

Abstract
Disease-modifying treatment strategies for Alzheimer disease (AD) are still under extensive research. Nowadays, only symptomatic treatments exist for this disease, all trying to counterbalance the neurotransmitter disturbance: 3 cholinesterase inhibitors and memantine. To block the progression of the disease, therapeutic agents are supposed to interfere with the pathogenic steps responsible for the clinical symptoms, classically including the deposition of extracellular amyloid β plaques and intracellular neurofibrillary tangle formation. Other underlying mechanisms are targeted by neuroprotective, anti-inflammatory, growth factor promotive, metabolic efficacious agents and stem cell therapies. Recent therapies have integrated multiple new features such as novel biomarkers, new neuropsychological outcomes, enrollment of earlier populations in the course of the disease, and innovative trial designs. In the near future different specific agents for every patient might be used in a “precision medicine” context, where aberrant biomarkers accompanied with a particular pattern of neuropsychological and neuroimaging findings could determine a specific treatment regimen within a customized therapeutic framework. In this review, we discuss potential disease-modifying therapies that are currently being studied and potential individualized therapeutic frameworks that can be proved beneficial for patients with AD.
Keywords: Alzheimer disease, disease-modifying drugs, anti-amyloid, anti-Tau, individualized therapeutic frameworks
Introduction
Alzheimer disease (AD) is one of the greatest medical care challenges of our century and is the main cause of dementia. In total, 40 million people are estimated to suffer from dementia throughout the world, and this number is supposed to become twice as much every 20 years, until approximately 2050.1
Because dementia occurs mostly in people older than 60 years, the growing expansion of lifespan, leading to a rapidly increasing number of patients with dementia,2 mainly AD, has led to an intensive growth in research focused on the treatment of the disease. However, despite all arduous research efforts, at the moment, there are no effective treatment options for the disease.3,4
The basic pathophysiology and neuropathology of AD that drives the current research suggests that the primary histopathologic lesions of AD are the extracellular amyloid plaques and the intracellular Tau neurofibrillary tangles (NFTs).5 The amyloid or senile plaques (SPs) are constituted chiefly of highly insoluble and proteolysis-resistant peptide fibrils produced by β-amyloid (Aβ) cleavage. Aβ peptides with Aβ38, Aβ40, and Aβ42 as the most common variants are produced after the sequential cleavage of the large precursor protein amyloid precursor protein (APP) by the 2 enzymes, β-secretase (BACE1) and γ-secretase. Nevertheless, Aβ is not formed if APP is first acted on and cleaved by the enzyme α-secretase instead of β-secretase.6 According to the “amyloid hypothesis” Aβ production in the brain initiates a cascade of events leading to the clinical syndrome of AD. It is the forming of amyloid oligomers to which neurotoxicity is mainly attributed and initiates the amyloid cascade. The elements of the cascade include local inflammation, oxidation, excitoxicity (excessive glutamate), and tau hyperphosphorylation.5 Tau protein is a microtubule-associated protein which binds microtubules in cells to facilitate the neuronal transport system. Microtubules also stabilize growing axons necessary for neuronal development and function. Abnormally hyperphosphorylated tau forms insoluble fibrils and folds into intraneuronic tangles. Consequently, it uncouples from microtubules, inhibits transport, and results in microtubule disassembly.6 Although in the amyloid hypothesis, tau hyperphosphorylation was thought to be a downstream event of Aβ deposition, it is equally probable that tau and Aβ act in parallel pathways causing AD and enhancing each other’s toxic effects.3 Progressive neuronal destruction leads to shortage and imbalance between various neurotransmitters (eg, acetylcholine, dopamine, serotonin) and to the cognitive deficiencies seen in AD.5
All of the already established treatments that are used today try to counterbalance the neurotransmitter imbalance of the disease. The acetylocholinesterase inhibitors (AChEIs) which are approved for the treatment of AD are donepezil, galantamine, and rivastigmine.4,5 Their development was based in the cholinergic hypothesis which suggests that the progressive loss of limbic and neocortical cholinergic innervation in AD is critically important for memory, learning, attention, and other higher brain functions decline. Furthermore, neurofibrillary degeneration in the basal forebrain is probably the primary cause for the dysfunction and death of cholinergic neurons in this region, giving rise to a widespread presynaptic cholinergic denervation. The AChEIs increase the availability of acetylcholine at synapses and have been proven clinically useful in delaying the cognitive decline in AD.7
A further therapeutic agent approved for moderate to severe AD is the low-to-moderate affinity, noncompetitive N-methyl-d-aspartate (NMDA) receptor antagonist memantine.4,5 Memantine binds preferentially to open NMDA receptor–operated calcium channels blocking NMDA-mediated ion flux and ameliorating the dangerous effects of pathologically elevated glutamate levels that lead to neuronal dysfunction.8
In clinical trials, both Aβ and tau are prime targets for disease-modifying treatments (DMTs) in AD. From this point of view, AD could be prevented or effectively treated by decreasing the production of Aβ and tau; preventing aggregation or misfolding of these proteins; neutralizing or removing the toxic aggregate or misfolded forms of these proteins; or a combination of these modalities.7
A number of additional pathogenic mechanisms have been described, possibly overlapping with Aβ plaques and NFT formation or induced by them, including inflammation, oxidative damage, iron deregulation, and cholesterol metabolism blood-brain barrier (BBB) dysfunction or α-synuclein toxicity.9-13
This article will review current nonpharmacological and pharmacological management of the cognitive and behavioral symptoms of AD, with a focus on the medications that are currently FDA (Food and Drug Administration)–approved for the treatment of the cognitive and functional deficits of AD.10 Pharmacological agents under research in phase 1, 2, and 3 clinical trials in AD will be summarized.11-13
Current management of AD
A multifactorial tailored management of AD is attempted nowadays based in the following components:
Open physician, caregiver, and patient communication: a sincere and successful conveying of information and feelings between them will offer opportune identifying of symptoms, exact evaluation and diagnosis, and suitable guidance.
- Behavioral approaches:
- Caregiver support:
- - Planned short rest periods for the caregiver;
- - Psychoeducation including preparing for effects of dementia on cognition, function and behaviors, expectations, avoiding situations that can worsen the symptoms or increasing the dangers for safety and well-being
- - Encouraging the development of support networks for the caregivers.10
Pharmacological interventions.
FDA-approved AD medications
The AChEIs donepezil, galantamine, rivastigmine, and the NMDA antagonist memantine are the only FDA-approved AD medications.10
AChEIs attempt at reducing the breakdown of acetylcholine levels in the brain of the patients with AD by inhibiting the responsible enzyme acetylcholinesterase in the synaptic cleft.5 Thus, AChEIs enhance central cholinergic neurotransmission and finally tend to mitigate decline in cognition at least during the first year of treatment. Further decline occurs, but even temporary discontinuation of these drugs results in rapid decline and is associated with greater risk of nursing home placement.16
Initiation of AChEI treatment as soon as possible after the diagnosis is preferred as patients who started the AChEI 6 months later showed more rapid cognitive decline than those who started the drug immediately.17 All 3 AChEIs have proved their treatment benefits in delaying decline, stabilizing, or even improving cognition and activities of daily living in randomized placebo-controlled trials up to 52 weeks duration.10,18 Longer term open-label extension studies support also longer term treatment benefits.10
Significant efficacy differences among the AChEIs have not been reported. Donepezil and rivastigmine have been approved by FDA for mild, moderate, and severe AD, whereas galantamine for mild and moderate AD.18
The most common adverse effects are triggered by the cholinomimetic action of the AChEIs on the gastrointestinal tract and often include diarrhea, nausea, and vomiting. Rapid eye movement sleep behavior disorder has been also remarked in some individuals. Administration of the drug after a meal in the morning can minimize all of these adverse effects. The transdermal patch of rivastigmine can induce rash at the site of application. Adverse effects affect usually a 5% to 20% of patients but are mostly transient and mild. The AChEIs may also trigger bradycardia and increase the risk of syncope. Thus, AChEIs are contraindicated in conditions including severe cardiac arrhythmias, especially bradycardia or syncope. They are also contraindicated in active peptic ulcer or gastrointestinal bleeding history and uncontrolled seizures. Slow titration over months to years to a maximal tolerated of the indicated dose is important for the safety of the patients.17,18
Pharmacokinetic characteristics differ among AChEIs: the primary route of elimination for donepezil and galantamine is hepatic metabolism, whereas for rivastigmine is liver and intestine metabolism. Donepezil and galantamine inhibit selectively and reversibly the acetylcholinesterase, whereas rivastigmine is a “pseudo-irreversible” inhibitor of acetylcholinesterase and butyrylcholinesterase. Donepezil has a long elimination half-life of 70 hours and galantamine of 6 to 8 hours. The elimination half-life of rivastigmine is very short (1-2 hours for oral and 3-4 hours for transdermal administration), but the duration of action is longer as acetylcholinesterase and butyrylcholinesterase are blocked for around 8.5 and 3.5 hours, respectively.10,17,18
Memantine is a noncompetitive low-affinity NMDA-receptor open-channel blocker and affects glutamatergic transmission.5 Its main elimination route is unchanged via the kidneys with a half-life of 70 hours. It has been approved by FDA for moderate and severe AD either as monotherapy or in combination with an AChEI.3 Memantine monotherapy has demonstrated short- and long-term benefits for patients with moderate to severe AD as assessed by different scales evaluating activities of daily living, cognition, and behavioral and psychological symptoms of dementia (BPSD).19
Memantine can be administered in combination with an AChEI, as they have complementary mechanisms of action. Their combination benefits patients with usually additive effects, without any increase in adverse effects.14,15
Duration and persistence of monotherapy or combination treatment with higher doses in moderate or even in advanced dementia are associated with better global function and outcomes.20
Medications for BPSD
Antipsychotics and antidepressants remain the main medications for BPSD. Selective serotonin reuptake inhibitors are preferred for treating depression and anxiety. Drugs with low anticholinergic effects and an acceptable tolerability, such as sertraline, citalopram, and escitalopram, are more appropriate. Antipsychotics should be administered only when a significant safety risk for the patient or for the caregivers by aggressive behaviors makes them necessary. Controversial and limited evidence cannot adequately support the use of benzodiazepines, anticonvulsants stimulants, or dextromethorphan/quinidine. Pharmacological approaches to managing BPSD are highly individualized and changeable, depending on patient’s comorbidities, stage of the disease, and symptoms’ severity.21
Removal of superfluous and deleterious medications
Polypharmacy in older patients with dementia is usual (with a prevalence of 25%-98%).22 Anticholinergics and sedatives are commonly used inappropriate medications. These drugs are prescribed despite strong evidence (Beers Criteria) that they should be avoided in cognitively vulnerable older persons because of their potential adverse cognitive effects.23 Estrogen is another commonly prescribed potentially inappropriate medication despite evidence that its use is associated with increased cognitive decline in postmenopausal women.24
Specific examples of usually prescribed potentially harmful medications in the elderly are diphenhydramine, often taken with acetaminophen for insomnia and pain, benzodiazepines for anxiety, anticholinergics (tolderodine, oxybutynin, tamsulosin) for urinary incontinence, biperiden, and pramipexole for extrapyramidal tremor25 and sedative/hypnotics for sleep disorders.26
Treating underlying medical conditions
Careful management of vascular risk factors (hyperlipidemia, diabetes, hypertension) is of paramount importance for patients with AD. Hydration, sleep, and nutrition status should also be closely monitored. Disorders in thyroid function or electrolytes, deficiencies in vitamin B12, folate, vitamin D, or systemic conditions and diseases that can affect cognition (infections, eg, urinary tract infection, pain, constipation) should be treated.27
Current Landscape in Treatment Research for AD
No new drug has been approved by FDA for AD since 2003 and there are no approved DMTs for AD, despite many long and expensive trials.22,28 As a matter of fact, more than 200 research projects in the last decade have failed or have been abandoned.10 Nevertheless, drug pipeline for AD is still full of agents with mechanisms of action (MOA) that target either disease modification or symptoms.4,10 Some of the recent failures of anti-amyloid agents in phase 3 clinical trials in patients with early-stage, mild, or mild-to-moderate stage AD were semagacestat,29 bapineuzumab,30 solanezumab31 and in similar trials of β-secretase inhibitors (BACE) lanabecestat,32 verubecestat,33 and atabecestat.34
The most popular and broadly accepted explanations for the multiple failures of clinical trials of DMT agents for AD include the too late starting of therapies in disease development, the inappropriate drug doses, the wrong main target of the treatment, and mainly an inadequate understanding of the pathophysiology of AD.35 A novel approach to the problem seems more technical and mathematical than biological and suggests that the selected trials’ clinical endpoint may be extremely premature, and additionally, the variability in diagnostic markers and end points may result in inaccurate diagnosis of patients’ disease state and is finally a definite source of errors.28 Given the fact that longer trial durations increase the probability of detecting a significant effect but at the same time increase tremendously the costs, the proposed solution seems to be the use of clinical trial simulators.28 These simulators are constructed with mathematical, computational, and statistical tools and can predict the likelihood that a strategy and clinical end point selection of a given trial are proper or not, before the initiation of the trial.36 They can also help in the perfecting of the design of the study; hence, they may augment the probability of success of estimated new drugs or save invaluable time and resources, by indicating earlier the forthcoming failure of any inappropriate therapy.37 Although the use of clinical trial simulators is not frequent in recent research,38 should this practice be abandoned, especially when potential treatments for diseases with slow progression and long duration, such as AD, are evaluated.37
At the same time, current research remains focused on the development of therapeutic approaches to slow or stop the disease progression, taking into consideration every new aspect in the biology of the disease, the diagnostic markers, and the precise diagnosis of disease state of every individual and the design of clinical trials. Furthermore, drug development research for AD has become more complicated as preclinical and prodromal AD populations are potentially included in current trials, as well as traditionally included populations of all the clinical stages of AD dementia.38 Consequently, current guidance provided by the FDA for AD clinical trials further includes use of fluid or neuroradiological biomarkers in disease staging for preclinical and prodromal AD trials and of a single primary outcome in prodromal AD trials. In addition, the use of clinical trial simulators, Bayesian statistics, and modifiable trial designs is strongly suggested.4
The National Institute on Aging and the Alzheimer’s Association (NIA-AA) proposed a new framework for research,39 which requires the application of amyloid, tau, and neurodegeneration biomarkers to clinical trials, succeeds in precise classification of patients in AD stages, and can be used to assist clinical trials design.
Tau positron emission tomography (tau PET), neurofilament light, and neurogranin are the new biomarkers that are increasingly used by clinical trials.40
The above-mentioned biological and statistical advances that are recently integrated in clinical trials may comprise the final assets for succeeding in drug development. The current clinical trials in AD in phases 1, 2, and 34,11-13 are briefly discussed. The tested agents in these trials are classified either as potentially modifying the disease or as symptomatic for the cognitive enhancement, and for the relief of neuropsychiatric symptoms. The new directions in AD clinical trials, such as agents with novel MOA, advanced immunotherapies, the involvement of biomarkers in drug development, and repurposed agents, are highlighted.
A search for phases 1, 2, and 3 “recruiting” or “active but not recruiting” clinical trials for AD in clinicaltrials.gov (accessed August 19, 2019) showed 165 outcomes. The last annual review of the drug development pipeline for AD examined clinicaltrials.gov in February, 2019 (132 agents in 156 trials) and provides information and conclusions available at that time: 28 drugs in 42 clinical trials in phase 3 trials, 74 drugs in 83 phase 2 trials, and 30 drugs in 31 phase 1 trials.4 The tested agents are classified as DMTs (73%), symptomatic cognitive enhancers (13%), and symptomatic for the treatment of BPSDs (11%).4 The DMT agents were further separated into small molecules or biologics (monoclonal antibodies [mAbs] and other immunotherapies). The DMT agents were also classified according to their potential MOA as amyloid targeting, as tau-related targeting, and as having other MOA such as anti-inflammatory or metabolic protection, neuroprotection, and growth factor support.4 The DMTs are suggested to be effective to delay or halt disease progression that would be expressed clinically with long-lasting benefits in cognition over many months to years. Symptomatic agents are supposed to show symptomatic benefits over weeks to many months in cognition improvement or BPSD elimination.10
In this review, agents currently studied as potential DMTs will be discussed. Furthermore, an approach to a future “precision medicine” multifactorial therapeutic model based on biomarkers profile, genetic analysis, neuropsychologic evaluation, and neuroimaging accomplished with risk factors restriction will be attempted.2,3
Currently studied DMTs for AD
Amyloid-related mechanisms—DMTs
The crucial step in AD pathogenesis is the production of amyloid (Aβ), which forms SPs (insoluble and proteolysis-resistant fibrils). The Aβ derives from a protein overexpressed in AD, APP through sequential proteolysis by β-secretase (BACE1) in the extracellular domain and γ-secretase in the transmembrane region. Full-length APP is first cleaved by α-secretase or β-secretase. The APP cleavage by α-secretase leads to nonamyloidogenic pathway, whereas APP cleavage by β-secretase (BACE1) leads to amyloidogenic pathway. Sequential cleavage of APP by BACE1 in the extracellular and γ-secretase in the transmembrane area results in the Aβ production. Major sites of γ-secretase cleavage usually occur in positions 40 and 42 of Aβ, thus Aβ40 and Aβ42 oligomers are the main products of the sequential APP cleavage, as the amyloidogenic pathway is favored in neurons because of the greater plentifulness of BACE1. On the contrary, the nonamyloidogenic processing is more favored in other cells without BACE1 predominance.5
“Amyloid hypothesis” suggests that Aβ production in the brain triggers a cascade of pathophysiologic events leading to the clinical expression of AD. Aβ is a protein consisting of 3 main isoforms: Aβ38, Aβ40, and Aβ42. Aβ42 is the most aggregation-prone form and has the tendency to cluster into oligomers. Oligomers can form Aβ-fibrils that will eventually form amyloid plaques. Aβ40 is somewhat aggregation-prone and it is mostly found in the cerebral vasculature as a main component of “cerebral amyloid angiopathy.” Aβ40 usually constitutes more than 50% of total detected Aβ. Aβ38 is soluble, present in the vasculature of patients with sporadic and familial AD. Neurotoxicity is mainly attributed to the forming of amyloid oligomers, which finally initiates the amyloid cascade.5
Oxidation, inflammation, excessive glutamate, and tau hyperphosphorylation are supposed to be the main pathophysiologic pillars of the cascade. Tau protein binds microtubules in cells to facilitate the neuronal transport system. Microtubules also stabilize growing axons. Hyperphosphorylated tau forms insoluble fibrils and folds into intraneuronic NFTs. Consequently, it inhibits neuronal transport and microtubule function.2 Although in the initial amyloid hypothesis, tau hyperphosphorylation was thought to be a downstream event of Aβ deposition, it is now equally probable that tau and Aβ act in parallel pathways causing AD and enhancing each other’s toxic effects.2 The result of massive neuronal destruction is the shortage and imbalance between neurotransmitters, such as acetylcholine, dopamine, serotonin, and to the cognitive and behavioral symptoms of AD.5
Consequently, anti-amyloid DMTs have focused on 3 major MOAs: (1) reduction of Aβ42 production (γ-secretase inhibitors, β-secretase inhibitors, α-secretase potentiation), (2) reduction of Aβ-plaque burden (aggregation inhibitors, drugs interfering with metals), and (3) promotion of Aβ clearance (active or passive immunotherapy).10
Reduction of Aβ42 production
γ-secretase inhibitors
According to the amyloid hypothesis, amyloidogenic pathway is promoted after the sequential cleavage of APP by BACE1 and γ-secretase. Consequently, the inhibition of these enzymes has been considered as a major therapeutic target. Unluckily, concerning γ-secretase, in addition to APP, this particular enzyme acts on many other substances and cleaves different transmembrane proteins. Notch receptor 1, which is essential for control of normal cell differentiation and communication, is among them.5 This fact is probably responsible for recent failures in clinical trials with γ-secretase inhibitors: semagacestat29 was associated with worsening of activities in daily leaving and increased rates of infections and skin cancer, avagacestat41 was associated with higher rate of cognitive decline and adverse dose-limiting effects (skin cancer) and tarenflurbil which showed low brain penetration.42 Serious safety concerns for γ-secretase inhibitors remove γ-secretase from the role of appropriate target for the treatment of AD43 until in depth studies on this key enzyme could help to therapeutically target γ-secretase in a safe way.44 No γ-secretase modulators are currently studied in phase 1-3 clinical trials.4
BACE inhibitors
Two BACE inhibitors are still elaborated: elenbecestat (E2609) in phase 2 and umibecestat (CNP520) in phase 3.4 The later agent is studied in asymptomatic individuals at risk of developing AD (APOE4 homozygotes or APOE4 heterozygotes with elevated amyloid, detected by cerebrospinal fluid [CSF] biomarkers or amyloid PET).45
Fluid and neuroimaging biomarkers indicative of AD pathology or neurodegeneration are integrated in this study.
However, the clinical trials with the BACE inhibitors lanabecestat,32 verubecestat,33 and atabecestat34 have been recently discontinued due to unexpected difficulties. The phase 3 lanabecestat trial was discontinued due to lack of efficacy, whereas verubecestat and atabecestat trials were ceased due to ineffectiveness, as well as safety reasons (rash, falls, liver toxicity, and neuropsychiatric symptoms).10,32-34 All agents showed significant and dose-dependent result of reducing CSF Aβ42, but without cognitive or functional benefit while many of them were poorly tolerated and some of them failed in subjects with prodromal AD. These results might support the suggestion that blocking the process of forming of Aβ may be not capable of halting the disease progression.46
α-secretase modulators
According to the amyloid hypothesis, nonamyloidogenic pathway is promoted after the cleavage of APP by α-secretase. Consequently, the modulation of the enzyme has been considered as a major therapeutic target. However, little is known of the main signaling pathways that could stimulate cleavage of APP by α-secretase. Restricted, nowadays, knowledge assumes that α-secretase activation is promoted through the phosphatidylinositol 3-kinase (PI3K)/Akt pathway and may be through γ-aminobutyric acid (GABA) receptor signaling; thus, agents that activate the PI3K/Akt pathway or act as selective GABA receptor modulators are suggested as potential therapeutic drugs for AD.47,48
Etazolate (EHT0202) stimulates the nonamyloidogenic α-secretase pathway acting as a selective modulator of GABA receptors. A previous, phase 2 trial has showed that the agent was safe and well tolerated in patients with mild to moderate AD. However, further evaluation of etazolate in phase 3 trials has not progressed.48 Etazolate is currently evaluated in animal studies for its preventive effect in post-traumatic stress disorder.49
Two α-secretase modulators that activate the PI3K/Akt pathway are studied in phase 2 clinical studies: APH-1105 and ID1201. APH-1105 is delivered intranasally and is assessed in mild to moderate AD.4 ID1201 is a fruit extract of melia toosendan and also induces α-secretase activation. It is evaluated in mild AD.47
Reduction of Aβ-plaque burden
Aggregation inhibitors (anti-amyloid aggregation agents)
Aggregation inhibitors interact directly with the Aβ peptide to inhibit Aβ42 fiber formation, thus they are considered potential therapeutic for AD.
The last Aβ42 aggregation inhibitor which was tested in humans was the oral agent scyllo-inositol (ELND005). A phase 2 clinical trial in patients with AD did not provide evidence to support a clinical benefit of ELND005 while severe toxicity issues (infections) forced the cessation of the study. Further development of the agent at a lower dose has not progressed in the last 8 years.50
Nowadays, specific agents in the form of peptidomimetics that inhibit and partially reverse the aggregation of Aβ42 are tested in transmission electron microscopic studies. KLVFF is a peptide sequence that resembles the hydrophobic central part of the Aβ and gradually replaces natural polypeptides. The KLVFF compound that mainly prevents the aggregation of Aβ42 and can also dissolve the oligomerics to a limited extend is the final compound 18, which is resilient in proteolytic decomposition.51
Another newly developed class of peptidomimetics are the “γ-AApeptides.”52 One of them, compound γ-AA26, seems almost 100-fold as efficient as the compound 18 of the KLVFF in the inhibition of the aggregation of Aβ42.52
In vivo animal studies will be developed to manifest the biological potential of peptidomimetics.
Reduction of Aβ-plaque burden via drugs interfering with metals
Abnormal accumulation or dyshomeostasis of metal ions such as iron, copper, and zinc has been associated with the pathophysiology of AD.5
Deferiprone is an iron chelating agent which is studied in phase 2 trials in participants with mild and prodromal AD.4,53
A metal protein–attenuating compound, PBT2, has recently progressed in phase 2 AD treatment trials, as it demonstrated promising efficacy data in preclinical studies.54 In a 3-month phase 2 study, PBT2 succeeded in a 13% reduction of CSF Aβ and an executive function improvement in a dose-related pattern in patients with early AD.55
Promotion of Aβ clearance (active or passive immunotherapy)
The 2 main immunotherapeutic approaches that intend to promote Aβ clearance and are currently tested in clinical and preclinical studies are active and passive immunization:56
Active immunization.
Aβ, phosphorylated tau (ptau) peptides, or specific artificial peptides such as polymerized British amyloidosis (ABri)-related peptide (pBri)57 are used as immunogens. ABri is a rare hereditary amyloidosis associated with a mutation that results in the production of a highly amyloidogenic protein with a unique carboxyl terminus that has no homology to any other human protein. The pBri peptide corresponds to this terminus and induces an immune response that recognizes Aβ and ptau.
Antigen-presenting cells present the immunogens to B cells. Use of Ab or ptau peptides will produce antibodies to Ab or ptau epitopes, respectively. Use of pBri will produce antibodies to both Aβ and ptau epitopes.56
Passive immunization.
Monoclonal Abs to Ab, ptau, or b sheet epitopes are systemically and adequately for BBB penetration infused. As antibodies cross the BBB, they act to clear, degrade, or alternatively disaggregate or neutralize their targets.56
Stimulation of innate immunity either by active or passive immunization also ameliorates the pathology of the disease by promoting microglia and macrophage function.56
Overall, Aβ-targeted strategies seem promising if used very early in the progression of the disease, before the presence of any symptoms; thus, they are developed in current trials in preclinical AD. Strategies that target tau pathology, although promising, bear the risk of toxicity at the moment. Nevertheless, it is hypothesized that, in sporadic late onset AD, ptau and Aβ pathologies could be evolved by separate pathways that can affect each other synergistically.58 Consequently, it is possible that effective AD immunotherapies must be able to simultaneously target both ptau and Aβ pathologies.56
Immunotherapeutic approaches have dominated in the past 15 years with negative results until now. However, lessons from these fails have altered the current immunotherapy development research for AD.56
Active Aβ immunotherapy
Six active immunotherapy agents are currently studied in phase 1, 2, and 3 clinical trials:
CAD106 is an active Aβ immunotherapeutic agent, is studied in preclinical AD under the umbrella of the Alzheimer prevention initiative generation program, which comprises 2 phase 3 studies that evaluate simultaneously the safety and efficacy of CAD106 and umibecestat in asymptomatic individuals at risk of developing AD (60-75 years of age, APOE4 homozygotes, or APOE4 heterozygotes with elevated amyloid in CSF or in amyloid PET).45
Subjects will be registered in generation study 1 (cohort 1: CAD106 or placebo, cohort 2: umibecestat or placebo) or generation study 2 (umibecestat 50 and 15 mg, or placebo).45
ABvac40 is evaluated in a phase 2 study, as the first active vaccine against the C-terminal end of Aβ40. A phase 1 study was conducted with patients with mild to moderate AD aged 50 to 85 years. Neither incident vasogenic edema nor microhemorrhages were identified. Specific anti-Aβ40 antibodies were developed in the 92% of the individuals receiving injections of ABvac40.59
GV1001 peptide (tertomotide) was previously studied as a vaccine against various cancers, whereas now it is evaluated in a phase 2 study for AD.60
ACC-001 (vanutide cridificar), an Aβ vaccine, was studied in phase 2a extension studies in subjects with mild to moderate AD. It was administered with QS-21 adjuvant. Long-term therapy with this combination was very well tolerated and produced the highest anti-Aβ IgG titers compared with other regimens.61
UB-311, a synthetic peptide used as Aβ vaccine, has been advanced into an ongoing phase 2 study in patients with mild and moderate AD. In phase 1, it induced a 100% responder rate in patients with AD. The usual adverse effects were swelling in the injection site and agitation. A slower cognitive decline rate was observed in patients with mild AD.62
Lu AF20513 epitope vaccine is estimated in a phase 1 study in mild AD.63
The occurrence of encephalitis in previous studies (AN-1792)64 led to the development of improved anti-Aβ active immunotherapy agents, more specific to Aβ sites less probable to activate T cells, currently studied in clinical trials.5,6
Passive Aβ immunotherapy—via mAbs
Passive Ab immunotherapy via mAbs is the most active and promising class. Cerebral microhemorrhages and vasogenic edema are the main drawbacks in this group of agents.5 Valuable learning gained from previous failed phase 3 trials of the first agents of this class, bapineuzumab65 and solanezumab,66 enlightened the mAbs’ research. Strict inclusion criteria were applied, such as biomarker proof of “amyloid positivity” and enrollment of individuals with preclinical stages of the disease. Furthermore, the design of the studies became more specific and targeted: the characteristics of amyloid-related imaging abnormalities were associated with the dose of antibodies and APOε4 genotyping, higher dosing necessity was recognized, and accurate measures for specific targets, such as reduction of Aβ plaque burden on amyloid PET, were required.10
Many ongoing mAbs trials are in phase 3, including aducanumab,67 gantenerumab,68 and BAN240169 in prodromal and very mild AD, and crenezumab,70 gantenerumab, and solanezumab71 in studies for preclinical or at-risk populations. First results from aducanumab and BAN2401 trials suggested, at first, a treatment-related result of reducing in cerebral amyloid burden in agreement to deceleration of cognitive decline in patients with prodromal and very mild AD.71,72 On the contrary, the initial trial of gantenerumab in prodromal AD was prematurely stopped for lack of efficacy, but exploratory analyses suggest that higher dosing of gantenerumab may be needed for clinical efficacy and an open-label extension for participating patients with mild AD is continued, simultaneously with a double-blind, placebo-controlled study in patients with mild AD.4,68 Similarly, until now, solanezumab did not delay rates of brain atrophy.73
Intravenous doses of LY3002813 (donanemab) and LY3372993 are studied in participants with mild cognitive impairment (MCI) and mild to moderate AD in separate phase 1 clinical studies.4
Passive Aβ immunotherapy—via immunoglobulins
Anti-Aβ antibodies are included in naturally occurring autoantibodies. In contrast to mAbs, blood-derived human anti-Aβ immunoglobulin G (IgG) Abs are polyclonal, with lower avidity for single Aβ molecules, and higher for a broader range of epitopes, especially in Aβ oligomers and fibrils. The natural presence of antibodies against Aβ has been reported in intravenous immunoglobulin (IVIg); thus, IVIg has been considered as a possible AD treatment. Intravenous immunoglobulin is obtained from plasma of healthy donors and is made up of human Abs mainly of the IgG-type.5,74
Nevertheless, the first completed phase 3 trial of IVIg as a treatment for AD demonstrated good tolerability but lack of efficacy of the agent on clinical stability or delay of cognitive or functional decline of participants with mild and moderate AD.74
Another strategy directed at diminishing the accumulation of Aβ in the brain is based in altering the transportation of Aβ through the BBB. A recent therapeutic method performs plasma exchange (PE) with albumin replacement, inducing the shifting of the existing dynamic equilibrium between plasma and brain Aβ. This therapeutic method is based in the following considerations: (1) high levels of Aβ aggregation in the brain are accompanied by low levels of Aβ in CSF in AD, (2) albumin is the main protein transporter in humans, (3) albumin binds around the 90% of the circulating Aβ, and (4) albumin has proved Aβ-binding ability. Consequently, it is suggested that PE-mediated possession of albumin-bound Aβ would increase the shift of free Aβ from CSF to plasma to correct the imbalance between brain and blood Aβ levels.75
A phase 3 trial called Alzheimer’s Management by Albumin Replacement (AMBAR) in mild and moderate AD assesses PE with several replacement volumes of albumin, with or without intravenous immunoglobulin.76
Furthermore, an ongoing phase 2 study evaluates IVIg Octagram 10% in mild and moderate AD.4
A novel immunotherapeutic strategy that targets simultaneously Aβ and tau is represented by the NPT088 agent. NPT088 is a mixture of the capsid protein of bacteriophage M13 (g3p) and human-IgG1-Fc. NPT088 reduced Aβ and ptau aggregates and improved cognition in aged Tg2576 mice. The agent is currently assessed in a phase 1 clinical study.77
Tau-related mechanisms—DMTs
Anti-phospho-tau approaches consist a major potential treatment strategy, even if there are yet no agents with this specific MOA advanced in phase 3 studies.
Only 1 agent with tau-related mechanism is evaluated in phase 2/3, whereas 10 agents that target tau as one of their mechanisms are evaluated in phase 2, and 5 more agents with tau-related mechanism are assessed in phase 1 studies.4
Prevention of ptau formation
The hyperphosphorylation of tau is induced by kinases.78 Thus, kinase inhibitors are examined as potential therapeutic approaches targeting tau. Glycogen synthase kinase 3 (GSK3β) has become prominent as a possible therapeutic target. The most studied GSK3 inhibitor is lithium chloride, a therapeutic agent for affective disorders, which seems to prevent phosphorylation of tau in mouse models. Lithium is currently reassessed within the novel framework for drug research.79
Another GSK-3 inhibitor, tideglusib, did not meet phase 2 clinical endpoints in patients with mild and moderate AD.80
ANAVEX 2-73 is evaluated in a phase 2 trial, for eligible subjects AD MCI or mild AD.81 ANAVEX 2-73 is also a GSK-3b inhibitor but additionally it is a high affinity sigma 1 receptor agonist and a low-affinity muscarinic agonist.4 Results presented at 2019 Alzheimer’s Association International Conference (AAIC) revealed that patients treated with ANAVEX 2-73 had high levels of 2 gut microbiota families, Ruminococcaceae and Porphyromonadaceae, which were associated with improved activities of daily living. The effect might potentially be reversal of the microbiota imbalances and might have a homeostatic effect on the brain-gut-microbiota axis.81
Inhibitors of tau aggregation
Methylene blue (MB), a known phenothiazine, is evaluated in AD studies as a potential tau aggregation inhibitor. The problem with this drug is that urine is colored blue, resulting in a lack of blinding. A monotherapy trial with MB on mild and moderate AD (NCT00515333) has demonstrated some clinical benefit in moderate, but not mild AD.82 However, the methodology of the study, as blinding is impossible, has been highly criticized.83
Methylene blue’s derivative TRx0237 (LMTX) which was studied in phase 3 failed finally to show efficacy, and based on the analysis of the results, a new phase 2/3 study named LUCIDITY was started 1 year ago in subjects with mild AD with a lower dose of the agent.84
Microtubule stabilizers
The microtubule-stabilizing agent davunetide was studied in a phase 2 trial but it did not meet the clinical end points.85
TPI-287 (abeotaxane), a small molecule derived from taxol, is a microtubule protein modulator. It was administered intravenously to patients with mild to moderate AD in a phase 1/2 study (NCT01966666). First results presented report that the agent was not well tolerated by the participants.84
IONIS MAPTRx (BIIB080), a microtubule-associated protein tau RNA inhibitor, an antisense oligonucleotide, is assessed in a phase 2 clinical study that is still in the recruiting process of patients with mild AD (NCT02623699).86
Targeting posttranslational modifications of Tau
Another tau modification that promotes aggregation besides phosphorylation is posttranslational modification by lysine acetylation. Thus, the use of inhibitors of tau acetylation is proposed as a possible therapeutic approach for AD.
Nilotinib is a c-Abl tyrosine kinase inhibitor which is used in patients with leukemia. It also appears to trigger intraneuronal autophagy to clear tau. It is now studied in a phase 2 trial in individuals with mild to moderate AD (NCT02947893).4,83
Promotion of Tau clearance—immunotherapy
Recently emerged evidence in various animal models strongly suggests that targeting ptau epitopes is a practical approach to induce antibody responses that are able to promote tau clearance.81 Hence, a number of active and passive immunotherapy projects have reached clinical trials for AD treatment.83
Active immunotherapy
AADvac1 contains a synthetic tau peptide and is currently studied in a phase 2 clinical study in mild to moderate AD (NCT02579252).4,10,83
Passive immunotherapy
ABBV-8E12 is a humanized anti-tau MAb assessed in a phase 2 clinical study in patients with early AD (NCT02880956).87
BIIB092 is a humanized IgG4 MAb against tau fragments derived from the stem cells of a patient with familial AD.84 A phase 2 clinical trial assesses the safety and efficacy of the agent in participants with AD MCI and mild AD.4
RO7105705 (MTAU9937 A) is an anti-tau MAb which is assessed in a phase 2 study in individuals with prodromal and mild AD (NCT03289143).83,88
Three other anti-tau mAbs (BIIB076, JNJ-63733657, and LY3303560) are currently assessed in phase 1 clinical trials.4
DMTs with other mechanisms
Neuroprotection
AGB101 (low-dose extended-release levetiracetam) is an SV2A modulator that is assessed in a phase 3 clinical trial as a repurposed agent (approved for use in another indication, not epilepsy but MCI due to AD). It is supposed to reduce neuronal hyperactivity induced by Aβ (NCT03486938) (Diagram 1).4
Diagram 1.
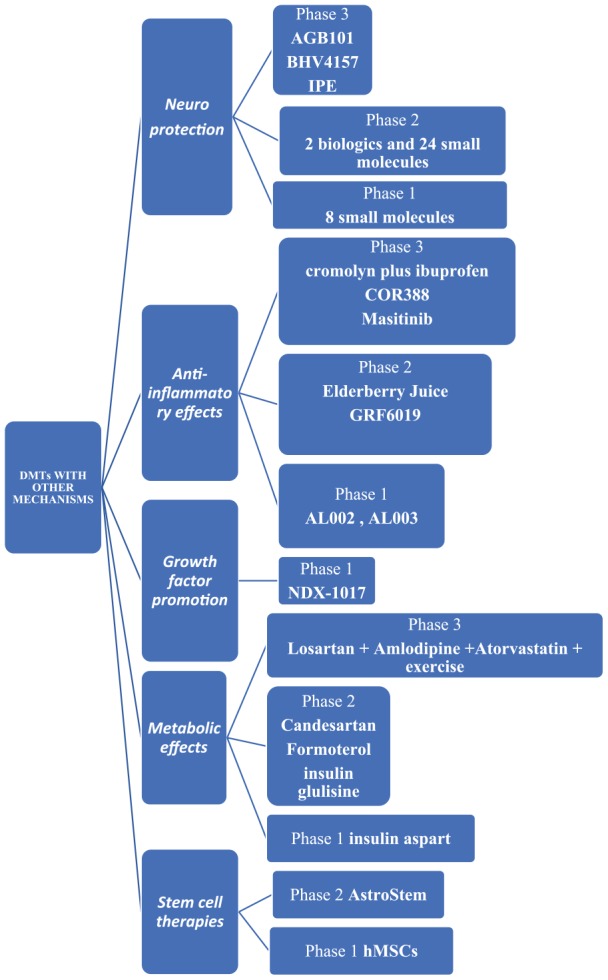
DMTs with other mechanisms. DMTs indicate disease-modifying therapies; hMSCs, human mesenchymal stem cells.
BHV4157 (troriluzole) is a glutamate modulator that reduces synaptic levels of glutamate and is assessed in a phase 3 clinical trial (NCT03605667).4
Icosapent ethyl is the eicosapentaenoic acid (EPA) omega-3 fatty acid in a purified form. It is supposed to protect neurons from disease pathology and is assessed in a phase 3 clinical trial (NCT02719327).4
There are also 2 biologics and 24 small molecules with neuroprotection as one of their mechanisms4 assessed in phase 2 clinical studies and 8 small molecules in phase 1 clinical trials.4
Anti-inflammatory effects
Although neuroinflammation has been proposed as a possible mechanism for the pathogenesis of AD more than 30 years ago, only recently research is spurred into neuroiflammation probably due to 2 enlightening discoveries: first, there is evidence that activated glial cells are involved in the formation of the brain lesions in AD and second, epidemiological studies revealed that patients with rheumatoid arthritis, who are treated with anti-inflammatory drugs for decades, are spared from AD.89 Further exploration of the inflammatory mechanisms in the disease showed that activation of glial cells, microglia, and astrocytes induces the production of inflammatory cytokines, mainly interleukin 1β (IL-1β) and tumor necrosis factor α (TNF-α). More specifically, TNF-α signaling has been proved to exacerbate both Aβ aggregation and tau phosphorylation in vivo,90 whereas its levels have been found elevated in brain and plasma of patients with AD.91
According to the previously mentioned neuroinflammatory mechanisms, it is established by multiple biomarker and epidemiological studies of Aβ levels in the CSF and the brain that nonsteroidal anti-inflammatory drugs, complement activation blockers, and other anti-inflammatory agents could postpone the clinical onset of AD if they are timely and for a long time applied, such as in rheumatoid arthritis.89
Furthermore, the already existing TNF-α inhibitors (TNFIs), which are FDA-approved biologic drugs (mAbs) for the treatment of rheumatoid arthritis, Crohn disease, psoriatic arthritis, and other peripheral inflammatory diseases, are studied as a potential therapeutic strategy for AD. The TNF-α–specific mAbs are the agents infliximab, adalimumab, golimumab, and certolizumab, whereas etanercept is a recombinant fusion protein, which is also a TNFI.91 The limited BBB penetration of these agents is the main drawback for their development. Peripheral targeting of TNF-α activity is the one proposed method for the tackling of the problem and reengineering of the TNFIs to enable BBB penetration is the other.91 To sum up, large-scale randomized controlled trials assessing the safety and the effectiveness of TNFIs on patients with AD are warranted.
The following are the anti-inflammatory agents currently assessed in phase 3 clinical trials:
ALZT-OP1a plus ALZT-OP1b (cromolyn plus ibuprofen) is a combination of a mast cell stabilizer and an anti-inflammatory agent, respectively, assessed in a phase 3 clinical trial (NCT02547818).4
COR388 targets a periodontal pathogen acting as bacterial protease inhibitor that reduces neuroinflammation and consequently hippocampal degeneration and is currently assessed in a phase 3 clinical trial (NCT03823404).4
Masitinib acts on mast cells as a selective tyrosine kinase inhibitor and a modulator of neuroinflammation. It is assessed in a phase 3 clinical trial (NCT01872598).4
The following are the anti-inflammatory agents studied in phase 2:
Elderberry Juice improves the mitochondrial function acting as powerful antioxidant rich in anthocyanins (NCT02414607) and GRF6019, a human plasma protein fraction administered with infusions, based on the hypothesis that brain neuroinflammation can be counteracted by young blood parabiosis (NCT03520998, NCT03765762).4
Anti-inflammatory agents studied in phase 1 are the mAbs AL002, AL003 (NCT03635047, NCT03822208).4
Growth factor promotion
NDX-1017 is an hepatocyte growth factor with the role to regenerate neurons, which is studied in a phase 1 clinical trial (NCT03298672).4
Metabolic effects
Losartan plus amlodipine plus atorvastatin plus exercise is a combination repurposed agent suggested to succeed significant reduction of the vascular risk capable of preserving cognitive function. It is assessed in a phase 3 clinical trial (NCT02913664).4
Candesartan, an angiotensin receptor blocker; formoterol, a β2 adrenergic receptor agonist; and intranasal insulin glulisine, which rises brain insulin signaling, are currently studied in phase 2 clinical trials (NCT02646982, NCT02500784, NCT02503501, respectively), whereas intranasal insulin aspart is assessed in a phase 1 clinical study.4
Stem cell therapies
AstroStem is a stem-cell-based treatment administered 10 times intravenously, which consists of stem cells derived from autologous adipose tissue. AstroStem is currently assessed in a phase 2 study (NCT03117738), whereas hMSCs (human mesenchymal stem cells) treatment is assessed in a phase 1 study (NCT02600130).4
Symptomatic agents
Symptomatic treatments are agents that target and improve the clinical symptoms of the disease, either cognitive or BPSD, without modifying the pathological steps leading to AD or acting on the evolution of the disease, as DMTs are supposed to do.
Overall, there are 33 symptomatic agents in current trials: 19 agents aim to improve cognition and 14 target BPSD.
Eleven of them are studied in phase 3: 3 cognitive intensifiers and 8 acting on BPSD.
Twenty symptomatic agents are in phase 2: 14 cognitive intensifiers and 6 acting on BPSD.
There are also 2 cognitive intensifiers being studied in phase 1.4
Discussion
Arduous research efforts persist to develop effective DMTs for AD, as well as symptomatic therapeutics. A plethora of continuing phase 1, 2, and 3 human studies are focused on various treatment targets in AD. Given the recent experience of a high proportion of lack of success in AD clinical trials on therapeutic agents, more recent trials appear robustly empowered by the integration of developments in biomarkers of AD, of the targeting of a single primary outcome, especially in prodromal AD studies, of the enrollment of earlier populations and the innovative trial designs.91-93
At the same time, innovative research targets the development of more sophisticated diagnostic tools (neuroimaging, fluid, proteomic, and genomic AD biomarkers), whereas prevention studies for the disease are also ongoing.10
If all these research efforts come to fruition, an effective “precision medicine” context could be applied in every patient with AD in the near future: risk factor elimination, comorbid disease treatment, and personalized advice for lifestyle modification will be provided. An AD biomarkers and neuropsychological evaluation profile will be outlined. Afterward, the patient may start a combination of DMTs tailored to meet his genetic, neuroimaging, biochemical, and neuropsychological requirements.3,94
Furthermore and beyond any DMT perspective, clinicians should always maintain a patient/caregiver-targeted dealing with AD. Establishing a strong therapeutic alliance with the patient and his or her caregivers with a holistic and realistic approach involving psychoeducation, behavioral, and environmental techniques; advanced planning for future care needs; and appropriate pharmaceutical treatment is not only an efficient but also an ethical care in AD.
Footnotes
Funding:The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests:The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions: SGP conceptualized the study, developed the proposal and coordinated the project. KGY completed initial data entry and analysis, and wrote the report. Both authors read and approved the final manuscript.
ORCID iD: Konstantina G Yiannopoulou  https://orcid.org/0000-0002-4876-4566
https://orcid.org/0000-0002-4876-4566
References
- 1. Prince M, Bryce R, Albanese E, Wimo A, Ribeiro W, Ferri CP. The global prevalence of dementia: a systematic review and metaanalysis. Alzheimers Dement. 2013;9:63-75. [DOI] [PubMed] [Google Scholar]
- 2. Kingston A, Comas-Herrera A, Jagger C. Forecasting the care needs of the older population in England over the next 20 years: estimates from the Population Ageing and Care Simulation (PACSim) modelling study. Lancet Public Health. 2018;3:e447-e455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Scheltens P, Blennow K, Breteler MMB, et al. Alzheimer’s disease. Lancet. 2016;388:505-517. [DOI] [PubMed] [Google Scholar]
- 4. Cummings J, Lee G, Ritter A, Sabbagh M, Zhong K. Alzheimer’s disease drug development pipeline: 2019. Alzheimers Dement. 2019;5:272-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yiannopoulou KG, Papageorgiou SG. Current and future treatments for Alzheimer’s disease. Ther Adv Neurol Disord. 2013;6:19-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Anand A, Patience A, Sharma N, Khurana N. The present and future of pharmacotherapy of Alzheimer’s disease: a comprehensive review. Eur J Pharmacol. 2017;815:364-375. [DOI] [PubMed] [Google Scholar]
- 7. Hampel H, Mesulam MM, Cuello AC, et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain. 2018;141:1917-1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Matsunaga S, Kishi T, Iwata N. Memantine monotherapy for Alzheimer’s disease: a systematic review and meta-analysis. PLoS ONE. 2015;10:e0123289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bondi MW, Edmonds EC, Salmon DP. Alzheimer’s disease: past, present, and future. J Int Neuropsychol Soc. 2017;23:818-831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Atri A. Current and future treatments in Alzheimer’s disease. Semin Neurol. 2019;39:227-240. [DOI] [PubMed] [Google Scholar]
- 11. Cummings J, Morstorf T, Lee G. Alzheimer’s disease drug development pipeline: 2016. Alzheimers Dement. 2016;2:222-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cummings J, Lee G, Mortsdorf T, Ritter A, Zhong K. Alzheimer’s disease drug development pipeline: 2017. Alzheimers Dement. 2017;3:367-384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cummings J, Lee G, Ritter A, Zhong K. Alzheimer’s disease drug development pipeline: 2018. Alzheimers Dement. 2018;4:195-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Na R, Yang JH, Yeom Y, et al. A systematic review and meta-analysis of nonpharmacological interventions for moderate to severe dementia. Psychiatry Investig. 2019;16:325-335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kishita N, Backhouse T, Mioshi E. Nonpharmacological interventions to improve depression, anxiety, and quality of life (QoL) in people with dementia: an overview of systematic reviews. J Geriatr Psychiatry Neurol. 2019;33:28-41. [DOI] [PubMed] [Google Scholar]
- 16. Howard R, McShane R, Lindesay J, et al. Nursing home placement in the donepezil and memantine in moderate to severe Alzheimer’s disease (DOMINO-AD) trial: secondary and post-hoc analyses. Lancet Neurol. 2015;14:1171-1181. [DOI] [PubMed] [Google Scholar]
- 17. Farlow M, Anand R, Messina J, Hartman R, Jr, Veach J. A 52-week study of the efficacy of rivastigmine in patients with mild to moderately severe Alzheimer’s disease. Eur Neurol. 2000;44:236-241. [DOI] [PubMed] [Google Scholar]
- 18. Rountree SD, Atri A, Lopez OL, Doody RS. Effectiveness of antidementia drugs in delaying Alzheimer disease progression. Alzheimers Dement. 2013;9:338-345. [DOI] [PubMed] [Google Scholar]
- 19. Tricco AC, Ashoor HM, Soobiah C, et al. Comparative effectiveness and safety of cognitive enhancers for treating Alzheimer’s disease: systematic review and network metaanalysis. J Am Geriatr Soc. 2018;66:170-178. [DOI] [PubMed] [Google Scholar]
- 20. Calvo-Perxas L, Turro-Garriga O, Vilalta-Franch J, et al. Trends in the prescription and long-term utilization of antidementia drugs among patients with Alzheimer’s disease in Spain: a cohort study using the registry of dementias of Girona. Drugs Aging. 2017;34:303-310. [DOI] [PubMed] [Google Scholar]
- 21. Bessey LJ, Walaszek A. Management of behavioral and psychological symptoms of dementia. Curr Psychiatry Rep. 2019;21:66. [DOI] [PubMed] [Google Scholar]
- 22. Hukins D, Macleod U, Boland JW. Identifying potentially inappropriate prescribing in older people with dementia: a systematic review. Eur J Clin Pharmacol. 2019;75:467-481. [DOI] [PubMed] [Google Scholar]
- 23. By the American Geriatrics Society 2015 Beers Criteria Update Expert Panel. American Geriatrics Society 2015 updated beers criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2015;63:2227-2246. [DOI] [PubMed] [Google Scholar]
- 24. Savolainen-Peltonen H, Rahkola-Soisalo P, Hoti F, et al. Use of postmenopausal hormone therapy and risk of Alzheimer’s disease in Finland: nationwide case-control study. BMJ. 2019;364:l665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gray SL, Anderson ML, Dublin S, et al. Cumulative use of strong anticholinergic medications and incident dementia. JAMA Intern Med. 2015;175:401-407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Levy HB. Polypharmacy reduction strategies: tips on incorporating American geriatrics society beers and screening tool of older people’s prescriptions criteria. Clin Geriatr Med. 2017;33:177-187. [DOI] [PubMed] [Google Scholar]
- 27. Blokh D, Stambler I, Lubart E, Mizrahi EH. The application of information theory for the estimation of old-age multimorbidity. Geroscience. 2017;39:551-556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Anderson RM, Hadjichrysanthou C, Evans S, Wong MM. Why do so many clinical trials of therapies for Alzheimer’s disease fail? Lancet. 2017;390:2327-2329. [DOI] [PubMed] [Google Scholar]
- 29. Doody RS, Raman R, Farlow M, et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N Engl J Med. 2013;369:341-350. [DOI] [PubMed] [Google Scholar]
- 30. Vandenberghe R, Rinne JO, Boada M, et al. Bapineuzumab for mild to moderate Alzheimer’s disease in two global, randomized, phase 3 trials. Alzheimers Res Ther. 2016;8:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. The Lancet Neurology. Solanezumab: too late in mild Alzheimer’s disease. Lancet Neurol. 2017;16:97. [DOI] [PubMed] [Google Scholar]
- 32. Burki T. Alzheimer’s disease research: the future of BACE inhibitors. Lancet. 2018;391:2486. [DOI] [PubMed] [Google Scholar]
- 33. Egan MF, Kost J, Voss T, et al. Randomized trial of verubecestat for prodromal Alzheimer’s disease. N Engl J Med. 2019;380:1408-1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Henley D, Raghavan N, Sperling R, Aisen P, Raman R, Romano G. Preliminary results of a trial of atabecestat in preclinical Alzheimer’s disease. N Engl J Med. 2019;380:1483-1485. [DOI] [PubMed] [Google Scholar]
- 35. Gauthier S, Albert M, Fox N, et al. Why has therapy development for dementia failed in the last two decades. Alzheimers Dement. 2016;12:60-64. [DOI] [PubMed] [Google Scholar]
- 36. Romero K, Ito K, Rogers JA, et al. The future is now: model-based clinical trial design for Alzheimer’s disease. Clin Pharmacol Ther. 2015;97:210-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hadjichrysanthou C, Ower AK, de Wolf F, Anderson RM. The development of a stochastic mathematical model of Alzheimer’s disease to help improve the design of clinical trials of potential treatments. PLoS ONE. 2018;13:e0190615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dubois B, Hampel H, Feldman HH, et al. Preclinical Alzheimer’s disease: definition, natural history, and diagnostic criteria. Alzheimers Dement. 2016;12:292-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jack CR, Jr, Bennett DA, Blennow K, et al. NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018;14:535-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Guzman-Martinez L, Maccioni RB, Farías GA, Fuentes P, Navarrete LP. Biomarkers for Alzheimer’s disease. Curr Alzheimer Res. 2019;16(6):518-528. [DOI] [PubMed] [Google Scholar]
- 41. Coric V, Salloway S, van Dyck CH, et al. Targeting prodromal Alzheimer disease with avagacestat: a randomized clinical trial. JAMA Neurol. 2015;72:1324-1333. [DOI] [PubMed] [Google Scholar]
- 42. Muntimadugu E, Dhommati R, Jain A, Challa VG, Shaheen M, Khan W. Intranasal delivery of nanoparticle encapsulated tarenflurbil: a potential brain targeting strategy for Alzheimer’s disease. Eur J Pharm Sci. 2016;92:224-234. [DOI] [PubMed] [Google Scholar]
- 43. Penninkilampi R, Brothers HM, Eslick GD. Pharmacological agents targeting γ-secretase increase risk of cancer and cognitive decline in Alzheimer’s disease patients: a systematic review and meta-analysis. J Alzheimers Dis. 2016;53:1395-1404. [DOI] [PubMed] [Google Scholar]
- 44. Steiner H, Fukumori A, Tagami S, Okochi M. Making the final cut: pathogenic amyloid-β peptide generation by γ-secretase. Cell Stress. 2018;2:292-310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lopez Lopez C, Tariot PN, Caputo A, et al. The Alzheimer’s Prevention Initiative Generation Program: study design of two randomized controlled trials for individuals at risk for clinical onset of Alzheimer’s disease. Alzheimers Dement;5:216-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Panza F, Lozupone M, Solfrizzi V, et al. BACE inhibitors in clinical development for the treatment of Alzheimer’s disease. Expert Rev Neurother. 2018;18:847-857. [DOI] [PubMed] [Google Scholar]
- 47. Cho WH, Park JC, Kim DH, et al. ID1201, the ethanolic extract of the fruit of Melia toosendan ameliorates impairments in spatial learning and reduces levels of amyloid beta in 5XFAD mice. Neurosci Lett. 2014;583:170-175. [DOI] [PubMed] [Google Scholar]
- 48. Vellas B, Sol O, Snyder PJ, et al. EHT0202 in Alzheimer’s disease: a 3-month, randomized, placebo-controlled, double-blind study. Curr Alzheimer Res. 2011;8:203-212. [DOI] [PubMed] [Google Scholar]
- 49. Alzoubi KH, Al Subeh ZY, Khabour OF. Molecular targets for the interactive effect of etazolate during post-traumatic stress disorder: role of oxidative stress, BDNF and histones. Behav Brain Res. 2019;369:111930. [DOI] [PubMed] [Google Scholar]
- 50. Salloway S, Sperling R, Keren R, et al. ; ELND005-AD201 Investigators. A phase 2 randomized trial of ELND005, scyllo-inositol, in mild to moderate Alzheimer disease. Neurology. 2011;77:1253-1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Stark T, Lieblein T, Pohland M, et al. Peptidomimetics that inhibit and partially reverse the aggregation of Aβ1-42. Biochemistry. 2017;56:4840-4849. [DOI] [PubMed] [Google Scholar]
- 52. Nimmagadda A, Shi Y, Cai J. γ-AApeptides as a new strategy for therapeutic development. Curr Med Chem. 2019;26:2313-2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Xu P, Zhang M, Sheng R, Ma Y, et al. Synthesis and biological evaluation of deferiprone-resveratrol hybrids as antioxidants, Aβ1-42aggregation inhibitors and metal-chelating agents for Alzheimer’s disease. Eur J Med Chem. 2017;15:174-186. [DOI] [PubMed] [Google Scholar]
- 54. Krishnan HS, Bernard-Gauthier V, Placzek MS. Metal protein-attenuating compound for PET neuroimaging: synthesis and preclinical evaluation of [11C]PBT2. Mol Pharm. 2018;15:695-702. [DOI] [PubMed] [Google Scholar]
- 55. Villemagne VL, Rowe CC, Barnham KJ, et al. A randomized, exploratory molecular imaging study targeting amyloid β with a novel 8-OH quinoline in Alzheimer’s disease: the PBT2-204 IMAGINE study. Alzheimers Dement. 2017;3:622-635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wisniewski T, Goñi F. Immunotherapeutic approaches for Alzheimer’s disease. Neuron. 2015;85:1162-1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Goñi F, Herline K, Peyser D, et al. Immunomodulation targeting of both Aβ and tau pathological conformers ameliorates Alzheimer’s disease pathology in TgSwDI and 3xTg mouse models. J Neuroinflammation. 2013;10:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yoshiyama Y, Lee VM, Trojanowski JQ. Therapeutic strategies for tau mediated neurodegeneration. J Neurol Neurosurg Psychiatry. 2013;84:784-795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lopez Lopez C, Tariot PN, Caputo A, et al. The Alzheimer’s Prevention Initiative Generation Program: study design of two randomized controlled trials for individuals at risk for clinical onset of Alzheimer’s disease. Alzheimers Dement. 2019;12:216-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lacosta AM, Pascual-Lucas M, Pesini P, et al. Safety, tolerability and immunogenicity of an active anti-Aβ40 vaccine (ABvac40) in patients with Alzheimer’s disease: a randomised, double-blind, placebocontrolled, phase I trial. Alzheimers Res Ther. 2018;10:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kim H, Seo EH, Lee SH, Kim BJ. The telomerase-derived anticancer peptide vaccine GV1001 as an extracellular heat shock protein-mediated cell-penetrating peptide. Int J Mol Sci. 2016;17: E2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hull M, Sadowsky C, Arai H, et al. Long-term extensions of randomized vaccination trials of ACC-001 and QS-21 in mild to moderate Alzheimer’s disease. Curr Alzheimer Res. 2017;14:696-708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wang CY, Wang PN, Chiu MJ, et al. UB-311, a novel UBITh® amyloid β peptide vaccine for mild Alzheimer’s disease. Alzheimers Dement. 2017;143:262-272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Davtyan H, Ghochikyan A, Petrushina I, et al. Immunogenicity, efficacy, safety, and mechanism of action of epitope vaccine (Lu AF20513) for Alzheimer’s disease: prelude to a clinical trial. J Neurosci. 2013;33:4923-4934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gilman S, Koller M, Black RS, et al. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;64:1553-1562. [DOI] [PubMed] [Google Scholar]
- 66. Salloway S, Sperling R, Fox NC, et al. ; Bapineuzumab 301 and 302 Clinical Trial Investigators. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370:322-333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Doody RS, Thomas RG, Farlow M, et al. ; Alzheimer’s Disease Cooperative Study Steering Committee; Solanezumab Study Group. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370:311-321. [DOI] [PubMed] [Google Scholar]
- 68. Sevigny J, Chiao P, Bussière T, et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature. 2016;537:50-56. [DOI] [PubMed] [Google Scholar]
- 69. Ostrowitzki S, Lasser RA, Dorflinger E, et al. A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimers Res Ther. 2017;9:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Logovinsky V, Satlin A, Lai R, et al. Safety and tolerability of BAN2401–a clinical study in Alzheimer’s disease with a protofibril selective Aβ antibody. Alzheimers Res Ther. 2016;8:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Yang T, Dang Y, Ostaszewski B, et al. Target engagement in an Alzheimer trial: crenezumab lowers amyloid β oligomers in cerebrospinal fluid. Ann Neurol. 2019;86:215-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Panza F, Seripa D, Lozupone M, et al. The potential of solanezumab and gantenerumab to prevent Alzheimer’s disease in people with inherited mutations that cause its early onset. Expert Opin Biol Ther. 2018;18:25-35. [DOI] [PubMed] [Google Scholar]
- 73. AAIC (July 2018. news). https://www.alzforum.org/therapeutics/ban2401.
- 74. Schwarz AJ, Sundell KL, Charil A, et al. Magnetic resonance imaging measures of brain atrophy from the EXPEDITION3 trial in mild Alzheimer’s disease. Alzheimers Dement. 2019;5:328-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Relkin NR, Thomas RG, Rissman RA, et al. A phase 3 trial of IV immunoglobulin for Alzheimer disease. Neurology. 2017;88:1768-1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Boada M, Lopez O, Nunez L, et al. Plasma exchange for Alzheimer’s disease Management by Albumin Replacement (AMBAR) trial: study design and progress. Alzheimers Dement. 2019;5:61-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Levenson JM, Schroeter S, Carroll JC, et al. NPT088 reduces both amyloid-β and tau pathologies in transgenic mice. Alzheimers Dement. 2016;2:141-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Yiannopoulou KG, Karydakis KD, Sakka P. Therapeutic targets in Alzheimer’s disease. Primary Psychiatry. 2009;16:29-36. [Google Scholar]
- 79. Hampel H, Lista S, Mango D, et al. Lithium as a treatment for Alzheimer’s disease: the systems pharmacology perspective. J Alzheimers Dis. 2019;69:615-629. [DOI] [PubMed] [Google Scholar]
- 80. Lovestone S, Boada M, Dubois B, et al. A phase II trial of tideglusib in Alzheimer’s disease. J Alzheimers Dis. 2015;45:75-88. [DOI] [PubMed] [Google Scholar]
- 81. Parmentier F, Etcheto A, Missling CU, et al. Exploring Gut Microbiota as a Source of Potential Biomarkers: Initial Results from the ANAVEX®2-73 Alzheimer’s Disease Clinical Study [O4-02-04]. AAIC; 2019. http://www.arianapharma.com/wp-content/uploads/2019/07/Anavex-Microbiota-Presentation-AAIC-July-2019-FINAL-1.pdf. [Google Scholar]
- 82. Wischik CM, Staff RT, Wischik DJ, et al. Tau aggregation inhibitor therapy: an exploratory phase 2 study in mild or moderate Alzheimer’s disease. J Alzheimers Dis. 2015;44:705-720. [DOI] [PubMed] [Google Scholar]
- 83. Medina M. An overview on the clinical development of tau-based therapeutics. Int J Mol Sci. 2018;19: E1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wilcock GK, Gauthier S, Frisoni GB, et al. Potential of low dose leuco-methylthioninium bis(Hydromethanesulphonate) (LMTM) monotherapy for treatment of mild Alzheimer’s disease: cohort analysis as modified primary outcome in a phase III clinical trial. J Alzheimers Dis. 2018;61:435-457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Morimoto BH, Schmechel D, Hirman J, Blackwell A, Keith J, Gold M. A double-blind, placebo-controlled, ascending-dose, randomized study to evaluate the safety, tolerability and effects on cognition of AL-108 after 12 weeks of intranasal administration in subjects with mild cognitive impairment. Dement Geriatr Cogn Disord. 2013;35:325-336. [DOI] [PubMed] [Google Scholar]
- 86. Lane RM, Kordasiewicz HB, Smith AE, et al. Rationale for and development of IONIS-MAPTRx, the first tau-lowering antisense oligonucleotide, in patients with mild AD (POSTER #M158). 142nd Annual Meeting of The American Neurological Association; October 15-17, San Diego, CA. [Google Scholar]
- 87. Budur KK, West T, Braunstein JB, et al. Results of a phase 1, single ascending dose, placebo-controlled study of ABBV-8E12 in patients with Progressive Supranuclear Palsy and phase 2 study design in early Alzheimer’s disease. Alzheimers Dement. 2017;13:599-600. [Google Scholar]
- 88. Doody R. Developing disease-modifying treatments in Alzheimer’s disease—a perspective from Roche and Genentech. J Prev Alzheimers Dis. 2017;4:264-272. [DOI] [PubMed] [Google Scholar]
- 89. McGeer PL, Rogers J, McGeer EG. Inflammation, antiinflammatory agents, and Alzheimer’s disease: the last 22 years. J Alzheimers Dis. 2016;54:853-857. [DOI] [PubMed] [Google Scholar]
- 90. Alam Q, Alam MZ, Mushtaq G, et al. Inflammatory process in Alzheimer’s and Parkinson’s diseases: central role of cytokines. Curr Pharm Des. 2016;22:541-548. [DOI] [PubMed] [Google Scholar]
- 91. Chang R, Yee KL, Sumbria RK. Tumor necrosis factor α inhibition for Alzheimer’s disease. J Cent Nerv Syst Dis. 2017;9:1179573517709278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. U.S. Food and Drug Administration, U.S. Department of Health and Human Services. Early Alzheimer’s Disease: Developing Drugs for Treatment. Washington, DC: Guidance for Industry; 2018. [Google Scholar]
- 93. U.S. Food and Drug Administration, U.S. Department of Health and Human Services. Adaptive Designs for Clinical Trials of Drugs and Biologics. Washington, DC: Draft Guidance for Industry; 2018. [Google Scholar]
- 94. Cummings J. The National Institute on Aging-Alzheimer’s Association framework on Alzheimer’s disease: application to clinical trials. Alzheimers Dement. 2018;15:172-178. [DOI] [PMC free article] [PubMed] [Google Scholar]