

Abstract
Background:
In addition to the reduced energy production, characteristic of mitochondrial disorders, nitric oxide (NO) deficiency can occur as well. The NO produced by vascular endothelial cells relaxes vascular smooth muscles, resulting in vasodilation that maintains the patency of small blood vessels and promotes blood flow through microvasculature. Endothelial dysfunction due to inability of vascular endothelium to generate enough NO to maintain adequate vasodilation can result in decreased perfusion in the microvasculature of various tissues, contributing to many complications seen in individuals with mitochondrial diseases. The amino acids arginine and citrulline are NO precursors: increasing their concentrations could potentially restore NO production.
Methods:
In this study, we assessed endothelial dysfunction in children and adolescents with mitochondrial diseases. We also investigated the effect of arginine and citrulline supplementation on endothelial dysfunction in these individuals. We used peripheral arterial tonometry to measure the reactive hyperemic index (RHI), which is low when there is endothelial dysfunction.
Results:
The results demonstrated low RHI in individuals with mitochondrial diseases, indicating endothelial dysfunction. RHI increased with arginine or citrulline supplementation suggesting that supplementation with NO precursors can improve endothelial dysfunction by enhancing NO production.
Conclusions:
This study is the first one to use peripheral arterial tonometry methodology in mitochondrial diseases. The results of this study provide evidence for endothelial dysfunction in mitochondrial diseases and demonstrate that arginine or citrulline supplementation can alleviate the endothelial dysfunction, providing more evidence for the potential therapeutic utility of these amino acids in mitochondrial diseases.
Keywords: Arginine, citrulline, peripheral arterial tonometry, endothelial dysfunction, mitochondrial disease, nitric oxide (NO)
Introduction
Mitochondria are organelles found in all nucleated human cells and perform several vital biological functions including energy generation, calcium homeostasis, and apoptosis regulation. Mitochondria have 2 membranes, with the inner membrane being folded into cristae. The large surface area of cristae allows the accommodation of electron transport chain (ETC) complexes that generate ATP via oxidative phosphorylation, a process incorporating electron transfer via complexes I to IV and ATP synthesis via complex V.1 It has been estimated that more than 1000 different mitochondria-localized proteins are needed for the structure and function of normal mitochondria.2 Mitochondrial DNA (mtDNA) encodes less than 1% of these proteins, whereas the others are encoded by nuclear DNA (nDNA), synthesized in cytoplasm, and imported into mitochondria.1
Primary mitochondrial diseases result from defects in mtDNA or nDNA genes encoding mitochondrial proteins that result in dysfunctional mitochondria. Mitochondrial dysfunction results in a wide range of cellular perturbations. These include aberrant calcium homeostasis, excessive reactive oxygen species (ROS) production, and dysregulated apoptosis. In addition, such mitochondria are unable to generate sufficient energy to meet the needs of various organs, particularly these with high energy demand, including the nervous system, skeletal and cardiac muscles, kidneys, liver, and endocrine system. As a result, mitochondrial diseases are clinically heterogeneous, often involving multiple organs and having variable manifestations.3 The nervous system is particularly vulnerable because its energy needs are very high. Neurological manifestations in mitochondrial diseases include cognitive impairment, epilepsy, peripheral nephropathy, sensorineural hearing loss, optic atrophy, encephalopathy, dementia, migraine, stroke-like episodes, ataxia, spasticity, chorea, and dementia.3
Nitric oxide (NO) deficiency can also occur in mitochondrial disorders and contribute to several complications observed in these diseases.4 NO produced by vascular endothelial cells relaxes vascular smooth muscles, resulting in vasodilation, maintaining patency of small blood vessels and blood flow through microvasculature.5,6 Endothelial dysfunction can result from reduction of endothelium-derived vasodilators (particularly NO), or from increase in endothelium-derived contracting factors including endothelin, angiotensin II, and ROS. The resulting imbalance between endothelium-derived relaxing and contracting factors leads to an impairment of endothelium-dependent vasodilation.7 NO deficiency in mitochondrial diseases can lead to endothelial dysfunction due to inability of vascular endothelium to generate enough NO to maintain adequate vasodilation and blood perfusion. Decreased perfusion in the microvasculature of various tissues can potentially contribute to many complications seen in mitochondrial diseases. In cerebral microvasculature, impaired blood flow can contribute to the pathogenesis of neurological complications such as stroke-like episodes and migraine headaches. Decreased muscular perfusion can contribute to the myopathic manifestations of mitochondrial diseases.4 Accordingly, individuals with mitochondrial diseases are expected to have endothelial dysfunction that could have important clinical consequences. To provide evidence for endothelial dysfunction in mitochondrial diseases, we undertook to assess endothelial dysfunction in children and adolescents with mitochondrial diseases.
NO is formed from arginine via the enzyme NO synthase, which catalyzes the conversion of arginine to citrulline. Citrulline can be converted back to arginine via argininosuccinate synthase and argininosuccinate lyase. The amino acids arginine and citrulline can restore NO production and therefore could treat NO deficiency-related manifestations of mitochondrial diseases.4,8 When NO production in individuals with mitochondrial diseases is enhanced with arginine or citrulline supplementation, endothelial dysfunction might improve because of increased vascular endothelium ability to produce NO. To investigate this theory, we needed to assess endothelial dysfunction before and after arginine or citrulline supplementation. Previous isotope infusion studies in subjects with the mitochondrial disease MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes) demonstrated that citrulline supplementation induced a greater increase in NO synthesis rate than that resulting from arginine supplementation.9,10 Therefore, it has been suggested that citrulline is a more effective NO precursor than arginine.9,10 We assessed that in this study by testing both arginine and citrulline, and comparing their effects on endothelial dysfunction.
Endothelial dysfunction has been classically assessed by the flow-mediated vasodilation (FMD) technique. FMD method uses ultrasound to quantitate the change in brachial artery diameter in response to increased flow after a period of vascular occlusion by a blood pressure cuff. When blood flow is restored, the reperfusion causes mechanical stress which results in endothelial cells releasing NO, leading to vasodilatation. A reduction in such FMD indicates endothelial dysfunction.11 Although FMD is known to be depressed in subjects with atherosclerosis and coronary artery disease,12,13 accurate assessment of FMD has been technically challenging to perform, highly user-dependent, and it requires appropriate training and validation.14,15 Peripheral arterial tonometry is a newer method to assess endothelial dysfunction that is easier to apply and less user-dependent, and demonstrates a good correlation with FMD.16,17 It measures pulse wave amplitude (PWA) to compute a reactive hyperemic index (RHI).18 Lower RHI indicates endothelial dysfunction.19-21 In this study, we used peripheral arterial tonometry to assess endothelial dysfunction. To the best of our knowledge, this is the first study to assess endothelial dysfunction in mitochondrial diseases using peripheral arterial tonometry methodology. The results of this study provide evidence that children and adolescents with mitochondrial diseases have endothelial dysfunction that improves with arginine or citrulline supplementation.
Subjects and Methods
Nine individuals with mitochondrial diseases were studied. The participants were children and adolescents less than 18 years of age who had multiorgan mitochondrial diseases that were confirmed molecularly by identifying mutations in genes known to be associated with mitochondrial diseases (Table 1). Nine healthy control individuals less than 18 years of age were also studied. The study was approved by Al Ain Medical District Human Research Ethics Committee (Protocol number 14/69-CRD 343-14). Informed consents were obtained from parents. All methods were performed in accordance with relevant guidelines and regulations.
Table 1.
Children with mitochondrial diseases who participated in the study.
| 1 | 8-year-old boy with TK2-related mitochondrial DNA depletion syndrome |
| 2 | 6-year-old girl with TK2-related mitochondrial DNA depletion syndrome |
| 3 | 7-year-old girl with MTTA-related mitochondrial myopathy |
| 4 | 17-year-old girl with TMEM70-related mitochondrial complex V deficiency |
| 5 | 9-year-old boy with NDUFS7-related Leigh syndrome |
| 6 | 6-year-old girl with POLG-related Alpers disease |
| 7 | 12-year-old boy with MTTK-related MERRF disease |
| 8 | 8-year-old boy with MTPAP-related spastic ataxia |
| 9 | 13-year-old boy with MTPAP-related spastic ataxia |
Abbreviation: MERRF, myoclonic epilepsy with ragged red fibers.
Baseline endothelial dysfunction was assessed in control individuals. Children and adolescents with mitochondrial diseases had endothelial dysfunction assessment 4 times. The first was at baseline. The second was after intervention: individuals with mitochondrial diseases were randomized to receive either oral arginine or citrulline (500 mg/kg/d if weight <20 kg and 10 g/m2 body surface area per day if weight ⩾20 kg divided in 3 doses) for 2 weeks after which an assessment of endothelial dysfunction was performed. The third was another baseline: the intervention was discontinued and after a 2-week washout period another baseline endothelial dysfunction assessment was performed. The fourth was a second intervention with the supplement not received previously with another assessment of endothelial dysfunction. Blood samples for plasma amino acid analysis were obtained with each endothelial dysfunction assessment to measure the plasma arginine and citrulline levels.
Endothelial dysfunction was assessed by measuring PWA using the EndoPAT instrument, which uses finger plethysmograph sensors to measure the PWA. Each assessment required 15 minutes: PWA was first measured for 5 minutes, then a pressure cuff on the upper arm was inflated to occlude systolic blood pressure for 5 minutes, and then the cuff was deflated to induce reactive hyperemia and the PWA was re-measured for 5 minutes. The RHI was calculated as the ratio of the average PWA during reactive hyperemia divided by the average PWA during the preocclusion period. Two plethysmograph sensors were placed on the index fingers, one for the measurement and the other was used as a control for concurrent nonendothelial dependent changes in vascular tone.18 Normal response is characterized by a distinct increase in the PWA after cuff release compared with the preocclusion value. Impairment of hyperemic response and lower RHI indicate endothelial dysfunction.19-21 Plasma amino acid concentrations were measured by standard high-performance liquid chromatography (HPLC).
The results of RHI and plasma arginine and citrulline levels were expressed as mean ± SD. The results of the 2 baseline assessments (before arginine and before citrulline supplementation) for individuals with mitochondrial diseases were averaged and compared with the values of the control individuals using the unpaired Student t test. Differences in individuals with mitochondrial diseases before and after arginine or citrulline supplementation were assessed by the paired Student t test. Tests were considered statistically significant if P < .05.
Results
Subjects with mitochondrial diseases ranged from 6 to 17 years (mean: 9.6 years) and controls from 3 to 15 years (mean: 9.4 years). Individuals with mitochondrial diseases had lower body weight and body mass index (BMI) (Table 2).
Table 2.
Characteristics of research subjects, presented as mean ± SD.
| Parameter | Children with mitochondrial diseases (n = 9) | Control children (n = 9) | P |
|---|---|---|---|
| Age, y | 9.6 ± 3.7 | 9.4 ± 4.1 | NS |
| Gender, male/female | 5/4 | 5/4 | NS |
| Weight, kg | 24.2 ± 8.5 | 35.7 ± 14.8 | <.5 |
| Height, cm | 126.1 ± 13.0 | 135.1 ± 21.7 | NS |
| BMI, kg/m2 | 14.9 ± 3.7 | 18.5 ± 3.4 | <.5 |
Abbreviations: BMI, body mass index; NS, not statistically significant.
When compared with controls, individuals with mitochondrial diseases had lower baseline RHI (1.02 ± 0.13 vs 1.43 ± 0.49, P < .05) (Figure 1). Baseline plasma arginine and citrulline were also lower in individuals with mitochondrial diseases. The difference in citrulline was statistically significant (22 ± 9 vs 31 ± 11 μmol/L, P < .05), whereas the difference in arginine did not reach a statistically significant level (65 ± 18 vs 81 ± 26 μmol/L, P = .09).
Figure 1.
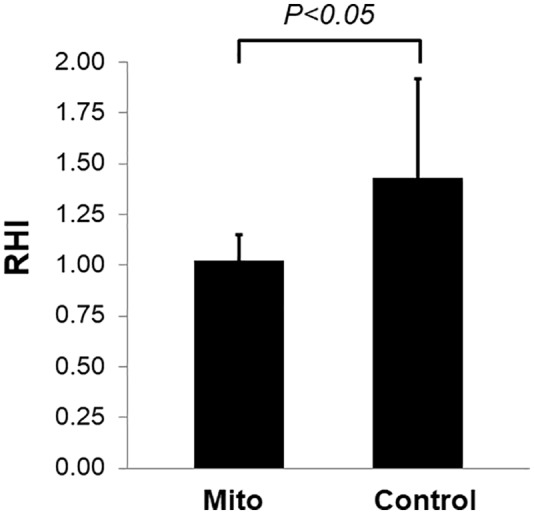
RHI in individuals with mitochondrial diseases and controls. The bars represent the mean (n = 9) and the vertical lines above the bars represent the positive SD. RHI indicates reactive hyperemic index.
When compared with baseline, RHI averages were higher after arginine or citrulline supplementation in individuals with mitochondrial diseases. With arginine supplementation, RHI increased from 1.04 ± 0.14 to 1.20 ± 0.15 (P < .05). With citrulline supplementation, RHI increased from 1.00 ± 0.15 to 1.18 ± 0.25 (P < .05) (Figure 2). Evaluating individual values revealed that 8 out of 9 individuals with mitochondrial diseases showed increased RHI with arginine supplementation that ranged from 3% to 46% (Figure 3A), whereas 7 out of 9 showed increased RHI with citrulline supplementation that ranged from 8% to 55% (Figure 3B). Average RHI increased 15% and 19% with arginine and citrulline supplementation, respectively (Figure 3A and B).
Figure 2.
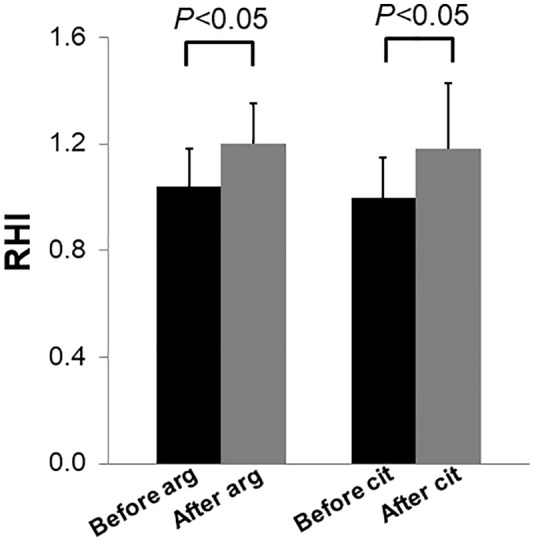
The effect of arginine and citrulline supplementation on RHI in individuals with mitochondrial diseases. The bars represent the mean (n = 9) and the vertical lines above the bars represent the positive SD. RHI indicates reactive hyperemic index.
Figure 3.

The changes in RHI after (A) arginine and (B) citrulline supplementations for individual subjects, with numbers indicated the individuals as listed in Table 1. The average RHI is represented with large dots and thick lines. RHI indicates reactive hyperemic index.
As expected, plasma arginine level increased with arginine supplementation (64 ± 20 → 178 ± 93 μmol/L, P < .05) and citrulline supplementation (66 ± 18 → 200 ± 72 μmol/L, P < .05). Citrulline also increased with arginine supplementation (19 ± 8 → 28 ± 15 μmol/L, P < .05) and citrulline supplementation (24 ± 13 → 161 ± 131 μmol/L, P < .05).
Discussion
The results of this study demonstrate lower RHI in children and adolescents with mitochondrial diseases. Reduced RHI provides evidence for endothelial dysfunction in mitochondrial diseases resulting from reduced NO availability. Several previous studies provided evidence for the occurrence of NO deficiency in mitochondrial diseases. In MELAS, lower concentrations of NO metabolites (nitrite and nitrate) were found during stroke-like episodes.22 In addition, the NO synthesis rate, measured by stable isotope infusion techniques, was low in adults and children with MELAS who were not experiencing acute stroke-like episodes.9,10 Furthermore, NO synthase activity was reduced in cytochrome c oxidase (COX)-deficient fibers of muscle biopsies obtained from individuals with variable mitochondrial diseases, including chronic progressive external ophthalmoplegia (CPEO), mitochondrial myopathy, and MELAS syndrome.23 FMD, which is a function of NO synthesized by endothelial cells in response to reperfusion, was also impaired in individuals with mitochondrial myopathy, MELAS, MERRF (myoclonic epilepsy with ragged red fibers), MIDD (maternally inherited diabetes and deafness), and CPEO, providing further evidence for NO deficiency in mitochondrial diseases.24,25 The finding of lower RHI reflecting endothelial dysfunction in this study provides more evidence for NO deficiency in mitochondrial diseases (Figure 1). Previous studies evaluated different mitochondrial diseases. This study included individuals with other mitochondrial diseases including Alpers, Leigh, MERRF, complex V deficiency, and mitochondrial myopathies (Table 1). Adding evidence for reduced NO availability in these mitochondrial diseases suggests that NO deficiency is not limited to the particular types of mitochondrial diseases studied previously, but is instead a common feature for many mitochondrial disorders.
NO is synthesized from arginine by the enzyme NO synthase which catalyzes the conversion of arginine to NO and citrulline. Citrulline can be converted back to arginine by the combined action of argininosuccinate synthase and argininosuccinate lyase. So both arginine and citrulline act as NO precursors in a wide variety of cells including vascular endothelium. The cause of NO deficiency in mitochondrial disorders is multifactorial due to impaired NO production and postproduction sequestration.4 Impaired NO production can result from decreased availability of NO precursors arginine and citrulline which has been observed in several mitochondrial diseases.9,10,26-28 Decreased NO production can also result from impaired NO synthase activity due to ROS overproduction (oxidative stress) resulting from the ETC impairment. Oxidative stress due to mitochondrial dysfunction may also result in increased asymmetric dimethylarginine (ADMA) which is an endogenous inhibitor of NO synthase.29 Postproduction NO sequestration can occur due to increased COX. Mitochondrial proliferation in endothelial cells in mitochondrial diseases can be associated with increased COX activity, which can react with and thus sequester NO. In addition, oxidative stress can result in decreased NO availability by shunting NO into reactive nitrogen species (RNS) formation.25 Previous studies demonstrated low plasma arginine and citrulline levels in individuals with different mitochondrial diseases including MELAS and NARP (neurogenic weakness, ataxia, and retinitis pigmentosa).9,10,26-28 In our study, arginine and citrulline were lower in individuals with different mitochondrial diseases. Therefore, the results of this study provide further evidence for the role of decreased arginine and citrulline availability in the pathogenesis of NO deficiency in mitochondrial diseases.
Peripheral arterial tonometry was used in this study to assess endothelial dysfunction in children and adolescents with mitochondrial diseases. This study is the first one to use this novel methodology in mitochondrial diseases. Results do not only provide evidence for endothelial dysfunction in mitochondrial diseases but also demonstrate that arginine and citrulline are capable of alleviating endothelial dysfunction (Figure 2). Because arginine and citrulline are NO precursors, the observed improvement in endothelial dysfunction with these supplements is believed to be due to increased ability to produce NO. This suggestion is further supported by previous isotope infusion studies demonstrating increments in NO synthesis resulting from arginine or citrulline supplementation in individuals with MELAS.9,10
Endothelial dysfunction due to impaired NO synthesis resulting in inadequate perfusion can contribute to several complications observed in mitochondrial diseases including stroke-like episodes, myopathy, diabetes, and lactic acidosis. The administration of the NO precursors, arginine and citrulline, could result in increased NO availability and hence may have therapeutic benefits in mitochondrial diseases.4 Arginine supplementation to individuals with MELAS was shown to result in an improvement in clinical symptoms associated with stroke-like episodes and a decrease in the frequency and severity of these episodes.26 However, there are no clinical studies evaluating the effect of arginine or citrulline supplementation on other mitochondrial diseases. This study showed increased RHI indicating improved endothelial dysfunction with arginine or citrulline supplementation in children and adolescents with mitochondrial diseases. The improvement in endothelial dysfunction suggests better vasodilation and blood perfusion in microvasculature which could lead to improvement in clinical complications observed in mitochondrial diseases. Therefore, the observed RHI increments with arginine and citrulline provide more support for their potential therapeutic utility in mitochondrial diseases.
Previous isotope infusion studies in subjects with MELAS demonstrated that citrulline supplementation induced a greater increase in NO synthesis rate than that resulting from arginine supplementation. Therefore, it has been suggested that citrulline is a more effective NO precursor than arginine.9,10 Although a higher increase in RHI was expected to be associated with citrulline supplementation, the RHI changes were comparable with arginine and citrulline supplementation without any significant differences. Although citrulline supplementation was not associated with higher increase in RHI when compared with arginine, both plasma arginine and citrulline levels showed higher increments with citrulline supplementation. This observation, which indicates a better citrulline bioavailability, was also noticed in previous studies.9,10
The average RHI increased with arginine and citrulline supplementation; however, when individual values are evaluated, RHI did not increase in 1 child who took arginine and 2 children who took citrulline (Figure 3A and B). This observation may indicate that arginine and citrulline are ineffective in a small subset of individuals with mitochondrial diseases. The average RHI increased from 1.04 to 1.20 (15%) with arginine supplementation and from 1.00 to 1.18 (19%) with citrulline supplementation. Even with these supplements, the RHI remains significantly lower than control (1.43). Clinical endpoints were not part of this study; therefore, the clinical significance of this partial correction of RHI is not known. Additional studies to evaluate clinical endpoints are needed to determine the potential clinical benefits of these supplements in mitochondrial diseases. Other potential limitations of this study include small sample size, relatively short duration of supplementation, and the heterogeneity of mitochondrial diseases.
In conclusion, the lower RHI in children and adolescents with mitochondrial diseases provides evidence for endothelial dysfunction due to inadequate NO production in mitochondrial diseases. The improvement in RHI with arginine or citrulline supplementation suggests that NO precursor supplementation can counter endothelial dysfunction through enhancing NO production. These results provide evidence for endothelial dysfunction in mitochondrial diseases and support the potential therapeutic utility of arginine and citrulline in mitochondrial diseases. Additional studies to assess the clinical effects of arginine and citrulline in mitochondrial diseases are needed to assess the clinical benefits of these potential therapeutic options.
Acknowledgments
We thank the participated children and their families. We also thank Michael M. Segal, MD, PhD, for his comments on the manuscript content and language.
Footnotes
Funding:The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by the National Research Foundation, Ministry of Higher Education & Scientific Research, United Arab Emirates (UIRCA 2014-696).
Declaration of conflicting interests:The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions: FAJ, NAZ, KA-T, AMAT, and JH participated in patient recruitment, conducting the clinical study procedure, and analyzing the data. AW E-H designed the study, reviewed and analyzed data, and wrote the initial draft. All co-authors participated in writing and reviewing the manuscript.
ORCID iD: Ayman W El-Hattab  https://orcid.org/0000-0002-5737-5271
https://orcid.org/0000-0002-5737-5271
References
- 1. El-Hattab AW, Scaglia F. Mitochondrial cytopathies. Cell Calcium. 2016;60:199-206. [DOI] [PubMed] [Google Scholar]
- 2. Calvo SE, Clauser KR, Mootha VK. MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016;44:D1251-D1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chinnery PF. Mitochondrial disorders overview. In: Adam MP, Ardinger HH, Pagon RA, et al. , eds. GeneReviews®. Seattle, WA: University of Washington, Seattle; 2000:1993-2018. http://www.ncbi.nlm.nih.gov/books/NBK1224/. Accessed June 25, 2018. [Google Scholar]
- 4. El-Hattab AW, Emrick LT, Craigen WJ, Scaglia F. Citrulline and arginine utility in treating nitric oxide deficiency in mitochondrial disorders. Mol Genet Metab. 2012;107:247-252. [DOI] [PubMed] [Google Scholar]
- 5. Green DJ, Maiorana A, O’Driscoll G, Taylor R. Effect of exercise training on endothelium-derived nitric oxide function in humans. J Physiol. 2004;561:1-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Toda N, Okamura T. The pharmacology of nitric oxide in the peripheral nervous system of blood vessels. Pharmacol Rev. 2003;55:271-324. [DOI] [PubMed] [Google Scholar]
- 7. Hadi HAR, Carr CS, Al Suwaidi J. Endothelial dysfunction: cardiovascular risk factors, therapy, and outcome. Vasc Health Risk Manag. 2005;1:183-198. [PMC free article] [PubMed] [Google Scholar]
- 8. El-Hattab AW, Almannai M, Scaglia F. Arginine and citrulline for the treatment of MELAS syndrome. J Inborn Errors Metab Screen. 2017;5:1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. El-Hattab AW, Hsu JW, Emrick LT, et al. Restoration of impaired nitric oxide production in MELAS syndrome with citrulline and arginine supplementation. Mol Genet Metab. 2012;105:607-614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. El-Hattab AW, Emrick LT, Hsu JW, et al. Impaired nitric oxide production in children with MELAS syndrome and the effect of arginine and citrulline supplementation. Mol Genet Metab. 2016;117:407-412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sorensen KE, Celermajer DS, Spiegelhalter DJ, et al. Non-invasive measurement of human endothelium dependent arterial responses: accuracy and reproducibility. Br Heart J. 1995;74:247-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Anderson TJ, Uehata A, Gerhard MD, et al. Close relation of endothelial function in the human coronary and peripheral circulations. J Am Coll Cardiol. 1995;26:1235-1241. [DOI] [PubMed] [Google Scholar]
- 13. Celermajer DS, Sorensen KE, Gooch VM, et al. Non-invasive detection of endothelial dysfunction in children and adults at risk of atherosclerosis. Lancet. 1992;340:1111-1115. [DOI] [PubMed] [Google Scholar]
- 14. Corretti MC, Anderson TJ, Benjamin EJ, et al. Guidelines for the ultrasound assessment of endothelial-dependent flow-mediated vasodilation of the brachial artery: a report of the International Brachial Artery Reactivity Task Force. J Am Coll Cardiol. 2002;39:257-265. [DOI] [PubMed] [Google Scholar]
- 15. Deanfield J, Donald A, Ferri C, et al. Endothelial function and dysfunction. Part I: methodological issues for assessment in the different vascular beds: a statement by the Working Group on endothelin and endothelial factors of the European Society of Hypertension. J Hypertens. 2005;23:7-17. [DOI] [PubMed] [Google Scholar]
- 16. Bonetti PO, Barsness GW, Keelan PC, et al. Enhanced external counterpulsation improves endothelial function in patients with symptomatic coronary artery disease. J Am Coll Cardiol. 2003;41:1761-1768. [DOI] [PubMed] [Google Scholar]
- 17. Kuvin JT, Patel AR, Sliney KA, et al. Assessment of peripheral vascular endothelial function with finger arterial pulse wave amplitude. Am Heart J. 2003;146: 168-174. [DOI] [PubMed] [Google Scholar]
- 18. Selamet Tierney ES, Newburger JW, Gauvreau K, et al. Endothelial pulse amplitude testing: feasibility and reproducibility in adolescents. J Pediatr. 2009;154:901-905. [DOI] [PubMed] [Google Scholar]
- 19. Hamburg NM, Keyes MJ, Larson MG, et al. Cross-sectional relations of digital vascular function to cardiovascular risk factors in the Framingham Heart Study. Circulation. 2008;117:2467-2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Goor DA, Sheffy J, Schnall RP, et al. Peripheral arterial tonometry: a diagnostic method for detection of myocardial ischemia induced during mental stress tests: a pilot study. Clin Cardiol. 2004;27:137-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bonetti PO, Pumper GM, Higano ST, Holmes DR, Kuvin JT, Lerman A. Noninvasive identification of patients with early coronary atherosclerosis by assessment of digital reactive hyperemia. J Am Coll Cardiol. 2004;44:2137-2141. [DOI] [PubMed] [Google Scholar]
- 22. Koga Y, Akita Y, Nishioka J, et al. L-arginine improves the symptoms of strokelike episodes in MELAS. Neurology. 2005;64:710-712. [DOI] [PubMed] [Google Scholar]
- 23. Tengan CH, Kiyomoto BH, Godinho RO, et al. The role of nitric oxide in muscle fibers with oxidative phosphorylation defects. Biochem Biophys Res Commun. 2007;359:771-777. [DOI] [PubMed] [Google Scholar]
- 24. Koga Y, Akita Y, Junko N, et al. Endothelial dysfunction in MELAS improved by l-arginine supplementation. Neurology. 2006;66:1766-1769. [DOI] [PubMed] [Google Scholar]
- 25. Vattemi G, Mechref Y, Marini M, et al. Increased protein nitration in mitochondrial diseases: evidence for vessel wall involvement. Mol Cell Proteomics. 2011; 10:M110002964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Koga Y, Akita Y, Nishioka J, et al. MELAS and L-arginine therapy. Mitochondrion. 2007;7:133-139. [DOI] [PubMed] [Google Scholar]
- 27. Parfait B, de Lonlay P, von Kleist-Retzow JC, et al. The neurogenic weakness, ataxia and retinitis pigmentosa (NARP) syndrome mtDNA mutation (T8993G) triggers muscle ATPase deficiency and hypocitrullinaemia. Eur J Pediatr. 1999; 158:55-58. [DOI] [PubMed] [Google Scholar]
- 28. Naini A, Kaufmann P, Shanske S, Engelstad K, De Vivo DC, Schon EA. Hypocitrullinemia in patients with MELAS: an insight into the “MELAS paradox.” J Neurol Sci. 2005;229-230:187-193. [DOI] [PubMed] [Google Scholar]
- 29. El-Hattab AW, Adesina AM, Jones J, Scaglia F. MELAS syndrome: clinical manifestations, pathogenesis, and treatment options. Mol Genet Metab. 2015;116:4-12. [DOI] [PubMed] [Google Scholar]