wassif C., Colaco A., Dardis A., Lloyd-Evans E., Bembi B., and Porter F. D. (2014) Disorders of cholesterol metabolism and their unanticipated convergent mechanisms of disease. Annu. Rev. Genomics Hum. Genet. 15, 173–194 10.1146/annurev-genom-091212-153412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Simons K., and Ehehalt R. (2002) Cholesterol, lipid rafts, and disease. J. Clin. Invest. 110, 597–603 10.1172/JCI0216390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Russell D. W. (2003) The enzymes, regulation, and genetics of bile acid synthesis. Annu. Rev. Biochem. 72, 137–174 10.1146/annurev.biochem.72.121801.161712 [DOI] [PubMed] [Google Scholar]
- 4. Payne A. H., and Hales D. B. (2004) Overview of steroidogenic enzymes in the pathway from cholesterol to active steroid hormones. Endocr. Rev. 25, 947–970 10.1210/er.2003-0030 [DOI] [PubMed] [Google Scholar]
- 5. Roux C., Wolf C., Mulliez N., Gaoua W., Cormier V., Chevy F., and Citadelle D. (2000) Role of cholesterol in embryonic development. Am. J. Clin. Nutr. 71, 1270S–1279S 10.1093/ajcn/71.5.1270s [DOI] [PubMed] [Google Scholar]
- 6. Prospective Studies Collaboration, Lewington S., Whitlock G., Clarke R., Sherliker P., Emberson J., Halsey J., Qizilbash N., Peto R., and Collins R. (2007) Blood cholesterol and vascular mortality by age, sex, and blood pressure: a meta-analysis of individual data from 61 prospective studies with 55,000 vascular deaths. Lancet 370, 1829–1839 10.1016/S0140-6736(07)61778-4 [DOI] [PubMed] [Google Scholar]
- 7. Sharpe L. J., Cook E. C. L., Zelcer N., and Brown A. J. (2014) The UPS and downs of cholesterol homeostasis. Trends Biochem. Sci. 39, 527–535 10.1016/j.tibs.2014.08.008 [DOI] [PubMed] [Google Scholar]
- 8. Prabhu A. V., Sharpe L. J., and Brown A. J. (2014) The sterol-based transcriptional control of human 7-dehydrocholesterol reductase (DHCR7): evidence of a cooperative regulatory program in cholesterol synthesis. Biochim. Biophys. Acta 1842, 1431–1439 10.1016/j.bbalip.2014.07.006 [DOI] [PubMed] [Google Scholar]
- 9. Zerenturk E. J., Sharpe L. J., and Brown A. J. (2014) DHCR24 associates strongly with the endoplasmic reticulum beyond predicted membrane domains: implications for the activities of this multi-functional enzyme. Biosci. Rep. 34, e00098 10.1042/BSR20130127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Prabhu A. V., Luu W., Sharpe L. J., and Brown A. J. (2016) Cholesterol-mediated degradation of 7-dehydrocholesterol reductase switches the balance from cholesterol to vitamin D synthesis. J. Biol. Chem. 291, 8363–8373 10.1074/jbc.M115.699546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Luu W., Hart-Smith G., Sharpe L. J., and Brown A. J. (2015) The terminal enzymes of cholesterol synthesis, DHCR24 and DHCR7, interact physically and functionally. J. Lipid Res. 56, 888–897 10.1194/jlr.M056986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Prabhu A. V., Luu W., Sharpe L. J., and Brown A. J. (2017) Phosphorylation regulates activity of 7-dehydrocholesterol reductase (DHCR7), a terminal enzyme of cholesterol synthesis. J. Steroid Biochem. Mol. Biol. 165, 363–368 10.1016/j.jsbmb.2016.08.003 [DOI] [PubMed] [Google Scholar]
- 13. Luu W., Zerenturk E. J., Kristiana I., Bucknall M. P., Sharpe L. J., and Brown A. J. (2014) Signaling regulates activity of DHCR24, the final enzyme in cholesterol synthesis. J. Lipid Res. 55, 410–420 10.1194/jlr.M043257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zerenturk E. J., Kristiana I., Gill S., and Brown A. J. (2012) The endogenous regulator 24(S),25-epoxycholesterol inhibits cholesterol synthesis at DHCR24 (Seladin-1). Biochim. Biophys. Acta 1821, 1269–1277 10.1016/j.bbalip.2011.11.009 [DOI] [PubMed] [Google Scholar]
- 15. Bennati A. M., Castelli M., Della Fazia M. A., Beccari T., Caruso D., Servillo G., and Roberti R. (2006) Sterol dependent regulation of human TM7SF2 gene expression: role of the encoded 3β-hydroxysterol Δ14-reductase in human cholesterol biosynthesis. Biochim. Biophys. Acta 1761, 677–685 10.1016/j.bbalip.2006.05.004 [DOI] [PubMed] [Google Scholar]
- 16. Schiavoni G., Bennati A. M., Castelli M., Della Fazia M. A., Beccari T., Servillo G., and Roberti R. (2010) Activation of TM7SF2 promoter by SREBP-2 depends on a new sterol regulatory element, a GC-box, and an inverted CCAAT-box. Biochim. Biophys. Acta 1801, 587–592 10.1016/j.bbalip.2010.01.013 [DOI] [PubMed] [Google Scholar]
- 17. Cohen T. V., Klarmann K. D., Sakchaisri K., Cooper J. P., Kuhns D., Anver M., Johnson P. F., Williams S. C., Keller J. R., and Stewart C. L. (2008) The lamin B receptor under transcriptional control of C/EBPϵ is required for morphological but not functional maturation of neutrophils. Hum. Mol. Genet. 17, 2921–2933 10.1093/hmg/ddn191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Byskov A. G., Andersen C. Y., Leonardsen L., and Baltsen M. (1999) Meiosis activating sterols (MAS) and fertility in mammals and man. J. Exp. Zool. 285, 237–242 [DOI] [PubMed] [Google Scholar]
- 19. Byskov A. G., Baltsen M., and Andersen C. Y. (1998) Meiosis-activating sterols: background, discovery, and possible use. J. Mol. Med. 76, 818–823 10.1007/s001090050286 [DOI] [PubMed] [Google Scholar]
- 20. Holmer L., Pezhman A., and Worman H. J. (1998) The human lamin B receptor/sterol reductase multigene family. Genomics 54, 469–476 10.1006/geno.1998.5615 [DOI] [PubMed] [Google Scholar]
- 21. Silve S., Dupuy P. H., Ferrara P., and Loison G. (1998) Human lamin B receptor exhibits sterol C14-reductase activity in Saccharomyces cerevisiae. Biochim. Biophys. Acta 1392, 233–244 10.1016/S0005-2760(98)00041-1 [DOI] [PubMed] [Google Scholar]
- 22. Waterham H. R., Koster J., Mooyer P., van Noort G., Kelley R. I., Wilcox W. R., Wanders R. J., Hennekam R. C., and Oosterwijk J. C. (2003) Autosomal recessive HEM/Greenberg skeletal dysplasia is caused by 3 β-hydroxysterol δ 14-reductase deficiency due to mutations in the lamin B receptor gene. Am. J. Hum. Genet. 72, 1013–1017 10.1086/373938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wassif C. A., Brownson K. E., Sterner A. L., Forlino A., Zerfas P. M., Wilson W. K., Starost M. F., and Porter F. D. (2007) HEM dysplasia and ichthyosis are likely laminopathies and not due to 3β-hydroxysterol Δ14-reductase deficiency. Hum. Mol. Genet. 16, 1176–1187 10.1093/hmg/ddm065 [DOI] [PubMed] [Google Scholar]
- 24. Tsai P.-L., Zhao C., Turner E., and Schlieker C. (2016) The Lamin B receptor is essential for cholesterol synthesis and perturbed by disease-causing mutations. Elife 5, e16011 10.7554/eLife.16011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bennati A. M., Schiavoni G., Franken S., Piobbico D., Della Fazia M. A., Caruso D., De Fabiani E., Benedetti L., Cusella De Angelis M. G., Gieselmann V., Servillo G., Beccari T., and Roberti R. (2008) Disruption of the gene encoding 3β-hydroxysterol Δ-reductase (Tm7sf2) in mice does not impair cholesterol biosynthesis. FEBS J. 275, 5034–5047 10.1111/j.1742-4658.2008.06637.x [DOI] [PubMed] [Google Scholar]
- 26. Soullam B., and Worman H. J. (1993) The amino-terminal domain of the lamin B receptor is a nuclear envelope targeting signal. J. Cell Biol. 120, 1093–1100 10.1083/jcb.120.5.1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liokatis S., Edlich C., Soupsana K., Giannios I., Panagiotidou P., Tripsianes K., Sattler M., Georgatos S. D., and Politou A. S. (2012) Solution structure and molecular interactions of lamin B receptor Tudor domain. J. Biol. Chem. 287, 1032–1042 10.1074/jbc.M111.281303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Prakash A., and Kasbekar D. P. (2002) Genes encoding chimeras of Neurospora crassa erg-3 and human TM7SF2 proteins fail to complement Neurospora and yeast sterol C-14 reductase mutants. J. Biosci. 27, 105–112 10.1007/BF02703766 [DOI] [PubMed] [Google Scholar]
- 29. Horton J. D., Shah N. A., Warrington J. A., Anderson N. N., Park S. W., Brown M. S., and Goldstein J. L. (2003) Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc. Natl. Acad. Sci. U.S.A. 100, 12027–12032 10.1073/pnas.1534923100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zerenturk E. J., Sharpe L. J., and Brown A. J. (2012) Sterols regulate 3β-hydroxysterol Δ24-reductase (DHCR24) via dual sterol regulatory elements: cooperative induction of key enzymes in lipid synthesis by sterol regulatory element binding proteins. Biochim. Biophys. Acta 1821, 1350–1360 10.1016/j.bbalip.2012.07.006 [DOI] [PubMed] [Google Scholar]
- 31. Metherall J. E., Goldstein J. L., Luskey K. L., and Brown M. S. (1989) Loss of transcriptional repression of three sterol-regulated genes in mutant hamster cells. J. Biol. Chem. 264, 15634–15641 [PubMed] [Google Scholar]
- 32. Roberti R., Bennati A. M., Galli G., Caruso D., Maras B., Aisa C., Beccari T., Della Fazia M. A., and Servillo G. (2002) Cloning and expression of sterol Δ14-reductase from bovine liver. Eur. J. Biochem. 269, 283–290 10.1046/j.0014-2956.2001.02646.x [DOI] [PubMed] [Google Scholar]
- 33. Clayton P., Fischer B., Mann A., Mansour S., Rossier E., Veen M., Lang C., Baasanjav S., Kieslich M., Brossuleit K., Gravemann S., Schnipper N., Karbasyian M., Demuth I., Zwerger M., Vaya A., Utermann G., Mundlos S., Stricker S., Sperling K., and Hoffmann K. (2010) Mutations causing Greenberg dysplasia but not Pelger anomaly uncouple enzymatic from structural functions of a nuclear membrane protein. Nucleus 1, 354–366 10.4161/nucl.1.4.12435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen L., Ma M.-Y., Sun M., Jiang L.-Y., Zhao X.-T., Fang X.-X., Lam S. M., Shui G.-H., Luo J., Shi X.-J., and Song B.-L. (2019) Endogenous sterol intermediates of the mevalonate pathway regulate HMG-CoA reductase degradation and SREBP-2 processing. J. Lipid Res. 60, 1765–1775 10.1194/jlr.RA119000201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gill S., Stevenson J., Kristiana I., and Brown A. J. (2011) Cholesterol-dependent degradation of squalene monooxygenase, a control point in cholesterol synthesis beyond HMG-CoA reductase. Cell Metab. 13, 260–273 10.1016/j.cmet.2011.01.015 [DOI] [PubMed] [Google Scholar]
- 36. Ravid T., Doolman R., Avner R., Harats D., and Roitelman J. (2000) The ubiquitin-proteasome pathway mediates the regulated degradation of mammalian 3-hydroxy-3-methylglutaryl-coenzyme A reductase. J. Biol. Chem. 275, 35840–35847 10.1074/jbc.M004793200 [DOI] [PubMed] [Google Scholar]
- 37. Zelcer N., Sharpe L. J., Loregger A., Kristiana I., Cook E. C., Phan L., Stevenson J., and Brown A. J. (2014) The E3 ubiquitin ligase MARCH6 degrades squalene monooxygenase and affects 3-hydroxy-3-methyl-glutaryl coenzyme A reductase and the cholesterol synthesis pathway. Mol. Cell Biol. 34, 1262–1270 10.1128/MCB.01140-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Meyer H., and Weihl C. C. (2014) The VCP/p97 system at a glance: connecting cellular function to disease pathogenesis. J. Cell Sci. 127, 3877–3883 10.1242/jcs.093831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Huang E. Y., To M., Tran E., Dionisio L. T. A., Cho H. J., Baney K. L. M., Pataki C. I., and Olzmann J. A. (2018) A VCP inhibitor substrate trapping approach (VISTA) enables proteomic profiling of endogenous ERAD substrates. Mol. Biol. Cell 29, 1021–1030 10.1091/mbc.E17-08-0514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chua N. K., Scott N. A., and Brown A. J. (2019) Valosin-containing protein mediates the ERAD of squalene monooxygenase and its cholesterol-responsive degron. Biochem. J. 476, 2545–2560 10.1042/BCJ20190418 [DOI] [PubMed] [Google Scholar]
- 41. Sharpe L. J., Howe V., Scott N. A., Luu W., Phan L., Berk J. M., Hochstrasser M., and Brown A. J. (2019) Cholesterol increases protein levels of the E3 ligase MARCH6 and thereby stimulates protein degradation. J. Biol. Chem. 294, 2436–2448 10.1074/jbc.RA118.005069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hornbeck P. V., Zhang B., Murray B., Kornhauser J. M., Latham V., and Skrzypek E. (2015) PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 43, D512–D520 10.1093/nar/gku1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jiang W., and Song B.-L. (2014) Ubiquitin ligases in cholesterol metabolism. Diabetes Metab. J. 38, 171–180 10.4093/dmj.2014.38.3.171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Howe V., Sharpe L. J., Prabhu A. V., and Brown A. J. (2017) New insights into cellular cholesterol acquisition: promoter analysis of human HMGCR and SQLE, two key control enzymes in cholesterol synthesis. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1862, 647–657 10.1016/j.bbalip.2017.03.009 [DOI] [PubMed] [Google Scholar]
- 45. Kim C. K., Jeon K. I., Lim D. M., Johng T. N., Trzaskos J. M., Gaylor J. L., and Paik Y. K. (1995) Cholesterol-biosynthesis from lanosterol—regulation and purification of rat hepatic sterol 14-reductase. Biochim. Biophys. Acta 1259, 39–48 10.1016/0005-2760(95)00128-Y [DOI] [PubMed] [Google Scholar]
- 46. Gale S. E., Westover E. J., Dudley N., Krishnan K., Merlin S., Scherrer D. E., Han X., Zhai X., Brockman H. L., Brown R. E., Covey D. F., Schaffer J. E., Schlesinger P., and Ory D. S. (2009) Side chain oxygenated cholesterol regulates cellular cholesterol homeostasis through direct sterol-membrane interactions. J. Biol. Chem. 284, 1755–1764 10.1074/jbc.M807210200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Martin-Serrano J., Eastman S. W., Chung W., and Bieniasz P. D. (2005) HECT ubiquitin ligases link viral and cellular PPXY motifs to the vacuolar protein-sorting pathway. J. Cell Biol. 168, 89–101 10.1083/jcb.200408155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Luu W., Sharpe L. J., Stevenson J., and Brown A. J. (2012) Akt acutely activates the cholesterogenic transcription factor SREBP-2. Biochim. Biophys. Acta 1823, 458–464 10.1016/j.bbamcr.2011.09.017 [DOI] [PubMed] [Google Scholar]
- 49. Goldstein J. L., Basu S. K., and Brown M. S. (1983) Receptor-mediated endocytosis of low-density lipoprotein in cultured cells. Methods Enzymol. 98, 241–260 10.1016/0076-6879(83)98152-1 [DOI] [PubMed] [Google Scholar]
- 50. Krycer J. R., Kristiana I., and Brown A. J. (2009) Cholesterol homeostasis in two commonly used human prostate cancer cell-lines, LNCaP and PC-3. PLoS ONE 4, e8496 10.1371/journal.pone.0008496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Brown A. J., Sun L., Feramisco J. D., Brown M. S., and Goldstein J. L. (2002) Cholesterol addition to ER membranes alters conformation of SCAP, the SREBP escort protein that regulates cholesterol metabolism. Mol. Cell 10, 237–245 10.1016/S1097-2765(02)00591-9 [DOI] [PubMed] [Google Scholar]
- 52. Shevchenko A., Wilm M., Vorm O., and Mann M. (1996) Mass spectrometric sequencing of proteins from silver-stained polyacrylamide gels. Anal. Chem. 68, 850–858 10.1021/ac950914h [DOI] [PubMed] [Google Scholar]
- 53. Hart-Smith G., and Raftery M. J. (2012) Detection and characterization of low abundance glycopeptides via higher-energy C-trap dissociation and orbitrap mass analysis. J. Am. Soc. Mass Spectrom. 23, 124–140 10.1007/s13361-011-0273-y [DOI] [PubMed] [Google Scholar]
- 54. Smith D. L., Götze M., Bartolec T. K., Hart-Smith G., and Wilkins M. R. (2018) Characterization of the interaction between arginine methyltransferase Hmt1 and its substrate Npl3: use of multiple cross-linkers, mass spectrometric approaches, and software platforms. Anal. Chem. 90, 9101–9108 10.1021/acs.analchem.8b01525 [DOI] [PubMed] [Google Scholar]
- 55. Chua N. K., Hart-Smith G., and Brown A. J. (2019) Non-canonical ubiquitination of the cholesterol-regulated degron of squalene monooxygenase. J. Biol. Chem. 294, 8134–8147 10.1074/jbc.RA119.007798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pruitt K. D., Brown G. R., Hiatt S. M., Thibaud-Nissen F., Astashyn A., Ermolaeva O., Farrell C. M., Hart J., Landrum M. J., McGarvey K. M., Murphy M. R., O'Leary N. A., Pujar S., Rajput B., Rangwala S. H., et al. (2014) RefSeq: an update on mammalian reference sequences. Nucleic Acids Res. 42, D756–D763 10.1093/nar/gkt1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D. J., Inuganti A., Griss J., Mayer G., Eisenacher M., Pérez E., Uszkoreit J., Pfeuffer J., Sachsenberg T., Yilmaz S., et al. (2019) The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47, D442–D450 10.1093/nar/gky1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sanchis J., Fernández L., Carballeira J. D., Drone J., Gumulya Y., Höbenreich H., Kahakeaw D., Kille S., Lohmer R., Peyralans J. J. P., Podtetenieff J., Prasad S., Soni P., Taglieber A., Wu S., et al. (2008) Improved PCR method for the creation of saturation mutagenesis libraries in directed evolution: application to difficult-to-amplify templates. Appl. Microbiol. Biotechnol. 81, 387–397 10.1007/s00253-008-1678-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chua N. K., Howe V., Jatana N., Thukral L., and Brown A. J. (2017) A conserved degron containing an amphipathic helix regulates the cholesterol-mediated turnover of human squalene monooxygenase, a rate-limiting enzyme in cholesterol synthesis. J. Biol. Chem. 292, 19959–19973 10.1074/jbc.M117.794230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Scott N. A., Sharpe L. J., Capell-Hattam I. M., Gullo S., Luu W., and Brown A. J. (2020) The cholesterol synthesis enzyme lanosterol 14α-demethylase is post-translationally regulated by the E3 ubiquitin ligase MARCH6. Biochem. 477, 541–555 10.1042/BCJ20190647 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.