

Abstract
Central nervous system (CNS) insulin resistance is associated with Alzheimer’s disease (AD). In addition, the apolipoprotein E4 (apoE4) isoform is a risk factor for AD. The connection between these two factors in relation to AD is being actively explored. We summarize this literature with a focus on the transport of insulin and apoE across the blood-brain barrier (BBB) and into the CNS, the impact of apoE and insulin on the BBB, and the interactions between apoE, insulin, and the insulin receptor once present in the CNS. We highlight how CNS insulin resistance is apparent in AD and potential ways to overcome this resistance by repurposing currently approved drugs, with apoE genotype taken into consideration as the treatment response following most interventions is apoE isoform-dependent. This review is part of a special issue focusing on apoE in AD and neurodegeneration.
Keywords: apolipoprotein E, central nervous system insulin, insulin receptor, blood-brain barrier, Alzheimer’s disease, amyloid beta
Introduction
Insulin has a dual role, being both a metabolic hormone and a mitogen. This multi-faceted fate of insulin is defined by the tissue in which it acts, in addition to the receptor activated. In the central nervous system (CNS), insulin contributes to the control of nutrient homeostasis, reproduction, olfaction, learning, and memory (Blazquez et al., 2014). Insulin in the CNS is primarily derived from blood insulin, which has been transported across the blood-brain barrier (BBB) (Banks et al., 1997a; Margolis and Altszuler, 1967; Woods and Porte, 1977). In the euglycemic state, insulin levels are approximately 14 fold less in the cerebrospinal fluid (CSF) levels than in the plasma (Mahley et al., 2009; Pitas et al., 1987).
Apolipoprotein E (apoE) plays an important role in the metabolism and redistribution of lipoproteins and cholesterol. In the brain, apoE is primarily produced by astrocytes (Kim et al., 2009), but also by neurons under stress (Xu et al., 1999) and is responsible for neuronal maintenance and repair (Holtzman et al., 2012). It was also recently reported that another major source of CNS apoE originates from the choroid plexus by glymphatic fluid transport (Achariyar et al., 2016). The peripheral pool of apoE is primarily produced by hepatocytes, and also by macrophages and adipocytes, each requiring tissue specific machinery for expression (Kockx et al., 2018). ApoE levels in the CSF are approximately 10-fold less than in the plasma (Chernick et al., 2019) but the correlation between apoE levels in these two pools is weak (Cruchaga et al., 2012). Regardless, apoE is important for BBB repair after injury (Bell et al., 2012; Donahue and Johanson, 2008; Fullerton et al., 2001).
Here, we review the literature supporting the link between CNS insulin and apoE. We first introduce how insulin enters and travels throughout the CNS and highlight some of the CNS-related roles. In this context, we touch on how CNS insulin resistance (IR) can lead to AD. We also present information regarding the role of apoE on the BBB and cerebrovascular blood flow and how insulin can modulate these interactions. Lastly, some recent clinical data is presented in ways to overcome CNS IR and challenges based on apoE genotype.
CNS access to insulin
Endogenous insulin is secreted by the pancreas and into the bloodstream where it can enter the brain via the BBB and blood-CSF barrier. Insulin is actively transported in a saturable manner across the BBB at levels below those needed to induce hypoglycemia (Banks et al., 1997a; Baura et al., 1993). It has been traditionally thought that a molecule the size of insulin would require a transcytotic transport mechanism to enter the brain. This can occur independent of the endothelial insulin receptor (Hersom et al., 2018; Rhea et al., 2018). Once insulin is transported across the BBB, efflux does not occur, even with cerebrospinal fluid (CSF) reabsorption, and is sequestered by the CNS (Cashion et al., 1996). Therefore, the transport protein present at the BBB responsible for allowing insulin to enter the CNS is unidirectional, moving insulin from the luminal side of the brain endothelial cell to the abluminal side. The average rate of insulin transport across the mouse BBB is approximately 0.55 μL/g-m (Banks et al., 2012). For reference, amyloid β (1–42) is transported across the BBB at a slower rate of 0.39 μL/g-min (Banks et al., 1991).
Exogenous insulin can be administered by multiple different routes to increase CNS insulin as recently reviewed by us (Rhea et al., 2019b). Administrations that increase serum insulin are always dangerous for the risk of hypoglycemia. Therefore, getting insulin directly to the CNS is optimal for therapeutic purposes. Distribution of insulin throughout the brain varies based on the delivery route. For example, following intravenous delivery (which likely reflects endogenous transport), the olfactory bulb and pons/medulla contain the greatest content, while following intranasal delivery, the olfactory bulb, striatum, hypothalamus, and midbrain contain the most (Rhea et al., 2017; Rhea et al., 2018). Following intracerebroventricular injection, the hypothalamus and hippocampus contain the greatest amount (Rhea et al., 2019b).
Insulin transport across the mouse BBB is fastest into the olfactory bulb and pons/medulla (Rhea et al., 2019b). It is unknown whether humans have the same regional rates of insulin transport into the brain. As human insulin has the same saturable effect on murine insulin and vice versa in mice (Banks et al., 1997a), it is likely the two transport systems are similar. Human insulin can also lower endogenous circulating murine insulin (Banks et al., 1997b). However, the primary evolutionary role for the olfactory bulb in the rodent is different than the primary role for it in humans. The mouse olfactory bulb also has the highest rate of insulin degradation (Banks et al., 1999). How sequestration and degradation compares to the human is difficult to determine.
Insulin at the BBB is not just present as an effector for regulating substrate entry, but its own transport can also be influenced by other conditions. As an effector, insulin can regulate not only a variety of substrate entry across the BBB but also neurotransmitter signaling. For example, insulin can regulate leptin (Kastin and Akerstrom, 2001) and insulin-like growth factor 1 (IGF-1) (Yu et al., 2006) transport into the brain. In contrast, there is no role of insulin in electrolyte (Thurston et al., 1976), GLP-1 (Kastin et al., 2002), or estrogen (May et al., 2016) transport into brain. Insulin can also regulate amino acid transport across the BBB. Tryptophan BBB transport is regulated by insulin (Cangiano et al., 1983), which can lead to multiple changes in CNS signaling pathways, especially in the serotonin pathway. In addition, dopamine and insulin signaling systems have a cross-regulatory interaction (Nash, 2017) in which insulin can regulate neuronal dopamine uptake and dopamine can regulate pancreatic insulin secretion. Therefore, insulin action just at the BBB can lead to a multitude of changes within the CNS. There are also different physiological states, including diabetes, obesity, and aging, that can impact the transport of insulin into the brain (Banks et al., 2012; Rhea and Banks, 2019).
As the above illustrates, insulin in the CNS has many roles and these differ from its metabolic role in the periphery. In addition, the receptors for insulin in the brain have a different structure and size compared to those expressed peripherally, consistent with a different evolutionary pathway that preserved more of ancestral insulin’s effects on growth (Banks et al., 2012; Chan et al., 1992). Insulin in the brain does not primarily affect glucose uptake as in the periphery since GLUT1 is the primary glucose BBB transporter and is non-responsive to insulin (Hasselbalch et al., 1999). However, there are some regions such as the hippocampus and hypothalamus that express GLUT4, an insulin-sensitive glucose transporter, localized to neurons (Alquier et al., 2006; Grillo et al., 2009; McEwen and Reagan, 2004). Here, GLUT4 translocates to the neuronal membrane downstream of insulin and plays an important role in hippocampal memory processes and brain insulin resistance (Duarte et al., 2012; McNay and Pearson-Leary, 2020). Peripheral insulin levels can regulate cerebellar GLUT4 expression (Vannucci et al., 1998). In addition, it has recently been shown that intranasal insulin can increase cerebral glucose uptake, as measured by [18F]-FDG uptake following traumatic brain injury in rats, although the mechanism for increased uptake was not examined (Duarte et al., 2012).
CNS insulin can have a direct, localized effect in addition to an indirect effect that elicits a change in the periphery (i.e. change in peripheral metabolism). CNS insulin can play a major role in reward, memory, and feeding behavior by impacting signal transduction pathways involved in neuronal survival, synaptic maintenance, dendritic arbor development, cognition, neuronal circuitry formation, and BBB transporter expression/localization (Banks, 2004; Banks et al., 2012; Schulingkamp et al., 2000; Stranahan et al., 2008). Areas within the brain that have high levels of insulin receptors, such as the hippocampus and hypothalamus, play important roles in cognition and regulation of peripheral metabolism, respectively (Ghasemi et al., 2013; Porte et al., 1998). This is consistent with evidence showing that insulin can influence cognition (Infante-Garcia et al., 2015). It was recently shown that insulin in the hippocampus could affect other signaling pathways besides the typical insulin receptor pathway (Frazier et al., 2019b; Rhea et al., 2019a). As an indirect mechanism, CNS insulin can act as a neuroregulatory peptide, regulating food intake and peripheral energy stores (Filippi et al., 2013; Gray et al., 2014). The impact of CNS insulin on food intake data should be interpreted with caution, however, as supraphysiological levels of insulin are often injected into the CNS. In such studies, the IGF-1 receptor and perhaps other receptors as well could be activated (Rechler and Nissley, 1986). CNS insulin infusion decreases serum insulin levels, which appears to be independent of a change in food intake (Foster et al., 1991).
Less studied is the role of insulin receptor signaling in other cell types of the neurovascular unit (astrocytes, pericytes, brain endothelial cells) and how this signaling might affect the BBB (Rhea and Banks, 2019). Many of the functions of CNS insulin have been well described, as discussed above. However, once insulin is transported across the BBB, it is unclear how this signaling peptide navigates the extracellular space and accesses its target. Whether insulin is released into the interstitium, packaged into exosomes, or transported directly to surrounding cells of the neurovascular unit, including pericytes, astrocytes, and neurons is not known. Brain extracellular diffusion patterns of similarly sized molecules including epidermal growth factor and nerve growth factor have been investigated (Stroh et al., 2003; Thorne et al., 2004), but research on insulin diffusion is limited. Importantly, diffusion parameters are known to change with age (Sykova, 2004). It is likely that these parameters also change with disease. It is also not clear whether insulin is sequestered by these cells and released as needed or if the CNS requirement for insulin is regulated by BBB transport, sequestration, and degradation. These concepts warrant further investigation to better understand the etiology of CNS IR.
Intranasal insulin is one way to therapeutically increase CNS insulin concentrations to overcome CNS IR. Clinical studies have shown a dose dependent benefit of intranasal insulin on cognition that is modulated by apoE genotype (Reger et al., 2006; Reger et al., 2008). Cerebrovascular function is critical for cognitive performance (Ozturk and Tan, 2018). Intranasal insulin can increase cerebral blood flow in young adults (Kullmann et al., 2017), older adults (Akintola et al., 2017), and cognitively impaired rats (Rajasekar et al., 2017). This therapeutic delivery of insulin will be brought up again later. The relation between cerebrovascular function, the BBB, and apoE isoforms will be discussed below.
ApoE and the BBB
The importance of cerebrovascular function for cognitive performance is already evident in the developing brain (Bakker et al., 2014). The BBB is also critical for cognitive performance. Brain capillary damage and BBB breakdown in the hippocampus are early biomarkers for cognitive dysfunction independent of amyloid β or tau (Nation et al., 2019) and reductions in microvascular length and decreased brain capillary density have been reported in several animal models of AD (Lee et al., 2005; Paris et al., 2004). BBB dysfunction can lead to microvascular impairments (Zlokovic, 2013) and is associated with cognitive decline in nondemented elders (Bowman et al., 2018). The importance of the BBB for cognitive performance is also evident in the developing brain, for example in the context of obstructive sleep apnea (Khalyfa et al., 2018). With the need to diagnose early and start long-term prevention interventions in neurodegenerative diseases such as AD (Yassine, 2017), cerebrovascular function and the BBB are attractive early biomarkers. As described below, apoE isoforms have differential effects on cerebrovascular function and the BBB. Therefore, it is important to understand these apoE isoform-dependent impacts in health and disease and in the developing and aging brain.
Transport of apoE across the BBB or blood-CSF barrier has not been reported to occur, as evident by independent studies (Liu et al., 2012; Zlokovic et al., 1994). However, apoE can affect the BBB directly in addition to altering transport of substrates, including amyloid β. Specifically, apoE4 disrupts amyloid β BBB efflux the most (Deane et al., 2008) and primarily impacts BBB integrity by contributing to BBB anomalies (Alata et al., 2015). Lastly, the diffusion of apoE throughout the CNS is isoform-dependent with diffusion being ranked as follows: apoE2 > apoE3 > apoeE4 (Achariyar et al., 2016).
In the developing brain, E4 carriers exhibit greater changes in regional activation than non-carriers in distinct regions of the temporal gyrus during a nonverbal memory task (Scarmeas et al., 2005). These effects and alterations in cerebral blood flow are not transient and are also seen in people during middle-age (Bookheimer et al., 2000). Consistent with these human data, vascular alterations are seen in mice at two weeks of age expressing E4 or mice deficient in murine apoE (KO) (Bell et al., 2012). The fact that this phenotype is also seen in apoE KO mice suggests that it is due to lack of trophic effects rather than a dominant negative effect of E4 as is seen for some other phenotypes (Raber, 2004; Raber et al., 2004), which in turn has implications for developing and testing therapeutic strategies. It also suggests that the interaction of this phenotype with age might be what contributes to increased risk of E4 carriers to develop age-related cognitive decline (Berteau-Pavy et al., 2007) and AD (Farrer et al., 1997). The BBB might play a critical role here (Zlokovic, 2011). Using a guinea-pig brain perfusion model, an anti-amyloidogenic role for the BBB involving transport of apoE and apolipoprotein J (apoJ) in AD and impairments in cerebrovascular function being an initiating event leading to neurodegeneration was proposed 23 years ago (Zlokovic, 1996). Environmental risk factors like a Western diet might also interact with this phenotype to further increase this risk. Importantly, both IR and E4 have been linked to vascular impairments. Insulin has many important actions in the brain as described above, and brain IR has been proposed to contribute to the progression of AD (de la Monte, 2012).
In the aging brain, cerebrovascular flow is lower in E4 carriers than non-carriers (Filippini et al., 2011). The aging brain of cognitively healthy E4 individuals show accelerated declines in regional cerebrovascular flow over time (Thambisetty et al., 2010). The relation between cognitive function and age-related changes in cerebrovascular flow is also modulated by apoE isoform (Wierenga et al., 2013). There are differences in blood oxygen level dependent (BOLD) functional magnetic resonance imaging (fMRI) signals, measures of functional brain activation, in middle-aged and aged E4 carriers (Scarmeas and Stern, 2006). E4 carriers, including cognitively healthy elderly and those at younger ages, show glucose hypometabolism (Altmann et al., 2015; Reiman et al., 2001; Reiman et al., 1996; Reiman et al., 2004; Small et al., 2000). This early onset of cerebrovascular and BBB phenotypes in E4 carriers is important as these phenotypes have been proposed as initiating events ultimately causing neurodegeneration associated with vascular disease (Snyder et al., 2015). APOE genotype itself has clear effects on several risk factors for dementia noted above, such as obesity (Arbones-Mainar et al., 2008; Arbones-Mainar et al., 2016; Elosua et al., 2003; Huebbe et al., 2015; Tejedor et al., 2014), hypercholesterolemia (Bennet et al., 2007; Wilson et al., 1996), and increasing in vivo and in vitro evidence also points to multiple cerebrovascular effects of APOE in the brain (Alata et al., 2015; Bell et al., 2012; Lin et al., 2017; Nishitsuji et al., 2011; Schilling et al., 2013; Wiesmann et al., 2016).
In general, this pattern is consistent with the proposed developmental origin of health and disease later in life (Heindel and Vandeberg, 2016; Roseboom et al., 2001). E4 isoform is associated with hyperactivity and hyperconnectivity (Koelewijn et al., 2019), increased BBB permeability, reductions in cerebral vascularization, thinner vessel walls, and reduced cerebrovascular blood flow (Alata et al., 2015; Bell et al., 2012). However, it is likely more complex since age of the E4 carriers results in lower (Filippini et al., 2011; Scarmeas et al., 2005; Thambisetty et al., 2010) and higher (Wierenga et al., 2013; Zlatar et al., 2014) cerebral blood flow. Since cerebral blood flow is decreased in people with age-related cognitive decline and AD (Celsis et al., 1997; Roher et al., 2012), it is conceivable that the complexity is partly due to the fact that cerebral hypoperfusion could be a result of diminished energy demands of the aging or AD brain or contributes more directly to neurodegeneration. So in E4 carriers with unaltered or higher cerebral blood flow, this might be part of a compensatory change and hypoperfusion might precede and even actively contribute to the risk to develop AD (Tai et al., 2016).
Consistent with the human data, aged E4 mice have a lower cerebrovascular flow than wild-type mice (Lin et al., 2017; Wiesmann et al., 2016). These changes are not necessarily associated with alterations in vessel morphology. While the cerebral blood volume was lower in in E4 than E3 mice, in one study this was observed in the absence of changes in vessel morphology (Johnson et al., 2019) while in another study they were associated with reductions in microvascular length (Bell et al., 2012).
Remarkably, one environmental factor that is front and center for the developmental origin of health and disease is nutrition and insulin sensitivity/resistance. In this context, it is extraordinary that type II diabetes, IR, and E4 carrier status are associated with a reduced ability to preserve a stable and sufficient cerebral blood flow (Jansen et al., 2016; Novak et al., 2014). This link between IR and cerebral blood flow is not surprising, as the microvasculature is sensitive to insulin and this sensitivity is impaired by IR (Belcik et al., 2015). Consistent with the human data, both E4 and a Western diet induce IR and reduce cerebral blood volume and glucose uptake, in an additive fashion, in apoE targeted replacement (TR) mice (Johnson et al., 2017). However, the cognitive, metabolic, and cerebrovascular responses to an acute glucose challenge revealed apoE isoform-dependent effects. E4, but not E3, mice benefitted from an acute increase in blood glucose levels (Johnson et al., 2017). Consistent with these mouse data, cognitively healthy people and cognitively impaired people carrying E4 have higher memory scores following a meal with a high glycemic index and high in saturated fat, while lower scores are seen in cognitively healthy people who do not carry E4, as compared to genotype-matched controls (Hanson et al., 2015). IR also affects cognitive performance (de la Monte, 2012) and might also contribute to the progression of AD. Administration of intranasal insulin enhances cognition and affects regional vasoreactivity in patients with IR as part of type II diabetes (Novak et al., 2014). Recently, nicotine addiction in rats was associated with a risk for type II diabetes by activation of a nicotinic acetylcholine receptor present in the habenula (Duncan et al., 2019). Nicotine consumption led to increased circulating glucagon and insulin levels and dysregulated glucose homeostasis via a direct connection from the habenula to the pancreas. An apoE isoform-dependent effect remains to be elucidated.
In E4 carriers, in addition to the central effects described above, there might also be indirect effects on cognitive function and brain metabolism (Pendse et al., 2009). E4 is associated with peripheral vascular disease (Johnson et al., 2011; Johnson et al., 2013; Minihane et al., 2007) and the peripheral response to acute or chronic glucose or a dietary challenge in E4 carriers might affect cognitive function (Hanson et al., 2016; Hanson et al., 2013; Hanson et al., 2015).
In studies involving special diets, it is important to consider potential effects of the diet on plasma and brain apoE levels. For example, a Western diet administered for 5 weeks starting at 2–3 months of age was shown to significantly reduce apoE levels in the hippocampus of E3 but not E4 TR mice (Lane-Donovan and Herz, 2016). Conversely, plasma levels of apoE were significantly increased in E4, but not E3, TR mice on a Western diet (Lane-Donovan and Herz, 2016). A ketogenic diet revealed an apoE-isoform dependent effect in plasma but did not affect hippocampal apoE levels. Like what was seen with a Western diet, a ketogenic diet caused elevated plasma apoE levels in E4 but not E3 TR mice (Lane-Donovan and Herz, 2016). Perhaps related to increased apoE levels, a ketogenic diet restored systemic insulin sensitivity and metabolic flexibility in a diabetic E4 carrying patient (Stoykovich and Gibas, 2019).
Interaction of insulin/insulin receptor with apoE
The differential effects of apoE isoforms in insulin receptor function could be due to differential binding to the receptor. In AD brain samples, more insulin receptor is precipitated by extracts from E3 brains than extracts from E4 brains (Chan et al., 2018). Consistent with these human data, E3 binds stronger than E4 to insulin receptor in brain extracts of 26- and 78-week old apoE TR mice crossed with APP J20 mice carrying a mutant human APP gene containing the Swedish (K670N/M671L) and Indiana (V717F) mutations (Chan et al., 2015). Interestingly, using hippocampal neuronal cultures prepared from postnatal day 0, E3 and E4 pups, there was no difference in insulin sensitivity except in the presence of 500 nM of amyloid β (1–42). Here, the insulin response was impaired only in hippocampal neuronal cultures from E4 pups (Chan et al., 2015). Amyloid β peptides can reduce insulin binding to the insulin receptor which decreases insulin signaling (Xie et al., 2002). E4 also has a stronger binding affinity to amyloid β than E3 (Chan et al., 2015; Saunders et al., 1993). More on the interaction of amyloid β and apoE is covered in another review in this special issue by Dr. Thomas Wisniewski.
The altered interaction of insulin with the insulin receptor might also related to the trapping of insulin receptor in the presence of E4 in endosomes contributing to impaired insulin signaling and impaired mitochondrial respiration and glycolysis by insulin (Zhao et al., 2017). Age might be able to replace the amyloid β challenge. In cortex and hippocampus of 22-month old E3 and E4 mice, insulin signaling was impaired in E4 mice compared to E3 mice (Zhao et al., 2017). This was not seen in the cortex at 3 or 12 months of age. Therefore, in aging brains, aggregation of E4 and endosomal dysfunction might drive this effect. A Western diet might be able to replace both amyloid β and aging. When E3 and E4 TR mice were put on a Western diet for 4 months starting at 8 months of age, insulin signaling was impaired in E4 mice, while the Western diet did not affect insulin signaling in the E3 mice (Zhao et al., 2017). Remarkably and consistent with the data described above, when neuronal-enriched extracellular vesicles were isolated from plasma of patients with amnestic mild cognitive impairment or probable AD receiving intranasal insulin, positive correlations in the relationship between insulin signaling mediators and measures of cognitive performance were only seen in non-E4 carriers (Mustapic et al., 2019). These data suggest a clear overlap and interaction between amyloid β, the insulin receptor, apoE, and insulin.
CNS Insulin Resistance in Alzheimer’s Disease
AD has been associated with a deficiency in insulin action within the CNS. Deficient action could be caused by either a decreased level of insulin in the brain or to a resistance to insulin actions at the level of its receptor. One of the earliest pieces of supporting evidence for defective insulin action was that there is decreased glucose utilization by the AD brain. However, neither glucose transport across the BBB nor glucose uptake by the vast majority of brain cells, with the exception of GLUT4-expressing neurons, is insulin-dependent. Four independent lines of evidence for a deficient insulin action in the AD brain are i) a decreased CSF/serum ratio, ii) insulin resistant diseases (obesity and diabetes) are risk factors for AD, iii) AD brains have impaired insulin signaling, and iv) insulin delivery to the CNS improves memory. These four lines of evidence are considered below.
i. Decreased CSF/serum ratio
A decreased CSF/serum ratio for insulin has been reported in aged and AD subjects (Craft et al., 1998; Sartorius et al., 2015). The reasoning that this is powerful presumptive evidence for a defect in insulin transport across the BBB relies on an understanding of the nonlinear pharmacokinetics of insulin transport across the BBB. As insulin transport across the BBB is saturable, the relation between CSF insulin and serum insulin is hyperbolic (Figure 1). That is, it is linear only at “physiologic” levels of insulin. As blood levels of insulin rise, CSF levels also rise, although to an ever lesser degree. In other words, the numerator (CSF insulin levels) rises less robustly than the denominator (blood insulin levels) and so the ratio (CSF/serum insulin ratios) declines as the serum insulin level rises.
Figure 1. Relation of CNS Levels and CNS/Blood Ratios to Blood Levels: Theoretical Data.

This figure illustrates how the nonlinear relation between CNS and Blood levels can result in increasing blood levels being accompanied by both increasing CNS levels and decreasing CNS/Blood Ratios. A less efficient transporter would shift the CNS curve downward/to the right so that CNS levels will be lower for any given Blood level.
This decrease in CSF/serum ratios could occur in two basic ways that have fundamentally different implications for the relation between central and peripheral IR, as well as the role of the BBB. First, serum levels of insulin could increase, as for example happens with peripheral IR. In this case, CSF insulin levels would be paradoxically elevated while CSF/serum insulin ratios would be decreased. Thus, a decrease in CNS insulin action would only result if there were an accompanying CNS IR; and the resistance would need to be greater than could be overcome by the increased CNS level of insulin. In this case, IR at both the CNS and peripheral levels could occur simultaneously while the insulin transporter at the BBB is functioning normally. A shifting to the right of the curve relating CSF and serum insulin levels is a second way that a decreased CSF/serum insulin ratio could occur. In this case, the amount of insulin transported across the BBB is lower than normal for any given level of insulin in the blood. As the insulin transporter is modulated by various factors (Banks et al., 2008; Urayama and Banks, 2008), such a shift is possible. CSF insulin levels are not necessarily increased and could even be decreased. In this case, CSF insulin levels could be lower than normal, CNS IR can occur independently of peripheral IR, and the BBB insulin transporter is impaired (Figure 2). Which of these dominate is unclear as CSF insulin levels in AD have been reported to be increased, decreased, and unchanged (Craft et al., 1998; de la Monte et al., 2019; Fugisawa et al., 1991; Gil-Bea et al., 2010; Molina et al., 2002).
Figure 2. Levels of Insulin Resistance.
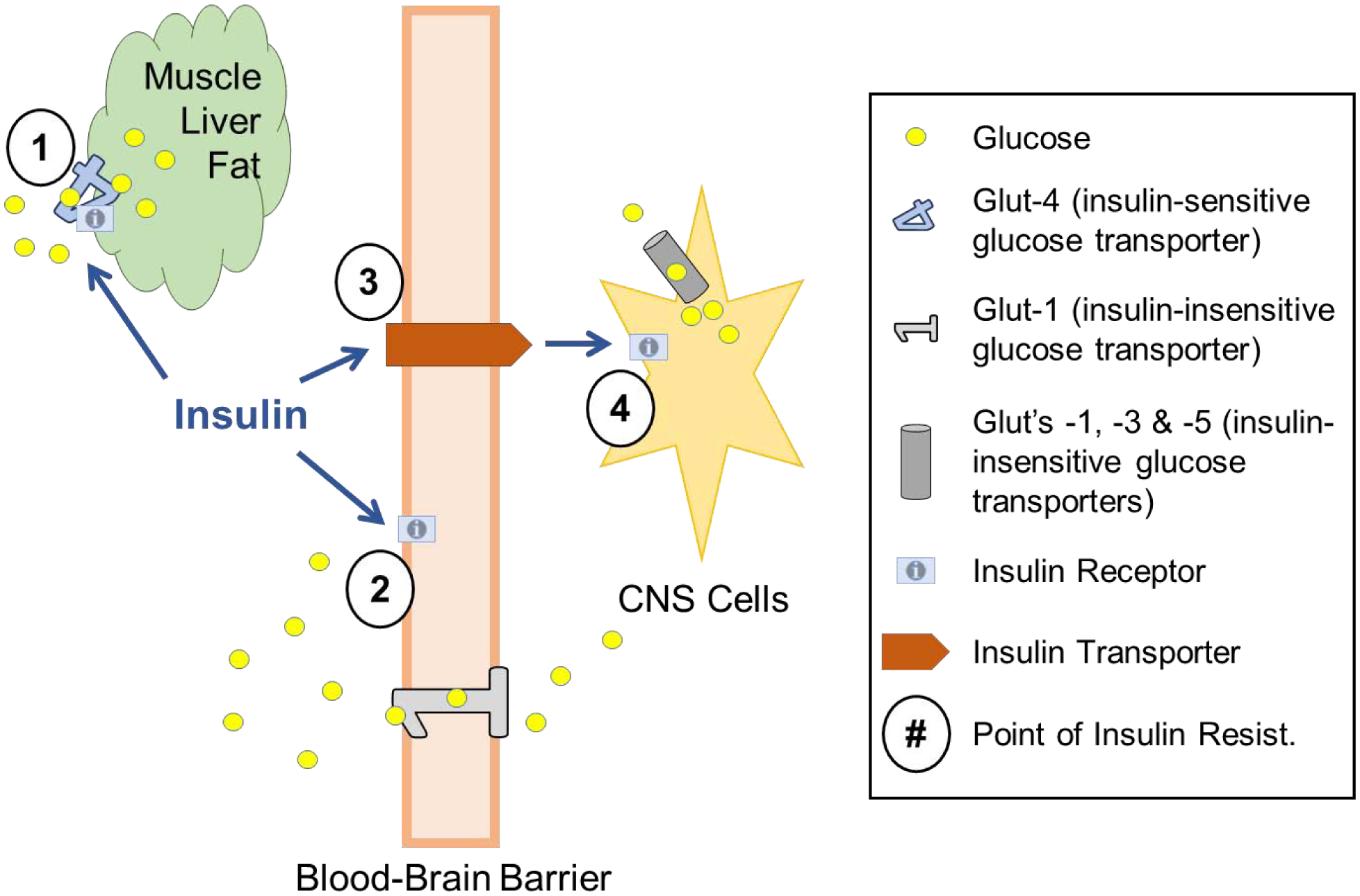
Glucose uptake is stimulated by tissues containing GLUT-4 (primarily muscle, liver, and adipose tissue), but not by those containing GLUT-1, 3, or 5 (such as brain endothelial cells, astrocytes, neurons, and microglia). Points of Insulin Resistance: 1) Insulin resistance at GLUT-4 dependent peripheral tissues results in hyperglycemia, unless countered by an elevation in insulin levels. 2) Brain endothelial cells respond to insulin in ways other than glucose uptake. 3) The insulin transporter at the BBB has a rate that can be modified; a decreased transport rate can result in less insulin action in the brain. 4) Insulin receptor resistance can occur on CNS cells.
Other explanations for a decrease in the CSF level or CSF/serum ratio of insulin are increased enzymatic activity in the CNS or increased efflux from the CNS.
ii. Insulin resistant diseases (obesity and diabetes) are risk factors for AD
This line of evidence involves several strengths and weaknesses in its support. First, diabetes mellitus and obesity can increase the incidence of cognitive deficits through a number of mechanisms that don’t depend on CNS insulin actions. Stroke and ischemia, multi-infarct dementia, inflammation, hypertension, dyslipidemia, and episodes of iatrogenic hypoglycemia all place the diabetic at risk of cognitive impairment (Elias et al., 2003; Manschot et al., 2006; Ott et al., 1999; Perlmuter et al., 1988; Rogers et al., 1989). Diabetes itself is associated with its own form of cognitive impairment, characterized by a decline in executive function (Elias et al., 2003; Manschot et al., 2006). On the one hand, these associations show how diabetes/obesity can be related to cognitive impairment without invoking CNS IR. On the other hand, peripheral IR is associated with factors that can affect CNS insulin actions. Hypertriglyceridemia is the signature dyslipidemia of peripheral IR, diabetes mellitus, and the metabolic syndrome. Triglycerides can cross the BBB to induce resistance at the CNS insulin receptor (Banks et al., 2018). A single high-fat meal can immediately induce cognitive impairment in subjects who are apoE2/3 positive and, paradoxically, cognitive improvement in those who carry apoE4 (Hanson et al., 2015).
Another early line of argument relied on the then untested assumption that IR is global; that is, if IR occurs in peripheral tissues, then it is also occurring in CNS tissues. However, for other receptor resistance syndromes such as resistance to thyroid hormone, resistance can occur in one tissue, such as the pituitary, but not in others (Refetoff, 1982). Furthermore, as the thyroid hormone transporter at the BBB is not the same as the thyroid hormone receptor, defects in transport of thyroid hormone across the BBB occur independently of thyroid receptor resistance (Bernal et al., 2015). An analogous situation is emerging for IR. The classic study of Talbot et al found that CNS IR can occur in AD patients that have no evidence of peripheral IR (Talbot et al., 2012). As the insulin transporter at the BBB is not the same protein as the insulin receptor (Rhea et al., 2018), it is probable that defects in CNS insulin receptor function do not necessarily imply defects in transport as well.
iii. Direct evidence that AD brains have impaired insulin signaling
Several studies involving post mortem tissue have revealed that the levels of protein or message for the insulin receptor and components of the insulin signaling pathway are decreased in AD brains (Chan et al., 2018; Liu et al., 2011; Rickle et al., 2004; Rivera et al., 2005; Steen et al., 2005). Talbot et al confirmed these changes and further showed that signaling as induced by insulin was impaired in post mortem brain tissue from individuals without diabetes (Talbot et al., 2012). In addition, alterations in insulin signaling do not appear to occur simultaneously in different brain areas. Instead, recent data suggest a progression from the hippocampus to the frontal cortex (Barone et al., 2019).
How IR could arise in the CNS in AD is not clear. That peripheral and central IRs interact is strongly suggested by the finding that the deficiency in insulin signaling in brain is more severe in patients who also have insulin resistant diabetes (Liu et al., 2011). Various studies have implicated amyloid β, tau, iron, and other factors (Bloom et al., 2018; Wan et al., 2019). In recent years, the role of the protein biliverdin reductase-A, a unique kinase involved in the regulation of insulin signaling (Lerner-Marmarosh et al., 2005), has been proposed to be involved in CNS insulin sensitivity (Barone et al., 2016; Barone et al., 2011; Sharma et al., 2019; Triani et al., 2018). Other conditions associated with CNS IR could offer clues. As mentioned above, hypertriglyceridemia, which is the classic dyslipidemia of diabetes mellitus and the metabolic syndrome, can induce CNS IR (Banks et al., 2018). Other conditions can affect CNS IR. Pregnancy is associated with resistance to the sympathoexcitatory effects of CNS insulin (Shi et al., 2019b), exercise can reverse the CNS IR associated with a high fat diet (Park et al., 2019), and antipsychotic drugs affect insulin signaling at various levels (Kowalchuk et al., 2019).
Another interesting question is whether CNS insulin levels are part of a negative feedback loop. In the periphery, blood insulin levels are locked into a negative feedback loop that tightly controls both of their levels: high blood glucose induces increased blood levels of insulin and high blood levels of insulin lower blood glucose. Hence, when IR occurs in the periphery, that is, when blood insulin is less potent in its ability to decrease blood glucose levels, the body responds by simply allowing serum insulin levels to rise until glucose levels return to normal. A question then is whether CNS insulin is part of a negative feedback loop, that is, can CNS insulin levels rise to overcome CNS IR? It is unlikely that any such feedback loop would involve an increase in peripheral insulin levels as this would risk hypoglycemia. Other control points would be at the BBB (this time shifting the CNS-serum relation to the left so that more insulin crossed into the brain at any given blood level), enzymatic degradation, and CNS-to-blood efflux rates of insulin. These control points could also keep CNS insulin levels from rising in the face of increased blood levels of insulin caused from peripheral resistance. Finally, although few cells in the CNS have been shown to be capable of secreting insulin, the question arises as to whether such secretion might be more widely inducible in brain.
iv). Insulin delivery to CNS improves cognition
Several studies have shown that insulin improves some aspects of cognition. An early study revealed an effect when the insulin was administered systemically (Craft et al., 1996). However, giving insulin by this route can induce hypoglycemia. Subsequently, a series of studies involved the intranasal route to deliver insulin to the brain as briefly touched on above. Substances delivered through the nares above the turbinates to the level of the cribriform plate show absorption by various routes into the brain (Lochhead and Thorne, 2012). Insulin given by this route shows a distribution pattern that differs from that of intravenous or intracerebroventricular injections (Rhea et al., 2019b). Cognitive improvement has been found after intranasal insulin in patients with mild cognitive impairment and AD as well as young healthy volunteers (Benedict et al., 2004; Craft et al., 1999a; Craft et al., 1999b; Craft et al., 2012; Reger et al., 2006; Reger et al., 2008; Schmid et al., 2018). Animal studies have found improvements with intranasal insulin in cognitively impaired rats (Sukhov et al., 2013) and mice (Marks et al., 2009; Salameh et al., 2015) by altering neuronal after-hyperpolarization (Maimaiti et al., 2016), cerebral blood flow (Anderson et al., 2017), amyloid β production (Mao et al., 2016), and tau hyperphosphorylation (Zhang et al., 2016). Most recently, results of animal studies investigating the molecular mechanism of intranasal insulin suggest that the beneficial effects may be independent of changes in insulin signaling and rather due to changes in inflammation and cell migration (Frazier et al., 2019b; Rhea et al., 2019a). This topic of alternative sources of insulin dysregulation has recently been reviewed by Frazier et al (Frazier et al., 2019a).
If high levels of insulin are therapeutic, why doesn’t the hyperinsulinemia of insulin resistant diabetes protect persons from AD? Interestingly, an early study investigating the relation between diabetes mellitus and AD concluded that these two conditions could not co-exist (Bucht et al., 1983). However, subsequently, diabetes mellitus has been established as a risk factor for AD. One reason the high blood insulin levels are not protective may have to do with the saturable nature of insulin transport across the BBB. As discussed above, diabetes mellitus is associated with many factors that can impact brain health, including association with other risk factors (e.g., peripheral IR, hyperglycemia, hypertension) and life styles (high-fat diets leading to hypothalamic inflammation (Thaler et al., 2013), for example). It may be that the risk factors associated with diabetes mellitus outweigh any therapeutic benefit of increased brain insulin levels.
Repurposing diabetes drugs for memory improvement
For the development of therapeutic strategies to improve cognitive performance, it is important to realize that there are apoE isoform differences in metabolic markers at baseline and in response to treatment. The relationship of these biomarkers with IR is also apoE isoform-dependent. When a dietary fatty acid (FA) intervention was used in patients with metabolic syndrome as part of a 12-week randomized trial, E4 carriers had higher plasma concentrations of total cholesterol (TC), low-density lipoprotein cholesterol (LDL-C) and apolipoprotein B (apo B) than E2 carriers and higher TC, LDL-C and apo B than E3 homozygotic carriers (Fallaize et al., 2017). In addition, while elevated plasma n-3 polyunsaturated FA (PUFA) levels were associated with a beneficially lower concentration of apo CIII in E2 carriers, a high proportion of plasma C16:0 was associated with IR in E4 carriers. Following the FA intervention, an increase in the proportion of plasma long chain (LC) n-3 PUFA was associated with an increase in triacylglycerol-rich lipoprotein (TRL)-C concentrations in E2 carriers, while a reduction in TRL-C was observed in the E3 homozygotic carriers. In E2 carriers, apo CII concentrations were reduced in E2 carriers following a reduction in plasma LC n-3 PUFA and increased when plasma LC n-3 PUFA was raised. In E4 carriers, the apo CII concentration was more increased in response to a reduction in the proportion of plasma LC n-3 PUFA than what was seen in E2 carriers. ApoE levels were also differentially affected by the intervention. Following an increase in plasma LC n-3 PUFA, E2 carriers showed increased apoE levels while E3/E3 and E4 carriers showed a decrease (Fallaize et al., 2017). Considering that the apoE levels in humans (Rezeli et al., 2015) and mice (Johnson et al., 2015), especially those expressing the human low-density lipoprotein receptor (LDL) receptor (Johnson et al., 2014) differ depending on apoE genotype and in apoE heterozygotic carriers the levels of the two apoE isoforms are not comparable either highlights the complex relationship between apoE isoform, apoE levels, and biomarkers of brain metabolism.
There is an increased interest in repurposing drugs (Pushpakom et al., 2018), including diabetes drugs, to improve cognitive function. Especially as there is an increased focus towards individualized medicine to improve medical treatments and strategies (Toplol, 2014), it is important to consider apoE genotypes in developing individual treatment strategies. Preclinical research underlines the importance of this consideration. When 13-month-old apoE TR mice were administered metformin (300 mg/kg/day) for 5 months, spatial memory of E3, but not E4, mice was improved and this was associated with enhanced insulin signaling in E3, but not, E4 mice (Zhang et al., 2019). Interestingly, tau phosphorylation was seen following metformin treatment in both E3 and E4 mice (Zhang et al., 2019), and recently apoE was shown to be critical in mediating tau pathology (Shi et al., 2019a). Interestingly, as described earlier, we found that E4 mice were benefitting from effects of an acute glucose administration on spatial memory (Johnson et al., 2017). Similarly, in a different apoE mouse model in which E3 or E4 was expressed at comparable levels in brains of mice deficient in murine apoE, E4 female mice benefitted from effects of testosterone and dihydrotestosterone on memory while E3 female mice did not (Raber et al., 2002). In addition, male E4 mice were more susceptible to effects of androgen receptor blockade than E3 mice and untreated E4 mice had lower cytosolic androgen receptor binding in cortex than E4 mice (Raber et al., 2002). We recognize that in all these studies a single dose was used for the treatments and it cannot be excluded yet that E3 and E4 carriers might need to receive a different dose based on apoE isoform-dependent dose-response curves. This is unlikely though as the protective effects of a healthy lifestyle on reducing dementia risk seems also apoE isoform-dependent and evident in non-E4 carriers only (Licher et al., 2019).
Conclusions
The purpose of this review was to present the evidence between the connection of apoE and cerebral insulin, implicated in the developmental origin of IR and AD (Figure 3). The majority of this evidence has been generated within the last decade, highlighting that this research area is growing and its importance. We know a lot more about the transport of insulin into the CNS than we do about the transport of apoE. Yet we do know quite a bit about how apoE might alter cerebrovascular blood flow and BBB function. We hope to uncover more about how these two might regulate BBB transport of one another. Increased understanding of how apoE and cerebral insulin might work together to impact CNS IR will facilitate the development of therapeutic strategies to overcome this deficit.
Figure 3. Developmental origin of Insulin Resistance, Mild Cognitive Impairment (MCI), and AD.

Genetic and environmental risk factors contribute to the detrimental brain impact that can lead to development of MCI and AD. Efficacy of intervention through either medication or life-style changes is also often apoE isoform-dependent.
Funding:
This work was supported by the UW Diabetes Research Center (EMR), National Institutes of Health [AG059088] (JR, WAB), and the Veterans Affairs R&D (EMR, WAB).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Achariyar TM, et al. , 2016. Glymphatic distribution of CSF-derived apoE into brain is isoform specific and suppressed during sleep deprivation. Mol Neurodegener. 11, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akintola AA, et al. , 2017. Effect of intranasally administered insulin on cerebral blood flow and perfusion; a randomized experiment in young and older adults. Aging (Albany NY). 9, 790–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alata W, et al. , 2015. Human apolipoprotein E varepsilon4 expression impairs cerebral vascularization and blood-brain barrier function in mice. J Cereb Blood Flow Metab. 35, 86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alquier T, et al. , 2006. Translocable Glucose Transporters in the Brain. Where Are We in 2006? 55, S131–S138. [Google Scholar]
- Altmann A, et al. , 2015. Regional brain hypometabolism is unrelated to regional amyloid plaque burden. Brain. 138, 3734–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KL, et al. , 2017. Impact of Single or Repeated Dose Intranasal Zinc-free Insulin in Young and Aged F344 Rats on Cognition, Signaling, and Brain Metabolism. J Gerontol A Biol Sci Med Sci. 72, 189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbones-Mainar JM, et al. , 2008. Differential modulation of diet-induced obesity and adipocyte functionality by human apolipoprotein E3 and E4 in mice. Int J Obes (Lond). 32, 1595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbones-Mainar JM, et al. , 2016. Metabolic shifts toward fatty-acid usage and increased thermogenesis are associated with impaired adipogenesis in mice expressing human APOE4. Int J Obes (Lond). 40, 1574–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker M, et al. , 2014. Cerebrovascular function and cognition in childhood: a systematic review of transcranial Doppler studies. BMC Neurol. 14, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks WA, 2004. The source of cerebral insulin. Eur J Pharmacol. 490, 5–12. [DOI] [PubMed] [Google Scholar]
- Banks WA, et al. , 2008. Nitric oxide isoenzymes regulate LPS-enhanced insulin transport across the blood-brain barrier. Endocrinology. 149, 1514–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks WA, et al. , 2018. Triglycerides cross the blood-brain barrier and induce central leptin and insulin receptor resistance. Int J Obes. 42, 391–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks WA, et al. , 1997a. Transport of insulin across the blood-brain barrier: saturability at euglycemic doses of insulin. Peptides. 18, 1423–9. [DOI] [PubMed] [Google Scholar]
- Banks WA, et al. , 1997b. Selective, physiological transport of insulin across the blood-brain barrier: novel demonstration by species-specific radioimmunoassays. Peptides. 18, 1257–62. [DOI] [PubMed] [Google Scholar]
- Banks WA, et al. , 1991. Lack of saturable transport across the blood-brain barrier in either direction for beta-amyloid1–28 (Alzheimer’s disease protein). Brain Res Bull. 27, 819–23. [DOI] [PubMed] [Google Scholar]
- Banks WA, et al. , 1999. Uptake and degradation of blood-borne insulin by the olfactory bulb. Peptides. 20, 373–8. [DOI] [PubMed] [Google Scholar]
- Banks WA, et al. , 2012. Insulin in the brain: there and back again. Pharmacol Ther. 136, 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barone E, et al. , 2016. Impairment of biliverdin reductase-A promotes brain insulin resistance in Alzheimer disease: A new paradigm. Free Radic Biol Med. 91, 127–42. [DOI] [PubMed] [Google Scholar]
- Barone E, et al. , 2011. Biliverdin reductase--a protein levels and activity in the brains of subjects with Alzheimer disease and mild cognitive impairment. Biochim Biophys Acta. 1812, 480–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barone E, et al. , 2019. Biliverdin Reductase-A Mediates the Beneficial Effects of Intranasal Insulin in Alzheimer Disease. Mol Neurobiol. 56, 2922–2943. [DOI] [PubMed] [Google Scholar]
- Baura GD, et al. , 1993. Saturable transport of insulin from plasma into the central nervous system of dogs in vivo. A mechanism for regulated insulin delivery to the brain. J Clin Invest. 92, 1824–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belcik JT, et al. , 2015. Contrast-enhanced ultrasound assessment of impaired adipose tissue and muscle perfusion in insulin-resistant mice. Circ Cardiovasc Imaging. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell RD, et al. , 2012. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 485, 512–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedict C, et al. , 2004. Intranasal insulin improves memory in humans. Psychoneuroendocrinology. 29, 1326–1334. [DOI] [PubMed] [Google Scholar]
- Bennet AM, et al. , 2007. Association of apolipoprotein E genotypes with lipid levels and coronary risk. JAMA. 298, 1300–11. [DOI] [PubMed] [Google Scholar]
- Bernal J, et al. , 2015. Thyroid hormone transporters - functions and clinical implications. Nature Reviews Endocrinology. 11, 405–416. [DOI] [PubMed] [Google Scholar]
- Berteau-Pavy F, et al. , 2007. Effects of sex and APOE e4 on object recognition and spatial navigation in the elderly. Neuroscience. 147, 6–17. [DOI] [PubMed] [Google Scholar]
- Blazquez E, et al. , 2014. Insulin in the brain: its pathophysiological implications for States related with central insulin resistance, type 2 diabetes and Alzheimer’s disease. Front Endocrinol (Lausanne). 5, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom GS, et al. , 2018. Reduced brain insulin signaling: A seminal process in Alzheimer’s disease. Neuropharmacology. 136, 192–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bookheimer SY, et al. , 2000. Patterns of brain activation in people at risk for Alzheimer’s disease.[see comment]. New England Journal of Medicine. 343, 450–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman G, et al. , 2018. Blood-brain barrier breakdown, neuroinflammation, and cognitive decline in older adults. Alzheimer’s Dementia. 14, 1640–1650. [DOI] [PubMed] [Google Scholar]
- Bucht G, et al. , 1983. Changes in blood glucose and insulin secretion in patients with senile dementia of Alzheimer type. Acta Med Scand. 387–392. [DOI] [PubMed] [Google Scholar]
- Cangiano C, et al. , 1983. On the stimulation by insulin of tryptophan transport across the blood-brain barrier. Biochem Int. 7, 617–27. [PubMed] [Google Scholar]
- Cashion MF, et al. , 1996. Sequestration of centrally administered insulin by the brain: effects of starvation, aluminum, and TNF-alpha. Horm Behav. 30, 280–6. [DOI] [PubMed] [Google Scholar]
- Celsis P, et al. , 1997. Age related cognitive decline: a clinical entity? A longitudinal study of cerebral blood flow and memory performance. J Neurol Neurosurg Psychiatry. 62, 601–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan E, et al. , 2015. Differential interaction of Apolipoprotein-E isoforms with insulin receptors modulates brain insulin signaling in mutant human amyloid precursor protein transgenic mice. Sci Rep. 5, 13842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan ES, et al. , 2018. Differential Binding of Human ApoE Isoforms to Insulin Receptor is Associated with Aberrant Insulin Signaling in AD Brain Samples. Neuromolecular Med. 20, 124–132. [DOI] [PubMed] [Google Scholar]
- Chan SJ, et al. , 1992. Structure and evolution of insulin and insulin-like growth factors in chordates. Prog Brain Res. 92, 15–24. [DOI] [PubMed] [Google Scholar]
- Chernick D, et al. , 2019. Peripheral versus central nervous system APOE in Alzheimer’s disease: Interplay across the blood-brain barrier. Neurosci Lett. 708, 134306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craft S, et al. , 1999a. Enhancement of memory in Alzheimer disease with insulin and somatostatin, but not glucose. Arch Gen Phychiatry. 56, 1135–1140. [DOI] [PubMed] [Google Scholar]
- Craft S, et al. , 1999b. Insulin metabolism in Alzheimer’s disease differs according to apolipoprotein E genotype and gender. Neuroendocrinology. 70, 146–152. [DOI] [PubMed] [Google Scholar]
- Craft S, et al. , 2012. Intranasal insulin therapy for Alzheimer’s disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch Neurol. 69, 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craft S, et al. , 1996. Memory improvement following induced hyperinsulinemia in Alzheimer’s disease. Neurobiology of Aging. 17, 123–130. [DOI] [PubMed] [Google Scholar]
- Craft S, et al. , 1998. Cerebrosinal fluid and plasma insulin levels in Alzheimer’s disease: relationship to severity of dementia and apolipoprotein E genotype. Neurology. 50, 164–168. [DOI] [PubMed] [Google Scholar]
- Cruchaga C, et al. , 2012. Cerebrospinal fluid APOE levels: an endophenotype for genetic studies for Alzheimer’s disease. Hum Mol Genet. 21, 4558–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Monte SM, 2012. Brain insulin resistance and deficiency as therapeutic targets in Alzheimer’s disease. Curr Alzheimer Res. 9, 35–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Monte SM, et al. , 2019. Early-stage Alzheimer’s disease is associated with simultaneous systemic and central nervous system dysregulation of insulin-linked metabolic pathways. J Alzheimer’s Dis. 68, 657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane R, et al. , 2008. apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J Clin Invest. 118, 4002–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donahue JE, Johanson CE, 2008. Apolipoprotein E, amyloid-beta, and blood-brain barrier permeability in Alzheimer disease. J Neuropathol Exp Neurol. 67, 261–70. [DOI] [PubMed] [Google Scholar]
- Duarte AI, et al. , 2012. Insulin in central nervous system: more than just a peripheral hormone. J Aging Res. 2012, 384017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan A, et al. , 2019. Habenular TCF7L2 links nicotine addiction to diabetes. Nature. 574, 372–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias MF, et al. , 2003. Lower cognitive function in the presence of obesity and hypertension: the Framingham heart study. Int.J.Obes.Relat Metab Disord. 27, 260–268. [DOI] [PubMed] [Google Scholar]
- Elosua R, et al. , 2003. Obesity modulates the association among APOE genotype, insulin, and glucose in men. Obes Res. 11, 1502–8. [DOI] [PubMed] [Google Scholar]
- Fallaize R, et al. , 2017. APOE genotype influences insulin resistance, apolipoprotein CII and CIII according to plasma fatty acid profile in the Metabolic Syndrome. Scientific Reports. 7, 6274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrer LA, et al. , 1997. Effects of age, sex, and ethnicity on the association between apol ipoprotein E genotype and Alzheimer disease. A meta-analysis. J. Am. Med. Assoc 278, 1349–1356. [PubMed] [Google Scholar]
- Filippi BM, et al. , 2013. Insulin and glucagon signaling in the central nervous system. Rev Endocr Metab Disord. 14, 365–75. [DOI] [PubMed] [Google Scholar]
- Filippini N, et al. , 2011. Differential effects of the APOE genotype on brain function across the lifespan. Neuroimage. 54, 602–10. [DOI] [PubMed] [Google Scholar]
- Foster LA, et al. , 1991. Food intake and serum insulin responses to intraventricular infusions of insulin and IGF-I. Physiol Behav. 50, 745–9. [DOI] [PubMed] [Google Scholar]
- Frazier HN, et al. , 2019a. Broadening the definition of brain insulin resistance in aging and Alzheimer’s disease. Exp Neurol. 313, 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazier HN, et al. , 2019b. Long-term intranasal insulin aspart: a profile of gene expression, memory, and insulin receptors in aged F344 rats. J Gerontol A Biol Sci Med Sci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fugisawa Y, et al. , 1991. Increased insulin levels after OGTT load in peripheral blood and cerebrospinal fluid of patients with dementia of the Alzheimer type. Biological Psychiatry. 30, 1219–1228. [DOI] [PubMed] [Google Scholar]
- Fullerton SM, et al. , 2001. Impairment of the blood-nerve and blood-brain barriers in apolipoprotein e knockout mice. Exp Neurol. 169, 13–22. [DOI] [PubMed] [Google Scholar]
- Ghasemi R, et al. , 2013. Insulin in the brain: sources, localization and functions. Mol Neurobiol. 47, 145–71. [DOI] [PubMed] [Google Scholar]
- Gil-Bea FJ, et al. , 2010. Insulin levels are decreased in the cerebrospinal fluid of women with prodomal Alzheimer’s disease. J Alzheimer’s Dis. 22, 405–413. [DOI] [PubMed] [Google Scholar]
- Gray SM, et al. , 2014. Insulin regulates brain function, but how does it get there? Diabetes. 63, 3992–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillo CA, et al. , 2009. Insulin-stimulated translocation of GLUT4 to the plasma membrane in rat hippocampus is PI3-kinase dependent. Brain Res. 1296, 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson AJ, et al. , 2016. Apolipoprotein E Genotype and Sex Influence Glucose Tolerance in Older Adults: A Cross-Sectional Study. Dement Geriatr Cogn Dis Extra. 6, 78–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson AJ, et al. , 2013. Effect of apolipoprotein E genotype and diet on apolipoprotein E lipidation and amyloid peptides: randomized clinical trial. JAMA Neurol. 70, 972–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson AJ, et al. , 2015. Differential Effects of Meal Challenges on Cognition, Metabolism, and Biomarkers for Apolipoprotein E varepsilon4 Carriers and Adults with Mild Cognitive Impairment. J Alzheimers Dis. 48, 205–18. [DOI] [PubMed] [Google Scholar]
- Hasselbalch SG, et al. , 1999. No effect of insulin on glucose blood-brain barrier transport and cerebral metabolism in humans. Diabetes. 48, 1915–21. [DOI] [PubMed] [Google Scholar]
- Heindel J, Vandeberg L, 2016. Developmental Origins of Health and Disease: A Paradigm for Understanding Disease Etiology and Prevention. Curr Opin Pediatr. 27, 248–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hersom M, et al. , 2018. The insulin receptor is expressed and functional in cultured blood-brain barrier endothelial cells, but does not mediate insulin entry from blood-to-brain. Am J Physiol Endocrinol Metab. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, et al. , 2012. Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb Perspect Med. 2, a006312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huebbe P, et al. , 2015. Apolipoprotein E (APOE) genotype regulates body weight and fatty acid utilization-Studies in gene-targeted replacement mice. Mol Nutr Food Res. 59, 334–43. [DOI] [PubMed] [Google Scholar]
- Infante-Garcia C, et al. , 2015. Long-term central pathology and cognitive impairment are exacerbated in a mixed model of Alzheimer’s disease and type 2 diabetes. Psychoneuroendocrinology. 65, 15–25. [DOI] [PubMed] [Google Scholar]
- Jansen JF, et al. , 2016. Cerebral blood flow, blood supply, and cognition in Type 2 Diabetes Mellitus. Sci Rep. 6, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson L, et al. , 2014. Apolipoprotein E–low density lipoprotein receptor interaction affects spatial memory retention and brain ApoE levels in an isoform-dependent manner. Neurobiol Disease. 64, 150–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson L, et al. , 2017. Apolipoprotein E4 mediates insulin resistance-associated cerebrovascular dysfunction and the post-prandial response. J Cereb Blood Flow Metab. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson L, et al. , 2015. ApoE2 exaggerates PTSD-related behavioral, cognitive, and neuroendocrine alterations. Neuropsychopharmacology. 40, 2443–2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LA, et al. , 2011. Apolipoprotein E4 exaggerates diabetic dyslipidemia and atherosclerosis in mice lacking the LDL receptor. Diabetes. 60, 2285–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LA, et al. , 2013. Diabetic atherosclerosis in APOE*4 mice: synergy between lipoprotein metabolism and vascular inflammation. J Lipid Res. 54, 386–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LA, et al. , 2019. Apolipoprotein E4 mediates insulin resistance-associated cerebrovascular dysfunction and the post-prandial response. J Cereb Blood Flow Metab. 39, 770–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastin AJ, Akerstrom V, 2001. Glucose and insulin increase the transport of leptin through the blood-brain barrier in normal mice but not in streptozotocin-diabetic mice. Neuroendocrinology. 73, 237–42. [DOI] [PubMed] [Google Scholar]
- Kastin AJ, et al. , 2002. Interactions of glucagon-like peptide-1 (GLP-1) with the blood-brain barrier. J Mol Neurosci. 18, 7–14. [DOI] [PubMed] [Google Scholar]
- Khalyfa A, et al. , 2018. Plasma Exosomes Disrupt the Blood–Brain Barrier in Children with Obstructive Sleep Apnea and Neurocognitive Deficits. Am J Respir Crit Care Med. 197, 1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, et al. , 2009. The role of apolipoprotein E in Alzheimer’s disease. Neuron. 63, 287–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kockx M, et al. , 2018. Cell-specific production, secretion, and function of apolipoprotein E. J Mol Med (Berl). 96, 361–371. [DOI] [PubMed] [Google Scholar]
- Koelewijn L, et al. , 2019. Oscillatory hyperactivity and hyperconnectivity in young APOE-varepsilon4 carriers and hypoconnectivity in Alzheimer’s disease. Elife. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalchuk C, et al. , 2019. Direct effects on antipsychotic drugs on insulin, energy sensing and inflammatory pathways in hypothalamic mouse neurons. Psychoneuroendocrinology. 109, 104400. [DOI] [PubMed] [Google Scholar]
- Kullmann S, et al. , 2017. Intranasal insulin enhances brain functional connectivity mediating the relationship between adiposity and subjective feeling of hunger. Sci Rep. 7, 1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane-Donovan C, Herz J, 2016. High-Fat Diet Changes Hippocampal Apolipoprotein E (ApoE) in a Genotype- and Carbohydrate-Dependent Manner in Mice . PLOSOne. 11, e0148099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee GD, et al. , 2005. Stereological analysis of microvascular parameters in a double transgenic model of Alzheimer’s disease. Brain Res Bull. 65, 317–22. [DOI] [PubMed] [Google Scholar]
- Lerner-Marmarosh N, et al. , 2005. Human biliverdin reductase: a member of the insulin receptor substrate family with serine/threonine/tyrosine kinase activity. Proc Natl Acad Sci U S A. 102, 7109–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licher S, et al. , 2019. Genetic predisposition, modifiable-risk factor profile and long-term dementia risk in the general population. . Nat Med. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin AL, et al. , 2017. Rapamycin rescues vascular, metabolic and learning deficits in apolipoprotein E4 transgenic mice with pre-symptomatic Alzheimer’s disease. J Cereb Blood Flow Metab. 37, 217–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, et al. , 2012. Apolipoprotein E does not cross the blood-cerebrospinal fluid barrier, as revealed by an improved technique for sampling CSF from mice. Am J Physiol Regul Integr Comp Physiol. 303, R903–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, et al. , 2011. Deficient brain insulin signalling pathway in Alzheimer’s disease and diabetes. J Pathol. 225, 54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochhead JJ, Thorne RG, 2012. Intranasal delivery of biologics to the central nervous system. Adv Drug Deliv Rev. 64, 614–628. [DOI] [PubMed] [Google Scholar]
- Mahley RW, et al. , 2009. Apolipoprotein E: structure determines function, from atherosclerosis to Alzheimer’s disease to AIDS. J Lipid Res. 50 Suppl, S183–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maimaiti S, et al. , 2016. Intranasal Insulin Improves Age-Related Cognitive Deficits and Reverses Electrophysiological Correlates of Brain Aging. J Gerontol A Biol Sci Med Sci. 71, 30–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manschot SM, et al. , 2006. Brain magnetic resonance imaging correlates of impaired cognition in patients with type 2 diabetes. Diabetes. 55, 1106–1113. [DOI] [PubMed] [Google Scholar]
- Mao YF, et al. , 2016. Intranasal insulin alleviates cognitive deficits and amyloid pathology in young adult APPswe/PS1dE9 mice. Aging Cell. 15, 893–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis RU, Altszuler N, 1967. Insulin in the cerebrospinal fluid. Nature. 215, 1375–6. [DOI] [PubMed] [Google Scholar]
- Marks DR, et al. , 2009. Awake intranasal insulin delivery modifies protein complexes and alters memory, anxiety, and olfactory behaviors. J Neurosci. 29, 6734–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May AA, et al. , 2016. Estrogen and insulin transport through the blood-brain barrier. Physiol Behav. 163, 312–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen BS, Reagan LP, 2004. Glucose transporter expression in the central nervous system: relationship to synaptic function. Eur J Pharmacol. 490, 13–24. [DOI] [PubMed] [Google Scholar]
- McNay EC, Pearson-Leary J, 2020. GluT4: A central player in hippocampal memory and brain insulin resistance. Exp Neurol. 323, 113076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minihane AM, et al. , 2007. ApoE genotype, cardiovascular risk and responsiveness to dietary fat manipulation. Proc Nutr Soc. 66, 183–97. [DOI] [PubMed] [Google Scholar]
- Molina JA, et al. , 2002. Cerebrospinal fluid levels of insulin in patients with Alzheimer’s disease. Acta Neurol Scand. 106, 347–350. [DOI] [PubMed] [Google Scholar]
- Mustapic M, et al. , 2019. Extracellular Vesicle Biomarkers Track Cognitive Changes Following Intranasal Insulin in Alzheimer’s Disease. J Alzheimer’s Dis. 69, 489–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash AI, 2017. Crosstalk between insulin and dopamine signaling: A basis for the metabolic effects of antipsychotic drugs. J Chem Neuroanat. 83–84, 59–68. [DOI] [PubMed] [Google Scholar]
- Nation D, et al. , 2019. Blood–brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat Med. 25, 270–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishitsuji K, et al. , 2011. Apolipoprotein E regulates the integrity of tight junctions in an isoform-dependent manner in an in vitro blood-brain barrier model. J Biol Chem. 286, 17536–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak V, et al. , 2014. Enhancement of vasoreactivity and cognition by intranasal insulin in type 2 diabetes. Diabetes Care. 37, 751–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott A, et al. , 1999. Diabetes and the risk of dementia: The Rotterdam Study. Neurology. 53, 1907–1909. [DOI] [PubMed] [Google Scholar]
- Ozturk E, Tan C, 2018. Human cerebrovascular function in health and disease: insights from integrative approaches. J Physiol Anthropol. 37, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paris D, et al. , 2004. Impaired angiogenesis in a transgenic mouse model of cerebral amyloidosis. Neurosci Lett. 366, 80–5. [DOI] [PubMed] [Google Scholar]
- Park HS, et al. , 2019. Exercise alleviates cognitive functions by enhancing hippocampal insulin signaling and neuroplasticity in high-fat diet-induced obesity. Nutrients. pii, E1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendse AA, et al. , 2009. Apolipoprotein E knock-out and knock-in mice: atherosclerosis, metabolic syndrome, and beyond. J Lipid Res. 50 Suppl, S178–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlmuter LC, et al. , 1988. Triglyceride levels affect cognitive function in noninsulin-dependent diabetics. J.Diabet.Complications. 2, 210–213. [DOI] [PubMed] [Google Scholar]
- Pitas RE, et al. , 1987. Lipoproteins and their receptors in the central nervous system. Characterization of the lipoproteins in cerebrospinal fluid and identification of apolipoprotein B,E(LDL) receptors in the brain. J Biol Chem. 262, 14352–60. [PubMed] [Google Scholar]
- Porte D Jr., et al. , 1998. Obesity, diabetes and the central nervous system. Diabetologia. 41, 863–81. [DOI] [PubMed] [Google Scholar]
- Pushpakom S, et al. , 2018. Drug repurposing: progress, cahllenges, and recommendations. Nat Rev Drug Discov. 18, 41–58. [DOI] [PubMed] [Google Scholar]
- Raber J, 2004. Differential gene actions of polymorphic alleles at the APOE locus; potential role of androgens and androgen receptor-mediated signaling. Science of Aging (SAGE) Knowledge Environment (KE). posted March 17, 2004. [Google Scholar]
- Raber J, et al. , 2002. Androgens protect against apolipoprotein E4-induced cognitive deficits. J Neurosci. 22, 5204–5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raber J, et al. , 2004. ApoE genotype accounts for the vast majority of AD risk and AD pathology. Neurobiol Aging. 25, 641–650. [DOI] [PubMed] [Google Scholar]
- Rajasekar N, et al. , 2017. Intranasal insulin improves cerebral blood flow, Nrf-2 expression and BDNF in STZ (ICV)-induced memory impaired rats. Life Sci. 173, 1–10. [DOI] [PubMed] [Google Scholar]
- Rechler MM, Nissley SP, 1986. Insulin-like growth factor (IGF)/somatomedin receptor subtypes: structure, function, and relationships to insulin receptors and IGF carrier proteins. Horm Res. 24, 152–9. [DOI] [PubMed] [Google Scholar]
- Refetoff S, 1982. Syndromes of thyroid hormone resistance. Am J Physiol. 243, E88–E98. [DOI] [PubMed] [Google Scholar]
- Reger MA, et al. , 2006. Effects of intranasal insulin on cognition in memory-impaired older adults: modulation by APOE genotype. Neurobiol Aging. 27, 451–8. [DOI] [PubMed] [Google Scholar]
- Reger MA, et al. , 2008. Intranasal insulin administration dose-dependently modulates verbal memory and plasma amyloid-beta in memory-impaired older adults. J Alzheimers Dis. 13, 323–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman EM, et al. , 2001. Declining brain activity in cognitively normal apolipoprotein E epsilon 4 heterozygotes: A foundation for using positron emission tomography to efficiently test treatments to prevent Alzheimer’s disease. Proc Natl Acad Sci U S A. 98, 3334–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman EM, et al. , 1996. Preclinical evidence of Alzheimer’s disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. N Engl J Med. 334, 752–8. [DOI] [PubMed] [Google Scholar]
- Reiman EM, et al. , 2004. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proc Natl Acad Sci U S A. 101, 284–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezeli M, et al. , 2015. Quantification of total apolipoprotein E and its specific isoforms in cerebrospinal fluid and blood in Alzheimer’s disease and other neurodegenerative diseases. EuPA Open Proteomics. 8, 137–143. [Google Scholar]
- Rhea EM, Banks WA, 2019. Role of the Blood-Brain Barrier in Central Nervous System Insulin Resistance. Frontiers in Neuroscience. 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhea EM, et al. , 2017. Intranasal Insulin Transport is Preserved in Aged SAMP8 Mice and is Altered by Albumin and Insulin Receptor Inhibition. J Alzheimers Dis. 57, 241–252. [DOI] [PubMed] [Google Scholar]
- Rhea EM, et al. , 2019a. Molecular Mechanisms of Intranasal Insulin in SAMP8 Mice. J Alzheimers Dis. 71, 1361–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhea EM, et al. , 2018. Insulin transport across the blood-brain barrier can occur independently of the insulin receptor. J Physiol. 596, 4753–4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhea EM, et al. , 2019b. Routes for the delivery of insulin to the central nervous system: A comparative review. Exp Neurol. 313, 10–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickle A, et al. , 2004. Akt activity in Alzheimer’s disease and other neurodegenerative disorders. NeuroReport. 15, 955–959. [DOI] [PubMed] [Google Scholar]
- Rivera EJ, et al. , 2005. Insulin and insulin-like growth factor expressin and function deteriorate with progression in Alzheimer’s disease: link to brain reductions in acetylcholine. J Alzheimer’s Dis. 8, 247–268. [DOI] [PubMed] [Google Scholar]
- Rogers RL, et al. , 1989. Reducing hypertriglyceridemia in elderly patients with cerebrovascular disease stabilizes or improves cognition and cerebral perfusion. Angiology. 40, 260–269. [PubMed] [Google Scholar]
- Roher AE, et al. , 2012. Cerebral blood flow in Alzheimer’s disease. Vasc Health Risk Manag. 8, 599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roseboom TJ, et al. , 2001. Effects of prenatal exposure to the Dutch famine on adult disease in later life: an overview. Twin Res. 4, 293–8. [DOI] [PubMed] [Google Scholar]
- Salameh TS, et al. , 2015. Central Nervous System Delivery of Intranasal Insulin: Mechanisms of Uptake and Effects on Cognition. J Alzheimers Dis. 47, 715–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartorius T, et al. , 2015. The brain response to peripheral insulin declines with age: a contribution of the blood-brain barrier? PLoS One. 10(5):e0126804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders AM, et al. , 1993. Association of apolipoprotein E allele e4 with late-onset familial and sporadic Alzheimer’s disease. Neurology. 43, 1467–1472. [DOI] [PubMed] [Google Scholar]
- Scarmeas N, et al. , 2005. APOE related alterations in cerebral activation even at college age. J Neurol Neurosurg Psychiatry. 76, 1440–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarmeas N, Stern Y, 2006. Imaging studies and APOE genotype in persons at risk for Alzheimer’s disease. Curr Psychiatry Rep. 8, 11–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling S, et al. , 2013. APOE genotype and MRI markers of cerebrovascular disease: systematic review and meta-analysis. Neurology. 81, 292–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid V, et al. , 2018. Safety of intranasal human insulin: A review. Diabetes Obes Metab. epub [DOI] [PubMed] [Google Scholar]
- Schulingkamp RJ, et al. , 2000. Insulin receptors and insulin action in the brain: review and clinical implications. Neurosci Biobehav Rev. 24, 855–72. [DOI] [PubMed] [Google Scholar]
- Sharma N, et al. , 2019. Loss of biliverdin reductase-A favors Tau hyper-phosphorylation in Alzheimer’s disease. Neurobiol Dis. 125, 176–189. [DOI] [PubMed] [Google Scholar]
- Shi Y, et al. , 2019a. Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J Exp Med. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z, et al. , 2019b. Resistance to the sympathoexcitatory effects of insulin and leptin in late pregnant rats. J. Physiology 597, 4087–4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small GW, et al. , 2000. Cerebral metabolic and cognitive decline in persons at genetic risk for Alzheimer’s disease. Proc Natl Acad Sci U S A. 97, 6037–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder HM, et al. , 2015. Vascular contributions to cognitive impairment and dementia including Alzheimer’s disease. Alzheimers Dement. 11, 710–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steen E, et al. , 2005. Imparied insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease - is this type 3 diabetes? J Alzheimer’s Dis. 7, 63–80. [DOI] [PubMed] [Google Scholar]
- Stoykovich S, Gibas K, 2019. APOE ε4, the door to insulin-resistant dyslipidemia and brain fog? A case study. Alzheimer Dement. 11, 264–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranahan AM, et al. , 2008. Diet-induced insulin resistance impairs hippocampal synaptic plasticity and cognition in middle-aged rats. Hippocampus. 18, 1085–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroh M, et al. , 2003. Diffusion of nerve growth factor in rat striatum as determined by multiphoton microscopy. Biophys J. 85, 581–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukhov IB, et al. , 2013. Long-term intranasal insulin administration improves spatial memory in male rats with prolonged type 1 diabetes mellitus and in healthy rats. Dokl Biol Sci. 453, 349–52. [DOI] [PubMed] [Google Scholar]
- Sykova E, 2004. Diffusion properties of the brain in health and disease. Neurochem Int. 45, 453–66. [DOI] [PubMed] [Google Scholar]
- Tai LM, et al. , 2016. The role of APOE in cerebrovascular dysfunction. Acta Neuropathol. 131, 709–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot K, et al. , 2012. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dyregulation, and cognitive decline. journal of Clinical Investigation. 122, 1316–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejedor MT, et al. , 2014. The apolipoprotein E polymorphism rs7412 associates with body fatness independently of plasma lipids in middle aged men. PLoS One. 9, e108605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaler JP, et al. , 2013. Hypothalamic inflammation: marker or mechanism of obesity pathogenesis? Diabetes. 62, 2629–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thambisetty M, et al. , 2010. APOE epsilon4 genotype and longitudinal changes in cerebral blood flow in normal aging. Arch Neurol. 67, 93–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorne RG, et al. , 2004. Diffusion of epidermal growth factor in rat brain extracellular space measured by integrative optical imaging. J Neurophysiol. 92, 3471–81. [DOI] [PubMed] [Google Scholar]
- Thurston JH, et al. , 1976. Insulin and brain metabolism. Absence of direct action of insulin on K+ and Na+ transport in mouse brain. Diabetes. 25, 758–63. [DOI] [PubMed] [Google Scholar]
- Toplol E, 2014. Individualized medicine from prewomb to tomb. Cell. 157, 241–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triani F, et al. , 2018. Biliverdin reductase-A impairment links brain insulin resistance with increased Abeta production in an animal model of aging: Implications for Alzheimer disease. Biochim Biophys Acta Mol Basis Dis. 1864, 3181–3194. [DOI] [PubMed] [Google Scholar]
- Urayama A, Banks WA, 2008. Starvation and triglycerides reverse the obesity-induced impairment of insulin transport at the blood-brain barrier. Endocrinology. 149, 3592–3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannucci SJ, et al. , 1998. GLUT4 glucose transporter expression in rodent brain: effect of diabetes. Brain Res. 797, 1–11. [DOI] [PubMed] [Google Scholar]
- Wan W, et al. , 2019. Iron deposition leads to hyperphosphorylation of tau and disrption of insulin signaling. Front Neurol. 10, 607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierenga CE, et al. , 2013. Interaction of age and APOE genotype on cerebral blood flow at rest. J Alzheimers Dis. 34, 921–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesmann M, et al. , 2016. A Dietary Treatment Improves Cerebral Blood Flow and Brain Connectivity in Aging apoE4 Mice. Neural Plast. 2016, 6846721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson PW, et al. , 1996. Apolipoprotein E alleles and risk of coronary disease. A meta-analysis. Arterioscler Thromb Vasc Biol. 16, 1250–5. [DOI] [PubMed] [Google Scholar]
- Woods SC, Porte D Jr., 1977. Relationship between plasma and cerebrospinal fluid insulin levels of dogs. Am J Physiol. 233, E331–4. [DOI] [PubMed] [Google Scholar]
- Xie L, et al. , 2002. Alzheimer’s beta-amyloid peptides compete for insulin binding to the insulin receptor. J Neurosci. 22, RC221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu PT, et al. , 1999. Specific regional transcription of apolipoprotein E in human brain neurons. Am J Pathol. 154, 601–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yassine H, 2017. Targeting prodromal Alzheimer’s disease: too late for prevention? Lancet Neurol. 16, 946–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, et al. , 2006. Reciprocal interactions of insulin and insulin-like growth factor I in receptor-mediated transport across the blood-brain barrier. Endocrinology. 147, 2611–5. [DOI] [PubMed] [Google Scholar]
- Zhang J, et al. , 2019. Metformin treatment improves the spatial memory of aged mice in an APOE genotype–dependent manner. FASEB J. 33, 7748–7757. [DOI] [PubMed] [Google Scholar]
- Zhang Y, et al. , 2016. Intranasal Insulin Prevents Anesthesia-Induced Spatial Learning and Memory Deficit in Mice. Sci Rep. 6, 21186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao N, et al. , 2017. Apolipoprotein E4 Impairs Neuronal Insulin Signaling by Trapping Insulin Receptor in the Endosomes. Neuron. 96, 115-+.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlatar ZZ, et al. , 2014. Increased hippocampal blood flow in sedentary older adults at genetic risk for Alzheimer’s disease. J Alzheimers Dis. 41, 809–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV, 1996. Cerebrovascular transport of Alzheimer’s amyloid b and apolipoproteins J and E: Possible anti-amyloidogenic role of the blood-brain barrier. Life Sci. 59, 1483–1497. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV, 2011. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci. 12, 723–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV, 2013. Cerebrovascular effects of apolipoprotein E: implications for Alzheimer disease. JAMA Neurol. 70, 440–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV, et al. , 1994. Brain uptake of circulating apolipoproteins J and E complexed to Alzheimer’s amyloid beta. Biochem Biophys Res Commun. 205, 1431–7. [DOI] [PubMed] [Google Scholar]