

Abstract
Loss of function of TREM2, a key receptor selectively expressed by microglia in the brain, contributes to the development of Alzheimer’s disease (AD). We therefore examined whether soluble TREM2 (sTREM2) concentrations in cerebrospinal fluid (CSF) were associated with reduced rates of cognitive decline and clinical progression in subjects with AD or mild cognitive impairment (MCI). We measured sTREM2 in CSF samples from 385 elderly subjects, including cognitively normal controls, individuals with MCI, and subjects with AD dementia (follow-up period: mean, 4 years; range 1.5 to 11.5 years). In subjects with AD defined by evidence of CSF Aβ1–42 (amyloid β-peptide 1 to 42; A+) and CSF p-tau181 (tau phosphorylated on amino acid residue 181; T+), higher sTREM2 concentrations in CSF at baseline were associated with attenuated decline in memory and cognition. When analyzed in clinical subgroups, an association between higher CSF sTREM2 concentrations and subsequent reduced memory decline was consistently observed in individuals with MCI or AD dementia, who were positive for CSF Aβ1–42 and CSF p-tau181 (A+T+). Regarding clinical progression, a higher ratio of CSF sTREM2 to CSF p-tau181 concentrations predicted slower conversion from cognitively normal to symptomatic stages or from MCI to AD dementia in the subjects who were positive for CSF Aβ1–42 and CSF p-tau181. These results suggest that sTREM2 is associated with attenuated cognitive and clinical decline, a finding with important implications for future clinical trials targeting the innate immune response in AD.
INTRODUCTION
Evidence from genome-wide association studies demonstrated that alterations of the innate immune system play a crucial role in the development of AD dementia (1, 2). In particular, rare missense variants in the gene encoding the triggering receptor expressed on myeloid cells 2 (TREM2) are associated with a two- to threefold increase in the odds ratio of developing AD (3, 4). This is comparable to that for the e4 allele of the gene encoding apolipoprotein E (ApoE), the strongest genetic risk factor for sporadic AD (5). TREM2 binds to the DNAX-activating protein of 12 kDa (DAP12), a receptor-signaling complex within the brain that is exclusively expressed by microglia (6, 7). TREM2 signaling serves as a key regulator that allows microglia to switch from a homeostatic to a disease-associated state (8, 9). Disease-associated microglia are characterized by up-regulated gene clusters triggering essential microglial functions such as chemotaxis, phagocytosis, and other functions that together may modulate amyloid plaque seeding and compaction (10–16). However, despite the evidence that TREM2 enhances the scavenger function of microglia and that disease-associated TREM2 variants are apparently loss-of-function mutations (13, 15), chronic microglial activation has been associated with the release of proinflammatory cytokines and may be detrimental (17). Therefore, an important question is whether TREM2-related microglial activation protects or exacerbates the development of dementia in patients with AD (18).
The translation of preclinical findings on TREM2 has recently been facilitated by the development of a soluble TREM2 (sTREM2) marker in cerebrospinal fluid (CSF) (13, 19–21). The extracellular domain of TREM2 protein is shed by ADAM10/17 cleavage and released in its soluble form into the interstitial fluid of the brain (13, 22, 23). Given that probably only full-length TREM2 expressed on the microglial surface is signaling competent and that TREM2 expression in microglia is greatly increased during neurodegeneration (9, 16), sTREM2 in biological fluids serves as a readout for TREM2-triggered microglial activity (21, 24–26). In cross-sectional studies of patients with late-onset AD, we and others have previously reported that CSF sTREM2 increases in a disease stage-dependent fashion (20, 21, 24, 25), reaching a peak during early symptomatic stages (21). This was confirmed in patients with autosomal dominant AD where we observed an increase in CSF sTREM2 about 5 years before the estimated onset of symptoms (20). A recent study assessed the association between CSF sTREM2 and regional gray matter volume in subjects with early-stage AD who had mild cognitive impairment (MCI) and CSF biomarker evidence of AD. The study reported that CSF sTREM2 was associated with higher gray matter volume in this group when controlled for CSF amyloid β-peptide (Aβ) and tau (27), suggesting protective effects. However, that study had a cross-sectional design and was not conclusive with regard to beneficial effects because higher gray matter volume might also have resulted from inflammation-related swelling and cognitive function was not investigated.
Here, we examined a large well-characterized sample of 385 elderly participants from the Alzheimer’s Disease Neuroimaging Initiative (ADNI). We investigated whether baseline CSF sTREM2 concentrations were predictive of subsequent changes in hippocampal volume, cognitive changes, and clinical progression to MCI or AD dementia. Our primary hypothesis was that in subjects with biomarker evidence of AD pathology, higher CSF sTREM2 might be associated with a slower rate of decline in episodic memory or cognition during an observation period of up to 11.5 years (mean, 4 years). We tested our hypothesis in subjects classified as AD according to the AT/N framework recently proposed by the National Institute on Aging and Alzheimer’s Association (NIA-AA) (28). Subjects were classified on the basis of biomarkers to determine abnormal deposition of Aβ (A) and pathological tau (T), regardless of clinical symptoms. We focused on subjects with AD based on CSF marker evidence of Aβ and tau pathology (A+T+). To assess whether any CSF sTREM2 effects varied by disease stage, we conducted an exploratory subanalysis of CSF sTREM2 concentrations in clinically defined subgroups of AD with mild cognitive impairment (A+T+) or AD with dementia (AD dementia A+T+) (29).
RESULTS
CSF sTREM2 concentrations are associated with reduced cognitive decline in AD
We assessed CSF sTREM2 concentrations in individuals who were cognitively normal (CN) with normal amounts of CSF Aβ and tau (CN A−T−) and in patients defined as having AD according to their CSF Aβ and tau concentrations (AD A+T+), regardless of their clinical symptoms (28). Characteristics of patients with AD are summarized in Table 1. In the AD A+T+ group, we found increased CSF sTREM2 concentrations (P = 0.002; Fig. 1A) and a decreased ratio of CSF sTREM2 to p-tau181 (P < 0.001; Fig. 1B) compared to the cognitively normal control group (CN A−T−).
Table 1. Subject characteristics for each group.
Values are shown as mean, with SD in parentheses.
| CN A−T− | All A+T+ | MCI A+T+ | AD dementia A+T+ | |
|---|---|---|---|---|
| Number | 100 | 285 | 184 | 66 |
| Age in years | 72.8 (5.36) | 73.53 (7.39) | 72.9 (7.11) | 73.6 (8.51) |
| Gender (F/M) | 45/55 | 126/159 | 77/107 | 32/34 |
| Years of education | 16.1 (2.80) | 15.91 (2.88) | 16.1 (2.81) | 15.2 (3.11)* |
| Clinical follow-up in years | 4.82 (2.40) | 3.69 (2.00)* | 4.05 (1.94)* | 2.18 (0.473)* |
| ADNI-MEM score | 1.11 (0.58) | −0.15 (0.74)* | −0.07 (0.57)* | −0.89 (0.5)* |
| ADAS13 score | 9.1 (4.32) | 20.19 (8.53)* | 18.67 (6.32)* | 29.66 (6.83)* |
| Aβ1–42 (pg/ml) | 1527 (311) | 639.62 (172.89)* | 657 (167)* | 552 (166)* |
| p-tau181 (pg/ml) | 16.4 (2.95) | 37.76 (13.12)* | 38.0 (13.5)* | 39.9 (13.5)* |
| Total tau (pg/ml) | 186.6 (33.3) | 370.9 (115.2)* | 374.6 (120.1)* | 386.9 (113.5)* |
| CSF sTREM2 (pg/ml) | 3762 (1841) | 4540 (2422)* | 4452 (2518)* | 4608 (2201)* |
Significantly (P < 0.05) different from cognitively normal controls (CN A−T−) via post hoc Tukey test.
Fig. 1. Baseline values of CSF sTREM2 concentrations and CSF sTREM2–to–p-tau181 ratio.
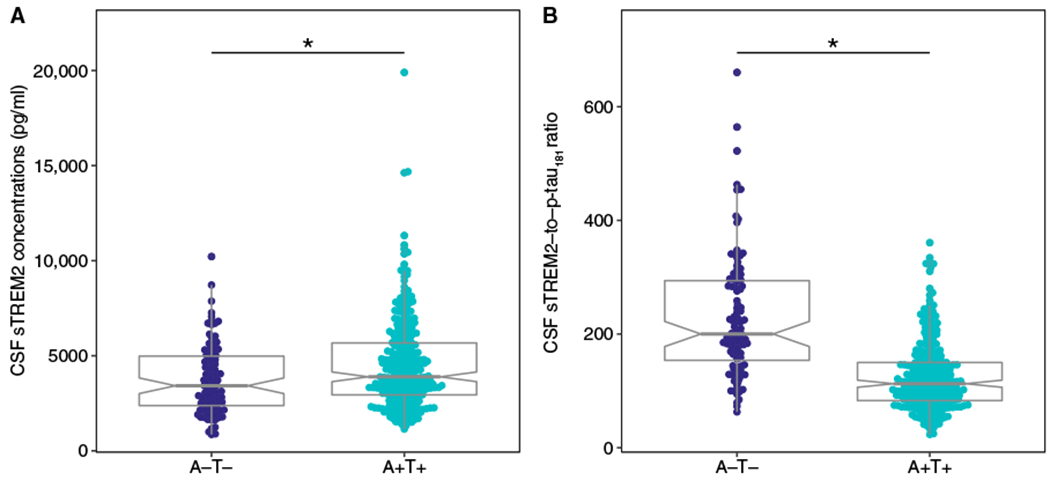
Box plots show CSF sTREM2 concentrations (A) and the ratio of concentrations of CSF sTREM2 to p-tau181 (B). CSF samples were obtained from 100 cognitively normal control subjects (CN), who were negative for CSF Aβ1–42 and CSF p-tau181 (A−T−). CSF samples were also obtained from 285 subjects who were positive for CSF Aβ1–42 and CSF p-tau181 (A+T+), including those who were cognitively normal or who had MCI or AD dementia. For both measurements, the group differences tested by ANCOVA were statistically significant, which remained after removal of the three outliers from the A+T+ group. Outliers were defined according to the Tukey criterion (66). *P < 0.01.
We next tested whether higher baseline CSF sTREM2 concentrations (used as a continuous measure) predicted attenuated future cognitive decline in the AD A+T+ group. To this end, we computed a linear mixed-effects regression analysis to test the interaction of CSF sTREM2 concentrations × time on cognitive scores. Note that in all regression models, the variable “time” refers to the follow-up duration of the neuropsychological testing. CSF sTREM2 concentrations were assessed only at baseline. We controlled for concentrations of CSF Aβ1–42 and CSF p-tau181 among other confounding variables including clinical syndrome (CN, MCI, or dementia), age, gender, and education. Our rationale was that CSF sTREM2 concentrations would increase in association with greater neurodegeneration, potentially reflecting higher microglial activity in response to neurotoxic amyloid and tau pathology (8, 9). Thus, we addressed the pivotal question of whether higher CSF sTREM2 concentrations at a given stage of AD pathology (measured by CSF Aβ1–42 and CSF p-tau181) were associated with less decline in memory and global cognition. Results of the regression analyses including the effect size d and P values from both parametric and nonparametric tests are displayed in Table 2. Specifically, for the prediction of the decline in episodic memory as measured by the composite score ADNI-Memory (MEM) (30), we observed a significant interaction effect of time × CSF sTREM2 concentration (β = 0.071, P = 0.001), where higher baseline CSF sTREM2 concentrations were associated with a slower decline in ADNI-MEM scores over time (Fig. 2A). For changes in global cognition as measured by the 13-tasks version of the AD assessment scale (ADAS13), higher CSF sTREM2 concentrations were associated with a slower increase in ADAS13 scores (β = −0.076, P = 0.034; Fig. 2B), suggesting a potential beneficial effect of CSF sTREM2 concentrations on cognition, although that P value missed the Bonferroni corrected threshold P < 0.0125.
Table 2. Regression analyses for the effects of CSF sTREM2 concentrations on the rate of change in the ADNI-MEM and ADAS-cog scores and in hippocampal volume.
All models are controlled for age, gender, education, CSF p-tau181 × time, CSF-Aβ1–42 × time, CSF sTREM2, CSF Aβ1–42, CSF p-tau181 (fixed effects) plus random slope (time) and intercept. Regular P values were assessed using Satterthwaite’s method. For nonparametric confirmation of results, P values were further assessed using permutation testing with 1000 iterations (P-perm.). B, regression coefficient; CI, confidence interval; T value, t test statistic; Cohen’s d, effect size d; P-perm., P value from permutation test.
| ADNI-MEM | Interaction term | B (SE) | 95% CI | T value | Cohen’s d | P | P-perm. |
|---|---|---|---|---|---|---|---|
| CN A−T− | CSF sTREM2 × time | 0.036 (0.046) | −0.052–0.125 | <1 | NA | 0.429 | 0.395 |
| CSF sTREM2/p-tau × time | 0.043 (0.043) | −0.042–0.127 | <1 | NA | 0.331 | 0.312 | |
| A11 A+T+ | CSF sTREM2 × time | 0.071 (0.022) | 0.028–0.113 | 3.258 | 0.43 | 0.001 | 0.020 |
| CSF sTREM2/p-tau × time | 0.088 (0.020) | 0.049–0.128 | 4.354 | 0.55 | <0.001 | <0.001 | |
| MCI A+T+ | CSF sTREM2 × time | 0.071 (0.028) | 0.017–0.125 | 2.581 | 0.41 | 0.011 | 0.046 |
| CSF sTREM2/p-tau × time | 0.092 (0.025) | 0.043–0.141 | 3.663 | 0.57 | <0.001 | 0.004 | |
| AD dementia A+T+ | CSF sTREM2 × time | 0.050 (0.023) | 0.006–0.094 | 2.194 | 0.62 | 0.033 | 0.027 |
| CSF sTREM2/p-tau × time | 0.059 (0.022) | 0.016–0.103 | 2.671 | 0.73 | 0.009 | 0.006 | |
| ADAS13 | Interaction term | B (SE) | 95% CI | T value | Cohen’s d | P | P-perm. |
| CN A−T− | CSF sTREM2 × time | 0.087 (0.047) | −0.006–0.179 | 1.849 | NA | 0.074 | 0.084 |
| CSF sTREM2/p-tau × time | 0.070 (0.043) | −0.015–0.157 | 1.626 | NA | 0.115 | 0.151 | |
| All A+T+ | CSF sTREM2 × time | −0.076 (0.035) | −0.145–−0.007 | −2.137 | 0.28 | 0.034 | 0.045 |
| CSF sTREM2/p-tau × time | −0.102 (0.033) | −0.167–−0.037 | −3.078 | 0.38 | 0.002 | 0.023 | |
| MCI A+T+ | CSF sTREM2 × time | −0.076 (0.046) | −0.165–0.013 | −1.672 | NA | 0.096 | 0.205 |
| CSF sTREM2/p-tau × time | −0.105 (0.042) | −0.186–−0.024 | −2.530 | 0.39 | 0.012 | 0.033 | |
| AD dementia A+T+ | CSF sTREM2 × time | −0.047 (0.054) | −0.151–0.056 | <1 | NA | 0.384 | 0.352 |
| CSF sTREM2/p-tau × time | −0.076 (0.054) | −0.181–0.029 | −1.402 | NA | 0.166 | 0.137 | |
| Hippocampal volume | Interaction term | B (SE) | 95% CI | T value | Cohen’s d | P | P-perm. |
| CN A−T− | CSF sTREM2 × time | −0.010 (0.013) | −0.046–0.016 | <1 | NA | 0.445 | 0.382 |
| CSF sTREM2/p-tau × time | −0.007 (0.013) | −0.032–0.018 | <1 | NA | 0.606 | 0.552 | |
| All A+T+ | CSF sTREM2 × time | 0.023 (0.010) | 0.004–0.043 | 2.318 | 0.39 | 0.022 | 0.024 |
| CSF sTREM2/p-tau × time | 0.023 (0.009) | 0.005–0.041 | 2.533 | 0.41 | 0.012 | 0.024 | |
| MCI A+T+ | CSF sTREM2 × time | 0.034 (0.013) | 0.008–0.060 | 2.563 | 0.49 | 0.012 | 0.010 |
| CSF sTREM2/p-tau × time | 0.030 (0.012) | 0.007–0.053 | 2.580 | 0.49 | 0.011 | 0.010 | |
| AD dementia A+T+ | CSF sTREM2 × time | −0.004 (0.023) | −0.047–0.040 | −0.159 | NA | 0.874 | 0.888 |
| CSF sTREM2/p-tau × time | 0.0001 (0.001) | −0.0001–0.0003 | <1 | NA | 0.999 | 0.999 |
Fig. 2. Effect of CSF sTREM2– and CSF sTREM2–to–p-tau181 ratio on changes in cognition.
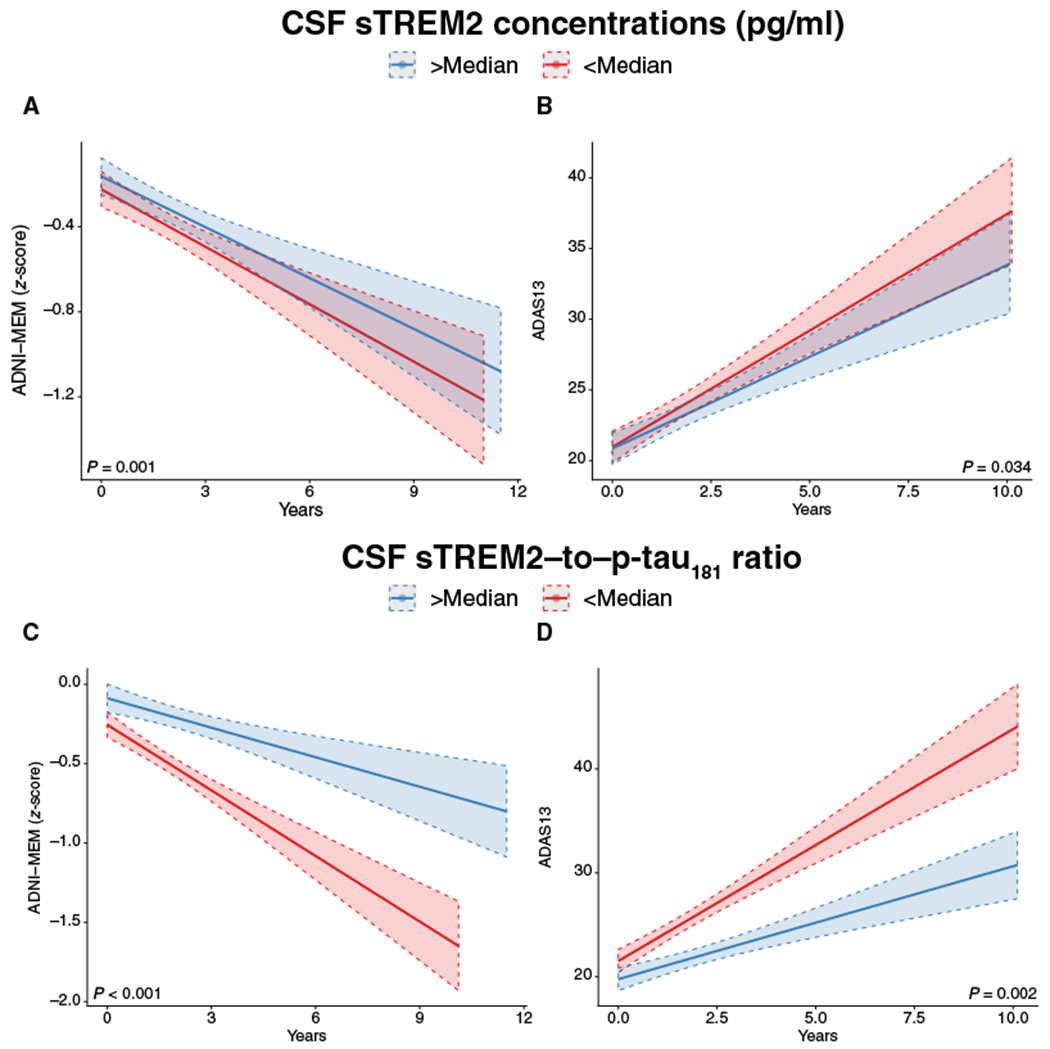
Regression plots show the change in episodic memory (A and C) and global cognition (B and D) as a function of CSF sTREM2 concentrations (top) or the CSF sTREM2–to–p-tau181 ratio (bottom) at different time points during cognitive follow-up in all subjects who were positive for CSF Aβ1–42 and CSF p-tau181 (A+T+). For illustration purposes, the regression lines are shown for subjects with high versus low values of CSF sTREM2 or the CSF sTREM–to–p-tau181 ratio (split at the median). In all regression analyses, CSF sTREM2 concentrations and CSF sTREM2–to–p-tau181 ratios were included as continuous measures. The full regression models included the interaction term of CSF sTREM2 (or CSF sTREM2–to–p-tau181 ratio) × time, controlled for CSF p-tau181 × time, and CSF Aβ1–42 × time interactions among other potentially confounding variables.
Next, we repeated the regression analyses using CSF sTREM2 and p-tau181 concentrations as indicators of an increase in CSF sTREM2 relative to tau pathology and as predictors of cognitive decline. Similar to the results for CSF sTREM2, the interaction of CSF sTREM2 and p-tau181 × time was significant for both ADNI-MEM scores (β = 0.088, P < 0.001; Fig. 2C) and ADAS13 scores (β = −0.102, P = 0.002; Fig. 2D). Higher baseline concentrations of CSF sTREM2 and p-tau181 were associated with slower rates of worsening performance on the ADNI-MEM and ADAS13 tests. All of these analyses survived the Bonferroni-corrected significance threshold (P < 0.0125). No significant effects of CSF sTREM2 concentrations or the ratio of CSF sTREM2 to p-tau181 on the rate of cognitive change were found in the CN control group (CN A−T−, P < 0.05). Summary statistics of the regression analyses including 95% confidence intervals and standardized effect sizes of interaction terms are shown in Table 2. In addition, we tested the predictive effects of CSF sTREM2 concentrations when controlling additionally for markers of neurodegeneration (the N category within the AT/N system) including CSF total tau concentrations and hippocampal volume measured by magnetic resonance imaging (MRI) in addition to the other covariates (CSF p-tau181, CSF Aβ1–42, age, gender, and education). The results of these analyses were congruent with our main results, regardless of the additional controls for CSF total tau concentrations (table S1) or hippocampal volume (table S2).
Effect of CSF sTREM2 on cognitive changes in clinically defined subgroups of A+T+ subjects
To assess whether the association between CSF sTREM2 concentrations and subsequent cognitive changes varied by clinical stage of AD, we performed subanalyses in the clinical subgroups of A+T+ subjects. These A+T+ subjects included those with MCI and AD dementia defined according to NIA-AA research criteria (28). The cognitively normal A+T+ subgroup yielded only 30 subjects and thus was not included in the subanalysis. CSF sTREM2 concentrations were significantly increased in the MCI A+T+ subgroup and the AD dementia A+T+ subgroup compared to cognitively normal subjects who were A−T− (CN A−T−). The ratio of CSF sTREM2 to p-tau181 was significantly reduced in the MCI A+T+ and AD dementia A+T+ subgroups compared to the CN A−T− group (P < 0.05; fig. S1). For the ADNI-MEM score, the interaction of CSF sTREM2 concentrations × time was significant in both the MCI A+T+ subgroup (β = 0.071, P = 0.011; fig. S3A) and the AD dementia A+T+ group (β = 0.050, P = 0.033; fig. S3B). For the ADAS13 score, the interaction effect did not reach significance in the MCI A+T+ subgroup (β = −0.076, P = 0.096) or AD dementia A+T+ subgroup (β = −0.047, P = 0.384). This may reflect the smaller effect size for the ADAS13 score (Table 2), which required a larger sample size for reliable detection. For the interaction of the CSF sTREM2 to p-tau181 × time, we found significant effects for the ADNI-MEM score in both the MCI A+T+ subgroup (β = 0.092, P < 0.001) and AD A+T+ group (β = 0.059, P = 0.009). For the ADAS13 score, the CSF sTREM2 to p-tau181 ratio × time interaction was significant in the MCI A+T+ subgroup (β = −0.105, P = 0.012) but not in the AD dementia A+T+ subgroup (Table 2). These results remained consistent when additionally controlling for baseline CSF total tau concentrations (table S1) or hippocampal volume (as a marker of neurodegeneration, N; table S2).
To assess whether the effect of a higher CSF sTREM2 to p-tau181 ratio on cognitive decline was as large as that of the core AD biomarkers CSF Aβ1–42 and p-tau181, we computed Cohen’s effect sizes d for the effects of CSF concentrations of sTREM2, CSF Aβ1–42, CSF p-tau181 on cognitive changes using the same regression equations as described above. The effect size d is a variance-standardized effect size measure where by convention d = 0.2, d = 0.5, and d = 0.8 are defined as “small,” “medium,” and “large,” respectively (31). The results for the different groups are shown in Fig. 3. With regard to predicting changes in the ADNI-MEM score, the effect sizes were d = 0.43 for CSF sTREM2, d = 0.27 for CSF Aβ1–42, and d = 0.47 for CSF p-tau181. With regard to predicting changes in the ADAS13 score, the effect sizes were d = 0.28 for CSF sTREM2, d = 0.33 for CSF Aβ1–42, and d = 0.45 for CSF p-tau181. Thus, regarding the change in episodic memory, the beneficial effect of higher CSF sTREM2 concentrations was of small to medium size and comparable in size to the detrimental effect of CSF p-tau181 concentrations on cognitive changes.
Fig. 3. Effect sizes for CSF sTREM2 and CSF p-tau181.
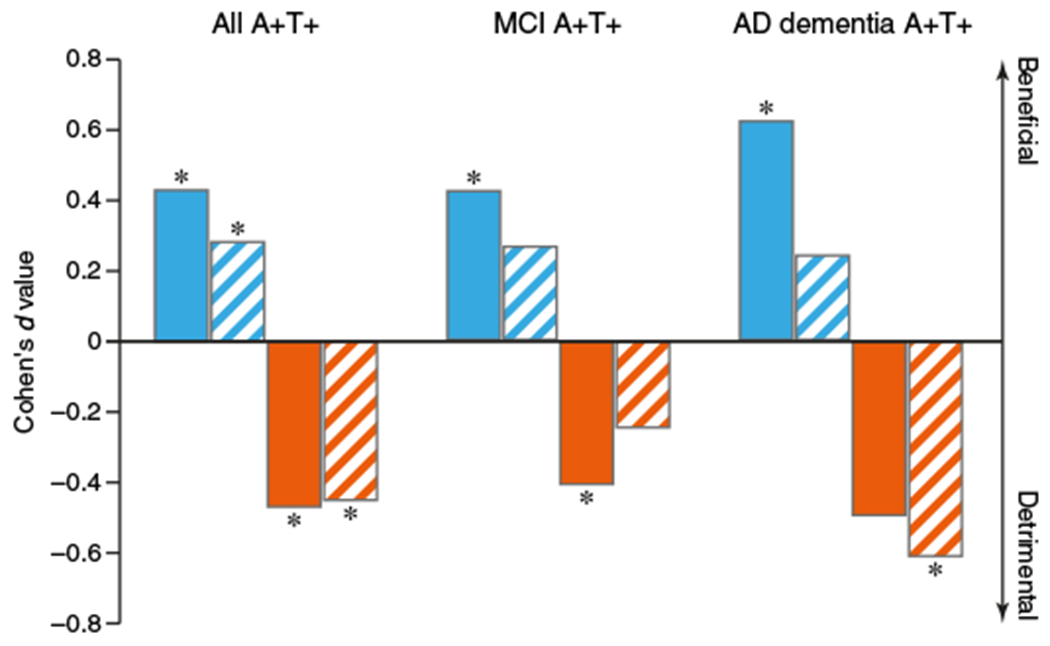
Bars show the effect size (Cohen’s d value) of the association between CSF sTREM2 concentrations (blue) or CSF p-tau181 concentrations (orange) on the rate of change in episodic memory (assessed by the ADNI-MEM score; solid bars) or global cognition (assessed by the ADAS13 score; striped bars). The effect sizes derived from linear mixed-effects analyses are shown for all subjects who were positive for CSF Aβ1–42 and CSF p-tau181 (A+T+) at different clinical stages (MCI, n = 184; AD dementia, n = 66) and after the data were pooled (n = 285). A positive d value means that higher CSF concentrations were associated with slower cognitive decline (less decrease in the ADNI MEM score and less increase in the ADAS13 score). A negative d value means that higher CSF concentrations were associated with worse cognitive decline as determined by mixed-effects regression analyses. *P < 0.05.
Higher CSF sTREM2 concentrations and CSF sTREM2 to p-tau181 ratios are associated with slower hippocampal atrophy
To assess whether higher CSF sTREM2 concentrations were associated with slower neurodegeneration, we used longitudinal hippocampal volumetric data acquired by MRI for subjects who had a minimum of 2 years of MRI follow-up. We used hippocampal volume as a marker of gray matter atrophy given that hippocampus atrophy is a hallmark of AD, with hippocampal volume declining throughout the course of AD (32). Using the same linear mixed models as described above, we found a significant CSF sTREM2 concentration × time interaction in the A+T+ subgroup with longitudinal MRI (n = 184) such that subjects with higher CSF sTREM2 concentrations (β = 0.023, P = 0.022; Fig. 4A) or a higher CSF sTREM2 to p-tau181 ratio (β = 0.023, P = 0.012; Fig. 4B) showed slower hippocampal atrophy rates over time. Clinical subgroup–specific analysis in the A+T+ subjects yielded a significant interaction for MCI A+T+ subjects (CSF sTREM2 × time: β = 0.034, P = 0.012; fig. S4A; CSF sTREM2–to–p-tau181 ratio × time β = 0.030, P = 0.011; fig. S4B) but not for AD dementia A+T+ subjects (CSF sTREM2 × time β = −0.004, P = 0.874; CSF sTREM2–to–p-tau181 ratio × time β = 0.0001, P = 0.999). These exploratory analyses did not survive Bonferroni correction though (P < 0.008). No significant interaction effects were detected in the cognitively normal control A−T− group, neither for CSF sTREM2 concentrations × time (β = −0.012, P = 0.390) nor CSF sTREM2–to–p-tau181 ratio × time (β = −0.007, P = 0.756). Model statistics are summarized in Table 2. In addition, controlling all analyses for CSF total tau concentrations yielded consistent results (table S1).
Fig. 4. Effect of CSF sTREM2– and CSF sTREM2–to–p-tau181 ratio on changes in hippocampal volume.
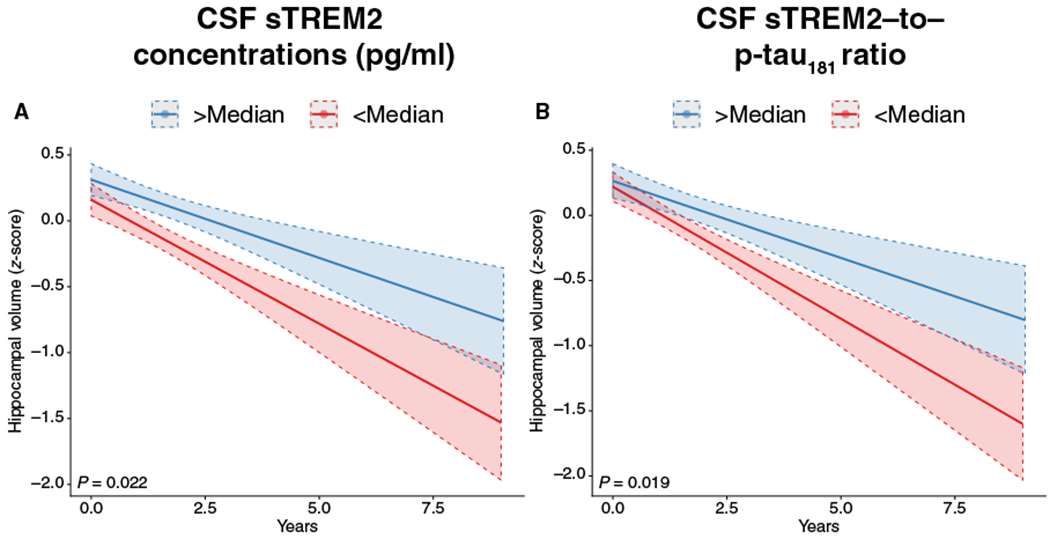
Regression plot shows the change in bilateral hippocampal volume measured by MRI as a function of CSF sTREM2 concentrations at different time points during follow-up (A) and CSF sTREM2–to–p-tau181 ratios (B) for subjects who were positive for CSF Aβ1–42 and CSF p-tau181 (A+T+) and for whom longitudinal hippocampal volume data were available (n = 285). For illustration purposes, the regression lines are shown for subjects with high versus low values for CSF sTREM2– or CSF sTREM–to–p-tau181 ratios (split at the median). All regression analyses included the interaction term of time × CSF sTREM2 concentrations (or CSF sTREM2–to–p-tau181 ratios) as continuous measures and controlled for the interactions of CSF p-tau181 × time and CSF Aβ1–42 × time among other potentially confounding variables.
A higher CSF sTREM2–to–p-tau181 concentration ratio is associated with slower clinical progression
Last, we assessed whether higher baseline CSF sTREM2 concentrations are predictive of the time to conversion to a worse clinical stage (MCI or AD dementia). We included all subjects of the A+T+ group who did not have dementia at baseline, that is, who were cognitively normal or who had MCI. Using Cox regression, we tested CSF sTREM2 concentrations (controlled for Aβ, p-tau181, age, gender, and education) as a predictor of conversion from cognitively normal to MCI or AD dementia or from MCI to AD dementia. For CSF sTREM2 concentrations, we did not detect significant effects on clinical progression (β = −0.113, P = 0.143). However, when testing the ratio of CSF sTREM2 to p-tau181 concentration as a predictor, we found significantly slower clinical progression in subjects with a higher CSF sTREM2–to–p-tau181 ratio (β = −0.486, P = 0.005; Fig. 5). Thus, a higher CSF sTREM2–to–p-tau181 ratio was associated with a lower risk to clinically progress for up to 11.5 years.
Fig. 5. Survival plot for conversion to MCI or AD dementia.
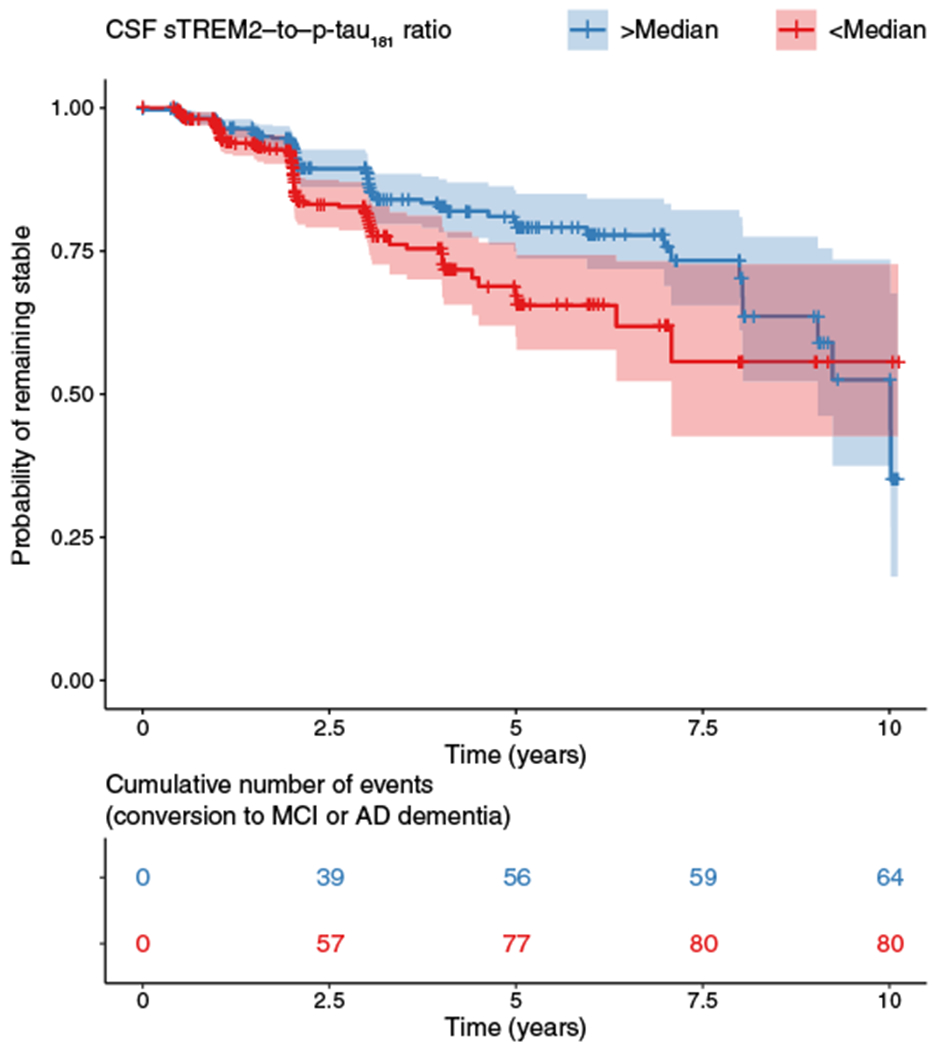
(Top) The Cox regression–derived survival plot shows that a higher ratio of the concentrations of CSF sTREM2 to p-tau181 (split by median) was associated with a higher probability of remaining clinically stable over time in the A+T+ group. (Bottom) The table indicates the number of subjects in the high and low CSF sTREM2–to–p-tau181 ratio subgroups at successive follow-up time points.
DISCUSSION
Here, we assessed whether baseline concentrations of CSF sTREM2 were associated with a slower or faster rate of cognitive decline and clinical worsening in AD. Our results show that higher CSF sTREM2 concentrations at baseline predicted a reduced rate of subsequent cognitive decline in subjects with a CSF biomarker profile of AD (A+T+ group), regardless of the clinical stage. Higher CSF sTREM2 concentrations were associated with less decline in both episodic memory and global cognition. For our secondary outcome parameters, higher CSF sTREM2 concentrations showed a trend of an association with attenuated decline in hippocampal volume. Furthermore, a higher CSF sTREM2–to–p-tau181 ratio predicted a slower rate of conversion from cognitively normal to MCI or AD dementia. Given that sTREM2 detected in the CSF likely reflects cerebral TREM2 expression and the amount of signaling-competent TREM2 on the surface of activated microglia, these results suggest that higher TREM2-related microglial function may be predictive of slower disease progression at the symptomatic stage of AD. Our results suggest that higher CSF sTREM2 is not only associated with less cognitive decline but may also be associated with neuroprotective effects as suggested by slower hippocampal volume changes, although the analyses of hippocampal volume changes were exploratory in nature and need to be confirmed in future studies. We further caution that although the current study examined the association between CSF sTREM2 and subsequent cognitive changes, a causative effect between microglial TREM2 function and clinical outcome is not necessarily implied. Nonetheless, our results demonstrate that in patients with biomarker evidence of underlying AD, higher CSF sTREM2 concentrations predict a more benign disease course in AD. A protective role of TREM2-related processes is also in line with the finding that several TREM2 variants are loss-of-function mutations (33). In the case of the TREM2 pT66M variant or complete loss of TREM2, microglia are prevented from adopting a disease-associated state, which would trigger phagocytosis, migration, chemotaxis, and microglial proliferation (10, 13, 15, 34).
Our clinical findings are based on a large sample of well-characterized subjects with biomarker evidence of AD pathology. For staging of AD, we used an AD biomarker–informed classification system in line with recently proposed NIA-AA research diagnostic guidelines for AD (28). Consistent with previous findings (35, 36), we showed that abnormal CSF concentrations of tau and Aβ were predictive of faster cognitive decline at each disease stage. In contrast, increases in CSF sTREM2 concentrations in A+T+ subjects were associated with more benign subsequent cognitive changes. The effect size of higher CSF sTREM2 concentrations as a predictor of reduced cognitive decline was comparable to that of higher CSF p-tau181 concentrations as a predictor of worse cognitive decline, suggesting that CSF sTREM2 concentrations may be relevant to future disease progression. The clinical relevance of altered CSF sTREM2 concentrations is supported by our finding of an association of a higher CSF sTREM2–to–p-tau181 ratio with attenuated rate of conversion from cognitively normal or MCI to AD dementia. The association between higher CSF sTREM2 and an attenuated rate of clinical worsening may be seen as counterintuitive considering that an increase in CSF sTREM2 concentrations is associated with higher CSF p-tau181 and total tau concentrations (20, 21, 24, 25). Mechanistically, it is likely that in response to amyloid pathology and neurofibrillary tangle degeneration (reflected by increased CSF p-tau181), microglial activation may be triggered by enhanced expression and surface localization of TREM2 (reflected by increased CSF sTREM2 concentrations). Thus, without controlling for AD pathology, increased CSF sTREM2 concentrations may simply reflect the presence of AD and correlate negatively with cognitive performance (37). The key question is whether stronger TREM2-related microglial activation at a given stage of AD pathology could be beneficial for slowing subsequent neurodegeneration and clinical progression. Therefore, we sought in our analyses to test the predictive value of higher CSF sTREM2 concentrations on future disease progression (cognitive, clinical, and hippocampal volume), and we controlled for biomarkers of AD pathology including CSF Aβ1–42 and p-tau181. Our results show that the higher the concentration of CSF sTREM2 for a given degree of neurofibrillary tangle degeneration (as measured by p-tau181), the better the clinical outcome.
Our findings are consistent with those of a recent study where higher CSF sTREM2 concentrations were associated with higher cortical gray matter integrity at the MCI stage (27), suggesting a potentially beneficial function of the TREM2-related immune response, particularly in the symptomatic phase of AD. Our results are also in general agreement with previous findings reporting increased microglial activation (based on PET imaging) in cognitively normal or MCI subjects who were amyloid positive on PET (38, 39) and a correlation between higher microglial activation on PET and higher tau on PET (40). Greater numbers of microglia based on PET imaging were associated with a slower subsequent cognitive decline in subjects with MCI (38). This is also in line with the observation that microglial-PET signals increase during physiological aging of wild-type mice, reflecting the accumulation of activated microglia around sites of neuronal damage. Notably, no such increase in microglial-PET signals nor in clustering of microglia during physiological aging has been observed upon loss of TREM2 function (13). Together, these results suggest a protective effect of greater microglial activation during disease progression in AD. The protective effects of higher TREM2 function have also been observed in transgenic mouse models of AD (16, 41, 42). A number of AD pathology studies in transgenic mouse models suggest enhanced amyloid plaque seeding and reduced amyloid plaque compaction combined with increased plaque-associated dystrophic neurites in the absence of TREM2 function (11, 16, 43). Increased TREM2 expression resulted in a reduction in dystrophic neurites in a transgenic mouse model of AD (44), a finding in line with our observation of the association between higher TREM2 concentrations and attenuated disease progression in human subjects.
For tauopathies, several studies have reported beneficial effects of higher TREM2 function, which was associated with reduced neuronal loss and improved spatial memory in the PS19 transgenic mouse model of tauopathy (45–49). However, a recent study reported beneficial effects of TREM2 deficiency in a transgenic mouse model of tauopathy (50). Differences between the studies may be explained by proinflammatory cytokine release, which was decreased in TREM2-deficient transgenic tauopathy mice (50) but was up-regulated or unaffected in those studies that reported protective effects of TREM2 (45–49). Thus, TREM2-related release of proinflammatory cytokines may play an important role in the beneficial effects of TREM2-triggered microglial activity in the presence of tau pathology (47, 51). In addition, a recent study reported that selective TREM2 haploin-sufficiency but not a complete loss of TREM2 increased tau pathology, which may suggest that some compensatory mechanisms occurred after the complete loss of TREM2 function (52). The findings regarding the effect of TREM2 in animal models of amyloid and tau pathology are heterogeneous, with both beneficial and detrimental effects being reported. Disease stage may be a moderating factor for TREM2-associated microglial activity (16, 53, 54), a factor that could also account for some inconsistencies observed in human subjects (39, 55). However, disease stage is a broad descriptive category; the key molecular events that are associated with TREM2 and that determine beneficial versus detrimental microglial activity remain to be established. It is tempting to speculate that any beneficial effects of increased TREM2 expression are associated with enhanced TREM2-mediated signaling. TREM2-triggered microglial activity facilitates chemotaxis and clustering of microglia around amyloid plaques, engulfment of amyloid seeds (16), compaction of amyloid plaques, removal of cellular debris (10, 15), and myelin repair (56) and potentially may also reduce pathological tau and neuroinflammation (48). Thus, increased TREM2 signaling may facilitate a protective response to neuronal injury. Studying the association between CSF sTREM2 concentrations and pro- and anti-inflammatory markers in future studies in patients with AD will be instrumental for understanding the reduced cognitive and clinical worsening associated with CSF sTREM2 concentrations (51, 54).
For the interpretation of the current study, some caveats should be considered. First, the current study included mostly subjects with dementia symptoms, but only a few subjects showed biomarker evidence of AD pathology and were still cognitively normal. Thus, it is unclear whether the current findings are also applicable to the pre-symptomatic stage of AD. Second, CSF sTREM2 concentrations are not a direct measure of TREM2-related microglial activity. Although TREM2 is cleaved by the secretase ADAM10 and thus may be released into the interstitial fluid in the brain, the relationship between sTREM2 in the CSF and higher microglial expression of TREM2 remains to be investigated. Third, although TREM2 is a key driver for switching microglia from a homeostatic to a disease-associated state, TREM2 receptor signaling acts in concert with a multitude of other microglia receptors, which were not assessed in the current study. Last, a causative effect between microglial TREM2 function and clinical outcome is not necessarily implied by our findings.
Together, we show that higher CSF sTREM2 concentrations are associated with a protective effect for cognitive changes at a similar magnitude to that found for the adverse effects of primary AD pathologies assessed by CSF Aβ1–42 and p-tau181 concentrations. Our findings have important implications for therapeutic attempts that aim to modulate microglial function, suggesting that stimulating TREM2-mediated microglial activity may be most beneficial within the early symptomatic phase of AD. From a clinical point of view, CSF sTREM2 concentrations could serve as a target engagement marker for treatments aimed at modulating microglial function. Despite the fact that the current study is observational in nature and therefore cause-effect conclusions cannot be drawn, our results support the notion that the modification of TREM2 function is a valuable target in AD. Our results suggest that drugs that up-regulate TREM2-related microglial activity may benefit patients with AD. Furthermore, our data indicate that the early symptomatic disease stage of AD is a preferred time window for therapeutic microglial modulation.
MATERIALS AND METHODS
Study design
In the current study, we examined cross-sectional CSF measurements of sTREM2, Aβ1–42, and p-tau181 concentrations as predictors of longitudinally assessed changes in cognition, clinical symptoms, and hippocampal volume. A total of 385 subjects were included from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). ADNI was launched in 2003 as a public-private partnership, with the primary goal to assess serial neuroimaging and biological markers for the prediction of disease progression in AD. The subjects studied herein were a subgroup of n = 1035 participants for whom CSF sTREM2 samples at any time point were available. Beyond the inclusion and exclusion criteria of ADNI, the following additional selection criteria were applied for the current study: (i) availability of baseline CSF sTREM2 values, (ii) no rare variants in the TREM2 gene, (iii) minimum of 1-year follow-up duration for neuropsychological or clinical testing to avoid strong censoring effects (4), CSF A+T+ profile or CSF A−T− profile for cognitively normal individuals (see fig. S2 for flow chart of subject selection). Application of these inclusion criteria led to the selection of n = 385 subjects, who did not differ in terms of age, gender, or education, or mini mental state exam (MMSE) score from the entire sample of 1035 subjects, suggesting that no selection bias was evident. All subjects were recruited between the years 2005 and 2013. Clinical diagnosis of amnestic MCI was defined according to the Mayo Clinic criteria (57). Neuropsychological and clinical testing was done annually over a period of up to 11.5 years (mean, 4 years; range, 1.5 to 11.5). AD dementia diagnosis was accomplished according to standard clinical criteria as described previously (58). The study was approved by the Institutional Review Board (IRB) of all participating centers in ADNI, as well as our local IRB at Ludwig-Maximilians University. All study participants (or their relatives) provided written informed consent.
Following the recently proposed NIA-AA guidelines (28), AD was defined as a biological construct and not by clinical symptoms. These criteria require both evidence of amyloid deposition (A+), defined in this study as abnormal values of CSF Aβ1–42, and of tau pathology (T+), defined in this study as abnormal values of CSF p-tau181 (28). On the basis of previously established cutoff points (59), the criterion for pathological CSF Aβ1–42 was defined as Aβ1–42 < 976.6 pg/ml, and for pathological p-tau181, it was defined as p-tau181 > 21.8 pg/ml. In contrast, we applied the traditional syndromal categories (CN, MCI, and AD dementia) for the clinical staging of AD (28).
CSF sTREM2 measurements were done at the laboratory of C. Haass at the DZNE Munich using ELISA with minor modifications to the procedure described previously (13, 21). The CSF concentrations of Aβ1–42 and p-tau181 were assessed by the ADNI biomarker core team at the University of Pennsylvania using the electrochemiluminiscence immunoassays Elecsys.
Measurements of CSF sTREM2, Aβ1–42, total tau, and p-tau181
The CSF sTREM2 assay is based on the MSD platform and has been comprehensively described previously (13, 20, 21). A detailed description of the CSF sTREM2 measurements in the ADNI samples, as well as the original data, can be downloaded from the LONI Image and Data Archive (https://ida.loni.usc.edu). Briefly, Streptavidin-coated 96-well plates (MSD Streptavidin Gold Plates, cat. no. L15SA) were first blocked overnight at 4°C in blocking buffer [3% bovine serum albumin (BSA) and 0.05% Tween 20 in PBS (pH 7.4)]. They were next incubated with the capture antibody (0.25 μg/ml; R&D Systems, cat. no. BAF1828), diluted in the antibody buffer [1% BSA and 0.05% Tween 20 in PBS (pH 7.4)] during 1 hour at room temperature, and subsequently washed four times with washing buffer (0.05% Tween 20 in PBS). Next, the recombinant human TREM2 protein (standard curve; Hölzel Diagnostika, cat. no. 11084-H08H), the blanks, the CSF samples (duplicates; dilution factor: 4), and four internal standards (IS) were diluted in assay buffer [0.25% BSA and 0.05% Tween 20 in PBS (pH 7.4)] supplemented with protease inhibitors (Sigma; cat. no. P8340) and incubated for 2 hours at RT. Plates were again washed three times and incubated for 1 hour at RT with the detection antibody (1 μg/ml; Santa Cruz Biotechnology, B-3, cat. no. sc373828). After three additional washing steps, plates were incubated with SULFO-tag conjugated secondary antibody (0.5 μg/ml; MSD, cat. no. R32AC) for 1 hour in the dark at RT, washed three times with wash buffer and two times with PBS. The electrochemical signal was developed with MSD Read buffer T (cat. no. R-92TC), and the light emission was measured using the MESO QuickPlex SQ 120. The mean intraplate CV was 3.1% (all duplicate measures had a CV < 15%) and the interplate CV for each of the IS were 11.4, 12.2, 10.5, and 7.1%. The CSF sTREM2 values used herein are those corrected on the basis of values of the four IS that were loaded on all plates (variable “MSD_sTREM2CORRECTED” in the ADNI database).
The CSF core biomarker measurements were performed by the ADNI biomarker core team at the University of Pennsylvania, using the electrochemiluminiscence immunoassays Elecsys Aβ1–42, p-tau181, and total tau on a fully automated Elecsys cobas e 601 instrument and a single lot of reagents for each of the three measured biomarkers (provided in UPENNBIOMK9.csv file available in the ADNI databank). The analyte measuring ranges (lower technical limit to upper technical limit) of these assays are as follows: 200 to 1700 pg/ml for Elecsys Aβ1–42, 80 to 1300 pg/ml for total tau CSF, and 8 to 120 pg/ml for p-tau181 CSF immunoassays.
Neuropsychological assessment
Annual assessment of neuropsychological tests was performed. We included a priori two measures of cognitive performance. Given that episodic memory is the cognitive domain primarily affected in early stages and by definition in amnestic MCI, we used the composite score of episodic memory called ADNI-MEM, which integrates memory scores of the Rey Auditory Verbal Learning Test, ADAS, as well as the Wechsler Logical Memory I and II and the word recall of the MMSE as previously described (30). The median follow-up time of neuropsychological and clinical testing was 4 years (range, 1.45 to 11.5 years). The Alzheimer’s Disease Assessment Scale Cognition 13-item scale (ADAS13) (60) was included to assess global cognition.
Measurement of hippocampal volume by MRI
Freesurfer-based (version 5.1) assessments of hippocampal volume were done on 3T MPRAGE MRI scans by the ADNI imaging core at UCSF (61) and were for the current study downloaded from the ADNI database (http://adni.loni.usc.edu/). For the longitudinal hippocampus volume measurement, images were processed using the Freesurfer-based longitudinal image processing framework (62). Briefly, for each subject, a median image is computed via robust registration of a subject’s images from all longitudinal time points to arrive at an unbiased subject-specific template. The subject-specific template informs the initialization of later algorithms including the surface reconstruction, nonlinear spatial normalization to atlas space, and parcellation to treat images from all time points in the same way and thus avoid a bias stemming from temporal order. All images undergo a series of preprocessing steps including intensity normalization, removal of nonbrain voxels, affine registration to Talairach space before the segmentation of subcortical white matter and subcortical nuclei, and a second intensity normalization. Subsequently, surface reconstruction was performed through a number of steps for all images, followed by nonlinear registration of the individual surface models to a spherical atlas and automated parcellation of the brain regions (62). Thus, the hippocampus volume for each time point and subject was extracted and adjusted for intracranial volume using linear regression, following previous recommendations (63). The individual rate of change in hippocampus volume was subsequently modeled in mixed-effect regression analysis (see “Statistical Analysis” section below). We caution that although the ADNI study protocol instructs that subject-specific baseline and follow-up images be acquired on the same scanner model, hardware and software upgrades could not be tracked in the ADNI data base. Such changes may introduce measurement variability but are unlikely to systematically bias the analysis of CSF sTREM2 as a predictor of hippocampal volume changes. Detailed descriptions of the longitudinal Freesurfer-based imaging pipelines applied to the ADNI data can be found online (http://adni.loni.usc.edu/) and in previous publications (64).
Statistical analysis
To test whether higher CSF sTREM2 was associated with slower cognitive decline in subjects with AD pathology, we conducted linear mixed-effects regression models in the pooled sample of A+T+ subjects regardless of cognitive status (28). The dependent variables were either ADNI-MEM or ADAS13. The fixed effects included CSF sTREM × time + CSF Aβ1–42 × time + CSF p-tau181 × time + CSF sTREM2 + CSF Aβ1–42 + CSF p-tau181 + clinical syndrome (CN, MCI, or dementia) + age + gender + years of education. Random effects included time and intercept. The variable time was defined as the follow-up duration of the neuropsychological testing in years. Note that we controlled in each analysis for the effects of CSF Aβ1–42 × time and CSF p-tau181 × time (in addition to the simple main effects of each variable), given that we were interested in whether higher concentrations of CSF sTREM2 relative to AD pathology (as assessed by CSF Aβ1–42 and p-tau181) were beneficial. As an alternative way to assess CSF sTREM2 concentrations relative to primary AD pathology, we used the ratio of CSF sTREM2 to p-tau181. Thus, a stronger increase of CSF sTREM2 relative to the increase in CSF p-tau181 would result in a larger ratio, whereas a weaker CSF sTREM2 increase relative to that of CSF p-tau181 would result in a smaller ratio. Ratio indices such as CSF p-tau181/Aβ1–42 have been previously found to be more predictive of AD than the linear combination of each term, and thus, ratio indices may represent biologically relevant quantities (65, 66). Therefore, we repeated the linear mixed-effects regression analyses on the rate of change of hippocampus volume or cognition (ADNI-MEM and ADAS13), this time using the novel ratio index of CSF sTREM2/p-tau181 instead of CSF sTREM2 as the predictor. We applied a Bonferroni-corrected significance threshold (P < 0.0125 accounting for four tests) to test our primary hypothesis in the pooled A+T+ group, i.e., higher CSF sTREM2 or CSF sTREM2/p-tau181 ratio is associated with slower rate of decline in cognition (as measured by ADNI-MEM or ADAS13). To ensure that our analyses were not confounded by potentially violated normality assumptions, we additionally derived two-sided P values from nonparametric permutation testing for all above listed analyses. Specifically, the values of CSF sTREM2 or sTREM2/p-tau181 were permuted 1000 times among the subjects to determine a distribution of the CSF sTREM2 × time or sTREM2/p-tau181 × time coefficient under the null hypothesis. Two-sided permutation P values were derived by assessing the percentage of permuted coefficients (i.e., absolute) that surpassed the absolute of the observed coefficient. For exploratory reasons, we further tested whether our results were robust when controlling for biomarker of neurodegeneration including CSF total tau or, alternatively, hippocampus volume. To this end, we repeated all models, this time also controlling for baseline CSF total tau levels or hippocampus volume, in addition to the other covariates (CSF p-tau181, CSF Aβ1–42, age, gender, and education). Next, we computed the effect size d for the effect of baseline CSF biomarkers on cognitive changes (31). Cohen’s effect size d was estimated from the linear mixed models using the lme.dscore command of the EMAtools R package (see https://cran.r-project.org/web/packages/EMAtools/EMAtools.pdf).
Using the same linear mixed model approach as described above, we assessed in a secondary exploratory analysis whether higher CSF sTREM2 levels were associated with slower progression of neurodegeneration, as assessed via longitudinally assessed hippocampal volume, using analogous regression analyses as described above. These exploratory analyses were all tested at P < 0.05 uncorrected. For predicting hippocampal volume changes, a total of six separate regression analyses were performed including either CFS sTREM2 or CSF sTREM/p-tau181 as predictors in either the pooled sample or the MCI A+T+ and AD dementia A+T+ groups, rendering a Bonferroni-corrected threshold of P < 0.008 as the more stringent significance threshold.
Next, we tested disease stage-specific effects of CSF sTREM2 on cognitive changes. We repeated the linear mixed-effects regression analyses for CSF sTREM2 and CSF sTREM2/p-tau181 stratified by diagnostic group including MCI A+T+ and AD dementia A+T+. For these exploratory analyses, we used P < 0.05 as the significance criterion.
Last, we tested whether higher CSF sTREM2 is associated with the conversion rate from MCI to AD. To this end, we used Cox Regression analyses, with CSF sTREM2 or the CSF sTREM2/p-tau181 ratio as main predictor, controlled for age, gender, Aβ1–42, and CSF p-tau181.
Supplementary Material
Fig. S1. CSF sTREM2 and CSF sTREM2 to p-tau181 ratio per group.
Fig. S2. Selection of the final subject group.
Fig. S3. Effect of CSF sTREM2 on changes in cognition in clinical subgroups.
Fig. S4. Effect of CSF sTREM2 measurements on hippocampal changes in MCI A+T+.
Table S1. Regression analyses with additional control for CSF total tau.
Table S2. Regression analyses with additional control for hippocampus volume.
Acknowledgments:
We thank J. L. Molinuevo, J. D. Gispert, and A.-L. Boulesteix for their helpful comments.
Funding: C.C. is supported by grants from the NIH (R01AG044546, P01AG003991, RF1AG053303, R01AG058501, and U01AG058922). This study was supported by the Deutsche Forschungsgemeinschaft (DFG) within the framework of the Munich Cluster for Systems Neurology (EXC 1010 SyNergy), a DFG funded Koselleck Project (HA1737/16-1 to C.H.), the Cure Alzheimer’s Fund (to C.H.), and the Association for Frontotemporal Degeneration (FTD-biomarker award to C.H.). M.S.-C. receives funding from the European Union’s Horizon 2020 Research and Innovation Program under the Marie Sklodowska-Curie action grant agreement #752310. Data collection and sharing for this project were funded by the ADNI (NIH grant no. U01 AG024904 and Department of Defense award no. W81XWH-12-2-0012). The investigators within the ADNI contributed to the design and implementation of ADNI or provided data but did not participate in analysis or writing of this study. A complete listing of ADNI investigators can be found at http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf. ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association, Alzheimer’s Drug Discovery Foundation, Araclon Biotech, BioClinica Inc., Biogen, Bristol-Myers Squibb Company, CereSpir Inc., Cogstate, Eisai Inc., Elan Pharmaceuticals Inc., Eli Lilly and Company, EuroImmun, F. Hoffmann–La Roche Ltd. and its affiliated company Genentech Inc., Fujirebio, GE Healthcare, IXICO Ltd., Janssen Alzheimer Immunotherapy Research and Development LLC., Johnson & Johnson Pharmaceutical Research & Development LLC., Lumosity, Lundbeck, Merck & Co. Inc., Meso Scale Diagnostics LLC., NeuroRx Research, Neurotrack Technologies, Novartis Pharmaceuticals Corporation, Pfizer Inc., Piramal Imaging, Servier, Takeda Pharmaceutical Company, and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the NIH (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for NeuroImaging at the University of Southern California.
Competing interests: C.H. is co-inventor on pending patent no. EP16180844 entitled “TREM2 cleavage modulators and uses thereof”. C.H. is an official collaboration partner of Denali Therapeutics and a chief consultant for ISAR Biosciences. C.C. receives research support from Biogen, EISAI, Alector, and Parabon and is a member of the advisory board of ADx Healthcare, Halia Therapeutics, and Vivid Genomics. M.W. has served on Advisory Boards for Eli Lilly, Cerecin/Accera, Roche, Alzheon Inc., Merck Sharp & Dohme Corp., Nestle/Nestec, PCORI/PPRN, Dolby Family Ventures, Brain Health Registry, and ADNI. He has consulted or has been a speaker for Cerecin/Accera Inc., Alzheimer’s Drug Discovery Foundation, Merck, BioClinica, Eli Lilly, Nestle/Nestec, Roche, Genentech, Lynch Group GLC, Health & Wellness Partners, and Bionest Partners. He holds stock options in Alzheon Inc.
Footnotes
SUPPLEMENTARY MATERIALS
Competing interests: The other authors declare that they have no competing interests.
Data and materials Availability: All data associated with this study are in the main text or the Supplementary Materials. All ADNI data including CSF TREM2 samples are available through the ADNI Data Archive. Interested scientists may apply for access via the ADNI-LONI website (http://adni.loni.usc.edu/).
REFERENCES AND NOTES
- 1.Jones L, Holmans PA, Hamshere ML, Harold D, Moskvina V, Ivanov D, Pocklington A, Abraham R, Hollingworth P, Sims R, Gerrish A, Pahwa JS, Jones N, Stretton A, Morgan AR, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Morgan K, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Love S, Kehoe PG, Mead S, Fox N, Rossor M, Collinge J, Maier W, Jessen F, Schurmann B, Heun R, Kolsch H, van den Bussche H, Heuser I, Peters O, Kornhuber J, Wiltfang J, Dichgans M, Frölich L, Hampel H, Hüll M, Rujescu D, Goate AM, Kauwe JS, Cruchaga C, Nowotny P, Morris JC, Mayo K, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Singleton AB, Guerreiro R, Mühleisen TW, Nöthen MM, Moebus S, Jockel KH, Klopp N, Wichmann HE, Ruther E, Carrasquillo MM, Pankratz VS, Younkin SG, Hardy J, O’Donovan MC, Owen MJ, Williams J, Genetic evidence implicates the immune system and cholesterol metabolism in the aetiology of Alzheimer’s disease. PLOS ONE 5, e13950 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heneka MT, Golenbock DT, Latz E, Innate immunity in Alzheimer’s disease. Nat. Immunol 16, 229–236 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ, Rujescu D, Hampel H, Giegling I, Andreassen OA, Engedal K, Ulstein I, Djurovic S, Ibrahim-Verbaas C, Hofman A, Ikram MA, van Duijn CM, Thorsteinsdottir U, Kong A, Stefansson K, Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med 368, 107–116 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guerreiro R, Hardy J, TREM2 and neurodegenerative disease. N. Engl. J. Med 369, 1569–1570 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE, Systematic meta-analyses of Alzheimer disease genetic association studies: The AlzGene database. Nat. Genet 39, 17–23 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Colonna M, TREMs in the immune system and beyond. Nat. Rev. Immunol 3, 445–453 (2003). [DOI] [PubMed] [Google Scholar]
- 7.Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang L.-c., Means TK, El Khoury J, The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci 16, 1896–1905 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, Itzkovitz S, Colonna M, Schwartz M, Amit I, A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276–1290.e17 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, Beckers L, O’Loughlin E, Xu Y, Fanek Z, Greco DJ, Smith ST, Tweet G, Humulock Z, Zrzavy T, Conde-Sanroman P, Gacias M, Weng Z, Chen H, Tjon E, Mazaheri F, Hartmann K, Madi A, Ulrich JD, Glatzel M, Worthmann A, Heeren J, Budnik B, Lemere C, Ikezu T, Heppner FL, Litvak V, Holtzman DM, Lassmann H, Weiner HL, Ochando J, Haass C, Butovsky O, The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47, 566–581.e9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mazaheri F, Snaidero N, Kleinberger G, Madore C, Daria A, Werner G, Krasemann S, Capell A, Trumbach D, Wurst W, Brunner B, Bultmann S, Tahirovic S, Kerschensteiner M, Misgeld T, Butovsky O, Haass C, TREM2 deficiency impairs chemotaxis and microglial responses to neuronal injury. EMBO Rep. 18, 1186–1198 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yuan P, Condello C, Keene CD, Wang Y, Bird TD, Paul SM, Luo W, Colonna M, Baddeley D, Grutzendler J, TREM2 haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron 90, 724–739 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takahashi K, Rochford CD, Neumann H, Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med 201, 647–657 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kleinberger G, Yamanishi Y, Suarez-Calvet M, Czirr E, Lohmann E, Cuyvers E, Struyfs H, Pettkus N, Wenninger-Weinzierl A, Mazaheri F, Tahirovic S, Lleo A, Alcolea D, Fortea J, Willem M, Lammich S, Molinuevo JL, Sanchez-Valle R, Antonell A, Ramirez A, Heneka MT, Sleegers K, van der Zee J, Martin JJ, Engelborghs S, Demirtas-Tatlidede A, Zetterberg H, Van Broeckhoven C, Gurvit H, Wyss-Coray T, Hardy J, Colonna M, Haass C, TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci. Transl. Med 6, 243ra86 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Yeh FL, Wang Y, Tom I, Gonzalez LC, Sheng M, TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron 91, 328–340 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Gilfillan S, Krishnan GM, Sudhakar S, Zinselmeyer BH, Holtzman DM, Cirrito JR, Colonna M, TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 160, 1061–1071 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parhizkar S, Arzberger T, Brendel M, Kleinberger G, Deussing M, Focke C, Nuscher B, Xiong C, Ghasemigharagoz A, Katzmarski N, Krasemann S, Lichtenthaler SF, Müller SA, Colombo A, Monasor LS, Tahirovic S, Willem M, Pettkus N, Butovsky O, Bartenstein P, Edbauer D, Rominger A, Ertürk A, Grathwohl SA, Neher JJ, Holtzman DM, Meyer-Luehmann M, Haass C, Loss of TREM2 function increases amyloid seeding but reduced plaque associated ApoE. Nat. Neurosci 22, 191–204 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T, Vitorica J, Ransohoff RM, Herrup K, Frautschy SA, Finsen B, Brown GC, Verkhratsky A, Yamanaka K, Koistinaho J, Latz E, Halle A, Petzold GC, Town T, Morgan D, Shinohara ML, Perry VH, Holmes C, Bazan NG, Brooks DJ, Hunot S, Joseph B, Deigendesch N, Garaschuk O, Boddeke E, Dinarello CA, Breitner JC, Cole GM, Golenbock DT, Kummer MP, Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tanzi RE, TREM2 and risk of Alzheimer’s disease—Friend or foe? N. Engl. J. Med 372, 2564–2565 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Piccio L, Buonsanti C, Cella M, Tassi I, Schmidt RE, Fenoglio C, Rinker II J, Naismith RT, Panina-Bordignon P, Passini N, Galimberti D, Scarpini E, Colonna M, Cross AH, Identification of soluble TREM-2 in the cerebrospinal fluid and its association with multiple sclerosis and CNS inflammation. Brain 131, 3081–3091 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suárez-Calvet M, Araque Caballero MÁ, Kleinberger G, Bateman RJ, Fagan AM, Morris JC, Levin J, Danek A, Ewers M, Haass C; Dominantly Inherited Alzheimer Network, Early changes in CSF sTREM2 in dominantly inherited Alzheimer’s disease occur after amyloid deposition and neuronal injury. Sci. Transl. Med 8, 369ra178 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suárez-Calvet M, Kleinberger G, Araque Caballero MÁ, Brendel M, Rominger A, Alcolea D, Fortea J, Lleó A, Blesa R, Gispert JD, Sanchez-Valle R, Antonell A, Rami L, Molinuevo JL, Brosseron F, Traschütz A, Heneka MT, Struyfs H, Engelborghs S, Sleegers K, Van Broeckhoven C, Zetterberg H, Nellgård B, Blennow K, Crispin A, Ewers M, Haass C, sTREM2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early-stage Alzheimer’s disease and associate with neuronal injury markers. EMBO Mol. Med 8, 466–476 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schlepckow K, Kleinberger G, Fukumori A, Feederle R, Lichtenthaler SF, Steiner H, Haass C, An Alzheimer-associated TREM2 variant occurs at the ADAM cleavage site and affects shedding and phagocytic function. EMBO Mol. Med 9, 1356–1365 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wunderlich P, Glebov K, Kemmerling N, Tien NT, Neumann H, Walter J, Sequential proteolytic processing of the triggering receptor expressed on myeloid cells-2 (TREM2) protein by ectodomain shedding and γ-secretase-dependent intramembranous cleavage. J. Biol. Chem 288, 33027–33036 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heslegrave A, Heywood W, Paterson R, Magdalinou N, Svensson J, Johansson P, Ohrfelt A, Blennow K, Hardy J, Schott J, Mills K, Zetterberg H, Increased cerebrospinal fluid soluble TREM2 concentration in Alzheimer’s disease. Mol. Neurodegen 11, 3 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Piccio L, Deming Y, Del-Águila JL, Ghezzi L, Holtzman DM, Fagan AM, Fenoglio C, Galimberti D, Borroni B, Cruchaga C, Cerebrospinal fluid soluble TREM2 is higher in Alzheimer disease and associated with mutation status. Acta Neuropathol. 131, 925–933 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Henjum K, Almdahl IS, Årskog V, Minthon L, Hansson O, Fladby T, Nilsson LNG, Cerebrospinal fluid soluble TREM2 in aging and Alzheimer’s disease. Alzheimer’s Res. Ther 8, 17 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gispert JD, Suarez-Calvet M, Monté GC, Tucholka A, Falcon C, Rojas S, Rami L, Sanchez-Valle R, Llado A, Kleinberger G, Haass C, Molinuevo JL, Cerebrospinal fluid sTREM2 levels are associated with gray matter volume increases and reduced diffusivity in early Alzheimer’s disease. Alzheimers Dement. 12, 1259–1272 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Jack CR Jr., Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, Holtzman DM, Jagust W, Jessen F, Karlawish J, Liu E, Molinuevo JL, Montine T, Phelps C, Rankin KP, Rowe CC, Scheltens P, Siemers E, Snyder HM, Sperling R; Contributors, NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 14, 535–562 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, Gamst A, Holtzman DM, Jagust WJ, Petersen RC, Snyder PJ, Carrillo MC, Thies B, Phelps CH, The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 7, 270–279 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crane PK, Carle A, Gibbons LE, Insel P, Mackin RS, Gross A, Jones R, Mukherjee S, Curtis SM, Harvey D, Weiner M, Mungas D; Alzheimer’s Disease Neuroimaging Initiative, Development and assessment of a composite score for memory in the Alzheimer’s Disease Neuroimaging Initiative (ADNI). Brain Imaging Behav. 6, 502–516 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cohen J, A power primer. Psychol. Bull 112, 155–159 (1992). [DOI] [PubMed] [Google Scholar]
- 32.Ewers M, Sperling RA, Klunk WE, Weiner MW, Hampel H, Neuroimaging markers for the prediction and early diagnosis of Alzheimer’s disease dementia. Trends Neurosci. 34, 430–442 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Colonna M, Wang Y, TREM2 variants: New keys to decipher Alzheimer disease pathogenesis. Nat. Rev. Neurosci 17, 201–207 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Kleinberger G, Brendel M, Mracsko E, Wefers B, Groeneweg L, Xiang X, Focke C, Deußing M, Suárez-Calvet M, Mazaheri F, Parhizkar S, Pettkus N, Wurst W, Feederle R, Bartenstein P, Mueggler T, Arzberger T, Knuesel I, Rominger A, Haass C, The FTD-like syndrome causing TREM2 T66M mutation impairs microglia function, brain perfusion, and glucose metabolism. EMBO J. 36, 1837–1853 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mattsson N, Zetterberg H, Hansson O, Andreasen N, Parnetti L, Jonsson M, Herukka SK, van der Flier WM, Blankenstein MA, Ewers M, Rich K, Kaiser E, Verbeek M, Tsolaki M, Mulugeta E, Rosen E, Aarsland D, Visser PJ, Schroder J, Marcusson J, de Leon M, Hampel H, Scheltens P, Pirttila T, Wallin A, Jonhagen ME, Minthon L, Winblad B, Blennow K, CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. JAMA 302, 385–393 (2009). [DOI] [PubMed] [Google Scholar]
- 36.Snider BJ, Fagan AM, Roe C, Shah AR, Grant EA, Xiong C, Morris JC, Holtzman DM, Cerebrospinal fluid biomarkers and rate of cognitive decline in very mild dementia of the Alzheimer type. Arch. Neurol 66, 638–645 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rauchmann B-S, Schneider-Axmann T, Alexopoulos P, Perneczky R; Alzheimer’s Disease Neuroimaging Initiative, CSF soluble TREM2 as a measure of immune response along the Alzheimer’s disease continuum. Neurobiol. Aging 74, 182–190 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hamelin L, Lagarde J, Dorothee G, Leroy C, Labit M, Comley RA, de Souza LC, Corne H, Dauphinot L, Bertoux M, Dubois B, Gervais P, Colliot O, Potier MC, Bottlaender M, Sarazin M; Clinical IMABio3 Team, Early and protective microglial activation in Alzheimer’s disease: A prospective study using 18F-DPA-714 PET imaging. Brain 139, 1252–1264 (2016). [DOI] [PubMed] [Google Scholar]
- 39.Fan Z, Brooks DJ, Okello A, Edison P, An early and late peak in microglial activation in Alzheimer’s disease trajectory. Brain 140, 792–803 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dani M, Wood M, Mizoguchi R, Fan Z, Walker Z, Morgan R, Hinz R, Biju M, Kuruvilla T, Brooks DJ, Edison P, Microglial activation correlates in vivo with both tau and amyloid in Alzheimer’s disease. Brain 141, 2740–2754 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Ulland TK, Song WM, Huang SC-C, Ulrich JD, Sergushichev A, Beatty WL, Loboda AA, Zhou Y, Cairns NJ, Kambal A, Loginicheva E, Gilfillan S, Cella M, Virgin HW, Unanue ER, Wang Y, Artyomov MN, Holtzman DM, Colonna M, TREM2 maintains microglial metabolic fitness in Alzheimer’s disease. Cell 170, 649–663.e13 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ulrich JD, Ulland TK, Colonna M, Holtzman DM, Elucidating the role of TREM2 in Alzheimer’s disease. Neuron 94, 237–248 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Wang Y, Ulland TK, Ulrich JD, Song W, Tzaferis JA, Hole JT, Yuan P, Mahan TE, Shi Y, Gilfillan S, Cella M, Grutzendler J, DeMattos RB, Cirrito JR, Holtzman DM, Colonna M, TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J. Exp. Med 213, 667–675 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee CYD, Daggett A, Gu X, Jiang L-L, Langfelder P, Li X, Wang N, Zhao Y, Park CS, Cooper Y, Ferando I, Mody I, Coppola G, Xu H, Yang XW, Elevated TREM2 gene dosage reprograms microglia responsivity and ameliorates pathological phenotypes in Alzheimer’s disease models. Neuron 97, 1032–1048.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiang T, Tan L, Zhu X-C, Zhou JS, Cao L, Tan M-S, Wang H-F, C hen Q, Zhang YD, Yu J-T, Silencing of TREM2 exacerbates tau pathology, neurodegenerative changes, and spatial learning deficits in P301S tau transgenic mice. Neurobiol. Aging 36, 3176–3186 (2015). [DOI] [PubMed] [Google Scholar]
- 46.Jiang T, Zhang Y-D, Chen Q, Gao Q, Zhu XC, Zhou J-S, Shi J-Q, Lu H, Tan L, Yu J-T, TREM2 modifies microglial phenotype and provides neuroprotection in P301S tau transgenic mice. Neuropharmacology 105, 196–206 (2016). [DOI] [PubMed] [Google Scholar]
- 47.Jiang T, Zhang Y-D, Gao Q, Ou Z, Gong P-Y, Shi J-Q, Wu L, Zhou J-S, TREM2 ameliorates neuronal tau pathology through suppression of microglial inflammatory response. Inflammation 41, 811–823 (2018). [DOI] [PubMed] [Google Scholar]
- 48.Bemiller SM, McCray TJ, Allan K, Formica SV, Xu G, Wilson G, Kokiko-Cochran ON, Crish SD, Lasagna-Reeves CA, Ransohoff RM, Landreth GE, Lamb BT, TREM2 deficiency exacerbates tau pathology through dysregulated kinase signaling in a mouse model of tauopathy. Mol. Neurodegen 12, 74 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jiang T, Tan L, Zhu X-C, Zhang Q-Q, Cao L, Tan M-S, Gu L-Z, Wang H-F, Ding Z-Z, Zhang Y-D, Yu J-T, Upregulation of TREM2 ameliorates neuropathology and rescues spatial cognitive impairment in a transgenic mouse model of Alzheimer’s disease. Neuropsychopharmacology 39, 2949–2962 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leyns CEG, Ulrich JD, Finn MB, Stewart FR, Koscal LJ, Remolina Serrano J, Robinson GO, Anderson E, Colonna M, Holtzman DM, TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc. Natl. Acad. Sci. U.S.A 114, 11524–11529 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jiang Y, Li Z, Ma H, Cao X, Liu F, Tian A, Sun X, Li X, Wang J, Upregulation of TREM2 ameliorates neuroinflammatory responses and improves cognitive deficits triggered by surgical trauma in Appswe/PS1dE9 mice. Cell. Physiol. Biochem 46, 1398–1411 (2018). [DOI] [PubMed] [Google Scholar]
- 52.Sayed FA, Telpoukhovskaia M, Kodama L, Li Y, Zhou Y, Le D, Hauduc A, Ludwig C, Gao F, Clelland C, Zhan L, Cooper YA, Davalos D, Akassoglou K, Coppola G, Gan L, Differential effects of partial and complete loss of TREM2 on microglial injury response and tauopathy. Proc. Natl. Acad. Sci. U.S.A 115, 10172–10177 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jay TR, Hirsch AM, Broihier ML, Miller CM, Neilson LE, Ransohoff RM, Lamb BT, Landreth GE, Disease progression-dependent effects of TREM2 deficiency in a mouse model of Alzheimer’s disease. J. Neurosci 37, 637–647 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jay TR, Miller CM, Cheng PJ, Graham LC, Bemiller S, Broihier ML, Xu G, Margevicius D, Karlo JC, Sousa GL, Cotleur AC, Butovsky O, Bekris L, Staugaitis SM, Leverenz JB, Pimplikar SW, Landreth GE, Howell GR, Ransohoff RM, Lamb BT, TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J. Exp. Med 212, 287–295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sarlus H, Heneka MT, Microglia in Alzheimer’s disease. J. Clin. Invest 127, 3240–3249 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Poliani PL, Wang Y, Fontana E, Robinette ML, Yamanishi Y, Gilfillan S, Colonna M, TREM2 sustains microglial expansion during aging and response to demyelination. J. Clin. Invest 125, 2161–2170 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Petersen RC, Mild cognitive impairment as a diagnostic entity. J. Intern. Med 256, 183–194 (2004). [DOI] [PubMed] [Google Scholar]
- 58.Petersen RC, Aisen PS, Beckett LA, Donohue MC, Gamst AC, Harvey DJ, Jack CR Jr., Jagust WJ, Shaw LM, Toga AW, Trojanowski JQ, Weiner MW, Alzheimer’s Disease Neuroimaging Initiative (ADNI): Clinical characterization. Neurology 74, 201–209 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hansson O, Seibyl J, Stomrud E, Zetterberg H, Trojanowski JQ, Bittner T, Lifke V, Corradini V, Eichenlaub U, Batrla R, Buck K, Zink K, Rabe C, Blennow K, Shaw LM; Swedish BioFINDER Study Group; Alzheimer’s Disease Neuroimaging Initiative, CSF biomarkers of Alzheimer’s disease concord with amyloid-β PET and predict clinical progression: A study of fully automated immunoassays in BioFINDER and ADNI cohorts. Alzheimers Dement. 14, 1470–1481 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Skinner J, Carvalho JO, Potter GG, Thames A, Zelinski E, Crane PK, Gibbons LE; Alzheimer’s Disease Neuroimaging Initiative, The Alzheimer’s Disease Assessment Scale-Cognitive-Plus (ADAS-Cog-Plus): An expansion of the ADAS-Cog to improve responsiveness in MCI. Brain Imaging Behav. 6, 489–501 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tosun D, Schuff N, Shaw LM, Trojanowski JQ, Weiner MW; Alzheimer’s Disease Neuroimaging Initiative, Relationship between CSF biomarkers of Alzheimer’s disease and rates of regional cortical thinning in ADNI data. J. Alzheimers Dis 26 (suppl. 3), 77–90 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Reuter M, Schmansky NJ, Rosas HD, Fischl B, Within-subject template estimation for unbiased longitudinal image analysis. Neuroimage 61, 1402–1418 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jack CR Jr., Wiste HJ, Weigand SD, Therneau TM, Lowe VJ, Knopman DS, Gunter JL, Senjem ML, Jones DT, Kantarci K, Machulda MM, Mielke MM, Roberts RO, Vemuri P, Reyes DA, Petersen RC, Defining imaging biomarker cut points for brain aging and Alzheimer’s disease. Alzheimers Dement. 13, 205–216 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jack CR Jr., Barnes J, Bernstein MA, Borowski BJ, Brewer J, Clegg S, Dale AM, Carmichael O, Ching C, DeCarli C, Desikan RS, Fennema-Notestine C, Fjell AM, Fletcher E, Fox NC, Gunter J, Gutman BA, Holland D, Hua X, Insel P, Kantarci K, Killiany RJ, Krueger G, Leung KK, Mackin S, Maillard P, Malone IB, Mattsson N, McEvoy L, Modat M, Mueller S, Nosheny R, Ourselin S, Schuff N, Senjem ML, Simonson A, Thompson PM, Rettmann D, Vemuri P, Walhovd K, Zhao Y, Zuk S, Weiner M, Magnetic resonance imaging in Alzheimer’s Disease Neuroimaging Initiative 2. Alzheimers Dement. 11, 740–756 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC, Blennow K, Soares H, Simon A, Lewczuk P, Dean R, Siemers E, Potter W, Lee VM-Y, Trojanowski JQ, Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann. Neurol 65, 403–413 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Landau SM, Harvey D, Madison CM, Reiman EM, Foster NL, Aisen PS, Petersen RC, Shaw LM, Trojanowski JQ, Jack CR Jr., Weiner MW, Jagust WJ; Alzheimer’s Disease Neuroimaging Initiative, Comparing predictors of conversion and decline in mild cognitive impairment. Neurology 75, 230–238 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. CSF sTREM2 and CSF sTREM2 to p-tau181 ratio per group.
Fig. S2. Selection of the final subject group.
Fig. S3. Effect of CSF sTREM2 on changes in cognition in clinical subgroups.
Fig. S4. Effect of CSF sTREM2 measurements on hippocampal changes in MCI A+T+.
Table S1. Regression analyses with additional control for CSF total tau.
Table S2. Regression analyses with additional control for hippocampus volume.