

Can a LC-MS/MS based omic antigen identification method provide reliable results and comply with pharmaceutical regulations? In this study, we performed several assays and evaluations to validate the immunopeptidomic pipeline according to FDA and EMA pharmaceutical guidelines. We performed a validation of the accuracy, precision, specificity, limit of detection and robustness of our LC-MS/MS system using an Orbitrap Fusion Lumos. We present different approaches that could be transferred to other MS-based discovery applications to enhance further personalized clinical therapies.
Keywords: Mass spectrometry, peptidomics, personalized medicine, cancer therapeutics, individualized medicine, GMP, immunopeptidome, LC-MS/MS, validation
Graphical Abstract

Highlights
Validation of an omic method for antigen identification using LC-MS/MS.
Validation of accuracy, precision, specificity, limit of detection and robustness.
Validation according to the current FDA and EMA guidelines.
Abstract
For more than two decades naturally presented, human leukocyte antigen (HLA)-restricted peptides (immunopeptidome) have been eluted and sequenced using liquid chromatography-tandem mass spectrometry (LC-MS/MS). Since, identified disease-associated HLA ligands have been characterized and evaluated as potential active substances. Treatments based on HLA-presented peptides have shown promising results in clinical application as personalized T cell-based immunotherapy. Peptide vaccination cocktails are produced as investigational medicinal products under GMP conditions. To support clinical trials based on HLA-presented tumor-associated antigens, in this study the sensitive LC-MS/MS HLA class I antigen identification pipeline was fully validated for our technical equipment according to the current US Food and Drug Administration (FDA) and European Medicines Agency (EMA) guidelines.
The immunopeptidomes of JY cells with or without spiked-in, isotope labeled peptides, of peripheral blood mononuclear cells of healthy volunteers as well as a chronic lymphocytic leukemia and a bladder cancer sample were reliably identified using a data-dependent acquisition method. As the LC-MS/MS pipeline is used for identification purposes, the validation parameters include accuracy, precision, specificity, limit of detection and robustness.
The immunopeptidome is a vast and diverse compilation of human leukocyte antigen (HLA)1-presented peptides (HLA ligands), which serve as a showcase of inter- and intracellular processes. T cells recognize presented peptides in the immunopeptidome, which is constantly modulated by gene expression, transcription, translation, posttranslational modification and antigen processing and presentation (1–4). Especially in tumor immunology, HLA ligands are used in many ways. They are suited as biomarkers, presenting intracellular abnormalities like malignant transformation and as active pharmaceuticals, activating cancer specific T cells (5).
Natural HLA ligands have been isolated and sequenced using LC-MS/MS for almost three decades (6–12). So far, the LC-MS/MS analysis is the only method to investigate the entirety of HLA presented peptides. However, based on these peptide data in silico prediction tools have been developed, which allow the prediction of possibly presented peptides from exome, RNA or whole genome sequencing data and have extended the toolbox even further (13–18).
Developing from such identifications, peptide vaccination cocktails have been produced as active pharmaceuticals under GMP conditions (19). The acceptance, safety and efficacy of peptide vaccinations have been investigated (19) and several clinical studies testing peptide vaccinations have been performed with our contribution (GAPVAC (5), NCT02149225, and NOA-16, NCT02454634) or are ongoing (iVAC-CLL01, NCT02802943). The procedures of active substance production, analysis, and batch release have been validated and reliably lead to reproducible products of desired quality. However, the initial antigen identification procedure using mass spectrometry-based immunopeptidomics has not been validated yet. In this study, the LC-MS/MS antigen identification procedure was fully validated for our technical equipment according to current FDA and EMA guidelines to support further clinical trials based on HLA-presented tumor-associated antigens. This validation should serve as a guidance that can be adapted to other LC-MS/MS platforms and samples.
Protocols for large-scale immunopeptidomics using LC-MS/MS and the identification of HLA ligands are established and have been published (20–22). To our knowledge, no validation of an omics method using LC-MS/MS-has been published so far (23, 24). This article presents an immunopeptidomics assay using LC-MS/MS, which is fully validated according to the latest US Food and Drug Administration (FDA) and European Medicines Agency (EMA) guidelines (23–27). We have to emphasize that such validations are specific only for dedicated equipment of one distinct laboratory. We provide a first protocol and template to enhance the validation of other laboratories with similar equipment and other omics fields using LC-MS/MS such as proteomics, metabolomics, and lipidomics.
EXPERIMENTAL PROCEDURES
Peptide Synthesis
The automated peptide synthesizer Liberty Blue (CEM, NC) was used to synthesize peptides following the 9-fluorenylmethyl-oxycarbonyl/tert-butyl (Fmoc/tBu) strategy. The identity and purity of the peptides were confirmed using a reversed-phase liquid chromatography (Alliance e2965, Waters, MA) and an uHPLC system (nanoUHPLC, UltiMate 3000 RSLCnano, Dionex, California) on-line coupled LTQ Orbitrap XL hybrid mass spectrometer (Thermo Fisher Scientific, MA) system. Synthesized peptides were employed in the validation of LC-MS/MS identifications. Peptide sequences used for the validation are listed in supplemental Table S1.
Tissue Samples
The EBV-transformed human B-cell line JY (ECACC, England, UK 94022533) was cultured in RPMI1640 with 10% heat-inactivated fetal bovine serum (FBS, Lonza, Basel, Switzerland) and 1% penicillin/streptomycin to a total number of 1×1011 cells, centrifuged at 1500 rpm for 15 min at 4 °C, washed two times with cold PBS and aliquots containing 75×106 cells were frozen and stored at −80 °C until use. The cells were tested negative for mycoplasma contamination via PCR.
The peripheral blood mononuclear cells (PBMC), chronic lymphocytic leukemia (CLL) and bladder cancer (BC) tissue samples were collected at the University Hospital of Tübingen with the informed consent of patients according to the principles of the Declaration of Helsinki. The local institutional review board (Ethics Committee at the Medical Faculty and the University Hospital of Tübingen) has approved the use of the patient samples.
Immunoaffinity Purification of HLA Ligands
HLA class I molecules were isolated using standard immunoaffinity purification as described (9, 20, 28, 29) using the HLA class I-specific monoclonal antibody W6/32 (30). First, the cell pellets were lysed in 10 mm CHAPS (Applichem, MO)/PBS (Lonza, Basel, Switzerland) containing protease inhibitors (Complete, Roche, Basel, Switzerland) and subsequently HLA molecules were purified using the pan-HLA class I-specific monoclonal W6/32 Ab (produced in-house) covalently linked to CNBr-activated Sepharose (GE Healthcare, England, UK). Repeated addition of 0.2% trifluoroacetic acid (Merck, MA) eluted HLA molecules and peptides. The peptides were isolated employing ultrafiltration with centrifugal filter units (Amicon, Merck Millipore), extracted and desalted using ZipTip C18 pipette tips (Merck Millipore), eluted in 35 μl acetonitrile (Merck Millipore)/0.1% trifluoroacetic acid, vacuum centrifuged to 5 μl, and resuspended in 25 μl of 1% acetonitrile/0.05% trifluoroacetic acid. Finally, the peptide solutions were stored at −20 °C until analysis by LC-MS/MS.
Analysis of HLA Ligands by LC-MS/MS
Peptides were separated by nanoflow high-performance liquid chromatography (nanoUHPLC, UltiMate 3000 RSLCnano, Dionex) and subsequently analyzed in an on-line coupled Orbitrap Fusion Lumos or LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific). Volumes of 5 μl peptide solution were injected onto a 75 μm × 2 cm trapping column (Acclaim PepMap RSLC, Dionex) at 4 μl/min for 5.75 min in five technical replicates. Subsequently, peptide separation was performed at 50 °C at a flow rate of 300 nl/min on a 50 μm × 25 cm separation column (Acclaim PepMap RSLC, Dionex) applying a gradient ranging from 2.4 to 32.0% of AcN over the course of 90 min. Eluted peptides were ionized by nanospray ionization and analyzed in the Orbitrap Fusion Lumos implementing top speed collision-induced dissociation (CID) fragmentation. Survey scans were performed at 120,000 resolution and fragment detection at 60,000 resolution in the Orbitrap.
To demonstrate a method transfer, the immunopeptidomics pipeline was transferred from the Orbitrap Fusion Lumos to a LTQ Orbitrap XL. In the LTQ Orbitrap XL peptides were analyzed using a top five CID method with survey scans at 60,000 resolution and fragment ion detection in the ion trap operated at normal scan speed. On both instruments, the mass range was limited to 400–650 m/z with precursors of charge states 2+ and 3+ eligible for fragmentation.
Maintenance and OQ of the LC-MS/MS system are performed annually (Thermo Fisher Scientific). A positive ion calibration using a Pierce™ LTQ Velos or LTQ ESI positive ion calibration solution (Thermo Fisher Scientific) and a system suitability test using natural HLA class I-presented peptides of JY cells is performed weekly.
Database Search and Spectral Annotation
Data was processed against the human proteome included in the Swiss-Prot database (http://www.uniprot.org, release September 27, 2013; containing 20,279 reviewed protein sequences) applying the Sequest algorithm (31) in the Proteome Discoverer (version 1.3, Thermo Fisher Scientific) software.
Precursor mass tolerance was set to 5 ppm, product ions mass tolerance was set to 0.02 Da for Orbitrap Fusion Lumos data and 0.5 Da for LTQ Orbitrap XL Data and oxidized methionine was allowed as the only dynamic modification with no restriction by enzymatic specificity. Percolator (32)-assisted false discovery rate (FDR) calculation was set at a target value of q ≤ 0.05 (1% FDR). Peptide-spectrum matches with q ≤ 0.05 were filtered according to additional orthogonal parameters to ensure spectral quality and validity. Peptide lengths were limited to 8–12 amino acids.
Validation Procedures
The validation of the immunopeptidomics procedure was done according to the OECD principles of Good Laboratory Practice (GLP) (33) and accuracy, precision, specificity, limit of detection and robustness were validated according to the FDA and EMA guidelines (25, 26). Clear definitions can be found in (25, 26, 33).
As the immunopeptidome LC-MS/MS system separates the peptides using the LC and subsequently identifies the peptide ions in MS mode and product ions in the MS/MS mode, we tried to consider these three parts for every validation parameter summarized in Table I and Fig. 1.
Table I. Acceptance criteria for the different parameters selected for LC-MS/MS validation.
| Characteristics | Specification/acceptance criteria |
|
|---|---|---|
| MS and MS/MS | LC | |
| Accuracy (comparison to theoretical masses and synthetic peptides) |
The median of the deviation of the theoretical masses (ΔM ppm) of
|
PC ≥ 95% |
| Precision (natural peptides)
|
S.D. of peptide number: ≤ 10% Recovery rate: 80% ± 20% |
PC ≥ 95% |
| Specificity (natural and synthetic peptides) |
The S.D. between the precursor ion and five top fragment masses of selected peptides: ≤ 0.001 Da. The peptide must be identified in two of three replicates | PC ≥ 95% |
| Selectivity of all identified peptides based on precursor mass combined with top five fragments | ||
| Limit of detection (synthetic peptides) |
50% (n = 31) of the peptides have to be identified Recovery rate: 80% ± 20% |
PC ≥ 95% |
| Robustness (natural peptides from three primary samples isolated by three different persons) |
Accuracy: as mentioned above Precision:
Specificity: as mentioned above |
PC ≥ 95% |
The acceptance criteria for the selected parameters are indicated for the mass spectrometer and the LC. Abbreviations: PC, Pearson correlation; S.D., standard deviation.
Fig. 1.
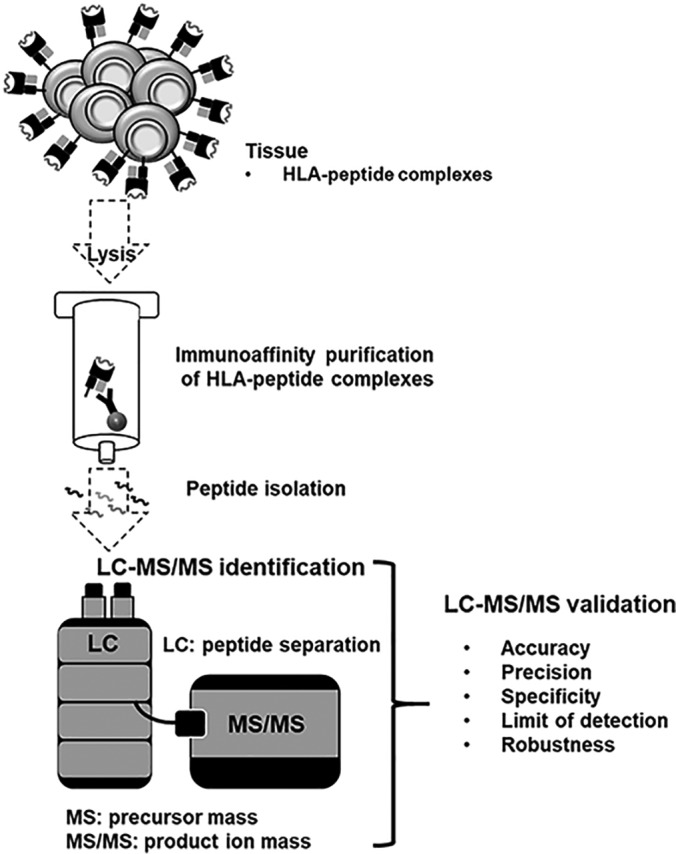
Schematic overview of the validation of the LC-MS/MS immunopeptidomics assay for the identification of HLA ligands suitable for pharmaceutical therapies. The LC-MS/MS pipeline is used for identification purposes, consequently the validation parameters accuracy, precision, specificity, limit of detection and robustness were validated according to current FDA and EMA guidelines.
Experimental Design and Statistical Rationale
A summary of the performed experiments, samples, technical replicates and MS RAW files is given in supplemental Table S2. Results were analyzed using GraphPad Prism (GraphPad Software, CA). The recovery rate was obtained by taking the average of the percentual overlapping peptides between the technical replicates normalized to the total number of peptides. The LC peptide retention times (RTs) were compared calculating the average of the Pearson correlations of the technical replicates.
RESULTS
Accuracy
To investigate the accuracy and specificity, the purified HLA-eluted peptides from one JY batch were spiked with 100 fmol isotope labeled synthetic peptides (supplemental Table S1) and analyzed in three separate analytical replicates (for identified peptides, see supplemental Table S3). The accuracy of the mass spectrometer did fulfill the acceptance criteria (Table I) with a deviation below 2 ppm between the median mass deviation from the theoretical mass of all identified natural (median ΔM: 0.05 ppm) and synthetic peptides (median ΔM: 0.19 ppm) in Fig. 2A. The peptide RTs between the replicates of all natural and all synthetic peptides do have a mean Pearson correlation above 95%, verifying the accuracy of the LC (Fig. 2B).
Fig. 2.

Validation of the accuracy using immunopeptidomes from JY cells and spiked isotope labeled synthetic peptides. Three replicates were analyzed. A, Mass deviation of the detected precursor mass from the theoretical mass (ΔM ppm) of all identified natural (n = 1648) and synthetic peptides (n = 62). B, Mean Pearson correlation of the peptide retention times. Abbreviations: PC, Pearson correlation; ppm, parts per million; ΔM, mass deviation.
Specificity
Based on our experience from the first series of analyses, the five peptides AIVDKVPSV, RPSGPGPEL, YLLPAIVHI, KVLEYVIKV, and SPSSILSTL are expected as natural HLA class I-presented peptides of JY cells. To prove the specificity, the mass spectrometer must fulfill the MS mode acceptance criteria for precursor ions and the MS/MS mode acceptance criteria for five selected top product ions of the expected five peptides (Table I). Here, we use two ways to select the five top product ions, we simply choose the top five most intensive fragments (last paragraph of specificity) or the most intensive fragments such as b and y fragments with the highest intensity and relevance (penultimate paragraph of specificity). Furthermore, in the LC separation, the correlation of the retention times of the natural and synthetic counterparts of the five peptides must fulfill the acceptance criteria.
The difference of the median of the mass deviation from the theoretical mass (ΔM ppm) of the five selected peptides AIVDKVPSV, RPSGPGPEL, YLLPAIVHI, KVLEYVIKV, and SPSSILSTL, which were identified as natural (median ΔM: 1.34 ppm) and synthetic peptides (median ΔM: 0.09 ppm), is below 2 ppm (Fig. 3A) (for identified peptides and product ions, see supplemental Table S4).
Fig. 3.

Validation of the specificity using immunopeptidomes from JY cells and spiked isotope labeled synthetic peptides. Three replicates were analyzed. A, Mean of the mass deviation from the theoretical precursor mass (ΔM ppm) of the five identified natural and synthetic peptides AIVDKVPSV, RPSGPGPEL, YLLPAIVHI, KVLEYVIKV and SPSSILSTL in three technical replicates. B, Mean Pearson correlation of the RTs of the five identified natural and synthetic peptides. Mass deviation as S.D. of the (C) precursor ion masses in MS mode and the (D) resulting five selected top fragments in MS/MS modes of the five identified natural and synthetic peptides. Abbreviations: PC, Pearson correlation; ppm, parts per million; ΔM, mass deviation; S.D., standard deviation.
The peptide RTs between the replicates of the five selected peptides AIVDKVPSV, RPSGPGPEL, YLLPAIVHI, KVLEYVIKV, and SPSSILSTL, which were identified as natural and synthetic peptides, do have a Pearson correlation above 95% (Fig. 3B).
The standard deviation (S.D.) of the mass accuracy of the precursor masses in MS mode (Fig. 3C) and of the five selected top product ions, selected based on intensity and relevance, in MS/MS mode (Fig. 3D, for MS/MS spectra, see supplemental Fig. S1) of the five selected peptides AIVDKVPSV, RPSGPGPEL, YLLPAIVHI, KVLEYVIKV, and SPSSILSTL is below 0.001 Da for both the natural and synthetic peptides.
As an additional step to prove the specificity of the LC-MS/MS system, we validated our manual quality control method, to distinguish all peptides based on the mass of a precursor ion combined with the masses of the top five most intensive product ions. As the S.D. of the mass accuracy deviates at four decimals, our peptide identity criteria of the quality control should enable specificity at a four-decimal level (Table I). Based on the precursor masses in MS mode at four-digit level there was an overlap of 7 to 12 natural peptide masses in the three replicates (Table IIA). These peptide masses could be separated using the top five masses with the highest intensity in MS/MS mode. In MS/MS mode the highest number of overlapping product ion masses were two duplicates in replicate two (Table IIB). All synthetic peptides could be separated based on the precursor masses at four decimals (Table IIC).
Table II. Validation of the specificity and suitability of the top five product ion peptide quality control using immunopeptidomes from JY cells and spiked isotope labeled synthetic peptides.
| (A) MS: natural peptides, precursor masses | |||
| Replicates | 1 | 2 | 3 |
| Unique | 1181 | 1260 | 1362 |
| Duplicate | 7 | 12 | 12 |
| (B) MSMS: natural peptides, five top-fragment masses | |||
| Replicates | 1 | 2 | 3 |
| Unique | 70 | 116 | 120 |
| Duplicate | 0 | 2 | 0 |
| (C) MS: synthetic peptides | |||
| Replicates | 1 | 2 | 3 |
| Unique | 61 | 62 | 61 |
| Duplicate | 0 | 0 | 0 |
(A) Overlap of the detected peptide precursor masses in MS mode of the natural peptides at four decimals. (B) Overlap of the measured top five product ion masses of the manifold peptide precursor masses from (A) in MS/MS mode of the natural peptides at four decimals. (C) Overlap of the measured 62 synthetic peptide precursor masses in MS mode at four decimals.
Limit of Detection
To determine the limit of detection (LOD), four aliquots of purified HLA-eluted peptides from JY cells were spiked with 0.1 fmol, 1 fmol, 10 fmol, or 100 fmol isotope labeled synthetic peptides (supplemental Table S1) and analyzed in three replicates leading to 12 separate analytical replicates (for identified peptides, see supplemental Table S3). Based on our experience with JY cells, in HLA ligandomic experiments an optimal setting enables a peptide recovery rate of 80 ± 20% between two replicates. Thus, there is no LOD where 100% of the peptides will be discovered, especially in data-dependent acquisition. Here, we set the LOD to the peptide concentration that enables an identification of at least 50% (n = 31) of the peptides per replicate, with a recovery rate of 80% ± 20% and a Pearson correlation of the peptide retention times above 95% between three replicates.
The JY sample spiked with 10 fmol synthetic peptides had the lowest peptide content enabling a reproducible identification of 50% of the 62 added isotope labeled peptides (Fig. 4A). At the LOD the recovery rate of peptides in a replicate mass spectrometric measurement is in the range of 80% ± 20% (Fig. 4B) and the mean Pearson correlation of the retention times of the synthetic peptides in the three technical replicates analyzed in the LC is above 95% (Fig. 4C).
Fig. 4.
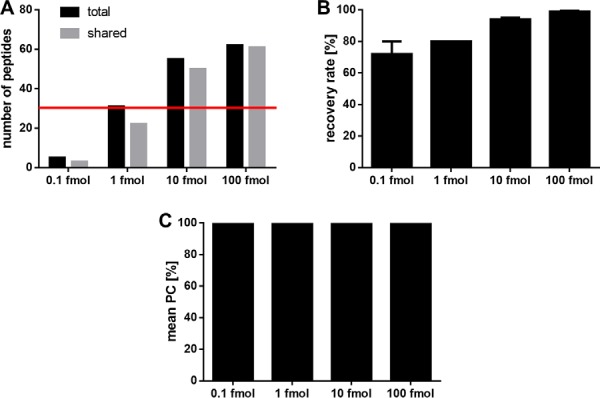
Validation of the limit of detection using spiked isotope labeled synthetic peptides. Three replicates of JY samples spiked with 0.1 fmol, 1 fmol, 10 fmol, and 100 fmol isotope labeled synthetic peptides were analyzed. A, Number of total identified isotope labeled peptides and shared peptides identified in the three replicates. The LOD of 50% (n = 31) of the spiked synthetic peptides is indicated with a red line. B, Recovery rate of synthetic peptides recovered between the three replicates of each condition. C, Mean Pearson correlation of the peptide retention times between the replicates. Abbreviation: PC, Pearson correlation.
Precision
The precision (also referred to as imprecision) was determined by assaying three aliquots of HLA-eluted peptides from JY samples in three technical replicates leading to nine separate analytical replicates. To prove the intermediate precision, the measurement series was repeated after 7 days (for identified peptides, see supplemental Table S5). The number of identified peptides fulfilled the acceptance criteria of ± 10% S.D. of the repeatability on the initial day and after 1 week (Fig. 5A, 5B). Furthermore, the acceptance criteria of the recovery rate with a recovery of 80 ± 20% of identified peptides in a repeated replicate were fulfilled on both measuring days (Fig. 5C). A closer look at the LC demonstrates that the mean Pearson correlation of the peptide retention times between all nine replicates was above 95% and fulfilled the criteria of the repeatability and intermediate precision (Fig. 5D).
Fig. 5.

Validation of the precision using immunopeptidomes from JY cells. A, Number of synthetic peptides identified at day 0 and day 7 in nine replicates, respectively. B, Standard deviation (S.D.) given in total peptide numbers and in percent. C, Recovery rate of peptides between the replicates. D, Mean Pearson correlation of the peptide retention times between the replicates. Abbreviation: S.D., standard deviation; PC, Pearson correlation.
Robustness of the Precision, Accuracy, and Specificity
The robustness was investigated performing a retrospective analysis of the purified HLA-eluted peptides of three primary samples: peripheral blood mononuclear cells from a healthy donor, a chronic lymphocytic leukemia sample, as well as a bladder cancer sample. The immunopeptidomes were isolated and analyzed by three different persons. To validate the robustness the specifications indicated in Table I should be fulfilled regarding accuracy, precision, and specificity.
The identified peptides of the three primary samples fulfill the acceptance criteria of the accuracy for the mass spectrometer and for the LC. The difference of the median of the mass deviation from the theoretical mass of all identified natural peptides (median ΔM: PBMC −0.06 ppm, CLL 0.05 ppm, BC 0.14 ppm) (Fig. 6A) and synthetic peptides (median ΔM: 0.19 ppm) is below 2 ppm (Fig. 6A). The peptide RTs between the replicates of all natural and all synthetic peptides have a Pearson correlation above 95% (Fig. 6B).
Fig. 6.

Validation of the accuracy using immunopeptidomes from primary PBMC, CLL, and BC samples. Three replicates were analyzed. A, Mass deviation from the theoretical precursor mass (ΔM ppm) to the theoretical mass of all identified natural peptides. B, Mean Pearson correlation of the peptide retention times between the replicates. Abbreviations: PC, Pearson correlation; ppm, parts per million; ΔM, mass deviation.
Regarding the precision, the three technical replicates of the three primary samples fulfill the acceptance criteria of the repeatability for both the mass spectrometer and the LC. The replicates have a percentage S.D. of identified peptide numbers below 10% (Fig. 7A, 7B) and the recovery rate is in the range of 80% ± 20% (Fig. 7C). The peptide RTs between the replicates have a Pearson correlation above 95% (Fig. 6B).
Fig. 7.

Validation of the repeatability using immunopeptidomes from primary PBMC, CLL, and BC samples. A, Total number of natural peptides identified in three technical replicates, respectively. B, Standard deviation (S.D.) given in total peptide numbers and in percent. C, Recovery rate of peptide identifications between the replicates. Abbreviation: S.D., standard deviation.
The specificity can be investigated, as all analyzed primary samples are expected to contain at least one of the previously used 62 synthetic peptides as natural HLA class I-presented peptide. The natural peptides should fulfill the acceptance criteria of the accuracy and specificity indicated in Table I for the mass spectrometer and the LC, respectively (for identified peptides and product ions, see supplemental Table S6).
The three replicates fulfill the acceptance criteria of the specificity for the mass spectrometer and for the LC. The difference of the median of the mass deviation from the theoretical mass (ΔM ppm) of the peptides AIVDKVPSV, YLLPAIVHI, GTYVSSVPR, RPSGPGPEL, SVINLVIVK, and RVYGGITTK, which were identified as natural and 100 fmol spiked synthetic peptides, is below 2 ppm (Fig. 8A).
Fig. 8.

Validation of the specificity using immunopeptidomes from primary PBMC, CLL, and BC samples and isotope labeled synthetic peptides spiked into JY. The samples were analyzed in three replicates. A, Mean mass deviation of the detected precursor masses from the theoretical masses (ΔM ppm) of the identified natural peptides AIVDKVPSV, YLLPAIVHI, GTYVSSVPR, RPSGPGPEL, SVINLVIVK and RVYGGITTK and 100 fmol synthetic peptides spiked into JY. B, Mass deviation as S.D. of the precursor masses detected in MS mode and (C) the resulting five selected top fragments in MS/MS modes of the identified natural and synthetic peptides. Abbreviations: ppm, parts per million; ΔM, mass deviation; S.D., standard deviation.
The S.D. of the mass accuracy of the precursor masses in MS mode and of the selected five top product ions in MS/MS mode of the peptides AIVDKVPSV, YLLPAIVHI, GTYVSSVPR, RPSGPGPEL, SVINLVIVK, and RVYGGITTK is below 0.001 Da for both the natural and synthetic peptides (Fig. 8B and 8C, for MS/MS spectra, see supplemental Fig. S1).
Transfer of the Method to Other LC-MS/MS Systems
In addition to the previous robustness analyses, the method was transferred to an LC-MS/MS system with a less sensitive LTQ Orbitrap XL and the HLA-eluted peptides from JY cells were analyzed. To demonstrate the method transfer to another LC-MS/MS system, the robustness measurements of the method were investigated with regard to accuracy, precision, and specificity on the LTQ Orbitrap XL containing LC-MS/MS system regardless of the specifications indicated in Table I set for an Orbitrap Fusion Lumos containing LC-MS/MS system. To increase the number of identified peptides, the MS/MS analysis is performed in the ion trap to enable a faster scanning throughput. Consequently, different peptide spectra are expected using adapted settings (described under Experimental Procedures) and therefore besides the JY samples also 500 fmol synthetic peptides of the five selected sequences AIVDKVPSV, RPSGPGPEL, YLLPAIVHI, KVLEYVIKV, and SPSSILSTL were spiked in JY matrix and measured on the LTQ Orbitrap XL for a spectral comparison. New five top product ions were selected based on intensity and relevance in MS/MS mode for the LTQ Orbitrap XL system. To investigate the accuracy, the purified HLA-eluted peptides from one JY batch were analyzed in three separate analytical replicates. Regarding the accuracy of the mass spectrometer in MS mode the median mass deviation from the theoretical mass of all identified natural peptides (median ΔM: −0.37 ppm) is below 2 ppm in Fig. 9A similar to the Orbitrap Fusion Lumos system. The peptide RTs between the replicates of all natural peptides do have a mean Pearson correlation above 95%, demonstrating the accuracy of the LC (Fig. 9B).
Fig. 9.

Investigation of the accuracy, repeatability and specificity using immunopeptidomes from JY cells analyzed after method transfer to a less sensitive LC-MS/MS system. Three replicates were analyzed. A, Mass deviation from the theoretical precursor mass (ΔM ppm) to the theoretical mass of all identified natural peptides and of the five identified natural peptides AIVDKVPSV, RPSGPGPEL, YLLPAIVHI, KVLEYVIKV and SPSSILSTL. B, Mean Pearson correlation of the peptide retention times between the replicates for all identified natural peptides and the five selected peptides. C, Total number of natural peptides identified in three technical replicates, respectively. D, Standard deviation (S.D.) given in total peptide numbers and in percent. E, Recovery rate of peptide identifications between the replicates. Abbreviation: S.D., standard deviation. Mass deviation as S.D. of the (F) precursor ion masses in MS mode and the (G) resulting five selected top fragments in MS/MS modes of the five selected natural and synthetic peptides. Abbreviations: PC, Pearson correlation; ppm, parts per million; ΔM, mass deviation; S.D., standard deviation.
The precision was determined by assaying the previously mentioned three technical replicates. The number of identified peptides was like the validated LC-MS/MS system below 10% S.D. with 5% S.D. of the repeatability on the initial day (Fig. 9C, 9D). However, the recovery rate with a recovery of 55 ± 4% of identified peptides in a repeated replicate is below the specifications of the Orbitrap Fusion Lumos system (Fig. 9E).
The specificity was again investigated using the mass deviation from the theoretical mass of the five selected peptides AIVDKVPSV, RPSGPGPEL, YLLPAIVHI, KVLEYVIKV, and SPSSILSTL. The median of the identified natural peptides is ΔM: −0.57 ppm (Fig. 9A) (for identified peptides and product ions, see supplemental Table S7).
The peptide RTs between the replicates of the five selected peptides identified as natural and synthetic peptides do have a Pearson correlation above 95% (Fig. 9B).
The standard deviation (S.D.) of the mass accuracy of the precursor masses in MS mode (Fig. 9F) and of the five selected top product ions in MS/MS mode (Fig. 9G, for MS/MS spectra, see supplemental Fig. S3), selected based on intensity and relevance, of the five selected peptides AIVDKVPSV, RPSGPGPEL, YLLPAIVHI, KVLEYVIKV, and SPSSILSTL is below 0.001 Da in MS Mode, analyzed in the Orbitrap, and below 0.1 in MS/MS mode, analyzed in the ion trap, for the natural peptides.
CONCLUSION/DISCUSSION
To provide reliable biomarker and patient-individual tumor-associated target antigen identification for clinical studies, the fast and sensitive LC-MS/MS assay for the identification of natural and synthetic HLA-restricted peptides was validated for the technical equipment of our laboratory, consisting of a nanoUHPLC, UltiMate 3000 RSLCnano on-line coupled to an Orbitrap Fusion Lumos mass spectrometer. The immunopeptidomics pipeline is used for identification and impurity detection, thus according to FDA and EMA guidelines a validation of the specificity and LOD is required. Additionally, we validated the accuracy, precision, and robustness to demonstrate the reliability of the pipeline.
The results of the JY samples spiked with isotope labeled synthetic peptides enabled verification of the accuracy of the LC-MS/MS system in terms of similarity of the analyzed natural peptides to the theoretical and synthetic peptide masses. With the same dataset we were able to identify five specific peptides, expected as natural and synthetic peptides, which fulfil the acceptance criteria of the accuracy and could prove the specificity. Furthermore, we could show that with five selected MS/MS product ions all identified peptides within one replicate can be distinguished. Instead of picking simply the five most intensive product ions for therapeutic peptide candidates, our quality control selects the top five product ions also according to meaningfulness (expert review: b- or y-ions are preferred), thus further increasing specificity. The validation of the precision showed a reliable identification of peptides with a uniform recovery rate proving the repeatability and intermediate precision after 1 week.
A major limitation of MS-based data-dependent acquisition (DDA) discovery approaches is the low recovery rate. In our immunopeptidomics experiments a recovery rate of 80% ± 20% was achieved for cell lines and tissue samples, owing to the tissue heterogeneity and high dynamic range. Because of the recovery rate, in our lab routinely triplicate measurements are performed. At the LOD a reasonable reliability should be provided with a recovery rate of 50%, when triplicate measurements are performed. A peptide content of 10 fmol synthetic peptides in JY matrix enabled a reliable identification of 50% of the peptides. An improvement of the recovery rate might be obtained with a replacement of the DDA analysis with data-independent acquisition, which has demonstrated a superior reproducibility (34–38).
To prove the robustness of the immunopeptidomics assay, we synthesized a large variety of known HLA ligands, with different length, mass, grand average of hydropathicity (GRAVY), theoretical isoelectric point (pI) and HLA allotype restriction. In addition, we employed several primary, clinically relevant samples in addition to the JY cell line and further analyzed soluble peripheral blood mononuclear cells from a healthy donor, a soluble chronic lymphocytic leukemia and a solid bladder cancer sample. Lastly, for the three primary samples the HLA immunoaffinity chromatography and immunopeptidomics analysis was performed from three different persons. We could successfully verify the specifications of the accuracy, precision, and specificity for both the mass spectrometer and LC, respectively.
Besides the robustness of the method, we exemplarily further investigated for precision, accuracy and specificity after the method transfer to a LC-MS/MS system using a LTQ Orbitrap XL. However, to obtain a high number of identified peptides, the MS/MS analysis is performed in the ion trap, instead of the Orbitrap, to enable a faster scanning throughput. Consequently, the mass accuracy of the product ions in the MS/MS analysis varies already at the second decimal and for the previously selected top five ions, two new ions had to be defined for SPSSILSTL and one new ion for the other peptides, except of AIVDKVPSV. Furthermore, the recovery-rate is much lower using the LTQ Orbitrap XL.
The specifications of our validated Orbitrap Fusion Lumos based LC-MS/MS system are not met by the LTQ Orbitrap XL. To fully validate the method on the LTQ Orbitrap XL for peptide identification according to the current FDA and EMA guidelines, at least the analyses regarding specificity and LOD must be performed or, ideally, all mentioned parameters should be investigated. The specifications of the precision, specificity and LOD must be adapted to the capabilities of the less sensitive mass spectrometer. However, the specifications should be tight enough to recognize immediately any functional problems with the LC-MS/MS system.
To enable standardization between different MS platforms and laboratories, the same immunopeptidome batch from one cell line should be used for immunopeptidomics. Here we present how the parameters accuracy, specificity, LOD, precision and robustness can be investigated for the validation of LC-MS/MS systems. However, every LC-MS/MS system in every laboratory needs to be validated independently with its own specifications (27). For the validation the specifications should be adapted as closely as possible to the optimal performance of the respective LC-MS/MS system, but should also consider the harmless device-related performance variations. Furthermore, it must be considered that more sensitive devices can detect peptides in lower quantities that less sensitive devices cannot identify. Here, the Orbitrap Fusion Lumos discovers twice the number of peptides. The validation is performed in a new process to show that the previously specified requirements (acceptance criteria) are reproducibly met in practical use and that the analytical method is appropriate for its intended use. After the validation a system suitability test (SST) is used to continuously monitor the performance of the instrument in different fixed intervals, to verify that an analytical method is suitable for the intended purpose on the day of analysis. Control peptides known from experience to be reliably identified in the respective immunopeptidome of the cell line in higher quantity should be selected for this purpose. The identification and the retention time of these peptides in the immunopeptidomes could be routinely checked in the LC-MS/MS analysis. The selected peptides can be standardized between different suitable LC-MS/MS systems and laboratories. The scope of each LC-MS/MS validation and the SSTs can prove the suitability and comparability of different laboratories for an analysis. The immunopeptidomic pipeline is currently in use for the identification of tumor-associated target antigens of multiple patients in the peptide vaccination study iVAC-CLL01 (NCT02802943) and for the preparation of further studies (e.g. PepIVAC01). Because of the peptide recovery rate of 80 ± 20% per replicate, patient samples are routinely analyzed in triplicates to ensure a high recovery. Finally, candidate peptide antigens are always verified using synthetic peptides to exclude false positives and artifacts. The identification procedure of tumor-specific HLA ligands using a comparison of ligand source proteins to established tumor antigens or ligand mapping on different malignant and benign tissues was carried out for cell lines and various tumor entities (8, 28, 39–43). During the last decades and in ongoing studies the immunopeptidomics pipeline has demonstrated its reliability and applicability.
In addition, we have now validated the accuracy, precision, specificity, LOD, and robustness in line with the current FDA and EMA guidelines. This validated pipeline enables the reliable identification of tumor-associated HLA-presented target antigens to support current and future clinical studies. Furthermore, different validation approaches are presented, that can be translated to other laboratories with similar equipment, or to other MS-based discovery approaches, such as proteomics, metabolomics, and lipidomics.
DATA AVAILABILITY
The mass spectrometry data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE (44) partner repository with the dataset identifier PXD012797 (http://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD012797).
Supplementary Material
Acknowledgments
We thank the Wirkstoffpeptidlabor, especially Patricia Hrstić, Ulrich Wulle, Nicole Bauer, Camille Supper, and Mirijam Bohn for expert peptide synthesis and quality control.
Footnotes
* This work was supported by the German Cancer Consortium (DKTK) and the Natural and Medical Sciences Institute at the University of Tübingen NMI. The authors declare that they have no conflicts of interest with the contents of this article.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- HLA
- Human leukocyte antigen
- AcN
- Acetonitrile
- BC
- Bladder cancer
- CLL
- Chronic lymphocytic leukemia
- EMA
- European Medicines Agency
- FDA
- Food and Drug Administration
- FDR
- False discovery rate
- GLP
- Good laboratory practice
- GMP
- Good manufacturing practice
- LOD
- Limit of detection
- OECD
- Organisation for Economic Co-operation and Development
- PBMC
- Peripheral blood mononuclear cells
- PPM
- Parts per million
- SD
- Standard deviation.
REFERENCES
- 1. Fortier M.-H., Caron E., Hardy M. P., Voisin G., Lemieux S., Perreault C., and Thibault P. (2008) The MHC class I peptide repertoire is molded by the transcriptome. J. Exp. Med. 205, 595–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mommen G. P. M., Frese C. K., Meiring H. D., van Gaans-van den Brink J., de Jong A. P. J. M., van Els C. A. C. M., and Heck A. J. R. (2014) Expanding the detectable HLA peptide repertoire using electron-transfer/higher-energy collision dissociation (EThcD). Proc. Natl. Acad. Sci. U.S.A. 111, 4507–4512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Caron E., Vincent K., Fortier M.-H., Laverdure J. P., Bramoulle A., Hardy M. P., Voisin G., Roux P. P., Lemieux S., Thibault P., and Perreault C. (2014) The MHC I immunopeptidome conveys to the cell surface an integrative view of cellular regulation. Mol. Syst. Biol. 7, 533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bassani-Sternberg M., Pletscher-Frankild S., Jensen L. J., and Mann M. (2015) Mass spectrometry of human leukocyte antigen class I peptidomes reveals strong effects of protein abundance and turnover on antigen presentation. Mol. Cell. Proteomics 14, 658–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hilf N., Kuttruff-Coqui S., Frenzel K., Bukur V., Stevanović S., Gouttefangeas C., Platten M., Tabatabai G., Dutoit V., van der Burg S. H., thor Straten P., Martínez-Ricarte F., Ponsati B., Okada H., Lassen U., Admon A., Ottensmeier C. H., Ulges A., Kreiter S., von Deimling A., Skardelly M., Migliorini D., Kroep J. R., Idorn M., Rodon J., Piró J., Poulsen H. S., Shraibman B., McCann K., Mendrzyk R., Löwer M., Stieglbauer M., Britten C. M., Capper D., Welters M. J. P., Sahuquillo J., Kiesel K., Derhovanessian E., Rusch E., Bunse L., Song C., Heesch S., Wagner C., Kemmer-Brück A., Ludwig J., Castle J. C., Schoor O., Tadmor A. D., Green E., Fritsche J., Meyer M., Pawlowski N., Dorner S., Hoffgaard F., Rössler B., Maurer D., Weinschenk T., Reinhardt C., Huber C., Rammensee H. G., Singh-Jasuja H., Sahin U., Dietrich P. Y., and Wick W. (2019) Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 565, 240–245 [DOI] [PubMed] [Google Scholar]
- 6. Hunt D. F., Henderson R. A., Shabanowitz J., Sakaguchi K., Michel H., Sevilir N., Cox A. L., Appella E., and Engelhard V. H. (1992) HLA-A2.1-associated peptides from a mutant cell line: a second pathway of antigen presentation. Science 255, 1264–1266 [DOI] [PubMed] [Google Scholar]
- 7. Lemmel C., Weik S., Eberle U., Dengjel J., Kratt T., Becker H. D., Rammensee H. G., and Stevanović S. (2004) Differential quantitative analysis of MHC ligands by mass spectrometry using stable isotope labeling. Nat. Biotechnol. 22, 450–454 [DOI] [PubMed] [Google Scholar]
- 8. Bassani-Sternberg M., Bräunlein E., Klar R., Engleitner T., Sinitcyn P., Audehm S., Straub M., Weber J., Slotta-Huspenina J., Specht K., Martignoni M. E., Werner A., Hein R., Busch H. D., Peschel C., Rad R., Cox J., Mann M., and Krackhardt A. M. (2016) Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat. Commun. 7, 13404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Falk K., Rötzschke O., Stevanović S., Jung G., and Rammensee H. G. (1991) Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature 351, 290–296 [DOI] [PubMed] [Google Scholar]
- 10. Abelin J. G., Keskin D. B., Sarkizova S., Hartigan C. R., Zhang W., Sidney J., Stevens J., Lane W., Zhang G. L., Eisenhaure T. M., Clauser K. R., Hacohen N., Rooney M. S., Carr S. A., and Wu C. J. (2017) Mass spectrometry profiling of HLA-associated peptidomes in mono-allelic cells enables more accurate epitope prediction. Immunity 46, 315–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bichmann L., Nelde A., Ghosh M., Heumos L., Mohr C., Peltzer A., Kuchenbecker L., Sachsenberg T., Walz J. S., Stevanović S., Rammensee H. G., and Kohlbacher O. (2019) MHCquant: automated and reproducible data analysis for immunopeptidomics. J. Proteome Res. acs.jproteome.9b00313 [DOI] [PubMed] [Google Scholar]
- 12. Bulik-Sullivan B., Busby J., Palmer C. D., Davis M. J., Murphy T., Clark A., Busby M., Duke F., Yang A., Young L., Ojo N. C., Caldwell K., Abhyankar J., Boucher T., Hart M. G., Makarov V., De Montpreville V. T., Mercier O., Chan T. A., Scagliotti G., Bironzo P., Novello S., Karachaliou N., Rosell R., Anderson I., Gabrail N., Hrom J., Limvarapuss C., Choquette K., Spira A., Rousseau R., Voong C., Rizvi N. A., Fadel E., Frattini M., Jooss K., Skoberne M., Francis J., and Yelensky R. (2019) Deep learning using tumor HLA peptide mass spectrometry datasets improves neoantigen identification. Nat. Biotechnol. 37, 55–63 [DOI] [PubMed] [Google Scholar]
- 13. Brown S. D., and Holt R. A. (2019) Neoantigen characteristics in the context of the complete predicted MHC class I self-immunopeptidome. Oncoimmunology 8, 1556080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yadav M., Jhunjhunwala S., Phung Q. T., Lupardus P., Tanguay J., Bumbaca S., Franci C., Cheung T. K., Fritsche J., Weinschenk T., Modrusan Z., Mellman I., Lill J. R., and Delamarre L. (2014) Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 515, 572–576 [DOI] [PubMed] [Google Scholar]
- 15. Backert L., and Kohlbacher O. (2015) Immunoinformatics and epitope prediction in the age of genomic medicine. Genome Med. 7, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Keskin D. B., Anandappa A. J., Sun J., Tirosh I., Mathewson N. D., Li S., Oliveira G., Giobbie-Hurder A., Felt K., Gjini E., Shukla S. A., Hu Z., Li L., Le P. M., Allesøe R. L., Richman A. R., Kowalczyk M. S., Abdelrahman S., Geduldig J. E., Charbonneau S., Pelton K., Iorgulescu J. B., Elagina L., Zhang W., Olive O., McCluskey C., Olsen L. R., Stevens J., Lane W. J., Salazar A. M., Daley H., Wen P. Y., Chiocca E. A., Harden M., Lennon N. J., Gabriel S., Getz G., Lander E. S., Regev A., Ritz J., Neuberg D., Rodig S. J., Ligon K. L., Suvà M. L., Wucherpfennig K. W., Hacohen N., Fritsch E. F., Livak K. J., Ott P. A., Wu C. J., and Reardon D. A. (2019) Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 565, 234–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ott P. A., Hu Z., Keskin D. B., Shukla S. A., Sun J., Bozym D. J., Zhang W., Luoma A., Giobbie-Hurder A., Peter L., Chen C., Olive O., Carter T. A., Li S., Lieb D. J., Eisenhaure T., Gjini E., Stevens J., Lane W. J., Javeri I., Nellaiappan K., Salazar A. M., Daley H., Seaman M., Buchbinder E. I., Yoon C. H., Harden M., Lennon N., Gabriel S., Rodig S. J., Barouch D. H., Aster J. C., Getz G., Wucherpfennig K., Neuberg D., Ritz J., Lander E. S., Fritsch E. F., Hacohen N., and Wu C. J. (2017) An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547, 217–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sahin U., Derhovanessian E., Miller M., Kloke B.-P., Simon P., Löwer M., Bukur V., Tadmor A. D., Luxemburger U., Schrörs B., Omokoko T., Vormehr M., Albrecht C., Paruzynski A., Kuhn A. N., Buck J., Heesch S., Schreeb K. H., Müller F., Ortseifer I., Vogler I., Godehardt E., Attig S., Rae R., Breitkreuz A., Tolliver C., Suchan M., Martic G., Hohberger A., Sorn P., Diekmann J., Ciesla J., Waksmann O., Brück A.-K., Witt M., Zillgen M., Rothermel A., Kasemann B., Langer D., Bolte S., Diken M., Kreiter S., Nemecek R., Gebhardt C., Grabbe S., Höller C., Utikal J., Huber C., Loquai C., and Türeci Ö. (2017) Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 547, 222–226 [DOI] [PubMed] [Google Scholar]
- 19. Walter S., Weinschenk T., Stenzl A., Zdrojowy R., Pluzanska A., Szczylik C., Staehler M., Brugger W., Dietrich P.-Y., Mendrzyk R., Hilf N., Schoor O., Fritsche J., Mahr A., Maurer D., Vass V., Trautwein C., Lewandrowski P., Flohr C., Pohla H., Stanczak J. J., Bronte V., Mandruzzato S., Biedermann T., Pawelec G., Derhovanessian E., Yamagishi H., Miki T., Hongo F., Takaha N., Hirakawa K., Tanaka H., Stevanovic S., Frisch J., Mayer-Mokler A., Kirner A., Rammensee H.-G., Reinhardt C., and Singh-Jasuja H. (2012) Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat. Med. 18, 1254–1261 [DOI] [PubMed] [Google Scholar]
- 20. Kowalewski D. J., and Stevanović S. (2013) Biochemical large-scale identification of MHC class I ligands. Methods Mol. Biol. 960, 145–157 [DOI] [PubMed] [Google Scholar]
- 21. Freudenmann L. K., Marcu A., and Stevanović S. (2018) Mapping the tumour human leukocyte antigen (HLA) ligandome by mass spectrometry. Immunology 154, 331–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nelde A., Kowalewski D. J., and Stevanović S. (2019) Purification and Identification of Naturally Presented MHC Class I and II Ligands. Methods Mol Biol. 1988, 123–136 [DOI] [PubMed] [Google Scholar]
- 23. Lathrop J. T., Jeffery D. A., Shea Y. R., Scholl P. F., and Chan M. M. (2016) US Food and Drug Administration Perspectives on Clinical Mass Spectrometry. Clin. Chem. 62, 41–47 [DOI] [PubMed] [Google Scholar]
- 24. Lynch K. L. (2016) CLSI C62-A: a new standard for clinical mass spectrometry. Clin. Chem. 62, 24–29 [DOI] [PubMed] [Google Scholar]
- 25. Food, U. S. (2018) Drug Administration Department of Health and Human Services Guidance for Industry: Bioanalytical Method Validation. at https://www.fda.gov/downloads/Drugs/../Guidances/ucm070107.pdf
- 26. European Medicines Agency Committee For Medicinal Products For Human Use Guideline on Bioanalytical Method Validation. (2018). at http://www.ema.europa.eu/docs/en%0AGB/document%0Alibrary/Scientific%0Aguideline/%0A2011/08/WC500109686.pdf
- 27. Vogeser M., and Seger C. (2016) Quality management in clinical application of mass spectrometry measurement systems. Clin. Biochem. 49, 947–954 [DOI] [PubMed] [Google Scholar]
- 28. Schuster H., Peper J. K., Bösmüller H.-C., Röhle K., Backert L., Bilich T., Ney B., Löffler M. W., Kowalewski D. J., Trautwein N., Rabsteyn A., Engler T., Braun S., Haen S. P., Walz J. S., Schmid-Horch B., Brucker S. Y., Wallwiener D., Kohlbacher O., Fend F., Rammensee H.-G., Stevanović S., Staebler A., and Wagner P. (2017) The immunopeptidomic landscape of ovarian carcinomas. Proc. Natl. Acad. Sci. U.S.A. 114, E9942–E9951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nelde A., Kowalewski D. J., Backert L., Schuster H., Werner J.-O., Klein R., Kohlbacher O., Kanz L., Salih H. R., Rammensee H.-G., Stevanović S., and Walz J. S. (2018) HLA ligandome analysis of primary chronic lymphocytic leukemia (CLL) cells under lenalidomide treatment confirms the suitability of lenalidomide for combination with T-cell-based immunotherapy. Oncoimmunology 7, e1316438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Barnstable C., Bodmer W. F., Brown G., Galfre G., Milstein C., Williams A. F., and Ziegler A. (1978) Production of monoclonal antibodies to group A erythrocytes, HLA and other human cell surface antigens-new tools for genetic analysis. Cell 14, 9–20 [DOI] [PubMed] [Google Scholar]
- 31. Eng J. K., McCormack A. L., and Yates J. R. (1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989 [DOI] [PubMed] [Google Scholar]
- 32. Käll L., Canterbury J. D., Weston J., Noble W. S., and MacCoss M. J. (2007) Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 4, 923–925 [DOI] [PubMed] [Google Scholar]
- 33. Organisation for economic co-operation and development. (1998) OECDPrinciples of Good Laboratory Practice. at http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=env/mc/chem(98)17&doclanguage=en
- 34. Panchaud A., Scherl A., Shaffer S. A., von Haller P. D., Kulasekara H. D., Miller S. I., and Goodlett D. R. (2009) Precursor acquisition independent from ion count: how to dive deeper into the proteomics ocean. Anal. Chem. 81, 6481–6488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Geiger T., Cox J., and Mann M. (2010) Proteomics on an Orbitrap benchtop mass spectrometer using all-ion fragmentation. Mol. Cell. Proteomics 9, 2252–2261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bern M., Finney G., Hoopmann M. R., Merrihew G., Toth M. J., and MacCoss M. J. (2010) Deconvolution of mixture spectra from ion-trap data-independent-acquisition tandem mass spectrometry. Anal. Chem. 82, 833–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Carvalho P. C., Han X., Xu T., Cociorva D., Carvalho M. da G., Barbosa V. C., and Yates J. R. (2010) XDIA: improving on the label-free data-independent analysis. Bioinformatics 26, 847–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Panchaud A., Jung S., Shaffer S. A., Aitchison J. D., and Goodlett D. R. (2011) Faster, quantitative, and accurate precursor acquisition independent from ion count. Anal. Chem. 83, 2250–2257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Di Marco M., Schuster H., Backert L., Ghosh M., Rammensee H.-G., and Stevanović S. (2017) Unveiling the peptide motifs of HLA-C and HLA-G from naturally presented peptides and generation of binding prediction matrices. J. Immunol. 199, 2639–2651 [DOI] [PubMed] [Google Scholar]
- 40. Walz S., Stickel J. S., Kowalewski D. J., Schuster H., Weisel K., Backert L., Kahn S., Nelde A., Stroh T., Handel M., Kohlbacher O., Kanz L., Salih H. R., Rammensee H.-G., and Stevanović S. (2015) The antigenic landscape of multiple myeloma: mass spectrometry (re)defines targets for T-cell-based immunotherapy. Blood 126, 1203–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kowalewski D. J., Schuster H., Backert L., Berlin C., Kahn S., Kanz L., Salih H. R., Rammensee H. G., Stevanovic S., and Stickel J. S. (2015) HLA ligandome analysis identifies the underlying specificities of spontaneous antileukemia immune responses in chronic lymphocytic leukemia (CLL). Proc. Natl. Acad. Sci. U.S.A. 112, E166–E175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Neidert M. C., Kowalewski D. J., Silginer M., Kapolou K., Backert L., Freudenmann L. K., Peper J. K., Marcu A., Wang S. S. Y., Walz J. S., Wolpert F., Rammensee H.-G., Henschler R., Lamszus K., Westphal M., Roth P., Regli L., Stevanović S., Weller M., and Eisele G. (2018) The natural HLA ligandome of glioblastoma stem-like cells: antigen discovery for T cell-based immunotherapy. Acta Neuropathol. 135, 923–938 [DOI] [PubMed] [Google Scholar]
- 43. Berlin C., Kowalewski D. J., Schuster H., Mirza N., Walz S., Handel M., Schmid-Horch B., Salih H. R., Kanz L., Rammensee H.-G., Stevanović S., and Stickel J. S. (2015) Mapping the HLA ligandome landscape of acute myeloid leukemia: a targeted approach toward peptide-based immunotherapy. Leukemia 29, 647–659 [DOI] [PubMed] [Google Scholar]
- 44. Vizcaíno J. A., Deutsch E. W., Wang R., Csordas A., Reisinger F., Ríos D., Dianes J. A., Sun Z., Farrah T., Bandeira N., Binz P. A., Xenarios I., Eisenacher M., Mayer G., Gatto L., Campos A., Chalkley R. J., Kraus H.-J., Albar J. P., Martinez-Bartolomé S., Apweiler R., Omenn G. S., Martens L., Jones A. R., and Hermjakob H. (2014) ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 32, 223–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE (44) partner repository with the dataset identifier PXD012797 (http://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD012797).