

Abstract
The orexin system, which consists of the two G protein-coupled receptors OX1 and OX2, activated by the neuropeptides OX-A and OX-B, is firmly established as a key regulator of behavioral arousal, sleep, and wakefulness and has been an area of intense research effort over the past two decades. X-ray structures of the receptors in complex with 10 new antagonist ligands from diverse chemotypes are presented, which complement the existing structural information for the system and highlight the critical importance of lipophilic hotspots and water molecules for these peptidergic GPCR targets. Learnings from the structural information regarding the utility of pharmacophore models and how selectivity between OX1 and OX2 can be achieved are discussed.
Introduction
Approximately 20 years ago, two research groups independently identified the orexin neuropeptides (orexin-A (OX-A) and orexin-B (OX-B), also referred to as hypocretin-1 and 2, respectively) as ligands for the pair of G protein-coupled receptors that are now known as OX1 and OX2.1,2 The receptors and their neuropeptide ligands are highly conserved across mammalian species,1,3 and over the past two decades, the orexin system has become firmly established as a key regulator of behavioral arousal, sleep, and wakefulness. Shortly after the discovery of the orexin system, a genetic link has been established with narcolepsy, a chronic sleep disorder characterized by excessive daytime sleepiness, fragmented sleep, and cataplexy,4 when a mutation in the OX2 receptor gene was demonstrated to be the cause of narcolepsy in canines.5 Together with a number of rodent knockout and transgenic studies that demonstrated a phenotype similar to human narcolepsy patients,6−8 this observation was the catalyst for the development of antagonists of the receptors for treatment of sleep disorders.9,10 A number of pre-clinical studies have concluded that antagonizing the OX2 receptor is efficacious in promoting sleep but that dual antagonism is more effective,11−13 although this view is not without debate in the literature.14,15 In line with this, a number of companies have been active in the development of dual orexin antagonist (DORA) molecules, with Actelion/GSK being the first to demonstrate clinical proof of concept with almorexant 1 (Chart 1). The molecule demonstrated a dose-dependent effect on sleep efficiency, together with reductions in wake after sleep onset (WASO) and latency to persistent sleep (LPS) as secondary endpoints,16 but development was terminated after clinical safety observations in a subsequent trial.17 A number of other DORA molecules subsequently progressed into the clinic, with Merck being the first to market with suvorexant 2 (Chart 1), which was approved by the FDA in 2014 for the treatment of primary insomnia.18,19 The most common adverse event observed for suvorexant was next-day somnolence, which trended higher at higher doses,9 contributing to approval by the FDA at a recommended dose of 10 mg per night, increasing to 20 mg if necessary.20 Several additional DORAs, including, filorexant 3 (MK-6096, Chart 1),21,22 lemborexant 4 (Chart 1),23,24 and daridorexant 5 (ACT-541468, formerly known as nemorexant, Chart 1),25,26 have since progressed into clinical trials for insomnia as well as a range of co-morbidities of sleep disorders, with an NDA submitted for lemborexant in January 2019 and Phase III trials currently underway for daridorexant.
Chart 1. Orexin Receptor Antagonists Almorexant 1, Suvorexant 2, Filorexant 3, Lemborexant 4, Daridorexant 5, MK-1064 6, Seltorexant 7, EMPA 8, SB-674042 9, GSK1059865 10, SB-334867 11, SB-408124 12, HTL6641 13, Pyridothiadiazinone Compound 14, ACT-462206 15, and Diazaspirodecane Compound 16.

Efforts to develop selective OX2 antagonists (2-SORAs) for sleep disorders have also been encouraging in recent years. Merck progressed a 2-SORA, MK-1064 6 (Chart 1),27 to Phase 1 clinical trials. Seltorexant 7 (JNJ-42847922, MIN-202, Chart 1) is being progressed by Minerva Neurosciences and Janssen for insomnia and as an adjunctive treatment for major depressive disorder; positive top-line clinical data from Phase 2b trials in these two indications was disclosed in mid-2019.28
In contrast, attempts to develop selective OX1 antagonists (1-SORAs) have been less successful to date despite evidence from several sources linking the OX1 receptor with addictive behaviors. A role for OX1 in substance seeking and craving was first demonstrated in 200529 using immunohistochemistry to demonstrate activation of orexinergic neurons in the lateral hypothalamus when conditioned animals received cues for cocaine, morphine, or food; in addition, when the reward-seeking behavior was extinguished, it could be reinstated by administration of orexin-A and could be blocked by an OX1 antagonist. A role for OX1 in seeking and craving of nicotine and alcohol has also been implicated.30,31 Several 1-SORAs have been disclosed, including GSK105986532−3510 and SB-33486736,3711 (Chart 1), which have been widely used as tool compounds and demonstrated efficacy in animal models of disease, but these two molecules have not progressed to clinical development. Idorsia and Janssen have recently progressed their 1-SORAs, ACT-539313 for psychiatric disorders and JNJ-61393215 for major depressive disorder and anxious distress, into clinical trials.
To date, all clinical stage orexin receptor antagonists have been discovered in the absence of structural knowledge of the OX1 and OX2 receptors. Hence, it is not surprising that development of DORAs has proved more successful than for 1-SORAs or 2-SORAs, where knowledge of the subtle differences in architecture between the receptor subtypes may be required to achieve selectivity in a molecule with properties suitable for development. The situation could now change as structures have recently been reported for both receptors, with the DORA suvorexant 2 (Chart 1) in both OX1 and OX2,38,39 the 2-SORA EMPA 8 (Chart 1) in OX2,40 and the 1-SORA SB-674042 9 (Chart 1) in OX1.39
As part of a program to discover 1-SORAs suitable for development as therapeutic agents for addictive disorders, we have determined the co-crystal structures of OX1 and OX2 with a diverse array of ligands displaying a range of selectivity profiles for these two receptors and, surprisingly, structurally diverse binding modes. Analysis of the structures, comparing and contrasting the binding modes of these small molecules, has led to several insights into the factors governing ligand recognition for these peptidergic receptors, which can then be deployed for the design of selective antagonists suitable for drug development.
Results and Discussion
Structure Determination of OX1 or OX2 in Complex with Diverse Ligands
The structures of OX1 and OX2 receptor complexes presented in this study were determined using thermostabilized receptors (StaRs). OX2 was thermostabilized in the presence of the OX2-selective radioligand [3H]-EMPA,41 which resulted in a StaR containing 12 mutations. Based on site-directed mutagenesis studies,42 the residue at position 3.33 (A1273.33 in OX1, T1353.33 in OX2) was identified as critical for subtype selectivity. We therefore mutated A1273.33 to T in OX1 and demonstrated that it was competent for EMPA binding. Consequently, we were able to use this as the template to thermostabilize OX1 in the presence of [3H]-EMPA. The resulting OX1 StaR contained eight thermostabilizing mutations in addition to the EMPA binding A127T3.33 mutation (hereafter referred to as OX1A127T StaR). For functional, biophysical, and structural studies, we reverted T1273.33 to A. The generic GPCR residue numbering system is used throughout this paper (see Experimental Section). Both orexin StaRs were further engineered to facilitate crystallization in vapor diffusion (VD; OX1) and lipidic cubic phase (LCP; OX2). In OX1, residues 1–27 (N-terminus), 254–285 (intracellular loop 3 (ICL3)), and 381–425 (C-terminus) were removed, the glycosylated residue N194 was mutated to A, and the palmitoylated residues C375 and C376 were both mutated to W. In OX2, the C-terminal residues 389–444 were removed, ICL3 residues 255–293 were replaced with residues 218 to 413 of Pyrococcus abyssi glycogen synthase,43 the glycosylated residues N14, N22, N30, and N202 were all mutated to D, and the potential palmitoylation sites C381, C382, and C383 were all mutated to W.
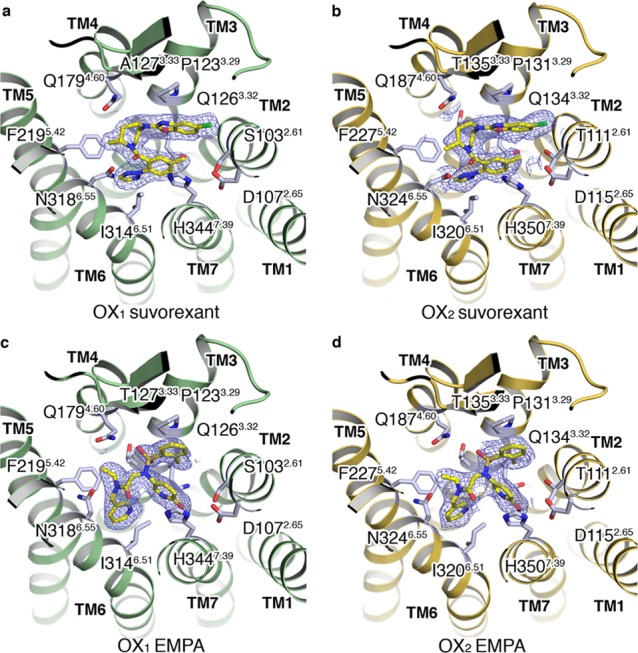
The OX1 StaR bound to suvorexant (2) was crystallized using the vapor diffusion method, and the structure was solved at 2.26 Å resolution by molecular replacement using the κ-opioid receptor structure (PDB ID: 4DJH) as the search model. The receptor structures of OX1 and OX2 display the canonical 7TM arrangement and the now widely recognized molecular hallmarks of the inactive receptor state (Figure 1 a,b,d,e). Following this, all additional OX1 and OX2 structures were solved by molecular replacement using the OX1–suvorexant coordinates as the search model. The superposition of the OX1–suvorexant, OX2–suvorexant, and OX2–EMPA structures generated using the StaR approach onto the literature (PDB ID: 4ZJ8, 4S0V, and 5WQC, respectively) structures results in root-mean-square deviations (RMSD) of main chain atoms lining the ligand binding pockets not exceeding 0.3 Å, allowing us to conclude that different approaches to GPCR crystallography for the same ligand/receptor pairing yield virtually identical results.
Figure 1.

Overview of the OX1 and OX2–antagonist complex crystal structures. (a–e) Overview of the OX1 and OX2 StaR structures in complex with suvorexant. (a) View from the extracellular space of OX1 in a surface representation (green) and suvorexant in a sphere representation with carbon, nitrogen, oxygen, and chlorine atoms colored yellow, blue, red, and green, respectively. (b) View of the OX1 receptor in a cartoon representation from a plane parallel to the membrane colored as in panel (a): approximate membrane boundaries are shown, and TM helices and loops are labeled. (c) Overlay of the OX1 and OX2 structures in a cartoon representation; receptors are colored green and gold. (d) View of the OX2 receptor in a cartoon representation from a plane parallel to the membrane colored as in panel (c): TM helices and loops are labeled, and suvorexant in a sphere representation is colored as in panel (a). (e) View from the extracellular space of OX2 in a surface representation (gold) and suvorexant in a sphere representation colored as in panel (a).
Table 1 summarizes the co-crystal X-ray structures of OX1 and OX2 presented in this manuscript, brief descriptions of the ligand–receptor interactions in each case are detailed in the following section, and the structures are presented in Figures 1–8. Details of data collection and refinement statistics for all structures are given in Supporting Information, Table S2.
Table 1. Summary of the OX1 and OX2 X-ray Crystal Structures Reported in This Study.
| receptor | ligand | pharmacological profilea | resolution |
|---|---|---|---|
| OX1 | suvorexant (2) | DORA | 2.26 Å |
| OX1 pKi 9.4, OX2 pKi 9.1 | |||
| EMPA (8) | 2-SORA | 2.11 Åb | |
| OX1 pKi 6.0, OX2 pKi 8.9 | |||
| lemborexant (4) | DORA | 2.22 Å | |
| OX1 pKi 8.6, OX2 pKi 9.3 | |||
| filorexant (3) | DORA | 2.34 Å | |
| OX1 pKi 9.2, OX2 pKi 9.7 | |||
| GSK1059865 (10) | 1-SORA | 2.16 Å | |
| OX1 pKi 8.7, OX2 pKi 7.2 | |||
| daridorexant (5) | DORA | 3.03 Å | |
| OX1 pKi 8.8, OX2 pKi 8.9 | |||
| Compound 14 | DORA | 2.55 Å | |
| OX1 pKi 7.8, OX2 pKi 7.3 | |||
| ACT-462206 (15) | DORA | 3.01 Å | |
| OX1 pKi 8.2, OX2 pKi 9.2 | |||
| Compound 16 | DORA | 2.30 Å | |
| OX1 pKi 7.1, OX2 pKi 7.8 | |||
| SB-334867 (11) | 1-SORA | 2.66 Å | |
| OX1 pKi 7.8, OX2 pKi 6.2 | |||
| SB-408124 (12) | 1-SORA | 2.66 Å | |
| OX1 pKi 7.6, OX2 pKi 6.1 | |||
| OX2 | suvorexant (2) | DORA | 2.76 Å |
| OX1 pKi 9.4, OX2 pKi 9.1 | |||
| EMPA (8) | 2-SORA | 2.74 Å | |
| OX1 pKi 8.9, OX2 pKi 6.0 | |||
| HTL6641 (13) | DORA | 2.61 Å | |
| OX1 pKi 7.5, OX2 pKi 8.3 |
Radioligand binding affinity data (see Supporting Information for assay details).
OX1 StaR harboring the A1273.33T mutation.
Figure 8.
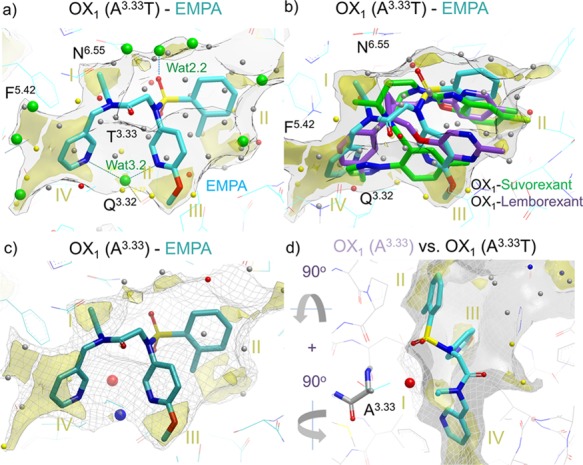
Role of water in EMPA (8) OX1/OX2 selectivity. (a) EMPA ligand in OX2 mimicking OX1 A1273.33T mutant with a WaterFLAP-computed network (small spheres, color-coded by energy) and X-ray crystallographic waters (large green spheres). The interstitial water hydrogen bonding to the two pyridine nitrogens and Q1263.32 can be clearly seen. (b) Suvorexant (2) and lemborexant (4) ligand poses from OX1 crystal structures overlaid with an EMPA water network in OX2. The carbon atoms of the ligands are colored cyan (EMPA), green (suvorexant), and purple (lemborexant). GRID maps are contoured (transparent solid) and colored in the following manner: C1 is the probe (lipophilic) in yellow at −2.8 kcal/mol, and the CH3 methyl group probe is in gray at 1 kcal/mol, which defines the pocket surface in terms of how close a ligand carbon atom can reside. WaterFLAP water networks calculated on the pseudo-apo structure (shown as large spheres) have been color-coded in red if predicted to have a free energy (ΔG) >3.5 kcal/mol, in yellow if ΔG is between 2.0 and 3.5 kcal/mol, in gray if ΔG is between −1.0 and 2.0 kcal/mol, and in blue if ΔG < −1.0 kcal/mol. All WaterFLAP free energy estimations are relative to bulk solvent. (c, d) Comparison of the binding site surfaces of the OX2 mimicking the OX1 A1273.33T mutant structure (solid) and back mutated T1273.33A/wild-type (WT) OX1 (dark gray mesh) indicates that in wild-type OX1, an energetically unhappy water molecule will be trapped by the OX2-selective EMPA antagonist. WaterMap water network calculations of the complex with OX1 (A1273.33) with a very unhappy (high relative energy to bulk solvent, 4 kcal/mol) water trapped in the larger OX1 binding site, shown as a large red sphere. The water stabilized by the two pyridines is also shown as a large blue sphere (stabilized, 2 kcal/mol); in the pseudo-apo structure, this water is calculated by WaterFLAP to be unstable relative to bulk water (small yellow sphere in panel (b)).
OX1–Suvorexant and OX2–Suvorexant
In the co-crystal structures of the OX1/OX2 StaR proteins complexed with suvorexant (2), the ligand adopts an intramolecular π-stacked horseshoe conformation, which is essentially the same as in the previously reported X-ray structures.38,39 The intramolecular π-stacking toluene and benzoxazole fragments of suvorexant are stabilized by offset π stacking with H7.39 and edge–face π stacking with W23.50, respectively, in both OX1 and OX2. The fragments sit in a hydrophobic pocket defined by A2.60, S2.61 (T in OX2), V2.64, I3.28, P3.29, Q3.32, and Y7.43. The chlorine substituent of the benzoxazole is positioned in a hydrophobic subpocket between A2.60, V2.64, and W23.50 (OX1/OX2), explaining the significant contribution of this functional group to OX1 and OX2 binding affinity.18 The homopiperazine ring sits under the salt bridges, E2.68–R7.28, D45.51–R6.59, and E45.52–H5.39, that stabilize the placement of the second extracellular loop (ECL2). The homopiperazine ring sits adjacent to A3.33 (T in OX2) and in direct contact with Q4.60 and F5.42. The carbonyl of the amide linker from homopiperazine makes a direct hydrogen bond with N6.55 and a water-mediated hydrogen bond to H7.39. In OX1, this water molecule (Wat 1.1) is involved in a water-mediated hydrogen bond network to another water (Wat 2.1), which in turn forms a hydrogen bond with the nitrogen on the benzoxazole (Figure 6). Binding site water molecules and other water molecules interacting with ligands in the different OX1 and OX2 structures are annotated in Figure 6. The N-linked triazole attached ortho to the toluene fragment makes edge–face π-stacking interactions with F5.42 and Y6.48 and hydrophobic contacts with V3.36 and I6.51; in addition, the triazole is in direct contact with a network of hydrogen bonding waters sitting in a cleft between transmembrane helices 5 and 6.
Figure 6.

Interaction analysis and water-mediated hydrogen bond networks in antagonist-bound OX1 and OX2 structures. (a) Polar interaction networks of suvorexant (2), filorexant (3), daridorexant (5), GSK1059865 (10), HTL6641 (13), pyridothiadiazinone compound 14, ACT-462206 (15), diazaspirodecane compound 16, lemborexant (4), EMPA (8), SB-334867 (11), and SB-408124 (12) in OX1, and OX2 crystal structures. Direct receptor–ligand hydrogen bond interactions, water–ligand interactions, and water–receptor hydrogen bond interactions are indicated by dashed red, blue, and gray lines, respectively. Residues and water molecules involved in hydrogen bond interactions are labeled in black, and residues involved in water-mediated interactions are labeled in gray. Water molecules involved in hydrogen bond interactions with ligands are located in four regions: (1) hydrogen bonded to H7.39 (Wat 1.1) and/or N6.55 (Wat 1.2); (2) hydrogen bonded to E45.52 (Wat 2.2) and/or close to ECL2 (Wat 2.1); (3) hydrogen bonded to Q3.32 (Wat 3.2) and/or deep in the binding pocket between TMs 2, 3, and 7 (Wat 3.1); (4) between TMs 5 and 6 (Wat 4). (b) Structural receptor–ligand and water–ligand interaction patterns in different OX1 and OX2 crystal structures, discriminating non-polar contacts (light gray), aromatic interactions (dark gray), and hydrogen bond interactions with receptor residues (red) and water molecules (blue). Water molecules are annotated as defined in panel (a). (c) Atoms in the chemical structures of the ligands depicted in panel (a) that only form direct hydrogen bond interactions with the receptor, only form water-mediated hydrogen interactions, or form hydrogen bond interactions with both the receptor and water molecules are colored red, blue, and magenta, respectively.
OX1–EMPA and OX2–EMPA
The X-ray structures of EMPA (8) bound to OX1 (bearing the additional A127T change described earlier to confer EMPA binding) and OX2 proteins demonstrate identical placements of the ligand within the binding sites relative to one another (Figure 2c,d). The aromatic rings flanking the sulfonamide linker are involved in an intramolecular π-stacking arrangement in a similar position to that seen in the suvorexant co-structures, although the EMPA substituents push the small molecule upward toward the extracellular surface, relative to suvorexant. The aromatic rings are stabilized by offset π stacking with H7.39 and edge–face π stacking with W23.50 and reside in a hydrophobic pocket defined by A2.60, S2.61, V2.64, I3.28, P3.29, Q3.32, and Y7.43 (Figure 6). The lone pair from the nitrogen of the methoxy-substituted pyridine makes a hydrogen bond via a water molecule (Wat 3.2) to Q3.32 (Figure 6). The same water molecule also makes another hydrogen bond across to the second pyridine ring that is sitting deeper in the pocket, face-to-edge π stacking with F5.42, and making hydrophobic contacts with Y6.48, V3.36, and I6.51. The amide linker from the deeper, unsubstituted pyridine to the sulfonamide appears to be in a π-stacking arrangement with N6.55, and the π electrons from the amide linker are stacking with the π electrons from the amide head group of the asparagine residue. The N-ethyl amide substituent makes hydrophobic contacts with F5.42, and a water-mediated hydrogen bond from the carbonyl oxygen of the amide across to H7.39 via water molecule Wat 1.1 can be seen, which is conserved in several other OX1/OX2 structures (Figure 6). The sulfonamide linker forms another water-mediated hydrogen bond (Wat 2.2, Figure 6).
Figure 2.
(a, b) Extracellular views of the OX1 and OX2 StaR structures in complex with suvorexant (2). (c, d) Extracellular views of the OX1 (A127T) and OX2 StaR structures in complex with EMPA (8). Ligands shown in a stick representation with carbon, nitrogen, oxygen, and chlorine atoms colored yellow, blue, red, and green, respectively. Ligand 2Fo–Fc electron density maps in blue mesh and contoured at 1.0σ.
OX1–Lemborexant
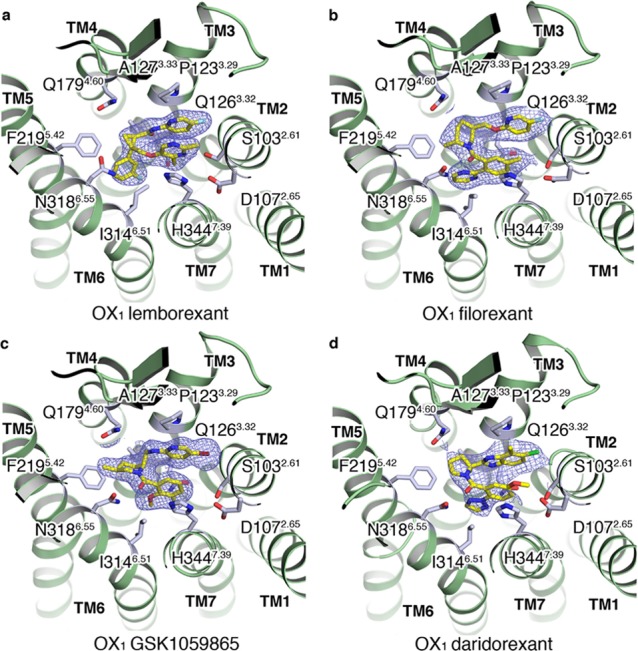
The co-structure of OX1 with lemborexant (4) bound in the orthosteric site shows the ligand adopting a horseshoe conformation with the amidopyridine and pyrimidine portions that are cis-substituted from the central cyclopropyl ring making intramolecular π-stacking interactions (Figure 3a). The intramolecular π-stacking is stabilized by edge–face π stacking between W11223.50 and the amidopyridine fragment and face–face π-stacking between H3447.39 and the pyrimidine fragment. The amidopyridine sits in an overlapping position to the benzoxazole of suvorexant, whereas the ether-linked dimethyl pyrimidine sits higher and offset in the pocket relative to the position of the amidotoluene fragment of suvorexant. These aromatic moieties occupy a hydrophobic pocket defined by A1022.60, S1032.61, V1062.64, I1223.28, P1233.29, Q1263.32, and Y3487.43. The 3-pyrimidine nitrogen is facing the hydroxyl moiety of S1032.61, and the 2-methyl and 5-fluoro substituents of the pyrimidine and pyridine groups, respectively, form tight fits with adjacent subpockets, consistent with subtle structure–affinity and OX1/OX2 selectivity relationships around this ring system (see later selectivity discussion).23 The central cyclopropyl ring does not appear to make any significant interactions with the receptor but instead is essential for providing the correct vectors for all the small molecule substituents. The phenyl meta fluoro substituent sits in a similar position to the triazole of suvorexant although the planes of the two ring systems differ by approximately 45°. This results in only one edge–face π-stacking interaction being observed with F2195.42, along with further hydrophobic contacts to V1303.36, Y3116.48, and I3146.51. Only two water molecules are seen within 5 Å of the ligand in the crystal structure, of which one forms bridging hydrogen bonds between N3186.55 and H3447.39 in contact with the ligand, yet no direct polar interaction is observed.
Figure 3.
(a–d) Extracellular views of the OX1 StaR structures in complex with lemborexant (4), filorexant (3), GSK1059865 (10), and daridorexant (5), respectively. Ligands shown in a stick representation with carbon, nitrogen, oxygen, chlorine, and fluorine atoms colored yellow, blue, red, green, and cyan, respectively. Ligand 2Fo–Fc electron density maps in blue mesh and contoured at 1.0σ.
OX1–Filorexant
The co-crystal structure of OX1 bound to filorexant (3) demonstrates that the ligand adopts an almost identical horseshoe conformation to that of suvorexant although in place of the suvorexant benzoxazole is an ether-linked fluoropyridine, forming an intramolecular π-stacking arrangement with the substituted benzamide portion (Figure 3). Similar π-stacking stabilizing interactions are seen with W11223.50 and H3447.39 along with the identical hydrophobic pocket defined by A1022.60, S1032.61, V1062.64, I1223.28, P1233.29, Q1263.32, and Y3487.43. The central piperidine ring features an axial methyl group adjacent to the amide, which enforces a trans-diaxial orientation of the methyl and aryloxymethyl substituents on the ring. The orientation, proposed by workers at Merck to be optimal for activity as it encourages the system to adopt a π-stacked horseshoe conformation, is consistent with significantly lower affinities for desmethyl analogues in the series and is confirmed by the X-ray structure.21 The piperidine sits in a similar position to the homopiperazine ring of suvorexant, adjacent to A1273.33 (T in OX2) and in direct contact with Q1794.60 and F2195.42 under the salt bridges that stabilize the placement of ECL2, E1102.68, R3337.28, D20345.51–R3226.59, and E20445.52–H2165.39. The amide carbonyl makes a direct hydrogen bond with N3186.55, and the pyrimidine substituent makes edge–face π stacking with F2195.42 and Y3116.48 and hydrophobic contacts with V1063.36 and I3146.51 and may form a weak hydrogen bond with Nε of Gln3.32, which sits 3.8–4 Å away. The amide carbonyl oxygen of filorexant forms a hydrogen bond with water molecule Wat 1.1, stabilized by a polar interaction network with N3186.55 and H3447.39 (Figure 6).
OX1–GSK1059865
In the co-crystal X-ray structure with OX1, the 1-SORA GSK1059865 (10) sits in a similar position to filorexant (Figure 3c). The bromopyridine portion intramolecularly π-stacks with the 2-methoxy-3-fluorophenyl ring, stabilized by offset π stacking with H3447.39 and edge–face π stacking with W11223.50, sitting within the hydrophobic pocket defined by A1022.60, S1032.61, V1062.64, I1223.28, P1233.29, Q1263.32, and Y3487.43. Interestingly, the related methyl-piperidine cores of filorexant and GSK1059865 sit in somewhat different orientations within the OX1 orthosteric binding site, which gives rise to the significantly different selectivity profile observed for GSK1059865. The axial attachment of the bromopyridine substituent causes the piperidine ring to extend deeply into the hydrophobic cleft adjacent to A1273.33 and make direct hydrophobic contacts. This space is occupied by T1353.33 in OX2, which occludes the binding of GSK1059865 and rationalizes the observed selectivity. Other hydrophobic contacts are also observed with Q1794.60 and F2195.42 under the E20445.52–H2165.39 salt bridge that stabilizes ECL2. The carbonyl of the amide linker from the piperidine ring makes a hydrogen bond to N3186.55, which connects through to the 2-methoxy-3-fluorophenyl ring, making deeper hydrophobic contacts with V1303.36, I3146.51, and V3477.42. Furthermore, GSK1059865 forms water-mediated hydrogen bond interactions with water molecules Wat 1.1 (stabilized by N3186.55 and H3447.39) and Wat 2.2 (stabilized by E20445.52, Figure 6).
OX1–Daridorexant
In the co-structure of OX1 bound to daridorexant (5), the 4-methyl-5-chloro-benzimidazole portion sits in a similar position to the substituted benzoxazole of suvorexant, making an intramolecular π-stacking arrangement to the 5-methoxy-2-triazolephenyl moiety (Figure 3d). This intramolecular interaction is stabilized by offset π stacking with H3447.39 and edge–face π stacking with W23.50 while sitting in a hydrophobic pocket defined by A1022.60, S1032.61, V1062.64, I1223.28, P1233.29, Q1263.32, and Y3487.43. The central (2S)-methylpyrrolidine ring connects the two aromatic π-stacking substituents and is located under the salt bridges (E1102.68–R3337.28, D20345.51–R3226.59, E20445.52–H2165.39), which stabilize the placement of ECL2, and is adjacent to A1273.33 and in direct contact with Q1794.60 and F2195.42. Additionally, the carbonyl oxygen of the pyrrolidine amide linker makes a direct hydrogen bond with N3186.55. Relative to the central homopiperazine ring of suvorexant, the smaller 2-methyl-pyrrolidine of daridorexant results in a tighter angle between the displayed heteroaryl groups on either side of the central core ring, and thus the substituted benzoyl portion sits higher in the orthosteric pocket relative to the analogous ring in suvorexant. Edge–face π stacking is observed with the side chain of Y3116.48 with the triazole substituent in addition to the aforementioned H3447.39 interactions. The substituted benzoyl portion resides in a hydrophobic pocket defined by V1303.36, I3146.51, and V3477.42 and is in contact with S1032.61.
OX2–HTL6641
DORA HTL6641 (13) is the lead compound from a series of molecules with a central benzo- or pyridothiadiazin-3-one 1,1-dioxide core44 and was co-crystallized with OX2 (Figure 4a). In a departure from a number of the dual antagonists described herein, HTL6641 binds in a different manner without a hydrophobic collapse of the molecule induced by intramolecular π stacking. Instead, the aromatic portion of the central core and the N-benzyl substituent effectively form an aromatic offset and edge–face π-stacking clamp around F2275.42. The trifluorobenzyl substituent forms hydrophobic contacts with V1383.36, Y3176.48, and I3206.51, explaining the published impact of hydrophobic substitution of this aromatic ring system on affinity.44 The aromatic core forms contacts with T1353.33 (A1273.33 in OX1), Q1874.60, and the ECL2-stabilizing salt bridge E21245.52–H2445.39. One of the sulfonamide oxygens of HTL6641 forms a direct hydrogen bond with Q1874.60, and the pyridine nitrogen forms a hydrogen bond with N3246.55. The carbonyl oxygen of the central thiadiazin-3-one ring appears to be making a water-mediated hydrogen bond across to H3507.39 via water molecule Wat 1.1, which is conserved in several of the OX1 and OX2 structures reported in this study (Figure 6). The dimethoxypyridyl group occupies the same region as the benzoxazole of suvorexant although the dimethoxy substitution appears to result in the ring sitting higher in the orthosteric pocket. There are hydrophobic interactions from this ring with A1102.60, T1112.61 (S1032.61 in OX1), V1142.64, I1303.28, P1313.28, and Q1343.32.
Figure 4.
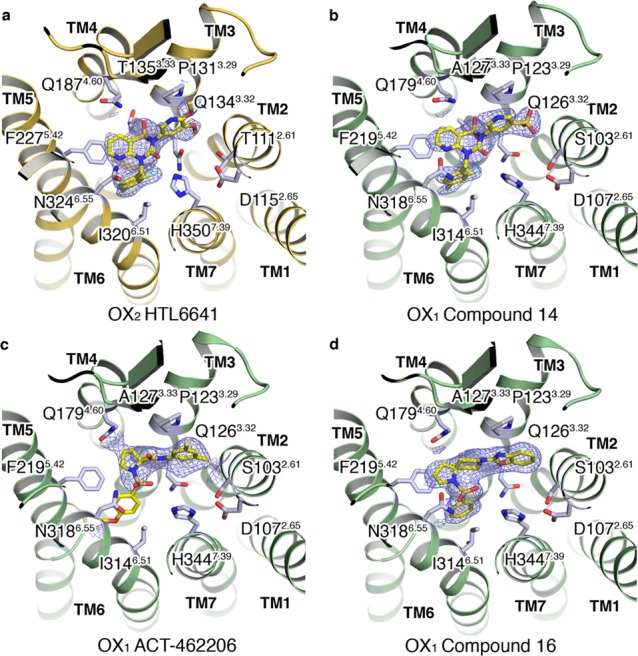
(a) Extracellular view of the OX2 StaR structure in complex with HTL6641 (13). (b–d) Extracellular views of the OX1 StaR structures in complex with compound 14, ACT-462206 (15), and compound 16, respectively. Ligands shown in a stick representation with carbon, nitrogen, oxygen, and fluorine atoms colored yellow, blue, red, and cyan, respectively. Ligand 2Fo–Fc electron density maps in blue mesh and contoured at 1.0σ.
OX1–Compound 14
A second example of an orexin antagonist with the pyridothiadiazin-3-one 1,1-dioxide core, the moderately OX1-selective 14, was subsequently crystallized, this time with OX1 (Figure 4b). In a similar fashion to HTL6641, the central aromatic core and the N-benzyl substituent form an aromatic offset and edge–face π-stacking clamp, respectively, around F2195.42. The trifluorobenzyl substituent forms hydrophobic contacts with V1303.36, Y3116.48, and I3146.51, and the aromatic core forms contacts with A1273.33 (T in OX2), Q1794.60, and the ECL2-stabilizing salt bridge, E20445.52–H2165.39. A water molecule located deep in the binding pocket (Wat 3.1) forms a hydrogen bond with the carbonyl oxygen of 14 (Figure 6). The sulfone-substituted pyridine ring occupies the same region as the benzoxazole of suvorexant and, as with HTL6641 substitution in this region, results in the ring sitting higher in the orthosteric pocket. Finally, the pyridine ring makes hydrophobic interactions with A1022.60, S1032.61 (T1112.61 in OX2), V1062.64, I1223.28, P1233.29, and Q1263.32.
OX1–ACT-462206
The X-ray structure of ACT-462206 (15) in complex with OX1 (Figure 4c) is unusual in that two conformations of the para-methoxyphenyl substituent off the sulfonamide linker are observed, which we term “collapsed” and “extended”. The collapsed, or intramolecular π-stacked horseshoe conformation, closely follows the shape of the portion of lemborexant minus the fluorophenyl group, whereas the extended conformation follows the shape of lemborexant minus the dimethylpyrimidine substituent. Both positive (and negative) peaks exist in the Fo–Fc electron density maps for the para-methoxyphenyl substituent in both the collapsed and extended conformations. Unfortunately, the conformation of this substituent could not be resolved by further employing atomic occupancy refinement. We conclude that the maps obtained reflect the para-methoxyphenyl substituent moving between both conformations and introduce a considerable degree of uncertainty for the precise position of this moiety. The 3,5-dimethylphenyl amide portion of 15 sits in a similar region to the suvorexant benzoxazole in a hydrophobic pocket defined by A1022.60, S1032.61, V1062.64, I1223.28, P1233.29, and Q1263.32, although the dimethyl substitution pushes the ring system slightly higher in the pocket resulting in an additional contact with C10245.50. The amide linker to the pyrrolidine ring has the carbonyl oxygen facing the extracellular surface, making a hydrogen bond to water (Wat 2.1) located at a similar location to one of the water molecules that suvorexant makes an interaction with in the co-crystal structure (Figure 6). The pyrrolidine ring sits adjacent to A1273.33 (T in OX2) and in direct contact with Q1794.60 and F2195.42, under the ECL2-stabilizing salt bridges defined by E1102.68–R3307.28, D20345.51–R3226.59, and E20445.52–H3165.39. One oxygen from the pyrrolidine sulfonamide linker resides in a hydrogen bonding distance to N3186.55. The para-methoxyphenyl ring density is too poorly defined to be confident of describing specific interactions in detail; however, we postulate that it is likely to be located in the vicinity of S1032.61 (T1112.61 in OX2), consistent with its published role as a determinant of OX1/OX2 selectivity (see later).45
OX1–Compound 16
Diazaspirodecane sulfonamide 16 was synthesized as a benchmark from the patent literature,46 as part of a strategy to provide a breadth of orexin chemotypes for structural biology evaluation and subsequently enable orexin antagonist SBDD approaches from diverse scaffolds. In our radioligand binding assays,4416 is a moderate affinity antagonist (OX1 pKi 7.1, OX2 pKi 7.8) but had sufficient affinity to allow elucidation of its binding mode in a complex with OX1 (Figure 4d). The antagonist sits in the OX1 binding site in an extended conformation that does not display any intramolecular π stacking. The benzoxazole portion, in a similar fashion to suvorexant, sits in the hydrophobic pocket defined by A1022.60, S1032.61, V1062.64, I1223.28, P1233.29, and Q1263.32. The 1,8-diazaspiro[4,5]decane core of the molecule sits adjacent to A1273.33 (T in OX2) and in direct contact with Q1794.60, F2195.42, and the ECL2-stabilizing salt bridge, E10445.52–H2165.39. One of the oxygens of the sulfonamide makes a hydrogen bond to N3146.55, in addition to a water-mediated hydrogen bond (Wat 1.1, see the discussion section on water molecules and Figure 6) across to H3447.39. This water additionally forms a hydrogen bond to another water positioned above it, which is involved in three hydrogen bonds, one back to the carbonyl oxygen of N3186.55 and the other two via the lone pairs from the oxygen to K3216.58 and R3226.59. The phenyl ring joined to the sulfonamide linker is sitting deep in the binding pocket, making edge–face π stacking with F2195.42 and is seen along with hydrophobic contacts to V1303.36, Y3116.48, and I3146.51.
OX1–SB-334867
The X-ray complex of the 1-SORA SB-334867 (11) in complex with OX1 is remarkable in that two antiparallel π-stacking orientations of the ligand can be clearly seen in the binding site, with ligand I sitting adjacent to helices 1, 2, 3, 4, and 5 and ligand II sitting adjacent to helices 5, 6, 7, and 1 (Figure 5a). Ligand I positions the 1,5-naphthyridine ring in a similar region to the benzoxazole of suvorexant, in a hydrophobic pocket defined by A1022.60, S1032.61, V1062.64, I1223.28, P1233.29, and Q1263.32, making an edge–face π-stacking interaction with W11223.50. The urea linker spans the region between the two heteroaryl systems, making van der Waals contacts with Q1263.32, with the 2-methyl-1,3-benzoxazole group forcing F2195.42 to rotate outward from the trans χ1 dihedral, seen in most OX1 structures, to a gauche +ve χ1 dihedral. The urea oxygen forms a hydrogen bond with water molecule Wat 2.2, stabilized by E20445.52 (Figure 6). The benzoxazole ring system makes direct contacts with V1303.36, M1764.57, Q1794.60, F2195.42, S2235.46, and most significantly A1273.33 (T1353.33 in OX2) from where the OX1 selectivity is derived, in addition to contacts with the ECL2-stabilizing salt bridge, E20445.52–H2165.39. The substituted benzoxazole ring system also π-stacks with the 1,5-naphthyridine ring system of ligand II. The 1,5-naphthyridine ring of ligand II forms π-stacking interactions with Y3116.48, hydrophobic contacts with F2205.43 and Y2245.47, and contacts with S2235.46, S3156.52, and N3186.55 and forms a water-mediated hydrogen bond with N3186.55 via water molecule Wat 4 (Figure 6). The urea linker in ligand II makes contact with I3146.51, while the benzoxazole π-stacks with H3447.39, makes contact with Y3407.35 and V1062.64, and makes a hydrogen bond from the lone pair oxygen to Q1263.32. Additionally, the benzoxazole ring system from ligand II also π-stacks with the 1,5-naphthyridine ring system of ligand I.
Figure 5.
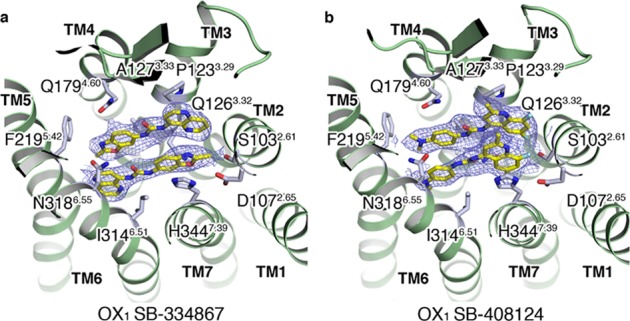
(a, b) Extracellular views of the OX1 StaR structures in complex with SB-334867 (11) and SB-408124 (12), respectively. Ligands shown in a stick representation with carbon, nitrogen, oxygen, and fluorine atoms colored yellow, blue, red, and cyan, respectively. Ligand 2Fo–Fc electron density maps in blue mesh and contoured at 1.0σ.
OX1–SB-408124
Intrigued by the observed binding mode of SB-334867, we sought to also investigate the binding mode in OX1 of a related selective urea antagonist, SB-408124 (12, Figure 5b). In a similar fashion to the OX1 SB-334867 complex, two ligands can also be seen in the complex between OX1 and SB-408124; however, in this case, the ligands are not arranged in an antiparallel π-stacking orientation but are instead parallel to one another, offset by ∼1 to 2 Å. The substituted quinoline rings are situated between helices 2, 3, and 7, parallel to the direction of the helices, with the 2-methyl substituents facing the extracellular surface. The quinoline rings of ligand I and ligand II are displayed in an offset π-stacking arrangement stabilized by offset π stacking with H3447.39 and edge–face π-stacking with W11223.50 in a hydrophobic pocket defined by A1022.60, S1032.61, V1062.64, I1223.29, P1233.28, Q1793.32, and Y3487.43. The urea linkers are slightly offset sitting in opposing directions to one another, with the ligand copy that is closer to TMs 6 and 7 having its carbonyl facing the intracellular side and the ligand adjacent to TM 3 having its urea carbonyl facing an extracellular direction. The urea of ligand II forms a water-mediated hydrogen bond across to N3186.55 via water molecule Wat 1.2 (Figure 6). The dimethylamino-substituted phenyl rings of the two ligands are again slightly offset and situated in a region defined by edge–face π-stacking with Y3116.48, hydrophobic contacts with A1273.33, V1313.37, M1764.57, F2205.43, and F2245.47, and contacts with T2235.46, S3156.52, and N3186.55.
Implications of the Receptor−Ligand Structures for Orexin Antagonist Drug Design
The 14 unique ligand-bound OX1 and OX2 co-crystal X-ray structures described in this paper comprise one of the most comprehensive structural sets currently available for a GPCR with a diverse range of chemotypes. Furthermore, they enable a protein structure-based view of how different ligands bind to their cognate receptors. The set is complemented by X-ray structures of four unique OX1 and OX2–ligand complex structures reported previously,38−40 including three complexes that were solved independently and are referenced accordingly in the current manuscript. Analysis of this data set allows features of ligand recognition to be elucidated, with implications for the design of selective orexin ligands, potentially extending to other GPCRs. A number of key findings are apparent including the recognition of limitations in using pharmacophore-based similarity principles for modeling receptor–ligand complexes of different chemotypes, the importance of lipophilic hotspots as drivers of GPCR druggability and ligand binding, and the variable role of direct polar receptor–ligand interactions. Furthermore, the structural set assembled and presented here highlights the key role of water molecules as determinants of GPCR–ligand binding and selectivity and demonstrate how subtle differences in local binding site electrostatics can be a determinant of selectivity between closely related receptors. These key points are expanded and discussed in depth in the following sections.
Lipophilic Hotspots as a Critical Determinant of Orexin Receptor–Ligand Binding
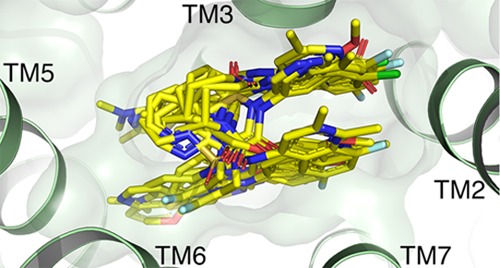
Lipophilic hotspots have been previously shown to be a key component of GPCR ligand binding and in characterizing a druggable binding site.47Figure 7 shows the integral role of these hotspots in the binding of orexin receptor ligands. Druggability assessment of the pseudo-apo antagonist binding pockets of orexin receptor structures using the GRID molecular interaction field (MIF),48,49 an analysis of energetically favorable regions for ligand interactions, together with WaterFLAP50,51 generation of complete water networks and relative energetic scoring shows that hydrophobic hotspots and the displacement of high-energy (relative to bulk solvent, “unhappy”) water molecules appear to drive ligand binding. Figure 7 shows how, despite their variability in binding modes and resultant structural ligand interaction patterns and water-mediated orexin receptor–ligand interactions (Figure 6), orexin receptor ligands target similarly located hydrophobic hotspots that drive ligand affinity. The co-crystal structures presented in this study add to the increasing wealth of GPCR ligand structures, and when analyzed in detail, illustrate how lipophilic interactions are key components of binding that must be considered alongside polar interactions.
Figure 7.
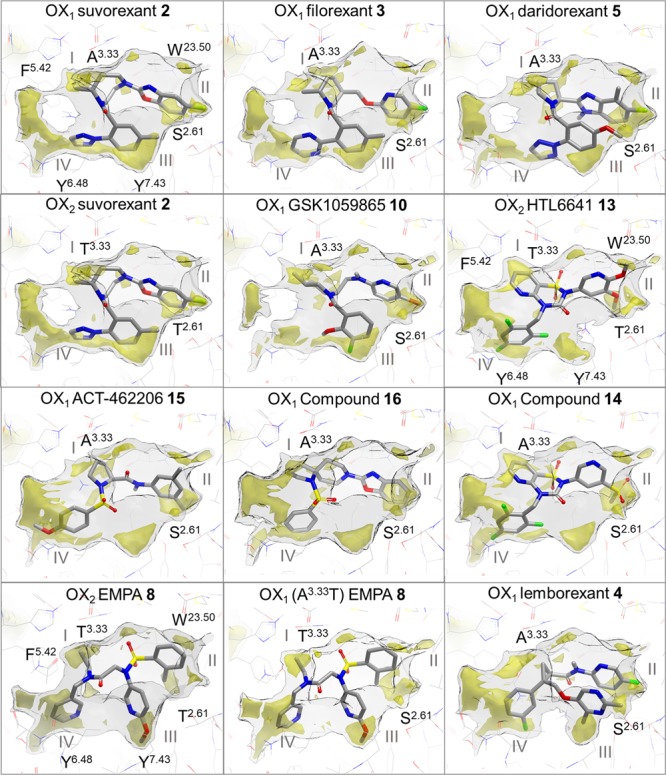
Ligand binding modes in conserved lipophilic hotspots in orexin receptor crystal structures. Comparison of binding modes of suvorexant (2), filorexant (3), daridorexant (5), GSK1059865 (10), pyridothiadiazinone compound 14, ACT-462206 (15), diazaspirodecane compound 16, lemborexant (4) in OX1, suvorexant (2), EMPA (8), HTL6641 (13) in OX2, and EMPA (8) in OX1 (A1273.33T mutant). GRID maps are contoured (transparent solid) and colored in the following manner: C1 is the probe (lipophilic) in yellow at −2.8 kcal/mol, and the CH3 methyl group probe is in gray at 1 kcal/mol, which defines the pocket surface in terms of how close a ligand carbon atom can reside. Positions of residues S1032.61, W11223.50, A1273.33, F2195.42, Y3116.48, and Y3487.43 in OX1 and of T1112.61, W12023.50, T1353.33, F2275.42, Y3176.48, and Y3547.43 in OX2 are provided as reference. Whereas direct polar interactions between the chemically diverse ligands and the orexin receptor binding site are limited and not conserved (see Figure 6), all ligands target at least three of the four lipophilic hotspot regions I–IV located between A/T3.33 and F5.42 (I), S/T2.61 and W23.50 (II), S/T2.61, Y6.48 and Y7.43 (III), and F5.42 and Y6.48 (IV).
Water-Mediated Polar Interaction Networks Provide a Basis for the Observed Diversity in Orexin Receptor–Ligand Binding Modes
All the ligands in these structures occupy the same general space within the interhelical cavity, with many distinct residues in contact (Figures 6 and 7). One feature readily apparent from the analyses in this study is the predominance of hydrophobic interactions between the ligands and the protein with very few direct polar interactions, albeit some being mediated by water molecules. This is also evident from the number of distinct hydrophobic patches in the ligand binding cavity (Figure 7), which are repeatedly utilized by the ligands presented in this study. It is thus not surprising that orexin antagonists are typically hydrophobic, with concomitant implications for physical properties including aqueous solubility, providing significant challenges for drug development. Another consequence is that the overlap of functional groups when comparing ligands is far less clear when considering the contribution of polar interactions to binding (Figures 6 and 7), partly due to the potential for diverse groups within each molecule to contribute to hydrophobic interactions that can be accessed from markedly different vectors. This is in contrast to a more limited subset of groups, which form a specific polar interaction, which then has a clear directional component. As a result, predicting the specific binding mode of each ligand is extremely difficult when matching polar pharmacophore features across chemical series as the hydrophobic interactions seem to dominate binding. In addition, the specific involvement of waters in hydrogen bonding between the protein(s) and each ligand class confounds a sensible comparison of the series. Overall, the large set of high-resolution orexin receptor–ligand structures presented here shows, for the first time, the degree of variability of water-mediated H-bond networks possible in GPCR–ligand interactions.
By way of illustration, the structures of EMPA with OX1 harboring the A1273.33T mutation and with OX2 (Figure 2 c-d) show the importance of interstitial waters in orexin–ligand binding and the consequent danger of ignoring them in any modeling studies, such as pharmacophore matching and ligand docking. The relatively polar ligand EMPA makes no direct H bonds to the receptor, and instead H bonding is mediated through two interstitial waters. Binding with direct H bonds to the receptor is sterically possible, and a plausible binding mode of EMPA based on a suvorexant X-ray structure has been proposed showing multiple direct H bonds between ligand and receptor.38 However, both the conformation and orientation of EMPA previously proposed are not observed experimentally, highlighting the key role of waters as a component of binding. In accordance with the previous published computational study, we have found that removing solvent molecules before docking the crystallized ligand back into its own structure (self-docking) does not identify the correct pose (observed experimentally) from the best scoring docked poses.
Understanding Orexin Ligand Selectivity
It is often the case for GPCRs with multiple subtypes that obtaining selectivity between receptor subtypes can be difficult due to the highly conserved nature of residues within the binding site. The only two residues that differ within the putative orthosteric binding sites of OX1 and OX2 in the interhelical cavity are S1032.61 (OX1) → T1112.61 (OX2) and A1273.33 (OX1) → T1353.33 (OX2) (Figure 6). The relatively subtle differences in size and electrostatics of these residues make targeting these regions for selectivity extremely difficult to rationalize without crystal structures of the two binding sites being available. Additionally, the overall RMSD of the two structures is only 0.5 Å2, and the 55 main chain atoms from the residues within 5 Å of suvorexant for both structures when aligned exhibit an RMSD of 0.3 Å2, highlighting the very similar shape of the two proteins and their corresponding binding sites. This similar shape, in addition to the lack of consistent direct protein/ligand interactions to the ligands and the reliance on targeting hydrophobic hotspots within the site, make this system challenging to pursue computationally. However, the basic principles of obtaining selectivity by optimizing interactions with the target of interest or by introducing unfavorable interactions at an off-target protein still apply, and both these possibilities can be deduced from the examples of OX1 and OX2-selective compounds described in this manuscript (Table 1). The examples described below involve subtle differences in direct polar interactions with the receptor and differential effects on water networks, including stabilization of favorable water-mediated H-bond interactions and the displacement or trapping of energetically unfavorable water molecules (Figures 6–8).
Displacement of high-energy water molecules that reside in lipophilic hotspots is likely to be a major component of ligand binding energy (Figure 8). It is also important to consider the perturbation of the energy of the remaining non-displaced waters and stabilization of the resulting water network, which may also play a role in ligand potency, selectivity, and kinetics. The A1273.33/T1353.33 difference (above) appears to account for the approximately 1000-fold OX2 selectivity of EMPA in a different way to the preceding examples. Somewhat surprisingly, EMPA is selective for the OX2 receptor with the larger residue (threonine) at this position, despite being sterically less demanding, and the X-ray structure demonstrates no direct interaction between the threonine hydroxyl group and the ligand, ruling out an H-bonding explanation. To investigate the interesting and surprisingly high selectivity of EMPA for OX2, a computational study of the water network energetics was performed using WaterFLAP.50Figure 8c, d shows a comparison of the binding site surfaces of OX1 and OX2. WaterFLAP calculations on the complexes are shown with waters as spheres. The water stabilized by the two pyridine groups is clearly seen (large blue sphere). In Figure 8d, the surfaces of both proteins with A1273.33 and T1273.33 are shown, and it can be clearly seen that a very unhappy water is trapped in the larger OX1 binding site; in the smaller OX2 binding site, this water would be clearly displaced, with EMPA making close contact to the surface. The trapped water explains the selectivity of EMPA. To confirm this result, WaterMap52,53 calculations were also run using a very different molecular dynamics approach, and the water was found to be very unstable. WaterMap calculations are less robust in complexes with trapped waters as only a first-order entropy calculation is used, so the more robust WaterFLAP results are shown in Figure 8. In summary, the computational analyses indicate that A1273.33 in OX1 creates a slightly larger site, which, if EMPA is bound, would trap a high-energy water into the highly lipophilic region II between A1273.33 and F2195.42 (Figure 7), a scenario that is energetically highly unfavorable (Figure 8). The gain of selectivity by trapping this putative water molecule in a larger counter-target binding site is an observation that may be useful in future ligand design, providing a strategy to gain selectivity without needing to make a higher-molecular-weight ligand, which may be undesirable, particularly for a CNS drug.
Compound 14 appears to achieve moderate OX1 selectivity by introduction of a detrimental interaction that would be present when complexed with OX2. The sulfone substituent on the pyridine ring has its lone pairs of electrons from the oxygens pointing toward the vacant region adjacent to S1032.61 in lipophilic pocket II (Figure 7). The region is vacant because the hydroxyl side chain from S1032.61 is found in a trans χ1 conformation, forming a hydrogen bond across i + 4 adjacent to D1072.65. T1112.61 in all OX2 structures to date has been found in a standard helical gauche +ve χ1 conformation, making an intramolecular hydrogen bond to the i – 4 residue C1072.57, which would place the lone pairs of the threonine hydroxyl oxygens in direct detrimental contact with the lone pairs from the sulfone oxygens of 14. We hypothesize that interactions at the T1112.61 residue (in OX2) are also responsible for enhanced OX2 selectivity in some cases due to introduction of a new H-bonding contact to certain ligands and/or adjusting the size and properties of the lipophilic pocket II between T1112.61, W12023.50, and P1313.29 (Figure 7).
Within the selection of X-ray structures discussed in this manuscript, we observe that selectivity can also be rationalized by optimizing favorable interactions at the target of interest. The OX1-selective compound GSK1059865 (10) emerged from what can be judged from the patent literature to be several years of research centered around a piperidine or piperazine core.54−56 The extensive work disclosed by GSK resulted in excellent OX1 selectivity that can now be post-rationalized in the context of structural information. The selectivity of the GSK1059865 we propose is due in part to excellent surface complementarity of the ligand with lipophilic pocket I between A1273.33 and F2195.42 in OX1, which is defined by T1353.33 and F2275.42 in OX2 (Figure 7). In this region, the T1353.33 in OX2 makes an intramolecular hydrogen bond to the i – 4 residue P1313.29, causing the γ-carbon to occupy additional space within the binding site. The A1273.33 residue in OX1 does not occupy the same volume as T1353.33 in OX2, and thus the central piperidine ring of GSK1059865 is able to sit within the hydrophobic cleft vacated by the T1353.33 γ-carbon giving rise to OX1 selectivity. This method of achieving OX1 selectivity by occupying the hydrophobic cleft adjacent to A1273.33 in OX1 also rationalizes the profile of a number of other scaffolds including those ranging from spiro-pyrrolidines57 to azabicyclo[4.1.0]heptanes.58
Pharmacophore- and Shape-Based Similarity Being Inaccurate Predictors of Orexin Receptor–Ligand Binding Modes
Another observation from the diverse set of orexin ligand–receptor X-ray structures presented herein relates to ligand-based pharmacophore modeling and design, often used for GPCRs due to the difficulty in obtaining X-ray structures. The issue is clearly illustrated in Figures 6–8 where the actual overlay of the ligands from their bound position in the co-crystal receptor structures demonstrates very little concordance of scaffolds and commonly used H-bond pharmacophoric points on the ligands. Overall, pharmacophoric models derived for orexin receptors would be unlikely to find the actual overlay observed experimentally and are thus likely to be misleading, particularly in informing any SAR learnings from one series to another. As discussed below, there are in fact direct lipophilic interactions of the ligands with at least three of the four lipophilic hotspot regions of the receptor binding site, and these are common between different ligands (see Figure 7). An emphasis on these hydrophobic interactions versus polar interactions could thus provide better models and docking results. However, even if in addition to the four common polar interaction types (H-bond acceptor, H-bond donor, basic, acidic) lipophilic hotspots are included in an analysis, they would likely only have a 1 in 3 to 1 in 5 (depending on the total number of pharmacophore types present in the ligands, 3 being the likely minimum of H-bond donors, acceptors, and lipophilic hotspots) influence on the model using standard pharmacophore identification methods (unless specially weighted, which the data indicates would be preferable). The inclusion of potential water-mediated interactions in pharmacophore modeling does not often occur, further complicating interpretation of a polar interaction-based model. In summary, a focus on the common lipophilic/hydrophobic interactions and consideration of water networks to aid the overlap of H-bonding interactions could yield improved results in defining ligand-based pharmacophores for the orexin system.
Unusual Binding Modes of Urea-Containing Orexin Ligands
The binding modes of ureas SB-334867 (11) and SB-408124 (12) are striking and unexpected (Figures 5 and 6). The relatively small and flat ligands occupy the large and hydrophobic OX1 binding pocket by binding of two copies of the ligand stacking against one another per receptor orthosteric site. The observation is consistent with our pharmacological characterization of the binding of radiolabeled 11 to WT OX1 and OX1 StaR proteins where saturation binding studies show a hill slope of ∼2 and indicate positive cooperation for ligand binding (Supporting Information, Figure S1). The overall ternary complexes contain features reminiscent of other larger ligands (described herein) that form internal aromatic stacking interactions by hydrophobic collapse conformations. These fascinating observations perhaps help to rationalize the steep and unpredictable SAR of this series of compounds and prompt the question of if multiple copies of ligands bind to other GPCR targets? Although unusual, the binding of more than one copy of a ligand to fulfill the binding interactions available in a protein binding site is not unprecedented.59,60 For example, Stornaiuolo et al. reported the binding of multiple ligand copies to acetylcholine-binding protein via similar assemblies of a π–π stacking of ligands.61 The authors note that, thanks to the plasticity of its ligand binding site, acetylcholine-binding protein can accommodate the formation of aromatic stacks of different sizes by simple loop repositioning and minimal adjustment of the interactions. The selectivity afforded by the urea ligands for OX1 is likely due to one of the ligand copies in the ligand dimer interacting with the A1273.33 residue in OX1. In the OX1 structures of 11 and 12, the benzoxazole fragment and benzene ring, respectively, sit in direct contact with A1273.33. The additional γ-carbon of the analogous T1353.33 residue in OX2 would occlude the ligands in question from binding, giving rise to the observed OX1 selectivity. Interestingly, these compounds also cause F2275.42 in OX1 to rotate outward from the trans χ1 dihedral, seen in most structures, to a gauche +ve χ1 dihedral, and it is not known if this also contributes to selectivity although no OX2 structures with F2275.42 rotated have been observed to date. Lastly, it is worth highlighting that despite the two urea ligands being similar in chemical structure, the ligand pairs do not stack in the same orientation when the X-ray structures are compared (Figure 5), further complicating the interpretation of these findings and the SAR of the series.
Conclusions
The structures presented in this report demonstrate a diverse range of binding modes for ligands in the orthosteric site of the two orexin receptors and demonstrate how they achieve selectivity in a variety of ways despite the receptors being very similar in their binding sites. The co-structures highlight the critical importance of lipophilic hotspots and also interactions with water molecules in controlling binding and selectivity for these peptidergic GPCRs. An observation is that selectivity can be driven by differences in ligand interactions with water molecules between the target and the counter target. Lipophilic hotspots and water molecules are often ignored or underestimated in pharmacophore-based approaches to ligand docking, which are then prone to gross inaccuracies, emphasizing the value of obtaining multiple experimental structures. Overall, the data presented suggests learnings that can be applied to other GPCR targets, and we would expect that these findings for the orexin system are of general relevance to GPCR drug discovery.
Experimental Section
Chemistry
Compounds 2–5, 8, and 10–16 were obtained from commercial sources or synthesized according to reported procedures, as detailed in Supporting Information, Table S1. Compounds were assessed for purity by LCMS and were ≥95%. LCMS data with electrospray ionization were generated under the following conditions: Instrument: Agilent 1260 Infinity LC with diode array detector, Agilent 6120B single quadrupole MS with API-ES source; Column: Phenomenex Gemini-NX C-18, 3 μm, 2.0 × 30 mm; Gradient [time (min)/solvent B in A (%)]: 0.00/2, 0.10/2, 8.40/95, 9.40/95, 9.50/2, 10.00/2; Solvents: solvent A = 2.5 L H2O + 2.5 mL 28% aqueous ammonia solution; solvent B = 2.5 L MeCN +129 mL H2O + 2.7 mL 28% aqueous ammonia solution). [3H]-EMPA was purchased from RC Tritec, Teufen, Switzerland.
StaR Generation, Cell Culture, and Thermostability Measurement
Full-length human WT OX2 (1–444) and human OX1 A127T (1–425) receptors were used as templates for the generation of conformationally thermostabilized receptors using a mutagenesis approach previously described.62 Mutants were analyzed for thermostability in the presence of the radioligand [3H]-EMPA. The OX2 StaR comprised 12 thermostabilizing mutations (E54A, Y91L, D100A, V142A, R170L, L206A, Y219A, M233A, A242L, L310V, L318A, T347A). The OX1 StaR comprised eight thermostabilizing mutations (E46A, I85L, V95A, R162L, L198A, Y211A, L304V, C339A). The final crystallography constructs are detailed in Supporting Information, Table S3.
HEK293T cells were cultured in DMEM supplemented with 10% (v/v) fetal bovine serum (FBS). Cells were transfected with template or mutant receptor constructs using GeneJuice (Merck Millipore) according to the manufacturer’s instructions and harvested after 48 h.
Transiently transfected HEK293T cells were harvested, and cell pellets were solubilized by incubation in 200 mM citric Acid/100 mM sodium phosphate pH 6, 150 mM NaCl, 1% (w/v) n-dodecyl-β-d-maltopyranoside (DDM) assay buffer supplemented with cOmplete protease inhibitor cocktail tablet (Roche) for 1 h rotating at 4 °C. Lysates were clarified by centrifugation at 16,000g for 15 min at 4 °C and incubated with 20 nM [3H]-EMPA for 1 h at 4 °C. Receptor thermostability was measured by incubation at varying temperatures for 30 min followed by separation of unbound radioligands by gel filtration. Levels of ligand-bound receptors were determined using a liquid scintillation counter, with thermal stability (Tm) defined as the temperature at which 50% ligand binding was retained.
Expression, Membrane Preparation, and Protein Purification of Crystallography Constructs
The truncated OX1–StaR(27–381) non-fusion construct with the ICL3 deletion between residues 254–285 and carrying the glycosylation and palmitoylation mutations (see main text and Supporting Information, Table S3) was expressed with a C-terminal decahistidine tag in Spodoptera frugiperda 21 cells using an ESF 921 medium (Expression Systems) supplemented with 10% (v/v) fetal bovine serum (Sigma-Aldrich) and 1% (v/v) penicillin/streptomycin (PAA Laboratories) with the Bac-to-Bac expression system (Invitrogen). Cells were infected at a density of 2 to 3 × 106 cells/mL with baculovirus at an approximate multiplicity of infection of 1. Cultures were grown at 27 °C with constant shaking and harvested by centrifugation 48 h post-infection.
The truncated OX2–StaR(27–389) with the Pyrococcus abyssi glycogen synthase fusion between residues 255–293 of ICL3 and carrying the glycosylation and palmitoylation mutations (see main text and Supporting Information, Table S3) was expressed with a C-terminal decahistidine tag in S. frugiperda 21 cells using an ESF 921 medium (Expression Systems) supplemented with 10% (v/v) fetal bovine serum (Sigma-Aldrich) and 1% (v/v) penicillin/streptomycin (PAA Laboratories) with the Bac-to-Bac expression system (Invitrogen). Cells were infected at a density of 2 to 3 × 106 cells/mL with baculovirus at an approximate multiplicity of infection of 1. Cultures were grown at 27 °C with constant shaking and harvested by centrifugation 48 h post-infection.
All subsequent steps were carried out at 4 °C unless otherwise stated. Membranes were prepared by resuspension of cells in PBS supplemented with cOmplete protease inhibitor cocktail tablets (Roche), 10 mM magnesium chloride, and 5 μg/mL DNaseI (Roche) followed by disruption using a microfluidizer at 60,000 PSI (M-110L Pneumatic, Microfluidics). Membranes were collected by ultracentrifugation at 204,700 g, resuspended in 50 mM Hepes–NaOH pH 7.5 and 200 mM NaCl with cOmplete protease inhibitor cocktail tablets (Roche), and stored at −80 °C until use.
To purify the OX1–StaR receptor, membranes were thawed at rt, incubated with 5 μM of any given ligand (e.g., suvorexant), and solubilized with 1.5% (w/v) n-decyl-β-d-maltopyranoside (DM) with an additional 0.5 mM phenylmethylsulfonyl fluoride (Sigma-Aldrich) and 50 mM NaCl for 90 min. The insoluble material was removed by ultracentrifugation at 204,700 g for 45 min with 7.5 mM imidazole subsequently directly added to the clarified solubilized material. The receptors were then immobilized by loading the clarified solubilized material onto a 5.0 mL prepacked NiNTA cartridge (Qiagen) at a flow rate of 0.25 mL/min. The resin was then washed with 40 column volumes of 50 mM Hepes–NaOH pH 7.5, 250 mM NaCl, 0.15% (w/v) n-decyl-β-d-maltopyranoside, and 30 mM imidazole followed by 35 column volumes of 50 mM Hepes–NaOH pH 7.5, 200 mM NaCl, and 0.3% (w/v) n-octyl-β-d-thioglucopyranoside (OTG) for complete detergent exchange. Elution was performed over three column volumes with 50 mM Hepes–NaOH pH 7.5, 200 mM NaCl, 500 mM imidazole, and 0.3% (w/v) n-octyl-β-d-thioglucopyranoside (OTG). All wash steps and the elution were run at a flow rate of 0.75 mL/min with 5 μM of any given ligand (e.g., suvorexant) added to all buffers at a temperature of 12 °C. The protein was then concentrated using an Amicon Ultra-15 centrifugal concentrator (MerckMillipore), MWCO 50 kDa, to a final volume of 650 μL before ultracentrifugation at 100,000 rpm for 20 min at 12 °C. The protein was then subjected to preparative size exclusion chromatography in 50 mM Hepes–NaOH pH 7.5, 200 mM NaCl, 0.3% (w/v) n-octyl-β-d-thioglucopyranoside and 5 μM of ligand on a Superdex 200 10/300 Increase column (GE Healthcare) at 12 °C. Receptor purity was analyzed by SDS-PAGE and LC–MS, and receptor monodispersity was assayed by analytical SEC. Fractions containing the pure monomeric receptor were concentrated to ∼5 mg/mL in an Amicon 4 regenerated cellulose centrifugal concentrator (MerckMillipore) at 12 °C. The protein concentration was determined using the receptor’s calculated extinction coefficient at 280 nm (ε280,calc = 83,100 M–1 cm–1) and confirmed by quantitative amino acid analysis. Prior to crystallization, the receptor was incubated with 0.5 mM (final concentration) of 1-hexadecanoyl-2-(9Z-octadecenoyl)-sn-glycero-3-phosphoglycerol (POPG) for 30 min followed by a final ultracentrifugation step at 50,000 rpm for 30 min at 12 °C.
To purify the OX2–StaR receptor, membranes were thawed at rt and incubated with 5 μM suvorexant (2), EMPA (8), or HTL6641 (13) for 30 min prior to solubilization followed by an additional 30 min of incubation with 2 mg/mL iodoacetamide. Membranes were solubilized with buffer containing 50 mM Hepes–NaOH pH 7.5, 200 mM NaCl, supplemented cOmplete EDTA-free protease inhibitor cocktail tablets (Roche), and 1.5% (w/v) n-decyl-β-d-maltopyranoside for 1 h at 4 °C. The insoluble material was removed by ultracentrifugation at 204,700 g for 1 h at 4 °C, and the receptors were then immobilized by batch binding to 5.0 mL of NiNTA resin (Qiagen). The resin was then packed into an Omnifit column (Kinesis) and washed with two column volumes of 20 mM Hepes–NaOH pH 7.5, 200 mM NaCl, 0.15% (w/v) n-decyl-β-d-maltopyranoside, and 5 μM suvorexant (2), EMPA (8), or HTL6641 (13) and then with four column volumes with the same buffer supplemented with 20 mM imidazole before the bound material was eluted in buffer containing 280 mM imidazole. All affinity chromatography steps were run at a flow rate of 0.75 mL/min. The eluted protein was then concentrated to ∼15 mg/mL using an Amicon Ultra-15 centrifugal concentrator (MerckMillipore), MWCO 50 kDa. Prior to preparative size exclusion chromatography, the protein was spun by ultracentrifugation at 40,000 rpm for 1 h at 4 °C to remove any aggregated material. The preparative size exclusion chromatography step was subsequently run in 50 mM Hepes–NaOH pH 7.5, 200 mM NaCl, 0.15% (w/v) n-decyl-β-d-maltopyranoside, and 5 μM suvorexant (2), EMPA (8), or HTL6641 (13) on a Superdex 200 10/300 Increase column (GE Healthcare). Receptor purity was analyzed by SDS-PAGE and LC–MS, and receptor monodispersity was assayed by analytical SEC. Fractions containing the pure monomeric receptor were concentrated to ∼30 mg/mL in a Vivaspin 500 centrifugal concentrator (Sartorius). Protein concentration was determined using the receptor’s calculated extinction coefficient at 280 nm (ε280,calc = 114,820 M–1 cm–1) and confirmed by quantitative amino acid analysis.
Crystallization of the OX1–StaR
OX1–StaR was crystallized using the vapor diffusion method at 10 °C. The concentrated protein at ∼5 mg/mL (which had been preincubated with 0.5 mM POPG) was dispensed onto 96-well sitting drop crystallization plates from Swissci (Molecular Dimensions) using a Mosquito from TTPLabtech and mixed with the mother liquor at a 1:1 ratio, resulting in final drop sizes of 100 nL. OX1–StaR crystals with a thick plate-like morphology grew to over 500 μm in size within 7 days in 100 mM trisodium citrate buffer at a pH range of 3.0–6.5, 50 mM sodium chloride, 50 mM lithium sulfate, and 15–34% (v/v) poly(ethylene glycol) 400 plus 20 μM of ligand. Single crystals were mounted for data collection and cryo-cooled in liquid nitrogen with cryoprotection performed at the pH of the trisodium citrate buffer they were grown in, plus 50 mM sodium chloride, 50 mM lithium sulfate, 32% (v/v) poly(ethylene glycol) 400, 0.5% (w/v) n-octyl-β-d-thioglucopyranoside, and 20 μM of ligand. The OX1–StaR crystals grown at low pH (3.3–5.5) belong to the monoclinic space group P21, whereas OX1–StaR crystals grown at higher pH (6.0–6.5) belong to the monoclinic space group I2. Complete datasets for each OX1–StaR co-structure were collected from two crystals on average.
Crystallization of the OX2–StaR
OX2–StaR was crystallized in lipidic cubic phase at 20 °C. The protein was concentrated to ∼30 mg/mL and mixed with monoolein (Nu-Check) supplemented with 10% (w/w) cholesterol (Sigma Aldrich) and 5 μM suvorexant (2), EMPA (8), or HTL6641 (13) using the twin-syringe method.63 The final protein/lipid ratio was 40:60 (w/w). Boli (30 nL) were dispensed on 96-well glass bases and overlaid with 750 nL of precipitant solution using a Mosquito LCP from TTPLabtech. Plate-shaped crystals of OX2–StaR 100 μm-thick were grown in 100 mM N-(2-acetamido)iminodiacetic acid (ADA) at a pH range of 6.0–7.0, 150–300 mM ammonium nitrate, and 28–43% (v/v) poly(ethylene glycol) 400 for suvorexant; in 100 mM trisodium citrate buffer at a pH range of 5.0–6.0, 150–300 mM sodium chloride, and 28–43% (v/v) poly(ethylene glycol) 400 for EMPA; and finally in 100 mM trisodium citrate buffer at a pH range of 5.0–6.0, 150–300 mM lithium nitrate or 150–300 mM potassium nitrate, and 28–43% (v/v) poly(ethylene glycol) 400 for HTL6641. Single crystals were mounted for data collection and cryo-cooled in liquid nitrogen without the addition of further cryoprotectants. Diffraction data from five crystals belonging to the C-centered orthorhombic space group C2221 was required to form a complete dataset for the OX2–EMPA co-structure at 2.74 Å. Diffraction data from one crystal belonging to the triclinic space group P1was required to form a complete dataset for the OX2–suvorexant co-structure at 2.76 Å. Diffraction data from eight crystals belonging to the C-centered orthorhombic space group C2221 was required to form a complete dataset for the OX2–HTL6641 co-structure at 2.61 Å.
Diffraction Data Collection and Processing
X-ray diffraction data for either OX1–StaR or OX2–StaR were measured on a Pilatus3 6 M detector at the Diamond Light Source beamline I24 or an Eiger1 16 M detector at the Swiss Light Source beamline X06SA. Crystals displayed moderately anisotropic diffraction at high resolution. For all diffraction datasets, the detector was set at a maximum resolution of 2.0 Å, the beam was attenuated to >20% of the full flux achievable, and data were collected using a fine slicing protocol (i.e., 0.1–0.2° oscillation per frame) exposing for 0.2 s per degree of oscillation on average. Data from individual crystals were integrated using XDS.64 Data merging and scaling was carried out using the program AIMLESS from the CCP4 suite65,66 and anisotropic correction using STARANISO from autoPROC.67 Data collection statistics are reported in Supporting Information, Table S2.
Structure Solution and Refinement
The structure of OX1–StaR bound to suvorexant was solved by molecular replacement (MR) with the program Phaser(68) using a truncated version of the kappa opioid receptor (PDB ID: 4DJH) as the search model looking for two copies in the A.S.U. All subsequent OX1–StaR and OX2–StaR co-structures utilized the OX1–StaR–suvorexant coordinates as the search model. Manual model building was performed in COOT(69) using sigma-A-weighted 2m|Fo|−|DFc|, m|Fo|−D|Fc| maps together with simulated annealing and simple composite omit maps calculated using Phenix.70 Initial refinement was carried out with REFMAC5(71) using maximum-likelihood restrained refinement in combination with the jelly-body protocol. Further and final stages of refinement were performed with either Phenix.refine(72) implementing positional and individual isotropic B-factor refinement or with refinement in Buster.73 The final refinement statistics are presented in Supporting Information, Table S2.
Generic GPCR Residue Numbering
The generic GPCR residue numbering system74 used throughout this paper is based on the Ballesteros–Weinstein residue numbering system,75 which includes two numbers (X.N), the first (1–7) denotes the transmembrane helix (TM) and the following number indicates the residue position relative to the most conserved amino acid in the helix (which is assigned the number 50). Conserved residue positions in extracellular loop 1 (EL1, between TM2 and TM3) and extracellular loop 2 (EL2, between TM4 and TM5) are defined as W23.50 and C45.50, respectively. For example, 3.33 indicates the residue 17 positions before the most conserved amino acid in class A GPCR TM3 (R3.50). If an amino acid is followed by its residue number, the generic GPCR residue numbering is included as a superscript (e.g., A1273.33).
Acknowledgments
Research reported in this manuscript was supported by the National Institute on Drug Addiction of the National Institutes of Health under award number R01DA039553. We express our gratitude to Giles A. Brown for assistance with radioligand and literature compound synthesis and to Cédric Fiez-Vandal and Robert K. Y. Cheng for assistance with receptor purification and crystallization.
Glossary
Abbreviations Used
- DORA
dual orexin receptor antagonist
- 1-SORA
OX1-selective antagonist
- 2-SORA
OX2-selective antagonist
- rcf
relative centrifugal force
- PMSF
phenylmethanesulfonyl fluoride
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b01787.
Accession Codes
Coordinates and structure factors have been deposited in the Protein Data Bank under the accession codes 6TO7 (2 in complex with OX1), 6TPJ (2 in complex with OX2), 6TP6 (3), 6TOT (4), 6TP3 (5), 6TOD (8 in complex with OX1), 6TPG (8 in complex with OX2), 6TOS (10), 6TQ7 (11), 6TQ9 (12), 6TPN (13), 6TQ6 (14), 6TP4 (15), and 6TQ4 (16). Authors will release the atomic coordinates and experimental data upon article publication.
Author Present Address
† Present address: Evotec (UK) Ltd., 114 Innovation Drive, Milton Park, Milton, Abingdon OX14 4RZ, U.K. (J.C.E.).
Author Present Address
§ Present address: OMass Therapeutics, The Schrödinger Building, Heatley Road, The Oxford Science Park, Oxford OX4 4GE, U.K. (A.J., B.G.T.).
Author Present Address
‡ Present address: MSD Discovery Research Centre, London Business Innovation Centre, 2 Royal College St, London NW1 0NH, U.K. (F.H.M., R.M.).
The authors declare no competing financial interest.
Supplementary Material
References
- Sakurai T.; Amemiya A.; Ishii M.; Matsuzaki I.; Chemelli R. M.; Tanaka H.; Williams S. C.; Richardson J. A.; Kozlowski G. P.; Wilson S.; Arch J. R. S.; Buckingham R. E.; Haynes A. C.; Carr S. A.; Annan R. S.; McNulty D. E.; Liu W.-S.; Terrett J. A.; Elshourbagy N. A.; Bergsma D. J.; Yanagisawa M. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein coupled receptors that regulate feeding behavior. Cell 1998, 92, 573–585. 10.1016/S0092-8674(00)80949-6. [DOI] [PubMed] [Google Scholar]
- de Lecea L.; Kilduff T. S.; Peyron C.; Gao X.-B.; Foye P. E.; Danielson P. E.; Fukuhara C.; Battenberg E. L. F.; Gautvik V. T.; Bartlett F. S. II; Frankel W. N.; van Den Pol A. N.; Bloom F. E.; Gautvik K. M.; Sutcliffe J. G. The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 322–327. 10.1073/pnas.95.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boss C.; Brisbare-Roch C.; Jenck F. Biomedical application of orexin/hypocretin receptor ligands in neuroscience. J. Med. Chem. 2009, 52, 891–903. 10.1021/jm801296d. [DOI] [PubMed] [Google Scholar]
- Tsujino N.; Sakurai T. Orexin/hypocretin: a neuropeptide at the interface of sleep, energy homeostasis, and reward system. Pharmacol. Rev. 2009, 61, 162–176. 10.1124/pr.109.001321. [DOI] [PubMed] [Google Scholar]
- Lin L.; Faraco J.; Li R.; Kadotani H.; Rogers W.; Lin X.; Qiu X.; de Jong P. J.; Nishino S.; Mignot E. The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell 1999, 98, 365–376. 10.1016/S0092-8674(00)81965-0. [DOI] [PubMed] [Google Scholar]
- Chemelli R. M.; Willie J. T.; Sinton C. M.; Elmquist J. K.; Scammell T.; Lee C.; Richardson J. A.; Williams S. C.; Xiong Y.; Kisanuki Y.; Fitch T. E.; Nakazato M.; Hammer R. E.; Saper C. B.; Yanagisawa M. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell 1999, 98, 437–451. 10.1016/s0092-8674(00)81973-x. [DOI] [PubMed] [Google Scholar]
- Beuckmann C.; Sinton C.; Williams S.; Richardson J.; Hammer R.; Sakurai T.; Yanagisawa M. Expression of a poly-glutamine-ataxin-3 transgene in orexin neurons induces narcolepsy-cataplexy in the rat. J. Neurosci. 2004, 24, 4469–4477. 10.1523/JNEUROSCI.5560-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara J.; Beuckmann C. T.; Nambu T.; Willie J. T.; Chemelli R. M.; Sinton C. M.; Sugiyama F.; Yagami K.-i.; Goto K.; Yanagisawa M.; Sakurai T. Genetic ablation of orexin neurons in mice results in narcolepsy, hypophagia, and obesity. Neuron 2001, 30, 345–354. 10.1016/S0896-6273(01)00293-8. [DOI] [PubMed] [Google Scholar]
- Roecker A. J.; Cox C. D.; Coleman P. J. Orexin receptor antagonists: new therapeutic agents for the treatment of insomnia. J. Med. Chem. 2016, 59, 504–530. 10.1021/acs.jmedchem.5b00832. [DOI] [PubMed] [Google Scholar]
- Andrews S. P.; Aves S. J.; Christopher J. A.; Nonoo R. Orexin receptor antagonists: historical perspectives and future opportunities. Curr. Top. Med. Chem. 2016, 16, 3438–3469. 10.2174/1568026616666150929111607. [DOI] [PubMed] [Google Scholar]
- Winrow C. J.; Gotter A. L.; Cox C. D.; Doran S. M.; Tannenbaum P. L.; Breslin M. J.; Garson S. L.; Fox S. V.; Harrell C. M.; Stevens J.; Reiss D. R.; Cui D.; Coleman P. J.; Renger J. J. Promotion of sleep by suvorexant a novel dual orexin receptor antagonist. J. Neurogenet. 2011, 25, 52–61. 10.3109/01677063.2011.566953. [DOI] [PubMed] [Google Scholar]
- Morairty S. R.; Revel F. G.; Malherbe P.; Moreau J.-L.; Valladao D.; Wettstein J. G.; Kilduff T. S.; Borroni E. Dual hypocretin receptor antagonism is more effective for sleep promotion than antagonism of either receptor alone. PLoS One 2012, 7, e39131 10.1371/journal.pone.0039131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willie J. T.; Chemelli R. M.; Sinton C. M.; Tokita S.; Williams S. C.; Kisanuki Y. Y.; Marcus J. N.; Lee C.; Elmquist J. K.; Kohlmeier K. A.; Leonard C. S.; Richardson J. A.; Hammer R. E.; Yanagisawa M. Distinct narcolepsy syndromes in orexin receptor-2 and orexin null mice: molecular genetic dissection of non-REM and REM sleep regulatory processes. Neuron 2003, 38, 715–730. 10.1016/S0896-6273(03)00330-1. [DOI] [PubMed] [Google Scholar]
- Gotter A. L.; Forman M. S.; Harrell C. M.; Stevens J.; Svetnik V.; Yee K. L.; Li X.; Roecker A. J.; Fox S. V.; Tannenbaum P. L.; Garson S. L.; De Lepeleire I.; Calder N.; Rosen L.; Struyk A.; Coleman P. J.; Herring W. J.; Renger J. J.; Winrow C. J. Orexin 2 receptor antagonism is sufficient to promote NREM and REM sleep from mouse to man. Sci. Rep. 2016, 6, 27147. 10.1038/srep27147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugovic C.; Shelton J. E.; Yun S.; Bonaventure P.; Shireman B. T.; Lovenberg T. W. Orexin-1 receptor blockade dysregulates REM sleep in the presence of orexin-2 receptor antagonism. Front. Neurosci. 2014, 8, 28. 10.3389/fnins.2014.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoever P.; Dorffner G.; Beneš H.; Penzel T.; Danker-Hopfe H.; Barbanoj M. J.; Pillar G.; Saletu B.; Polo O.; Kunz D.; Zeitlhofer J.; Berg S.; Partinen M.; Bassetti C. L.; Högl B.; Ebrahim I. O.; Holsboer-Trachsler E.; Bengtsson H.; Peker Y.; Hemmeter U.-M.; Chiossi E.; Hajak G.; Dingemanse J. Orexin receptor antagonism, a new sleep enabling paradigm: a proof of concept clinical trial. Clin. Pharmacol. Ther. 2012, 91, 975–985. 10.1038/clpt.2011.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GlaxoSmithKline press release. GSK and Actelion discontinue clinical development of almorexant. https://www.gsk.com/en-gb/media/press-releases/gsk-and-actelion-discontinue-clinical-development-of-almorexant (accessed October 20, 2019).
- Cox C. D.; Breslin M. J.; Whitman D. B.; Schreier J. D.; McGaughey G. B.; Bogusky M. J.; Roecker A. J.; Mercer S. P.; Bednar R. A.; Lemaire W.; Bruno J. G.; Reiss D. R.; Harrell C. M.; Murphy K. L.; Garson S. L.; Doran S. M.; Prueksaritanont T.; Anderson W. B.; Tang C.; Roller S.; Cabalu T. D.; Cui D.; Hartman G. D.; Young S. D.; Koblan K. S.; Winrow C. J.; Renger J. J.; Coleman P. J. Discovery of the dual orexin receptor antagonist [(7R)-4-(5-chloro-1,3-benzoxazol-2-yl)-7-methyl-1,4-diazepan-1-yl][5-methyl-2-(2H-1,2,3-triazol-2-yl)phenyl]methanone (MK-4305) for the treatment of insomnia. J. Med. Chem. 2010, 53, 5320–5332. 10.1021/jm100541c. [DOI] [PubMed] [Google Scholar]
- Merck press release. FDA Approves BELSOMRA® (suvorexant) for the Treatment of Insomnia http://www.mrknewsroom.com/news-release/prescription-medicine-news/fda-approves-belsomra-suvorexant-treatment-insomnia (accessed October 20, 2019).
- Belsomra® prescribing information. https://www.merck.com/product/usa/pi_circulars/b/belsomra/belsomra_pi.pdf (Accessed October 20, 2019).
- Coleman P. J.; Schreier J. D.; Cox C. D.; Breslin M. J.; Whitman D. B.; Bogusky M. J.; McGaughey G. B.; Bednar R. A.; Lemaire W.; Doran S. M.; Fox S. V.; Garson S. L.; Gotter A. L.; Harrell C. M.; Reiss D. R.; Cabalu T. D.; Cui D.; Prueksaritanont T.; Stevens J.; Tannenbaum P. L.; Ball R. G.; Stellabott J.; Young S. D.; Hartman G. D.; Winrow C. J.; Renger J. J. Discovery of [(2R,5R)-5-{[(5-fluoropyridin-2-yl)oxy]methyl}-2-methylpiperidin-1-yl][5-methyl-2-(pyrimidin-2-yl)phenyl]methanone (MK-6096): a dual orexin receptor antagonist with potent sleep-promoting properties. ChemMedChem 2012, 7, 415–424. 10.1002/cmdc.201200025. [DOI] [PubMed] [Google Scholar]
- Winrow C. J.; Gotter A. L.; Cox C. D.; Tannenbaum P. L.; Garson S. L.; Doran S. M.; Breslin M. J.; Schreier J. D.; Fox S. V.; Harrell C. M.; Stevens J.; Reiss D. R.; Cui D.; Coleman P. J.; Renger J. J. Pharmacological characterization of MK-6096 – a dual orexin receptor antagonist for insomnia. Neuropharmacology 2012, 62, 978–987. 10.1016/j.neuropharm.2011.10.003. [DOI] [PubMed] [Google Scholar]
- Yoshida Y.; Naoe Y.; Terauchi T.; Ozaki F.; Doko T.; Takemura A.; Tanaka T.; Sorimachi K.; Beuckmann C. T.; Suzuki M.; Ueno T.; Ozaki S.; Yonaga M. Discovery of (1R,2S)-2-{[(2,4-dimethylpyrimidin-5-yl)oxy]methyl}-2-(3-fluorophenyl)-N-(5-fluoropyridin-2-yl)cyclopropanecarboxamide (E2006): a potent and efficacious oral orexin receptor antagonist. J. Med. Chem. 2015, 58, 4648–4664. 10.1021/acs.jmedchem.5b00217. [DOI] [PubMed] [Google Scholar]
- Murphy P.; Moline M.; Mayleben D.; Rosenberg R.; Zammit G.; Pinner K.; Dhadda S.; Hong Q.; Giorgi L.; Satlin A. Lemborexant, a dual orexin receptor antagonist (DORA) for the treatment of insomnia disorder: results from a bayesian, adaptive, randomized, double-blind, placebo-controlled study. J. Clin. Sleep. Med. 2017, 13, 1289–1299. 10.5664/jcsm.6800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treiber A.; de Kanter R.; Roch C.; Gatfield J.; Boss C.; von Raumer M.; Schindelholz B.; Muehlan C.; van Gerven J.; Jenck F. The use of physiology-based PK and PD modeling in the discovery of the dual orexin receptor antagonist ACT-541468. J. Pharmacol. Exp. Ther. 2017, 362, 489–503. 10.1124/jpet.117.241596. [DOI] [PubMed] [Google Scholar]
- https://www.idorsia.com/about-idorsia/idorsia-today/our-pipeline
- Roecker A. J.; Mercer S. P.; Schreier J. D.; Cox C. D.; Fraley M. E.; Steen J. T.; Lemaire W.; Bruno J. G.; Harrell C. M.; Garson S. L.; Gotter A. L.; Fox S. V.; Stevens J.; Tannenbaum P. L.; Prueksaritanont T.; Cabalu T. D.; Cui D.; Stellabott J.; Hartman G. D.; Young S. D.; Winrow C. J.; Renger J. J.; Coleman P. J. Discovery of 5-chloro-N-[(5,6-dimethoxypyridin-2-yl)methyl]-2,2″:5″,3-terpyridine-3′′-carboxamide (MK-1064): a selective orexin 2 receptor antagonist (2-SORA) for the treatment of insomnia. ChemMedChem 2014, 9, 311–322. 10.1002/cmdc.201300447. [DOI] [PubMed] [Google Scholar]
- Minerva Neurosciences pipeline. http://www.minervaneurosciences.com/innovation-pipeline/min-202/ (Accessed October 20, 2019).
- Harris G. C.; Wimmer M.; Aston-Jones G. A role for lateral hypothalamic orexin neurons in reward seeking. Nature 2005, 437, 556–559. 10.1038/nature04071. [DOI] [PubMed] [Google Scholar]
- Hollander J. A.; Lu Q.; Cameron M. D.; Kamenecka T. M.; Kenny P. J. Insular hypocretin transmission regulates nicotine reward. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 19480–19485. 10.1073/pnas.0808023105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence A. J.; Cowen M. S.; Yang H. J.; Chen F.; Oldfield B. The orexin system regulates alcohol-seeking in rats. Br. J. Pharmacol. 2006, 148, 752–759. 10.1038/sj.bjp.0706789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez M. F.; Moorman D. E.; Aston-Jones G.; Becker H. C. The highly selective orexin/hypocretin 1 receptor antagonist GSK1059865 potently reduces ethanol drinking in ethanol dependent mice. Brain Res. 2016, 1636, 74–80. 10.1016/j.brainres.2016.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccoli L.; Di Bonaventura M. V. M.; Cifani C.; Costantini V. J. A.; Massagrande M.; Montanari D.; Martinelli P.; Antolini M.; Ciccocioppo R.; Massi M.; Merlo-Pich E.; Di Fabio R.; Corsi M. Role of orexin-1 receptor mechanisms on compulsive food consumption in a model of binge eating in female rats. Neuropsychopharmacology 2012, 37, 1999–2011. 10.1038/npp.2012.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlo Pich E.; Melotto S. Orexin 1 receptor antagonists in compulsive behavior and anxiety: possible therapeutic use. Front. Neurosci. 2014, 8, 26. 10.3389/fnins.2014.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozzi A.; Turrini G.; Piccoli L.; Massagrande M.; Amantini D.; Antolini M.; Martinelli P.; Cesari N.; Montanari D.; Tessari M.; Corsi M.; Bifone A. Functional magnetic resonance imaging reveals different neural substrates for the effects of orexin-1 and orexin-2 receptor antagonists. PLoS One 2011, 6, e16406 10.1371/journal.pone.0016406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart D.; Sabido-David C.; Brough S. J.; Jewitt F.; Johns A.; Porter R. A.; Jerman J. C. SB-334867-A: The first selective orexin-1 receptor antagonist. Br. J. Pharmacol. 2001, 132, 1179–1182. 10.1038/sj.bjp.0703953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter R. A.; Chan W. N.; Coulton S.; Johns A.; Hadley M. S.; Widdowson K.; Jerman J. C.; Brough S. J.; Coldwell M.; Smart D.; Jewitt F.; Jeffrey P.; Austin N. 1,3-Biarylureas as selective non-peptide antagonists of the orexin-1 receptor. Bioorg. Med. Chem. Lett. 2001, 11, 1907–1910. 10.1016/S0960-894X(01)00343-2. [DOI] [PubMed] [Google Scholar]
- Yin J.; Mobarec J. C.; Kolb P.; Rosenbaum D. M. Crystal structure of the human OX2 orexin receptor bound to the insomnia drug suvorexant. Nature 2015, 519, 247–250. 10.1038/nature14035. [DOI] [PubMed] [Google Scholar]
- Yin J.; Babaoglu K.; Brautigam C. A.; Clark L.; Shao Z.; Scheuermann T. H.; Harrell C. M.; Gotter A. L.; Roecker A. J.; Winrow C. J.; Renger J. J.; Coleman P. J.; Rosenbaum D. M. Structure and ligand-binding mechanism of the human OX1 and OX2 orexin receptors. Nat. Struct. Mol. Biol. 2016, 23, 293–299. 10.1038/nsmb.3183. [DOI] [PubMed] [Google Scholar]
- Suno R.; Kimura K. T.; Nakane T.; Yamashita K.; Wang J.; Fujiwara T.; Yamanaka Y.; Im D.; Horita S.; Tsujimoto H.; Tawaramoto M. S.; Hirokawa T.; Nango E.; Tono K.; Kameshima T.; Hatsui T.; Joti Y.; Yabashi M.; Shimamoto K.; Yamamoto M.; Rosenbaum D. M.; Iwata S.; Shimamura T.; Kobayashi T. Crystal structures of human orexin 2 receptor bound to the subtype-selective antagonist EMPA. Structure 2018, 26, 7–19.e5. 10.1016/j.str.2017.11.005. [DOI] [PubMed] [Google Scholar]
- Malherbe P.; Borroni E.; Gobbi L.; Knust H.; Nettekoven M.; Pinard E.; Roche O.; Rogers-Evans M.; Wettstein J. G.; Moreau J.-L. Biochemical and behavioural characterization of EMPA, a novel high-affinity, selective antagonist for the OX2 receptor. Br. J. Pharmacol. 2009, 156, 1326–1341. 10.1111/j.1476-5381.2009.00127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malherbe P.; Roche O.; Marcuz A.; Kratzeisen C.; Wettstein J. G.; Bissantz C. Mapping the binding pocket of dual antagonist almorexant to human orexin 1 and orexin 2 receptors: comparison with the selective OX1 antagonist SB-674042 and the selective OX2 antagonist N-ethyl-2-[(6-methoxy-pyridin-3-yl)-(toluene-2-sulfonyl)-amino]-N-pyridin-3-ylmethyl-acetamide (EMPA). Mol. Pharmacol. 2010, 78, 81–93. 10.1124/mol.110.064584. [DOI] [PubMed] [Google Scholar]
- Horcajada C.; Guinovart J. J.; Fita I.; Ferrer J. C. Crystal structure of an archaeal glycogen synthase: insights into oligomerization and substrate binding of eukaryotic glycogen synthases. J. Biol. Chem. 2006, 281, 2923–2931. 10.1074/jbc.M507394200. [DOI] [PubMed] [Google Scholar]
- Christopher J. A.; Aves S. J.; Brown J.; Errey J. C.; Klair S. S.; Langmead C. J.; Mace O. J.; Mould R.; Patel J. C.; Tehan B. G.; Zhukov A.; Marshall F. H.; Congreve M. Discovery of HTL6641, a dual orexin receptor antagonist with differentiated pharmacodynamic properties. Med. Chem. Commun. 2015, 6, 947–955. 10.1039/C5MD00027K. [DOI] [Google Scholar]
- Boss C.; Roch-Brisbare C.; Steiner M. A.; Treiber A.; Dietrich H.; Jenck F.; von Raumer M.; Sifferlen T.; Brotschi C.; Heidmann B.; Williams J. T.; Aissaoui H.; Siegrist R.; Gatfield J. Structure–activity relationship, biological, and pharmacological characterization of the proline sulfonamide ACT-462206: a potent, brain-penetrant dual orexin 1/orexin 2 receptor antagonist. ChemMedChem 2014, 9, 2486–2496. 10.1002/cmdc.201402258. [DOI] [PubMed] [Google Scholar]
- Breslin M. J.; Cox C. D.; Whitman D. B. Diazaspirodecane Orexin Receptor Antagonists. PCT Patent Appl. WO2007/025069 A2, Mar 1, 2007.
- Mason J. S.; Bortolato A.; Congreve M.; Marshall F. H. New insights from structural biology into the druggability of G protein-coupled receptors. Trends Pharmacol. Sci. 2012, 33, 249–260. 10.1016/j.tips.2012.02.005. [DOI] [PubMed] [Google Scholar]
- Goodford P. J. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J. Med. Chem. 1985, 28, 849–857. 10.1021/jm00145a002. [DOI] [PubMed] [Google Scholar]
- Sciabola S.; Stanton R. V.; Mills J. E.; Flocco M. M.; Baroni M.; Cruciani G.; Perruccio F.; Mason J. S. High-throughput virtual screening of proteins using GRID molecular interaction fields. J. Chem. Inf. Model. 2010, 50, 155–169. 10.1021/ci9003317. [DOI] [PubMed] [Google Scholar]
- FLAP, WaterFLAP. Molecular Discovery Ltd., Borehamwood, U.K.
- Bortolato A.; Tehan B. G.; Smith R. T.; Mason J. S. Methodologies for the Examination of Water in GPCRs. In: Computational Methods for GPCR Drug Discovery. Methods in Molecular Biology; Heifetz A., Ed.; Humana Press: New York, NY, 2017; Vol. 1705, pp 207–232. [DOI] [PubMed] [Google Scholar]
- Abel R.; Salam N. K.; Shelley J.; Farid R.; Friesner R. A.; Sherman W. Contribution of explicit solvent effects to the binding affinity of small-molecule inhibitors in blood coagulation factor serine proteases. ChemMedChem 2011, 6, 1049–1066. 10.1002/cmdc.201000533. [DOI] [PubMed] [Google Scholar]
- WaterMap. Schrödinger, LLC., New York, NY, U.S.A.
- Alvaro G.; Amantini D.; Stasi L. P.. Pyridine Derivatives Used to Treat Orexin Related Disorders. PCT Patent Appl. WO2009/124956 A1, Oct 15, 2009.
- Cai J.; Cooke F. E.; Sherborne B. S. Antagonists of the orexin receptors. Expert Opin. Ther. Pat. 2006, 16, 631–646. 10.1517/13543776.16.5.631. [DOI] [Google Scholar]
- Coleman P. J.; Renger J. J. Orexin receptor antagonists: a review of promising compounds patented since 2006. Expert Opin. Ther. Pat. 2010, 20, 307–324. 10.1517/13543770903567085. [DOI] [PubMed] [Google Scholar]
- Stasi L. P.; Rovati L.. Spiro Amino Compounds Suitable for the Treatment of Inter Alia Sleep Disorders and Drug Addiction. PCT Patent Appl. WO2011/006960 A1, Jan 20, 2011.
- Stasi L. P.; Rovati L.. Chemical Compounds. PCT Patent Appl. WO2013/139730 A1, Sept 26, 2013.
- Ren J.; Zhao Y.; Fry E. E.; Stuart D. I. Target identification and mode of action of four chemically divergent drugs against ebolavirus infection. J. Med. Chem. 2018, 61, 724–733. 10.1021/acs.jmedchem.7b01249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speranzini V.; Rotili D.; Ciossani G.; Pilotto S.; Marrocco B.; Forgione M.; Lucidi A.; Forneris F.; Mehdipour P.; Velankar S.; Mai A.; Mattevi A. Polymyxins and quinazolines are LSD1/KDM1A inhibitors with unusual structural features. Sci. Adv. 2016, 2, e1601017 10.1126/sciadv.1601017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stornaiuolo M.; De Kloe G. E.; Rucktooa P.; Fish A.; van Elk R.; Edink E. S.; Bertrand D.; Smit A. B.; de Esch I. J. P.; Sixma T. K. Assembly of a π– π stack of ligands in the binding site of an acetylcholine-binding protein. Nat. Commun. 2013, 4, 1875. 10.1038/ncomms2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson N.; Jazayeri A.; Errey J.; Baig A.; Hurrell E.; Zhukov A.; Langmead C. J.; Weir M.; Marshall F. H. The properties of thermostabilised G protein-coupled receptors (StaRs) and their use in drug discovery. Neuropharmacology 2011, 60, 36–44. 10.1016/j.neuropharm.2010.07.001. [DOI] [PubMed] [Google Scholar]
- Caffrey M.; Cherezov V. Crystallizing membrane proteins using lipidic mesophases. Nat. Protoc. 2009, 4, 706–731. 10.1038/nprot.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr., D: Biol. Crystallogr. 2010, 66, 133–144. 10.1107/S0907444909047374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. R.; Murshudov G. N. How good are my data and what is the resolution?. Acta Crystallogr., D: Biol. Crystallogr. 2013, 69, 1204–1214. 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The CCP4 suite: programs for protein crystallography. Acta Crystallogr., D: Biol. Crystallogr 1994, 50, 760–763. 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Tickle I. J.; Flensburg C.; Keller P.; Paciorek W.; Sharff A.; Vonrhein C.; Bricogne G.. STARANISO; Cambridge, United Kingdom: Global Phasing Ltd. (2018). [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and development of Coot. Acta Crystallogr., D: Biol. Crystallogr. 2010, 66, 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkóczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L.-W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 213–221. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N.; Skubák P.; Lebedev A. A.; Pannu N. S.; Steiner R. A.; Nicholls R. A.; Winn M. D.; Long F.; Vagin A. A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr., D: Biol. Crystallogr. 2011, 67, 355–367. 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine P. V.; Grosse-Kunstleve R. W.; Echols N.; Headd J. J.; Moriarty N. W.; Mustyakimov M.; Terwilliger T. C.; Urzhumtsev A.; Zwart P. H.; Adams P. D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr., D: Biol. Crystallogr. 2012, 68, 352–367. 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricogne G.; Blanc E.; Brandl M.; Flensburg C.; Keller P.; Paciorek W.; Roversi P.; Sharff A.; Smart O. S.; Vonrhein C.; Womack T. O.. BUSTER version 2.11.7. Global Phasing Ltd.: Cambridge, United Kingdom: (2017).
- Isberg V.; de Graaf C.; Bortolato A.; Cherezov V.; Katritch V.; Marshall F. H.; Mordalski S.; Pin J. P.; Stevens R. C.; Vriend G.; Gloriam D. E. Generic GPCR residue numbers – aligning topology maps while minding the gaps. Trends Pharmacol. Sci. 2015, 36, 22–31. 10.1016/j.tips.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros J. A.; Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure–function relations in G protein-coupled receptors. Methods Neurosci. 1995, 25, 366–428. 10.1016/S1043-9471(05)80049-7. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.