

Abstract
Microfibril-associated glycoprotein-1 (MAGP-1) is a component of vertebrate extracellular matrix (ECM) microfibrils that, together with the fibrillins, contributes to microfibril function. Many of the phenotypes associated with MAGP-1 gene inactivation are consistent with dysregulation of the transforming growth factor β (TGFβ)/bone morphogenetic protein (BMP) signaling system. We have previously shown that full-length MAGP-1 binds active TGFβ-1 and some BMPs. The work presented here further defines the growth factor–binding domain of MAGP-1. Using recombinant domains and synthetic peptides, along with surface plasmon resonance analysis to measure the kinetics of the MAGP-1–TGFβ-1 interaction, we localized the TGFβ- and BMP-binding site in MAGP-1 to a 19-amino acid–long, highly acidic sequence near the N terminus. This domain was specific for binding active, but not latent, TGFβ-1. Growth factor activity experiments revealed that TGFβ-1 retains signaling activity when complexed with MAGP-1. Furthermore, when bound to fibrillin, MAGP-1 retained the ability to interact with TGFβ-1, and active TGFβ-1 did not bind fibrillin in the absence of MAGP-1. The absence of MAGP was sufficient to raise the amount of total TGFβ stored in the ECM of cultured cells, suggesting that the MAGPs compete with the TGFβ large latent complex for binding to microfibrils. Together, these results indicate that MAGP-1 plays an active role in TGFβ signaling in the ECM.
Keywords: extracellular matrix, transforming growth factor beta (TGFβ), cell signaling, bone morphogenetic protein (BMP), growth factor, fibrillar, growth factor signaling, MFAP2, microfibril, microfibril associated glycoprotein (MAGP), TGFβ, fibrillin, extracellular cellular matrix (ECM), protein-protein interaction
Introduction
The extracellular matrix (ECM)2 is a composite biomaterial that contains informational signals that influence cell phenotypes. The changing composition of the ECM during development and tissue repair provides necessary feedback signals used by cells to build functional tissues. Information from the ECM is transmitted to cells through direct interaction with specific receptors (e.g. integrins), through mechanical signals that reflect the material properties of the ECM and cellular microenvironment, and through regulation of growth factor accessibility and signaling. Many ECM proteins bind growth factors and regulate their activity by providing a pericellular substrate for presenting growth factors to specific receptors on cells or by sequestering active and latent forms away from the cell for later utilization (1). Growth factors bind to discrete domains and motifs in ECM proteins, and there is remarkable specificity to these binding interactions. As the ECM changes during development, so does the repertoire of ECM-associated growth factors that influence cell phenotypes.
The microfibril is an extracellular matrix structure made up of fibrillin as the core unit. Fibrillin appeared early in evolution and, over time, developed both structural and signaling functions (2). Fibrillin monomers assemble into fibers of 12–15 nm diameter that provide strength to tissues by forming fiber bundles. An example includes the ciliary zonule in the eye, where microfibrils form the suspensory ligaments that support the lens (3, 4). Microfibrils are also an important structural component of primitive vertebrate and invertebrate arteries, where they provide a degree of elastic recoil (5–7).
Another key function of microfibrils is modulation of growth factor signaling through fibrillin interactions with the latent TGFβ-binding proteins (LTBPs). LTBPs covalently bind the small latent complex of TGFβ to form the large latent TGFβ complex, which binds to fibrillin (8, 9). By sequestering latent TGFβ in the matrix, microfibrils indirectly regulate TGFβ signaling by providing a store of inactive growth factor that can be utilized through activation by proteolysis or by mechanical strain (10).
With the arrival of vertebrates in evolution, new proteins appeared that interact with fibrillin and modify microfibrillar function. These include elastin and microfibril-associated glycoprotein-1 and -2 (MAGP-1 and MAGP-2). Elastic fiber formation requires microfibrils (11) and, because MAGP-1 can bind both tropoelastin (12, 13) and fibrillin (14), the MAGPs were initially thought to promote the tropoelastin–microfibril interaction necessary for elastin assembly. However, inactivation of the genes for MAGP-1 and MAGP-2 in mice did not affect elastic fiber formation. Instead, mice lacking these proteins had abnormalities in bone, fat, hematopoiesis, and wound healing that were associated with abnormal TGFβ signaling (15–17). We have previously documented a direct binding interaction between TGFβ family growth factors and the MAGPs (18), but the sequence within MAGP that mediates this interaction was unknown. In this report, we localize the growth factor–binding domain in MAGP-1 to a sequence near the protein's N terminus and address the functional consequences of the TGFβ–MAGP interaction.
Results
Characterization of recombinant purified full-length MAGP-1 and MAGP-1 fragments
Full-length mouse MAGP-1 (exons 3–9) was expressed in Escherichia coli along with a series of N-terminal truncation mutants containing an N-terminal His6 tag. The truncations were based on the exon structure of the gene encoding the mature molecule shown in Fig. 1A (the protein sequence for mouse MAGP-1 can be found in Fig. S1). The gene for MAGP-1 contains nine exons, but only seven encode the secreted full-length protein. Exon 1 is untranslated, and exon 2 encodes the majority of the signal peptide, which is absent from the mature molecule. After confirming the correct sequence for each construct by DNA sequence analysis, the constructs were expressed, and the proteins purified. The results of the purification are shown in Fig. 1B. The full-length protein has a predicted mass of 20.6 kDa, but by SDS-PAGE analysis migrates ∼36 kDa. Similarly, the bacterially expressed domain 3–4 construct has a predicted mass of ∼7.5 kDa but runs at ∼16 kDa. Purified domain 4–9 runs slightly higher than expected (19 kDa compared with the predicted 17.5 kDa), and the domain 5–9 protein runs true to the predicted mass of ∼16 kDa. Because the MAGP-1 protein and fragments are expressed in bacteria, the irregular migration is not because of posttranslational modifications. Anomalous migration of full-length MAGP-1 on SDS-PAGE has been described (19). Our results suggest that the sequence within domains 3–4 is responsible for this irregular behavior.
Figure 1.
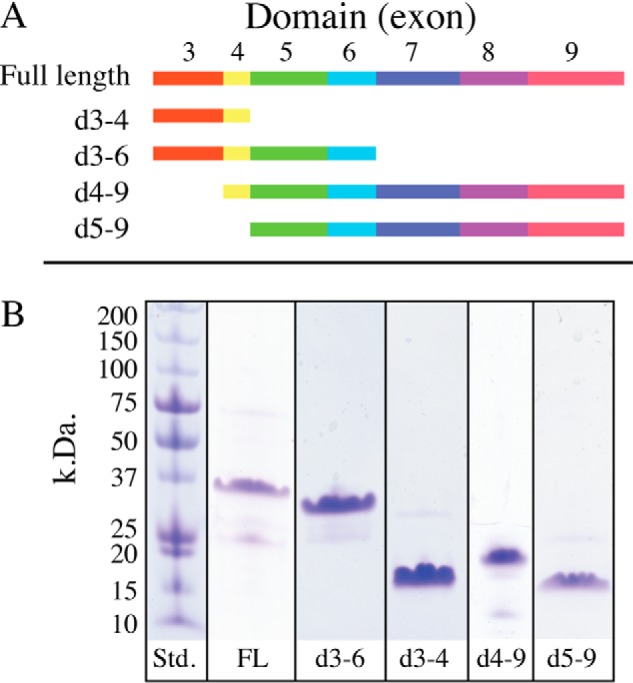
Domain structure of MAGP-1 and purity of expressed peptides. A, full-length, secreted, MAGP-1 protein is encoded by seven exons (exons 3–9). In this report, each exon defines a protein domain, and truncations used for functional analysis were constructed based on exon boundaries. The schematic shows the domains included in the recombinant fragments (d followed by exon numbers). B, SDS-PAGE analysis of recombinant full-length MAGP-1 (FL) and purified truncated peptides shown in panel A. Proteins were separated on 8–25% Phastgels under reducing conditions and detected with Coomassie Brilliant Blue. The observed and expected molecular weights of each fragment are described in “Results.”
TGFβ binding to MAGP-1 fragments
Our goal in this study was to localize the region of MAGP-1 that interacts with TGFβ growth factors. To this end, we performed a set of surface plasmon resonance (SPR) experiments where purified TGFβ-1 was injected over immobilized full-length MAGP-1 or over the purified truncation fragments coupled to CM-5 sensor chips. Fig. 2A shows binding isotherms for soluble TGFβ-1 interacting with immobilized full-length MAGP-1 in the SPR-based assay. The affinity constant calculated using 1:1 Langmuir binding was 15 nm. When tested for binding to the MAGP-1 fragments, TGFβ-1 bound the domain 3–4–coupled sensor chip with a slightly higher affinity than the full-length protein, as seen in Fig. 2B. The affinity constant calculated using 1:1 Langmuir binding was 3 nm. Binding of TGFβ-1 to domain 4–9 showed substantially lower affinity (190 nm), and there was no TGFβ-1 binding to a chip coated with domain 5–9 (Fig. 2, C and D, respectively). These results suggest that the primary binding site for TGFβ-1 is located in domain 4 of MAGP-1 with an affinity contribution from domain 3.
Figure 2.
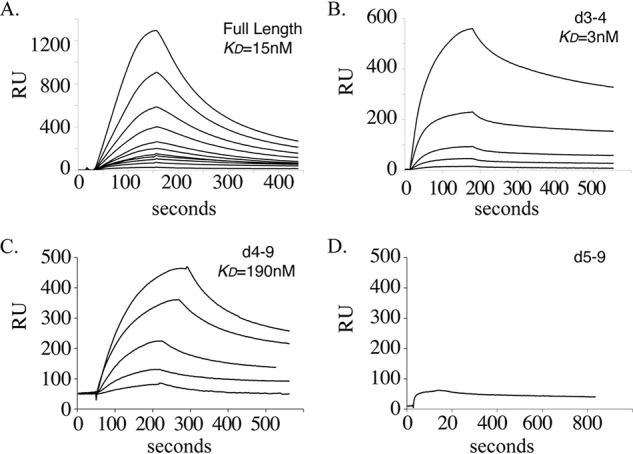
Binding of TGFβ-1 to purified full-length MAGP-1 or MAGP-1 fragments. A, graph of isotherms from the binding of TGFβ-1 to a CM-5 chip coated with full-length MAGP-1 (2200 resonance units coupled). TGFβ-1 concentrations were, from the top, 100, 66.7, 44.4, 29.6, 19.8, 13.2, 8.8, 5.9, 3.9, 2.6, and 1.7 nm. B–D, interaction of TGFβ-1 (400, 200, 80, 67, and 27 nm) with 440 resonance units of MAGP-1 domain protein 3–4 (B), with 4300 resonance units of MAGP-1 domain 4–9 (TGFβ-1 concentrations of 120, 60, 48, 24,12 nm) (C), and 400 nm TGFβ-1 with 2140 resonance units of MAGP-1 domain 5–9 (D).
TGFβ binds to a 19-residue peptide sequence spanning domains 3 and 4
Fine mapping of the MAGP-1–binding sequence for TGFβ-1 was done using overlapping synthetic peptides that spanned the sequence encoded by exons 3 and 4. Each peptide was purified by HPLC, and the sequence confirmed by MS (data not shown). Surprisingly, TGFβ-1 failed to bind to a CM-5 sensor chip coated with a synthetic peptide made from the sequence of domain 3–4. The sequence was the same as the bacterially expressed domain 3–4 peptide except for an N-terminal His6 tag on the bacterially expressed protein. The His6 tag itself was not an active binding sequence because the expressed peptide 5–9 did not bind TGFβ-1 even though it also had a His6 tag at the N terminus.
Although unable to bind TGFβ-1 when bound to a solid substrate, domain 3–4 synthetic peptide (p1–35) did inhibit binding of TGFβ-1 to a full-length MAGP-1–coated sensor chip, thereby establishing its activity (Fig. S2A). Therefore, for subsequent mapping studies, we used equilibrium titration experiments to assess the inhibitory activity of a series of peptides summarized in Fig. 3A. The peptide based on the first 18 residues of domain 3 (p1–18) showed no inhibitory activity. A peptide derived from residues 22–36 (p22–36) and a domain 4 peptide (p26–35) both failed to inhibit TGFβ-1 binding to full-length MAGP-1 (the inhibitory curve for peptide p22–36 is shown in Fig. S2B as representative of no binding inhibition). A peptide with residues 12–37 (not shown) and a smaller peptide spanning residues 17–35 (p17–35) were both inhibitory.
Figure 3.

Fine mapping of TGFβ-1 binding activity in domains 3–4. A, summary of the inhibitory activity, the sequence, and the location of the peptides tested for inhibition of TGFβ-1 (50 nm) binding to 120 resonance units (RU) of full-length MAGP-1 coupled to a carboxymethyl dextran hydrogel surface sensor chip (MAGP-1 chip). B, a graph of isotherms showing p17–35 dose-dependent inhibition of TGFβ-1 binding to the MAGP-1–coated chip. C, relates the maximal binding of TGFβ-1 (at 100 s) to the MAGP-1 chip in the presence of increasing concentrations of peptide p17–35 (taken from panel B). D, isotherms show that the control peptide p17–35pro (100, 50, 25, and 0 μm) has no inhibitory effect on TGFβ-1 binding to MAGP-1.
A detailed investigation into the dose response of the smallest peptide with inhibitory activity (p17–35) is shown in Fig. 3B. There is a dose-dependent decrease in TGFβ-1 binding to full-length MAGP-1, with half-maximal inhibition at ∼30 μm p17–35 (Fig. 3C). These results are consistent with data from studies with recombinant MAGP-1 fragments and show that the TGFβ-1 binding activity of MAGP-1 is defined by a 19-amino acid sequence in domains 3 and 4. A control peptide with conformational restraints containing four proline residues (p17–35pro) failed to inhibit TGFβ-1 binding to full-length MAGP-1 (Fig. 3D).
Secondary structure predictions for the MAGP-1 p17–35 peptide indicated that the sequence does not have extensive secondary structure elements, with most residues predicted to be a random coil (data not shown). IUPred results predicted a high degree of disorder from the primary sequence. PEP-FOLD models tended to be elongated or compact coils with little secondary structure. This region of the MAGP-1 protein is likely intrinsically disordered and cannot adopt an ordered state without binding to a binding partner such as a member of the TGFβ superfamily.
MAGP-1 binds active, but not latent, TGFβ-1
TGFβ is produced as a disulfide-linked homodimer that is inactive because of the presence of the N-terminal latency-associated peptide. This small latent TGFβ complex is usually associated with LTBP1 to produce the large latent TGFβ complex, which is covalently bound to fibrillin-containing microfibrils in the ECM (9). To determine whether MAGP-1 interacts with latent TGFβ-1, we injected the small latent complex form of TGFβ-1 over a sensor chip coated with full-length MAGP-1. As shown in Fig. 4, latent TGFβ-1 at 15 nm failed to bind to the full-length protein. However, after acid activation for 15 min, the same latent TGFβ-1 preparation showed a robust interaction with MAGP-1.
Figure 4.
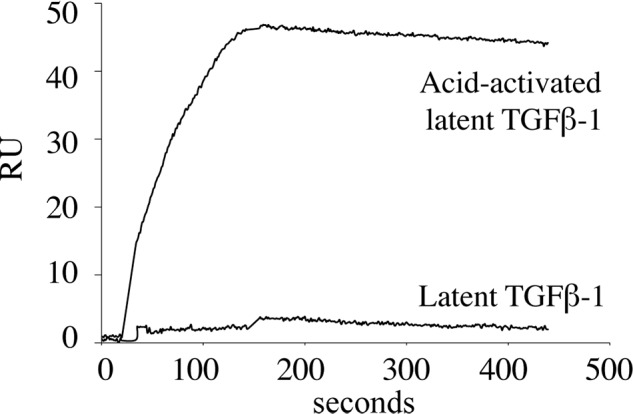
MAGP-1 does not bind latent TGFβ-1. Latent TGFβ-1 (15 nm) was injected onto a MAGP-1 chip, and no interaction was detected. A second sample of latent TGFβ-1 was treated with 100 mm HCl for 10 min at room temperature, neutralized with 100 mm NaOH, and then diluted to a final concentration of 15 nm. The binding isotherm shows that the acid-activated TGFβ-1 bound with high affinity to the MAGP-1 chip. The injection volume was 125 μl, and the flow rate was 50 μl/min for both injections.
Neither MAGP-1 nor p17–35 inhibits TGFβ-1 signaling
To investigate whether TGFβ-1 can still interact with its cellular receptor when complexed with MAGP-1, we pre-mixed MAGP-1 or MAGP-1 fragments with active TGFβ-1 and then added the complex to the reporter cell line MFB-F11. MFB-F11 cells are embryonic fibroblasts from Tgfb1−/− embryos stably transfected with a reporter plasmid consisting of TGFβ-responsive Smad-binding elements coupled to a secreted alkaline phosphatase reporter gene (20). The secreted alkaline phosphatase can be detected with a chemiluminescent substrate and provides a sensitive assay for TGFβ activity. We found that full-length MAGP-1 does not influence TGFβ-1–stimulated alkaline phosphatase release from the MFB-F11 reporter cells when tested over a wide range of MAGP-1 concentrations (Fig. 5A). Similarly, MAGP-1 peptide p17–35 that blocks binding of TGFβ-1 to full-length MAGP-1, presumably by binding to the MAGP-1–binding site on the growth factor, does not affect the signaling potential of TGFβ-1 (Fig. 5B).
Figure 5.

MAGP-1 and p17–35 do not block TGFβ-1 signaling. A, active TGFβ-1 (80 pm) was mixed with the indicated concentrations of MAGP-1, incubated for 15 min, and then added to MFB-F11 cells for 24 h. The culture medium was then assayed for alkaline phosphatase activity, which provides a measure of TGFβ activity. Shown are mean ± S.D., n = 8. B, active TGFβ-1 (80 pm) was mixed with 10 μm or 100 μm p17–35, p17–35pro, or no peptide for 15 min and added to MFB-F11 cells for 24 h. A control with no added TGFβ-1 was included. The medium was collected and assayed for alkaline phosphatase activity. Shown are mean ± S.D., n = 4. One-way analysis of variance showed no differences between samples in panels A or B. C, increasing concentrations of soluble MAGP-1 were pre-incubated for 1 h with 50 pm TGFβ-1 and the complex incubated with RFL-6 cell (a fibroblast line that does not make MAGP-1) for 15 min. Cell extracts were then analyzed by Western blotting for phospho-Smad2 (pSmad-2) (top) and for total Smad2 (tSmad2) (bottom). The ratio of pSmad2 to tSmad2 is shown in the bottom graph. D, Smad2 phosphorylation in RFL-6 cells is TGFB-1 dose dependent with minimal baseline phosphorylation. RFL-6 cells were treated with the indicated dose of TGFβ-1 and levels of phosphorylated Smad-2 (top panel) or total Smad-2 (bottom panel) were determined from cell extracts by Western blot analysis. The ratio of pSmad2 to tSmad2 is shown in the bottom graph.
The effects of MAGP-1 on TGFβ activity were also investigated by assessing changes in the SMAD signaling pathway in RFL-6 cells. In agreement with the MFB-F11 reporter cell assay, pre-incubating TGFβ-1 with soluble MAGP-1 did not inhibit RFL-6 TGFβ-1–stimulated Smad-2 phosphorylation (Fig. 5C). Fig. 5D shows that Smad-2 phosphorylation is TGFβ dose dependent in RFL-6 cells with minimal pSmad2 detected in the absence of TGFβ.
Predicted MAGP-1–binding site on TGFβ
Molecular docking experiments consistently placed the MAGP-1 peptide p17–35 in contact with the finger region of TGFβ-1 (Fig. 6). The peptide prefers to dock near a cluster of positive residues in this region (Arg-94, Lys-95, and Lys-97) that are solvent-exposed and, hence, in a position to attract and bind the negatively charged MAGP-1 peptide. The MAGP-1 peptide remains a highly flexible coil even when bound, suggesting that binding occurs through a promiscuous charge– interaction between the highly positive region of TGFβ-1 and the negative residues of MAGP-1. The critical positively charged residues are inaccessible in the inactive TGFβ-LAP complex, because of the presence of the latency lasso in the straight-jacket region of the large latency-associated complex (21). Masking of the MAGP-1 binding site by the latency peptide explains why MAGP-1 binds active TGFβ but not the small latent complex.
Figure 6.
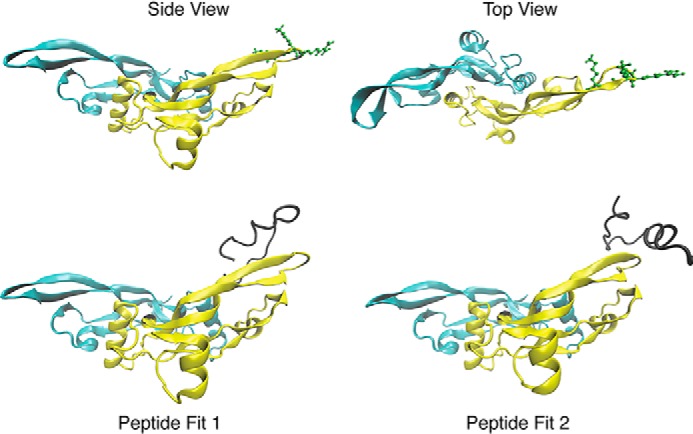
The structure of TGFβ-1 with and without the MAGP-1 p17–35 peptide. Top, side and top views of the TGFβ-1 dimer with solvent exposed residues Arg-94, Lys-95, and Lys-97 shown in green. Bottom, representative binding of the MAGP-1 p17–35 peptide (shown in black) to TGFβ-1. The MAGP-1 peptide remains largely disordered while consistently interacting with the positive residues in the TGFβ-1 finger region.
MAGP forms a ternary complex with fibrillin-2 and TGFβ-1
Fibrillin interacts with LTBP1 to covalently anchor the TGFβ large latent complex into the ECM (9, 22). Whether active TGFβ-1 can bind to fibrillin is unclear. To explore a possible binding interaction between the two proteins, we injected 50 nm of active TGFβ-1 over full-length fibrillin-2 immobilized on an SPR chip. Fibrillin-2 was purified from a baculoviral expression system and showed a single band by SDS-PAGE (Fig. S3). Fig. 7A shows that no significant interaction was detected between TGFβ-1 and fibrillin-2. Soluble MAGP-1, however, bound tightly to fibrillin-2 with an apparent affinity constant of 100 pm (Fig. 7B). The binding curve fits 1:1 Langmuir binding with a noticeable slow dissociation rate of −2.64 × 104s−1 and a half-life of 2600 s. We took advantage of this slow release rate to follow MAGP-1 injection onto the fibrillin-2 chip with 50 nm of TGFβ-1 and found robust binding of the growth factor to the fibrillin-2–MAGP-1 complex (Fig. 7C). That MAGP-1 mediated the interaction was established by inhibiting TGFβ binding with the active MAGP-1 peptide p17–35 (Fig. 7C). The binding of TGFβ-1 to the MAGP-1–fibrillin-2 complex showed similar binding kinetics to TGFβ-1 binding directly to a MAGP-1–coated chip (compare the top curve in Fig. 3B to Fig. 7C).
Figure 7.
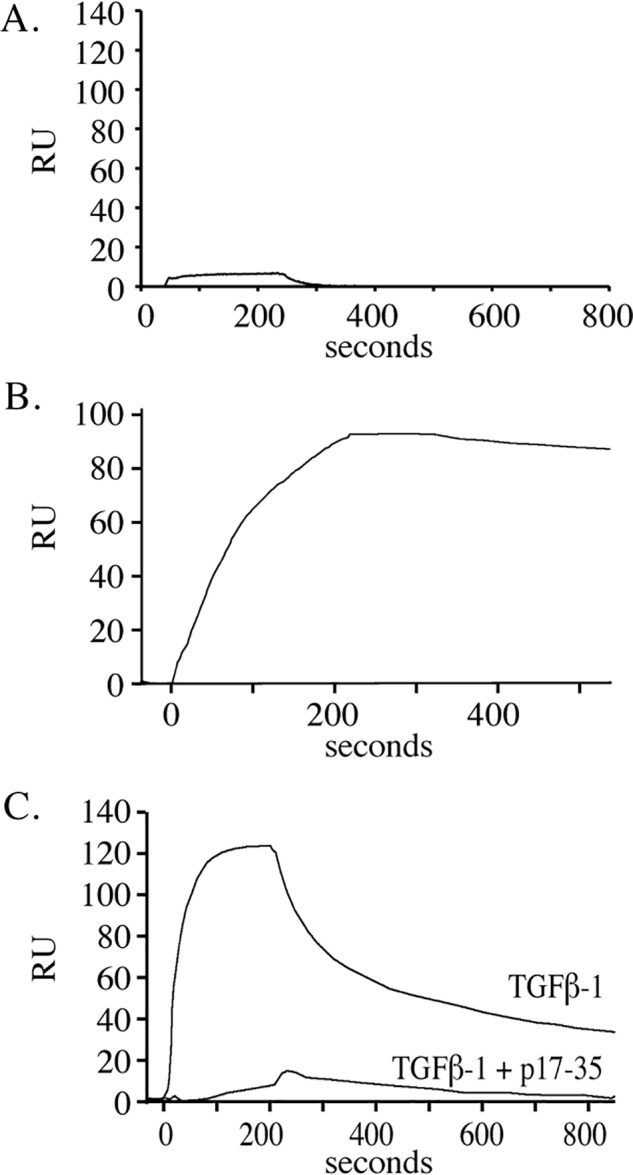
TGFβ-1 binds to fibrillin-2 only in the presence of MAGP-1. A, purified full-length fibrillin-2 was covalently coupled (3000 resonance units) to a carboxymethyl dextran hydrogel surface sensor chip (fibrillin-2 chip), and TGFβ-1 was injected at a concentration of 50 nm at 25 μl per minute for 3.5 min. Data show no measurable interaction between the two proteins. B, MAGP-1 (6.2 nm for 3.5 min at 25 μl/min) bound to fibrillin-2 with an apparent affinity of 100 pm. C, TGFβ-1 (50 nm for 3.5 min at 25 μl/min) bound to the MAGP-1-fibrillin-2 complex whereas TGFβ-1 pre-incubated with 50 μm p17–35 peptide failed to bind to the complex.
BMP-2 binds to the same MAGP-1 sequence as TGFβ
Our previous studies found that MAGP-1 bound to BMP-7 with an affinity approximately equal to TGFβ (18). To determine whether other BMP family members interact with MAGP-1, we performed SPR experiments with BMP-2. Fig. 8A shows BMP-2 binding to MAGP-1 with an affinity of 25 nm. The binding was inhibited by the active MAGP-1 peptide p17–35, suggesting that TGFβs and BMPs recognize the same sequence on the MAGP-1 protein. The control peptide p17–35pro (Fig. 8B) and the N-terminal peptide p1–18 (not shown) both failed to inhibit BMP-2 binding to full-length MAGP-1. We were unable to determine whether BMP-2 could form a ternary complex with fibrillin-2 and MAGP-1 because BMP-2 bound directly to fibrillin-2 with an affinity of 8 nm (Fig. S4).
Figure 8.
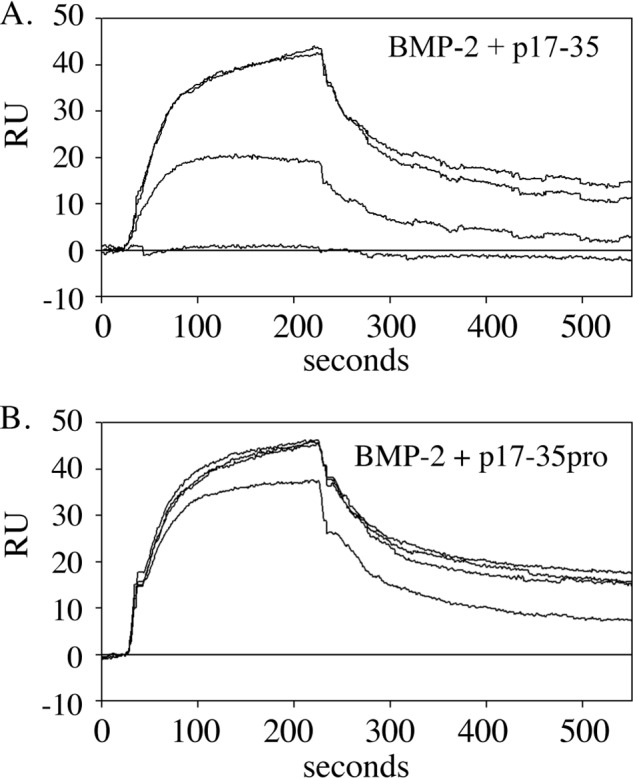
BMP-2 binding localizes to the same domain on MAGP-1 as TGFβ-1. A, isotherms documenting binding of 50 nm BMP-2 to a MAGP-1-coated chip. B, binding was inhibited by preincubation of BMP-2 with peptide p17–35 from the top 0, 5, 25, and 50 uM) but not the control peptide p17–35pro (0, 5, 25, and 50 uM).
Matrix devoid of MAGPs has higher TGFβ incorporation
The majority of TGFβ in the ECM is in the form of the large latent complex bound to fibrillin. MAGP-1 and the large latent complex bind to the same region of fibrillin (23), suggesting a possible competition between the two proteins for binding to microfibrils. To determine whether the absence of MAGP allows for more latent complex to accrue in the ECM, we compared total and active TGFβ levels in cultures of fibroblasts from WT and MAGP knockout cells. Because MAGP-2 can also bind TGFβ (16) and might compensate in the absence of MAGP-1, we evaluated TGFβ levels in MAGP-1 and MAGP-2 knockout MEFs as well as MEFs from MAGP-1:MAGP-2 double knockout mice. After 14 days of culture, there was two to three times more total (latent plus active) TGFβ activity in acid-activated cell-ECM extracts of all MAGP knockout cells compared with WT (Fig. 9, black bars). TGFβ activity detected without acid activation (indicative of active TGFβ) was also increased in the single knockout cell lines but was significantly lower in the double knockout MEFs (Fig. 9, gray bars). Quantitative PCR confirmed that TGFβ expression was similar for WT and MAGP knockout cells. Cell number was also equivalent for all cell types (Fig. S5).
Figure 9.
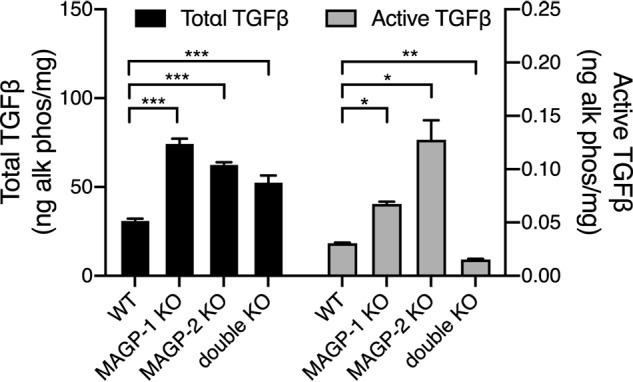
MAGP 1 and MAGP-2 modulate TGFB levels in fibroblast ECM. Comparison of total TGFβ levels (black bars) in 14-day post confluent WT and MAGP knockout cell layers showing significant increases in total TGFβ in MAGP-1 knockout (**, p > 0.01, n = 3), MAGP-2 knockout (*, p > 0.05, n = 3), and MAGP-1:MAGP-2 double knockout (**, p > 0.01) fibroblasts. Similarly, active TGFβ levels (gray bars) are significantly increased in MAGP-1 knockout (*, p > 0.05, n = 2) and MAGP-2 knockout (*, p > 0.05, n = 3) cells, but are decreased (**, p > 0.01 n = 3) in the double knockouts.
Discussion
The bioavailability of active TGFβ is tightly regulated at multiple levels, both inside and outside the cell. It is a crucial regulator of ECM production (24) yet, in turn, is influenced by the ECM itself in ways that provide spatiotemporal control of growth factor activity (1, 25, 26). In the ECM, TGFβ family growth factors bind avidly to sulfated sugars on proteoglycans as well as to ECM proteins (1, 26). Fibrillin-containing microfibrils are particularly important in modulating TGFβ signaling and the mechanisms whereby microfibril-associated proteins regulate growth factor availability and activity are complex. We and others have shown that the fibrillin-binding partners MAGP-1 and MAGP-2 work together with fibrillin to modulate TGFβ availability in the ECM (16, 27–29). Mutations in MAGP genes lead to phenotypes in mice and humans that are associated with abnormal growth factor signaling (15, 16, 18, 30–33).
The MAGPs are functionally defined by the amino acid composition of their front and back halves. The back half of MAGP-1 contains 13 cysteine residues, the first 7 of which define the matrix-binding domain that mediates interactions with fibrillin (34, 35). The matrix-binding region is the only conserved structural motif shared with MAGP-2 and is conserved in all species of MAGP-1 and MAGP-2. The cysteine-free N-terminal half of MAGP-1 is acidic, enriched in proline, and contains tyrosine residues that undergo sulfation and threonine residues that are sites for O-glycosylation. Sequences in this region also mediate noncovalent interactions with tropoelastin (12, 13), the α3 chain of collagen VI (13), decorin (36), and biglycan (37). Although secondary structure prediction algorithms suggest a random configuration for the N-terminal sequences, the absence of cysteine residues, the high negative charge density, and the binding of numerous proteins to this area of the molecule suggests a structure that is accessible and adaptable.
In this report, we used an overlapping peptide mapping strategy to localize the growth factor–binding domain to 19 amino acids in the N-terminal region of MAGP-1. The TGFβ binding site is contained in domain 4 but requires a part of domain 3 for full affinity (Fig. 2). The binding sequence is highly acidic because of four aspartic acid residues, one glutamic acid, and three tyrosine residues that are sites for sulfation (with the sulfate group adding more negative charge). The acidic residues form a structural repeat, with each separated by two nonpolar amino acids. The functional significance of this repeating structure is not known. The high charge density at this site resembles characteristics of heparin, which binds TGFβ with high affinity through negatively charged sulfated sugars that interact with lysine residues on TGFβ (38). In fact, the predicted MAGP-1–binding site on TGFβ is the same as that postulated for heparin (38, 39). Although the charged residues in MAGP-1 are important for growth factor binding, there are conformational and flexibility requirements as well, because the biologically active p17–35 peptide no longer interacts with TGFβ when prolines are inserted to introduce structural constraints.
MAGP-1 is one of the few matrix proteins that binds active TGFβ without the involvement of glycosaminoglycans. Some ECM proteins, such as fibronectin, thrombospondin, fibulins, tenascins, and fibrillins, bind the large latent complex form of TGFβ through interaction sites on the LTBPs (9, 40–43), although several bind the active isoform as well. In general, binding sites on matrix proteins for active TGFβ are not well-characterized, but the properties of the MAGP-1 growth factor–binding domain may be instructive for identifying active sequences in these as well as other proteins. The microfibrillar protein emilin-1, for example, binds specifically to the proTGFβ precursor and prevents its maturation by furin convertases (44). Growth factor–binding activity was mapped to the protein's EMI domain, but the precise sequence was not identified nor is it clear whether the interaction is with the active or latent regions of the growth factor. Similarly, TGFβ-binding activity in the matricellular protein SPARC was localized in one study to the last 37 amino acids in the C-terminal EC domain (45), but other work found that SPARC interacted with TGFβ only when bound to its cognate type II receptor (46). Interestingly, both emilin-1 and SPARC have clusters of acidic amino acids in the putative TGFβ-binding regions that create a negatively charged sequence similar to the growth factor–binding domain in MAGP-1.
Our mapping studies create a clearer picture of the functionality of the MAGP-1 N terminus. The first four amino acids in the TGFβ-binding peptide p17–35 overlap with the last four amino acids of a peptide used to define the binding site for tropoelastin and collagen VI (amino acids 12–21 in our numbering scheme) (13). It is not known, however, if TE or collagen VI can affect the binding of TGFβ. Although this N-terminal region seems to be available to interact with numerous proteins noncovalently, there is a specificity to the binding because MAGP-1 does not bind TGFβ-3; collagens I, II, or V; FGF9; FGF21; or the cytokines TNFα; IL-4; IL-13; and RANKL (13, 17).
Our finding that MAGP-1 binds active TGFβ but not the small latent precursor form of the growth factor suggests that MAGP-containing microfibrils have the potential to store active TGFβ in addition to the large latent form that covalently binds to fibrillin. The association of TGFβ with MAGP-1 does not alter the growth factor's signaling potential, nor does an association between MAGP-1 and fibrillin alter MAGP-1's ability to bind TGFβ. Thus, activated TGFβ bound to MAGP-1 on microfibrils is capable of signaling when interacting with its receptor on cells and does not require an additional activation step to do so.
It is noteworthy that the absence of MAGP-1 or MAGP-2 (or both) is sufficient to raise the amount of total TGFβ stored in the ECM of cultured cells. This finding is consistent with studies by Massam-Wu et al. (23), who showed the MAGP-1 could directly compete with LTBP1 for binding to fibrillin. In the absence of MAGP, one would, therefore, expect more total TGFβ to be associated with the ECM, which is what we found using MAGP knockout cells. Importantly, the presence of latent but not active TGFβ in the ECM of MAGP-1:MAGP-2 double knockout cells suggests that in the absence of the MAGPs, active TGFβ is not retained in the matrix.
The mechanistic and functional mapping studies described in this report, together with our previous characterization of MAGP knockout mouse phenotypes indicating increased TGFβ activity, provide strong evidence for regulation of the TGFβ pathway as one of MAGP's major functional roles in tissues.
Experimental procedures
Materials
Carrier-free active TGFβ-1 (7666-MB-CF), BMP-2 (355-BM-010/CF), and latent TGFβ-1 (299-LT) were purchased from R&D Systems (Minneapolis, MN). Antibodies to MAGP-1 and fibrillin-2 have been described (36, 47, 48). SimplyBlue Coomassie G-250 stain was obtained from Thermo Fisher Scientific. Antibodies to Smad2/3 (5678S) and phospho-Smad2 (18338S) were from Cell Signaling Technology (Danvers, MA).
Cell culture and TGFβ-1 activity assay
The TGFβ reporter cell line MFB-F11 was a generous gift from Tony Wyss-Coray, Stanford University. The cells were routinely cultured in DMEM supplemented with nonessential amino acid, 10% fetal calf serum, and 100 μg/ml hygromycin B (Invitrogen). On the afternoon before an assay, the cells were split into a 96-well assay plate (4 × 104 cells per well) and cultured overnight in the absence of hygromycin. For the assay, cells were rinsed in DMEM then incubated for 1 h in DMEM-BSA (DMEM + 1.0 mg/ml heat-denatured BSA). To determine whether MAGP-1 or the p17–35 peptide inhibits TGFβ signaling, TGFβ (80 pm) was mixed with varying concentrations of MAGP-1 protein or p17–35 peptide in DMEM-BSA for 15 min at room temperature then incubated with the reporter cells for 24 h. Soluble alkaline phosphatase released into the media in response to TGFβ was measured using the Phospha-LightTM SEAP Reporter Gene Assay System (Life Technologies).
MEF and MFB-F11 co-culture
Levels of active and total TGFβ in the ECM of cultured MEFs were assessed using established methods (20, 49). Embryonic fibroblasts from WT and MAGP-1 knockout mice were generated from explants as described previously (18). Early passage cells (passage 4 or less) were maintained in DMEM with 10% fetal calf serum supplemented with nonessential amino acids and routinely passaged 1:3 at confluence. For growth factor measurements, cells were plated at confluence in replicate 12-well tissue culture dishes and maintained for 14 days prior to TGFβ assays. The cells were rinsed with PBS and incubated for 1 h in DMEM containing 1 mg/ml of heat-denatured BSA. For measurement of active TGFβ levels, ∼1 × 105 MFB-F11 reporter cells in DMEM heat-denatured BSA were added to the cultured MEF cells and incubated overnight. Supernatant from this co-culture was assayed for alkaline phosphatase using the Phospha-LightTM SEAP Reporter Gene Assay System (Life Technologies).
To determine total protein and total TGFβ, MEF cells were rinsed with PBS and the cell layer incubated in 250 μl 0.2 m HCl per well for 1 h on ice. An aliquot was removed for total protein determination and the remaining solution neutralized by the addition of an equimolar amount of NaOH. The cell-layer extract was diluted 1:100 in DMEM containing 1 mg/ml heat-denatured BSA and assayed for TGFβ using MFB-F11 cells as described above. The samples taken for protein determination were dried and hydrolyzed in 6N HCl for 48 h, and amine content was measured with ninhydrin (50).
Smad2 phosphorylation
RFL-6 cells were plated at a confluent density and incubated overnight in Ham's F12 medium containing 0.5% horse serum followed by a 1-hour incubation with Ham's F12 containing 0.1 mg/ml heat denatured BSA to decrease background TGFβ activity. TGFβ-1 (50 pm) was mixed with 0, 0.4, 20, or 1000 nm MAGP-1 in Ham's F-12 containing 0.1 mg/ml heat-denatured BSA for 1 h at 37 °C then added to the RFL-6 cells for 15 min. Cells were lysed in buffer containing 20 mm Tris, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% Triton X-100, 2.5 mm sodium pyrophosphate, 1 mm β-glycerophosphate, 1 mm Na3VO4, 1 mm NaF 1× protease inhibitor (Roche Applied Science). Lysates were cleared by centrifugation, and protein concentration determined. Eight μg of lysate was run per lane on 10% SDS-PAGE gels then transferred to nitrocellulose membranes. Membranes were blocked in 1% casein then incubated overnight with the indicated primary antibodies. Antibodies were detected with secondary antibodies conjugated to horseradish peroxidase and visualized using chemiluminescent substrate (Millipore WBKLS0100). The standards used were Precision Plus Dual Color protein standards (Bio-Rad 161-0374).
MAGP-1 recombinant expression constructs
Mouse MAGP-1 cDNA was amplified from a full-length clone (IMAGE Consortium Clone:5249342) using a forward primer with a SphI restriction site added to the 5′end (full-length forward primer with Sph1) and a reverse primer with SacI restriction site added to 3′ end (full-length reverse primer with SacI). The resultant amplification product along with pQE31 was digested with SphI and SacI, gel purified, and appropriate bands ligated. The ligation mix was used to transform the M15 strain of E. coli, and a bacterial colony containing a plasmid with the appropriate sequence was used for expression.
A fragment of MAGP-1 representing domains 3–4 was amplified from the full-length pQE31 construct using a forward primer (truncation forward) from pQE31 sequence and a reverse primer from the MAGP-1 sequence containing a SacI restriction site (exon 4 reverse with SacI). The amplification product, along with pQE31, was digested with EcoRI and SacI, gel purified, and appropriate bands extracted and ligated. Fragments for domain 4–9 and 5–9 truncations were amplified from the full-length construct using a reverse primer from pQE31 (truncation reverse) sequence and a forward primer from MAGP-1 sequence containing a BamHI restriction site (exon 4 forward with BamH1 or exon 5 forward with BamHI). The amplification products were digested along with pQE31 with BamHI and SacI, gel purified, and the appropriate bands ligated. The ligation mix from each construct was used to transform M15 E. coli. Colonies were picked from the transformations and sequence verified. The primer sequences used to construct His6 full-length and truncated MAGP-1 are as follows: full-length forward primer with Sph1, TAGCATGCTCAGGGCCAATATGACCTGGA; full-length reverse primer with SacI, TAGAGCTCCTAGCAGCCCCCACAGCTCCTG; truncation forward, CACACAGAATTCATTAAAGAGGAG; exon 4 reverse with SacI, CAGAGCTCTCTTGGTAGTCATAGTAGTCTG; exon 4 forward with BamH1, CAGGATCCTGACAACGCAGACTACTATG; exon 5 forward with BamH1, CAGGATCCAGAAGTGAGTCCTCGG; truncation reverse, TTAAGCTTGGCTGCAGGT.
Peptides, protein expression, and purification
His6-tagged fusion proteins were expressed and purified using the Qiagen Expression System (Valencia, CA). Bacteria expressing full-length or truncated protein were expanded, and while in log-phase, induced with isopropyl β-d-thiogalactopyranoside at 1 mm final concentration for 4 h at 30 °C. The bacteria were collected by centrifugation, extracted overnight at 4 °C with gentle agitation in TRIS-phosphate buffered urea (8 m, pH8.0) containing protease inhibitors. After clarifying the extract by centrifugation (12,000 × g for 20 min), recombinant His6–containing proteins were isolated with nickel resin chromatography. Proteins were refolded on the column (51) and eluted with a 0–300 mm imidazole gradient in 50 mm TRIS pH7.4. Fractions were analyzed by SDS-PAGE, and fractions containing protein were dialyzed against water and lyophilized. The fusion proteins were further purified using HPLC over a PRP-3 reverse-phase column using a 0–30% acetonitrile gradient in 0.05% triethylamine. The protein content of purified proteins was determined by absorbance at 280 nm using an extinction coefficient based on the protein sequence (DNASTAR, Madison WI). The purity of the expressed proteins was assessed using SDS-PAGE with reducing (10 mm DTT) sample buffer on 8–25% gradient Phastgels (GE Healthcare Life Sciences) and stained with SimplyBlue (Invitrogen). Peptides were synthesized by GenScript (Piscataway, NJ) and further purified using HPLC over a PRP-3 reverse-phase column using a 0–30% acetonitrile gradient in 0.05% triethylamine. Fractions were dried and analyzed by amino acid analysis.
RNA isolation and quantitative PCR
RNA was isolated using a RNeasy kit (Qiagen 74104) after cell homogenization using QIAshredder (Qiagen 79656). cDNA was generated using the high-capacity RNA-to-cDNA kit (Applied Biosystems 4387406). Quantitative PCR was performed on a ViiA7 (Applied Biosystems) using reagents and TaqMan probes supplied by Applied Biosystems for GAPDH (Mm99999915G1) and TGFβ (Mm01178820_m1). Quadruplicate measurements from 100 ng RNA were performed for each sample.
Purification of fibrillin-2
Fifty ml of serum-free conditioned medium from SF-9 cells expressing full-length fibrillin-23 was applied at a flow rate of 4 ml/min onto a 1.0 × 9.5 cm Source 15Q strong anion exchange column (GE Healthcare) at room temperature using a Pharmacia FPLC system. The column was washed with 50 ml buffer A followed by a 10 ml gradient from 0–25% buffer B. Fibrillin-2, which remained bound to the resin, was eluted with a 50-ml 25–50% linear buffer B gradient before the column was recycled with 15 ml 100% buffer B and 100 ml buffer A. The elution stream was monitored for protein at 228 nm and NaCl content using conductivity. Each fraction was assessed for fibrillin-2 using an anti-fibrillin-2 antibody via dot-blot and densitometry with ImageJ. The three peak fractions containing fibrillin-2 were pooled and further purified on a 1.6 cm × 1.6 m Sephacryl S-500 column (fractionation range 40–20,000 kDa) equilibrated in 25 mm HEPES, 125 mm NaCl, 0.1 mm CaCl2, pH 7.4. The column flow rate was 1 ml/min at 4° C, and 6 ml fractions were collected. The elution stream was monitored for protein at 280 nm and fibrillin-2 content for each fraction was determined by dot-blot. The final product was analyzed by SDS-PAGE on a 4% polyacrylamide gel with Laemmli buffer in the presence and absence of a reducing agent. Duplicate gels were either stained with SimplyBlue Coomassie G-250 or transferred to nitrocellulose and stained for fibrillin-2 by Western blotting. The purity of the fibrillin-2 is shown in Fig. S3. The standards used were Precision plus dual color protein standards (Bio-Rad). The Western blotting detection used a secondary donkey anti-rabbit antibody-HRP conjugate (Amersham Biosciences Inc.) that was visualized on film using a chemiluminescent substrate (EMD Millipore).
Surface plasmon resonance
Initial mapping of the TGFβ-1–binding site in MAGP-1 was accomplished using a Biacore X to measure the change in surface plasmon resonance because of the interaction of TGFβ-1 in solution with expressed full-length MAGP-1 or truncated MAGP-1 fragments coupled to a CM-5 chip. For coupling, the protein was dissolved at 20 μg/ml in 5 mm acetate buffer, pH4.5, and injected for 10 min onto a CM-5 sensor chip previously activated for 10 min with 0.05 m N-hydroxysuccinimide–0.2 m EDC. Reactive sites on the chip were quenched with 100 μl of 100 mm ethanolamine, pH 8.0, then the chip was equilibrated in HBST running buffer (10 mm HEPES 150 mm NaCl, pH 7.4, containing 0.01% Triton-100). Varying concentrations of TGFβ-1 diluted in running buffer were then injected. The volume of TGFβ-1 injected was 60 μl, and the flow rate was 20 μl/min. The precise localization of the growth factor–binding site in MAGP-1 was determined using inhibition of TGFβ-1 binding to full-length MAGP-1 by synthetic peptides. These experiments and the latent TGFβ binding studies were performed on a Reichert SR7000DC SPR system. Full-length MAGP-1 was coupled to a carboxymethyl dextran hydrogel surface sensor chip using the same coupling conditions used for the Biacore CM-5 chip. TGFβ-1 or BMP-2 (50 nm final concentration) was mixed with varying concentrations of MAGP-1 synthetic peptide at room temperature for at least 15 min prior to injection. The flow rate for the localization studies was 50 μl/min, and the injection volume was 100 μl.
Latent TGFβ-1 (5 μm stock solution) was diluted to 15 nm in HBST and injected over the MAGP-1 chip. To activate the latent TGFβ-1, HCl was added to the latent TGFβ-1 stock solution to a final concentration of 100 mm for 15 min at room temperature followed by neutralization with the addition of an equimolar concentration of NaOH. The activated TGFβ-1 was diluted to 15 nm in HBST and injected over the full-length MAGP-1 chip.
In ternary complex studies, fibrillin-2 was diluted 1:5 in 5 mm sodium acetate and coupled with repeat 250 μl injections onto a CM-5 activated and blocked as described above. Approximately 4000uRU were coupled to the chip. Direct binding of TGFβ-1 and BMP-2 to fibrillin-2 was initially assessed prior to loading MAGP-1 onto the fibrillin-2 chip. After direct binding experiments, the chip was recycled, and MAGP-1 was loaded onto the chip with duplicate MAGP-1 injections (100 μl injection at 500 nm) where ∼130 RU bound. Binding of TGFβ-1 and BMP-2 to the complex of fibrillin-2 and MAGP-1 was then re-assessed. No recycling was performed between injections on the fibrillin-2 MAGP-1 complex. HBST was used as the sample dilution buffer as the mobile phase. Unless otherwise noted, chips were recycled with 50 μl injections of 0.2 m glycine pH, 2.3.
MAGP peptide structure prediction
The MAGP-1 p17–35 region that was experimentally determined to bind to TGFβ was analyzed for potential secondary structure and disorder. Secondary structure was predicted using Jpred4 (52), Psipred (53), and DISOPRED (54). Additionally, the sequence was analyzed using IUPred2A (55). Structural predictions were performed using PEP-FOLD and PEP-FOLD3 (56, 57)
MAGP–TGFβ interaction predictions
TGFβ family structures were downloaded from the Protein Data Bank: 1TGK (58), 4KV5 (59), 1KLA, 1KLC, and 1KLD (60). Structures were edited to delete any additional ligands or binding partners. When appropriate, structures were subjected to energy minimization in NAMD (61) to produce a relaxed structure. Based on experimental SPR results from the lab, the MAGP-1 p17–35 sequences were chosen as docking candidates. The sequences were treated as flexible peptides and were docked to the TGFβ structures using GalaxyDock (62) and CABS-dock (63). Predicted structures were evaluated by score and feasibility. Structures were manually compared with structures of known TGFβ-containing complexes, specifically receptor-bound TGFβ (PDB ID 3KFD) (64) and TGFβ in the large latent complex (PDB IDs) 3RJR (21) and 5VQP (65). Structures were visualized using VMD (66) and VMD was used to generate all images.
Author contributions
T. J. B. data curation; T. J. B. and N. K. B. investigation; T. J. B. and R. P. M. methodology; T. J. B. writing-original draft; N. K. B. formal analysis; N. K. B. visualization; R. P. M. conceptualization; R. P. M. funding acquisition; R. P. M. project administration; R. P. M. writing-review and editing.
Supplementary Material
Acknowledgments
We thank Dr. Tony Wyss-Coray, Department of Neurology and Neurological Sciences, Stanford University, for providing the TGFβ reporter cell line MFB-F11.
This work was supported by NHLBI, National Institutes of Health Grants R01 HL53325 and R01 HL105314 (to R. P. M.) and by National Institutes of Health Training Grants T32 HL125241 and T32 HL07317 (to N. K. B.), and also by the Ines Mandl Research Foundation (to R. P. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S5.
T.J. Broekelmann, N.K. Bodmer, and R.P. Mecham, manuscript in preparation.
- ECM
- extracellular matrix
- LTBP
- latent TGFβ-binding proteins
- SPR
- surface plasmon resonance
- MEF
- mouse embryonic fibroblast
- buffer A
- 25 mm HEPES, 0.1 mm CaCl2, pH 7.4
- buffer B
- 25 mm HEPES, 0.1 mm CaCl2, 1.0 m NaCl, pH7.4
- HBST
- HEPES buffered saline containing Triton X-100
- EDC
- N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide.
References
- 1. Hynes R. (2009) The extracellular matrix: Not just pretty fibrils. Science 326, 1216–1219 10.1126/science.1176009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Robertson I., Jensen S., and Handford P. (2011) TB domain proteins: Evolutionary insights into the multifaceted roles of fibrillins and LTBPs. Biochem. J. 433, 263–276 10.1042/BJ20101320 [DOI] [PubMed] [Google Scholar]
- 3. Davis E. C., Roth R. A., Heuser J. E., and Mecham R. P. (2002) Ultrastructural properties of ciliary zonule microfibrils. J. Struct. Biol. 139, 65–75 10.1016/S1047-8477(02)00559-2 [DOI] [PubMed] [Google Scholar]
- 4. Goldfischer S., Coltoff-Schiller B., and Goldfischer M. (1985) Microfibrils, elastic anchoring components of the extracellular matrix, are associated with fibronectin in the zonule of Zinn and aorta. Tissue Cell 17, 441–450 10.1016/0040-8166(85)90023-0 [DOI] [PubMed] [Google Scholar]
- 5. Wagenseil J. E., and Mecham R. P. (2009) Vascular extracellular matrix and arterial mechanics. Physiol. Rev. 89, 957–989 10.1152/physrev.00041.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McConnell C. J., DeMont M. E., and Wright G. M. (1997) Microfibrils provide non-linear elastic behaviour in the abdominal artery of the lobster Homarus americanus. J. Physiol. 499 513–526 10.1113/jphysiol.1997.sp021945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Davison I. G., Wright G. M., and DeMont M. E. (1995) The structure and physical properties of invertebrate and primitive vertebrate arteries. J. Exp. Biol. 198, 2185–2196 [DOI] [PubMed] [Google Scholar]
- 8. Isogai Z., Ono R. N., Ushiro S., Keene D. R., Chen Y., Mazzieri R., Charbonneau N. L., Reinhardt D. P., Rifkin D. B., and Sakai L. Y. (2003) Latent transforming growth factor β-binding protein 1 interacts with fibrillin and is a microfibril-associated protein. J. Biol. Chem. 278, 2750–2757 10.1074/jbc.M209256200 [DOI] [PubMed] [Google Scholar]
- 9. Robertson I. B., Horiguchi M., Zilberberg L., Dabovic B., Hadjiolova K., and Rifkin D. B. (2015) Latent TGF-β-binding proteins. Matrix Biol. 47, 44–53 10.1016/j.matbio.2015.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Travis M. A., and Sheppard D. (2014) TGF-β activation and function in immunity. Annu. Rev. Immunol. 32, 51–82 10.1146/annurev-immunol-032713-120257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Carta L., Pereira L., Arteaga-SoliS E., Lee-Arteaga S. Y., Lenart B., Starcher B., Merkel C. A., Sukoyan M., Kerkis A., Hazeki N., Keene D. R., Sakai L. Y., and Ramirez F. (2006) Fibrillins 1 and 2 perform partially overlapping functions during aortic development. J. Biol. Chem. 281, 8016–8023 10.1074/jbc.M511599200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brown-Augsburger P., Broekelmann T., Mecham L., Mercer R., Gibson M. A., Cleary E. G., Abrams W. R., Rosenbloom J., and Mecham R. P. (1994) Microfibril-associated glycoprotein binds to the carboxy-terminal domain of tropoelastin and is a substrate for transglutaminase. J. Biol. Chem. 269, 28443–28449 [PubMed] [Google Scholar]
- 13. Finnis M. L., and Gibson M. A. (1997) Microfibril-associated glycoprotein-1 (MAGP-1) binds to the pepsin-resistant domain of the α3(VI) chain of type VI collagen. J. Biol. Chem. 272, 22817–22823 10.1074/jbc.272.36.22817 [DOI] [PubMed] [Google Scholar]
- 14. Werneck C. C., Trask B. C., Broekelmann T. J., Trask T. M., Ritty T. M., Segade F., and Mecham R. P. (2004) Identification of a major microfibril-associated glycoprotein-1-binding domain in fibrillin-2. J. Biol. Chem. 279, 23045–23051 10.1074/jbc.M402656200 [DOI] [PubMed] [Google Scholar]
- 15. Craft C. S. Pietka T. A., Schappe T., Coleman T., Combs M. D., Klein S., Abumrad N. A., and Mecham R. P. (2014) The extracellular matrix protein MAGP1 supports thermogenesis and protects against obesity and diabetes through regulation of TGF-β. Diabetes 63, 1920–1932 10.2337/db13-1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Combs M. D., Knutsen R. H., Broekelmann T. J., Toennies H. M., Brett T. J., Miller C. A., Kober D. L., Craft C. S., Atkinson J. J., Shipley J. M., Trask B. C., and Mecham R. P. (2013) Microfibril-associated glycoprotein 2 (MAGP2) loss of function has pleiotropic effects in vivo. J. Biol. Chem. 288, 28869–28880 10.1074/jbc.M113.497727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Craft C. S., Broekelmann T. J., Zou W., Chappel J. C., Teitelbaum S. L., and Mecham R. P. (2012) Oophorectomy-induced bone loss is attenuated in MAGP1-deficient mice. J. Cell. Biochem. 113, 93–99 10.1002/jcb.23331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Weinbaum J. S., Broekelmann T. J., Pierce R. A., Werneck C. C., Segade F., Craft C. S., Knutsen R. H., and Mecham R. P. (2008) Deficiency in microfibril-associated glycoprotein-1 leads to complex phenotypes in multiple organ systems. J. Biol. Chem. 283, 25533–25543 10.1074/jbc.M709962200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Trask B. C., Broekelmann T. J., Ritty T. M., Trask T. M., Tisdale C., and Mecham R. P. (2001) Posttranslational modifications of microfibril associated glycoprotein-1 (MAGP-1). Biochemistry 40, 4372–4380 10.1021/bi002738z [DOI] [PubMed] [Google Scholar]
- 20. Tesseur I., Zou K., Berber E., Zhang H., and Wyss-Coray T. (2006) Highly sensitive and specific bioassay for measuring bioactive TGF-β. BMC Cell Biol. 7, 15 10.1186/1471-2121-7-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shi M., Zhu J., Wang R., Chen X., Mi L., Walz T., and Springer T. (2011) Latent TGF-β structure and activation. Nature 474, 343–349 10.1038/nature10152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kanzaki T., Olofsson A., Morén A., Wernstedt C., Hellman U., Miyazono K., Claesson-Welsh L., and Heldin C. H. (1990) TGF-β1 binding protein: A component of the large latent complex of TGF-β1 with multiple repeat sequences. Cell 61, 1051–1061 10.1016/0092-8674(90)90069-Q [DOI] [PubMed] [Google Scholar]
- 23. Massam-Wu T., Chiu M., Choudhury R., Chaudhry S. S., Baldwin A. K., McGovern A., Baldock C., Shuttleworth C. A., and Kielty C. M. (2010) Assembly of fibrillin microfibrils governs extracellular deposition of latent TGF β. J. Cell Sci. 123, 3006–3018 10.1242/jcs.073437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Broekelmann T. J., Limper A. H., Colby T. V., and McDonald J. A. (1991) Transforming growth factor β 1 is present at sites of extracellular matrix gene expression in human pulmonary fibrosis. Proc. Natl. Acad. Sci. U.S.A. 88, 6642–6646 10.1073/pnas.88.15.6642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hinz B. (2015) The extracellular matrix and transforming growth factor-β1: Tale of a strained relationship. Matrix Biol. 47, 54–65 10.1016/j.matbio.2015.05.006 [DOI] [PubMed] [Google Scholar]
- 26. ten Dijke P., and Arthur H. (2007) Extracellular control of TGFβ signalling in vascular development and disease. Nat. Rev. Mol. Cell Biol. 8, 857–869 10.1038/nrm2262 [DOI] [PubMed] [Google Scholar]
- 27. Mecham R. P., and Gibson M. A. (2015) The microfibril-associated glycoproteins (MAGPs) and the microfibrillar niche. Matrix Biol. 47, 13–33 10.1016/j.matbio.2015.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Craft C. S., Broekelmann T. J., and Mecham R. P. (2018) Microfibril-associated glycoproteins MAGP-1 and MAGP-2 in disease. Matrix Biol. 71–72, 100–111 10.1016/j.matbio.2018.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Villain G., Lelievre E., Broekelmann T., Gayet O., Havet C., Werkmeister E., Mecham R., Dusetti N., Soncin F., and Mattot V. (2018) MAGP-1 and fibronectin control EGFL7 functions by driving its deposition into distinct endothelial extracellular matrix locations. FEBS J. 285, 4394–4412 10.1111/febs.14680 [DOI] [PubMed] [Google Scholar]
- 30. Craft C. S., Zou W., Watkins M., Grimston S., Brodt M. D., Broekelmann T. J., Weinbaum J. S., Teitelbaum S. L., Pierce R. A., Civitelli R., Silva M. J., and Mecham R. P. (2010) Microfibril-associated glycoprotein-1, an extracellular matrix regulator of bone remodeling. J. Biol. Chem. 285, 23858–23867 10.1074/jbc.M110.113019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Walji T. A., Turecamo S. E., Sanchez A. C., Anthony B. A., Abou-Ezzi G., Scheller E. L., Link D. C., Mecham R. P., and Craft C. S. (2016) Marrow adipose tissue expansion coincides with insulin resistance in MAGP1-deficient mice. Front. Endocrinol. 7, 87 10.3389/fendo.2016.00087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu Y. J., Xu F. H., Shen H., Liu Y. Z., Deng H. Y., Zhao L. J., Huang Q. Y., Dvornyk V., Conway T., Davies K. M., Li J. L., Recker R. R., and Deng H. W. (2004) A follow-up linkage study for quantitative trait loci contributing to obesity-related phenotypes. J. Clin. Endocrinol. Metab. 89, 875–882 10.1210/jc.2003-030774 [DOI] [PubMed] [Google Scholar]
- 33. Barbier M., Gross M. S., Aubart M., Hanna N., Kessler K., Guo D. C., Tosolini L., Ho-Tin-Noe B., Regalado E., Varret M., Abifadel M., Milleron O., Odent S., Dupuis-Girod S., Faivre L., et al. (2014) MFAP5 loss-of-function mutations underscore the involvement of matrix alteration in the pathogenesis of familial thoracic aortic aneurysms and dissections. Am. J. Hum. Genet. 95, 736–743 10.1016/j.ajhg.2014.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Segade F., Trask B. C., Broekelmann T. J., Pierce R. A., and Mecham R. P. (2002) Identification of a matrix binding domain in MAGP1 and 2 and intracellular localization of alternative splice forms. J. Biol. Chem. 277, 11050–11057 10.1074/jbc.M110347200 [DOI] [PubMed] [Google Scholar]
- 35. Weinbaum J. S., Tranquillo R. T., and Mecham R. P. (2010) The matrix-binding domain of microfibril-associated glycoprotein-1 targets active connective tissue growth factor to a fibroblast-produced extracellular matrix. Macromol. Biosci. 10, 1338–1344 10.1002/mabi.201000121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Trask B. C., Trask T. M., Broekelmann T., and Mecham R. P. (2000) The microfibrillar proteins MAGP-1 and fibrillin-1 form a ternary complex with the chondroitin sulfate proteoglycan decorin. Molec. Biol. Cell 11, 1499–1507 10.1091/mbc.11.5.1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Reinboth B., Hanssen E., Cleary E. G., and Gibson M. A. (2002) Molecular interactions of biglycan and decorin with elastic fiber components: Biglycan forms a ternary complex with tropoelastin and microfibril-associated glycoprotein 1. J. Biol. Chem. 277, 3950–3957 10.1074/jbc.M109540200 [DOI] [PubMed] [Google Scholar]
- 38. Lee J., Wee S., Gunaratne J., Chua R. J., Smith R. A., Ling L., Fernig D. G., Swaminathan K., Nurcombe V., and Cool S. M. (2015) Structural determinants of heparin-transforming growth factor-β1 interactions and their effects on signaling. Glycobiology 25, 1491–1504 10.1093/glycob/cwv064 [DOI] [PubMed] [Google Scholar]
- 39. Lyon M., Rushton G., and Gallagher J. T. (1997) The interaction of the transforming growth factor-βs with heparin/heparan sulfate is isoform-specific. J. Biol. Chem. 272, 18000–18006 10.1074/jbc.272.29.18000 [DOI] [PubMed] [Google Scholar]
- 40. Smaldone S., Clayton N. P., del Solar M., Pascual G., Cheng S. H., Wentworth B. M., Schaffler M. B., and Ramirez F. (2016) Fibrillin-1 regulates skeletal stem cell differentiation by modulating TGFβ activity within the marrow niche. J Bone Miner Res. 31, 86–97 10.1002/jbmr.2598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dallas S. L., Sivakumar P., Jones C. J., Chen Q., Peters D. M., Mosher D. F., Humphries M. J., and Kielty C. M. (2005) Fibronectin regulates latent transforming growth factor-β (TGF β) by controlling matrix assembly of latent TGF β-binding protein-1. J. Biol. Chem. 280, 18871–18880 10.1074/jbc.M410762200 [DOI] [PubMed] [Google Scholar]
- 42. Murphy-Ullrich J. E., and Suto M. J. (2018) Thrombospondin-1 regulation of latent TGF-β activation: A therapeutic target for fibrotic disease. Matrix Biol. 68–69, 28–43 10.1016/j.matbio.2017.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Alcaraz L. B., Exposito J. Y., Chuvin N., Pommier R. M., Cluzel C., Martel S., Sentis S., Bartholin L., Lethias C., and Valcourt U. (2014) Tenascin-X promotes epithelial-to-mesenchymal transition by activating latent TGF-β. J. Cell Biol. 205, 409–428 10.1083/jcb.201308031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zacchigna L., Vecchione C., Notte A., Cordenonsi M., Dupont S., Maretto S., Cifelli G., Ferrari A., Maffei A., Fabbro C., Braghetta P., Marino G., Selvetella G., Aretini A., Colonnese C., et al. (2006) Emilin1 links TGF-β maturation to blood pressure homeostasis. Cell 124, 929–942 10.1016/j.cell.2005.12.035 [DOI] [PubMed] [Google Scholar]
- 45. Tumbarello D. A., Andrews M. R., and Brenton J. D. (2016) SPARC regulates transforming growth factor β induced (TGFBI) extracellular matrix deposition and paclitaxel response in ovarian cancer cells. PLoS One 11, e0162698 10.1371/journal.pone.0162698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Francki A., McClure T. D., Brekken R. A., Motamed K., Murri C., Wang T., and Sage E. H. (2004) SPARC regulates TGF-β1-dependent signaling in primary glomerular mesangial cells. J. Cell. Biochem. 91, 915–925 10.1002/jcb.20008 [DOI] [PubMed] [Google Scholar]
- 47. Brown-Augsburger P. B., Broekelmann T., Rosenbloom J., and Mecham R. P. (1996) Functional domains on elastin and MAGP involved in elastic fiber assembly. Biochem. J. 318, 149–155 10.1042/bj3180149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Trask T., Ritty T., Broekelmann T., Tisdale C., and Mecham R. (1999) N-terminal domains of fibrillin 1 and fibrillin 2 direct the formation of homodimers: A possible first step in microfibril assembly. Biochem. J. 340, 693–701 [PMC free article] [PubMed] [Google Scholar]
- 49. Mazzieri R., Munger J. S., and Rifkin D. B. (2000) Measurement of active TGF-β generated by cultured cells. Methods Mol. Biol. 142, 13–27 10.1385/1-59259-053-5:13 [DOI] [PubMed] [Google Scholar]
- 50. Stoilov I., Starcher B. C., Mecham R. P., and Broekelmann T. J. (2018) Measurement of elastin, collagen, and total protein levels in tissues. Methods Cell Biol. 143, 133–146 10.1016/bs.mcb.2017.08.008 [DOI] [PubMed] [Google Scholar]
- 51. Hutchinson M. H., and Chase H. A. (2006) Adsorptive refolding of histidine-tagged glutathione S-transferase using metal affinity chromatography. J. Chromatogr. A 1128, 125–132 10.1016/j.chroma.2006.06.050 [DOI] [PubMed] [Google Scholar]
- 52. Drozdetskiy A., Cole C., Procter J., and Barton G. J. (2015) JPred4: A protein secondary structure prediction server. Nucleic Acids Res. 43, W389–W394 10.1093/nar/gkv332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Buchan D. W. A., and Jones D. T. (2019) The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Res. 47, W402–W407 10.1093/nar/gkz297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jones D. T., and Cozzetto D. (2015) DISOPRED3: Precise disordered region predictions with annotated protein-binding activity. Bioinformatics 31, 857–863 10.1093/bioinformatics/btu744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mészáros B., Erds G., and Dosztányi Z. (2018) IUPred2A: Context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 46, W329–W337 10.1093/nar/gky384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shen Y., Maupetit J., Derreumaux P., and Tufféry P. (2014) Improved PEP-FOLD approach for peptide and miniprotein structure prediction. J. Chem. Theory Comput. 10, 4745–4758 10.1021/ct500592m [DOI] [PubMed] [Google Scholar]
- 57. Thévenet P., Shen Y., Maupetit J., Guyon F., Derreumaux P., and Tufféry P. (2012) PEP-FOLD: An updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides. Nucleic Acids Res. 40, W288–W293 10.1093/nar/gks419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mittl P. R., Priestle J. P., Cox D. A., McMaster G., Cerletti N., and Grütter M. G. (1996) The crystal structure of TGF-β 3 and comparison to TGF-β 2: Implications for receptor binding. Protein Sci. 5, 1261–1271 10.1002/pro.5560050705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Moulin A., Mathieu M., Lawrence C., Bigelow R., Levine M., Hamel C., Marquette J.-P., Le Parc J., Loux C., Ferrari P., Capdevila C., Dumas J., Dumas B., Rak A., Bird J., Qiu H., Pan C. Q., Edmunds T., and Wei R. R. (2014) Structures of a pan-specific antagonist antibody complexed to different isoforms of TGFβ reveal structural plasticity of antibody–antigen interactions. Protein Sci. 23, 1698–1707 10.1002/pro.2548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hinck A. P., Archer S. J., Qian S. W., Roberts A. B., Sporn M. B., Weatherbee J. A., Tsang M. L. S., Lucas R., Zhang B.-L., Wenker J., and Torchia D. A. (1996) Transforming growth factor β1: Three-dimensional structure in solution and comparison with the x-ray structure of transforming growth factor β2. Biochemistry 35, 8517–8534 10.1021/bi9604946 [DOI] [PubMed] [Google Scholar]
- 61. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., and Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 10.1002/jcc.20289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lee H., Heo L., Lee M. S., and Seok C. (2015) GalaxyPepDock: A protein–peptide docking tool based on interaction similarity and energy optimization. Nucleic Acids Res. 43, W431–W435 10.1093/nar/gkv495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kurcinski M., Pawel Ciemny M., Oleniecki T., Kuriata A., Badaczewska-Dawid A. E., Kolinski A., and Kmiecik S. (2019) CABS-dock standalone: A toolbox for flexible protein–peptide docking. Bioinformatics 35, 4170–4172 10.1093/bioinformatics/btz185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Radaev S., Zou Z., Huang T., Lafer E. M., Hinck A. P., and Sun P. D. (2010) Ternary complex of transforming growth factor-β1 reveals isoform-specific ligand recognition and receptor recruitment in the superfamily. J. Biol. Chem. 285, 14806–14814 10.1074/jbc.M109.079921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhao B., Xu S., Dong X., Lu C., and Springer T. A. (2017) Prodomain–growth factor swapping in the structure of pro-TGF-β1. J. Biol. Chem. 293, 1579–1589 10.1074/jbc.M117.809657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Humphrey W., Dalke A., and Schulten K. (1996) VMD: Visual molecular dynamics. J. Mol. Graph. 14, 33–38 10.1016/0263-7855(96)00018-5 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.