

Summary
Background:
The small GTPase Rap1 and its guanine nucleotide exchange factor, CalDAG-GEFI (CDGI), are critical for platelet function and hemostatic plug formation. CDGI function is regulated by a calcium binding EF hand regulatory domain and an atypical C1 domain with unknown function.
Objective:
Here, we investigated whether the C1 domain controls CDGI subcellular localization, both in vitro and in vivo.
Methods:
CDGI interaction with phosphoinositides was studied by lipid co-sedimentation assays and molecular dynamics (MD) simulations. Cellular localization of CDGI was studied in heterologous cells by immunofluorescence and subcellular fractionation assays.
Results:
Lipid co-sedimentation studies demonstrated that the CDGI C1 domain associates with membranes through exclusive recognition of phosphoinositides, phosphatidylinositol (4,5)-biphosphate (PIP2) and phosphatidylinositol (3,4,5)-triphosphate (PIP3). MD simulations identified a phospholipid recognition motif consisting of residues exclusive to the CDGI C1 domain. Mutation of those residues abolished co-sedimentation of the C1 domain with lipid vesicles and impaired membrane localization of CDGI in heterologous cells.
Conclusion:
Our studies identify a novel interaction between an atypical C1 domain and PIP2/PIP3 in cellular membranes, which is critical for Rap1 signaling in health and disease.
Keywords: C1 domain, CalDAG-GEF, membrane, platelet, thrombosis
Introduction
Platelets are essential for primary hemostasis and repair of the endothelium, but they also play a key role in the development of acute coronary syndromes and contribute to cerebrovascular events1. To fulfill their role in hemostatic plug formation, platelets need to sense disruptions in the vascular lining and rapidly change from an anti-adhesive to a pro-adhesive state. The latter depends on a powerful intracellular signaling machinery that facilitates the inside-out activation of integrin receptors on the platelet surface, in particular the fibrinogen receptor, αIIbβ3. The final step in this inside-out activation is mediated by the assembly of the integrin activation complex, consisting of the cytoskeletal adapter proteins, talin and kindlin3, and the small GTPase Rap1.
Rap1 is a member of the Ras subclass of Ras superfamily GTPases2. It is a small (~21 kD) guanine nucleotide binding protein which functions as a highly regulated molecular switch that cycles between inactive GDP and active GTP bound states to control cell signaling processes. The high affinity for guanine nucleotides and the low intrinsic GTPase activity necessitate the action of regulatory factors to control its activity. Guanine nucleotide exchange factors (GEFs) facilitate exchange of bound GDP for GTP to promote activation, whereas inactivation is achieved by GTPase Activating Proteins (GAPs) that stimulate GTP hydrolysis. Rap1 undergoes lipid modification, specifically geranyl-geranylation, to facilitate membrane association3. In platelets, both Rap1 isoforms, Rap1A and Rap1B, contribute to integrin-mediated adhesion and hemostatic plug formation, with Rap1B being the dominant isoform4,5. Platelet Rap1B activity is controlled by calcium and diacylglycerol-regulated guanine nucleotide exchange factor 1, CalDAG-GEFI (CDGI), also known as RasGRP2, and a GAP, Rasa3 (Gap1IP4bp)6,7,8. We and others have shown previously that CDGI is critical for the near-immediate activation of Rap1 and integrin αIIbβ3 in platelets stimulated with physiological agonists9,10,11. Mice lacking CDGI exhibit a marked protection from arterial thrombosis and prolonged bleeding after vascular injury6,12,13. In humans, mutations in CDGI also cause impaired platelet function and a moderate to severe bleeding phenotype10,14,15,16,17,18,19. Interestingly, in vitro and in vivo platelet function is markedly impaired in mice and humans expressing a truncated CDGI lacking the atypical C1 regulatory domain13,16. The role of this C1 domain in CDGI function, however, is not understood.
The various members of the CalDAG-GEF (RasGRP) family of GEFs are important activators of Ras and Rap GTPases20. They are multi-domain proteins containing a Ras Exchange Motif (REM), Cdc25 catalytic domain, EF hand and C1 regulatory domains. The Cdc25 domain contains a helical hairpin that removes GDP when Ras or Rap binds to the catalytic pocket, thereby facilitating exchange of GDP for GTP to promote GTPase activation21. Our recent biochemical and biophysical work demonstrated that calcium binding to the CDGI EF hand domain leads to release of autoinhibition and induction of nucleotide exchange activity22. The C1 domains comprise a family of ~60 members, and contain two β-sheets and an α-helix that promote zinc coordination via two sets of three cysteines and one histidine23. C1 domains were originally characterized as diacylglycerol (DAG) binding motifs in Protein Kinase C (PKC) proteins where they facilitate activation by translocation of the enzyme to the membrane and by release of autoinhibitory interactions. In addition to lipid binding, C1 domains can serve as protein interaction sites24. While CDGII (RasGRP1), CDGIII (RasGRP3) and CDGIV (RasGRP4) proteins all contain a typical C1 domain for regulation by DAG, CDGI contains an atypical, non-canonical C1 domain with very low capacity to bind and respond to DAG25.
In this work, we present evidence that the CDGI C1 domain rather mediates interaction with phosphoinositides in the plasma membrane and plays a key role in membrane localization of CDGI to promote Rap1B activation. We demonstrate that the isolated C1 domain and intact CDGI co-sediment with liposomes containing common plasma membrane phosphoinositides, such as PIP2 or PIP3. However, CDGIΔC1, that lacks the C1 domain, is unable to associate with membranes containing these phosphoinositides. Computational modeling in the presence of model membrane systems shows specific interactions with both PIP2 and PIP3 through a subset of C1 domain residues, namely R508, R513 and R530. Mutations within the CDGI C1 domain predicted to disrupt phosphoinositide interactions exhibit a dramatic reduction in its association with PIP2-containing liposomes and impair membrane localization of CDGI in heterologous cells, thus validating key interacting residues indicated by our computational models. A particular residue exclusively present in the C1 domain of CDGI, namely R513, appears to play a crucial coordinating role within the cluster of interacting residues for phosphoinositide binding.
Results
Atypical C1 domain is critical for CDGI-Rap1 signaling in platelets.
Like other CDG (RasGRP) proteins, CDGI contains typical REM, GEF/Cdc25 and EF hand domains, but an atypical C1 domain (Figure 1A). Our previous work identified a significant defect in platelet adhesion to collagen under physiological flow conditions in chimeric mice expressing a truncated version of CDGI lacking the C1 domain in blood cells13. Consistent with a defect in the inside-out activation of αIIbβ3, the main integrin receptor expressed on the platelet surface, CDGIΔC1 platelets showed a defect in their in vitro aggregation response (Figure 1B) and Rap1 activation (Figure 1C) when stimulated with convulxin, a selective agonist for the collagen receptor GPVI. These studies suggest a critical role for the C1 domain in CDGI function. To test whether deletion of the C1 domain affects the catalytic activity of CDGI, we next tested purified CDGI protein containing all functional domains (consisting of residues 1–551, referred to simply as CDGI) and the C1-truncated protein (consisting of residues 1–499, referred to as CDGIΔC1) for catalytic activity towards nucleotide exchange in Rap1B22. CDG1ΔC1 lacking the C1 domain showed similar nucleotide exchange activity towards Rap1B when compared to CDG1 (Figure 1D). These studies demonstrate that the C1 domain is critical for CDGI-Rap1 signaling in platelets, and that it contributes to CDGI function by a mechanism other than the regulation of nucleotide exchange activity.
Figure 1: The atypical C1 domain plays an important role for CDGI/Rap1-mediated platelet aggregation.

(A) Schematic representation of CDGI domain structure. Shown are full-length CDGI protein (609 residues), a C-terminally truncated CDGI that contains all functional domains (1–551), CDGI lacking the C1 domain (CDGIΔC1; 1–499) and the isolated C1 domain (492–551). (B, C) Aggregation and Rap1 activation response in platelets expressing CDGIΔC1. Platelets isolated from wild-type (+/+), CDGI heterozygous (+/−), CDGI knockout (−/−) and chimeric mice expressing CDGIΔC1 (ΔC1)13 were activated with convulxin (GPVI agonist) and aggregation was recorded over time (B). At the indicated time points, platelets were lysed and Rap1-GTP levels were determined (C). Results are representative of 3 independent experiments. (D) Nucleotide exchange activity assay. Purified CDGI or CDGIΔC1 protein was added to Rap1B in the presence of BODIPY-GDP. The change in fluorescence intensity due to binding of BODIPY-GDP to Rap1B was measured over time. Results are representative of 5 independent experiments.
The C1 domain facilitates CDGI membrane localization.
We next tested whether the C1 domain plays a role in controlling the subcellular localization of CDGI. Green fluorescence protein (GFP)-tagged CDGI was expressed in GripTite™ 293 MSR human embryonic kidney cells and subcellular localization was determined by confocal microscopy (Figure 2A). Compared to CDGI-GFP, which showed strong membrane localization in adherent GripTite™ 293 MSR cells, CDGIΔC1-GFP showed a more cytosolic expression pattern. To confirm that the C1 domain mediates CDGI localization to membranes, we performed subcellular fractionation studies in cells expressing (HA)-tagged CDGI and/or CDGIΔC1 (Figure 2B). Samples were probed for sTransferrin receptor and GAPDH as positive controls for membrane/microsome and cytosolic localization. The protein ratio between the membrane and cytosolic fraction was determined (Figure 2C). Compared to CDGI, CDGIΔC1 exhibited a significantly lower membrane/cytosol ratio, confirming that the C1 domain targets CDGI to membranes.
Figure 2: Subcellular localization of full-length CDGI and CDGIΔC1.

(A) GFP-tagged CDGI or CDGIΔC1 were transfected into GripTite™ 293 MSR cells and subcellular localization was determined by epifluorescence microscopy. Results are representative of 3 independent experiments. (B) HA-tagged CDGI (CDGI-HA; ~75 kDa), HA-only vector (empty vector), or HA-tagged CDGIΔC1 (CDGIΔC1-HA; ~60 kDa) were transfected into GripTite™ 293 MSR cells and assayed for membrane and cytosolic localization by subcellular fractionation. GAPDH (~40 kDa) and sTransferrin receptor (~110 kDa) were included as predominantly cytosolic and membrane proteins, respectively. (C) Quantification of studies described in (B). Band intensities were determined and expressed as the ratio of membrane fraction to cytosolic fraction (data are mean ± standard deviation (SD); n=4).
The C1 domain of CDGI preferentially interacts with phosphoinositides.
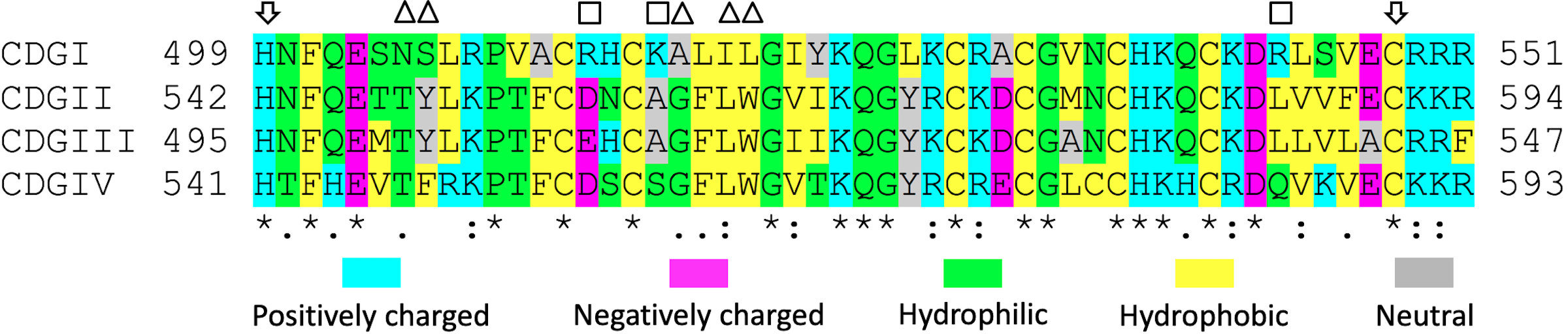
Sequence comparison of CDGI with the other three CDG family members highlights key differences in residues important for DAG/phorbol ester binding (Figure 3A, Supplemental Figure S1), consistent with previous reports showing very low affinity of the CDGI C1 domain for DAG25. Interestingly, this domain contains several positively charged residues that are unique to CDGI, suggesting that one or more of these residues could confer binding to negatively charged phospholipids in the plasma membrane. To test this hypothesis, we conducted co-sedimentation studies of CDGI with plasma membrane mimetic liposomes containing phosphatidylcholine (PC) and phosphatidylethanolamine (PE), negatively charged phospholipid phosphatidylserine (PS) as a control as well as PS substituted by compensating amounts of either PIP2 or PIP3, to probe specificity for these phosphoinositides. A dose-dependent interaction between CDGI and PIP2 or PIP3 was observed (Figure 3B). Interestingly, similar interaction profiles were obtained for both PIP2 and PIP3. A dramatic reduction in binding to either PIP2 or PIP3 was observed for CDGIΔC1 compared to CDGI (Figure 3C), indicating a role of the C1 domain in PIP2/PIP3 recognition. Next, we compared the PIP2 binding specificity between the isolated C1 domains of CDGI and CDGII, using the Vinculin tail (Vt) domain, a protein known to preferentially interact with PIP226,27 as a positive control. While very limited co-sedimentation with PIP2 was observed for the typical, DAG-binding C1 domain of CDGII, co-sedimentation of Vt and the C1 domain of CDGI demonstrated association with PIP2 (Figure 3D). Taken together, these lipid binding studies demonstrate that CDGI preferentially interacts with phosphoinositides, PIP2 and PIP3, and that its atypical C1 domain plays a key role in phosphoinositide specificity.
Figure 3: The C1 domain of CDGI associates with the phosphoinositides, PIP2 and PIP3.

(A) Sequence comparison of the human CDG isoforms. Numbers correspond to the residue positions in the full-length proteins. Gray shading highlights the C1 domain boundaries. Complete (*) or partial (: and .) conservation of the residues are indicated. Blue shading indicates critical residues needed for DAG/phorbol ester binding by typical C1 domains of CDGII, CDGIII and CDGIV28,29. Green shading indicates positively charged residues (R513, K516 and R543) present only in the atypical C1 domain of CDGI. (B) Co-sedimentation assays using liposomes, formed from PC (60%) and PE (25%) plus molar equivalent compensating amounts of PS between (15% to 0%) and PIP2 or PIP3 (0% to 15%), show concentration dependent CDGI association profiles for both PIP2 and PIP3. The amount (%) of bound CDGI corresponds to the dynamic range based normalized values obtained by subtracting the scaled basal level binding by non-PIP2/PIP3 liposome components. The error bars correspond to SD, n=3. (C) Association of CDGI with either PIP2 or PIP3 is significantly reduced upon C1 domain deletion. Data are shown for liposomes containing 15% PIP2 or PIP3 along with 60% PC and 25% PE, but lacking PS. The amount (%) of bound protein corresponds to the normalized values obtained by subtracting the scaled basal level binding by non-PIP2/PIP3 components. The error bars correspond to SD (6 experimental replicates). (D) Concentration dependent association of PIP2 is observed for the Vinculin tail (Vt) domain (positive control) and the C1 domain of CDGI, but not that of CDGII. The amount (%) of bound protein corresponds to the normalized values obtained by subtracting the scaled basal level binding by non-PIP2 liposome components. The error bars correspond to SD (3 experimental replicates for Vt, 6 experimental replicates for C1(CDGI) and C1(CDGII)).
Molecular dynamics simulations identify key C1 domain residues involved in PIP2-PIP3 interaction.
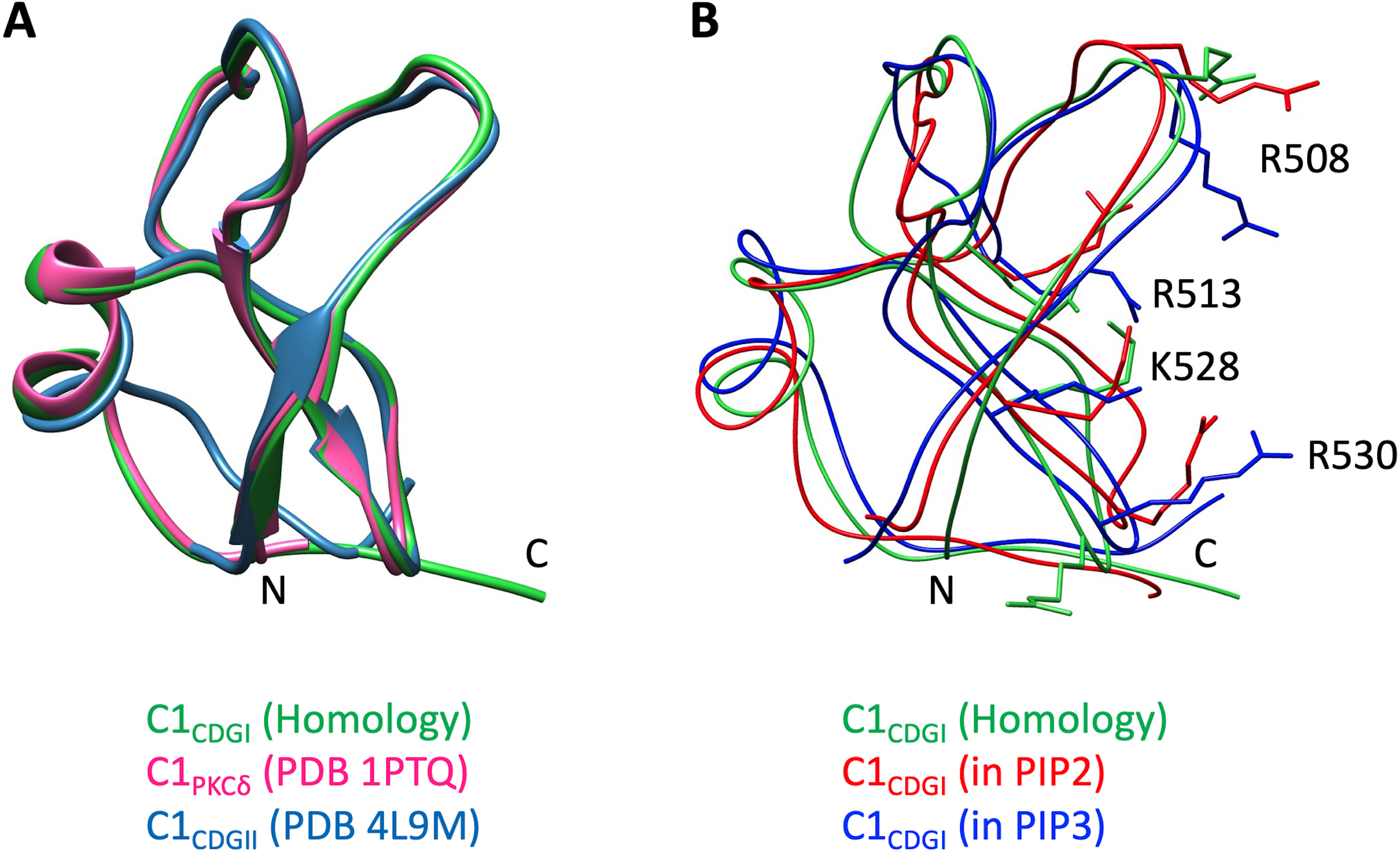
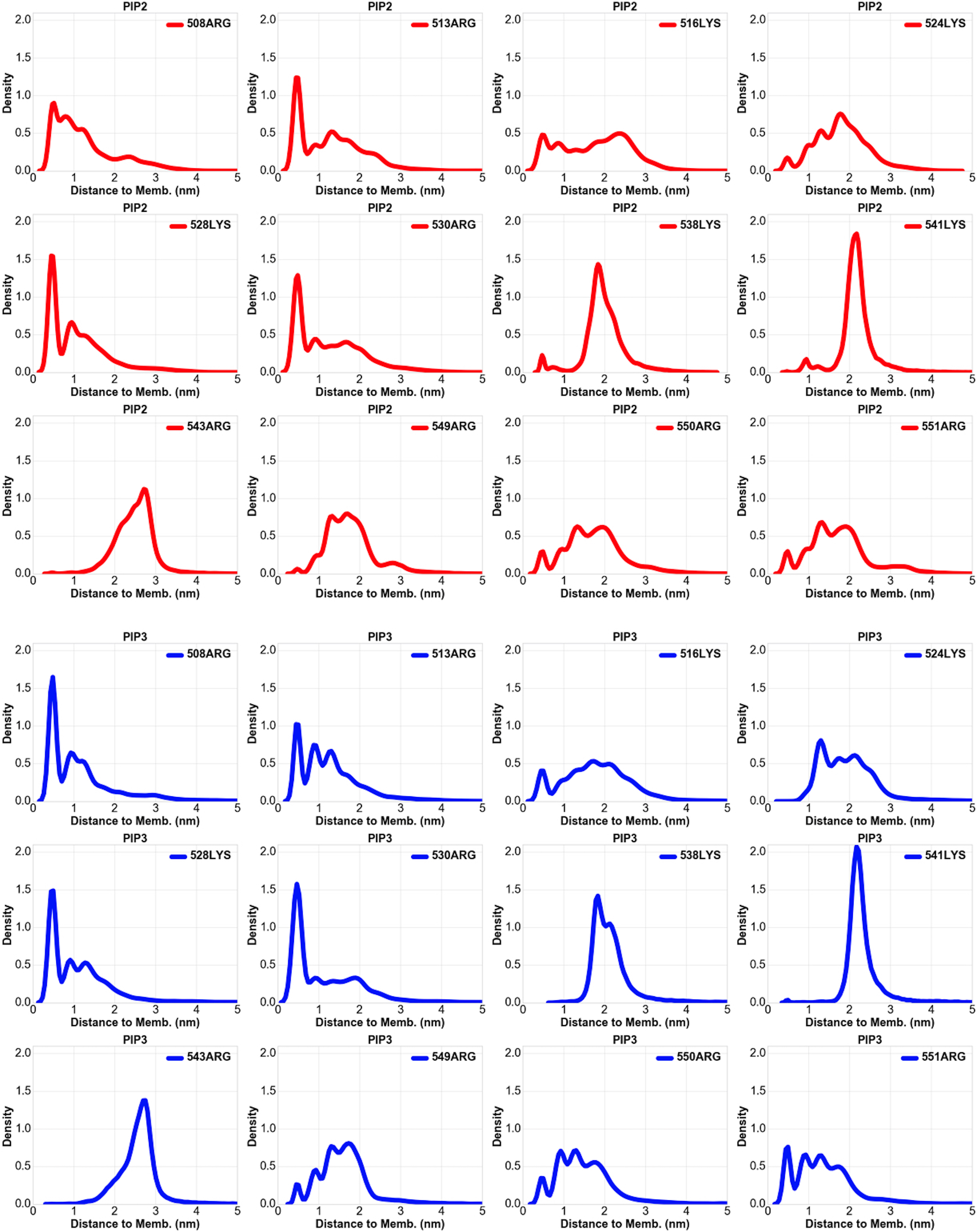
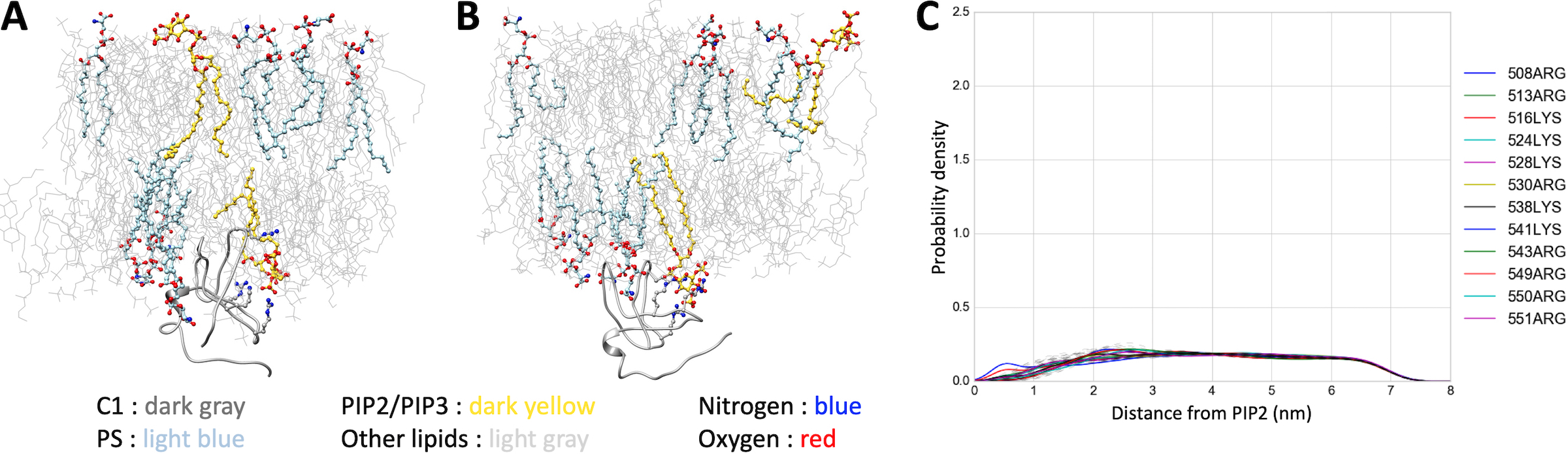
Following experimental validation that the C1 domain of CDGI provides specificity for phosphoinositide binding in model liposome membranes, we next performed molecular dynamics (MD) simulations to gain molecular insight into the observed phosphoinositide specificity. We generated a homology model of the CDGI C1 domain (residues 499–551) using the C1 domain of PKCδ as the modeling template (PDB ID 1PTQ, 44% sequence identity)30 (Supplemental Figure S2). Mimicking physiological conditions, our model membranes contained 45% 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 21% 1-palmitoyl-2-oleoyl-sn-glycero-phosphoethanolamine (POPE), 25% cholesterol, 7% 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine (POPS) and either 2% PIP2 or 2% PIP3, while an additional membrane lacking PIP2 or PIP3 was employed as a negative control (Figure 4 A,B). Kernel density estimation for distances between all of the arginine and lysine residues showed interaction proximity between the headgroups of PIP2 or PIP3 and a subset of the C1 domain residues consisting of R508, R513, K528 and R530 (Figure 4 C,D, Supplemental Figure S3). Interestingly, R513 is the only residue within this subset that is exclusive to the C1 domain of CDGI, while the other two exclusive residues, namely R516 and R543, were not found to be involved in interactions with PIP2/PIP3 (Supplemental Figure S3). Notably, residues R508, R513, K528 and R530 did not interact with negatively charged headgroups of PS despite being in 3.5-fold excess (Supplemental Figure S4).
Figure 4: Computational modeling identified specific residues mediating the interaction of the CDGI C1 domain with inositol phospholipids PIP2 and PIP3 in model membranes.

Lipid membranes used in the simulations mimic cellular membranes and are composed of zwitterionic phospholipids PC (45%) and PE (21%), cholesterol (25%), and acidic phospholipids PS (7%) and PIP2 (2%) (A) or PIP3 (2%) (B). The C1 domain is represented by yellow ribbon and PIP2/PIP3 by spheres with red representing phosphate groups. Kernel density estimation for distances between the C1 domain residues and the headgroups of PIP2 (C) and PIP3 (D) were generated for the coarse grain simulations prior to all atom simulation. The control PS system did not show residue specific interactions (Supplemental Figure S4). Both PIP2 and PIP3 exhibited preferential interactions with a subset of C1 domain basic residues, namely, R508, R513, K528 and R530, as the probability of finding these residues within 0.5 nm was significantly increased.
Site-directed mutagenesis confirms key residues involved in C1 domain PIP2/PIP3 interaction.
As the MD simulations indicated that the CDGI C1 domain specifically recognizes PIP2 and PIP3 via residues R508, R513, K528 and R530 (Figure 5 A,B), we conducted mutagenesis experiments to test these observations. Liposome co-sedimentation studies demonstrated substantial reduction in PIP2-membrane binding by the R508A+R530A mutant compared to the wild-type (WT) C1 domain. Notably, binding was almost fully abolished for the R508A+R530A+R513A mutant (Figure 5C). Circular dichroism (CD) spectroscopy revealed that both the double and triple C1 mutants retain similar conformation when compared to the WT control (Figure 5D), indicating that the PIP2/PIP3 association defect was due to loss of these key residues rather than a major structural perturbation.
Figure 5: The C1 domain of CDGI interacts with PIP2 and PIP3 acidic headgroups through basic residues R508, R513, K528 and R530.

(A, B) Corresponding zoomed in regions from MD simulations (Figure 4) in PIP2 and PIP3 containing bilayer systems, respectively. Residues R508, R513, K528 and R530 do not interact with acidic headgroups of PS despite being in 3.5-fold excess (Supplemental Figure S3), supporting predictions from MD simulations that the CDGI C1 domain specifically recognizes inositol phospholipids via these residues. (C) Liposome co-sedimentation studies demonstrated substantial reduction in PIP2-membrane binding by the R508A+R530A C1 mutant compared to the wild-type (WT) C1 domain, while binding was almost fully abolished for the R508A+R530A+R513A C1 mutant, consistent with interactions predicted by MD simulations. (D) Comparison of mean residue ellipticity CD spectra performed on WT and mutant C1 domains, indicate that the C1 mutants retain the protein fold.
Subcellular fractionation demonstrates role of specific C1 residues in membrane association.
Our lipid co-sedimentation studies identified residues R508, R513 and R530 as critical for C1 domain binding to phosphoinositides. To confirm the importance of these residues for CDGI interaction with membranes in a cellular context, we expressed a CDGI variant containing mutations R508A/R513A/R530A in GripTite™ 293 MSR cells, and conducted subcellular fractionation studies (Figure 6). Comparable expression levels of the different CDGI variants (CDGI, CDGIΔC1 and CDGI R308A/R513A/R530A) were observed in transfected cells. The membrane to cytosol ratio of CDGI R308A/R513A/R530A was significantly reduced when compared to CDGI control. The change in subcellular localization observed for the triple mutant was comparable to that of CDGIΔC1. In combination with the lipid co-sedimentation data, these results provide strong evidence for a specific interation of R508, R513 and R530 with phosphoinositides in the cell membrane.
Figure 6: Arginines 508, 513, and 530 are critical for the interaction of CDGI with cellular membranes.

. (A) HA-tagged CDGI (Lane 1; CDGI-HA; ~75 kDa), HA tagged CDGIΔC1 (Lane 2; CDGIΔC1-HA; ~60 kD), or HA-tagged CDGI R508A/R513A/R530A triple mutant (Lane 3; CDGI(R508A/R513A/R530A)-HA) were transfected into GripTite™ 293 MSR cells and assayed for microsome/membrane or cytosolic localization by subcellular fractionation. GAPDH (~40 kDa) and sTransferrin receptor (~110 kDa) were included as a predominantly cytosolic and membrane protein, respectively. (B) Quantification of studies described in (A). Band intensities were determined and expressed as the ratio of membrane fraction to cytosolic fraction (data are mean ± SD; n=3).
Discussion
Platelet adhesion at sites of vascular injury is dependent on a powerful intracellular signaling machinery that facilitates the near-immediate activation of integrin receptors on the cell surface. The CDGI/Rap1 signaling module plays a central role in this process8. Critical to CDGI catalytic activity regulation is a pair of EF hands in its regulatory domain, which have high affinity for calcium21,22. In this study, we demonstrate that an atypical C1 regulatory domain controls CDGI subcellular localization by interacting with phosphoinositides in cellular membranes. Deletion of the C1 domain or disruption of the C1-PIP2/PIP3 interface leads to translocation of CDGI to the cytosol, thereby limiting its ability to activate membrane-bound Rap1. Loss of the C1 domain in CDGI markedly impairs the adhesive function of platelets in mice and humans13,16 demonstrating the physiological importance of this novel interaction between an atypical C1 domain and phosphoinositides in cellular membranes.
The integrin activation complex requires its components, namely Rap1, talin and kindlin 3, to assemble in close proximity of the integrin cytoplasmic tail. In platelets, deficiency in either of these proteins leads to almost complete inhibition of αIIβ3 integrin activation and platelet aggregation5,31,32,33. Both talin and kindlin3 directly interact with αIIbβ3 via distinct binding sites within the β3 tail, and both proteins are critical for the inside-out activation of integrin receptors34. The molecular mechanisms by which Rap1 interacts with the talin-kindlin3-integrin complex are not fully understood at the moment. Studies in mutant mice demonstrated that Rap1-interacting molecule 1, Riam1, an important adapter for Rap1 and talin in leukocytes, is not important in platelet integrin signaling35,36. Recent studies provided evidence that Rap1 can directly interact with talin, and that disrupting this interaction leads to partially impaired integrin activation in murine platelets37,38,39.
The head domain of talin also harbors binding sites for phosphoinositides within its F2F3 domain. In its autoinhibited confirmation, the interaction of talin with anionic lipids is inhibited by the talin rod domain. However, PIP2 was shown to bind to autoinhibited talin, suggesting that PIP2 binding may recruit talin to the plasma membrane and also contribute to its activation at focal adhesions40. Geranylgeranylated Rap1 is already found in the plasma membrane and may be recruited to areas of phosphoinositide enrichment via its interaction with talin. However, Rap1 activity is under the control of both positive (GEFs) and negative (GAPs) regulators, which need to be recruited, and potentially also sequestered away from Rap1 in order to facilitate its activation and inhibition. We recently identified the RapGAP, Rasa3, as an important inhibitor of Rap1 signaling in platelets7. Rasa3 contains a unique PH/Btk regulatory domain that facilitates its interaction with PIP2 and PIP3, and in circulating platelets Rasa3 is found associated with the plasma membrane41. The studies presented here provide direct evidence that CDGI, a main activator of platelet Rap1, uses an atypical C1 domain to interact with PIP2 and PIP3. Thus, both regulators of Rap1 activity and talin interact with the same phospholipids in the plasma membrane. Our work provides a foundation for future studies to uncover how these interactions are spatio-temporally regulated for Rap1 signaling to occur.
CDGI is not the only GEF expressed in platelets that is regulated by its phosphoinositides. For example, the Rac-GEF, guanine nucleotide exchange factor phosphatidylinositol 3,4,5-trisphosphate-dependent Rac exchanger 1 (P-Rex1) protein, contains a pleckstrin homology (PH) domain that is required for PIP3 binding. In addition to its role in protein targeting, the PIP3-PH interaction is also thought to contribute to P-Rex activation via release of autoinhibition42,43. The activity of another platelet Rac-GEF, Vav, is also upregulated by PIP3 binding to its PH domain44,45. While PH domains are known phosphoinositide binders, our observations that the isolated C1 domain of CDG1 specifically recognizes phosphoinositides required for membrane association of CDG1 is novel. C1 domains are classified into two subtypes, typical and atypical. Typical C1 domains bind DAG whereas atypical do not46. C1 domain function is perhaps best characterized for a subset of PKC isoforms, where they play a key role in membrane association and kinase activation47. Our findings establish the atypical C1 domain in CDGI as an important domain that mediates membrane localization. These findings are consistent with studies showing that atypical C1 domains contained in other proteins interact with lipids distinct from DAG, including negatively charged phospholipids23. In addition to lipids, atypical C1 domains can also recognize proteins24. For example, the atypical Raf kinase C1 domain binds PS and RAS, and may form intramolecular interactions that control kinase activity48,49. Interestingly, the inactive conformation of CDGII is stabilized by a C1-dimer interface which prevents membrane localization. This homodimer formation is stabilized by a C-terminal coiled coil sequence21. However, both homodimerization and the coiled coil sequence are missing in CDGI. Future studies will be required to determine if, in addition to phosphoinositide-dependent membrane association, the C1 domain of CDGI forms intramolecular interactions or facilitates interaction with new binding partners that may contribute to CDGI activity modulation.
Our studies have important clinical implications. Mutations in RasGRP2, the gene encoding CDGI, have recently been recogized as a new class of platelet function disorder19,50. While most variants lead to impaired expression of CDGI in patient platelets, some impair the function of an expressed protein. Interestingly, several patients with a frame shift mutation just upstream of the CDGI C1 domain were identified16,19 and expression of a truncated protein in platelets was reported for one of these families16. Our studies provide a molecular mechanism for why CDGI/Rap1-mediated platelet adhesion is impaired in these patients. As shown in our cell- and liposome-free activity assay, CDGIΔC1 retains its ability to promote nucleotide exchange on Rap1 when both proteins are combined in solution. The mutant protein, however, is impaired in its ability to interact with phosphoinositides in liposomes and cellular membranes, a defect that indirectly reduces GTP loading of Rap1. Consistent with this residual activity, defects in Rap1 activation and integrin-mediated platelet adhesion were significantly milder in mice expressing CDGIΔC1 when compared to mice lacking the protein13. Patients expressing a reduced level of truncated CDGI present with a moderate to severe bleeding diathesis, with the severity of bleeding decreasing during adulthood16.
The relatively mild bleeding defect in mice and adult human patients expressing CDGI lacking the C1 domain provides a rationale for targeting the C1-phosphoinositide interaction in the prevention of atherothrombosis. Our studies in mice expressing CDGIΔC1 and mice expressing low levels of CDGI demonstrated strong protection from experimental thrombosis, including models of arterial thrombosis and immune complex-mediated thrombocytopenia and thrombosis13,51. On a mechanistic level the protection from arterial thrombosis is most likely explained by a delay and reduction in integrin activation, the key event in platelet aggregation. CDGI plays a critical role in the near-immediate activation of αIIbβ38, with deficiency in the protein leading to markedly delayed platelet aggregation in mice and humans. Impaired targeting of CDGI to the plasma membrane also leads to delayed platelet aggregation, but the defect is more subtle than in knockout mice. Under high shear stress conditions, such as found in arteries and arterioles, even this subtle delay in integrin activation cannot be compensated for by the alternative, slower pathway leading to Rap1 activation. Under lower shear stress conditions, however, small hemostatic plugs can form. Thus, targeting specifically the interaction between the CDGI C1 domain and membrane phosphoinositides may be a powerful new strategy to prevent platelet-driven arterial thrombosis while preserving hemostasis.
In summary, we here identify a key role for the C1 domain of CDGI in membrane association. Moreover, the CDGI C1 domain provides a novel example of an atypical C1 domain that interacts with PIP2 and PIP3 in membranes. In the Rap GTPase activator, CDGI, specific C1 residues R508, R513, and R530 contribute to phosphoinositide specificity, facilitating its recruitment to Rap1 and the integrin activation complex in the platelet membrane. Deletion of the CDGI C1 domain leads to impaired platelet adhesive function and reduced hemostatic/thrombotic plug formation in mice and humans.
Methods
Plasmid constructs
Plasmid for the full-length human CDGI containing residues 1–609 was purchased from the Human ORFeome v5.1 of Harvard University. The gene for the C-terminal truncated CDGI containing residues 1–551 was PCR-amplified and sub-cloned into a p15LIC2 vector with a 6-His tag at the N-terminus following the ligation-independent cloning protocol52. The C1 domain deletion, CDGIΔC1 containing residues 1–499, was prepared by introducing a stop codon at position 500, using Quikchange site-directed mutagenesis kit (Stratagene) following the manufacturer’s protocol. The C1 domain construct of CDGI encoding residues 492–551 was modified with BamHI and XhoI cut sites for digestion and insert ligation and cloned into a pET-SUMO vector. The R508A+R530A C1 mutant was generated by PCR mutagenesis. R513A and R508A+R530A+R513A mutants of the C1 domain of CDGI were purchased from GenScript. Human Rap1B containing residues 1–181 (with C181S mutation) was cloned into pProEXHtb vector.
Protein Expression and Purification
Truncated CDGI and CDGIΔC1 vectors were transformed into the Rosetta E. coli strain (Novagen). Cells were first grown at 37 °C to an optical density between 0.6–0.8 (600 nm) in lysogeny broth medium supplemented with 1 μM ZnSO4, 100 μg/ml ampicillin and 35 μg/ml chloramphenicol. Protein expression was then induced by addition of 0.5 mM isopropyl-D-1-thiogalactopyranoside (IPTG). Cells were then grown at 18 °C overnight and harvested by centrifugation (4500 RPM, 30 min, 4 °C). Cell pellets were resuspended in lysis buffer (20 mM PIPES (pH 6.8), 300 mM NaCl, 1 μM ZnSO4, 10 mM imidazole, 10 mM β-mercaptoethanol, 5% glycerol and protease inhibitor cocktail (Roche)) and lysed by sonication. The soluble fraction was separated from the particulate fraction by centrifugation (15000 RPM, 45 min, 4 °C). Filtered supernatant was then applied to nickel-nitrilotriacetic acid (Ni-NTA)-agarose beads (Qiagen) equilibrated with wash buffer (20 mM PIPES (pH 6.8), 300 mM NaCl, 10 mM imidazole and 5% glycerol). Ni-bound protein was eluted with elution buffer (20 mM PIPES (pH 6.8), 300 mM NaCl, 400 mM imidazole and 5% glycerol). For His-tag removal, the eluted volume was dialyzed into tobacco etch virus (TEV) cleavage buffer (20 mM PIPES (pH 6.8), 300 mM NaCl, 10 mM imidazole and 5% glycerol) overnight at 4 °C in the presence of TEV. Cleaved His-tag and TEV protease were removed by passing the dialysate over Ni-NTA affinity column. Flow-through fractions were analyzed by SDS-PAGE and desired protein containing fractions were concentrated using Amicon Ultra 15 centrifugal concentrator (Millipore) with 10 kD MWCO filter. The concentrated protein was further purified by size exclusion chromatography using an S100 column in FPLC buffer (40 mM HEPES (pH 7.0), 100 mM NaCl, 15 μM ZnSO4 and 1 mM DTT).
All C1 domain constructs were expressed in BL21 (DE3) RIPL E. coli strain (Agilent in a similar manner described earlier. Purification steps were also similar. The cells were lysed in lysis buffer (20 mM HEPES (pH 7.0), 250 mM NaCl, 1 μM ZnSO4, 5 mM β-mercaptoethanol, 10 mM imidazole and protease inhibitor cocktail (Roche)), wash buffer (20 mM HEPES (pH 7.0), 250 mM NaCl, 1 μM ZnSO4, 40 mM imidazole and 5 mM β-mercaptoethanol) and elution buffer (20 mM HEPES (pH 7.0), 250 mM NaCl, 1 μM ZnSO4, 400 mM imidazole and 5 mM β-mercaptoethanol). The N-terminal His-Sumo tag was cleaved by dialyzing Ni-NTA eluent with 1 ml of 2 mg/ml ubiquitin-like-specific protease 1 (ULP1) overnight in cleavage buffer (20 mM HEPES (pH 7.0), 250 mM NaCl, 1 μM ZnSO4, 10 mM imidazole and 5 mM β-mercaptoethanol) at 4°C. The cleaved His-sumo tag was removed by a second round of loading to Ni-NTA column. Flow-through fractions were concentrated using Amicon Ultra centrifugal concentrator (Millipore) with 3 kD MWCO filter and loaded onto S75 size exclusion column equilibrated with FPLC buffer (40 mM HEPES (pH 7.0), 100 mM NaCl, 15 μM ZnSO4 and 1 mM DTT) for final purification. Rap1B was expressed in E. coli strain Rosetta 2 BL21 (Novagen), induced with 200 μM IPTG for 10 hours at 20°C. Protein purification was done using the same chromatographic steps as for CDGI proteins, with buffer modifications substituting the PIPES buffer with Tri-HCl (pH 7.0), 5 mM MgCl2 for 1 μM ZnSO4 and addition of 50 μM GDP to the protein lysis buffer. All purified proteins were pooled, concentrated, aliquoted, flash frozen in liquid nitrogen, and stored at −80°C.
Nucleotide exchange assay
Nucleotide exchange was measured using a fluorescence-based assay as previously described22. Purified Rap1B (1μM) and BODIPY-GDP (1μM; Life technologies) were incubated in 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 5 mM MgCl2, 1 mM DTT, 5% glycerol, and 0.004% NP-40, prior to addition of 400 nM CDGI proteins for a final volume of 100 μl using a black bottom 96 well plate (Corning). Fluorescent signal was measured using a Pherastar (BMG LABTECH; λex/em = 480/520 nm; 1 nm slits).
Mice
CDGI−/−6 and chimeric mice expressing CDGIΔC1 in blood cells only13 have been described previously. Experimental procedures were approved by the Animal Care and Use Committee of Thomas Jefferson University (Philadelphia, USA) and of the University of North Carolina (Chapel Hill, USA).
Preparation of platelet-rich plasma of chimeric mice
Blood was drawn from the retro-orbital plexus into heparinized tubes. Platelet-rich plasma (PRP) was obtained by centrifugation at 100g for 5 min. When necessary, PRP was diluted in modified Tyrode’s Buffer (137 mM NaCl, 0.3 mM Na2HPO4 (pH 7.3), 2mM KCl, 12 mM NaHCO3, 5 mM N-2-hydroxyethylpiperazine-N’−2-ethanesulfonic acid, 5 mM glucose) to ensure that each sample had a final platelet count of 3 × 108 platelets/ml.
Aggregometry and Rap1 activation assay
Platelets were incubated for 10 minutes with 100 μM 2-MeSAMP (P2Y12 inhibitor) and then stimulated with 650 ng/ml convulxin (specific agonist for the collagen receptor GPVI). The experiment was performed at 37°C and under stirring conditions (1200 rpm) and light transmission was recorded on a Chrono-log 4-channel optical aggregation system (Chrono-log, Havertown, PA). Reactions were stopped after 1 or 10 minutes with ice-cold 2x lysis buffer (100 mM Tris-HCl (pH 7.4), 400 mM NaCl, 5 mM MgCl2, 2% Nonidet P-40, 20% glycerol and protease inhibitor cocktail lacking ethylenediaminetetraacetic acid). Cell lysis was completed on ice for 15 minutes. The cell lysates were incubated for 45 minutes with RalGDS-RBD beads (Millipore, Billerica, MA) to pull-down Rap1-GTP. After three washing steps the pellets were solubilized in sample buffer for the detection of active Rap1 by immunoblot.
Subcellular fractionation
GripTiteTM 293 MSR cells (ThermoFischer) were transfected with the vectors containing the different CDGI variants (500 ng) using PolyJet transfection reagent (SignaGen Laboratories). Thirty-six hours after transfection, cells were scrapped, pelleted, washed and lyzed using a potter in a high sucrose (0.25 mM) hypotonic lysis buffer (0.2 mM KCl, 10mM Tris-HCl (pH 7.4), 1mM EGTA) complemented with protease inhibitor cocktail (Roche) and phosphatase inhibitors (1mM Na3VO4 and 1mM NaF, Sigma-Aldrich). Crude cell lysates were sequentially centrifuged at 1000 RCF for 10 min, 15000 RCF for 10 min and 100000 RCF for 1h at 4° C. After each centrifugation pellets were suspended in 50 μl of RIPA lysis buffer containing protease and phosphatase inhibitors and a sample of each supernatant was collected. Total protein amounts contained in the samples were assayed using the Bicinchoninic Acid kit (Sigma-Aldrich) and the samples were further used for immunoblot analysis as described in the next section.
Immunoblot analysis
For GripTiteTM 293 MSR cells, proteins in the samples (10 μg protein) were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a polyvinylidene difluoride membrane. Individual proteins were detected with antibodies against HA-probe (clone Y-11; Santa Cruz), human Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH; clone 6C5; Millipore), and human transferrin receptor (clone H8.4, ThermoFischer). Secondary antibodies were goat anti–rabbit or anti–mouse horseradish peroxidase coupled (Bio- Rad Laboratories). Proteins were visualized by chemiluminescence on an ImageQuant LAS 400 mini (G&E Healthcare Life Sciences). Densitometry analysis was performed off-line using the Gels plugin on ImageJ software (National Institutes of Health). For platelets, proteins were separated by SDS-PAGE on 10–20% gradient gels and transferred to polyvinylidene fluoride membranes. Standard western blotting procedures were used. Rap1 was detected with the Odyssey Infrared Imaging System (Li-Cor Biosystems).
Immunocytochemistry
GripTiteTM 293 MSR cells plated on glass coverslips were transfected with the vectors containing the different CDGI variants (500 ng) using PolyJet transfection reagent (SignaGen Laboratories). Thirty-six hours after transfection, cells were washed and fixed with a 1% PFA PBS solution. Cells were permeabilized using 1% triton X-100 in PBS. HA-CDGI was detected using a HA-probe antibody (clone Y-11; Santa Cruz) followed by an anti-mouse alexa-488-coupled secondary antibody (ThermoFischer). F-actin was stained using Alexa-647-coupled phalloidin and cell nuclei with DAPI. Stained cells were visualized with an Axio-Imager M1 microscope (40X or 100X oil objective). Digital images were recorded using an Axiocam MRm camera (Carl Zeiss, Inc.) and analyzed off-line using ImageJ software (National Institutes of Health).
Lipid co-sedimentation
Phospholipid binding to full-length CDGI, CDGIΔC1 and the C1 domain wild-type and mutant proteins were assessed by co-sedimentation with liposomes. The liposomes were prepared using phosphatidylcholine (PC), phosphatidylethnolamine (PE), phosphatidylserine (PS), and phosphatidyl inositol (4,5) bisphosphate (PIP2) or phosphatidyl inositol (3,4,5) triphosphate (PIP3) purchased from Avanti Polar Lipids (Alabaster, AL) as chloroform stocks. All liposomes contained 60% PC and 25% PE. In the negative control PS liposomes, the remaining 15% was PS. The PIP2 or PIP3 liposomes substituted PS by 1.25, 2.5, 5, 10 and 15%. Required quantities of lipids for individual liposome systems were mixed in a glass tube and chloroform evaporated under the flow of nitrogen gas in fume hood. The lipid mixture was dried overnight in vacuum and then resuspended in co-sedimentation buffer (40 mM HEPES (pH 6.9), 100 mM NaCl, 15 μM ZnSO4, 1 mM DTT). Liposomes were prepared using Avanti’s mini extruder kit with a 100 nm polycarbonate membrane. Each liposome sample contained 250 μg of lipid (90 μL) and 10 μM of protein (10 μL of a 100 μM stock) in a final volume of 100 μL. Samples were nutated at 4 °C for 1 h and then centrifuged at 100000 RCF for 1 h. The supernatant was separated and the pellet was resuspended in co-sedimentation buffer to equal volume. Supernatant and pellet samples were analyzed by 15% SDS-PAGE and the lipid-bound protein was quantified from the fraction present in the pellet. Densitometry was performed using the software ImageJ53. Statistical significances (p values) was determined using the Microsoft Excel t-Test function.
Molecular dynamics simulations
Homology model of the C1 domain (residues 499–551) was generated using Modeller v9.954 (with the C1 domain of PKCδ as the modeling template30. The martinize.py script55 was used to construct the course grained (CG) C1 starting model for molecular dynamics (MD) simulations. The starting model of the C1 protein carries a positive charge of +9. During the CG simulations, the backbone of the protein was attached to a harmonic potential thus keeping it relatively fixed. Parameters for the membrane were taken from the MARTINI force field55,56,57,58. Using the insane.py script59, two leaflets of membrane were built with 45% POPC, 21% POPE, 25% Cholesterol, 7% POPS (charge −1) and either 2% PIP2 (charge −5) or 2% PIP3 (charge −7). An additional negative control membrane was used without any PIP2 or PIP3. The membrane system contained a total of 124 lipid molecules and approximately 16000 polarizable water molecules60. Required number of Na+ or Cl− ions were added to neutralize the system in each simulation. For Lennard-Jones interactions, a cut-off of 1.2 nm was used and electrostatics were treated using a shift scheme with a cut-ff of 1.2 nm. The time step for the integration was 20 fs and the dielectric constant was set to 2.5. Initially, the membranes were equilibrated for 1 μs at 320 K. After that, C1 protein was inserted into the simulation box in the solution, near the surface of the membrane. In order to enhance sampling, replica exchange molecular dynamics was performed where five replicas covering the range of temperatures from 320 to 400 K were simulated for 500 ns each (i.e., total of 2.5 μs), for all three membrane-protein systems. All five trajectories for each system were merged and analyzed to generate histograms that were transformed to densities using the Gaussian kernel density estimation (KDE) method for distances between Arg and Lys side-chain beads (SC2) of the C1 domain and PS (PO4), PIP2 (P1 and P2) or PIP3 (P1, P2, and P3) headgroups. Histograms were normalized based on the total number of events to provide a probability density of residue presence with respect to the membrane. Following the analysis of the CG trajectories, desirable frames from the PIP2 and PIP3 systems were selected and transformed to all atom (AA) CHARMM force field61,62 using the backward tool63 with water molecules treated using the TIP3P64 model. The short range non-bonded interactions were calculated with a cut-off of 1.2 nm. For long range electrostatics, the particle mesh Ewald (PME) algorithm was used65,66. The time step for the integration was 2 fs. Each of the frames were simulated for 25 ns of equilibration following the observation of no further changes of the system’s potential energy. The simulations were performed using GROMACS 5.1.267,68,69,70,71 and the trajectories analyzed using VMD72 software.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Essentials.
CalDAG-GEFI, a guanine nucleotide exchange factor for Rap1, is essential for platelet function.
CalDAG-GEFI contains an atypical C1 domain that plays a key role in membrane localization.
Interaction with phosphoinositides is dependent on residues exclusive to this atypical C1 domain.
Disruption of CalDAG-GEFI C1 domain function impairs hemostasis in mice and humans.
Acknowledgements
We thank Peter M. Thompson for his assistance in preliminary studies. This work was supported by the NIH grants (PO1 CA203657 and 1R01GM115597 to S.L.C, 1R35 HL144976-01 to WB and P30 CA016086 to B.R.T.).
Footnotes
Declaration of Interests
The authors declare no competing interests.
References
- 1.Broos K, Feys HB, De Meyer SF, Vanhoorelbeke K & Deckmyn H Platelets at work in primary hemostasis. Blood Rev 25, 155–167 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Bos JL, de Rooij J & Reedquist KA Rap1 signalling: adhering to new models. Nat. Rev. Mol. Cell Biol 2, 369–377 (2001). [DOI] [PubMed] [Google Scholar]
- 3.Gloerich M & Bos JL Regulating Rap small G-proteins in time and space. Trends Cell Biol 21, 615–623 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Chrzanowska-Wodnicka M, Smyth SS, Schoenwaelder SM, Fischer TH & White GC Rap1b is required for normal platelet function and hemostasis in mice. J. Clin. Invest 115, 680–687 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stefanini L et al. Functional redundancy between RAP1 isoforms in murine platelet production and function. Blood 132, 1951–1962 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crittenden JR et al. CalDAG-GEFI integrates signaling for platelet aggregation and thrombus formation. Nat. Med 10, 982–986 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Stefanini L et al. RASA3 is a critical inhibitor of RAP1-dependent platelet activation. J. Clin. Invest 125, 1419–1432 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stefanini L & Bergmeier W RAP1-GTPase signaling and platelet function. J. Mol. Med. (Berl). 94, 13–19 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cifuni SM, Wagner DD & Bergmeier W CalDAG-GEFI and protein kinase C represent alternative pathways leading to activation of integrin alphaIIbbeta3 in platelets. Blood 112, 1696–1703 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kato H et al. Human CalDAG-GEFI deficiency increases bleeding and delays IIb 3 activation. Blood 128, 2729–2733 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Stefanini L, Roden RC & Bergmeier W CalDAG-GEFI is at the nexus of calcium-dependent platelet activation. Blood 114, 2506–2514 (2009). [DOI] [PubMed] [Google Scholar]
- 12.Bergmeier W et al. Mice lacking the signaling molecule CalDAG-GEFI represent a model for leukocyte adhesion deficiency type III. J. Clin. Invest 117, 1699–1707 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stolla M et al. The kinetics of αIIbβ3 activation determines the size and stability of thrombi in mice: implications for antiplatelet therapy. Blood 117, 1005–1013 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bermejo E et al. Marked bleeding diathesis in patients with platelet dysfunction due to a novel mutation in RASGRP2, encoding CalDAG-GEFI (p.Gly305Asp). Platelets 29, 84–86 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Canault M et al. Human CalDAG-GEFI gene (RASGRP2) mutation affects platelet function and causes severe bleeding. J. Exp. Med 211, 1349–1362 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Desai A et al. Phenotype analysis and clinical management in a large family with a novel truncating mutation in RASGRP2, the CalDAG-GEFI encoding gene. Res. Pract. Thromb. Haemost 1, 128–133 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lozano ML et al. Novel mutations in RASGRP2, which encodes CalDAG-GEFI, abrogate Rap1 activation, causing platelet dysfunction. Blood 128, 1282–1289 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sevivas T et al. Identification of two novel mutations in RASGRP2 affecting platelet CalDAG-GEFI expression and function in patients with bleeding diathesis. Platelets 29, 192–195 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Westbury SK et al. Expanded repertoire ofRASGRP2variants responsible for platelet dysfunction and severe bleeding. Blood 130, 1026–1030 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ksionda O, Limnander A & Roose JP RasGRP Ras guanine nucleotide exchange factors in cancer. Front. Biol. (Beijing). 8, 508–532 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iwig JS et al. Structural analysis of autoinhibition in the Ras-specific exchange factor RasGRP1. Elife 2, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cook AA et al. Calcium-induced structural rearrangements release autoinhibition in the Rap-GEF CalDAG-GEFI. J. Biol. Chem 293, 8521–8529 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Das J & Rahman GM C1 domains: structure and ligand-binding properties. Chem. Rev 114, 12108–12131 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Colon-Gonzalez F & Kazanietz MG C1 domains exposed: from diacylglycerol binding to protein-protein interactions. Biochim. Biophys. Acta 1761, 827–837 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Johnson JE et al. Differential membrane binding and diacylglycerol recognition by C1 domains of RasGRPs. Biochem. J 406, 223–236 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Palmer SM, Playford MP, Craig SW, Schaller MD & Campbell SL Lipid Binding to the Tail Domain of Vinculin. J. Biol. Chem 284, 7223–7231 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thompson PM et al. A Structural Model for Vinculin Insertion into PIP2-Containing Membranes and the Effect of Insertion on Vinculin Activation and Localization. Structure 25, 264–275 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Czikora A et al. Structural Basis for the Failure of the C1 Domain of Ras Guanine Nucleotide Releasing Protein 2 (RasGRP2) to Bind Phorbol Ester with High Affinity. J. Biol. Chem 291, 11133–11147 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dries DR, Gallegos LL & Newton AC A Single Residue in the C1 Domain Sensitizes Novel Protein Kinase C Isoforms to Cellular Diacylglycerol Production. J. Biol. Chem 282, 826–830 (2006). [DOI] [PubMed] [Google Scholar]
- 30.Zhang G, Kazanietz MG, Blumberg PM & Hurley JH Crystal structure of the Cys2 activator-binding domain of protein kinase Cδ in complex with phorbol ester. Cell 81, 917–924 (1995). [DOI] [PubMed] [Google Scholar]
- 31.Moser M, Nieswandt B, Ussar S, Pozgajova M & Fässler R Kindlin-3 is essential for integrin activation and platelet aggregation. Nat. Med 14, 325–330 (2008). [DOI] [PubMed] [Google Scholar]
- 32.Nieswandt B et al. Loss of talin1 in platelets abrogates integrin activation, platelet aggregation, and thrombus formation in vitro and in vivo. J. Exp. Med 204, 3113–3118 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Petrich BG et al. Talin is required for integrin-mediated platelet function in hemostasis and thrombosis. J. Exp. Med 204, 3103–3111 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun Z, Costell M & Fässler R Integrin activation by talin, kindlin and mechanical forces. Nat. Cell Biol 21, 25–31 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Stritt S et al. Rap1-GTP-interacting adaptor molecule (RIAM) is dispensable for platelet integrin activation and function in mice. Blood 125, 219–222 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Su W et al. Rap1 and its effector RIAM are required for lymphocyte trafficking. Blood 126, 2695–2703 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bromberger T et al. Direct Rap1/Talin1 interaction regulates platelet and neutrophil integrin activity in mice. Blood 132, 2754–2762 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lagarrigue F et al. Rap1 binding to the talin 1 F0 domain makes a minimal contribution to murine platelet GPIIb-IIIa activation. Blood Adv 2, 2358–2368 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gingras AR et al. Rap1 binding and a lipid-dependent helix in talin F1 domain promote integrin activation in tandem. J. Cell Biol (2019). doi: 10.1083/jcb.201810061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ye X, McLean MA & Sligar SG Phosphatidylinositol 4,5-Bisphosphate Modulates the Affinity of Talin-1 for Phospholipid Bilayers and Activates Its Autoinhibited Form. Biochemistry 55, 5038–5048 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cozier GE et al. GAP1IP4BP contains a novel group I pleckstrin homology domain that directs constitutive plasma membrane association. J. Biol. Chem 275, 28261–28268 (2000). [DOI] [PubMed] [Google Scholar]
- 42.Qian F et al. Role for the guanine nucleotide exchange factor phosphatidylinositol-3,4,5-trisphosphate-dependent rac exchanger 1 in platelet secretion and aggregation. Arterioscler. Thromb. Vasc. Biol 32, 768–777 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Srijakotre N et al. P-Rex1 and P-Rex2 RacGEFs and cancer. Biochem. Soc. Trans 45, 963–977 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Insall RH & Weiner OD PIP3, PIP2, and cell movement--similar messages, different meanings? Dev. Cell 1, 743–747 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pearce AC et al. Vav1 and vav3 have critical but redundant roles in mediating platelet activation by collagen. J. Biol. Chem 279, 53955–53962 (2004). [DOI] [PubMed] [Google Scholar]
- 46.Hurley JH, Newton AC, Parker PJ, Blumberg PM & Nishizuka Y Taxonomy and function of C1 protein kinase C homology domains. Protein Sci 6, 477–480 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leonard TA & Hurley JH Regulation of protein kinases by lipids. Curr. Opin. Struct. Biol 21, 785–791 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Travers T et al. Molecular recognition of RAS/RAF complex at the membrane: Role of RAF cysteine-rich domain. Sci. Rep 8, 8461 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mott HR et al. The solution structure of the Raf-1 cysteine-rich domain: a novel ras and phospholipid binding site. Proc. Natl. Acad. Sci 93, 8312–8317 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Palma-Barqueros V et al. RASGRP2 gene variations associated with platelet dysfunction and bleeding. Platelets 30, 535–539 (2019). [DOI] [PubMed] [Google Scholar]
- 51.Piatt R et al. Mice Expressing Low Levels of CalDAG-GEFI Exhibit Markedly Impaired Platelet Activation With Minor Impact on Hemostasis. Arterioscler. Thromb. Vasc. Biol 36, 1838–1846 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rojas RJ, Kimple RJ, Rossman KL, Siderovski DP & Sondek J Established and emerging fluorescence-based assays for G-protein function: Ras-superfamily GTPases. Comb. Chem. High Throughput Screen 6, 409–418 (2003). [DOI] [PubMed] [Google Scholar]
- 53.Schneider CA, Rasband WS & Eliceiri KW NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marti-Renom MA et al. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct 29, 291–325 (2000). [DOI] [PubMed] [Google Scholar]
- 55.de Jong DH et al. Improved Parameters for the Martini Coarse-Grained Protein Force Field. J. Chem. Theory Comput 9, 687–697 (2013). [DOI] [PubMed] [Google Scholar]
- 56.Marrink SJ, Risselada HJ, Yefimov S, Tieleman DP & de Vries AH The MARTINI force field: coarse grained model for biomolecular simulations. J. Phys. Chem. B 111, 7812–7824 (2007). [DOI] [PubMed] [Google Scholar]
- 57.Monticelli L et al. The MARTINI Coarse-Grained Force Field: Extension to Proteins. J. Chem. Theory Comput 4, 819–834 (2008). [DOI] [PubMed] [Google Scholar]
- 58.Marrink SJ & Tieleman DP Perspective on the Martini model. Chem. Soc. Rev 42, 6801–6822 (2013). [DOI] [PubMed] [Google Scholar]
- 59.Wassenaar TA, Ingolfsson HI, Bockmann RA, Tieleman DP & Marrink SJ Computational Lipidomics with insane: A Versatile Tool for Generating Custom Membranes for Molecular Simulations. J. Chem. Theory Comput 11, 2144–2155 (2015). [DOI] [PubMed] [Google Scholar]
- 60.Yesylevskyy SO, Schafer LV, Sengupta D & Marrink SJ Polarizable water model for the coarse-grained MARTINI force field. PLoS Comput. Biol 6, e1000810 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brooks BR et al. CHARMM: the biomolecular simulation program. J. Comput. Chem 30, 1545–1614 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bjelkmar P, Larsson P, Cuendet MA, Hess B & Lindahl E Implementation of the CHARMM Force Field in GROMACS: Analysis of Protein Stability Effects from Correction Maps, Virtual Interaction Sites, and Water Models. J. Chem. Theory Comput 6, 459–466 (2010). [DOI] [PubMed] [Google Scholar]
- 63.Wassenaar TA, Pluhackova K, Bockmann RA, Marrink SJ & Tieleman DP Going Backward: A Flexible Geometric Approach to Reverse Transformation from Coarse Grained to Atomistic Models. J. Chem. Theory Comput 10, 676–690 (2014). [DOI] [PubMed] [Google Scholar]
- 64.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW & Klein ML Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 79, 926–935 (1983). [Google Scholar]
- 65.Darden T, York D & Pedersen L Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys 98, 10089–10092 (1993). [Google Scholar]
- 66.Essmann U et al. A smooth particle mesh Ewald method. J. Chem. Phys 103, 8577–8593 (1995). [Google Scholar]
- 67.Berendsen HJC, van der Spoel D & van Drunen R GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun 91, 43–56 (1995). [Google Scholar]
- 68.Lindahl E, Hess B & van der Spoel D GROMACS 3.0: a package for molecular simulation and trajectory analysis. Mol. Model. Annu 7, 306–317 (2001). [Google Scholar]
- 69.Van Der Spoel D et al. GROMACS: fast, flexible, and free. J. Comput. Chem 26, 1701–1718 (2005). [DOI] [PubMed] [Google Scholar]
- 70.Hess B, Kutzner C, van der Spoel D & Lindahl E GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput 4, 435–447 (2008). [DOI] [PubMed] [Google Scholar]
- 71.Abraham MJ et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25 (2015). [Google Scholar]
- 72.Humphrey W, Dalke A & Schulten K VMD: visual molecular dynamics. J. Mol. Graph 14, 27–28, 33–38 (1996). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.