

Abstract
Background
Definitive chemoradiation therapy (dCRT) is the standard treatment for patients with nonsurgical esophageal squamous cell carcinoma (ESCC), yet patients have demonstrated great variations in their responses to dCRT and inevitably progressed following treatment.
Methods
To identify prognostic biomarkers, we performed targeted next‐generation sequencing of 416 cancer‐related genes on primary tumors from 47 nonsurgical ESCC patients prior to dCRT treatment. The association between genetic alterations and patients' local recurrence‐free survival (LRFS), progression‐free survival (PFS), and overall survival (OS) was analyzed.
Results
TP53 (78% of patients), NOTCH1 (32%), ARID1A (13%), FAT1 (13%), and CDKN2A (13%) were commonly mutated in ESCC patients, while gene amplifications frequently occurred in MCL1 (36%), FGF19 (34%), MYC (32%), CCND1 (27%), ZNF217 (15%), CDKN2A (13%), and YAP1 (11%). Univariate and multivariate analyses of clinical factors and genetic alterations indicated that sex is an independent prognostic factor, with males tending to have better LRFS (hazard ratio [HR], 0.25; 95%CI, 0.08‐0.77, P = .015) and progression‐free survival (PFS) (HR, 0.35; 95%CI, 0.13‐0.93, P = .030) following dCRT. Meanwhile, YAP1 amplification (n = 7) was an adverse prognostic factor, and patients with this alteration demonstrated a tendency toward worse outcomes with shorter LRFS (HR, 4.06; 95%CI, 1.26‐13.14, P = .019) and OS (HR, 2.78; 95%CI, 0.95‐8.17, P = .062). In a subgroup analysis, while sex and M‐stage were controlled, a much stronger negative effect of YAP1 amplification vs wild‐type in LRFS was observed (log‐rank P = .0067).
Conclusion
The results suggested that YAP1 amplification is a potentially useful biomarker for predicting treatment outcomes and identifying patients with a high risk of relapse who should be closely monitored.
Keywords: chemoradiation therapy, esophageal squamous cell carcinoma, local recurrence‐free survival, next‐generation sequencing, YAP1 amplification
We found that in patients with nonsurgical esophageal squamous cell carcinoma, YAP1 amplification is an adverse prognostic factor associated with significantly shorter local recurrent‐free survival (LRFS) after definitive chemoradiotherapy.

1. INTRODUCTION
Esophageal cancer is one of the most common malignancies worldwide, accounting for 4% of cancer‐related deaths in the US and 13% in China.1, 2, 3 Its two major histological subtypes, adenocarcinoma and squamous cell carcinoma (SCC), are distributed differently between different ethnic groups. Adenocarcinoma is the dominant subtype in Europe and North America, while ESCC is more prevalent in Asia, Africa, and South America.4 Approximately 40%‐60% of patients with ESCC are inoperable at the time of diagnosis due to locally advanced disease, distant metastasis, poor performance status, and the existence of comorbidities that increase surgical risks.5 For those patients, definitive chemoradiation therapy (dCRT) is the standard curative therapy and has demonstrated efficacy in improving overall survival (OS) time and locoregional control rate over radiotherapy alone, or the sequential use of radiotherapy and chemotherapy.6, 7, 8, 9 Although dCRT results in relatively high response rates and favorable short‐term survival, the locoregional recurrence rate of ESCC is high (40%‐55%) within 5 years of treatment. Distant recurrence also occurred in 28% ESCC patients.10, 11 For patients who developed local recurrence, 50% experienced the first recurrence within 6 months after dCRT, which was highly correlated with a poorer prognosis.11, 12
Previous studies have identified several prognostic factors for poor treatment outcomes of dCRT, such as lymph node metastasis,13 pretreatment weight loss,14 history of heavy smoking,15 and an increased number of circulating tumor cells prior to chemoradiotherapy.16 However, none of the previous studies explored the genetic biomarkers that could possibly correlate with the prognosis of dCRT. To identify potential biomarkers for discriminating patients at a heightened risk of early failure and poor prognosis, we conducted this retrospective study by performing targeted next‐generation sequencing (NGS) of 416 cancer‐related genes on pre‐dCRT tumor biopsies in 47 unresectable ESCCs, and correlating those genetic alterations to the treatment outcomes following dCRT.
2. MATERIALS AND METHODS
2.1. Patients
The study included 47 patients who were diagnosed with ESCC and underwent dCRT in the Oncology Center of Shandong Provincial Hospital. The inclusion criteria were as follows: (a) all patients had histologically proven primary ESCC; (b) the diseases were classified as stage II‐III and IV with only supraclavicular lymph node metastasis, according to the 7th edition of the American Joint Committee on Cancer [AJCC] staging system for ESCC17; (c) all patients were precluded from surgery and received dCRT; (d) the Eastern Cooperative Oncology Group (ECOG) performance score ranged from 0 to 2; and (e) patients had adequate bone marrow, renal, and hepatic functions to endure the treatment. The study was approved by the Ethical Review Board of the Oncology Center of Shandong Provincial Hospital, and informed written consent was obtained from all participants or their next of kin, if the patient was diseased.
2.2. dCRT treatment approaches
All patients received standard dCRT. A median of two cycles of fluorouracil and cisplatin were administrated concurrently with radiotherapy. For radiotherapy, three‐dimensional conformal radiotherapy (3DCRT) and intensity‐modulated radiotherapy (IMRT) were used, and the target volume was based on the clinical target volume (CTV), which included an expansion of 3‐4 cm above and below the gross tumor and corresponding lymphatic drainage area. The planning target volume (PTV) was defined as the CTV plus a 0.8 cm expansion margin. All patients received a total radiation dose of 60‐66 Gy with 2 Gy per fraction, 5 days per week.
2.3. Evaluations and outcomes
The follow‐up of all patients was conducted 1 month after radiotherapy, and every 3 months thereafter during the first year. After the first year, patients were followed up with every 3‐6 months. Disease responses were evaluated according to the revised RECIST (Response Evaluation Criteria in Solid Tumors) guideline, version 1.1.
Treatment responses were monitored by esophagography and CT imaging at each follow‐up evaluation, and compared to the images obtained prior to dCRT or those from the preceding follow‐up. Suspected esophageal recurrences were confirmed by histological or cytological testing of tumor biopsies. Lymph node recurrences were diagnosed when lymph nodes reappeared after complete disappearance, or became enlarged after remaining stable, or newly enlarged lymph nodes were detected. Suspected supraclavicular lymph node recurrences were confirmed by fine‐needle aspiration and pathological examination. Disease recurrence was classified as locoregional and/or distant. Locoregional recurrence included tumor recurrences at the site of the primary tumor or locoregional lymph nodes. Distant recurrence included nonregional lymph node recurrences, systemic metastases, malignant pleural effusions, and peritoneal metastases.18
OS in this study was defined from the beginning of radiotherapy to the time of death from any cause, or the date of last follow‐up evaluation. PFS was defined from the beginning of radiotherapy to the time of tumor recurrence, or death in the absence of tumor recurrence, or the date of last follow‐up evaluation. LRFS was defined from the beginning of radiotherapy to the time of tumor recurrences at the site of the primary tumor or locoregional lymph nodes recurrence, or death in the absence of primary tumor or locoregional lymph nodes recurrence, or the date of the last follow‐up evaluation.
2.4. DNA extraction and sequencing library preparation
For each patient, archived formalin‐fixed and paraffin‐embedded (FFPE) blocks of tumor biopsies that were collected through endoscopic inspection before the dCRT were used for DNA extraction. The tumor content of all samples was confirmed to be at least 10% by pathologists. Eight 10‐µm FFPE sections were de‐paraffinized with xylene, from which genomic DNA was extracted using the QIAamp DNA FFPE Tissue Kit (Qiagen) according to the manufacturer's protocol. The quantity and quality of the extracted DNA were evaluated using a Qubit 3.0 fluorometer and Nanodrop 2000, respectively (Thermo Fisher Scientific). The DNA was fragmented using a Covaris M220 sonication system to obtain 350 bp fragments and purified using Agencourt AMPure XP beads (Beckman Coulter).
Library preparations of the fragmented DNA were performed using the KAPA hyper library preparation kit (KAPA Biosystems), following the manufacturer's protocol. Libraries with different indices were pooled for targeted enrichment with IDT xGen Lockdown Reagents, and a customized enrichment panel (IDT) covering the exonic regions of 416 genes and the introns of 16 fusion genes. The captured library was further amplified using Illumina p5 (5' AAT GAT ACG GCG ACC ACC GA 3') and p7 (5' CAA GCA GAA GAC GGC ATA CGA GAT 3') primers in the KAPA Hifi HotStart ReadyMix (KAPA Biosystems), and purified with Agencourt AMPure XP beads. Sequencing libraries were quantified by qPCR using the KAPA Library Quantification kit (KAPA Biosystems) and the size distribution was examined on a Bioanalyzer 2100 (Agilent Technologies). The final libraries were sequenced on an Illumina Hiseq 4000 platform to a mean coverage depth of at least 250×, following the manufacturer's instructions.
2.5. Sequencing data analysis
The sequencing data were analyzed by a validated automation pipeline with the main steps being performed as previously described.19 Data cleaning was performed in bck2fastq for demultiplexing and then Trimmomatic20 was used for FASTQ file quality control (QC). Leading/trailing low quality (base phred score below 30) or N bases were removed. Read mapping to the reference human genome, hg19, was conducted in the Burrows‐Wheeler Aligner (BWA‐mem, v0.7.12).21 PCR duplicates were removed by Picard. The Genome Analysis Toolkit (GATK 3.4.0) was employed to apply local realignment around indels and recalibrate the base quality score. VarScan2 was employed for the detection of single‐nucleotide variations (SNVs) and insertion/deletion mutations with the following parameters: minimum read depth = 20, minimum base quality = 25, minimum variant allele frequency (VAF) = 0.03, minimum variant supporting reads = 3, variant supporting reads mapped to both strands, and strand bias no greater than 10%.22
A comprehensive assay validation was performed for the copy number variation (CNV) pipeline using 38 samples against droplet digital polymerase chain reaction (ddPCR) results as the “gold standard.” We reduced system noise in copy number data by principal component analysis of 100 normal samples sequenced in the same batch. This analysis pipeline is capable of detecting 0.4‐fold copy number loss at ≥50% tumor cell content, and fourfold amplification at ≥10% tumor cell content. A ≥ 1.6‐fold change in DNA copy number was set as the cutoff for amplification, while a ≤ 0.6‐fold change was used as the cutoff for deletion.
2.6. Variant filtering and annotation
The vcf files containing both single‐nucleotide polymorphism (SNP) and small insertions/deletions (indels) were annotated by ANNOVAR against the following databases: dbSNP (v138), 1000 Genome, ExAC, COSMIC (v70), ClinVAR, and SIFT. Mutations were removed if they were present in a > 1% population frequency in the 1000 Genomes Project or 65000 exomes project (ExAC). The resulting mutation lists were filtered through an internally collected list of recurrent sequencing errors on the same sequencing platform, which was compiled from the sequencing results of 53 normal samples with a minimum average sequencing depth of 700×. Specifically, if a variant was detected (ie, ≥3 mutant reads and > 1% VAF) in > 20% of the normal samples, it was considered a likely artifact and was removed. Mutations occurring within the repeat masked regions were also removed. In an additional filtering step, a mutation was only called out when the VAF was above 2% with a minimum of three mutant reads for COSMIC mutations, or above 3% with a minimum of five mutant reads for non‐COSMIC mutations.
2.7. Statistical analysis
The median and 95% confidence intervals were calculated using the Wilcoxon signed‐rank test. Univariate and multivariate analyses using the Cox model were performed to calculate the associations between patients' clinical characteristics—including age, sex, disease stage, tumor length, and smoking history—and LRFS, OS, and PFS. The Kaplan‐Meier method was used to calculate survival rates, and the log‐rank test was used to analyze differences between groups. A statistically significant difference was set at P < .05.
3. RESULTS
3.1. Patient characteristics and treatment outcomes
The clinical characteristics of the 47 patients included in this study are shown in Table 1. The median age of the patients was 64 years, and ranged from 41 to 83 years. Thirty‐eight patients (80.9%) were male and nine (19.1%) were female. The majority of patients had stage II (15, 31.9%) or III (28, 59.6%) disease, and only four patients had stage IV disease with supraclavicular lymph node metastasis. Heavy smokers (smoke ≥ 20/day or ≥ 20 packs/year) accounted for 68.1% of all patients (n = 32).
Table 1.
Clinical characteristics and treatment outcomes of the 47 ESCC patients
| Characteristics | Value or No. of Patients (%) | |
|---|---|---|
|
Age (year) Median (range) 64 (41‐83) |
||
| 41‐50 | 7 (14.9) | |
| 51‐60 | 12 (25.5) | |
| 61‐70 | 20 (42.6) | |
| >70 | 8 (17.0) | |
| Sex | ||
| Male | 38 (80.9) | |
| Female | 9 (19.1) | |
| Smoking history | ||
| Heavy smoker | 32 (68.1) | |
| Nonsmoker | 15 (31.9) | |
| TNM stage (2009 AJCC) | ||
| T‐stage | 2 | 33 (70.2) |
| 3 | 5 (10.6) | |
| 4 | 9 (19.1) | |
| N‐stage | 0 | 16 (34.0) |
| 1‐3 | 31 (66.0) | |
| M‐stage | 0 | 43 (91.5) |
| 1 | 4 (8.5) | |
| Tumor length (cm) | ||
| ≤3 | 8 (17.0) | |
| >3 | 39 (83.0) | |
| Failure categories | ||
| Local failure | 20 (42.6) | |
| Distant failure | 10 (21.3) | |
| No evidence of relapse | 17 (36.2) | |
| Overall best response | ||
| CR | 25 (53.2) | |
| PR | 10 (21.3) | |
| SD | 8 (17.0) | |
| PD | 4 (8.5) | |
Abbreviations: CR, complete response; PD, progression disease; PR, partial response; SD, stable disease.
All patients were followed until death or the end of the study, which ranged from 5.9 to 106.5 months, with a median of 19.4 months (Table 1). At the time of the last follow‐up, 16 (34%) patients were alive. Of all patients, 20 (42.6%) had local recurrences, 10 (21.3%) had distant diseases, and 17 (36.2%) had no detectable tumor recurrences. The median PFS time was 9.8 months with the occurrence of either local or distance relapse (Table 1).
3.2. Gene amplifications are the predominant features of ESCC
We performed targeted NGS of 416 cancer‐related genes on treatment‐naïve tumor biopsies from 47 ESCC patients. Genes that were mutated in more than five cases (>10% patients) included TP53 (79%), NOTCH1 (32%), ARID2A (13%), FAT1 (13%), and CDKN2A (11%), which were consistent with a previous report.23 TP53 mutations were identified in 38 patients, among which 20 patients had frameshift or nonsense mutations (Table S1). Our analysis showed that (CNVs) were prevalent in the cohort, with 46 (98%) of patients having CNVs in one or more genes (Figure 1). The most common genes with CNVs were located on chromosomes 3q26 (TERC, SOX2, PIK3CA), 11q13 (CCND1, FGF19), 11q22 (YAP1), 1q21 (MCL1), and 8q24 (MYC, PTK2, RECQL4). TERC, which encodes an RNA component of telomerase and serves as the template of the telomere, was amplified in 35 patients (74%), while SOX2, another gene on chromosome 3q26, was amplified in 23 patients (47%), and 23 patients exhibited amplification of both genes (Figure 1). Other frequently amplified genes included MCL1 (66% of patients), MYC (62%), CCND1 (45%), ZNF217 (38%), CDK6 (17%), and YAP1 (15%), which are all modulators of cell cycle,24 apoptosis,25 proliferation, or the cytoskeleton.26, 27, 28
Figure 1.
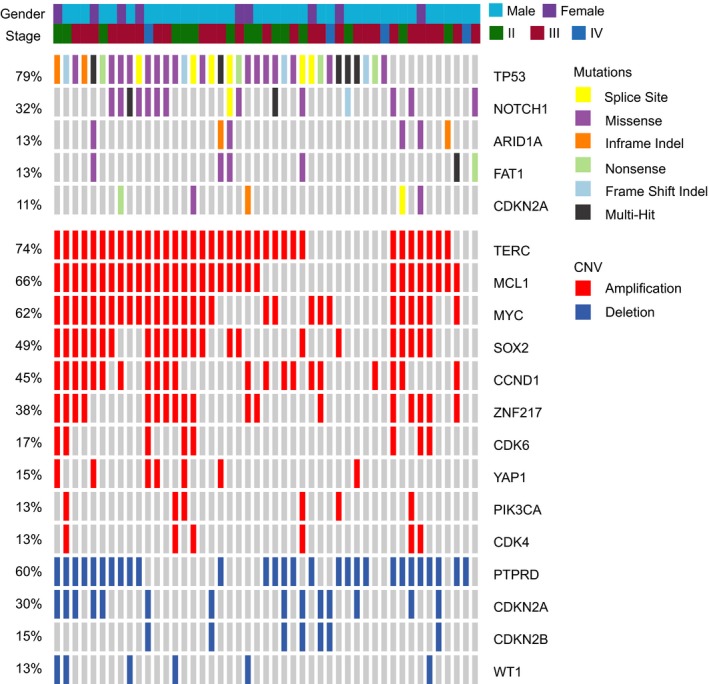
Mutation and copy number variation (CNV) plot for all patients. Each column represents one patient, and only genes that have alterations in > 5 patients are shown
3.3. The prognostic role of genetic alterations in disease recurrence
Clinical factors and genetic alterations are all potential predictors of prognosis in dCRT treatment. Therefore, we examined the association of the top gene alterations in Figure 1 with patients' LRFS, PFS, and OS using the univariate Cox regression model. Baseline clinical characteristics, including age, sex, TNM stages, tumor length, and smoking history, were examined together. Variables with P < .2 were then used for multivariate analyses. Sex was found to be an independent predictor of LRFS (HR 0.25, P = .015) and PFS (HR 0.35, P = .030), with poorer outcomes in females compared to males. Meanwhile, advanced M‐stage was associated with poorer LRFS (HR 4.35, P = .030, Table 2) than early stage disease. Multivariate analyses revealed that YAP1 amplification was the only genetic biomarker found to be significantly associated with shorter LRFS (HR 4.06, P = .019). A nearly significant association was also found between YAP1 amplification and shorter OS (HR 2.78, P = .062), while a nonsignificant association was observed with PFS (HR 1.56, P = .11, Table 2).
Table 2.
Univariate and multivariate Cox regression analyses of prognostic parameters. Variables with a P‐value < 0.2 in univariate tests were selected for multivariate tests
| Univariate | Multivariate | ||||||
|---|---|---|---|---|---|---|---|
| HR | 95% CI | P‐value | HR | 95% CI | P‐value | ||
| LRFS | Sex (male) | 0.35 | 0.13‐0.96 | .042b | 0.25 | 0.08‐0.77 | .015b |
| M‐stage | 3.6 | 1‐13 | .043b | 4.35 | 1.15‐16.46 | .030b | |
| CCND1 amp | 0.48 | 0.18‐1.3 | .14 | 0.53 | 0.16‐1.70 | .284 | |
| YAP1 amp | 2.9 | 1‐8.3 | .046b | 4.06 | 1.26‐13.14 | .019b | |
| ZNF217 amp | 0.45 | 0.16‐1.3 | .13 | 0.45 | 0.14‐1.50 | .195 | |
| PFS | Age | 0.97 | 0.93‐1.00 | .08 | 0.96 | 0.92‐1.00 | .031b |
| Sex (male) | 0.49 | 0.20‐1.20 | .11 | 0.35 | 0.13‐0.93 | .030b | |
| YAP1 amp | 2.10 | 0.85‐5.30 | .11 | 1.56 | 0.62‐4.02 | .303 | |
| OS | Sex (male) | 0.48 | 0.2‐1.1 | .1 | 0.43 | 0.18‐1.06 | .067a |
| FAT1 mutation | 1.9 | 0.73‐5.1 | .19 | 1.46 | 0.47‐4.47 | .513 | |
| YAP1 amp | 2.3 | 0.91‐5.8 | .078 | 2.78 | 0.95‐8.17 | .062a | |
| ZNF217 amp | 0.59 | 0.27‐1.3 | .18 | 0.49 | 0.21‐1.16 | .105 | |
Abbreviation: amp, amplification.
Close to statistically significant
Statistically significant.
3.4. The predictive role of YAP1 amplification in ESCC prognosis
The prognostic effect of YAP1 CNV on LRFS, PFS, and OS was illustrated by Kaplan‐Meier curves (Figure 2). The data showed that patients with wild‐type (wt) YAP1 had a significantly better LRFS (median: 55.4 months), compared to patients with YAP1 amplification (median: 8.6 months, P = .037, Figure 2A). The 2‐year survival probability was also different between the two groups (wt vs amplification: 65.8% vs 21.4%). Moreover, favorable PFS and OS were observed in patients with wt YAP1, despite that such differences were not significant (PFS P = .097; OS P = .072; Figure 2B,C). The best overall responses (BORs) were also different between the two groups, with a lower complete response (CR, 28.6% vs 57.5%) in the YAP1 amplification group, compared to YAP1 wt group, which might be associated with increased locoregional recurrence following dCRT (Figure 2D).
Figure 2.
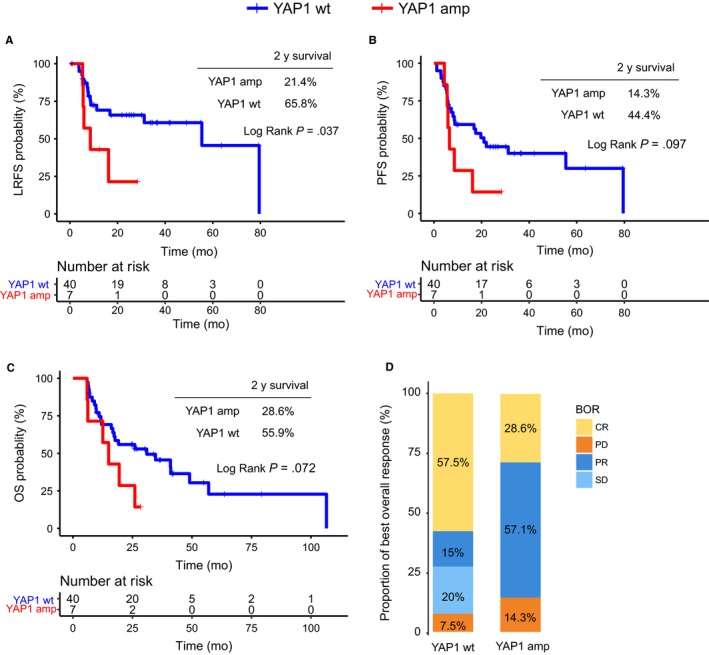
Survival analysis and best overall responses (BORs) in patients with YAP1 amplification. A‐C, Kaplan‐Meier survival curves for local recurrence‐free survival (LRFS), progression‐free survival (PFS), and overall survival (OS) for the 47 ESCC patients. D, The BOR of all patients in the YAP1 wt and amplification groups
Given that both M staging and sex were associated with LRFS in multivariate analyses (Table 2), we compared the survival times of male patients with stage II‐III disease between the YAP1 amplification and wt groups. As illustrated in Figure 3A, after controlling for clinical stage and sex, the survival advantage become even more pronounced for YAP1 wt patients. Such patients demonstrated significant improvements in LRFS (wt vs amplification median: 79.5 vs 10.9 months, P = .0067, Figure 3A), and visually better PFS (31.2 vs 10.9 months, P = .22, Figure 3B) and OS (40.9 vs 17.2 months, P = .17, Figure 3C), compared to YAP1 amplification patients. The prognostic role of YAP1 amplification in female patients was also examined. However, due to the limited number of patients (n = 2), we only observed a trend for better OS in YAP1 wt patients vs YAP1 amplification patients, while the difference in LRFS and PFS could not be discriminated between the groups (Figure S1).
Figure 3.
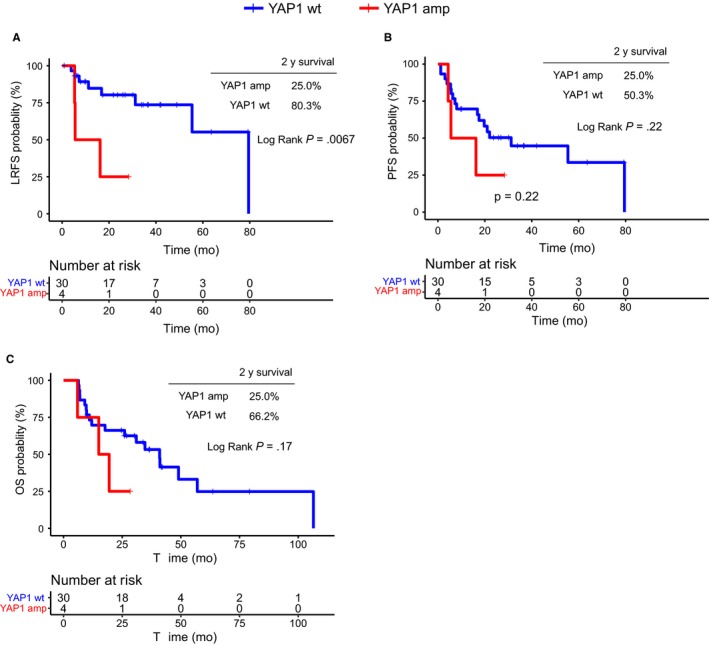
Kaplan‐Meier survival curves for local recurrence‐free survival (LRFS), progression‐free survival (PFS), and overall survival (OS) in male patients with stage II‐III disease (n = 34)
4. DISCUSSION
In this study, using a pan‐cancer NGS panel, we retrospectively inspected the cancer genomes of 47 patients with inoperable ESCC, with the aim of identifying prognostic genetic biomarkers for dCRT. Baseline clinical features were accounted for in the analysis and sex appeared to be an independent prognostic factor in LRFS and PFS, with increased HR in females. The lower impact of sex on OS is consistent with the SCOPE1 trial in which females did not show a pronounced difference in OS from males following dCRT treatment.29 Other reports suggested a longer OS for females than males,30 but under different treatments.31, 32
Genomic profiling of ESCC tumors found 98% patients having amplification in one or more cancer‐relevant genes, as well as the co‐amplification of genes at adjacent chromosome locations, such as CCND1/FGF19 and TERC/SOX2/PIK3CA. The amplification of TERC and SOX2 was frequently observed in a variety of SCC, including oral and oropharyngeal,33 lung, esophagus,34 and cervical.35 However, despite their prevalence in cancers, those genes were not correlated with any of the survival parameters in this study.
Interestingly, YAP1 amplification was significantly correlated with shorter LRFS, but not PFS and OS. In a subgroup of patients (male patients with stage II/III disease), of which the impact of sex and clinical stages was controlled, the difference in LRFS between YAP1 amplification and wt groups was more pronounced, suggesting that YAP1 is an independent prognostic marker for dCRT. Although the underlying mechanisms were not fully elucidated, our current understanding of the physical and pathological roles of YAP1 is supportive of this finding. YAP1 is the main downstream target of the Hippo pathway and acts as a transcriptional coactivator to regulate organ growth,36, 37 tissue homeostasis,38 and neoplasia,28, 39, 40, 41 primarily by interacting with TEAD transcription factors, which subsequently target a series of oncogenes and tumor suppressors to promote cell proliferation, transformation, migration, and invasion.42, 43 YAP1 overexpression has been identified in non‐small cell lung cancer, urothelial carcinoma of the bladder, and pancreatic cancer, and is consistently correlated with unfavorable clinical outcomes.44, 45, 46 YAP1 amplification was also reported in 4%‐6% of ESCC patients,23, 47 and was suggested by independent in vitro studies to mediate chemo‐ and radioresistance by downregulating PTEN,48 and upregulating EGFR and CDK6 expression.49, 50 While local relapse is a major challenge for patients receiving dCRT, it is important to identify high‐risk patient groups for close monitoring. Our study identified that a subgroup of patients with YAP1 CNV amplification tended to have earlier local relapse, and therefore, may require more intensive clinical interventions. It is necessary to validate this finding in a larger clinical cohort, and improve the treatment benefits for such patients.
AUTHOR CONTRIBUTIONS
Honghai Dai: Conceptualization, supervision, project administration, funding acquisition, methodology, investigation, and manuscript preparation, review, and editing. Yang W. Shao: Methodology, software, formal analysis, data curation, and manuscript preparation, review, and editing. Xiaoling Tong: Formal analysis, investigation, and manuscript preparation, review, and editing. Xue Wu: Investigation, and manuscript preparation, review, and editing. Jiaohui Pang: Investigation and manuscript preparation, review, and editing. Alei Feng: Resources, investigation, and manuscript preparation, review, and editing. Zhe Yang: Conceptualization, supervision, project administration, funding acquisition, methodology, investigation, and manuscript preparation, review, and editing.
Supporting information
Dai H, Shao YW, Tong X, et al. YAP1 amplification as a prognostic factor of definitive chemoradiotherapy in nonsurgical esophageal squamous cell carcinoma. Cancer Med. 2020;9:1628–1637. 10.1002/cam4.2761
Funding information
The study was funded by the National Nature Science Foundation of China (Funding ID: 81502508).
DATA AVAILABILITY STATEMENT
Genetic sequencing data are available upon request, certain restrictions may apply.
REFERENCES
- 1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet‐Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87‐108. [DOI] [PubMed] [Google Scholar]
- 2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5‐29. [DOI] [PubMed] [Google Scholar]
- 3. Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115‐132. [DOI] [PubMed] [Google Scholar]
- 4. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359‐386. [DOI] [PubMed] [Google Scholar]
- 5. Tachibana M, Kinugasa S, Hirahara N, Yoshimura H. Lymph node classification of esophageal squamous cell carcinoma and adenocarcinoma. Eur J Cardiothorac Surg. 2008;34:427‐431. [DOI] [PubMed] [Google Scholar]
- 6. Cooper JS, Guo MD, Herskovic A, et al. Chemoradiotherapy of locally advanced esophageal cancer: long‐term follow‐up of a prospective randomized trial (RTOG 85–01). Radiation Therapy Oncology Group. JAMA. 1999;281:1623‐1627. [DOI] [PubMed] [Google Scholar]
- 7. Minsky BD, Pajak TF, Ginsberg RJ, et al. INT 0123 (Radiation Therapy Oncology Group 94–05) phase III trial of combined‐modality therapy for esophageal cancer: high‐dose versus standard‐dose radiation therapy. J Clin Oncol. 2002;20:1167‐1174. [DOI] [PubMed] [Google Scholar]
- 8. Tang H‐R, Ma H‐F, An S‐M, et al. A phase II study of concurrent chemoradiotherapy with paclitaxel and cisplatin for inoperable esophageal squamous cell carcinoma. Am J Clin Oncol. 2016;39:350‐354. [DOI] [PubMed] [Google Scholar]
- 9. Yao B, Tan B, Wang C, et al. Comparison of definitive chemoradiotherapy in locally advanced esophageal squamous cell carcinoma. Ann Surg Oncol. 2016;23:2367‐2372. [DOI] [PubMed] [Google Scholar]
- 10. Al‐Sarraf M, Martz K, Herskovic A, et al. Progress report of combined chemoradiotherapy versus radiotherapy alone in patients with esophageal cancer: an intergroup study. J Clin Oncol. 1997;15:277‐284. [DOI] [PubMed] [Google Scholar]
- 11. Xi M, Xu C, Liao Z, et al. The impact of histology on recurrence patterns in esophageal cancer treated with definitive chemoradiotherapy. Radiother Oncol. 2017;124:318‐324. [DOI] [PubMed] [Google Scholar]
- 12. Welsh J, Settle SH, Amini A, et al. Failure patterns in patients with esophageal cancer treated with definitive chemoradiation. Cancer. 2012;118:2632‐2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Aiko S, Yoshizumi Y, Ishizuka T, et al. Reduction rate of lymph node metastasis as a significant prognostic factor in esophageal cancer patients treated with neoadjuvant chemoradiation therapy. Dis Esophagus. 2007;20:94‐101. [DOI] [PubMed] [Google Scholar]
- 14. Park JW, Kim JH, Choi EK, et al. Prognosis of esophageal cancer patients with pathologic complete response after preoperative concurrent chemoradiotherapy. Int J Radiat Oncol Biol Phys. 2011;81:691‐697. [DOI] [PubMed] [Google Scholar]
- 15. Shitara K, Matsuo K, Hatooka S, et al. Heavy smoking history interacts with chemoradiotherapy for esophageal cancer prognosis: a retrospective study. Cancer Sci. 2010;101:1001‐1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Su P‐J, Wu M‐H, Wang H‐M, et al. Circulating tumour cells as an independent prognostic factor in patients with advanced oesophageal squamous cell carcinoma undergoing chemoradiotherapy. Sci Rep. 2016;6:31423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. NCCN . Clinical Practice Guidelines in Oncology. Esophageal and Esophagogastric Junction Cancers. Available at http://www.nccn.org/professionals/physician_gls/pdf/esophageal.pdf.Version2. 2016; Accessed: September 10, 2017.
- 18. Oppedijk V, van der Gaast A, van Lanschot JJB, et al. Patterns of recurrence after surgery alone versus preoperative chemoradiotherapy and surgery in the CROSS trials. J Clin Oncol. 2014;32:385‐391. [DOI] [PubMed] [Google Scholar]
- 19. Lu H, Yang S, Zhu H, et al. Targeted next generation sequencing identified clinically actionable mutations in patients with esophageal sarcomatoid carcinoma. BMC Cancer. 2018;18:251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114‐2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li H, Durbin R. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics. 2009;25:1754‐1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Koboldt DC, Zhang Q, Larson DE, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568‐576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gao Y‐B, Chen Z‐L, Li J‐G, et al. Genetic landscape of esophageal squamous cell carcinoma. Nat Genet. 2014;46:1097‐1102. [DOI] [PubMed] [Google Scholar]
- 24. Fujise K, Zhang D, Liu J, Yeh ET. Regulation of apoptosis and cell cycle progression by MCL1. Differential role of proliferating cell nuclear antigen. J Biol Chem. 2000;275:39458‐39465. [DOI] [PubMed] [Google Scholar]
- 25. Nilsson JA, Cleveland JL. Myc pathways provoking cell suicide and cancer. Oncogene. 2003;22:9007‐9021. [DOI] [PubMed] [Google Scholar]
- 26. Gastonguay A, Berg T, Hauser AD, Schuld N, Lorimer E, Williams CL. The role of Rac1 in the regulation of NF‐kappaB activity, cell proliferation, and cell migration in non‐small cell lung carcinoma. Cancer Biol Ther. 2012;13:647‐656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Slorach EM, Chou J, Werb Z. Zeppo1 is a novel metastasis promoter that represses E‐cadherin expression and regulates p120‐catenin isoform expression and localization. Genes Dev. 2011;25:471‐484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhao B, Wei X, Li W, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21:2747‐2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cox S, Powell C, Carter B, Hurt C, Mukherjee S, Crosby TD. Role of nutritional status and intervention in oesophageal cancer treated with definitive chemoradiotherapy: outcomes from SCOPE1. Br J Cancer. 2016;115:172‐177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bohanes P, Yang D, Chhibar RS, et al. Influence of sex on the survival of patients with esophageal cancer. J Clin Oncol. 2012;30:2265‐2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Trivers K, Deroos A, Gammon M, et al. Demographic and lifestyle predictors of survival in patients with esophageal or gastric cancers. Clin Gastroenterol Hepatol. 2005;3:225‐230. [DOI] [PubMed] [Google Scholar]
- 32. Morita M, Yoshida R, Ikeda K, et al. Advances in esophageal cancer surgery in Japan: an analysis of 1000 consecutive patients treated at a single institute. Surgery. 2008;143:499‐508. [DOI] [PubMed] [Google Scholar]
- 33. Kokalj Vokac N, Cizmarevic B, Zagorac A, Zagradisnik B, Lanisnik B. An evaluation of SOX2 and hTERC gene amplifications as screening markers in oral and oropharyngeal squamous cell carcinomas. Mol Cytogenet. 2014;7:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bass AJ, Watanabe H, Mermel CH, et al. SOX2 is an amplified lineage‐survival oncogene in lung and esophageal squamous cell carcinomas. Nat Genet. 2009;41:1238‐1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kokalj‐Vokac N, Kodric T, Erjavec‐Skerget A, Zagorac A, Takac I. Screening of TERC gene amplification as an additional genetic diagnostic test in detection of cervical preneoplastic lesions. Cancer Genet Cytogenet. 2009;195:19‐22. [DOI] [PubMed] [Google Scholar]
- 36. Lee K‐P, Lee J‐H, Kim T‐S, et al. The Hippo‐Salvador pathway restrains hepatic oval cell proliferation, liver size, and liver tumorigenesis. Proc Natl Acad Sci U S A. 2010;107:8248‐8253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lu L, Li Y, Kim SM, et al. Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc Natl Acad Sci USA. 2010;107:1437‐1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. He C, Lv X, Huang C, et al. YAP1‐LATS2 feedback loop dictates senescent or malignant cell fate to maintain tissue homeostasis. EMBO Rep. 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shao D, Xue W, Krall E, et al. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell. 2014;158:171‐184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kapoor A, Yao W, Ying H, et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell. 2014;158:185‐197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Barry ER, Morikawa T, Butler BL, et al. Restriction of intestinal stem cell expansion and the regenerative response by YAP. Nature. 2013;493:106‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lamar JM, Stern P, Liu H, Schindler JW, Jiang ZG, Hynes RO. The Hippo pathway target, YAP, promotes metastasis through its TEAD‐interaction domain. Proc Natl Acad Sci USA. 2012;109:E2441‐2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yu M, Chen Y, Li X, et al. YAP1 contributes to NSCLC invasion and migration by promoting Slug transcription via the transcription co‐factor TEAD. Cell Death Dis. 2018;9:464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang Y, Dong Q, Zhang Q, Li Z, Wang E, Qiu X. Overexpression of yes‐associated protein contributes to progression and poor prognosis of non‐small‐cell lung cancer. Cancer Sci. 2010;101:1279‐1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liu J‐Y, Li Y‐H, Lin H‐X, et al. Overexpression of YAP 1 contributes to progressive features and poor prognosis of human urothelial carcinoma of the bladder. BMC Cancer. 2013;13:349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Salcedo Allende MT, Zeron‐Medina J, Hernandez J, et al. Overexpression of yes associated Protein 1, an independent prognostic marker in patients with pancreatic ductal adenocarcinoma, correlated with liver metastasis and poor prognosis. Pancreas. 2017;46:913‐920. [DOI] [PubMed] [Google Scholar]
- 47. Sawada G, Niida A, Uchi R, et al. Genomic landscape of esophageal squamous cell carcinoma in a Japanese population. Gastroenterology. 2016;150:1171‐1182. [DOI] [PubMed] [Google Scholar]
- 48. Tumaneng K, Schlegelmilch K, Russell RC, et al. YAP mediates crosstalk between the Hippo and PI(3)K‐TOR pathways by suppressing PTEN via miR‐29. Nat Cell Biol. 2012;14:1322‐1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Song S, Honjo S, Jin J, et al. The Hippo coactivator YAP1 mediates EGFR overexpression and confers chemoresistance in esophageal cancer. Clin Cancer Res. 2015;21:2580‐2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li F, Xu Y, Liu B, et al. YAP1‐mediated CDK6 activation confers radiation resistance in esophageal cancer ‐ rationale for the combination of YAP1 and CDK4/6 inhibitors in esophageal cancer. Clin Cancer Res. 2018;25:2264‐2277. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Genetic sequencing data are available upon request, certain restrictions may apply.