

Abstract
[2,3]-Sigmatropic rearrangements (Wittig rearrangements) of α-alkoxy oxazolidinone enolates are described. Whereas alkali metal enolates fail owing to facile deacylation, boron enolates generated from di-n-butylboron triflate and triethylamine rearranged in good yields and high selectivities with exceptions noted. IR and NMR spectroscopies show the boron was chelated by the α-alkoxy group rather than the more distal oxazolidinone carbonyl in the complex and enolate. The rearrangement product contained a boron alkoxide that remained unchelated by either carbonyl. Optimization was guided by density functional theory computations suggesting that valine-derived oxazolidinones would be superior to the phenylalanine-derived analogs.
Graphical Abstract

Introduction
Wittig rearrangements of allyloxy-substituted enolates and related carbanions have enjoyed considerable popularity (eq 1).1 Several aspects of this reaction captured our imagination. First, the aggregation of lithium enolates presents the possibility of rearranging within an aggregate, especially given mounting evidence that enolate aggregates equilibrate slowly on laboratory timescales.2a The consequences to stereochemistry would be considerable and pose some challenging but interesting questions. Second, our interest in the chemistry of oxazolidinone-derived Evans enolates3 previously revealed that, while the lithium enolates in THF are highly aggregated,2b TMEDA-solvated sodium enolates are monomeric,2c raising the prospect of probing aggregation effects from two extremes. Lastly, there are no reports that describe the use of the Evans oxazolidinone auxiliaries for such [2,3]-sigmatropic rearrangements.4
 |
(1) |
Ultimately, the allure of employing sodium and lithium Evans enolates in the [2,3]-sigmatropic rearrangement proved for naught: both substrates failed to rearrange owing to competing deacylation pathways (eq 2).2c While this explained the absence of such rearrangements, it also brought into focus a more basic question: How would one carry out such a rearrangement?
After briefly exploring titanium enolates with little luck,3 we were naturally drawn to boron enolates owing to reports of boron-based [2,3]-sigmatropic rearrangements on simpler systems,5 boron-based aldol additions of Evans enolates including α-alkoxy cases,3d,6 and our previous structural and mechanistic studies of the boron-based Evans enolates.2d,7
 |
(2) |
We describe herein structural, rate, and computational studies of the [2,3]-sigmatropic rearrangement outlined in eq 3. Although our interests are largely structural and mechanistic, we offer select examples that illustrate efficacy, applications, and logical steps toward optimization.
Results and Discussion
Methods.
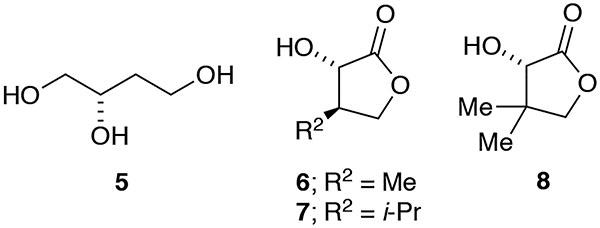
Emblematic results are illustrated in eq 3 and Table 1, with several more specialized examples discussed below (eqs 8–11). Dichloromethane (CH2Cl2) and CDCl3 are used interchangeably. Substrates 1a–1f were prepared from the alkoxy-substituted acids using standard protocols as described by eq 4.8,9,10 The absolute and relative stereochemistries were determined by converting 2a to triol 5,11 2d to lactone 6,12 and 2e to lactone 7.12a A quaternization (23, vide infra) was correlated with γ-lactone 8.13 The stereochemistries of the remaining products were inferred by analogy. Stereocontrol in the rearrangement is discussed at greater length in a section below.
 |
(3) |
 |
(4) |
Table 1.
[2,3]-Sigmatropic rearrangements of oxazolidinone-derived boron enolates.
| entry | substrate | R1 | R2 | temp | 2:3 | Yield |
|---|---|---|---|---|---|---|
| 1 | 1a | Bn | H | 0 °C to rt | 2:1 | 90% |
| 2 | 1b | i-Pr | H | 0 °C to rt | 3:1 | 81% |
| 3 | 1c | Bn | Me | 0 °C to rt | 5:1 | 90% |
| 4 | 1d | i-Pr | Me | 0 °C to rt | >15:1a | 82% |
| 5 | 1e | i-Pr | i-Pr | 0 °C to rt | >30:1 | 71% |
| 6 | 1f | i-Pr | Ph | −78 °C to rt | >30:1 | 91% |
Starting substrate 1d contains 6% cis isomer, which affords an anti isomer.
Previous studies showed that boron NMR spectroscopy would not be useful for studying oxazolidinone-derived intermediates 9–11,2d whereas IR spectroscopy proved critical.14 In the current study we again rely on IR spectroscopy and augment the assignments with standard two-dimensional 1H and 13C NMR spectroscopies (COSY, HSQC, HMBC, and ROESY). Comparisons of key intermediates with propionate-derived species 9–11 reveal the relative importance of the alkoxy and oxazolidinone carbonyl as ligands for boron.

Density functional theory (DFT) calculations were carried out at the B3LYP/6–31G(d) level with single-point calculations at the MP2/6–31G(d) level of theory.15 Transition structures display single negative frequencies. Allusions to results without further elaboration are documented in the Supporting Information.
Oxazolidinone-Boron Complex.
We first studied the [2,3] Wittig rearrangement of the simplest substrate, 1a. IR spectroscopy showed that treatment of 1a with 1.0–2.0 equiv of n-Bu2BOTf at 0 °C causes oxazolidinone and carboxamide carbonyl absorbances of 1a at 1783 cm−1 and 1719 cm−1 (Figure 1a) to be replaced by absorbances at 1825 cm−1 and 1613 cm−1, respectively (Figure 1b). Given that the carboxamide and oxazolidinone carbonyls of propionate-derived complex 9 both appear at 1727 cm−1, we conclude that complex 13, with a complexed carboxamide and a relatively unperturbed oxazolidinone carbonyl, is formed to the exclusion of 12, in which the oxazolidinone is complexed (eq 5). The full complement of 2D NMR spectroscopies show correlations, including those derived from the B-CH2 and allyloxy O-CH2 protons, that are consistent with the assignment of this intermediate as 13. DFT computations show an 11.9 kcal/mol preference for 13, which seems large but probably a consequence of the net charge. The computed structure of 13 shows a marked 180° rotation of the oxazolidinone moiety about the C–N bond relative to 12.
 |
(5) |
Figure 1.

IR spectra of 0.030 M 1a in CH2Cl2 recorded at 0 °C with (a) no additive, (b) 0.060 M n-Bu2BOTf affording 13, (c) 0.060 M n-Bu2BOTf and 0.10 M Et3N affording 15, and (d) product 17 after warming to 25 °C.
Boron Enolate.
Treatment of complex 13 with Et3N at −30 °C causes immediate formation of boron enolate 15, with absorbances for the oxazolidinone and enolate at 1781 cm−1 and 1713 cm−1, respectively (Figure 1c). Once again, the minor shift of the oxazolidinone absorbance when compared with enolate 10 (1706 cm−1) in the propionate series is consistent with ether-based chelate 15 and an uncomplexed oxazolidinone, rather than 14. Two-dimensional NMR spectroscopies show a single Z-isomeric enolate 15. DFT computations predict a 1.6 kcal/mol preference for 15 versus 14.
 |
(6) |
Sigmatropic Rearrangement.
Warming enolate 15 to 25 °C causes rearrangement following a clean, first-order decay (Figure 2) with formation of alkoxide 17 as a 2:1 mixture of diastereomers (confirmed after workup) in 90% isolated yield. The absorbances for 17 at 1779 cm−1 and 1711 cm−1, respectively (Figure 1d), show that the boron alkoxide is not chelated by either carbonyl. DFT computations predict a 4.9 kcal/mol preference for open (unchelated) three-coordinate alkoxide 17 (analogous to 11) relative to the chelate 16.
 |
(7) |
Figure 2.
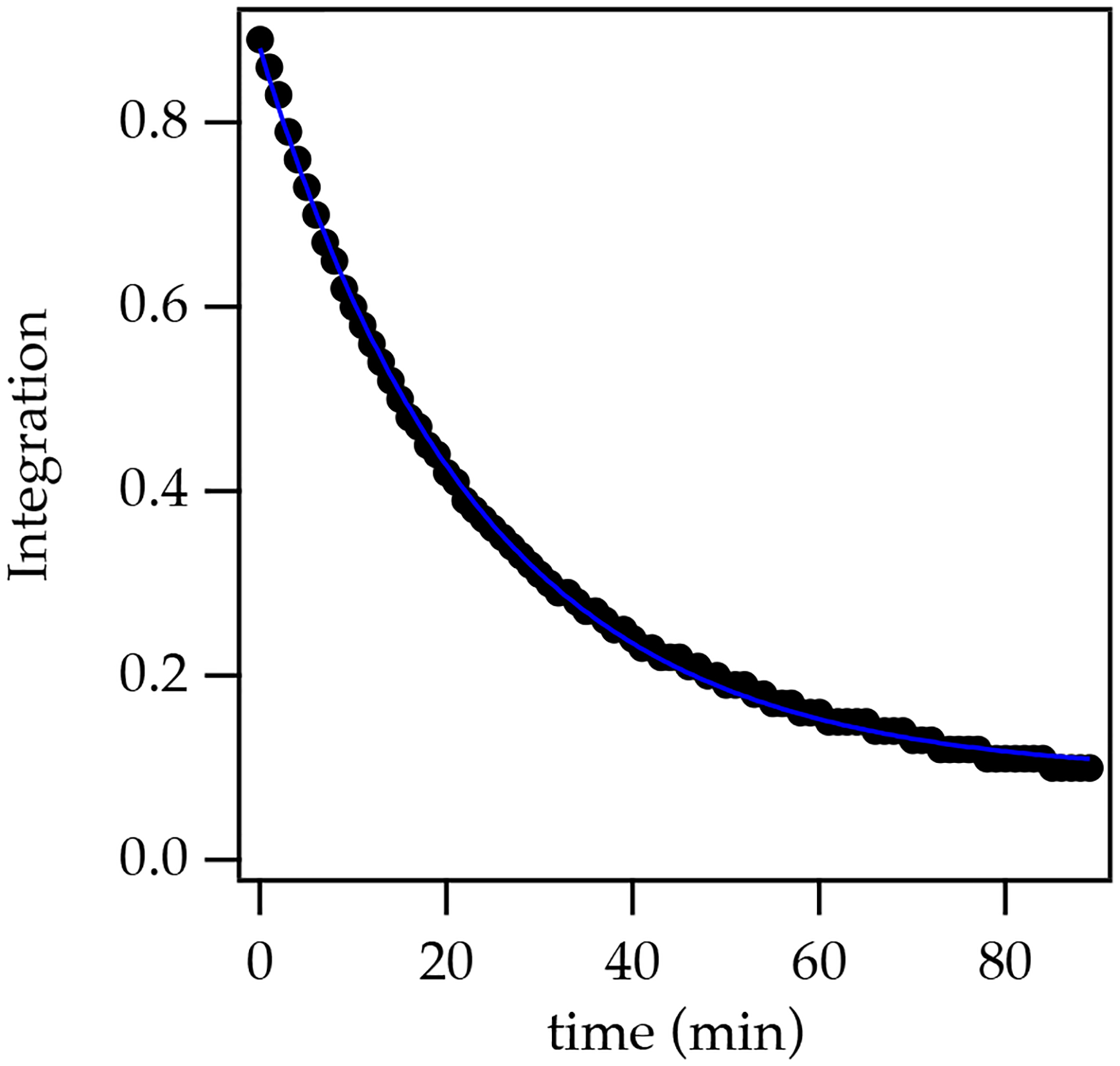
First-order decay of the rearrangement of enolate 15 to give alkoxide 17.
The rearrangement in eq 7 represents the simplest mechanism we have ever studied. The first-order decay is operationally the entire rate law. The first-order rate constant is independent of initial concentration and unaffected by excess Et3N. DFT computations were carried out on four isomeric transition structures 18 (Scheme 1). The barrier from the pre-complex 15 to transition structure 18A is 12.2 kcal/mol. The benzyl moiety appears to not only be an inadequate stereochemical determinant in this simple case but seemed likely to remain so even with substituents placed on the allyloxy fragment. A significant preference for half-chair conformers 18A and 18B relative to 18C and 18D appears to derive from unfavorable OCH–HCB interactions (see arrow) in the latter two. The cleaving C–O bond and forming C–C bond are 2.0–2.1 Å and 2.8 Å, respectively, for all the transition structures described herein.
Scheme 1.

DFT computed stereoisomers of transition structure 18 using methyls as surrogates for n-butyls for the rearrangement of 1a to major and minor products 2a and 3a (Table 1, entry 1).
The chelation serves several purposes. In theory it provides organization for stereocontrol, which bore no fruit for substrate 1a owing to an absence of stereochemically determining interactions. It also, however, allows for a smooth transition from enolate 15 to alkoxide 16 with minimal atomic movement or charge development. Contrast this with the developing charge in transition structure 19 derived from chelate 14, which causes a large spike in its computed energy. The ether-based chelate also may competitively inhibit the boron-assisted deacylation via a transition structure such as 20.

Optimizing Stereoselectivity.
The near stereorandom rearrangement of 1a and computational support for this observed stereorandomness amounted to an inauspicious first effort. It would be more expedient to carry out a simple allylation of α-alkoxy Evans enolates via direct allylation.16 However, inspection of the cyclic transition structures (Scheme 1), along with follow-up computational studies, led us to hypothesize that improved stereocontrol might be achieved through installation of a vinylic substituent at the 3-position (R2) and replacement of the oxazolidinone benzyl group with an isopropyl moiety.
Table 1 (entries 2–4) confirmed this supposition. Thus, trans-crotyl ether 1c rearranges to yield a modest 5:1 mixture of 2c and 3c to the exclusion of two other possible isomers (entry 3). Rearrangement of the valine-derived variant 1d occurs with significantly improved 15:1 selectivity (entry 4). The origin of this enhanced selectivity is reflected in the DFT computations (Scheme 2). The most stable isomeric transition structure, 21A, corresponds to the major product (2d) while the second most stable isomer, 21B, corresponds to the syn product 3d (eq 1). A 2.6 Å CH3–CH3 contact in 21B (see arrow) absent in 21A appears to be the source of the facial selectivity.
Scheme 2.

DFT computed stereoisomers of transition structure 21 using methyls as surrogates for n-butyls for the rearrangement of 1d to the major and minor products 2d and 3d (Table 1, entry 4).
Rearrangement of the i-propyl and phenyl-substituted allyl ethers gave excellent results (entries 5 and 6). In the latter case, it was essential to maintain the reaction temperature at −78 °C until after the addition of Et3N to avoid decomposition of the cinnamyl ether-based substrate 1f by an apparent solvolysis.
We pressed our luck with a few additional substrates not shown in Table 1. The cis-crotyl ether 1g (eq 8) rearranged to give two dominant anti isomers albeit with a poor 1.7:1.0:0.5:0.1 stereocontrol. DFT computations are consistent with the modest reversal. Quaternization of the α carbon was largely a disaster (eq 9). Quaternization of the β carbon afforded 23 in 10:1 selectivity in high yield (eq 10),17 but that did not translate particularly well to a stereochemically controlled quaternization (eq 11).
 |
(8) |
 |
(9) |
 |
(10) |
 |
(11) |
Conclusion
The Evans-boron-enolate-based [2,3]-sigmatropic reaction is an effective protocol using trans allylic ethers and showed some promise for the 2,2-disubstituted allylic ethers. A combination of spectroscopic and computational studies helped us understand the structures of the intermediates along the reaction coordinate (Scheme 3) and guided us to a functional protocol where none had seemed to exist in the early stages. The intermediate enolate, chelated by the alkoxy moiety rather than the oxazolidinone carbonyl, is common to a number of boron-based aldol additions of such α-alkoxy Evans enolates.3,6
Scheme 3.

Summary of the boron-enolate based [2,3]-sigmatropic rearrangement.
Experimental
Reagents and Solvents.
CH2Cl2, CHCl3, and CDCl3 were distilled from molecular sieves. Trialkylamines were distilled from sodium benzophenone ketyl. n-Bu2BOTf was used from a commercial 1.0 M n-Bu2BOTf solution in CH2Cl2. Air- and moisture-sensitive materials were manipulated under argon using standard glovebox, vacuum line, and syringe techniques. While the reported yields of rearranged products were optimized, those of the starting materials and correlation products were not.
NMR Spectroscopy.
An NMR tube under vacuum was flame-dried on a Schlenk line and allowed to return to room temperature, backfilled with argon, and placed in a −78 °C dry ice/acetone bath. The appropriate amounts of oxazolidinone, n-Bu2BOTf, and Et3N in CDCl3 were added sequentially via syringe. The tube was flame-sealed under partial vacuum, mixed on a vortex mixer three times for ~10 s with cooling between each vortexing, and stored in a freezer at −80 °C. Standard 1H and 13C NMR spectra were recorded on a 500 MHz spectrometer at 500 and 125 MHz, respectively. The 1H and 13C resonances are referenced to CDCl3 (CHCl3 7.26 and CDCl3 77.2 ppm).
IR spectroscopic analyses.
IR spectra were recorded with an in situ IR spectrometer fitted with a 30-bounce, silicon-tipped probe. The spectra were acquired in 16 scans at a gain of 1 and a resolution of 4 cm−1. A representative reaction was carried out as follows: The IR probe was inserted through a nylon adapter and O-ring seal into an oven-dried, cylindrical flask fitted with a magnetic stir bar and a T-joint. The T-joint was capped with a septum for injections and a nitrogen line. After evacuation under full vacuum, heating, and flushing with nitrogen, the flask was charged with CH2Cl2 and cooled in a 0 °C ice bath. After a background spectrum was recorded, oxazolidinone 1a (41.3 mg, 0.15 mmol) was added as a 1.0 M solution in CH2Cl2 with stirring, followed by 1.0 M n-Bu2OTf (0.30 mL, 0.30 mmol), and neat Et3N (70 μL, 0.50 mmol). IR spectra were recorded every 15 s with monitoring of the absorbance at 1783 cm−1 and 1825 cm−1 over the course of the reaction.
Mass Spectrometry.
The high-resolution mass spectra (HRMS) were measured using a DART-Orbitrap. The α-alkoxy oxazolidinones showed relatively minor parent ions (corresponding to i below) and numerous fragmentation products. The structures listed below are emblematic.

(S,E)-3-(2-(but-2-en-1-yloxy)acetyl)-4-isopropyloxazolidin-2-one (1d).
To a solution of NaHMDS (40 mmol, 7.3 g) in THF (20 mL) was added crotyl alcohol (20 mmol, 1.7 mL, 15:1 trans:cis) followed by stirring under argon for 15 min at rt. A solution of α-bromo acetic acid (18 mmol, 2.5 g) in THF (10 mL) was added. After stirring for 12 h, the reaction was quenched by KOH solution (1.0 M, 20 mL) and extracted three times with KOH solution. The combined aqueous layers were acidified to pH = 1 using concentrated HCl at 0 °C and extracted six times with CH2Cl2. The organic extracts were dried over MgSO4 and concentrated in vacuo. Flash chromatography (30% ethyl acetate/hexanes) afforded the acid 4c as a light yellow oil (1.3 g, 56% yield). 1H NMR (500 MHz, CDCl3) δ 5.77 (dqt, J = 15.4, 6.6, 1.2 Hz, 1H), 5.58 (dtq, J = 15.0, 6.6, 1.6 Hz, 1H), 4.10 (d, J = 1.4 Hz, 2H), 4.04 (dp, J = 6.6, 1.2 Hz, 2H), 1.73 (ddt, J = 6.5, 1.9, 1.1 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 177.2, 131.8, 126.0, 72.3, 66.6, 17.8. To a solution of 4c (4.0 mmol, 464.5 mg) in THF (5.0 mL) was added triethylamine (4.4 mmol, 613 μL) followed by stirring for 5 min at −78 °C. Trimethylacetyl chloride (4.4 mmol, 542 μL) was added. The mixture was warmed to rt and stirred for additional 30 min to generate the mixed anhydride. To a solution of (S)-4-isopropyl-2-oxazolidinone (4.0 mmol, 516.6 mg) in THF (30 mL) was added n-BuLi (4.0 mmol, 2.5 mL) as a 1.6 M solution in hexane followed by stirring under argon for 15 min at −78 °C. The mixed anhydride solution was cooled to −78 °C, and the solution containing the lithiated oxazolidinone was added by cannula. The reaction was warmed to rt and stirred for additional 30 min. The mixture was quenched with saturated NH4Cl solution and extracted three times with diethyl ether. The organic extracts were dried over MgSO4 and concentrated in vacuo. Flash chromatography (25% ethyl acetate/hexanes) afforded 1d as a colorless oil (673 mg, 70% yield, 15:1 trans:cis). 1H NMR (500 MHz, CDCl3) δ 5.80 – 5.72 (m, 1H), 5.62 (dtd, J = 15.2, 6.5, 1.7 Hz, 1H), 4.64 (d, J = 2.3 Hz, 2H), 4.45 (dt, J = 8.5, 3.6 Hz, 1H), 4.34 (t, J = 8.7 Hz, 1H), 4.26 (dd, J = 9.1, 3.1 Hz, 1H), 4.09 – 4.00 (m, 2H), 2.43 (heptd, J = 7.3, 3.1 Hz, 1H), 1.72 (d, J = 6.6, 3H), 0.92 (d, J = 7.1 Hz, 3H), 0.88 (d, J = 6.9 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 170.4, 154.2, 131.1, 126.8, 72.3, 69.3, 64.6, 58.3, 28.4, 18.0, 17.9, 14.8. HRMS (DART-Orbitrap) m/z [M+H]+ calcd for C12H20NO4 242.1386, found 242.1399.
(S)-3-(2-(allyloxy)acetyl)-4-benzyloxazolidin-2-one (1a).
Following the procedure of 1d using allyl alcohol and (S)-4-benzyl-2-oxazolidinone afforded alkoxy acid 4a as a yellow liquid (273.1 mg, 47% yield). 1H NMR (500 MHz, CDCl3) δ 5.91 (ddt, J = 17.2, 10.3, 5.9 Hz, 1H), 5.33 (dq, J = 17.2, 1.6 Hz, 1H), 5.27 (dq, J = 10.4, 1.3 Hz, 1H), 4.15 – 4.10 (m, 4H). 13C{1H} NMR (126 MHz, CDCl3) δ 174.7, 133.3, 119.01, 72.7, 66.7. Further conversion of 4a affords 1a as a colorless oil (520.3 mg, 86% yield). 1H NMR (500 MHz, CDCl3) δ 7.34 (tt, J = 6.9, 1.1 Hz, 2H), 7.31 – 7.27 (m, 1H), 7.24 – 7.18 (m, 2H), 5.97 (ddt, J = 17.2, 10.4, 5.8 Hz, 1H), 5.35 (dq, J = 17.2, 1.6 Hz, 1H), 5.26 (dq, J = 10.4, 1.3 Hz, 1H), 4.74 – 4.64 (m, 3H), 4.33 – 4.26 (m, 1H), 4.24 (dd, J = 9.1, 3.0 Hz, 1H), 4.21 – 4.13 (m, 2H), 3.34 (dd, J = 13.5, 3.3 Hz, 1H), 2.82 (dd, J = 13.5, 9.4 Hz, 1H). 13C{1H} NMR (126 MHz, CDCl3) δ 170.4, 153.6, 135.1, 134.0, 129.6, 129.2, 127.6, 118.4, 72.7, 69.7, 67.4, 55.0, 37.9. HRMS (DART-Orbitrap) m/z [M+H]+ calcd for C15H18NO4 276.1230, found 276.1244.
(S)-3-(2-(allyloxy)acetyl)-4-isopropyloxazolidin-2-one (1b).
Following the procedure of 1d using allyl acohol afforded 1b as a colorless oil (543.8 mg, 60% yield). 1H NMR (500 MHz, CDCl3) δ 5.95 (ddt, J = 16.4, 10.3, 5.8 Hz, 1H), 5.32 (dt, J = 17.3, 1.6 Hz, 1H), 5.27 – 5.20 (m, 1H), 4.68 (d, J = 3.5 Hz, 2H), 4.45 (dt, J = 8.3, 3.5 Hz, 1H), 4.35 (t, J = 8.8 Hz, 1H), 4.27 (dd, J = 9.2, 3.1 Hz, 1H), 4.18 – 4.08 (m, 2H), 2.43 (hd, J = 7.0, 3.9 Hz, 1H), 0.93 (d, J = 7.0 Hz, 3H), 0.88 (d, J = 6.9 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 170.3, 154.2, 134.0, 77.4, 72.7, 69.7, 64.6, 58.4, 28.4, 18.1, 14.8. HRMS (DART-Orbitrap) m/z [M+H]+ calcd for C11H18NO4 228.1230, found 228.1242.
(S,E)-4-benzyl-3-(2-(but-2-en-1-yloxy)acetyl)oxazolidin-2-one (1c).
Following the procedure to prepare 1d using (S)-4-benzyl-2-oxazolidinone afforded 1c as a colorless oil (504.3 mg, 83% yield). 1H NMR (500 MHz, CDCl3) δ 7.34 (dd, J = 8.1, 6.5 Hz, 2H), 7.31 – 7.26 (m, 1H), 7.21 (dd, J = 7.0, 1.8 Hz, 2H), 5.84 – 5.75 (m, 1H), 5.64 (dtq, J = 14.7, 6.3, 1.6 Hz, 1H), 4.73 – 4.67 (m, 1H), 4.65 (d, J = 4.3 Hz, 2H), 4.29 (dd, J = 9.2, 7.8 Hz, 1H), 4.23 (dd, J = 9.1, 3.0 Hz, 1H), 4.12 – 4.04 (m, 2H), 3.33 (dd, J = 13.4, 3.3 Hz, 1H), 2.82 (dd, J = 13.4, 9.5 Hz, 1H), 1.74 (dq, J = 6.5, 1.2 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 170.5, 153.6, 135.1, 131.1, 129.6, 129.2, 127.6, 126.8, 72.4, 69.4, 67.4, 55.0, 37.9, 18.0. HRMS (DART-Orbitrap) m/z [M+H]+ calcd for C16H20NO4 290.1387, found 290.1397.
(S,E)-4-isopropyl-3-(2-((4-methylpent-2-en-1-yl)oxy)acetyl)oxazolidin-2-one (1e).
trans-4-Methylpent-2-en-1-ol was prepared by a literature procedure as follows.18 To a solution of ethyl (triphenylphosphoranylidene)acetate (20 mmol, 6.97 g) in CH2Cl2 (40 mL) was added isobutyraldehyde (25 mmol, 2.3 mL) and stirred at rt overnight. The mixture was concentrated in vacuo, dissolved in hexanes, and filtered. The solution was concentrated in vacuo to afford the ester precursor as a colorless liquid (1.2 g, 42% yield). 1H NMR (500 MHz, CDCl3) δ 6.94 (dd, J = 15.7, 6.6 Hz, 1H), 5.76 (dd, J = 15.7, 1.5 Hz, 1H), 4.18 (q, J = 7.1 Hz, 2H), 2.45 (dpd, J = 13.5, 6.8, 1.5 Hz, 1H), 1.29 (t, J = 7.1 Hz, 3H), 1.06 (d, J = 6.8 Hz, 6H). 13C{1H} NMR (126 MHz, CDCl3) δ 167.2, 155.6, 118.8, 60.3, 31.1, 21.4, 14.4. To a solution of the ester (8.5 mmol, 1.2 g) in toluene (20 mL) was added diisobutylaluminum hydride (20 mmol, 20 mL) as 1.0 M solution in toluene at −78 °C. The reaction was warmed to rt and stirred for additional 12 h. The reaction was quenched with saturated NH4Cl and acidified with concentrated HCl until all solid dissolved. The mixture was extracted three times with diethyl ether. The organic extracts were dried over MgSO4 and concentrated in vacuo to afford the alcohol precursor as a colorless liquid (648 mg, 76% yield). 1H NMR (500 MHz, CDCl3) δ 5.67 (ddt, J = 15.4, 6.2, 1.2 Hz, 1H), 5.59 (dtd, J = 15.5, 5.8, 1.2 Hz, 1H), 4.09 (dt, J = 6.0, 1.1 Hz, 2H), 2.36 (s, 1H), 2.34 – 2.26 (m, 1H), 1.00 (dd, J = 6.7, 0.8 Hz, 6H). Following the procedure of 1d, trans-4-Methylpent-2-en-1-ol was converted to alkoxy acid 4e as a light yellow liquid (312 mg, 32% yield). 1H NMR (500 MHz, CDCl3) δ 5.73 (ddt, J = 15.5, 6.5, 1.2 Hz, 1H), 5.50 (dtd, J = 15.5, 6.5, 1.4 Hz, 1H), 4.09 (s, 2H), 4.06 (dt, J = 6.6, 1.0 Hz, 2H), 2.38 – 2.27 (m, 1H), 1.00 (d, J = 6.7 Hz, 6H). 13C{1H} NMR (126 MHz, CDCl3) δ 171.45, 144.01, 121.83, 72.62, 66.33, 30.94, 22.21. Alkoxy acid 4e was converted to 1e as a colorless liquid (295 mg, 52% yield). 1H NMR (500 MHz, CDCl3) δ 5.75 (ddt, J = 15.5, 6.4, 1.2 Hz, 1H), 5.57 (dtd, J = 15.6, 6.4, 1.4 Hz, 1H), 4.67 (d, J = 1.8 Hz, 2H), 4.50 – 4.45 (m, 1H), 4.37 (t, J = 8.8 Hz, 1H), 4.29 (dd, J = 9.1, 3.1 Hz, 1H), 4.12 – 4.05 (m, 2H), 2.46 (heptd, J = 7.0, 3.9 Hz, 1H), 2.40 – 2.29 (m, 1H), 1.02 (d, J = 6.8 Hz, 6H), 0.95 (d, J = 7.0 Hz, 3H), 0.90 (d, J = 7.0 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 170.3, 154.1, 143.0, 122.4, 72.4, 69.3, 64.4, 58.2, 30.8, 28.2, 22.1, 22.1, 17.9, 14.7. HRMS (DART-Orbitrap) m/z [M+H]+ calcd for C14H24NO4 270.1700, found 270.1707.
(S)-3-(2-(cinnamyloxy)acetyl)-4-isopropyloxazolidin-2-one (1f).
Following the procedure of 1d using cinnamyl alcohol afforded 4f as a yellow solid (488 mg, 51% yield). 1H NMR (500 MHz, CDCl3) δ 7.43 – 7.37 (m, 2H), 7.35 – 7.31 (m, 2H), 7.29 – 7.27 (m, 1H), 6.67 – 6.62 (m, 1H), 6.28 (dt, J = 15.9, 6.3 Hz, 1H), 4.29 (dd, J = 6.4, 1.3 Hz, 2H), 4.17 (s, 2H). 13C{1H} NMR (126 MHz, CDCl3) δ 171.4, 136.2, 134.6, 128.8, 128.3, 126.8, 124.2, 72.4, 66.6. And 1f as a light yellow solid (283 mg, 37% yield). 1H NMR (500 MHz, CDCl3) δ 7.39 (dd, J = 7.4, 1.8 Hz, 2H), 7.32 (td, J = 7.6, 1.7 Hz, 2H), 7.24 (dd, J = 7.8, 1.6 Hz, 1H), 6.64 (d, J = 15.9 Hz, 1H), 6.33 (dtd, J = 15.9, 6.3, 1.7 Hz, 1H), 4.72 (d, J = 2.1 Hz, 2H), 4.45 (dt, J = 8.1, 3.3 Hz, 1H), 4.34 – 4.24 (m, 4H), 2.47 – 2.37 (m, 1H), 0.92 (dd, J = 7.1, 1.6 Hz, 3H), 0.86 (dd, J = 6.9, 1.6 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 170.2, 154.1, 136.4, 133.6, 128.6, 127.9, 126.6, 125.0, 72.2, 69.5, 64.4, 58.2, 28.3, 17.9, 14.6. HRMS (DART-Orbitrap) m/z [M+H]+ calcd for C17H22NO4 304.1543, found 304.1557.
(S,Z)-3-(2-(but-2-en-1-yloxy)acetyl)-4-isopropyloxazolidin-2-one (1g).
Following the procedure of 1d except using 2-butyn-1-ol afforded the alkoxy acid 4g as a light yellow liquid (805 mg, 33% yield). 1H NMR (500 MHz, CDCl3) δ 4.27 (q, J = 2.4 Hz, 2H), 4.23 (s, 2H), 1.86 (t, J = 2.3 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 174.3, 84.6, 73.6, 65.8, 59.3, 3.7. Alkoxy acid 4g was converted to the acylated oxazolidinone as a colorless oil (987 mg, 71% yield). 1H NMR (500 MHz, CDCl3) δ 4.76 (s, 2H), 4.48 – 4.42 (m, 1H), 4.35 (t, J = 8.7 Hz, 1H), 4.30 – 4.24 (m, 3H), 2.42 (heptd, J = 7.0, 4.0 Hz, 1H), 1.85 (t, J = 2.3 Hz, 3H), 0.93 (d, J = 7.0 Hz, 3H), 0.89 (d, J = 7.0 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 169.95, 154.2, 83.9, 74.2, 68.6, 64.6, 59.1, 58.4, 28.4, 18.0, 14.8, 3.8. Lindlar hydrogenation was carried out according to a modified literature procedure as follows.19 To a solution of acylated oxazolidinone (2 mmol, 479 mg) in MeOH (10 mL) was added Lindlar catalyst (0.10 mmol, 212 mg) and stirred under H2 (1.0 atm) at rt for 18 hr. The reaction was filtered through Celite and concentrated in vacuo. Flash chromatography (25% ethyl acetate/hexanes, Rf = 0.30) afforded 1g as colorless oil (409 mg, 85%, 12:1 cis:trans). 1H NMR (500 MHz, CDCl3) δ 5.73 (dqt, J = 10.9, 6.9, 1.3 Hz, 1H), 5.65 – 5.58 (m, 1H), 4.66 (d, J = 4.5 Hz, 2H), 4.46 (ddd, J = 8.3, 4.0, 3.1 Hz, 1H), 4.34 (dd, J = 9.2, 8.5 Hz, 1H), 4.26 (dd, J = 9.2, 3.0 Hz, 1H), 4.24 – 4.15 (m, 2H), 2.44 (dtt, J = 14.0, 7.0, 3.5 Hz, 1H), 0.93 (d, J = 7.1 Hz, 3H), 0.88 (d, J = 7.0 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 170.4, 154.2, 129.3, 126.0, 69.6, 66.7, 64.6, 58.3, 28.4, 18.0, 14.8. HRMS (DART-Orbitrap) m/z [M+H]+ calcd for C12H20NO4 242.1387, found 242.1396.
(4S)-3-(2-(allyloxy)propanoyl)-4-isopropyloxazolidin-2-one (1h).
To a solution of 1b (0.50 mmol, 114 mg) in THF (3 mL) was added NaHMDS (0.6 mmol, 110 mg) in THF (2 mL) under argon at −78 °C. After stirring for 15 min, methyl iodide (1.0 mmol, 62 μL) was added and the mixture was warmed to 0 °C and stirred for another 30 min. The reaction was quenched with NH4Cl and extracted three times with diethyl ether. The organic extracts were dried over MgSO4 and concentrated in vacuo. Flash chromatography (25% ethyl acetate/hexanes, Rf = 0.30) afforded 1h as a colorless oil (65 mg, 54%). 1H NMR (500 MHz, CDCl3) δ 5.93 (ddt, J = 17.2, 10.3, 5.8 Hz, 1H), 5.29 (dq, J = 17.2, 1.6 Hz, 1H), 5.19 (dq, J = 10.3, 1.3 Hz, 1H), 5.11 (q, J = 6.6 Hz, 1H), 4.51 (ddd, J = 8.5, 4.1, 3.2 Hz, 1H), 4.34 (t, J = 8.8 Hz, 1H), 4.25 (dd, J = 9.2, 3.2 Hz, 1H), 4.07 (ddt, J = 12.3, 5.8, 1.4 Hz, 1H), 3.92 (ddt, J = 12.3, 6.0, 1.3 Hz, 1H), 2.34 (pd, J = 6.9, 4.1 Hz, 1H), 1.49 (d, J = 6.7 Hz, 3H), 0.92 (d, J = 7.0 Hz, 3H), 0.88 (d, J = 6.9 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 173.7, 153.7, 134.4, 117.9, 73.7, 71.3, 64.3, 58.3, 28.6, 19.0, 18.0, 15.0. HRMS (DART-Orbitrap) m/z [M+H]+ calcd for C12H20NO4 242.1387, found 242.1399.
(S)-4-isopropyl-3-(2-((3-methylbut-2-en-1-yl)oxy)acetyl)oxazolidin-2-one (1i).
Following the procedure of 1d except using 3-methyl-2-buten-1-ol afforded 4i as a pale oil (573 mg, 40% yield). 1H NMR (500 MHz, CDCl3) δ 5.35 (tp, J = 7.1, 1.4 Hz, 1H), 4.11 (d, J = 7.2 Hz, 2H), 4.09 (s, 2H), 1.77 (d, J = 1.5 Hz, 3H), 1.70 (d, J = 1.4 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 174.64, 139.50, 119.59, 67.89, 66.44, 25.96, 18.17. And 1i as a white solid (183.5 mg, 36% yield). 1H NMR (500 MHz, CDCl3) δ 5.40 (tdt, J = 5.7, 2.8, 1.5 Hz, 1H), 4.65 (d, J = 2.9 Hz, 2H), 4.46 (dt, J = 8.3, 3.5 Hz, 1H), 4.34 (t, J = 8.8 Hz, 1H), 4.26 (dd, J = 9.1, 3.1 Hz, 1H), 4.12 (qd, J = 11.4, 7.1 Hz, 3H), 2.44 (pd, J = 7.0, 3.9 Hz, 1H), 1.76 (d, J = 1.5 Hz, 3H), 1.70 (d, J = 1.4 Hz, 3H), 0.93 (d, J = 7.0 Hz, 3H), 0.88 (d, J = 6.9 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 170.5, 154.2, 138.6, 120.3, 69.5, 67.9, 64.5, 58.3, 28.4, 26.0, 18.2, 18.1, 14.8. HRMS (DART-Orbitrap) m/z [M+H]+ calcd for C13H22NO4 256.1543, found 256.1556.
(S,E)-3-(2-((3,7-dimethylocta-2,6-dien-1-yl)oxy)acetyl)-4-isopropyloxazolidin-2-one (1j).
Following the procedure of 1d using geraniol afforded 4j as a colorless oil (835 mg, 39% yield). 1H NMR (500 MHz, CDCl3) δ 5.34 (t, J = 7.1 Hz, 1H), 5.08 (t, J = 7.1 Hz, 1H), 4.14 (d, J = 7.1 Hz, 2H), 4.08 (s, 2H), 2.11 (t, J = 7.1 Hz, 2H), 2.08 – 2.04 (m, 2H), 1.68 (d, J = 2.3 Hz, 6H), 1.60 (s, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 173.7, 142.9, 132.1, 123.8, 119.2, 67.9, 66.3, 39.7, 26.4, 25.8, 17.8, 16.6. Conversion of 4j to 1j afforded a colorless oil (468 mg, 38% yield). 1H NMR (500 MHz, CDCl3) δ 5.43 (ddt, J = 8.3, 7.0, 1.3 Hz, 1H), 5.15 – 5.08 (m, 1H), 4.68 (d, J = 2.6 Hz, 2H), 4.48 (dt, J = 8.3, 3.5 Hz, 1H), 4.37 (t, J = 8.8 Hz, 1H), 4.29 (dd, J = 9.2, 3.1 Hz, 1H), 4.17 (qd, J = 11.5, 6.9 Hz, 2H), 2.46 (heptd, J = 7.1, 3.9 Hz, 1H), 2.17 – 2.10 (m, 2H), 2.07 (dd, J = 9.4, 6.1 Hz, 2H), 1.71 (d, J = 1.4 Hz, 3H), 1.70 (d, J = 1.4 Hz, 3H), 1.62 (s, 3H), 0.95 (d, J = 7.0 Hz, 3H), 0.90 (d, J = 6.9 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 170.5, 154.2, 141.8, 131.9, 124.0, 120.0, 69.5, 68.0, 64.5, 58.3, 39.8, 28.4, 26.5, 25.8, 18.1, 17.8, 16.7, 14.8. HRMS (DART-Orbitrap) m/z [M+H]+ calcd for C18H30NO4 324.2169, found 324.2185.
(S)-3-((2S,3R)-2-hydroxy-3-methylpent-4-enoyl)-4-isopropyloxazolidin-2-one (2d).
To a solution of 1d (0.40 mmol, 96.4 mg) in CH2Cl2 (1.0 mL) was added n-Bu2BOTf (0.80 mmol, 800 μL) as 1.0 M solution in CH2Cl2 followed by stirring at 0 °C for 5 min. Triethyamine (1.2 mmol, 168 μL) was added. The reaction was warmed to rt and stirred for an additional 3 h. The reaction was quenched with saturated NH4Cl and extracted three times with CH2Cl2. The organic extracts were dried over MgSO4 and concentrated in vacuo. Flash chromatography (20% ethyl acetate/hexanes) afforded 2d and its minor isomer as colorless oil (78.9 mg, 82% yield, 15:1 selectivity). 1H NMR (500 MHz, CDCl3) δ 5.86 (ddd, J = 17.6, 10.3, 7.6 Hz, 1H), 5.13 – 5.02 (m, 3H), 4.38 (dt, J = 7.2, 3.4 Hz, 1H), 4.30 (t, J = 8.4 Hz, 1H), 4.26 (dd, J = 9.1, 2.8 Hz, 1H), 3.31 (d, J = 8.9 Hz, 1H), 2.61 – 2.54 (m, 1H), 2.44 (heptd, J = 7.2, 3.7 Hz, 1H), 1.03 (d, J = 6.8 Hz, 3H), 0.92 (d, J = 7.0 Hz, 3H), 0.88 (d, J = 7.0 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 174.3, 153.9, 140.0, 115.5, 73.8, 64.2, 59.2, 41.8, 28.4, 18.1, 14.6, 13.9. HRMS (DART-Orbitrap) m/z [M+H]+ calcd for C12H20NO4 242.1387, found 242.1399.
(S)-4-benzyl-3-((S)-2-hydroxypent-4-enoyl)oxazolidin-2-one (2a).
Following the procedure for rearrangement of 1d, 1a afforded 2a and its minor isomer as colorless oil (90% yield, 2:1 selectivity). Major product 1H NMR (500 MHz, CDCl3) δ 7.35 (dd, J = 8.1, 6.5 Hz, 2H), 7.32 – 7.27 (m, 1H), 7.24 – 7.19 (m, 2H), 5.87 (ddt, J = 17.2, 10.2, 7.1 Hz, 1H), 5.21 – 5.09 (m, 3H), 4.66 (ddt, J = 10.0, 6.7, 3.3 Hz, 1H), 4.31 – 4.23 (m, 2H), 3.55 (d, J = 7.9 Hz, 1H), 3.32 (dd, J = 13.5, 3.3 Hz, 1H), 2.84 (dd, J = 13.5, 9.4 Hz, 1H), 2.62 (dddt, J = 14.3, 7.1, 4.7, 1.3 Hz, 1H), 2.46 (dtt, J = 14.2, 7.1, 1.2 Hz, 1H). 13C{1H} NMR (126 MHz, CDCl3) δ 174.1, 153.4, 134.9, 133.0, 129.6, 129.2, 127.7, 118.8, 70.4, 67.1, 55.7, 38.4, 37.7. Minor product 1H NMR (500 MHz, CDCl3) δ 7.35 (dd, J = 8.1, 6.5 Hz, 2H), 7.32 – 7.27 (m, 1H), 7.24 – 7.19 (m, 2H), 5.87 (ddt, J = 17.1, 10.1, 7.1 Hz, 1H), 5.23 – 5.14 (m, 3H), 4.75 (ddt, J = 9.8, 8.1, 3.5 Hz, 1H), 4.31 (t, J = 8.6 Hz, 1H), 4.24 (dd, J = 9.2, 3.4 Hz, 1H), 3.35 – 3.25 (m, 2H), 2.74 (dd, J = 13.4, 9.8 Hz, 1H), 2.67 (dddd, J = 11.3, 5.8, 2.5, 1.2 Hz, 1H), 2.53 – 2.45 (m, 1H). 13C{1H} NMR (126 MHz, CDCl3) δ 174.7, 153.1, 134.9, 132.6, 129.5, 129.2, 127.7, 119.1, 70.3, 67.3, 55.2, 39.1, 38.3. HRMS (DART-Orbitrap) m/z [M+H]+ calcd for C15H18NO4 276.1230, found 276.1243.
(S)-3-((S)-2-hydroxypent-4-enoyl)-4-isopropyloxazolidin-2-one (2b).
Following the procedure for rearrangement of 1d, 1b afforded 2b and its minor isomer (81% yield, 3:1 selectivity). 1H NMR (500 MHz, CDCl3) δ 5.85 (ddt, J = 17.2, 10.2, 7.1 Hz, 1H), 5.19 – 5.06 (m, 3H), 4.41 (dt, J = 7.7, 3.3 Hz, 1H), 4.34 (t, J = 8.6 Hz, 1H), 4.29 (dd, J = 9.1, 2.9 Hz, 1H), 3.57 (d, J = 8.0 Hz, 1H), 2.59 (dddt, J = 14.3, 7.1, 4.9, 1.3 Hz, 1H), 2.48 – 2.38 (m, 2H), 0.94 (d, J = 7.0 Hz, 3H), 0.90 (d, J = 6.9 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 174.0, 154.1, 133.1, 118.6, 70.4, 64.4, 59.1, 38.4, 28.4, 18.1, 14.7. HRMS (DART-Orbitrap) m/z [M+H]+ calcd for C11H18NO4 228.1230, found 228.1243.
(S)-4-benzyl-3-((2S,3R)-2-hydroxy-3-methylpent-4-enoyl)oxazolidin-2-one (2c).
Following the procedure for 1d using 1c afforded 2c and its minor isomer as colorless oil (90% yield, 5:1 selectivity). 1H NMR (500 MHz, CDCl3) δ 7.34 (dd, J = 8.1, 6.5 Hz, 2H), 7.32 – 7.27 (m, 1H), 7.24 – 7.20 (m, 2H), 5.89 (ddd, J = 17.5, 10.3, 7.5 Hz, 1H), 5.16 – 5.06 (m, 3H), 4.64 (ddt, J = 9.6, 6.4, 3.5 Hz, 1H), 4.26 – 4.23 (m, 2H), 3.37 – 3.30 (m, 2H), 2.82 (dd, J = 13.5, 9.6 Hz, 1H), 2.66 – 2.58 (m, 1H), 1.06 (d, J = 6.9 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 174.4, 153.2, 139.9, 135.0, 129.6, 129.2, 127.7, 115.6, 73.9, 67.0, 55.8, 41.8, 37.7, 13.9. HRMS (DART-Orbitrap) m/z [M+H]+ calcd for C16H20NO4 290.1386, found 290.1398.
(S)-3-((2S,3R)-2-hydroxy-3-isopropylpent-4-enoyl)-4-isopropyloxazolidin-2-one (2e).
Following the procedure for rearrangement of 1d, 1e afforded 2e (71% yield, >30:1 selectivity). 1H NMR (500 MHz, CDCl3) δ 5.70 (dt, J = 17.1, 10.1 Hz, 1H), 5.23 (t, J = 9.8 Hz, 1H), 5.08 (dd, J = 10.3, 2.2 Hz, 1H), 5.02 (dd, J = 17.1, 2.2 Hz, 1H), 4.33 (q, J = 4.7 Hz, 1H), 4.24 (d, J = 4.8 Hz, 2H), 3.18 (d, J = 10.2 Hz, 1H), 2.39 (heptd, J = 7.0, 3.7 Hz, 1H), 2.24 (dddd, J = 19.2, 13.1, 8.3, 3.5 Hz, 2H), 0.92 (dd, J = 6.9, 1.4 Hz, 6H), 0.89 (d, J = 6.9 Hz, 6H). 13C{1H} NMR (126 MHz, CDCl3) δ 175.0, 154.4, 134.5, 118.8, 71.9, 64.3, 59.1, 56.5, 28.7, 26.9, 21.5, 18.2, 16.7, 14.8. HRMS (DART-Orbitrap) m/z [M+H]+ calcd for C14H24NO4 270.1670, found 270.1712.
(S)-3-((2S,3S)-2-hydroxy-3-phenylpent-4-enoyl)-4-isopropyloxazolidin-2-one (2f).
To a solution of 1f (0.040 mmol, 12.1 mg) in CH2Cl2 (1.0 mL) was added n-Bu2BOTf (0.080 mmol, 80 μL) as 1.0 M solution in CH2Cl2 followed by stirring at −78 °C for 5 min. Triethylamine (0.12 mmol, 17 μL) was added. The reaction was warmed to rt and stirred for additional 3 h. The reaction was quenched with saturated NH4Cl and extracted three times with CH2Cl2. The organic extracts were dried over MgSO4 and concentrated in vacuo. Flash chromatography (20% ethyl acetate/hexanes) afforded 2f and its minor isomer (11.2 mg, 91% yield, >30:1 selectivity). 1H NMR (500 MHz, CDCl3) δ 7.35 – 7.29 (m, 2H), 7.29 – 7.22 (m, 3H), 6.19 (ddd, J = 17.1, 10.2, 8.2 Hz, 1H), 5.58 (dd, J = 9.2, 6.0 Hz, 1H), 5.20 (dt, J = 17.1, 1.3 Hz, 1H), 5.17 (dt, J = 10.2, 1.2 Hz, 1H), 4.33 – 4.23 (m, 3H), 3.78 – 3.71 (m, 1H), 3.21 (d, J = 9.2 Hz, 1H), 2.40 (heptd, J = 7.0, 3.5 Hz, 1H), 0.90 (d, J = 7.0 Hz, 3H), 0.88 (d, J = 6.9 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 173.7, 154.0, 138.9, 137.4, 128.7, 128.6, 127.5, 117.3, 73.7, 64.3, 59.3, 54.5, 28.4, 18.1, 14.7. HRMS (DART-Orbitrap) m/z [M+H]+ calcd for C17H22NO4 304.1543, found 304.1558.
(4S)-3-(2-hydroxy-3-methylpent-4-enoyl)-4-isopropyloxazolidin-2-one (22).
Following the procedure for rearrangement of 1d, 1g afforded 22 and its minor isomers (90% yield, 1.7:1:0.5:0.1 selectivity). Major product 1H NMR (500 MHz, CDCl3) δ 5.75 (ddd, J = 17.1, 10.4, 8.0 Hz, 1H), 5.11 – 5.05 (m, 3H), 4.38 – 4.35 (m, 1H), 4.33 – 4.30 (m, 1H), 4.30 – 4.28 (m, 1H), 3.19 (d, J = 8.6 Hz, 1H), 2.69 – 2.62 (m, 1H), 2.48 (dtd, J = 14.1, 7.0, 3.6 Hz, 1H), 1.21 (d, J = 6.9 Hz, 3H), 0.93 (dd, J = 7.1, 1.6 Hz, 3H), 0.89 (dd, J = 6.9, 1.2 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 174.3, 153.8, 137.6, 116.6, 74.4, 64.3, 59.4, 41.4, 28.3, 18.1, 17.0, 14.6. Minor product 1H NMR (500 MHz, CDCl3) δ 5.77 – 5.70 (m, 1H), 5.10 – 5.06 (m, 2H), 5.04 (ddd, J = 17.2, 1.9, 1.2 Hz, 1H), 4.52 (dt, J = 8.6, 3.4 Hz, 1H), 4.37 – 4.34 (m, 1H), 4.29 – 4.26 (m, 1H), 3.01 (d, J = 8.9 Hz, 1H), 2.78 (dtdd, J = 9.4, 8.3, 6.5, 2.6 Hz, 1H), 2.26 (heptd, J = 6.8, 3.4 Hz, 1H), 1.26 (d, J = 6.9 Hz, 3H), 0.94 – 0.92 (m, 3H), 0.90 – 0.89 (m, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 174.8, 153.5, 136.9, 117.1, 74.1, 64.2, 58.3, 42.0, 28.7, 18.1, 17.7, 14.9. HRMS (DART-Orbitrap) m/z [M+H]+ calcd for C12H20NO4 242.1387, found 242.1399.
(S)-3-((S)-2-hydroxy-3,3-dimethylpent-4-enoyl)-4-isopropyloxazolidin-2-one (23).
To a solution of 1i (0.10 mmol, 25.5 mg) in CH2Cl2 (1.0 mL) was added n-Bu2BOTf (0.15 mmol, 150 μL) as 1.0 M solution in CH2Cl2 followed by stirring at −78 °C for 5 min. Triethylamine (0.30 mmol, 42 μL) was added. The reaction was warmed to rt and stirred for additional 12 h. The reaction was quenched with saturated NH4Cl and extracted three times with CH2Cl2. The organic extracts were dried over MgSO4 and concentrated in vacuo. Flash chromatography (20% ethyl acetate/hexanes) afforded 23 and its minor isomer (21.3 mg, 84% yield, 10:1 selectivity). 1H NMR (500 MHz, CDCl3) δ 5.94 (dd, J = 17.5, 10.8 Hz, 1H), 5.22 (d, J = 10.0 Hz, 1H), 5.06 (dd, J = 10.8, 1.3 Hz, 1H), 5.02 (dd, J = 17.5, 1.3 Hz, 1H), 4.33 (ddd, J = 7.1, 3.9, 2.9 Hz, 1H), 4.26 – 4.20 (m, 2H), 3.10 (d, J = 10.0 Hz, 1H), 2.43 (heptd, J = 7.0, 3.8 Hz, 1H), 1.15 (s, 3H), 1.08 (s, 3H), 0.93 (d, J = 7.0 Hz, 3H), 0.89 (d, J = 7.0 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 173.7, 154.1, 143.7, 113.6, 74.7, 64.0, 59.4, 42.3, 28.7, 24.9, 20.7, 18.2, 14.8. HRMS (DART-Orbitrap) m/z [M+H]+ calcd for C13H22NO4 256.1543, found 256.1556.
(S)-3-((2S,3R)-2-hydroxy-3,7-dimethyl-3-vinyloct-6-enoyl)-4-isopropyloxazolidin-2-one (24).
Following the procedure of 1i using 1j afforded 24 (85% yield, 5:1:0.7 selectivity). Major 1H NMR (500 MHz, CDCl3) δ 5.79 – 5.73 (m, 1H), 5.27 (dd, J = 10.8, 0.8 Hz, 1H), 5.19 (ddd, J = 17.5, 4.1, 0.9 Hz, 1H), 5.08 (tdt, J = 7.1, 2.8, 1.4 Hz, 1H), 4.58 (s, 1H), 4.04 (dt, J = 14.8, 7.5 Hz, 1H), 3.84 (dd, J = 11.8, 3.1 Hz, 1H), 3.75 (ddd, J = 10.1, 8.1, 3.1 Hz, 1H), 2.37 (dp, J = 10.2, 6.7 Hz, 1H), 2.02 – 1.88 (m, 2H), 1.74 (ddd, J = 13.6, 11.9, 4.8 Hz, 1H), 1.67 (t, J = 1.3 Hz, 3H), 1.59 (d, J = 1.3 Hz, 3H), 1.58 – 1.54 (m, 1H), 1.21 (s, 3H), 1.03 (d, J = 6.7 Hz, 3H), 0.90 – 0.87 (m, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 174.0, 154.1, 142.4, 131.7, 124.5, 114.9, 74.2, 64.0, 59.5, 46.0, 38.6, 28.8, 25.8, 22.7, 18.2, 17.8, 15.9, 14.9. Minor 1H NMR (500 MHz, CDCl3) δ 5.94 (dd, J = 17.6, 10.9 Hz, 1H), 5.32 (d, J = 10.2 Hz, 1H), 5.13 (dd, J = 10.9, 1.4 Hz, 1H), 5.10 – 5.05 (m, 1H), 5.00 (dd, J = 17.6, 1.5 Hz, 1H), 4.29 (ddd, J = 7.8, 3.9, 2.4 Hz, 1H), 4.22 (dd, J = 9.1, 2.4 Hz, 1H), 4.19 (dd, J = 9.1, 7.8 Hz, 1H), 3.01 (d, J = 10.3 Hz, 1H), 2.42 (pd, J = 6.9, 3.8 Hz, 1H), 1.90 (dq, J = 11.8, 6.2 Hz, 2H), 1.70 – 1.65 (m, 5H), 1.57 (d, J = 1.3 Hz, 3H), 1.03 (s, 3H), 0.92 (d, J = 7.0 Hz, 3H), 0.89 (d, J = 6.9 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 172.7, 155.9, 138.9, 132.3, 123.8, 117.5, 84.2, 61.7, 61.4, 43.8, 36.0, 26.6, 25.8, 22.3, 20.1, 19.9, 18.8, 17.8. HRMS (DART-Orbitrap) m/z [M+H]+ calcd for C18H30NO4 324.2169, found 324.2182.
(3S,4R)-3-hydroxy-4-methyldihydrofuran-2(3H)-one (6).
To a solution of 2d (0.21 mmol, 50.8 mg) in CH2Cl2 cooled to −78 °C, O3 was bubbled through until the solution turned blue. Dimethyl sulfide (13.5 mmol, 1.0 mL) was added gradually until the blue color disappeared. The mixture was warmed to rt and stirred for another 30 min to ensure complete quenching. The reaction was washed three times with brine, dried over MgSO4, and concentrated in vacuo. Flash chromatography (40% ethyl acetate/hexanes) afforded the corresponding aldehyde (21.3 mg, 42% yield). 1H NMR (500 MHz, CDCl3) δ 9.79 (s, 1H), 5.32 (dd, J = 7.0, 5.3 Hz, 1H), 4.44 (dt, J = 3.8, 2.3 Hz, 1H), 4.39 (t, J = 8.7 Hz, 1H), 4.32 (dd, J = 9.2, 2.6 Hz, 1H), 3.96 (d, J = 7.0 Hz, 1H), 2.95 (qd, J = 7.3, 5.4 Hz, 1H), 2.42 (dtq, J = 11.0, 7.2, 4.0, 3.4 Hz, 1H), 1.15 (d, J = 7.2 Hz, 3H), 0.94 (d, J = 6.9 Hz, 3H), 0.90 (d, J = 6.9 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 201.8, 172.2, 154.5, 70.3, 64.7, 59.1, 48.1, 28.4, 18.0, 14.7, 8.5. NaBH4 (1.0 mmol, 37.8 mg) was added to acetic acid (0.5 mL) at 0 °C, warmed to rt, then stirred for 1.0 hr. The aldehyde in acetic acid (0.20 mL) was added and the mixture stirred for an additional 30 min. The reaction was quenched by saturated NaHCO3 solution and extracted three times with CH2Cl2. The organic extracts were dried over MgSO4 and concentrated in vacuo. Flash chromatography (40% ethyl acetate/hexanes) afforded the cyclized lactone 6 (2 mg, 20%), which was compared to literature data.12 1H NMR (500 MHz, CDCl3) δ 4.42 (dd, J = 9.1, 7.9 Hz, 1H), 4.01 (dd, J = 10.6, 2.9 Hz, 1H), 3.80 (dd, J = 10.7, 9.1 Hz, 1H), 2.59 – 2.48 (m, 2H), 1.26 (d, J = 6.6 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 177.4, 73.9, 70.7, 39.1, 14.5. [a]D20 −40° (c 1.0, CDCl3). HRMS (DART-Orbitrap) m/z [M+H–H2O]+. calcd for C5H7O2 99.0440, found 99.0446.
(3S,4R)-3-hydroxy-4-isopropyldihydrofuran-2(3H)-one (7).
Following the procedure converting 2d to lactone 6, 2e afforded the intermediate aldehyde as a colorless liquid (15 mg, 42% yield). 1H NMR (500 MHz, CDCl3) δ 9.86 (d, J = 1.5 Hz, 1H), 5.10 – 5.06 (m, 1H), 4.38 (t, J = 8.1 Hz, 1H), 4.30 (d, J = 6.4 Hz, 2H), 3.09 (ddd, J = 9.8, 4.0, 1.5 Hz, 1H), 2.44 (tt, J = 7.3, 3.7 Hz, 1H), 2.32 (dqd, J = 12.0, 6.6, 3.1 Hz, 1H), 1.21 (d, J = 7.3 Hz, 3H), 1.00 (d, J = 7.0 Hz, 3H), 0.94 (dd, J = 7.2, 1.8 Hz, 3H), 0.89 (d, J = 7.0 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 203.9, 171.6, 155.3, 68.7, 64.9, 59.0, 57.4, 28.7, 26.2, 21.2, 18.6, 18.0, 14.9. And the cyclized lactone 7 as a colorless liquid (2 mg, 28% yield), which was compared to literature data.12a 1H NMR (500 MHz, CDCl3) δ 4.43 (t, J = 8.7 Hz, 1H), 4.15 (dd, J = 10.4, 2.4 Hz, 1H), 3.89 (dd, J = 10.7, 9.2 Hz, 1H), 2.50 (d, J = 2.5 Hz, 1H), 2.23 (tt, J = 10.5, 8.5 Hz, 1H), 1.78 (ddt, J = 14.8, 13.0, 6.6 Hz, 1H), 1.11 (d, J = 6.7 Hz, 3H), 0.95 (d, J = 6.7 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 178.0, 71.8, 68.9, 50.2, 30.9, 20.7, 20.3. HRMS (DART-Orbitrap) m/z [M+H–H2O]+ calcd for C7H11O2 127.0754, found 127.0761.
(S)-3-hydroxy-4,4-dimethyldihydrofuran-2(3H)-one (8).
Following the procedure of 6 using 23 afforded the intermediate aldehyde as a white crystal (6.1 mg, 23% yield). 1H NMR (500 MHz, CDCl3) δ 9.69 (s, 1H), 4.94 (d, J = 6.9 Hz, 1H), 4.44 (ddd, J = 8.3, 4.1, 2.9 Hz, 1H), 4.37 (dd, J = 9.1, 8.3 Hz, 1H), 4.31 – 4.28 (m, 2H), 2.33 (hd, J = 6.9, 4.0 Hz, 1H), 1.25 (s, 3H), 1.22 (s, 3H), 0.92 (d, J = 7.0 Hz, 3H), 0.90 (d, J = 6.9 Hz, 3H). And the cyclized lactone 8 as a colorless oil (3 mg, 96% yield) shown to be identical to a known sample.13 1H NMR (500 MHz, CDCl3) δ 4.10 (d, J = 3.3 Hz, 1H), 4.03 (d, J = 9.0 Hz, 1H), 3.94 (d, J = 8.9 Hz, 1H), 2.37 (d, J = 3.1 Hz, 1H), 1.24 (s, 3H), 1.08 (s, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 176.8, 76.5, 75.9, 41.1, 23.1, 18.9. HRMS (DART-Orbitrap) m/z [M+H]+ calcd for C6H12O3 131.0703, found 131.0704. [a.D20 −33.3° (c 1.2, CDCl3).
(S)-butane-1,2,4-triol (5).
To a solution of 2a (0.07 mmol, 18.5 mg) in CH2Cl2 cooled to −78 °C, O3 was bubbled through until the solution turned blue. Dimethyl sulfide (13.5 mmol, 1.0 mL) was added gradually until the blue color disappeared and the mixture was warmed to rt and stirred for another 30 min to ensure the complete quenching. The reaction was extracted three times with brine, dried over MgSO4, and concentrated in vacuo. Flash chromatography (50% ethyl acetate/hexanes) afforded the aldehyde as a colorless liquid (5.8 mg, 31% yield). 1H NMR (500 MHz, CDCl3) δ 9.81 (d, J = 0.9 Hz, 1H), 7.37 – 7.31 (m, 2H), 7.31 – 7.27 (m, 1H), 7.21 (dd, J = 7.0, 1.7 Hz, 2H), 4.81 (d, J = 4.6 Hz, 1H), 4.69 (ddt, J = 10.4, 9.2, 3.0 Hz, 1H), 4.31 (dd, J = 9.2, 8.0 Hz, 1H), 4.27 – 4.24 (m, 2H), 3.85 – 3.65 (m, 1H), 3.33 (dd, J = 13.5, 3.5 Hz, 1H), 2.85 – 2.81 (m, 1H). To a solution of the aldehyde in MeOH (0.10 mL) was added NaBH4 (0.05 mmol, 2 mg) at 0 °C and stirred for 1 hr after warming to rt. The reaction was concentrated in vacuo. Flash chromatography (ethyl acetate) afforded the triol 5 as a colorless liquid (1.9 mg, 85%), which was compared with literature data.11 1H NMR (500 MHz, D2O) δ 3.75 (ddt, J = 8.5, 6.8, 4.0 Hz, 1H), 3.64 (ddd, J = 7.6, 5.9, 1.6 Hz, 2H), 3.52 (dd, J = 11.8, 3.8 Hz, 1H), 3.41 (dd, J = 11.7, 6.8 Hz, 1H), 1.66 (dtd, J = 14.6, 7.3, 4.3 Hz, 1H), 1.56 (ddt, J = 14.5, 8.9, 6.0 Hz, 1H). {1H}13C NMR (126 MHz, D2O) δ 69.0, 65.6, 58.4, 34.7. [a D20 −31.5° (c 1.0, D2O). HRMS (DART-Orbitrap) m/z [M+H–HOD]+ calcd for C4H7D2O2 91.0723, found 91.0728.
Supplementary Material
Acknowledgments.
We thank the National Institutes of Health (GM131713) for support.
Footnotes
Supporting Information: spectroscopic, rate, and computational data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interests.
References and Footnotes
- 1.(a) Tomooka K; Komine N; Nakai T External Chiral Ligand-Induced Enantioselective Versions of the [2,3]-Wittig Sigmatropic Rearrangement. Chirality 2000, 12, 505. [DOI] [PubMed] [Google Scholar]; (b) Nakai T; Mikami K The [2,3]-Wittig Rearrangement. Org. React 1994, 46. [Google Scholar]; (c) Knochel P; Molander GA The Wittig rearrangement In Comprehensive Organic Synthesis (2nd Ed.), 2014, Vol. 3, p. 1038. [Google Scholar]
- 2.(a) Jermaks J; Tallmadge EH; Keresztes I; Collum DB Lithium Amino Alkoxide–Evans Enolate Mixed Aggregates: Aldol Addition with Matched and Mismatched Stereocontrol. J. Am. Chem. Soc 2018, 140, 3077. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tallmadge EH; Jermaks J; Collum DB Structure–Reactivity Relationships in Lithiated Evans Enolates: Influence of Aggregation and Solvation on the Stereochemistry and Mechanism of Aldol Additions. J. Am. Chem. Soc 2016, 138, 345. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhang Z; Collum DB Structures and Reactivities of Sodiated Evans Enolates: Role of Solvation and Mixed Aggregation on the Stereochemistry and Mechanism of Alkylations. J. Am. Chem. Soc 2019, 141, 388. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zhang Z; Collum DB Evans Enolates: Structures and Mechanisms Underlying the Aldol Addition of Oxazolidinone-Derived Boron Enolates. J. Org. Chem 2017, 82, 7595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Modern Aldol Reactions, Mahrwald R, Ed.; Wiley-VCH: Weinheim, 2004, Vol. 1 and 2. [Google Scholar]; (b) Lin G-Q; Li Y-M; Chan ASC Principles and Applications of Asymmetric Synthesis, Wiley & Sons: New York, 2001; p. 135. [Google Scholar]; (c) Mahrwald R in Aldol Reactions, Springer: New York, 2009. [Google Scholar]; (d) Evans DA; Kim AS; Skrydstrup T; Taaning RH (S)-4-Benzyl-2-oxazolidinone In e-EROS Encyclopedia of Reagents for Organic Synthesis; John Wiley & Sons: New York, 2007, pp. 1–18. [Google Scholar]
- 4.For structurally similar auxiliaries undergoing [2,3] sigmatropic rearrangements, see:; (a) Mikami K; Takahashi O; Kasuga T; Nakai T Asymmetric [2,3] Wittig Sigmatropic Rearrangements Involving Chiral Amide Enolates as the Migrating Termini. Chem. Lett 1985, 1729. [Google Scholar]; (b) Peňaška T; Mojzes MM; Filo J; Jurdáková H; Mečiarová M; Šebesta R Organocatalysts Effect on the Stereoselectivity of [2,3]-Wittig Rearrangement Eur . J. Org. Chem 2019, 605. [Google Scholar]; (c) Kress MH; Yang C; Yasuda N; Grabowski EJJ Stereoselective [2,3]-Wittig Rearrangement of (1S,2R)-l-Amino-lndan-2-ol Derived Amide Enolates. Tetrahedron Lett. 1997, 38, 2633; [Google Scholar]; (d) Uchikawa M; Hanamoto T; Katsuki T; Yamaguchi M Asymmetric [2,3] Wittig Rearrangement of 2’-Alkenyloxyacetamide Bearing trans-2,5-bis(Methoxymethoxymethyl)pyrrolidine Moiety as a Chiral Auxiliary. Tetrahedron Lett. 1986, 27, 4577. [Google Scholar]
- 5.(a) Oh T; Wrobel Z; Rubenstein SM [2,3]-Wittig Rearrangement of Allylic Glycolate Esters via Boron and Tin Enolates. Tetrahedron Lett. 1991, 32, 4647. [Google Scholar]; (b) Fujimoto K; Nakai T Enantioselective [2,3]Wittig Rearrangement Involving a Chiral Boron Enolate Terminus Tetrahedron Lett. 1994, 35, 5019. [Google Scholar]
- 6.(a) Emmadi NR; Bingi C; Kumar CG; Yedla P; Atmakur K Stereoselective Total Synthesis of epi-Phomopsolide B. Synthesis 2014, 46, 2945. [Google Scholar]; (b) Evans DA; Adams DJ; Kwan EE Progress Toward the Syntheses of (+)-GB 13, (+)-Himgaline, and Himandridine. New Insights into Intramolecular Imine/Enamine Aldol Cyclizations. J. Am. Chem. Soc 2012, 134, 8162. [DOI] [PubMed] [Google Scholar]; (c) Cowden CJ; Paterson I Asymmetric Aldol Reactions Using Boron Enolates. Org. React 1997, 51, 1. [Google Scholar]
- 7.For other examples of physical studies of Evans enolates, see:; (a) Shinisha CB; Sunoj RB Transition State Models for Probing Stereoinduction in Evans Chiral Auxiliary-Based Asymmetric Aldol Reactions. J. Am. Chem. Soc 2010, 132, 12319. [DOI] [PubMed] [Google Scholar]; (b) Sreenithya A; Sunoj RB Noninnocent Role of N-Methyl Pyrrolidinone in Thiazolidinethione-Promoted Asymmetric Aldol Reactions. Org. Lett 2012, 14, 5752. [DOI] [PubMed] [Google Scholar]; (c) Shinisha CB; Sunoj RB The Pivotal Role of Chelation as a Stereochemical Control Element in Non-Evans Anti Aldol Product Formation. Org. Lett 2010, 12, 2868. [DOI] [PubMed] [Google Scholar]; (d) Goodman JM; Paton RS Enantioselectivity in the Boron Aldol Reactions of Methyl Ketones. Chem. Commun 2007, 2124. [DOI] [PubMed] [Google Scholar]; (e) Baringhaus KH; Matter H; Kurz M Solution Structure of a Chiral Dialkylboron Enolate by NMR Spectroscopy and Simulated Annealing: Unusual Stabilization of This Intermediate by a Boron-Nitrogen Interaction. J. Org. Chem 2000, 65, 5031. [DOI] [PubMed] [Google Scholar]; (f) Kimball DB; Michalczyk R; Moody E; Ollivault-Shiflett M; De Jesus K; Silks LA III Determining the Solution State Orientation of a Ti Enolate via Stable Isotope Labeling, NMR Spectroscopy, and Modeling Studies. J. Am. Chem. Soc 2003, 125, 14666. [DOI] [PubMed] [Google Scholar]
- 8.Taillier C; Hameury T; Bellosta V; Cossy J Synthesis of 3-Oxooxa- and 3-Oxoazacycloalk-4-Enes by Ring Closing Metathesis. Application to the Synthesis of an Inhibitor of Cathepsin K. Tetrahedron 2007, 63, 4472. [Google Scholar]
- 9.Crimmins MT; King BW; Zuercher WJ; Choy AL An Efficient, General Asymmetric Synthesis of Carbocyclic Nucleosides: Application of an Asymmetric Aldol/Ring-Closing Metathesis Strategy. J. Org. Chem 2000, 65, 8499. [DOI] [PubMed] [Google Scholar]
- 10.A more convergent synthesis based on alkoxide displacement of an α-bromoacetyl-containing oxazolidinone failed owing to competitive deacylation.
- 11.Liu Q; Xiong F-J; He Q-Q; Chen F-E Development of an Efficient Process for the Decomposition of the Borate Complexes Formed During the Large-Scale Synthesis of (S)-1,2,4-Butanetriol. Org. Process Res. Dev 2013, 17, 1540. [Google Scholar]
- 12.(a) Scheffler U; Mahrwald R Histidine-Catalyzed Asymmetric Aldol Addition of Enolizable Aldehydes: Insights into its Mechanism. J. Org. Chem 2012, 77, 2310. [DOI] [PubMed] [Google Scholar]; (b) Akita H; Matsukura H; Oishi T Synthesis of Useful Chiral Synthons (2R, 3R)-2-Methylmalate and its Congeners via Microbial Asymmetric Reduction. Chem. Pharm. Bull 1986, 34, 2656. [Google Scholar]
- 13.(a) Heidlindemann M; Hammel M; Scheffler U; Mahrwald R; Hummel W; Berkessel A; Groger H Chemoenzymatic Synthesis of Vitamin B5-Intermediate (R)-Pantolactone via Combined Asymmetric Organo- and Biocatalysis. J. Org. Chem 2015, 80, 3387. [DOI] [PubMed] [Google Scholar]; (b) Shimizu T; Hiranuma S; Nakata T Efficient Method for Inversion of Secondary Alcohols by Reaction of Chloromethanesulfonates with Cesium Acetate. Tetrahedron Letters, 1996, 37, 6145. [Google Scholar]
- 14.Rein AJ; Donahue SM; Pavlosky MA In Situ FTIR Reaction Analysis of Pharmaceutical-Related Chemistry and Processes. Curr. Opin. Drug Discov. Dev 2000, 3, 734. [PubMed] [Google Scholar]
- 15.Gaussian 09, Revision A.02, Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich A, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams-Young D, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery JA Jr., Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Millam JM, Klene M, Adamo C, Cammi R, Ochterski JW, Martin RL, Morokuma K, Farkas O, Foresman JB, and Fox DJ, Gaussian, Inc., Wallingford CT, 2016. [Google Scholar]
- 16.Stevens JM; Parra-Rivera AC; Dixon DD; Beutner GL; DelMonte AJ; Frantz DE; Janey JM; Paulson J; Talley MR Direct Lewis Acid Catalyzed Conversion of Enantioenriched N-Acyloxazolidinones to Chiral Esters, Amides, and Acids. J. Org. Chem 2018, 83, 14245. [DOI] [PubMed] [Google Scholar]
- 17.Ozonolysis of 23 afforded an aldehyde characterized crystallographically. We thank Dr. Samantha N. MacMillan for her help.
- 18.Morgan JB; Morken JP Platinum-catalyzed Tandem Diboration/Asymmetric Allylboration: Access to Nonracemic Functionalized 1,3-diols. Org. Lett 2003, 5, 2573. [DOI] [PubMed] [Google Scholar]
- 19.Adam W; Peters K; Peters E-M; Schambony SB Efficient Control of the Diastereoselectivity and Regioselectivity in the Singlet-Oxygen Ene Reaction of Chiral Oxazolidinone-Substituted Alkenes by a Remote Urea NH Functionality. Comparison with Dimethyldioxirane and m-Chlorobenzoic Acid Epoxidations. J. Am. Chem. Soc 2001, 123, 7228. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.