

Abstract
Many proteins in living organisms are glycosylated. As their glycan patterns exhibit protein‐, cell‐, and tissue‐specific heterogeneity, changes in the glycosylation levels could serve as useful indicators of various pathological and physiological states. Thus, the identification of glycoprotein biomarkers from specific changes in the glycan profiles of glycoproteins is a trending field. Lectin microarrays provide a new glycan analysis platform, which enables rapid and sensitive analysis of complex glycans without requiring the release of glycans from the protein. Recent developments in lectin microarray technology enable high‐throughput analysis of glycans in complex biological samples. In this review, we will discuss the basic concepts and recent progress in lectin microarray technology, the application of lectin microarrays in biomarker discovery, and the challenges and future development of this technology. Given the tremendous technical advancements that have been made, lectin microarrays will become an indispensable tool for the discovery of glycoprotein biomarkers.
Keywords: biomarkers, glycans, glycoproteomics, glycomics, lectin microarrays
A powerful tool to find novel biomarkers! Lectin microarray technology is a fast, high‐throughput, and inexpensive tool for the discovery of glycol‐biomarkers in complex biological samples. This review summarizes the recent progress and application of lectin microarrays in biomarker discovery under different physiopathological states.
1. Introduction
A biomarker is a biological indicator that objectively measures and evaluates normal and pathological biological processes, or responses to therapy. Biomarkers can be used for the diagnosis and classification of diseases, efficacy monitoring, disease risk prediction, and screening of high‐risk groups.1 They can be divided into clinical, imaging, and biochemical and genetic markers.2a, 2b Proteins and their posttranslationally modified forms are involved in almost all biological processes including gene expression regulation, cytoskeleton formation, material transport, and metabolism. Thus, proteins and their modifications can potentially serve as objective indicators to evaluate normal physiological function or pathological status.
Glycosylation is the most common posttranslational modification, wherein over half of proteins are glycosylated.3 Glycosylation plays essential roles in various biological processes such as cell proliferation and differentiation, intercellular communication and signaling, cell−cell and protein−protein interactions, cell adhesion, and immune responses.4a, 4b Moreover, glycosylation patterns exhibit protein‐, cell‐ and tissue‐specific heterogeneity, thus enabling the assessment of changes in various pathological and physiological states such as tissue origin, tissue development, tumorigenesis, and the degree of malignancy.5a, 5b For instance, the core fucosylation of α‐Fetoprotein L3 is a common diagnostic glycoprotein biomarker for hepatocarcinoma; and the glycan antigen sialyl−Lea is a common marker for gastrointestinal cancer.6a, 6b, 6c, 6d Additionally, many tumors have been reported to show significantly increased expression of truncated O‐glycans and N‐Acetyl‐D‐glucosamine‐branching N‐glycan.7a, 7b Therefore, glycomic profiling of tissues under different physiopathological states may contribute to the discovery of biomarkers that are associated with tissue‐ or disease‐specific alterations. Over the past decade, mass spectrometry (MS)‐based methods have been traditionally used for glycomic analysis.8 Although MS‐based methods can reliably identify the structure, linkage, and position of glycans, enzymatic or chemical stripping of glycans from proteins prior to MS profiling prevents accurate detection and identification of total glycans. Moreover, these methods are usually time‐consuming and require complex sample preparation procedures.9a, 9b
A technique called lectin microarray, which was developed in 2005,10a, 10b, 10c, 10d has gained increasing popularity for high‐throughput analysis of glycans. Lectins are a group of carbohydrate‐binding proteins that specifically bind different glycans. The advantages of using lectin microarray over traditional MS‐based methods include the simplicity and high sensitivity of the method that supports direct global glycomic profiling, the lower stringency of initial sample purity (crude glycoprotein samples can be analyzed), and the comparatively simple sample preparation procedure (without glycan‐release and purification).9a As protein fragmentation or glycan liberation is not required during sample preparation, the sampled glycoproteins can retain their intact natural conformations and abundance. Thus, lectin microarrays are suitable for analyzing the differential glycomic profiles of biological samples. However, this technique is not quantitative and does not allow complete determination of glycan structures like MS. Instead, lectin microarrays are more appropriate for comparative purposes, such as for analyzing differences between glycomic profiles.11 The sensitivity, simplicity, and robustness of lectin microarrays require further improvement to broaden their applications.
Currently, lectin microarray is widely used to assess tumor characteristics and to screen for novel diagnostic cancer biomarkers.12 In this paper, we will review the recent advances in lectin microarray technology and focus on the recent progress and application of lectin microarrays in biomarker discovery under various pathological and physiological states.
2. Lectin microarray strategies for biomarker discovery
2.1. Direct Assay
Lectins with known specificity were first immobilized through either covalent bonding or physical adsorption before incubation with the fluorescently‐tagged samples. Subsequently, the binding event can be monitored through fluorescence detection (Figure 1A).10d, 13 This assay can be used to analyze differential glycosylation patterns of normal versus disease samples or to investigate the effect of various treatment conditions. Traditional lectin arrays can only show the bulk glycosylation levels of abundant proteins, which have a relatively higher representation in the protein mixture. To minimize the bias towards identification of abundant proteins, Etxebarria et al. developed a fluorescence‐based method for the rapid analysis of protein glycosylation in biofluids.14 In this method, fluorescently‐tagged glycoproteins that have been transferred to a lectin‐coated slide retain their relative positions on the SDS‐PAGE gel.14, 15 The individual lectin binding profiles for all separated proteins, independent of abundance, can thus be obtained.
Figure 1.
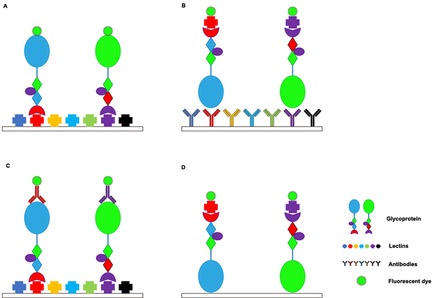
Lectin microarray strategies. (A) Direct assay. (B) Lectin‐overlay antibody sandwich array. (C) Antibody‐overlay lectin sandwich array. (D) Glycoprotein−lectin array.
2.2. Lectin‐Overlay Antibody Sandwich Array
This method developed by Chen et al. involves the use of antibody microarray capture of multiple proteins followed by detection using various biotinylated lectin probes (Figure 1B).16 Chemical derivatization of glycans on captured antibodies is an essential preliminary step in this method to prevent the binding of lectin probes to the glycans. Derivatized antibodies are immobilized on nitrocellulose slides, and unprocessed samples can be directly loaded onto the array. Chen et al. successfully identified cancer‐associated glycan alterations on MUC1 and CEA proteins in the serum of pancreatic cancer patients using this method. Thus, this method is thought to be particularly useful for profiling variations of specific glycans on multiple proteins.16 To measure the amount of both glycans and glycoproteins in a crude sample, Yue et al. used a dual antibody sandwich assay to determine protein levels and a lectin‐overlay antibody sandwich array to characterize glycan levels.17 This method may potentially be useful for characterizing the rate of alterations to the glycans and the relationship between glycans and their carrier proteins.
2.3. Antibody‐Overlay Lectin Sandwich Array
This assay was initially developed by Rosenfeld et al. in 2007.18 As a brief summary, the lectins spotted on nitrocellulose membrane‐coated glass slides are used to capture the target glycoproteins. The selectively‐bound glycoproteins are then probed with either directly labeled or indirectly labeled secondary antibodies. The arrays are scanned and analyzed to generate a characteristic fingerprint that mirrors alterations in the protein‘s glycan composition (Figure 1C). Rosenfeld et al. developed Qproteome™ GlycoArray kits based on this method, which enables rapid analysis (within 4–6 hours) of the glycosylation profiles of intact glycoproteins.18 Kuno et al. established antibody‐assisted lectin profiling to detect glycosylation changes in low‐abundance target molecules.19 This method involves enrichment of target proteins with specific antibodies by immunoprecipitation followed by quantification with immunoblotting. An accurate glycan profile of the target glycoprotein can be obtained at the sub‐picomolar level from a trace amount of crude samples in a highly reproducible and high‐throughput manner.19 Although weak lectin−glycan interactions can be detected using this method, a specialized evanescent‐field fluorescence scanner is necessary to increase sensitivity.10d, 19 An improved version of the antibody‐overlay lectin microarray method involving the integration of tyramide signal amplification (TSA), a horseradish peroxidase (HRP)‐mediated signal amplification method, was developed by Meany et al. to further boost sensitivity by over 100 times.20 In addition, this improved method does not require specialized instruments for detecting weak lectin−glycan interactions. Thus, this enables glycan profiling at the sub‐nanogram level.20
2.4. Glycoprotein−Lectin Array
In this reversed‐phase lectin microarray, glycoproteins are first enriched using a general lectin column and are then separated by reversed‐phase high‐performance liquid chromatography (HPLC). The purified glycoproteins are then immobilized on the slide surface and probed with labeled lectins with a wide range of glycan binding specificities (Figure 1D). This method enables the profiling of glycan distribution in the human glycoproteome and the monitoring of individual glycosylation alterations at a global scale. This array could successfully identify serum biomarkers in pancreatic diseases.21 However, the high effective sample concentration on one spot can increase the risk of spurious interactions.22
3. Factors Influencing Lectin Microarrays for Biomarker Discovery
3.1. Types of Lectins
3.1.1. Natural Lectins
Natural lectins are lectins that are purified from plants, animals, bacteria, or fungi. Among them, plant lectins, also known as (phyto)hemagglutinins, are the most widely used lectins for glycan profiling due to their availability, stability, and varied binding specificities.23a, 23b For example, the families of B‐chain‐like lectins, legume lectins and ricin, can bind to a wide range of glycans.23b The jacalin‐related lectins are both Man‐ and Gal‐specific,24 but monocot‐derived lectins are only specific for Man25 and hevein‐type lectins only for GlcNAc.26 Disadvantages of natural lectins include inherent glycosylation, batch‐to‐batch variation, and inconsistencies in activity due to differences arising from purification procedures.23b, 27 Nonetheless, advancements in genome technology as well as glycotechnology28 are expected to support the development of more useful natural lectins with unique glycan‐binding specificities. The origin, monosaccharide specificity, and preferred glycan structure as well as specificity profile of natural lectins mentioned in this review are summarized in Table 1.
Table 1.
Specificity profile of natural lectins.
|
Lectin name |
Origin |
Monosaccharide specificity a) |
Preferred glycan structure |
Refs. |
|---|---|---|---|---|
|
Fungal lectins | ||||
|
AAL |
Aleuria aurantia |
Fuc |
Fucα6GlcNAc (core Fuc), Fucα3(Galβ4) GlcNAc (Lex) |
|
|
ABA |
Agaricus bisporus |
Gal, GlcNAc |
Galβ3GalNAc, GlcNAc |
|
|
ACG |
Agrocybe cylindracea |
Gal |
Siaα3Galβ4GlcNAc |
|
|
Plant lectins | ||||
|
ACA |
Amaranthus caudatus |
Gal |
Galβ3GalNAc |
|
|
ACL |
Amaranthus caudatus |
Gal |
Galβ1‐3GalNAc |
|
|
BPL |
Bauhinia purpurea |
Gal |
Galβ3GalNAc, GalNAc |
|
|
BS−I |
Bandeiraea simplicifolia |
Gal, GalNAc |
αGal, αGalNAc |
|
|
ConA |
Canavalia ensiformis |
Man |
High‐Man including Manα6(Manα3) Man |
|
|
DBA |
Dolichos biflorus |
GalNAc |
GalNAcα3GalNAc |
|
|
DSA |
Datura stramonium |
GlcNAc |
(GlcNAcβ4) n, triantennary, tetraantennary N‐glycans |
|
|
ECA |
Erythrina cristagalli |
Gal |
Galβ4GlcNAc |
|
|
GNA |
Galanthus nivalis |
Man |
High‐Man including Manα3Man |
|
|
GSL−I−B4 |
Griffonia simplicifolia |
Gal |
α Gal |
|
|
GSL−II |
Griffonia simplicifolia |
GlcNAc |
Agalactosylated tri/tetra antennary glycans, GlcNAc |
|
|
HHL |
Hippeastrum Hybrid |
Man |
High‐Man including Mana3Man or Mana6Man |
|
|
Jacalin |
Artocarpus integrifolia |
Gal |
Galβ3GalNAc, αGalNAc (6O‐unsubstituted) |
|
|
LCA |
Lens culinaris |
Fuc/Man |
Fucα6GlcNAc, High‐Man |
|
|
LEL |
Lycopersicon esculentum |
GlcNAc |
(GlcNAcβ4) n, (Galβ4GlcNAc) n (polylactosamine) |
|
|
LTL |
Lotus tetragonolobus |
Fuc |
Fucα3(Galβ4) GlcNAc (Lex), Fucα2Galβ4GlcNAc (H‐type 2) |
|
|
MPL |
Maclura pomifera |
GalNAc |
αGalNAc |
|
|
NPA |
Narcissus pseudonarcissus |
Man |
High‐Man including Manα6Man |
|
|
PHA−E |
Phaseolus vulgaris |
Gal |
N‐glycans with outer Gal and bisecting GlcNAc |
|
|
PHA−L |
Phaseolus vulgaris |
Complex |
Tri/tetra‐antennary complex‐type N‐glycan |
|
|
PNA |
Arachis hypogaea |
Gal |
Galβ3GalNAc |
|
|
PSA |
Pisum sativum |
Fuc/Man |
Fucα6GlcNAc, High‐Man |
|
|
PTL−I |
Psophocarpus tetragonolobus |
GalNAc |
α GalNAc |
|
|
PWM |
Phytolacca Americana |
GlcNAc |
(GlcNAcβ4) n |
|
|
RCA−I |
Ricinus communis |
Gal |
Galβ4GlcNAc |
|
|
SBA |
Dolichos biflorus |
GalNAc |
GalNAc, GalNAcα3Gal |
|
|
SNA |
Sambucus nigra |
Sia |
Siaα2‐6Gal/GalNAc |
|
|
SSA |
Sambucus sieboldiana |
Sia |
Siaα2‐6Gal/GalNAc |
|
|
STL |
Solanum tuberosum |
GlcNAc |
(GlcNAcβ) n, (GlcNAcβ4MurNAc) n (peptidoglycan backbone) |
|
|
TJA−I |
Tanthes japonica |
Sia |
Siaα2‐6Gal/GalNAc |
|
|
UDA |
Urtica dioica |
GlcNAc |
GlcNAcβ4GlcNAc, Man5∼ Man9 |
|
|
UEA−I |
Ulex europaeus |
Fuc |
Fucα2Galβ4GlcNAc (H‐type 2) |
|
|
VVA |
Vicia villosa |
GalNAc |
α GalNAc, GalNAcα3Gal |
|
|
WFA |
Wisteria floribunda |
GalNAc |
GalNAcβ4GlcNAc, Galβ3(‐6) GalNAc |
|
a) Fuc, fucose; Gal, galactose; GlcNAc, N‐acetyl‐D‐glucosamine; Man, mannose; GalNAc, N‐Acetyl‐D‐galactosamine; Sia, sialic acid
3.1.2. Recombinant Lectins
Recombinant lectins, particularly those of plant and animal origin, are produced from microorganism expression systems by recombinant technology.55 Bacteria and yeasts, which have simple gene expression systems and good recombinant protein yield, are the preferred tools for recombinant lectin generation.55 As recombinant lectins can overcome the limitations of natural lectins, they have been increasingly used for glycan profiling.56a, 56b The source organism, organism used for clonal expansion, and specificity profile of recombinant lectins mentioned in this review are summarized in Table 2.
Table 2.
Specificity profile of recombinant lectins.
|
Lectin |
Source organism |
Organism used for clonal expansion |
Specificity |
Refs. |
|---|---|---|---|---|
|
MBL |
Gallus Gallus |
HeLa R19 Cells |
Man |
|
|
GafD |
Escherichia coli |
Escherichia coli |
β‐GlcNAc |
|
|
PA−IL |
Pseudomonas aeruginosa |
Escherichia coli |
Gal |
|
|
PA−IIL |
Pseudomonas aeruginosa |
Escherichia coli |
Fuc/Man |
|
|
PapGII |
Escherichia coli |
Escherichia coli |
GbO4 |
|
|
PapGIII |
Escherichia coli |
Escherichia coli |
GbO5 |
|
|
RS−IIL |
Ralstonia solanacearum |
Escherichia coli |
Man/Fuc |
|
|
Mutated MAH |
Maackia amurensis |
Escherichia coli |
Complex |
|
|
EW29 |
Earthworm |
Escherichia coli |
α2‐6Sia |
|
|
EW29Ch |
Earthworm |
Escherichia coli |
6‐sulfo‐Gal |
Hsu et al. was the first to develop the protocol and demonstrate the use of recombinant lectins in microarrays. They expressed bacterially‐derived lectins in Escherichia coli as fusion proteins and then purified them using glutathione (GSH) affinity chromatography. Lectin activity and glycan‐binding specificities (glycopatterns) of the purified lectins for both proteins and cell samples could be determined using carbohydrate microarray and ELISA.57a Increasing the accessibility of the carbohydrate‐binding site can enhance the sensitivity of the lectin microarray. Indeed, Propheter et al. confirmed this by developing an oriented lectin microarray based on the interactions between GSH and glutathione‐S‐transferase (GST)‐tagged recombinant lectins.65 Advantages of this method include simple one‐step GST‐tagged protein sample loading, specific orientation of the tagged proteins on NHS‐activated slides with the GSH scaffold, increased analytical capacity, and maintenance of lectin diversity.66
To develop new glycan recognition patterns and customize the glycan specificities of recombinant lectins, multiple mutations have been genetically engineered at the carbohydrate‐recognition domain (CRD) of natural lectins. Maenuma et al. mutated two positions (Gly131 and Ser133) in the CRD of wild‐type Maackiaamurensis hemagglutinin to obtain thirty‐five lectin variants that showed unique carbohydrate‐binding specificities. The lectin variant library is useful for profiling various cells according to variations in their surface glycans.62 Yabe et al. described a novel recombinant lectin generated by the “natural evolution‐mimicry” strategy, wherein the Gal‐binding lectin is randomly mutated by error‐prone PCR to create a novel sialic acid (Sia)‐binding α2‐6Sia‐recognition protein (Sia‐recognition EW29Ch; SRC). However, the SRC had a lower affinity for α2‐6Sia compared with natural lectins.63 They then engineered another construct of SRC tandem repeats with higher selectivity for branched N‐glycans conjugated with multiple α2‐6Sia residues but no apparent hemagglutinating activity. This construct could be used to detect and isolate α2‐6Sia‐containing glycoconjugates.49 Hu et al. also reported customized novel lectins that showed increased binding affinity for 6′‐Sulfo‐LN (6‐O‐sulfo‐Galβ 1‐4GlcNAc). These mutants could potentially discriminate cells containing different amounts of 6S‐Gal‐terminated glycans.64
3.2. Detection Techniques
3.2.1. Label‐Based Techniques
Fluorescence, chemiluminescence, and radioactivity are three popular label‐based techniques for detecting a target amidst a complex background via direct or indirect labeling.67 The advantages of this method are that it is conveniently applied with commonly available reagents and uses a simple experimental procedure. Fluorescent labels, such as Cyanine 3 (Cy3) or 5 (Cy5), are commonly used in lectin microarray detection.
Direct labeling is the most common method for identifying lectin−glycan interactions. Lectins or glycoproteins in samples, such as formalin‐fixed paraffin‐embedded tumor tissues, serum, and urine, are labeled directly with a fluorescent dye and are subsequently washed and detected with a fluorescence scanner.68a, 68b, 68c, 68d Although direct labeling is often the preferred labeling method, the drawbacks include the requirement for a relatively high amount of glycoproteins, low sensitivity, and a potential disruption of interactions between glycoproteins and lectins.12
The single‐color lectin microarray is a direct labeling method that has been established to study the glycoprofiling of mammalian cells. This method has multiple flaws in the protocol, such as no reliable quality control, poor reproducibility, and disregard of the effects of cellular glycolipids.69 Pilobello et al. then developed a two‐color lectin microarray approach that can rapidly determine the difference in glycoprofiles among heterogeneous mammalian samples. Either Cy3−NHS or Cy5−NHS dye molecules are conjugated with the lysines within proteins in this two‐color direct labeling method. The Cy3‐ and Cy5‐labeled samples are mixed in a 1 : 1 ratio and are hybridized to each lectin microarray. This method may be applied for the systematic evaluation of glycan information in complex systems.70 However, careful consideration should be exercised during the quantitative comparison of lectin signals for the two‐color labeling method due to potential competition between immobilized lectins for various glycans.71a, 71b
The indirect labeling method is generally used in the sandwich format in assays, wherein the lectin−glycoprotein interaction is detected using a biotinylated antibody and a corresponding fluorescent dye (HRP)‐conjugated streptavidin.5b, 72 Meany et al. included Cy3‐labeled streptavidin into this system to increase the sensitivity of targeted glycan profiling. Cy3‐labeled streptavidin further amplified the signal from biotin that have conjugated with HRP‐conjugated streptavidin by over 100 times.20 Cao et al. introduced a lectin multimerization approach to increase lectin avidity to targeted glycans, wherein several biotinylated lectins are conjugated through streptavidin interactions. Proteins in biological samples are captured by immobilized antibodies on arrays, and the glycans on the captured proteins are probed with biotinylated lectins. Unlike the conventional non‐multimerization method wherein primary and secondary detection reagents are added consecutively, these reagents are added in a single step for the multimerization method. Single‐step addition may enable enhanced binding through multivalent interactions. Thus, the multimerization method can potentially broaden the range of glycan structures that can be detected and provide more and different information compared to monomeric detection.73 Because of low background labeling and signal amplification, the indirect labeling methods have advantages of high specificity and sensitivity.
Confocal fluorescence has been widely used for lectin microarray detection. However, the glycan−lectin interaction formed on the microarray may be disrupted due to the washing step after the binding reaction. Multicolor confocal fluorescence detection using, for example, Cy3 and Cy5 dyes can be used to analyze relatively weakly bound glycan on fluorescent dye‐labeled glycoproteins.74 Kuno et al. introduced an evanescent‐field fluorescence detection method for real‐time detection of interactions under equilibrium conditions without washing on a lectin microarray after a probing reaction.10d Koshi et al. developed a bimolecular fluorescence quenching and recovery method for glycan detection in a lectin microarray. Fluorescently labeled lectins are non‐covalently fixed under semi‐wet conditions and recognize and bind to specific glycans. Labeling of target glycans is unnecessary because of high selectivity and affinity of glycans for the immobilized lectins.75
3.2.2. Label‐Free Techniques
Label‐free methods, such as surface plasmon resonance (SPR), optical microscopy, and MALDI‐TOF MS, are used to determine the inherent properties (dielectric or optical properties and mass) of molecules bound to lectin microarrays. SPR, which monitors biomolecular interactions in real‐time through measuring changes in the light reflected on the underside of the metal, is a common method for evaluating the affinity, kinetics, specificity, and concentration of biomolecules.76 Nand et al. coupled SPR imaging with a lectin microarray to discriminate between the different stem cells.77 Geuien et al. established a lectin microarray with multiplex SPR capable of accurately quantifying the relative sialylation levels of erythropoietin.78 Since optical methods are sensitive and non‐destructive, they have been used to measure lectin−glycan interactions on a solid surface. Optical microscopy can be used to observe distinct binding patterns of glycans on a lectin array.10c, 79 Chen et al. used this approach to detect differences in glycoprofiles between normal and tumorigenic human breast cell lines, as well as determine the metastatic potential of different sublines.80 Unlike MS, a lectin microarray cannot provide in‐depth structural information on glycans. However, combining lectin microarrays with MS enables high‐throughput glycoprotein biomarker screening, accurate mass measurement, and resolution of glycoproteins that bind to each immobilized lectin on the array.81 Of note, MALDI‐MS detection of captured proteins on the array allows quantitative measurement of glycosylation levels as well as detection of non‐specific binding.81 Indeed, Hu et al. validated this method of coupling MALDI‐TOF‐MS with a lectin microarray on polydimethylsiloxane for the differential analysis of serum glycoproteins in oral cancer versus healthy control subjects.81
4. Application of Lectin Microarrays for Biomarker Discovery
4.1. Cancer Biomarker Discovery
4.1.1. Lung Cancer
Lung cancer is the most common disease worldwide. Small‐cell lung cancer (SCLC) and non‐small‐cell lung cancer (NSCLC) are the two common types. The subtypes of NSCLCs include lung adenocarcinoma (ADC), squamous cell lung cancer (SQLC), and large‐cell carcinoma.82 As current diagnostic methods such as computed tomography, X‐ray, and sputum cytology are lacking in detecting early stages of lung cancer, it is thus important to elucidate serum biomarkers for early cancer detection, cancer staging determination, and for monitoring the response to therapeutic strategies.82 Although several serum proteins, including carcinoembryonic antigen, carbohydrate antigen 125, cytokeratin 19, and neuron‐specific enolase, have been proposed as potential biomarkers of lung cancer, low specificity and sensitivity limit their clinical use.83a, 83b, 83c Glycan profiling may offer an alternative approach to identify specific biomarkers for lung cancer.
Shi et al. showed the distinct glycoprotein profiles of cells in pleural effusions from lung cancer patients (carcinoma cells) versus those with benign lung disease (reactive mesothelial cells) using lectin microarrays.84 Analysis of serum N‐ and O‐glycan profiles in NSCLC patients versus healthy controls by Liang et al. using lectin microarrays revealed 18 lectins (e. g. Aleuriaaurantia lectin [AAL], Jacalin, Griffoniasimplicifolia Lectin I [GSL−I], and Dolichosbiflorus [DBA]) in lung adenocarcinoma and 16 lectins (e. g., Jacalin, Hippeastrumhybridlectin [HHL], Phaseolus vulgaris erythroagglutinin [PHA−E], and Phaseolus vulgaris leucoagglutinin [PHA−L]) in SQLC with significantly altered serum glycopatterns. Notably, the majority of lectins that showed altered expression profiles were found in patients with early stage adenocarcinoma and SQLC (Table 3).85
Table 3.
Application of lectin microarrays for biomarker discovery of various diseases.
|
Diseases |
Sample type |
Glycoprotein marker |
Specific lectin (target glycan) b) |
Microarray Strategy |
Potential applications |
Refs. |
|---|---|---|---|---|---|---|
|
Cancer | ||||||
|
Lung cancer |
Serum |
Not identified |
AAL(Fucα1‐6GlcNAc); Jacalin [Galβ1‐3GalNAcα]; GSL−I(GlcNAc and αGal); DBA (GalNAcα and GalNAcα1‐3Gal); HHL(High‐Man, Manα1‐3Man, Manα1‐6Man, Man5‐GlcNAc2); PHA−E+L (the bisecting GlcNAc and biantennary N‐glycans) |
Direct assay |
Early diagnosis of lung adenocarcinoma and squamous cell lung cancer |
|
|
Serum |
α‐1‐antitrypsin |
BS−I (αGal and αGalNAc); AAL [Fucα1‐6GlcNAc and Fucα1‐3(Galβ1‐4)GlcNAc]; PWM (Branched (LacNAc)n) |
Direct assay |
Distinguish non‐small‐cell lung cancer, lung adenocarcinoma and small‐cell lung cancer from benign pulmonary diseases |
||
|
Tissues and cell culture supernatants |
Fibronectin |
PNA(Galβ1‐3GalNAc) |
Antibody‐overlay lectin sandwich array |
Biomarker of non‐small‐cell lung cancer |
||
|
Gastric cancer |
Tissues |
Not identified |
MPL (αGalNAc); VVA (GalNAc and GalNAcα) |
Direct assay |
Distinguish gastric cancer from ulcer |
|
|
Serum |
Not identified |
LEL and STL (GlcNAc) |
Direct assay |
Diagnostic marker for early gastric cancer |
||
|
Colorectal cancer |
Plasma |
Complement C3; histidine‐rich glycoprotein; kininogen‐1 |
AAL and SNA (sialylation and fucosylation) |
Glycoprotein‐lectin array |
Distinguish colorectal cancer from adenoma and normal |
|
|
Cancer tissues and adjacent normal tissues |
HSP90β and Annexin A1 |
STL (GlcNAc) |
Direct assay |
Diagnostic marker for colorectal cancer |
||
|
Formalin‐fixedtissues |
No identified |
ABA (Galβ1‐3GalNAcα) |
Direct assay |
A predictive biomarker for recurrence of colorectal cancer |
||
|
Serum |
Alpha‐2‐macroglobulin |
SNA (terminal α2,6 Sia bound to Gal or GalNAc); PHA−E (bi/tri‐antennary complex type N‐glycans with terminal Gal and bisecting GlcNAc); ConA [High‐Man type N‐glycans, Man α1,6(Manα1,3) Man] |
Glycoprotein−lectin array |
Diagnostic marker colorectal cancer |
||
|
Cell lines |
Not identified |
UEA−I (α‐1,2‐fucosylation) |
Direct assay |
Lectin biomarker for colorectal cancer |
||
|
Hepatocellular carcinoma |
Cell lines |
Not identified |
ACL, BPL, JAC, MPL (Galβ1‐3GalNAcα and GalNAcα); PHA−E (NA2 and bisecting GlcNAc); SNA (Siaα2‐6Gal/GalNAc); SBA (terminal α or βGalNAc) |
Direct assay |
Metastasis‐specific glycan markers |
|
|
Serum |
Immunoglobulin G |
PSA, LCA, and AAL [core (α‐1,6) fucosylation)]; SNA−I [(α‐2,6) sialylation] |
Direct assay |
Disease diagnosis |
||
|
Serum |
GP73 |
AAL [Fucα6GlcNAc (core Fuc), Fucα3(Galβ4) GlcNAc (Lex)]; LCA (Fucα6GlcNAc, High‐Man); PSA (Fucα6GlcNAc, High‐Man) |
Antibody‐overlay lectin sandwich array |
Distinguish HCC from liver cirrhosis |
||
|
Serum |
Not identified |
PHA−L (β1,6‐GlcNAc) |
Direct assay |
Metastasis‐related marker |
||
|
Tissues |
Not identified |
HHL and NPA(Manα1‐6Man) |
Direct assay |
Disease diagnosis |
||
|
Cell lines |
Annexin A2; Heat shock protein 90 beta family member 1 |
LCA (Fucα1‐6GlcNAc, α‐D‐Man) |
Direct assay |
Diagnostic biomarkers for hepatoma cells after HCV infection |
||
|
Breast cancer |
Cell lines |
POTE ankyrin domain family member F |
RCA−I (Lac/LacNAc, Terminal Galβ 1‐4 GlcNAcβl) |
Direct assay |
Marker for metastatic triple‐negative breast cancer cells |
|
|
Pancreatic cancer |
Cell lines |
Lysosome‐associated membrane glycoprotein 1;hypoxia upregulatedprotein 1 |
PSA (α‐1‐6 core Fuc); DSA and PHAE (Galβ1‐4GlcNAc); NPA, GNA, and HHL(High‐Man) |
Direct assay |
Disease diagnosis |
|
|
Cell lines |
Cytokeratin 8, integrin β1, ICAM1, and ribophorin 2 |
UEA‐1 (Fuc); DBA (Gal) |
Direct assay |
Prognostic markers for pancreatic cancer |
||
|
Ascites fluids |
CD133 |
SNA (NeuAcα2‐3); STL (Glc‐NAc β1‐4GlcNAc); UDA (GlcNAcβ1‐4GlcNAc and Man) |
Direct assay |
Prognostic biomarker for advanced pancreatic cancer |
||
|
Bladder cancer |
Cell lines |
Not identified |
LTL (Sialyl Lewis X); PTL−II (terminal GalNAc and Gal) |
Direct assay |
Related to bladder cancer progression |
|
|
Ovarian cancer |
Ascites fluids and culture supernatants of cell lines |
Ceruloplasmin |
WFA [GalNAcβ1,4GlcNAc (LacdiNAc)] |
Direct assay |
Disease diagnosis |
|
|
Inflammatory diseases | ||||||
|
Rheumatoid arthritis |
Serum |
Matrix metalloproteinase‐3 |
ACG (Siaα2‐3Galβ1‐4GlcNAc); ABA (Galβ1‐3GalNAc); ACA (Galβ1‐3GalNAc) |
Antibody‐overlay lectin sandwich array |
Assess disease activity |
|
|
Pneumonia |
Serum |
Haptoglobin‐related protein |
SNA−I (α2‐6 linked Sia) |
Direct assay |
Distinguish non‐bacterial pneumonia from bacterial pneumonia |
|
|
Chronic hepatitis |
Serum |
Mac‐2 binding protein |
WFA[GalNAc β1‐4GlcNAc, Galβ1‐3(‐6)GalNAc] |
Direct assay |
Related to liver fibrosis progression |
|
|
Crohn's disease |
Serum |
Immunoglobulin G |
ABA and GSL−II (agalactosyl N‐linked oligosaccharides) |
Direct assay |
Correlated with disease activity and predictability of therapeutic outcomes |
|
|
Other diseases | ||||||
|
Diabetic nephropathy |
Urine |
Fetuin−A |
SSA (Siaα2‐6Gal/GalNAc) |
Direct assay |
Predict the progression diabetic nephropathy |
|
|
Major depressive disorder |
Plasma |
No identified |
TJA−I, SNA, and SSA(Sia‐α2‐6Gal/GalNAc) |
Direct assay |
Clinical diagnosis and monitoring |
|
|
Aging |
Plasma |
Haptoglobin |
ECA (Galβ1‐4GlcNAc) |
Direct assay |
Characterized as human longevity and healthy aging |
|
|
Subfertility |
Sperm |
Not identified |
ABA (Galβ1‐3GalNAc); MPL (Galβ1‐3GalNAcα and GalNAcα) |
Direct assay |
Clinical diagnosis of subfertility |
|
a) NeuAc, N‐acetylneuraminic acid; LacNAc, N‐Acetyl‐D‐lactosamine; Lac, lactose
Recent studies reported an increase of human α‐1‐antitrypsin (A1AT; a serum glycoprotein with three potential glycosylation sites) levels in various cancers including lung, prostate, and breast cancers. Elevated A1AT levels were also noted in certain benign pulmonary diseases.115a, 115b Liang et al. examined A1AT glycosylation alterations in ADC, SQLC, and SCLC serum samples using lectin microarrays coupled with ELISA. Three markers were identified. Galactosylated A1AT was identified as a marker capable of differentiating NSCLC from benign pulmonary diseases; fucosylated A1AT as a marker to distinguish ADC from benign diseases or other lung cancer subtypes; and A1AT containing poly−LacNAc as a marker to differentiate SCLC from benign diseases (Table 3).86 Due to the small sample size in the Liang et al. study (12 samples for each group), further validation of the potential role of glycosylated A1AT as a lung cancer biomarker is necessary using larger sample sizes.
Hirao et al. developed an integrated glycoproteomics approach to identify a glycoprotein biomarker that is capable of differentiating NSCLC from SCLC. Tissue extracts and cell culture supernatants were added to the lectin microarray to identify NSCLC‐specific lectin probes. Next, isotope‐coded glycosylation site‐specific tagging‐LC‐MS‐based screening of glycobiomarker candidate molecules present in the supernatants from SCLC and NSCLC cell lines was performed. The preliminary candidates were then examined using a Western blot and immunoprecipitation analyses before final reevaluation using an antibody‐overlay lectin microarray. A successful example of this strategy is the identification of NSCLC‐specific biomarkers like fibronectin that have fucose‐ and oligomannose‐modified N‐glycans (Table 3).87
4.1.2. Gastric cancer
Gastric cancer is the most common epithelial tumor worldwide and ranks second in cancer‐related mortality rates. The relationship between stomach ulcers and cancer has long been controversial.116 Huang et al. observed higher glycosylation levels in gastric cancer tissues than in gastric ulcer tissues from a 37‐lectin microarray. Notably, two lectins, Maclurapomifera lectin (MPL) and Viciavillosa (VVA), have been identified and validated as specific gastric cancer biomarkers (Table 3).88
Shu et al. found 15 lectins (e. g., Pisum sativum [PSA], PHA−E, and Erythrina cristagalli [ECA]) that showed significantly altered salivary protein glycosylation in gastric cancer (adenocarcinoma of stage I/II/III) and atrophic gastritis patients versus healthy volunteers using lectin microarrays. Outer‐arm fucosylation and core‐fucosylation expression levels of glycans specific for AAL and PSA lectins were downregulated, while that of Gal and GalNAc structures recognized by Peanut agglutinin (PNA), Euonymus europaeus lectin (EEL), MPL, GSL−I, Bandeiraea (Griffonia) simplicifolia lectin−I (BSI), ECA, Glycine max (SBA), and VVA were upregulated in the saliva of gastric cancer patients. They could construct the diagnostic models of gastric cancer and atrophic gastritis with high diagnostic accuracy with the 15 selected lectins.117
Li et al. observed altered glycan expression profiles in serum samples of early gastric cancer patients using a 50‐tumor‐associated‐lectin microarray. GlcNAc, GalNAc, Tri/tetra‐antennary N‐glycan, β‐1,6‐GlcNAc branched structure, α‐linked fucose residues, and Tn antigen were upregulated while N‐acetyl‐D‐galactosamine structure and (α‐1,3) Man residues were downregulated. GlcNAc, which is highly expressed, may serve as a potential diagnostic marker for early gastric cancer (Table 3).89
4.1.3. Colorectal Cancer
Colorectal cancer is the third most common cancer worldwide.118 The most common serum glycoprotein biomarker for colorectal cancer detection, carcinoembryonic antigen (CEA), has poor sensitivity and specificity and cannot be used for early cancer detection.119 Similarly, other potential colorectal cancer serum glycoprotein markers, such as CA 19‐9, CA 242, CA‐195, CA 50, CA 74‐2, and tissue metalloproteinase inhibitor 1,120a, 120b, 120c, 120d also have low sensitivity and specificity and are inadequate for screening or diagnostic applications.
Qiu et al. developed lectin glycoarrays capable of identifying plasma markers from normal, adenoma, and colorectal cancer patients. Abundant plasma proteins are first subjected to immunodepletion, and then plasma N‐linked glycoproteins are enriched using lectin affinity chromatography and nonporous silica reversed‐phase HPLC. The enriched glycoproteins are then added to lectin microarrays to determine glycopatterns. Notably, complement C3, histidine‐rich glycoprotein, and kininogen‐1, which showed elevated sialylation and fucosylation levels, were identified as potential colorectal cancer markers (Table 3).90
Li et al. established an integrated approach involving lectin microarrays and MS quantification for identifying candidate colon cancer biomarkers. Candidates with differential glycan profiles in colon cancer tissues versus adjacent normal colon tissues were first identified using a lectin microarray, and the shortlisted proteins are verified with lectin histochemistry. Next, enrichment and identification of specific lectins were performed using label‐free MS. HSP90b and Annexin A1 proteins were found to be GlcNAcylated and their expression levels were upregulated in colon cancer tissues (Table 3).91
Distant recurrence markers from formalin‐fixed, paraffin‐embedded tumor specimens and normal epithelium from 53 consecutive curatively resected stage I–III colorectal cancer patients (identified using lectin microarray in a study by Nakajima et al.) were validated with an additional 55 curatively resected stage II colorectal cancer cases. Notably, Agaricusbisporus (ABA) lectin, with high lectin−glycan interaction (LGI) values in cancer tissues and significant statistical association with distant recurrence, has been proposed as a novel biomarker for distant recurrence of curatively resected colorectal cancer (Table 3).92
Sunderic et al. presented a lectin‐based protein microarray to distinguish changes in alpha‐2‐macroglobulin (α2M) sera glycosylation levels between healthy individuals versus colorectal cancer patients. Target proteins are isolated through immunoprecipitation and then spotted onto the lectin microarray. The lectin‐based protein microarray revealed a higher content of α2,6 sialic acid, N‐acetylglucosamine and Man residues, and tri‐/tetraantennary complex type high‐Man N‐glycans for α2M molecules isolated from the sera of colorectal cancer patients. Thus, the α2M glycopattern may be a potential colorectal cancer biomarker (Table 3).93
The polyacrylamide hydrogel‐based lectin microarray introduced by Tian et al. can be used to screen for colorectal cancer cells that express high levels of surface glycans. Multivalent lectins that are immobilized on the polyacrylamide hydrogel bind to the glycans with increased binding affinity and selectivity. Differentially expressed glycans with D‐Gal, D‐glucose, and sialic acid residues, and UelxEuropaeus Agglutinin−I (UEA−I) in SW480 colorectal cancer cells were identified from a 27‐lectin‐microarray screening. Further, in vitro and in vivo experiments confirmed UEA−I as a colorectal cancer biomarker (Table 3).94
4.1.4. Hepatocellular Carcinoma
Hepatocellular carcinoma (HCC) is the most prevalent primary liver cancer and ranks third in cancer‐related deaths worldwide. HCC usually occurs in patients with an underlying chronic liver disease, such as cirrhosis or a chronic hepatitis B virus (HBV) infection.121 Changes in glycosylation levels are known to occur during HCC development.122
Li et al. conducted glycoprofiling of intact Huh7 HCC cell surface glycoproteins in the epithelial mesenchymal transition (EMT) model using a lectin microarray to identify HCC metastasis‐specific glycans. Decreased levels of T/Tn‐antigen, NA2, bisecting GlcNAc, Siaα2‐6Gal/GalNAc, terminal α, or βGalNAc structures, and increased levels of terminal αFuc and ±Sia‐Le, α‐or β‐linked GalNAc, core fucose, β1,6 GlcNAc branching structures, and tetraantennary complex oligosaccharides were noted (Table 3).95
Wang et al. developed a multiplex assay to analyze the glycopatterns of HCC‐associated immunoglobulin G (IgG) and found increased (α‐1,6) fucosylation and (α‐2,6) sialylation levels in the IgG from the sera of HCC patients (Table 3).96 In this assay, a double‐laser fluorescence system is used to identify biotin‐labeled glycoproteins that bind to the immobilized lectins that are conjugated to fluorescent dye‐coated microbeads. This system enables the three‐dimensional interaction between lectins and specific glycans as well as the simultaneous detection of multiple glycan epitopes in a single reaction vessel.
Jiang et al. identified serum GP73, a resident Golgi type II membrane protein with three potential N‐glycosylation sites, as a HCC diagnostic biomarker using antibody‐overlay lectin microarray and a lectin blot. They found significantly higher fucosylated GP73 levels in liver cirrhosis patients compared to that in HCC patients. In addition, they showed that the combined detection of fucosylated GP73 and α‐fetoprotein‐L3 increased the sensitivity and specificity of a HCC diagnosis (Table 3).97
Liu et al. performed a lectin microarray, lectin affinity chromatography, and MS to elucidate biomarkers for diagnosing early hepatic encephalopathy (HE) in patients with terminal HCC using serum from HCC patients with or without early HE. As PHA−E levels showed a significant decrease in HCC patients with early HE, they concluded that 26 PHA−E‐associated glycoproteins might be involved in the occurrence of early HE.123 In another study by Liu et al., 11 PHA−L reactive glycoproteins with a significantly altered N‐glycosite occupancy (β1,6‐GlcNAc branched N‐glycan) were identified in HCC patients with metastasis, using a lectin microarray, lectin affinity chromatography, and stable isotope labeling coupled with LC‐MS, which suggested their involvement in HCC metastasis (Table 3).98
HBV and hepatitis C viral (HCV) infection‐induced liver diseases are closely related to HCC. Analysis of the differential expression profiles of liver glycoproteins in normal pericardial tissues, liver cirrhosis, and tumor tissues induced by HBV using a lectin microarray revealed increased high‐Man type glycans during cirrhosis‐tumor progression in normal pericardial tissues, increased Manα1‐3Man (GNA) in cirrhotic and tumor tissue, and increased Manα1‐6Man (HHL and Narcissus pseudonarcissus [NPA]) only in tumor tissue (Table 3).99 Xiang et al. identified fucosylated annexin A2 and heat shock protein 90 beta family member 1, which showed significantly increased levels in HCV‐infected Huh7.5.1 human liver cells, as potential markers using a combined lectin microarray with MS and a lectin pull‐down assay (Table 3).100
4.1.5. Breast Cancer
Arndt et al. showed that MDA‐MB435 breast cancer cells bind to lectin ECA, Limax flavus (LFA) and Canavaliaensiformis (ConA) using a lectin microarray.124 Similarly, the 91‐lectin microarray screening by Zhou et al. revealed that increased binding of Ricinus communis agglutinin I (RCA−I) to triple‐negative breast cancer (TNBC) cells increases TNBC metastatic capacity. In addition, they found a differential galactosylation expression pattern of POTE ankyrin domain family member F, which is capable of binding RCA−I, in high/low metastatic TNBC cells (Table 3).101
Fry et al. found 6 lectins (GSL−II, Phytolaccaamericana [PWM], PNA, Psophocarpustetragonolobus [PTL−I], GSL−I−B4, and jacalin) as potential metastatic primary breast tumor biomarkers using 45‐lectin N‐ and O‐linked glycan‐specific microarrays coupled with evanescent‐field activated fluorescence detection, glycomic analysis of primary breast tumors, and the serum and urine of patients with metastatic breast cancer. Increased binding to Aspergillus oryzae l‐fucose‐specific lectin (AOL) and Galanthusnivalis agglutinin (GNA) lectins but decreased binding to Ricinus communis (RCA120) and PHA−E lectins were noted for the sera of metastatic patients. Three lectins, Trichosanthes japonica agglutinin I (TJA−I), RCA120, and Bauhinia purpurea lectin (BPL), displayed significantly increased levels in the urine of metastatic patients.68a
Guo et al. showed that conditioned medium (CM) derived from malignant breast cancer cells (MDA‐MB‐231 and MDA‐MB‐453) exhibit an altered N‐glycan profile and induced an EMT‐like process in non‐tumorigenic normal mammary epithelial cells (MCF10A).125 Moreover, reduced levels of bisecting GlcNAc proteins, and the corresponding MGAT3 glycosyltransferase were observed in a hypoxia‐induced EMT model using MCF7 and MDA‐MB‐231 breast cancer cell lines.126
4.1.6. Pancreatic Cancer
Using a lectin microarray and LC‐MS/MS, Tian et al. showed that the sialoglycoproteins LAMP1 and ORP150 were overexpressed in the SW1990 human pancreatic cancer line (Table 3).102 Using the same strategy, Zhu et al. identified differentially expressed glycoproteins in pancreatic cancer CD24+CD44+stem‐like cells and observed significantly increased fucosylated and galactosylated glycoproteins, such as cytokeratin 8/CK8, integrin β1/CD29, ICAM1/CD54, and ribophorin 2/RPN2, in CD24+CD44+cells (Table 3).103 Resistance to current pancreatic cancer therapies is heavily attributed to cancer stem cells (CSC). Terao et al. showed that fucosylation, a common modification in pancreatic cancer CSC‐like cells from lectin microarray analysis, could serve as a novel biomarker to determine anticancer drug resistance.127 A lectin microarray performed by Sakaue et al. revealed that the CSC marker CD133 in ascites‐derived exosomes from patients with unresectable pancreatic cancer is commonly glycosylated by sialic acids. Thus, sialylated CD133 could potentially serve as a prognostic biomarker for advanced pancreatic cancer (Table 3).104
4.1.7. Bladder Cancer
Using combined lectin microarray analysis, MALDI‐TOF‐MS and glycogene microarray analysis, Guo et al. showed decreased biantennary N‐glycan structures and tetra‐antennary complex‐type N‐glycan (recognized by PHA−E+L) levels during transforming growth factor‐beta (TGF‐β)‐induced EMT in non‐malignant bladder transitional epithelium HCV29 cells. This led to their conclusion of the involvement of α‐mannosidase 2 and Type 1 α‐L‐fucosidase in TGF‐β‐induced EMT.128 Integrating lectin microarray and MS methods, Yang et al. found highly expressed core‐fucosylated N‐glycans but lowly expressed terminally fucosylated N‐glycans in four bladder cancer cell lines (KK47, YTS1, J82, and T24) versus a normal bladder mucosa cell line (HCV29), suggesting their direct correlation with bladder cancer progression (Table 3).105
4.1.8. Ovarian Cancer
Epithelial ovarian cancer (EOC), which accounts for 90 % of all ovarian cancers, is usually asymptomatic and has a poor prognosis. The conventional marker CA125 often outputs false‐negative results. Sogabe et al. discovered glycobiomarker candidates using a lectin microarray coupled with IGOT‐LC/MS analysis. The cancer‐associated glycopeptides are first enriched from ascites and culture supernatants of cancer cell lines using the lectin AAL before subsequent identification by IGOT‐LC/MS. The EOC‐specific lectin Wisteria floribunda agglutinin (WFA) was used for subsequent Western blot analysis to elucidate glycobiomarker candidates from the WFA‐bound fraction of ascites fluids. The WFA‐reactive ceruloplasmin generated higher signals in the ascites fluids of EOC patients (Table 3).106 Zhao et al. performed lectin microarray‐MS glycomic analysis of ovarian cancer side population cells to determine antigens associated with cancer recurrence and drug resistance. They observed increased core fucosylated N‐glycan and tumor‐associated Tn, T, and sT antigen levels but decreased hybrid glycan, α2,3‐linked sialic glycan, and multivalent sialyl−glycan levels in side population cells.129
4.2. Inflammatory Diseases Biomarker Discovery
The common serological biomarker for rheumatoid arthritis matrix metalloproteinase‐3 (MMP‐3) lacks specificity and accuracy. An antibody‐overlay lectin microarray of MMP‐3, immunoprecipitated from the sera of rheumatoid arthritis patients, led to the identification of altered glycoprofiles of sialic acid‐binding lectin ACG and O‐glycan‐binding lectins (Jacalin, ABA, and Amaranthus caudatus agglutinin [ACA]). Thus, changes in MMP‐3 glycosylation levels could serve as a potential rheumatoid arthritis‐specific biomarker (Table 3).130
Yang et al. found increased lectin SNA−I signal in the mycoplasma and viral pneumonia groups using lectin microarray coupled with LC‐MS/MS. They identified haptoglobin‐related protein (HPR) from serum samples of patients with mycoplasma pneumonia by a SNA−I pull‐down assay and further confirmed elevated SNA−I expression in the mycoplasma pneumonia and viral pneumonia groups versus the bacterial pneumonia group (Table 3).131
The serum levels of Mac‐2 binding protein (M2BP) and M2BPGi, a specific glycoform recognized by WFA, are positively correlated with liver fibrosis progression. Narimatsu et al. found that WFA lectin exhibits significantly high specificity for M2BP but did not bind to most serum proteins in normal serum samples using a lectin microarray. The M2BPGi assay kit was then established and was validated using more than 8,000 samples. Thus, M2BPGi levels are an excellent diagnostic marker for chronic hepatitis and cirrhosis (Table 3).132
Human inflammatory bowel disease (IBD), Crohn's disease (CD), and ulcerative colitis (UC) are characterized by chronic recurrence and remission of digestive tract inflammation. Significantly higher agalactosyl fraction among fucosylated oligosaccharides of serum IgG was reported in CD and UC patients. A lectin microarray screening revealed that ABA and GSL−II lectins exhibit higher affinity for serum agalactosyl IgG from IBD and especially CD patients. The observation of higher agalactosyl IgG levels in CD patients was confirmed with a ABA or GSL−II lectin−enzyme immunoassay. Thus, agalactosyl IgG levels could serve as a novel biomarker for IBD (Table 3).133
4.3. Other Diseases Biomarker Discovery
Diabetic nephropathy (DN), a serious complication of diabetes, is the main cause of chronic and terminal kidney disease worldwide. Glycoprofile alterations in the urine can be used to predict and monitor DN. A lectin microarray of urine samples from DN patients revealed increased Siaα2‐6Gal/GalNAc‐binding lectin (SNA, SSA, TJA−I) signals in the urine samples of DN patients. Fetuin‐A glycoprotein was identified as the prognostic biomarker for DN progression (Table 3).134 Another study confirmed the positive correlation between Siaα2‐6Gal/GalNAc (recognized by SNA lectin) expression levels and DN progression and suggested its application in differentiating DN from nondiabetic renal disease (NDRD).135 However, Yang et al. showed that another glycan, (β‐1,4)‐linked GlcNAc, recognized by the lectin Datura stramonium agglutinin (DSA), could be a biomarker capable of differentiating DN from NDRD.136 Thus, further research to confirm the candidates for DN biomarkers is necessary to resolve these inconsistencies.
Yamagata et al. noted common changes in Sia‐α2‐6Gal/GalNAc glycan in both depression mice models and major depressive disorder (MDD) patients using a 45‐lectin microarray. Moreover, decreased ST6GALNAC2 expression levels were noted in the leukocytes from MDD patients. Thus, they concluded that Sia‐α2‐6GalNAc glycan in plasma protein and ST6GALNAC2 glycan in peripheral leukocytes may serve as potential biomarkers for MDD clinical diagnosis and monitoring (Table 3).137
Glycans are emerging as aging biomarkers. A lectin microarray analysis showed the differential expression of α2‐6sialylated and α2‐3sialylated O‐glycan glycans during cellular senescence between elderly‐ (86‐year old and 97‐year old subjects) versus fetus‐derived human skin diploid fibroblast cells.138 In contrast to those membrane glycoproteins that decrease with age, α2‐3/2‐6sialylated intracellular glycoproteins, except for some α2‐3sialylated O‐glycans, increase with age.107 Lectin microarrays coupled with LC‐MS of plasma proteins from Japanese semi‐supercentenarians (106–109 years), aged controls (70–88 years), and young controls (20–38 years) revealed increased binding to ECA lectins. Abundant tri‐antennary, and sialylated N‐glycans of haptoglobin at Asn207 and Asn211 sites, which are abundant in semi‐supercentenarians, are signatures of extreme human longevity (Table 3).108
The glycocalyx coating on the sperm surface is vital for sperm motility, maturation, and fertilization. Comparing the binding abilities of multiple lectins in seminal plasma of fertile men with that in infertile men, using a lectin microarray, revealed that lectin reactivity is positively associated with fertility.109 β‐defensin 126 (DEFB126) contributes to sialylation on the sperm surface. Homozygous DEFB126 mutations can lead to male subfertility as evident from decreased binding affinity for 6 lectins (Jacalin/AIA, Gossypium hirsutum agglutinin [GHA], Amaranthus caudatus lectin [ACL], Maclurapomifera lectin [MPL], Viciavillosa lectin [VVL], and ABA). Of the 6 lectins, ABA and MPL lectins were validated as potential DEFB126 homozygous mutant male subfertility biomarkers (Table 3).110
4.4. Stem Cells Biomarker Discovery
Since glycan profiles undergo characteristic changes during development, they are used as stem/progenitor cell markers. Glycoprotein profiling of three different pluripotent stem cells (mouse embryonic stem cells [ESCs], mouse‐induced pluripotent stem cells [iPSCs], and mouse embryonic fibroblast stem cells [MEFs]) versus non‐pluripotent cells elucidated 8 lectins (DBA, Maackiaamurensis [MAL], PHA_E, PHA_L,EEL, AAL, PNA, and Sambucusnigra [SNA]) as potential pluripotency markers for MEFs77 and 3 lectins (EEL, MAL and PHA−L) as differentiation potency markers for human ESCs and iPSCs.111 Tateno et al. developed a high‐density 96‐lectin microarray with broader glycome coverage and found Burkholderiacenocepacia (rBC2LCN) can distinguish undifferentiated iPSCs/ESCs from differentiated SCs.139 Podocalyxin, a heavily glycosylated type 1 transmembrane protein, was identified as a glycoprotein ligand of rBC2LCN on human iPSCs and ESCs using an antibody‐overlay lectin microarray. Moreover, significant affinity of rBC2LCN for a branched O‐glycan with a H type 3 structure in human iPSCs suggest its potential as a pluripotency marker.140 Furthermore, rBC2LCN showed strong specificity for human iPSC‐derived extracellular vesicles (EVs) but not for non‐human iPSC‐derived EVs.112 Recombinant lectins from diverse lectin families were engineered to ensure broader coverage of glycan‐binding specificities. A lectin microarray with 38 recombinant lectins revealed increased expression of α2‐6Sia, α1‐2Fuc, and type 1 LacNAc in undifferentiated human iPSCs. Moreover, increased expression levels of corresponding glycosyltransferase genes ST6Gal1, FUT1/2, and B3GalT5 were observed in human iPSCs versus that in somatic cells.141
Tateno et al. combined lectin microarray technology with flow cytometry analysis and anion‐exchange chromatography and found that α2‐6Sia‐specific lectins showed higher binding affinity for human mesenchymal stem cells (hMSCs) with differentiation potential, suggesting that α2‐6Sia.α2‐6sialylation of integrinα5 could function as a marker for differentiation potency of stem cells including adipose‐derived hMSCs, bone marrow‐derived hMSCs, and cartilage tissue‐derived chondrocytes.113, 142 A lectin microarray showed increased GlcNAc protein modification and α‐1‐2‐fucosylation but decreased α‐1‐6‐fucosylation, α‐2‐6‐sialylation, and α‐1‐6‐mannosylation during ESC adipogenesis.143
CSCs have been reported to drive tumor initiation and growth. Lectin microarray coupled with FACS analysis revealed that sialylated glycan‐recognizing lectins, MAL−I, SNA, Sambucussieboldiana (SSA), TJA−I, Agrocybecylindracea (ACG), ABA, and Maackiaamurensis (MAH) displayed higher affinity to CSCs in CD133+CD13+ Huh7 human liver cancer cells than to CSCs in CD133+cells. Subsequent validation led to the proposal of SSA lectin as a candidate marker for CSC recognition from heterogeneous cell types.114
5. Conclusions and Outlook
Glycoproteomics is an emerging field in post‐genome science. Glycosylation is the most abundant and complex posttranslational modification that plays a primary role in regulating lipids, proteins, and cell functions. Lectin has been found to be very useful for detecting specific glycosyl structures. Biomarker discovery is a new exciting application of lectin microarray technology. This can potentially contribute to fundamental biological and clinical applications including the discovery of cancer diagnostic biomarkers and new drugs. We have discussed in the present review about lectin microarray as a potent tool for the discovery of glycosylation‐related biomarkers. However, the current limitations of this technology must be addressed to expand its applicability.
Firstly, since most of the current natural lectins are derived from plants, it is difficult to obtain a comprehensive repertoire that is representative of glycome complexity. Thus, the repertoire of unique lectin probes on microarrays must be continuously expanded to cope with the number of glycosyl epitopes present in humans. Specifically, the discovery of novel natural lectins and their biochemical properties, cloning, and purification of all known and predicted lectin or lectin‐like proteins, as well as the rational design and development of new recombinant lectins, are necessary to continue expanding the list of known and available lectins. The scope of lectin research should be extended to humans. Most of the known and predicted human lectins are ligands and receptors in cell membranes or body fluids, which participate in regulating cell functions, protein levels, and host‐pathogen interactions. Thus, human lectin‐based microarrays have broad application prospects in disease biomarker research.
Secondly, due to a limitation in sample quantity in many clinical situations, a normal basic lectin microarray is not sensitive enough to detect low abundance target cells or proteins. Thus, the current format of lectin microarrays must be substantially improved.
Thirdly, information on glycan structure and details of glycosylation patterns cannot be obtained from lectin microarray analysis. Thus, a lectin microarray is coupled with MS to satisfy the requirements of a fast, low‐cost, accurate, and high‐throughput method.
Fourthly, our goal is to elucidate highly sensitive and specific clinically relevant glycan diagnostic disease biomarkers. The sera of advanced cancer patients often contain complex non‐cancer‐related protein patterns. Unfortunately, the histopathological status is often overlooked during analysis of sera from advanced cancer patients. Advancement in technology facilitated the screening and elucidation of potential candidate biomarkers. However, useful disease markers account for less than 1 percent of the identified markers. Since proteins that exhibit either specific or preferential expression in cancer cells can be potential biomarkers, high‐quality and high‐content sample collection is a prerequisite to ensure successful glycan‐based biomarker identification via lectin microarray screening.
Lectin microarrays provide a fast, high‐throughput, and inexpensive tool that can support the discovery of glycol‐biomarkers as well as obtain better understanding of glycans’ structure and function in various biological processes and diseases.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Dr. Kai Dang obtained his PhD from Northwest University (Xi'an, China) in 2016 and is currently an assistant researcher at School of Life Sciences, Northwestern Polytechnical University (Xi'an, China). His research interests are functional carbohydrate, glycomics and animal physiology.

Biographical Information
Dr. Wenjuan Zhang was born in 1985 in China, and received her Ph.D. degree under Pro. Yonghua Wang from Northwest A&F University in 2016. She is currently a lecturer at Northwestern Polytechnical University. Her research interests are in chemical biology, bioinformatics, systems biology.

Biographical Information
Dr. Shanfeng jiang obtained his PhD from Northwest University (China), and currently served as a assistant researcher in Northwestern Polytechnical University. His research interests are in animal physiology and bioinformatics, particularly muscle physiology, bone physiology, and comparative genomics. Dr Jiang is a hibernation physiology specialist with 10+years of experience.

Biographical Information
Dr. Xiao Lin, Ph.D., pursued her Ph.D. in cell biology from Tsinghua University and is currently an assistant professor at School of Life Sciences, Northwestern Polytechnical University (Xi'an, China). Her research interests are bone cell biology research, tissue engineering, pharmacology and application.

Biographical Information
Prof. Dr. Airong Qian was born in Gansu Province in 1973. She received the Ph.D. degree in Pathology from Fourth Military Medical University, Xi'an, China, in 2003. Her major field of study is mechanobiology, space cell biology and space biotechnology and tumor biology. She is currently a Professor in Key Laboratory for Space Biosciences and Biotechnology, Northwestern Polytechnical University, Xi'an. She has authored or coauthored more than 100 papers and applied more than 20 patents. Her current research interests include the mechanism of cell sensing mechanical stimulation, research and development of noncoding RNA drugs, and the mechanism and countermeasure of skeletal muscle metabolism disorders.

Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 31570940; 81700784).
K. Dang, W. Zhang, S. Jiang, X. Lin, A. Qian, ChemistryOpen 2020, 9, 285.
References
- 1. Clin Pharmacol Ther 2001, 69, 89–95.
- 2.
- 2a. Wu Y., Le W., Jankovic J., Arch. Neurol. 2011, 68, 22–30; [DOI] [PubMed] [Google Scholar]
- 2b. Le W., Dong J., Li S., Korczyn A. D., Neurosci. Bull. 2017, 33, 535–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Apweiler R., Hermjakob H., Sharon N., Biochim. Biophys. Acta 1999, 1473, 4–8. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Hart G. W., Copeland R. J., Cell. 2010, 143, 672–676; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Feizi T., Glycoconjugate J. 2000, 17, 553–565. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Pinho S. S., Reis C. A., Nat. Rev. Cancer 2015, 15, 540–555; [DOI] [PubMed] [Google Scholar]
- 5b. Kuno A., Matsuda A., Unno S., Tan B., Hirabayashi J., Narimatsu H., Methods Mol. Biol. 2014, 1200, 265–285. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Aoyagi Y., Isemura M., Suzuki Y., Sekine C., Soga K., Ozaki T., Ichida F., Lancet. 1985, 2, 1353–1354; [DOI] [PubMed] [Google Scholar]
- 6b. Kobayashi M., Kuroiwa T., Suda T., Tamura Y., Kawai H., Igarashi M., Fukuhara Y., Aoyagi Y., Hepatology Res. 2007, 37, 914–922; [DOI] [PubMed] [Google Scholar]
- 6c. Magnani J. L., Steplewski Z., Koprowski H., Ginsburg V., Cancer Res. 1983, 43, 5489–5492; [PubMed] [Google Scholar]
- 6d. Narimatsu H., Iwasaki H., Nakayama F., Ikehara Y., Kudo T., Nishihara S., Sugano K., Okura H., Fujita S., Hirohashi S., Cancer Res. 1998, 58, 512–518. [PubMed] [Google Scholar]
- 7.
- 7a. Dennis J. W., Laferte S., Waghorne C., Breitman M. L., Kerbel R. S., Science. 1987, 236, 582–585; [DOI] [PubMed] [Google Scholar]
- 7b. Iwai T., Kudo T., Kawamoto R., Kubota T., Togayachi A., Hiruma T., Okada T., Kawamoto T., Morozumi K., Narimatsu H., Proc. Natl. Acad. Sci. USA 2005, 102, 4572–4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Satomaa T., Heiskanen A., Leonardsson I., Angstrom J., Olonen A., Blomqvist M., Salovuori N., Haglund C., Teneberg S., Natunen J., Carpen O., Saarinen J., Cancer Res. 2009, 69, 5811–5819. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Zhang L., Luo S., Zhang B., MAbs. 2016, 8, 205–215; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. Zhang L., Luo S., Zhang B., MAbs. 2016, 8, 524–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.
- 10a. Angeloni S., Ridet J. L., Kusy N., Gao H., Crevoisier F., Guinchard S., Kochhar S., Sigrist H., Sprenger N., Glycobiology. 2005, 15, 31–41; [DOI] [PubMed] [Google Scholar]
- 10b. Pilobello K. T., Krishnamoorthy L., Slawek D., Mahal L. K., ChemBioChem 2005, 6, 985–989; [DOI] [PubMed] [Google Scholar]
- 10c. Zheng T., Peelen D., Smith L. M., J. Am. Chem. Soc. 2005, 127, 9982–9983; [DOI] [PubMed] [Google Scholar]
- 10d. Kuno A., Uchiyama N., Koseki-Kuno S., Ebe Y., Takashima S., Yamada M., Hirabayashi J., Nat. Methods 2005, 2, 851–856. [DOI] [PubMed] [Google Scholar]
- 11. Zou X., Yoshida M., Nagai-Okatani C., Iwaki J., Matsuda A., Tan B., Hagiwara K., Sato T., Itakura Y., Noro E., Kaji H., Toyoda M., Zhang Y., Narimatsu H., Kuno A., Sci. Rep. 2017, 7, 43560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Syed P., Gidwani K., Kekki H., Leivo J., Pettersson K., Lamminmaki U., Proteomics. 2016, 16, 1257–1265. [DOI] [PubMed] [Google Scholar]
- 13. Krishnamoorthy L., J. W. Bess, Jr. , Preston A. B., Nagashima K., Mahal L. K., Nat. Chem. Biol. 2009, 5, 244–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Etxebarria J., Calvo J., Martin-Lomas M., Reichardt N. C., ACS Chem. Biol. 2012, 7, 1729–1737. [DOI] [PubMed] [Google Scholar]
- 15. Pazos R., Echevarria J., Hernandez A., Reichardt N. C., Curr. Protoc. Cell. Biol. 2017, 76, 6 12 11–16 12 12. [DOI] [PubMed] [Google Scholar]
- 16. Chen S., LaRoche T., Hamelinck D., Bergsma D., Brenner D., Simeone D., Brand R. E., Haab B. B., Nat. Methods 2007, 4, 437–444. [DOI] [PubMed] [Google Scholar]
- 17. Yue T., Goldstein I. J., Hollingsworth M. A., Kaul K., Brand R. E., Haab B. B., Mol. Cell. Proteomics 2009, 8, 1697–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rosenfeld R., Bangio H., Gerwig G. J., Rosenberg R., Aloni R., Cohen Y., Amor Y., Plaschkes I., Kamerling J. P., Maya R. B., J. Biochem. Biophys. Methods 2007, 70, 415–426. [DOI] [PubMed] [Google Scholar]
- 19. Kuno A., Kato Y., Matsuda A., Kaneko M. K., Ito H., Amano K., Chiba Y., Narimatsu H., Hirabayashi J., Mol. Cell. Proteomics 2009, 8, 99–108. [DOI] [PubMed] [Google Scholar]
- 20. Meany D. L., L. Hackler, Jr. , Zhang H., Chan D. W., J. Proteome Res. 2011, 10, 1425–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Patwa T. H., Zhao J., Anderson M. A., Simeone D. M., Lubman D. M., Anal. Chem. 2006, 78, 6411–6421. [DOI] [PubMed] [Google Scholar]
- 22. Zhao J., Patwa T. H., Qiu W., Shedden K., Hinderer R., Misek D. E., Anderson M. A., Simeone D. M., Lubman D. M., J. Proteome Res. 2007, 6, 1864–1874. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. Rudiger H., Gabius H. J., Glycoconjugate J. 2001, 18, 589–613; [DOI] [PubMed] [Google Scholar]
- 23b. Hirabayashi J., Yamada M., Kuno A., Tateno H., Chem. Soc. Rev. 2013, 42, 4443–4458. [DOI] [PubMed] [Google Scholar]
- 24. Houles Astoul C., Peumans W. J., van Damme E. J., Barre A., Bourne Y., Rouge P., Biochem. J. 2002, 367, 817–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Barre A., Bourne Y., Van Damme E. J., Peumans W. J., Rouge P., Biochimie. 2001, 83, 645–651. [DOI] [PubMed] [Google Scholar]
- 26. Wright H. T., Sandrasegaram G., Wright C. S., J. Mol. Evol. 1991, 33, 283–294. [DOI] [PubMed] [Google Scholar]
- 27. Zhou S. M., Cheng L., Guo S. J., Zhu H., Tao S. C., Comb. Chem. High Throughput Screening 2011, 14, 711–719. [DOI] [PubMed] [Google Scholar]
- 28. Hirabayashi J., J. Biochem. 2008, 144, 139–147. [DOI] [PubMed] [Google Scholar]
- 29. Matsumura K., Higashida K., Hata Y., Kominami J., Nakamura-Tsuruta S., Hirabayashi J., Anal. Biochem. 2009, 386, 217–221. [DOI] [PubMed] [Google Scholar]
- 30. Nakamura-Tsuruta S., Kominami J., Kuno A., Hirabayashi J., Biochem. Biophys. Res. Commun. 2006, 347, 215–220. [DOI] [PubMed] [Google Scholar]
- 31. Imamura K., Takeuchi H., Yabe R., Tateno H., Hirabayashi J., J. Biochem. 2011, 150, 545–552. [DOI] [PubMed] [Google Scholar]
- 32. Rinderle S. J., Goldstein I. J., Matta K. L., Ratcliffe R. M., J. Biol. Chem. 1989, 264, 16123–16131. [PubMed] [Google Scholar]
- 33. Pilobello K. T., Agrawal P., Rouse R., Mahal L. K., Curr. Protoc. Chem. Biol. 2013, 5, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Iwaki J., Tateno H., Nishi N., Minamisawa T., Nakamura-Tsuruta S., Itakura Y., Kominami J., Urashima T., Nakamura T., Hirabayashi J., Biochim. Biophys. Acta 2011, 1810, 643–651. [DOI] [PubMed] [Google Scholar]
- 35. Ohyama Y., Kasai K., Nomoto H., Inoue Y., J. Biol. Chem. 1985, 260, 6882–6887. [PubMed] [Google Scholar]
- 36. Knibbs R. N., Goldstein I. J., Ratcliffe R. M., Shibuya N., J. Biol. Chem. 1991, 266, 83–88. [PubMed] [Google Scholar]
- 37. Crowley J. F., Goldstein I. J., Arnarp J., Lonngren J., Arch. Biochem. Biophys. 1984, 231, 524–533. [DOI] [PubMed] [Google Scholar]
- 38. Itakura Y., Nakamura-Tsuruta S., Kominami J., Sharon N., Kasai K., Hirabayashi J., J. Biochem. 2007, 142, 459–469. [DOI] [PubMed] [Google Scholar]
- 39. Fouquaert E., Smith D. F., Peumans W. J., Proost P., Balzarini J., Savvides S. N., Damme E. J., Biochem. Biophys. Res. Commun. 2009, 380, 260–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nakamura-Tsuruta S., Kominami J., Kamei M., Koyama Y., Suzuki T., Isemura M., Hirabayashi J., J. Biochem. 2006, 140, 285–291. [DOI] [PubMed] [Google Scholar]
- 41. Kaku H., Van Damme E. J., Peumans W. J., Goldstein I. J., Arch. Biochem. Biophys. 1990, 279, 298–304. [DOI] [PubMed] [Google Scholar]
- 42. Nakamura-Tsuruta S., Uchiyama N., Peumans W. J., Van Damme E. J., Totani K., Ito Y., Hirabayashi J., FEBS J. 2008, 275, 1227–1239. [DOI] [PubMed] [Google Scholar]
- 43. Tateno H., Nakamura-Tsuruta S., Hirabayashi J., Glycobiology. 2009, 19, 527–536. [DOI] [PubMed] [Google Scholar]
- 44. Stowell S. R., Dias-Baruffi M., Penttila L., Renkonen O., Nyame A. K., Cummings R. D., Glycobiology. 2004, 14, 157–167. [DOI] [PubMed] [Google Scholar]
- 45. Yan L., Wilkins P. P., Alvarez-Manilla G., Do S. I., Smith D. F., Cummings R. D., Glycoconjugate J. 1997, 14, 45–55. [DOI] [PubMed] [Google Scholar]
- 46. Green E. D., Baenziger J. U., J. Biol. Chem. 1987, 262, 12018–12029. [PubMed] [Google Scholar]
- 47. Matsuda T., Kabat E. A., Surolia A., Mol. Immunol. 1989, 26, 189–195. [DOI] [PubMed] [Google Scholar]
- 48. Yokoyama K., Terao T., Osawa T., Biochim. Biophys. Acta 1978, 538, 384–396. [DOI] [PubMed] [Google Scholar]
- 49. Yabe R., Itakura Y., Nakamura-Tsuruta S., Iwaki J., Kuno A., Hirabayashi J., Biochem. Biophys. Res. Commun. 2009, 384, 204–209. [DOI] [PubMed] [Google Scholar]
- 50. Debray H., Decout D., Strecker G., Spik G., Montreuil J., Eur. J. Biochem. 1981, 117, 41–55. [DOI] [PubMed] [Google Scholar]
- 51. Yamashita K., Umetsu K., Suzuki T., Ohkura T., Biochemistry. 1992, 31, 11647–11650. [DOI] [PubMed] [Google Scholar]
- 52. Shibuya N., Goldstein I. J., Shafer J. A., Peumans W. J., Broekaert W. F., Arch. Biochem. Biophys. 1986, 249, 215–224. [DOI] [PubMed] [Google Scholar]
- 53. Baldus S. E., Thiele J., Park Y. O., Hanisch F. G., Bara J., Fischer R., Glycoconjugate J. 1996, 13, 585–590. [DOI] [PubMed] [Google Scholar]
- 54. Puri K. D., Gopalakrishnan B., Surolia A., FEBS Lett. 1992, 312, 208–212. [DOI] [PubMed] [Google Scholar]
- 55. Martinez-Alarcon D., Blanco-Labra A., Garcia-Gasca T., Int. J. Mol. Sci. 2018, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.
- 56a. Oliveira C., Teixeira J. A., Domingues L., Front. Plant Sci. 2014, 5, 390; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56b. Zhang W., van Eijk M., Guo H., van Dijk A., Bleijerveld O. B., Verheije M. H., Wang G., Haagsman H. P., Veldhuizen E. J., Immunobiology. 2017, 222, 518–528. [DOI] [PubMed] [Google Scholar]
- 57.
- 57a. Hsu K. L., Gildersleeve J. C., Mahal L. K., Mol. BioSyst. 2008, 4, 654–662; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57b. Saarela S., Taira S., Nurmiaho-Lassila E. L., Makkonen A., Rhen M., J. Bacteriol. 1995, 177, 1477–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Avichezer D., Katcoff D. J., Garber N. C., Gilboa-Garber N., J. Biol. Chem. 1992, 267, 23023–23027. [PubMed] [Google Scholar]
- 59. Mitchell E. P., Sabin C., Snajdrova L., Pokorna M., Perret S., Gautier C., Hofr C., Gilboa-Garber N., Koca J., Wimmerova M., Imberty A., Proteins. 2005, 58, 735–746. [DOI] [PubMed] [Google Scholar]
- 60. Haslam D. B., Boren T., Falk P., Ilver D., Chou A., Xu Z., Normark S., Mol. Microbiol. 1994, 14, 399–409. [DOI] [PubMed] [Google Scholar]
- 61. Sudakevitz D., Kostlanova N., Blatman-Jan G., Mitchell E. P., Lerrer B., Wimmerova M., Katcoff D. J., Imberty A., Gilboa-Garber N., Mol. Microbiol. 2004, 52, 691–700. [DOI] [PubMed] [Google Scholar]
- 62. Maenuma K., Yim M., Komatsu K., Hoshino M., Takahashi Y., Bovin N., Irimura T., Proteomics. 2008, 8, 3274–3283. [DOI] [PubMed] [Google Scholar]
- 63. Yabe R., Suzuki R., Kuno A., Fujimoto Z., Jigami Y., Hirabayashi J., J. Biochem. 2007, 141, 389–399. [DOI] [PubMed] [Google Scholar]
- 64. Hu D., Tateno H., Kuno A., Yabe R., Hirabayashi J., J. Biol. Chem. 2012, 287, 20313–20320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Propheter D. C., Hsu K. L., Mahal L. K., ChemBioChem 2010, 11, 1203–1207. [DOI] [PubMed] [Google Scholar]
- 66. Propheter D. C., Mahal L. K., Mol. BioSyst. 2011, 7, 2114–2117. [DOI] [PubMed] [Google Scholar]
- 67. Sang S., Wang Y., Feng Q., Wei Y., Ji J., Zhang W., Crit. Rev. Biotechnol. 2015, 36, 465–481. [DOI] [PubMed] [Google Scholar]
- 68.
- 68a. Fry S. A., Afrough B., Lomax-Browne H. J., Timms J. F., Velentzis L. S., Leathem A. J., Glycobiology. 2011, 21, 1060–1070; [DOI] [PubMed] [Google Scholar]
- 68b. Roy B., Chattopadhyay G., Mishra D., Das T., Chakraborty S., Maiti T. K., Biomicrofluidics. 2014, 8, 034107; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68c. Matsuda A., Kuno A., Nakagawa T., Ikehara Y., Irimura T., Yamamoto M., Nakanuma Y., Miyoshi E., Nakamori S., Nakanishi H., Viwatthanasittiphong C., Srivatanakul P., Miwa M., Shoda J., Narimatsu H., Anal. Chem. 2015, 87, 7274–7281; [DOI] [PubMed] [Google Scholar]
- 68d. Chan K., Ng T. B., Protein Pept. Lett. 2010, 17, 1417–1425. [DOI] [PubMed] [Google Scholar]
- 69. Ebe Y., Kuno A., Uchiyama N., Koseki-Kuno S., Yamada M., Sato T., Narimatsu H., Hirabayashi J., J. Biochem. 2006, 139, 323–327. [DOI] [PubMed] [Google Scholar]
- 70. Pilobello K. T., Slawek D. E., Mahal L. K., Proc. Natl. Acad. Sci. USA 2007, 104, 11534–11539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.
- 71a. Hirabayashi J., Kuno A., Tateno H., Electrophoresis. 2011, 32, 1118–1128; [DOI] [PubMed] [Google Scholar]
- 71b. Batista B. S., Eng W. S., Pilobello K. T., Hendricks-Munoz K. D., Mahal L. K., J. Proteome Res. 2011, 10, 4624–4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kuno A., Ikehara Y., Tanaka Y., Angata T., Unno S., Sogabe M., Ozaki H., Ito K., Hirabayashi J., Mizokami M., Narimatsu H., Clin. Chem. 2011, 57, 48–56. [DOI] [PubMed] [Google Scholar]
- 73. Cao Z., Partyka K., McDonald M., Brouhard E., Hincapie M., Brand R. E., Hancock W. S., Haab B. B., Anal. Chem. 2013, 85, 1689–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hirabayashi J., Kuno A., Tateno H., Top. Curr. Chem. 2015, 367, 105–124. [DOI] [PubMed] [Google Scholar]
- 75. Koshi Y., Nakata E., Yamane H., Hamachi I., J. Am. Chem. Soc. 2006, 128, 10413–10422. [DOI] [PubMed] [Google Scholar]
- 76. Duverger E., Lamerant-Fayel N., Frison N., Monsigny M., Methods Mol. Biol. 2010, 627, 157–178. [DOI] [PubMed] [Google Scholar]
- 77. Nand A., Singh V., Wang P., Na J., Zhu J., Anal. Biochem. 2014, 465, 114–120. [DOI] [PubMed] [Google Scholar]
- 78. Geuijen K. P., Halim L. A., Schellekens H., Schasfoort R. B., Wijffels R. H., Eppink M. H., Anal. Chem. 2015, 87, 8115–8122. [DOI] [PubMed] [Google Scholar]
- 79. Peelen D., Kodoyianni V., Lee J., Zheng T., Shortreed M. R., Smith L. M., J. Proteome Res. 2006, 5, 1580–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Chen S., Zheng T., Shortreed M. R., Alexander C., Smith L. M., Anal. Chem. 2007, 79, 5698–5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hu S., Wong D. T., Proteomics Clin. Appl. 2009, 3, 148–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Pirozynski M., Respir. Med. 2006, 100, 2073–2084. [DOI] [PubMed] [Google Scholar]
- 83.
- 83a. Bharti A., Ma P. C., Salgia R., Mass Spectrom. Rev. 2007, 26, 451–466; [DOI] [PubMed] [Google Scholar]
- 83b. Li X., Asmitananda T., Gao L., Gai D., Song Z., Zhang Y., Ren H., Yang T., Chen T., Chen M., Neoplasma. 2012, 59, 500–507; [DOI] [PubMed] [Google Scholar]
- 83c. Li X., Lu J., Ren H., Chen T., Gao L., Di L., Song Z., Zhang Y., Yang T., Thakur A., Zhou S. F., Yin Y., Chen M., Mol. Clin. Oncol. 2013, 1, 153–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Shi Y. Q., He Q., Zhao Y. J., Wang E. H., Wu G. P., Cytotechnology. 2013, 65, 355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Liang Y., Han P., Wang T., Ren H., Gao L., Shi P., Zhang S., Yang A., Li Z., Chen M., Clin. Proteomics 2019, 16, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Liang Y., Ma T., Thakur A., Yu H., Gao L., Shi P., Li X., Ren H., Jia L., Zhang S., Li Z., Chen M., Glycobiology. 2015, 25, 331–340. [DOI] [PubMed] [Google Scholar]
- 87. Hirao Y., Matsuzaki H., Iwaki J., Kuno A., Kaji H., Ohkura T., Togayachi A., Abe M., Nomura M., Noguchi M., Ikehara Y., Narimatsu H., J. Proteome Res. 2014, 13, 4705–4716. [DOI] [PubMed] [Google Scholar]
- 88. Huang W. L., Li Y. G., Lv Y. C., Guan X. H., Ji H. F., Chi B. R., World J. Gastroenterol. 2014, 20, 5474–5482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Li T., Mo C., Qin X., Li S., Liu Y., Liu Z., Clin. Lab. 2018, 64, 153–161. [DOI] [PubMed] [Google Scholar]
- 90. Qiu Y., Patwa T. H., Xu L., Shedden K., Misek D. E., Tuck M., Jin G., Ruffin M. T., Turgeon D. K., Synal S., Bresalier R., Marcon N., Brenner D. E., Lubman D. M., J. Proteome Res. 2008, 7, 1693–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Li Y., Wen T., Zhu M., Li L., Wei J., Wu X., Guo M., Liu S., Zhao H., Xia S., Huang W., Wang P., Wu Z., Zhao L., Shui W., Li Z., Yin Z., Mol. BioSyst. 2013, 9, 1877–1887. [DOI] [PubMed] [Google Scholar]
- 92. Nakajima K., Inomata M., Iha H., Hiratsuka T., Etoh T., Shiraishi N., Kashima K., Kitano S., Cancer Med. 2015, 4, 293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Sunderic M., Sediva A., Robajac D., Miljus G., Gemeiner P., Nedic O., Katrlik J., Biotechnol. Appl. Biochem. 2016, 63, 457–464. [DOI] [PubMed] [Google Scholar]
- 94. Tian R., Zhang H., Chen H., Liu G., Wang Z., Adv Sci (Weinh). 2018, 5, 1800214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Li S., Mo C., Peng Q., Kang X., Sun C., Jiang K., Huang L., Lu Y., Sui J., Qin X., Liu Y., PLoS One. 2013, 8, e71273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Wang H., Li H., Zhang W., Wei L., Yu H., Yang P., Proteomics. 2014, 14, 78–86. [DOI] [PubMed] [Google Scholar]
- 97. Jiang K., Shang S., Li W., Guo K., Qin X., Zhang S., Liu Y., Glycoconjugate J. 2015, 32, 657–664. [DOI] [PubMed] [Google Scholar]
- 98. Liu T., Shang S., Li W., Qin X., Sun L., Zhang S., Liu Y., Front Physiol. 2017, 8, 472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Qin Y., Zhong Y., Ma T., Wu F., Wu H., Yu H., Huang C., Li Z., Glycoconjugate J. 2016, 33, 125–136. [DOI] [PubMed] [Google Scholar]
- 100. Xiang T., Yang G., Liu X., Zhou Y., Fu Z., Lu F., Gu J., Taniguchi N., Tan Z., Chen X., Xie Y., Guan F., Zhang X. L., Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1036–1045. [DOI] [PubMed] [Google Scholar]
- 101. Zhou S. M., Cheng L., Guo S. J., Wang Y., Czajkowsky D. M., Gao H., Hu X. F., Tao S. C., Breast Cancer Res. 2015, 17, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Tian Y., Almaraz R. T., Choi C. H., Li Q. K., Saeui C., Li D., Shah P., Bhattacharya R., Yarema K. J., Zhang H., Clin. Proteomics 2015, 12, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Zhu J., He J., Liu Y., Simeone D. M., Lubman D. M., J. Proteome Res. 2012, 11, 2272–2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.T. Sakaue, H. Koga, H. Iwamoto, T. Nakamura, Y. Ikezono, M. Abe, F. Wada, A. Masuda, T. Tanaka, M. Fukahori, T. Ushijima, Y. Mihara, Y. Naitou, Y. Okabe, T. Kakuma, K. Ohta, K. I. Nakamura, T. Torimura, Med Mol Morphol 2019. [DOI] [PubMed]
- 105. Yang G., Tan Z., Lu W., Guo J., Yu H., Yu J., Sun C., Qi X., Li Z., Guan F., J. Proteome Res. 2015, 14, 639–653. [DOI] [PubMed] [Google Scholar]
- 106. Sogabe M., Nozaki H., Tanaka N., Kubota T., Kaji H., Kuno A., Togayachi A., Gotoh M., Nakanishi H., Nakanishi T., Mikami M., Suzuki N., Kiguchi K., Ikehara Y., Narimatsu H., J. Proteome Res. 2014, 13, 1624–1635. [DOI] [PubMed] [Google Scholar]
- 107. Itakura Y., Sasaki N., Toyoda M., Aging (Albany NY). 2018, 10, 2190–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Miura Y., Hashii N., Ohta Y., Itakura Y., Tsumoto H., Suzuki J., Takakura D., Abe Y., Arai Y., Toyoda M., Kawasaki N., Hirose N., Endo T., Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 1462–1471. [DOI] [PubMed] [Google Scholar]
- 109. Kolodziejczyk J., Blixt O., Olejnik B., Zimmer M., Ferens-Sieczkowska M., Andrologia. 2018, 50, e13018. [DOI] [PubMed] [Google Scholar]
- 110. Xin A., Cheng L., Diao H., Wu Y., Zhou S., Shi C., Sun Y., Wang P., Duan S., Zheng J., Wu B., Yuan Y., Gu Y., Chen G., Sun X., Shi H., Tao S., Zhang Y., Sci. Rep. 2016, 6, 20249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Toyoda M., Yamazaki-Inoue M., Itakura Y., Kuno A., Ogawa T., Yamada M., Akutsu H., Takahashi Y., Kanzaki S., Narimatsu H., Hirabayashi J., Umezawa A., Genes Cells. 2011, 16, 1–11. [DOI] [PubMed] [Google Scholar]
- 112. Saito S., Hiemori K., Kiyoi K., Tateno H., Sci. Rep. 2018, 8, 3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Hasehira K., Hirabayashi J., Tateno H., Glycoconjugate J. 2017, 34, 797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Moriwaki K., Okudo K., Haraguchi N., Takeishi S., Sawaki H., Narimatsu H., Tanemura M., Ishii H., Mori M., Miyoshi E., Cancer Sci. 2011, 102, 1164–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.
- 115a. El-Akawi Z. J., Abu-Awad A. M., Sharara A. M., Khader Y., Neuroendocrinol. Lett. 2010, 31, 113–116; [PubMed] [Google Scholar]
- 115b. El-Akawi Z. J., Al-Hindawi F. K., Bashir N. A., Neuroendocrinol. Lett. 2008, 29, 482–484. [PubMed] [Google Scholar]
- 116. Hicks S., Br J. Nurs. 2001, 10, 529–536. [DOI] [PubMed] [Google Scholar]
- 117. Shu J., Yu H., Li X., Zhang D., Liu X., Du H., Zhang J., Yang Z., Xie H., Li Z., Oncotarget. 2017, 8, 35718–35727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Parkin D. M., Bray F., Ferlay J., Pisani P., Ca-Cancer J. Clin. 2005, 55, 74–108. [DOI] [PubMed] [Google Scholar]
- 119. Fletcher R. H., Ann. Intern. Med. 1986, 104, 66–73. [DOI] [PubMed] [Google Scholar]
- 120.
- 120a. Duffy M. J., Ann. Clin. Biochem. 1998, 35, 364–370; [DOI] [PubMed] [Google Scholar]
- 120b. Kuusela P., Haglund C., Roberts P. J., Br. J. Cancer 1991, 63, 636–640; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120c. Ward U., Primrose J. N., Finan P. J., Perren T. J., Selby P., Purves D. A., Cooper E. H., Br. J. Cancer 1993, 67, 1132–1135; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120d. Holten-Andersen M. N., Christensen I. J., Nielsen H. J., Stephens R. W., Jensen V., Nielsen O. H., Sorensen S., Overgaard J., Lilja H., Harris A., Murphy G., Brunner N., Clin. Cancer Res. 2002, 8, 156–164. [PubMed] [Google Scholar]
- 121. Bertuccio P., Turati F., Carioli G., Rodriguez T., La Vecchia C., Malvezzi M., Negri E., J. Hepatol. 2017, 67, 302–309. [DOI] [PubMed] [Google Scholar]
- 122. Dai Z., Zhou J., Qiu S. J., Liu Y. K., Fan J., Electrophoresis. 2009, 30, 2957–2966. [DOI] [PubMed] [Google Scholar]
- 123. Liu T. H., Liu D. H., Mo C. J., Sun L., Liu X. X., Li W., Zhang S., Liu Y. K., Guo K., Am. J. Transl Res. 2016, 8, 4250–4264. [PMC free article] [PubMed] [Google Scholar]
- 124. Arndt N. X., Tiralongo J., Madge P. D., von Itzstein M., Day C. J., J. Cell. Biochem. 2011, 112, 2230–2240. [DOI] [PubMed] [Google Scholar]
- 125. Guo J., Liu C., Zhou X., Xu X., Deng L., Li X., Guan F., Int. J. Mol. Sci. 2017, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Tan Z., Wang C., Li X., Guan F., Front. Physiol. 2018, 9, 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Terao N., Takamatsu S., Minehira T., Sobajima T., Nakayama K., Kamada Y., Miyoshi E., World J. Gastroenterol. 2015, 21, 3876–3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Guo J., Li X., Tan Z., Lu W., Yang G., Guan F., Molecules. 2014, 19, 20073–20090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Zhao R., Liu X., Wang Y., Jie X., Qin R., Qin W., Zhang M., Tai H., Yang C., Li L., Peng P., Shao M., Zhang X., Wu H., Ruan Y., Xu C., Ren S., Gu J., Clin. Proteomics 2016, 13, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Takeshita M., Kuno A., Suzuki K., Matsuda A., Shimazaki H., Nakagawa T., Otomo Y., Kabe Y., Suematsu M., Narimatsu H., Takeuchi T., Arthritis Res. Ther. 2016, 18, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Yang L., Yang Z., Cheng L., Cheng J., Sun Y., Li W., Song K., Huang W., Yin Y., Tao S., Zhang Q., Proteomics Clin Appl. 2018, 12, e1800030. [DOI] [PubMed] [Google Scholar]
- 132. Narimatsu H., Expert Rev. Proteomics 2015, 12, 683–693. [DOI] [PubMed] [Google Scholar]
- 133. Shinzaki S., Kuroki E., Iijima H., Tatsunaka N., Ishii M., Fujii H., Kamada Y., Kobayashi T., Shibukawa N., Inoue T., Tsujii M., Takeishi S., Mizushima T., Ogata A., Naka T., Plevy S. E., Takehara T., Miyoshi E., Inflamm Bowel Dis. 2013, 19, 321–331. [DOI] [PubMed] [Google Scholar]
- 134. Inoue K., Wada J., Eguchi J., Nakatsuka A., Teshigawara S., Murakami K., Ogawa D., Terami T., Katayama A., Tone A., Iseda I., Hida K., Yamada M., Ogawa T., Makino H., PLoS One. 2013, 8, e77118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Zhu H., Liu M., Yu H., Liu X., Zhong Y., Shu J., Fu X., Cai G., Chen X., Geng W., Yang X., Wu M., Li Z., Zhang D., J. Diabetes Res. 2017, 2017, 5728087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Yang X. L., Yu H. J., Zhu H. Y., Zheng Y., Han Q. X., Cai G. Y., Chen X. M., Chin. Med. J. (Engl). 2018, 131, 180–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Yamagata H., Uchida S., Matsuo K., Harada K., Kobayashi A., Nakashima M., Higuchi F., Watanuki T., Matsubara T., Watanabe Y., J. Affective Disord. 2017, 233, 79–85. [DOI] [PubMed] [Google Scholar]
- 138. Itakura Y., Sasaki N., Kami D., Gojo S., Umezawa A., Toyoda M., Cell Biosci. 2016, 6, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Tateno H., Toyota M., Saito S., Onuma Y., Ito Y., Hiemori K., Fukumura M., Matsushima A., Nakanishi M., Ohnuma K., Akutsu H., Umezawa A., Horimoto K., Hirabayashi J., Asashima M., J. Biol. Chem. 2011, 286, 20345–20353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Tateno H., Matsushima A., Hiemori K., Onuma Y., Ito Y., Hasehira K., Nishimura K., Ohtaka M., Takayasu S., Nakanishi M., Ikehara Y., Ohnuma K., Chan T., Toyoda M., Akutsu H., Umezawa A., Asashima M., Hirabayashi J., Stem Cells Transl. Med. 2013, 2, 265–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Hirabayashi J., Tateno H., Onuma Y., Ito Y., Adv. Healthcare Mater. 2015, 4, 2520–2529. [DOI] [PubMed] [Google Scholar]
- 142. Tateno H., Saito S., Hiemori K., Kiyoi K., Hasehira K., Toyoda M., Onuma Y., Ito Y., Akutsu H., Hirabayashi J., Glycobiology. 2016, 26, 1328–1337. [DOI] [PubMed] [Google Scholar]
- 143. Liu W., Yan X., Wang Y., Rao Y., Yu H., Cui J., Xie X., Sun M., Yin L., Li H., Chen F., Int. J. Mol. Med. 2018, 41, 293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]